

Abstract
Monoclonal antibodies (mAbs) have long provided powerful research tools for virologists to understand the mechanisms of virus entry into host cells and of antiviral immunity. Even so, commercial development of human (or humanized) mAbs for the prophylaxis, preemptive and acute treatment of viral infections has been slow. This is surprising, as new antibody discovery tools have increased the speed and precision with which potent neutralizing human antiviral mAbs can be identified. As longstanding barriers to antiviral mAb development, such as antigenic variability of circulating viral strains and the ability of viruses to undergo neutralization escape, are being overcome, deeper insight into the mechanisms of mAb action and engineering of effector functions are also improving the efficacy of antiviral mAbs. These successes, in both industrial and academic laboratories, coupled with ongoing changes in the biomedical and regulatory environments, herald an era when the commercial development of human antiviral mAb therapies will likely surge.
Main
In the 1890s, pioneering studies from the laboratory of Robert Koch demonstrated that administration of sheep antiserum against diphtheria toxin to a girl dying from diphtheria led to her rapid recovery within hours, and ultimate survival1. As early as 1907, sera from individuals recovering from rubeola (measles) was used to prevent infection from this highly contagious virus2. Since then, the strategy of administering sera from survivors of various viral epidemics for the treatment of acute cytopathic viral disease has been widely practiced (Table 1). During the 1940s, improvements in the quality of fractionated human immunoglobulin led to the widespread use of intramuscular injections of pooled human immunoglobulin to prevent and treat viral infections, such as rubella and hepatitis A, with better protection and less severe reactions than sera. By the early 1980s, several immunoglobulin G (IgG) preparations were licensed for intravenous use to prevent and treat viral diseases, permitting as much as 10- to 20-fold increases in the amounts of immunoglobulin administered3. Many of these products are still on the market today and include generic pooled human IgG for prevention of measles and hepatitis A, as well as hyperimmune human IgG preparations against cytomegalovirus (CMV), respiratory syncytial virus (RSV), hepatitis B virus (HBV), hepatitis C virus (HCV), rabies, vaccinia, varicella-zoster virus (VZV) and West Nile virus (WNV) (Box 1 and Table 2).
Table 1.
Passive immunotherapy with convalescent human serum
| Year of study | Disease | Prophylaxis or treatment | Number of study subjects | Trend in benefit | Reference |
|---|---|---|---|---|---|
| 1907 | Measles (Rubeola) | Prophylaxis | Unknown | Prevention. | 2 |
| 1918 | Measlesa | Prophylaxis | 1 | One child in a family of four children was given serum from the first infected child and was protected; the other two contracted measles. | 96 |
| 1918 | Measles | Prophylaxis | 4 | Prophylaxis was effective. | 96 |
| 1918 | 1918 Pandemic flu | Treatment | 56 | Early administration generally resulted in distinct improvement in clinical symptoms. | 97 98 |
| 1923 | Varicella-Zoster virus | Prophylaxis | 42 | Seven contracted a mild form of the disease, 35 escaped without symptoms. | 99 |
| 1963b | Bolivian hemorrhagic fever | Treatment | 4 | Individuals recovered after 6–8 weeks. | 100 |
| 1959–1983 | Argentine hemorrhagic fever | Treatment | 4,433 | Mortality rate of 3.29% (versus 42.85% in individuals treated before convalescent plasma was used). | 101 |
| 1974–1978 | Argentine hemorrhagic fever | Treatment | 217 | 1.1% mortality rate of those treated with immune plasma. | 102 |
| 1969 | Lassa fever | Treatment | 1 | The individual recovered. | 103 |
| 1984 | Lassa fever | Prophylaxis and treatment | 27 | All study subjects given plasma on or before the 10th day survived with a rapid response to therapy. | 104 |
| 1995 | Ebola hemorrhagic fever | Treatment | 8 | 12.5% fatality rate (versus overall case fatality rate of 80%); inconclusive regarding neutralizing antibodies in convalescent blood. | 105 |
| 1993 | HIV-1 | Treatment of stage IV AIDS individuals | 63 | Randomized double-blind controlled trial. Study subjects were given 250 ml of HIV-immune plasma every 4 weeks. No significant toxicity and effect were found. | 106 |
| 1995 | HIV-1 | Treatment of symptomatic HIV infection | 86 | Randomized double-blind controlled trial. Study subjects were given 300 ml of plasma rich in anti-HIV-1 antibody every 14 days for 1 year. Clinical benefit was observed. | 107 |
| 2002c | HIV-1 | Prevention of vertical transmission in Uganda | 60 | Phase 1/2 trial showed it is safe, well tolerated and similar pharmacokinetic property as other immunoglobulin products. | 108 |
| 2003 | SARS | Treatment | 1 | Fever decreased after administration of convalescent plasma. | 109 |
| 2007 | Influenza A (H5N1) | Treatment | 1 | Viral load was reduced after infusion of plasma; the individual recovered. | 110 |
aOther studies refer to ref. 1.
bImmune BHF gamma globulin was used.
cHIV hyperimmune globulin was used.
Table 2.
Antiviral immunoglobulin products on the US market
| Type of product | Brand name | Manufacturer (location) | Indications |
|---|---|---|---|
| Intramuscular rabies immunoglobulin | IMOGAM Rabies–HT | Aventis Pasteur (Paris) | |
| BayRab | Talecris Biotherapeutics (Research Triangle Park, NC, USA) for Bayer (Leverkusen, Germany) | Post-exposure prophylaxis and treatment, used along with rabies vaccine to prevent infection caused by the rabies virus. | |
| HyperRab S/D | Talecris Biotherapeutics | ||
| Intravenous RSV immunoglobulin | Respigam | MedImmune (Gaithersburg, MD, USA; now part of Astra Zeneca) | Prevention of serious lower respiratory tract infection caused by RSV in children under 24 months of age with bronchopulmonary dysplasia or a history of premature birth. |
| Intravenous CMV immunoglobulin | Cytogam | MedImmune | Prophylaxis of CMV associated with transplantation of kidney, lung, liver, pancreas and heart. |
| Intravenous HCV immunoglobulin | Civacira | Nabi Pharmaceuticals (Boca Raton, FL, USA) | Prevention of re-infection with HCV in HCV-positive liver transplant individuals. |
| Intravenous HBV immunoglobulin | HepaGamB | Cangene (Winnipeg, Canada) | Treatment of acute exposure to blood containing HBsAg. |
| NabiHB | Nabi | ||
| Intravenous vaccinia immunoglobulin | VIGIV | DynPort Vaccine Company (Frederick, MD, USA) | Treatment of infections caused by the vaccinia virus; treat rare adverse reactions to smallpox vaccination. |
| Intramuscular VZV immunoglobulin | VariZIG | Massachusetts Public Health Biologic Laboratoriesb (Boston) and Cangenec | Prevents severe varicella zoster infection. |
| Human immunoglobulind | Omr-IgG-am | Omrix Biopharmaceuticals (New York) | WNV-related encephalitis or for those at risk of developing encephalitis. |
| Intramuscular generic immunoglobulin | Gamastan S/D | Talecris BioTherapeutics | Post-exposure prophylaxis of Hepatitis A; Measles; modification of Varicella; prophylaxis of Rubella. |
| BayGam | Bayer | ||
| Generic subcutaneous immunoglobulin | Vivaglobin | CSL Behring (King of Prussia, PA, USA) | The only human immunoglobulin approved by FDA for subcutaneous use. Home treatment. Prevents infection in individuals with primary immune deficiency diseases. |
| Gammagard | Baxter (Deerfield, IL, USA) | Used to reduce the risk of infection in individuals with HIV and other immunodeficiency diseases. | |
| Generic intravenous immunoglobuline | Sandoglobulin | CSL Bioplasma (Parkville, Australia) | |
| Gamimune N | Bayer |
aFDA fast track, license in progress.
bMBL has discontinued production of VZV Ig.
cAvailable under an investigational new drug application (IND) protocol.
dPhase 1/2 trial sponsored by NIAID.
eDeveloped from the plasma of Israeli donors who have high levels of antibodies to WNV, which is endemic in Israel.
As polyclonal antibody responses are generated against numerous epitopes on different viral antigens during infection, serum-derived polyclonal antibody preparations contain a large and diverse population of antibodies, which include neutralizing antibodies (nAbs) against multiple epitopes. Different nAbs within these polyclonal antibody preparations can bind to these distinct epitopes and provide strong antiviral activity due to additive or even synergistic effects on neutralization. However, a disadvantage associated with polyclonal antibody products is that the vast majority of their constituent virus-specific antibodies are non-neutralizing; some of them are directed against epitopes on native viral surface proteins for which antibody binding does not interfere with viral infection, whereas others are directed against internal, denatured or incompletely translated/assembled proteins released from infected cells. Non-native viral surface proteins or misfolded proteins that are incorporated onto the virus or shed into the circulation can also elicit non-neutralizing antibodies. Other disadvantages of using polyclonal antibody products from immune serum include batch-to-batch variation and risk of pathogen transmission.
Several excellent reviews on passive antibody therapies for infectious diseases are available3,4,5. This article focuses on recent advances in human mAb engineering that have enabled isolation and characterization of potent human antiviral antibodies at unprecedented speed. We also describe how the perceived limitations of antiviral mAb therapies resulting from antigenic variability of circulating strains and the ability of viruses to undergo neutralization escape are being overcome. These advances—combined with a greater understanding of their antiviral mechanisms of action, Fc effector engineering and improved 'humanized' mouse models of infectious diseases—will accelerate translational research of candidate antiviral mAbs and foster a new era of passive immunotherapy.
Platforms for generating mAbs
Antibodies can bind their viral targets with high affinity and exquisite specificity. The rationale behind using antiviral mAb immunotherapies is that they provide a more potent product with better activity than their polyclonal counterparts. Virus-neutralizing human mAbs have been isolated from both non-immune and immune sources using a wide range of recently developed antibody isolation technologies. Among these approaches are the display technologies in which different microorganisms—including phage, yeast, bacteria and viruses—are used to display repertoires of single-chain variable antibody fragments (scFvs), antigen-binding fragments (Fabs) or domain antibodies (Dabs) on their surfaces, from where they can be enriched and isolated through iterative cycles of panning (Fig. 1a)6,7. Other display technologies, including ribosome and mammalian cell display, have also been reported7,8. Although phage display–based selections are currently used most often for development of human mAbs, these selection methods are often complementary, as fundamental selection biases are difficult to control during panning. For example, a recent study demonstrated that an HIV-1 immune scFv library displayed on yeast was more fully expressed than on phage and selected not only all of the scFvs identified by phage but also twice as many novel antibodies9. The common observation that different nAbs can be identified using different display technologies suggests that antibody folding efficiency, post-translational modification and epitope accessibility may all influence the outcome of selection. In addition, the method of presentation of antigen as bait for panning may affect the number and distribution of available neutralizing epitopes. Purified envelope glycoproteins in either monomeric or oligomeric forms and viral particles are two types of antigen that are commonly used as bait for panning.
Figure 1. Human antibody techniques.
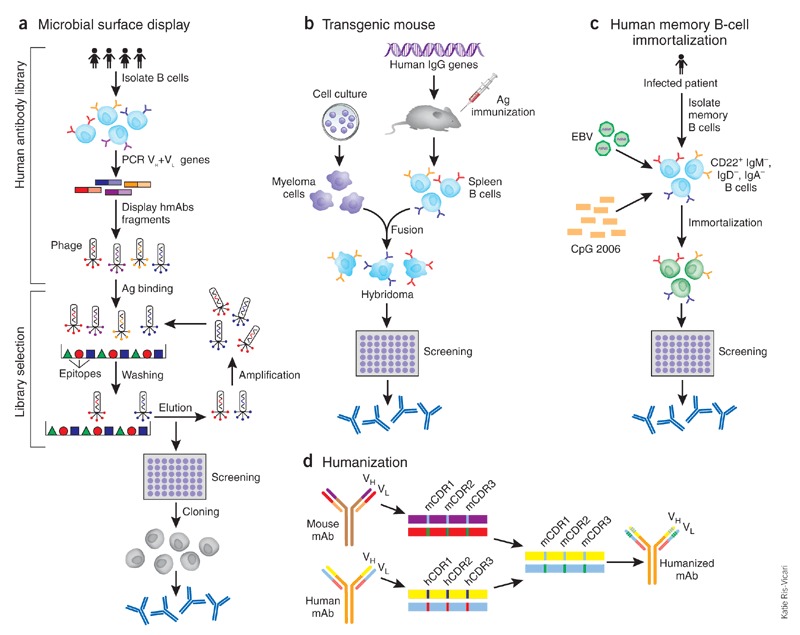
(a) Phage display exemplifies human antibody library display techniques (phage, bacteria, yeast, mammalian cell and ribosome). Three steps are included in this technique: antibody library construction and display onto the phage surface, selection by panning the library against antigen (Ag) targets, and screening for desired specificity. Diverse human immunoglobulin-variable-region gene segments (as scFv or Fab fragments) are amplified from human B cells of immune or non-immune sources to construct the antibody library. The library is then cloned for display on the surface of the phage. Selection against the desired target is then performed using the phage display library; antibodies that do not bind are washed away and the binders are eluted and amplified by infection of Escherichia coli. After several rounds of such selection, desired specificity can be screened using enzyme-linked immunosorbent assay (ELISA) or techniques such as fluorescent-activated cell sorting (FACS) if a cell-membrane bound protein is the target. Once the desired specificity is obtained, the genes of antibody variable regions can be cloned into whole human IgG expression vectors and transfected into cell lines to produce fully human mAbs (hmAbs). (b) Transgenic mouse. The mouse immunoglobulin genes have been genetically knocked out and replaced with human counterparts. The transgenic mouse will make human antibodies after foreign antigen immunization. The B cells harvested from immunized mice are immortalized by fusion with a myeloma cell line, as in traditional hybridoma technology. The hybridomas are then screened for desired specificity. (c) Memory B-cell immortalization. Memory B cells (CD22+ IgM−, IgD−, IgA−) are isolated from peripheral blood mononuclear cells (PBMCs). They are immortalized by EBV in the presence of a CpG oligodeoxynucleotide and irradiated allogeneic PBMCs. The culture supernatants are then screened directly for specific antibodies. Positive cultures are further cloned by limiting dilution and fully human mAbs can then be produced from the cloned B cells. (d) CDR grafting exemplifies humanization. CDR residues from variable region of a mouse mAb are transferred to human antibody frameworks that have high sequence homology with the mouse counterparts.
Katie Ris-Vicari
Other technologies, such as human immunoglobulin transgenic mice, have been used for the generation of human mAbs against severe acute respiratory syndrome coronavirus (SARS-CoV) and rabies10,11,12 (Fig. 1b). This technique involves cloning human mAb-expressing B cells isolated from immunized transgenic mice in which a human mini-immunoglobulin gene locus has been knocked-in to replace the mouse immunoglobulin gene locus. As the immune response in transgenic mice is sometimes less robust than in strains that are used to generate mouse mAbs, more immunizations or antibody screens are often required. Another technique involves the use of human memory B-cells as a source of human mAbs13(Fig. 1c). Memory B-cells are obtained from the blood of an individual recovering from a virus infection. These cells are then transformed with Epstein-Barr virus (EBV) in the presence of polyclonal memory B-cell–activating agents, such as irradiated mononuclear cells and CpG oligonucleotides, which are TLR4 (Toll-like receptor 4) ligands that greatly increase transformation efficiency14, thereby obviating the need for a fusion step to generate hybrid cells, as in the hybridoma technique. Memory B-cells isolated from the blood of individuals who have recovered from SARS-CoV and H5N1 avian influenza infections have been immortalized using this method to produce nAbs against these viruses14,15.
In addition to isolation and screening technologies, increased access to different immune repertoires will likely increase the rescue of therapeutic antibodies that make only a minor contribution to a typical antibody response. For example, memory B-cell libraries may provide a richer source of antibodies against viral pathogens than using bone marrow aspirates and peripheral blood B-cells for constructing both non-immune and immune libraries16.
Lastly, 'humanization' of murine mAbs —the oldest tool for generating therapeutic human mAbs—is still used, particularly when the murine mAbs have been extensively characterized and have desirable antiviral properties and/or superior fully human mAbs are hard to obtain. By grafting complementarity determining region (CDR) residues together with minimal key framework residues from variable regions of a rodent donor mAb onto acceptor human antibody frameworks, humanization allows the binding affinity and specificity of the parental mAb to be retained with reduced risk of immunogenicity (Fig. 1d). This viable approach is the classic and simplest strategy of humanization among many other available strategies, such as grafting specificity determining residues17 and is supported by advances in genetic engineering and three-dimensional modeling of protein structures. The fact that nearly half of all US Food and Drug Administration (FDA)-approved therapeutic mAbs are humanized antibodies testifies to their safety and tolerance by humans.
Mechanisms and models of virus neutralization
Viral neutralization—the ability of an antibody to bind to and inactivate virus—is tested in vitro under defined experimental conditions. Most nAbs identified in this manner also protect animals in vivo, although host protection is complex and involves the interaction of antibodies with cells and molecules of the innate immune system18. Therefore, a better understanding of viral neutralization mechanisms will definitely enrich the antiviral strategies under development (the reader is referred to several excellent reviews on viral neutralization mechanisms and models, which are described only briefly below18,19,20,21,22,23).
Viruses can be categorized as enveloped or non-enveloped according to whether they possess a lipid membrane. Although their entry mechanisms differ, both enveloped and non-enveloped viruses share the same main steps of virus entry, beginning with attachment to cell-surface receptors and ending with delivery of the viral genetic material into the cytoplasm24. Fusion of viral and cellular membranes is a basic entry mode for enveloped viruses (influenza virus and HIV-1 have been extensively studied25), whereas non-enveloped viruses penetrate cells either by lysing a membrane or by creating a pore-like structure in a membrane. Although the mechanisms for membrane penetration of non-enveloped viruses are poorly understood, conformational changes in the viral entry proteins are important for both enveloped and non-enveloped viruses to enter into cells. The triggers for conformational changes include receptor binding, receptor/coreceptor binding, and exposure to the low pH within the endocytic pathway26,27.
MAbs can inhibit viral infection by several different mechanisms in parallel with the steps viruses take to enter into cells (Fig. 2). They can directly block virus attachment to target cells by interfering with virus-receptor interactions (Fig. 2a), such as for nAbs against the CD4-binding site on HIV-1 gp120 (ref. 22), the receptor-binding site of influenza hemagglutinin28 or the receptor binding domain of SARS-CoV (Tables 3 and 4). This same goal can also be achieved by directing the antibodies to the virus receptor and/or coreceptor, as exemplified by anti-CD4 and/or anti-CCR5/CXCR4 (CC-motif receptor 5/CXC-motif receptor 4) mAbs, respectively, under development for the prophylaxis and treatment of HIV-1/AIDS (Fig. 2b and Table 3). In the case of viruses that require internalization by endocytosis and conformational changes in viral proteins triggered by the low pH in the endosome to activate viral and endosomal membrane fusion, mAbs may block conformational changes and/or the requisite interactions between the viral and endosomal membranes required for fusion (Fig. 2c). As an example, some anti-hemagglutinin mAbs that lack hemagglutinin inhibition activity (by blocking receptor binding) neutralize infectivity of influenza virus by interfering with the low pH–induced conformational change in the hemagglutinin molecule, resulting in the inhibition of fusion during viral replication29,30. MAbs can also block fusion at the cell membrane at the post-binding/pre-fusion stage (Fig. 2d). For some viruses, binding to a receptor or both receptor and coreceptor serves as an attachment point as well as a trigger of conformational changes that allow membrane fusion. For example, the interaction of HIV-1 gp120 with CD4 triggers conformational changes in the envelope, resulting in exposure of a transient binding site for coreceptor CCR5 or CXCR4; this in turn promotes additional conformational changes in gp41 that allow it to insert its fusion peptide into the target cell membrane to initiate membrane fusion and viral entry into host cells. MAbs against the external proximal membrane region of gp41 can interfere with conformational changes needed for membrane fusion31,32,33. Inhibition of the release of progeny virus is another mechanism of action; for example, mAbs to influenza A virion surface neuraminidase prevent the release of virions from the infected cell surface34 (Fig. 2e).
Figure 2. Mechanisms of viral neutralization.
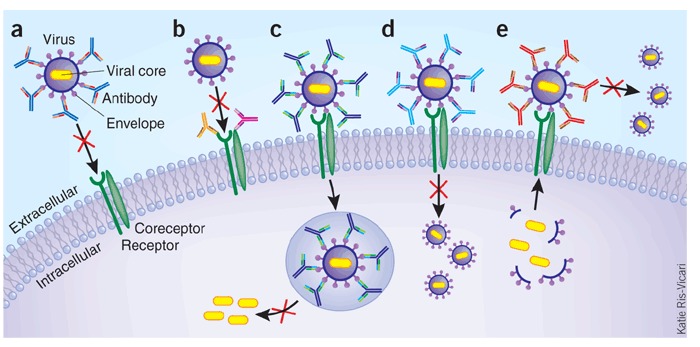
(a) Antibodies block receptor engagement by binding to spikes on an enveloped virus. (b) Antibody blocks virus entry by binding to a viral cellular receptor (or coreceptor) on cell surface. (c) Post-binding/pre-fusion neutralization occurring inside endosome. For some viruses, conformational changes in viral proteins required for fusion are triggered by the low pH in the endosome. MAbs that block the requisite interactions between viral and endosomal membrane proteins would delay or prevent the penetration of the viral core into the target-cell cytoplasm. (d) Post-binding/pre-fusion neutralization occurring at the cell membrane. Antibodies binding to non-receptor binding regions of the viral envelope can neutralize viral infection through interfering with conformational changes that are required for membrane fusion. (e) Inhibition of the release of progeny virus. For example, mAbs to influenza A virion surface neuraminidase prevent the release of virions from the infected cell surface. Not shown in the figure are neutralizing effects of antibodies on the virus before cell binding, which include antibody-mediated virus aggregation to reduce the number of infectious particles.
Katie Ris-Vicari
Table 3.
Commercial development of antiviral mAbsa
| Virus | Stage of development | mAb | Isoform | Target | Development technology | Company (location) | Indication | Reference | |
|---|---|---|---|---|---|---|---|---|---|
| Acute cytopathic | RSV | Approved | Synagis (Palivizumab; MEDI-493) | IgG1 | Glycoprotein F | Humanized | MedImmune | Prophylaxis in high risk infantsb | 111 |
| RSV | Phase 3 | Numax (Motavizumab; MEDI-524) | IgG1 | Glycoprotein F | Humanized and affinity matured from palivizumab | MedImmune | Prophylaxis in high risk infants | 53 112 | |
| Rabies | Phase 1 | CR4098 | IgG1 | Glycoprotein antigenic site III | Immune Ab phage display library | Crucell (Leiden, The Netherlands) | Post-exposure prophylaxis; use in combination | 57 c | |
| CR 57 | Glycoprotein antigenic site I | EBV immortalization | |||||||
| Rabies | Preclinical (clinical trials to start in India in 2008) | 17C7 | IgG1 | Glycoprotein, either antigenic site III or minor site A | Transgenic HuMab-Mouse (Medarex) | Massachusetts Biologic Laboratories | Post-exposure prophylaxis | 10 | |
| WNVd | Phase I | hE16 (MGAWN1) | IgG1 | Envelope (E) protein, domain III | Humanized | Macrogenics (Rockville, MD, USA) | A potential therapy for diseases associated with severe West Nile Virus infection | 113 | |
| WNV | Pre-clinical | CR4374 | IgG1 | E protein, domain III | Immune human Ab phage display library | Crucell | Protected mice from infection | 114 | |
| SARS | Pre-clinical | CR3014 CR3022 | IgG1 | S1-RBD | Immune phage display Ab library | Crucell | CR3014 protected ferrets CR3022 neutralized CR3014 escape viruses; mixture of CR3014/3022 showed synergistic effect | 116 | |
| Acute cytopathic/latent reactivation | CMV | Phase 2 | TI-23 | IgG1 | Envelope glycoprotein gb | Human hybridoma | Teijin Pharma (Tokyo, Japan) | Treatment of CMV Retinitis in HIV individuals / CMV pneumonia in bone marrow transplantation—individuals | 117 |
| CMV | Information not available | HCMV37 | IgG1 | Envelope glycoprotein gb | Humanized | Scotgen Biopharmaceuticals (Aberdeen, UK; closed doors in 1997) | HCMV infections in immunocompromised individuals | 118 | |
| EBV | Phase 2 | Rituxan (Rituximab) | IgG1 | CD20 | Chimeric | Genentech (S. San Francisco, CA, USA) | Treatment, EBV-associated post-transplant lymphoproliferative disorders (interstitial pneumonia) | 119 | |
| VZV | Preclinical | TI-57 | IgG1 | Envelope glycoprotein III | Human hybridoma | Teijin Pharma | Treatment of Varicella Zoster | 120 | |
| Chronic, non-cytopathic | HIV | Phase 2 | TNX-355 | IgG4 | CD4 | Humanized | Genentech | Treatment | 121 |
| HIV | Phase 1 | KD-247 | IgG1 | gp120-V3 tip | Humanized | Kaketsuken-Chemo-Sero-Therapeutic Research Institute (Kumamoto, Japan) | Treatment | 84 | |
| HIV | Phase 1 | PRO140 | IgG4 | CCR5 | Humanized | Progenics Pharmaceuticals (Tarrytown, NY, USA) | Treatment | 84 | |
| HIV | Phase 1 Preclinical | HGS004 HGS101 | IgG4 IgG1 | CCR5 | Abgenix XenoMouse technology | Human Genome Sciences (Rockville, MD, USA) | Treatment 5.5-fold greater potency and a broader range of activity against HIV-1 viral strains than HGS004 | 84 e | |
| HIV | Preclinical | Tarvacin (Bavituximab) | IgG1 | Aminophospholipids exposed on the surface of cells | Chimeric | Peregrine Pharmaceuticals (Tustin, CA, USA) | Treatment | f | |
| HCV | Phase 1 | Tarvacin (Bavituximab) | IgG1 | Aminophospholipid, binds to phospholipids which are derived from the host cell | Chimeric | Peregrine Pharmaceuticals | Treatment of HCV and HIV co-infection | g | |
| HCV | Phase 1 | HepeX-C (XTL-6865, cocktail of mAbs AB68 and AB65) | IgG1 | Envelope protein E2 | AB68, human hybridoma; AB65, EBV immortalization | XTL Biopharmaceuticals (Rehovot, Israel) | Treatment | 122 | |
| HCV | Preclinical | HuMax-HepC | IgG1 | Envelope protein E2 | EBV immortalization | Genmab (Copenhagen) | Prophylaxis | 123 |
aInformation collected from medical literature, manufacturers' websites and press releases.
bFor prevention and treatment of RSV infection in high risk pediatric patients, the current recommendations are Palivizumab at 15 mg/kg of body weight as an intramuscular injection monthly throughout the RSV season, generally November through April in North America. In addition, the American Academy of Pediatrics (AAP) currently recommends palivizumab prophylaxis for children who present to the emergency department with bronchiolitis.
chttp://www.crucell.com/R_and_D-Clincal_Development-Rabies_Antibody_Product.
dWNV establishes persistent infection in central nervous system. Neurons are infected, and destruction of bystander nerve cells and immune-mediated damage contribute to neurologic disease. Different clinical syndromes of WNV neuroinvasive disease have been described124.
e http://www.progenics.com/pro140.cfm
f http://www.peregrineinc.com/content.php?mi=MTk=
ghttp://www.peregrineinc.com/content.php?mi=Mzg= and http://www.rxpgnews.com/article_4411.shtml
Table 4.
Antiviral human mAb research in academic laboratoriesa
| Viral family | Virus | Target | Antibody type/ development technology | First publication date | mAbb | Latest stage of study | Referencec |
|---|---|---|---|---|---|---|---|
| Envelope glycoprotein G2 | Human, hybridoma | 2000 | 1 nAb | In vitro | 125 | ||
| Puumala hantavirus (PUUV) | |||||||
| Envelope glycoprotein G1 | Human, phage display antibody library from splenic lymphocytes of a splenectomized individual with PUUV | 2000 | 1 nAb | in vitro | 126 | ||
| Bunyaviridae | |||||||
| Hantaan virus (HTNV) | Envelope glycoprotein G2 | Human, phage display antibody library from PBLs of four reconvalescent HFRS patients 2–3.5 months after onset | 2000 | 5 nAbs | in vitro | 127 | |
| Envelope glycoprotein G1 | Human, phage display antibody library from PBLs of a convalescent individual | 2003 | 2 Fabs | in vitro | 128 | ||
| Human, phage display antibody library from non-immune donors | 2004 | 80R | in vivo d | 62 64 | |||
| Coronaviridae | SARS | Spike protein | Human, EBV+CpG 2006 immortalization of memory B cells | 2004 | S3.1 | in vivo d | 14 |
| Human, transgenic HuMab-Mouse | 2005 | mAb 201 | in vivo d | 12 129 | |||
| Human, phage display antibody library from non-immune donors | 2007 | m396 | in vivo d | 60 130 | |||
| Human, XenoMouse hybridoma | 2007 | 8 nAbs | in vitro | 11 | |||
| Filoviridae | |||||||
| Ebola | GP | Human, phage display antibody library from bone marrow of 2 donors recovered from infection | 1999 | KZ52 | in vivo e | 55 131 | |
| WNV | DI/DII of E protein | Human, phage display antibody library from non-immune donors | 2005 | mAb no. 11 | in vivo d | 132 | |
| Yellow fever virus | Envelope protein E | Human, phage display antibody library from individuals with yellow fever | 2005 | 3 scFvs | in vitro | 133 | |
| Flaviviridae | |||||||
| Hepatitis C virus (HCV) | Envelope protein E2 | Human, phage display antibody library from bone marrow of an individual chronically infected with HCV | 2000 | mAb 1:7 and A8 | in vitro | 134 135 | |
| Envelope protein E1 | Human, hybridoma | 2004 | H-111 | In vitro | 136 | ||
| Hepadnaviridae | Hepatitis B virus (HBV) | Pre-S1 Surface antigen (HBsAg) | Human, phage display antibody library from non-immune donors | 2005 | G10 | in vitro | 137 |
| Human, phage display antibody library from vaccinated volunteers | 2002 | 5 Fabs | in vitro | 138 | |||
| Human, hybridoma | 2000 | 3 nAbs | in vitro | 139 | |||
| Herpesviridae | VZV | Glycoprotein E | Human, phage display antibody library from PBLs of convalescent individual | 2004 | 2 nAbs | in vitro | 140 |
| Orthomyxoviridae | Avian Influenza H5N1 | Hemagglutinin | Human, EBV+CpG 2006 immortalization of memory B cells | 2007 | 4 nAbs | in vivo d | 15 |
| Chimeric | 2006 | 2 nAbs | in vivo d | 141 | |||
| RSV | F protein | Humanized | 1995 | RSHZ19 | in vivo d | 142 | |
| Hendra (HeV) and Nipah virus (NiV) | Envelope glycoprotein | Human, phage display antibody library from non-immune donors | 2006 | 7 nAbs | in vitro | 143 | |
| Paramyxoviridae | |||||||
| Measles | Hemogglutinin (H) protein | Human, phage display antibody library from bone marrow or splenic lymphocytes of three MV-immune individuals | 2002 | 3 Fabs | In vitro | 144 | |
| Parvoviridae | Parvovirus B19 | Minor capsid protein VP1, major capsid protein VP2 or non-structural protein NS1 | Human, hybridoma | 1999 | 4 nAbs | in vitro | 145 |
| Picornaviridae | Hepatitis A virus (HAV) | Viral structure protein VP1 and VP3 | Human, phage display antibody library from PBLs of 7 HAV-immune donors | 2004 | HA6 and HA9 | in vitro | 146 |
| Vaccinia virus (B5) | Envelope B5 protein | Chimpanzee/human, phage display antibody library from immunized chimpanzee bone marrow-derived lymphocytes | 2006 | 2 nAbs | in vivo d | 147 | |
| Poxviridae | Vaccinia virus | Unidentified | Human, phage antibody display library from PBLs of vaccinia virus (VACV)-immune donor | 1999 | 6 Fabs | in vitro | 148 |
| Vaccinia virus (A33) | Envelope A33 protein | Chimpanzee/human, phage display antibody library from immunized chimpanzee bone marrow-derived lymphocytes | 2007 | 4 nAbs | in vivo d | 149 | |
| Reoviridae | Rotavirus | Viral outer capsid protein 4 and 7 (VP4 and VP7) | Human, phage display antibody library from B cells of tonsils, umbilical cord blood, peripheral blood, and bone marrow | 2004 | 3 nAbs | in vitro | 150 |
| CD4-binding site on gp120 | Human, phage display antibody library from bone marrow of an asymptomatic individual (HIV+ for 6 years). | 1991 | b12 | in vivo e | 151 152 | ||
| Glycan cluster on gp120 | Human, hybridoma | 1994 | 2G12 | Phase I and proof-of-principle efficacy study in human | 153 155 | ||
| HIV-1f | |||||||
| Retroviridae | Membrane proximal region (MPR) on gp41 | Human, hybridoma | 1993 | 2F5 | Same as 2G12 | 154 156 | |
| MPR on gp41 | Human, hybridoma | 1994 | 4E10 | Same as 2G12 | 153 155 | ||
| Envelope glycoprotein gp 120 or gp 41 | Human, phage display antibody library from a donor with long term nonprogressive HIV-1 infection | 2007 | 5 nAbs | in vitro | 157 | ||
| HTLV-1 | Gp46 of HTLV-1 and 2 | Human, hybridoma | 1997 | 8 Abs | in vitro | 158 | |
| Rhabdoviridae | Rabies | Glycoprotein | Human, hybridoma | 2000 | 8 nAbs | in vitro | 159 |
| Human, phage display antibody library from PBLs of 6 vaccine-boosted donors | 2005 | 2 Fabs | In vitro | 160 |
aIt covers literatures published during 1995-2007 except for Abs against HIV. It may not be a complete list.
bNamer of nAb or number of nAb(whole IgG) or antibody fragment being developed is listed.
cFirst and latest publications were listed.
dAnimal study in rodent.
eAnimal study in non-human primate.
fhMabs against HIV-1 have been extensively studied, only well-known broadly neutralizing antibodies (bcnAbs) and recently discovered bcnAbs are listed here.
Several models for the neutralization of various viruses by antibodies have been proposed18,19,20,21,22,23. One is the simple 'occupancy or coating' model that has generally corroborated a multi-hit neutralization hypothesis and is sometimes referred to as the 'multi-hit' model19,35. According to this model, neutralization occurs when a sufficient number of available and functional proteins on the virion are occupied by antibody, leading to inhibition of viral attachment or interference with the fusion process. It is supported by neutralization studies of various viruses, which show a linear relationship between virus size and the number of antibodies required for neutralization21. A recent study dissecting the stoichiometry of antibody neutralization of WNV showed that at lower occupancy, antibody-dependent enhancement (ADE; Box 2) of virus infection dominated, whereas at higher concentrations, neutralization ensued. This study also supports the occupancy model by showing that the cumulative functional outcome is determined by the interplay between antibody affinity and epitope accessibility36,37.
Another proposed model of neutralization is 'the critical binding site' model. According to this model, antibody coating of the virion alone is not the only requisite for neutralization; certain binding sites are critical for neutralization, whereas others are not. This model is compatible with both a single- or multi-hit theory of neutralization. It should be noted, however, that considerable disagreement in the area remains; there is no commonality that can be applied to all virus–antibody neutralizing interactions. Despite this, predictions from these models could lead to a better design of therapeutic antibodies. For example, the occupancy model predicts that larger virions or those possessing more available epitopes will require more bound antibodies to neutralize them. More importantly, a higher affinity antibody will neutralize with greater efficacy. Regardless of the mechanism, the best strategy is to develop potent nAbs with ultra-high binding affinity that target the most critical neutralization site. The hierarchy of which neutralization site(s) to target can be determined by analyzing epitope accessibility and number, ease of neutralization escape and variability of circulating strains.
Immune system–mediated clearance of viruses and infected cells
As natural products of the immune system, antibodies can interact with other immune cells and proteins to fulfill antiviral functions. Numerous immune-mediated mechanisms of virus clearance have been described23,38. For example, the Fc region can mediate complement binding to and deposition on free virions, which can cause a direct virotoxic effect or inhibit virus binding to cells (Fig. 3). Fcγ and complement receptors can bind to antibody- and/or complement-coated virions, which leads to phagocytosis and subsequently inactivation of the virion in an intracellular compartment of the phagocyte. Antibody-dependent cell-mediated cytotoxicity (ADCC) by natural killer cells is another mechanism by which infected cells can be cleared. The relative contributions of these different immune-mediated clearance mechanisms have been studied in transgenic mice that lack complement or complement receptors39. A recent study demonstrated that the Fcγ receptor, but not complement binding, is important in antibody-mediated protection against HIV-1 in vivo40.
Figure 3. Antiviral mechanisms mediated by antibody Fc.
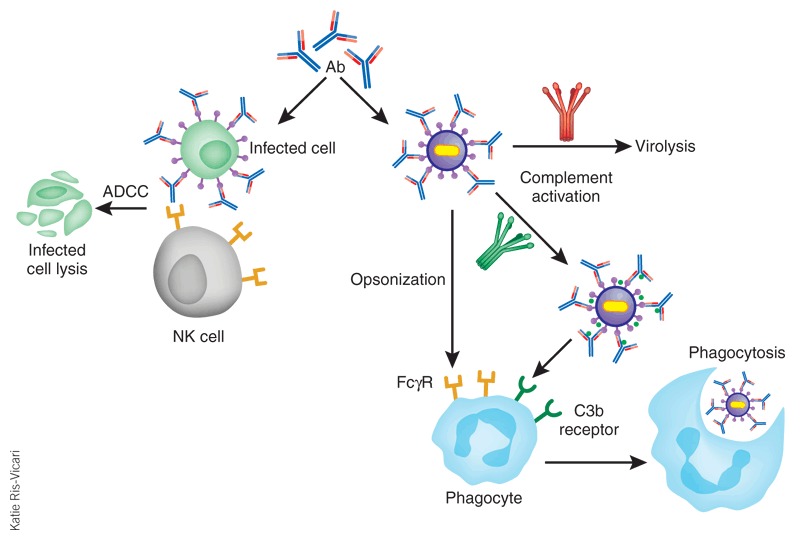
Antibody-dependent cell-mediated cytotoxicity (ADCC) is shown on the left. Antibody opsonization and activation of the complement leads to antibody-dependent, complement-mediated virolysis, phagocytosis directed against free virions or infected cells (only the free virus is shown) are shown on the right. NK, natural killer.
Katie Ris-Vicari
Finally, augmentation of immune responses is also a goal in the control of chronic viral infections. Molecules, such as glucocorticoid-induced tumor necrosis factor (TNF) receptor, which are expressed on subsets of T cells, including regulatory T cells and effector T cells, can suppress effector T cell–function in response to viral antigens. MAbs against glucocorticoid-induced TNF receptor blunt virus replication by blocking their inhibition of effector T-cell–function and/or decreasing the threshold of effector T cells that respond to viral antigens41,42,43. Likewise, recent studies have demonstrated that the pathway PD-1/PD-1L, which are members of the B7-CD28 family of inhibitory immunoregulatory molecules, plays a major role in regulating CD8+ T-cell exhaustion during chronic lymphocytic choriomeningitis virus (LCMV) and HIV-1 infection44,45,46,47,48. When antibodies were used to block this pathway in vivo during chronic LCMV infection, virus-specific CD8+ T-cell responses were enhanced48. Another inhibitory regulator in the B7-CD28 family, cytotoxic T lymphocyte antigen 4 (CTLA-4) is selectively upregulated in HIV-specific CD4+ T-cells and coexpressed with PD-1. The finding that anti-CTLA4 mAb blockade in vitro augmented HIV-specific CD4+ T-cell function suggests that mAb-mediated immune modulation of this target may also provide a clinical benefit in individuals infected with HIV49. Despite the therapeutic potential of mAb-mediated augmentation of immune response for the control of chronic viral infection, it is worth noting that the recent tragic human trial of TGN1412, a mAb against human CD28, has highlighted potential risks associated with manipulation of immunological responses. More carefully planned preclinical studies in non-human primates are clearly required to identify potential problems and to prevent the premature testing of these potent mAbs in human studies50,51.
Commercial development of antiviral mAbs
Table 3 summarizes the antiviral mAbs that are currently under clinical development and describes the technologies used to isolate them. The viruses are grouped as acute cytopathic, acute cytopathic/latent reactivation and chronic non-cytopathic38. Acute cytopathic viruses cause excessive damage in infected tissues and must be controlled rapidly for host survival. Herpes viruses remain latent after primary infection and reactivation can lead to acute cytopathic effects and tissue damage, particularly in immunocompromised individuals, and are thus characterized as 'acute cytopathic/latent reactivation'. Poorly or non-cytopathic viruses usually persist over a lifetime of the host. The first category is especially noteworthy because in 1998, the FDA approved the licensing of palivizumab (Synagis)—a neutralizing 'humanized' murine mAb—for the prevention of serious RSV pulmonary infections in high-risk pediatric individuals. Reports of the safety and efficacy of palivizumab in clinical trials involving infants with bronchopulmonary dysplasia, infants born prematurely (≤35 weeks gestational age) and children with a hemodynamically significant congenital heart disease were favorable52. This humanized murine mAb remains the only FDA-approved antiviral (and anti-infectious agent) antibody on the market.
Motavizumab, a second generation ultra-high affinity mature variant of palivizumab, markedly reduced lung RSV titers of cotton rats and reduces RSV titers in the human upper respiratory track more effectively than its predecessor53. Compared with palivizumab, motavizumab has a 70-fold greater binding avidity for RSV F protein, with a sixfold faster association rate and an 11-fold slower dissociation rate. The improvement in potency of motavizumab is thought to be largely due to the fact that its association rate is faster than that of palivizumab53. A recent phase 3 placebo-controlled study showed that motavizumab reduced both RSV hospitalizations (83%) and lower respiratory infections requiring outpatient management (71%)54.
Two other companies have anti-rabies mAbs in development for post-exposure prophylaxis against infection, one of which has entered phase 1 studies. Two other firms are conducting phase 1 studies with mAbs against WNV for the treatment of neurological disease; anti-SARS-CoV mAbs are in earlier stages of development (Table 3).
Among the acute cytopathic/latent reactivation group, two mAbs are under commercial development for treating CMV disease. Phase 2 studies are underway for one of these antibodies for treating CMV retinitis in individuals infected with HIV-1 and CMV pneumonia in bone marrow transplantation–recipients. A mAb against the envelope glycoprotein III of VZV is also in preclinical development for the treatment of VZV disease. Rituximab, which is directed against CD20 and is approved by FDA for treating non-Hodgkin's lymphoma and rheumatoid arthritis, is in phase 2 studies for treating EBV-associated post-transplant lymphoproliferative disease with interstitial pneumonia.
Clinical development in the category of chronic, non-cytopathic viruses is more extensive and includes mAbs directed against HCV and HIV-1. The most clinically advanced is an anti-CD4 mAb, currently in phase 2 trials for the treatment of HIV-1 with optimized background therapy. Three other mAbs in development are directed against the HIV-1 coreceptor CCR5. The anti-CD4 and two of three anti-CCR5 mAbs are of the IgG4 subtype. As the Fc region of IgG4 interacts poorly with complement and Fcγ receptors expressed on immune cells, using IgG4 should allow the antibodies to act as HIV-1 receptor/coreceptor blocking agents without the elimination of the target cells by immune-mediated clearance mechanisms. Another interesting mAb, under development for treatment of HCV and HIV-1 co-infection, is directed against anionic phospholipids, principally phosphatidylserine that becomes exposed on the surfaces of virus infected cells.
Human antiviral mAb research in academic laboratories
A vast effort, spanning >30 academic laboratories worldwide and involving >20 different viruses from at least 14 virus families, is underway to develop human antiviral mAbs (Table 4). Notably, the majority of academic publications were reported since 2000. Most of this research, involving a broad range of screening and isolation technologies, is directed toward the isolation and characterization of nAbs against envelope proteins. The immune repertoires used to isolate the antibodies vary considerably. In many cases, these antibodies are being used as research tools to better understand mechanisms of virus entry and neutralization, but the studies also aim to identify and characterize neutralizing epitopes on these glycoproteins.
Several laboratories have performed pre-clinical prophylaxis and post-viral challenge mAb protection studies in rodents, whereas similar pre-clinical studies with Ebola and HIV-1 involved non-human primates. Both sterile protection and virus infection with disease modification have been seen in the animals after virus challenge and treatment with high concentrations of administered antibodies23. Important mechanisms of disease pathogenesis are being uncovered by these efforts, as exemplified by the failure of a nAb to affect the course of Ebola virus infection in monkeys, in contrast to its protective effects in guinea pigs and potent neutralizing activity in vitro55. The results of neutralizing mAb studies are being used extensively to guide vaccine design, with particular effort directed toward HIV-1. These mAbs will likely fuel the clinical development pipeline as these research efforts continue.
Cocktails of mAbs
Numerous studies on HBV, RSV and other viruses have demonstrated synergistic and/or additive effects of combinations of two or more mAbs on virus neutralization35. Accordingly, one of the most exciting recent advancements in this field is in the development of mAb cocktails for the prophylaxis and treatment of viral infections. This addresses on several levels one of the major risks of targeting only one epitope on a viral pathogen through a single mAb. For example, the 'occupancy' model (multi-hit theory) of neutralization proposes that obtaining sufficient antibody density on the surface of a viral pathogen is the most critical factor—often more important than the epitope recognized—needed for viral neutralization and elimination21. This may not be possible in all cases when a single epitope is recognized, but can be achieved by having more than one epitope occupied on the virus surface.
Equally important is that the target epitope may not be conserved on all strains of the pathogen; even if an epitope is conserved, selection pressure by the antibody may drive 'neutralization escape' of the virus through point mutations (Fig. 4). A combination of two mAbs against two neutralizing epitopes (shown in Fig. 4 as non-overlapping) can cover that vast majority of circulating strains and prevent neutralization escape. The most clinically advanced mAbs in this area are those against rabies56,57,58 (Box 3). The fact that a phase 1 clinical trial is currently underway with this CR57/CR4098 mAb cocktail without prior clinical testing of each mAb separately underscores the FDA's recognition that mAb cocktails may provide equal or better antiviral activity than the equivalent available commercial immunoglobulin products derived from human hyperimmune serum58. Monoclonal antibody cocktails may also fill a needed gap in the global shortage of hyperimmune serum, whose supply and availability is sometimes limited. A technology to develop recombinant human polyclonal antibodies against viral pathogens has been reported that combines cloning of antibodies from single antigen-specific immune B-cells and site-specific integration into antibody-producing cells. This results in a more uniform master cell bank for manufacturing of polyclonal antibodies with defined specificity35.
Figure 4. Human mAb combinational therapy.
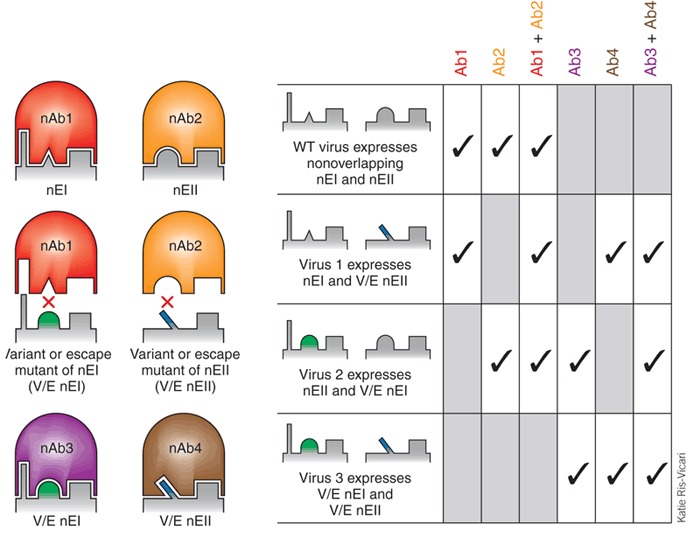
Two separate neutralizing epitopes that are prevalent on the envelope glycoprotein of a circulating virus are shown. Neutralizing antibody nAb1 and nAb2 can recognize the neutralizing epitope EI (nEI) and EII (nEII) on the circulating strain (row 1). The combination of nAb1 and nAb2 will provide broad neutralization activity and can prevent neutralization escape. A natural variant of nEII or neutralization escape mutant of nAb2 is still recognized by nAb1 (row 2) and nAb4. Likewise, a natural variant of nEI or neutralization escape mutant of nAb1 is still recognized by nAb2 (row 3) and nAb3. Both nAb3 and nAb4 are available and can be used to maintain two nAbs against circulating variant viruses. Only rarely would a combination of nAb3 and nAb4 be required (row 4).
Katie Ris-Vicari
Antiviral mAb therapies in biodefense and viral outbreak settings
A major advantage of antiviral mAb therapies is their long serum half-life, often ∼21 days, which confers prolonged in vivo protection. Animal studies have demonstrated efficacy against infection and disease progression from a number of viral pathogens when a single dose of mAb has been given prophylactically or as early treatment after infection, respectively14,15,59,60,61,62. A single-dose treatment strategy may be ideal for the prophylaxis or treatment of acute cytopathic respiratory virus infections, such as SARS-CoV, H5N1 avian influenza and even smallpox, provided that sufficient serum nAb levels can be achieved by intravenous, intramuscular or subcutaneous administration. Human antiviral mAb inhalation therapy is another intriguing possibility. In several animal studies, the correlation between the prophylactically administrated mAb concentration in the serum and in vitro neutralization titer of the serum has been quantitatively measured (e.g., protection required a TCID90 (90% tissue culture infective dose) in vitro neutralization titer at 1:400 dilution of serum23,63). This type of quantitative analysis could be established in phase 1/2 studies for each therapeutic antiviral mAb.
The first consideration in using antiviral mAb immunotherapy in an outbreak setting is to determine whether the circulating virus is sensitive to neutralization by the mAb(s) at hand. An approach proposed by our laboratory in response to a potential future SARS outbreak involves obtaining an early biological specimen from a suspected case (e.g., nasal-pharyngeal aspirate or respiratory secretions) and then rapidly performing PCR amplification and DNA sequencing of the viral protein gene segment that encodes the mAb epitope. These data would then be compared against a pre-constructed sensitivity/resistance map of the mAb based on analyses of naturally occurring as well as focused or random mutagenesis of amino acids within the epitope and in vitro neutralization studies of mutant viral protein pseudotyped reporter viruses64. In this way, guided antiviral mAb passive immunotherapy could be instituted with a more realistic expectation of clinical efficacy. Furthermore, in a respiratory virus outbreak setting, preventative strategies could be stratified according to high-risk exposure, such as prioritizing treatment of family members, health-care workers and first responders. The goal here would be to provide immediate protection, so that even in virus-infected individuals, additional time could be obtained to mount an effective native antiviral immune response and/or to administer antiviral drugs that inhibit virus replication at the post-entry phase. Immunocompromised individuals, the elderly and individuals with comorbid medical risk factors for serious disease—all of whom may have weakened immune systems—are also likely to be good candidates for passive immunotherapy. Epidemic control strategies, including 'passive' ring immunization, could be applied to decrease transmissibility and reduce contacts65,66.
Perspective
Although the past century witnessed tremendous progress in the development of polyclonal antiviral immunotherapies, the pace of antiviral mAb discovery has never been greater. These advances are fueled by new antibody screening and isolation technologies, a better understanding of how to construct and use non-immune and immune antibody repertoires, and growing interest in virology. The emergence of new viral pathogens has also kept the field ablaze, in part because the globalization of world travel has increased the risk for rapid transmission and dissemination of new viral diseases. There is also a growing realization that for some viral pathogens, optimal disease prevention and management may not be achieved through traditional vaccination of the population at risk, but rather through instituting local, regional and international public health-care measures early to prevent and contain the infections, as was successfully demonstrated in the case of SARS67. In situations where the frequency of viral infection is low, protection could be provided by passive immunotherapy during the period of high-risk exposure, avoiding the potential side effects of vaccination that can cause untoward morbidity and even rare mortality, as exemplified by yellow fever vaccine–associated viscerotropic disease68. Moreover, for some antiviral vaccines, high titers of broadly neutralizing antibodies are either not elicited, or their titers wane over time. In either case, the nAb titers may not be protective at the time of viral challenge, which may occur years after vaccination. Another possible risk is ADE for vaccines against viruses for which enhanced disease-associated clinical syndromes have been demonstrated and/or postulated to occur when antibody titers fall below their neutralization threshold (Box 2). In other cases, dominant antibody responses may be stronger against the vaccine strain than a secondary viral challenge stain, a phenomenon known as 'original antigenic sin'69. In all of the above examples, the use of short-term passive immunotherapy where high titer of nAbs can be readily achieved may provide some tangible advantages over vaccination.
The paramount importance of technological improvements to the development of antiviral mAb therapeutics cannot be understated. Antibody-envelope cocrystallographic studies have provided important structural details at the atomic level that serve as a guide to understand mechanisms of neutralization and neutralization escape. When combined with genetic mutagenesis, in silico modeling of mAb binding can improve mAb affinity and neutralization activity. Other technological improvements on the horizon include the incorporation of neutralization assays earlier in the screening platforms, as well as broadening the specificity of an existing nAb to both prevent neutralization escape and generate broad cross-viral-strain neutralizing activities. Further improvement in antiviral activity will also be seen when modifications in Fc effector functions are introduced, such as through Fc and glycoengineering to improve FcRγ−binding and ADCC activity70,71,72,73,74. In certain viral illnesses, such as Dengue virus infection, altering the Fc region has been explored as a way to reduce or eliminate ADE75. The development of newer generations of humanized mice hosting Fcγ-expressing human immune cells will likely allow a better understanding of the complex role of Fc effector functions in viral clearance76. Whether Fab arm exchange of IgG4 subclass can be used to enhance the antiviral activity of mAbs in vivo through cross-linking of neutralizing epitopes and/or enabling the bi-specific targeting of viruses to immune effector cells should be explored77,78.
A particularly exciting area currently being explored is the use of human antibodies against viral receptors and coreceptors, of which several have been recently discovered. These include transferrin receptor 1, a receptor for New World hemorrhagic fever arenaviruses, and claudin-1, a coreceptor for HCV24,79,80. The potential advantage of directly blocking the cellular receptor/coreceptor is that the mAb is more likely to block virus entry of genetically diverse circulating strains. For example, anti-TfR1 antibodies can block several different New World arenaviruses, and anti-CCR5 mAbs can block a broad range of R-tropic viruses expressing different envelope glycoproteins. Nevertheless, antibody blockade of cell surface receptors may pose unforeseen risks. For example, although CCR5 promotes leukocyte trafficking to the brain and improves survival following WNV infection, a deficiency in CCR5 increases the risk of symptomatic WNV infection81,82.
Despite this progress, several formidable challenges remain. Although there has been explosive growth in commercial exploitation of therapeutic antibodies5,6,83, their application in the prevention and/or treatment of infectious diseases and viral infections has lagged behind other disease-types84. Scientific challenges specific to the development of mAbs as anti-infectious agents, as well as clinical and market challenges have clearly tempered commercial interest in this area. Some of the potential commercial obstacles include the concern of antigenic escape and viral variability (which can now be managed by engineering higher-affinity mAbs and using mAb cocktails), the low prevalence of treatable viral diseases, a growing pipeline of small-molecule inhibitors of virus replication and competition with effective antiviral vaccines entering the market that could rapidly replace the need for passively infused mAbs. Another potential obstacle is the relatively short duration of viral illnesses, which makes them less attractive commercial targets than such diseases as cancer or chronic inflammatory or autoimmune diseases that require longer mAb treatment cycles. Even so, the prevalence of commercialized antiviral mAb treatments may increase if more potent mAbs with optimum effector capabilities to combat them are identified. In addition, it is likely that antiviral mAbs will not only be used on their own, but also with increasing frequency in combination with small-molecule drugs that inhibit post-entry steps of the virus life cycle. Combination therapy is certainly a commercial path being taken with antibodies that block HCV and HIV infection. Chronic monthly treatment with mAbs as part of a drug regimen would provide a strong incentive for participation of biopharmaceutical companies in development of antiviral mAb products. The abundance of antiviral mAbs in the pipeline, their established track record with manufacturing and clinical safety and the need for companies to compete in the marketplace all suggest that antibody drugs are likely to become an increasing part of any clinician's armamentarium.
Notwithstanding our sanguine view of the growth and potential of antiviral antibody therapeutics, this zeal must be tempered by the reality of the costs of intensive mAb production to an already overtaxed health-care system. We have provided a strategic prophylaxis and treatment stratification plan on how passive immunotherapy could be used in a virus outbreak setting. The plan would substantially limit the number of doses that would need to be stockpiled. For other viral diseases, treatment stratifications will not be possible. However, costs of mAb production are decreasing and mAb manufacturing facilities with capabilities for large-scale production are on the rise. The productivity of mammalian cells cultivated in bioreactors is now one gram per liter and in several cases, a more than 100-fold improvement over titers for similar processes in the mid-1980s has been realized85. Newer methods of mAb manufacturing, which improve productivity through advances in DNA delivery and integration, host cell engineering, medium optimization, more efficient selection, gene amplification and cell-line screening systems will accelerate this trend. Plant biopharming of mAbs also offers promise in ensuring cheaper mass production of mAbs for global health–related viral diseases, such as rabies86,87. Indeed, the vision of large-scale biopharming of antiviral mAbs in plants throughout the developing world could support growth in this vital area in more ways than one.
Box 1: Immunoglobulin subclasses and oligosaccharide modifications.
Differences in the amino acid sequences of different subclasses of IgG (IgG1 to IgG4 in humans) can affect their binding affinity for different classes of activating (pro-inflammatory) or inhibitory (anti-inflammatory) Fcγ receptors88. Likewise, differences in the sequences of attached oligosaccharides may be critical for antibody function with the removal of terminal sialic acid resulting in increased anti-inflammatory activity89,90. Refinements of commercial immunoglobulin preparations with regard to IgG subclasses and oligosaccharide content may result in polyclonal immunoglobulin products with improved antiviral activity.
Box 2: Antibody-dependent enhancement.
ADE is a mechanism by which virus-specific antibodies enhance viral entry into—and in some cases, replication in—monocytes/macrophages and granulocytic cells through interaction with Fcγ and/or complement receptors. ADE has been demonstrated in vitro using anti-dengue and anti-WNV antibodies and cells bearing Fcγ receptors, as well as in monkeys infused with anti-dengue antibodies36,75,91,92,93. ADE is the proposed mechanism responsible for Dengue hemorrhagic fever and Dengue shock syndrome, two clinical conditions that are frequently seen in patients infected with a second heterotropic infection or in infants with maternally transferred anti-Dengue antibodies. Infection with Dengue virus or any other flavivirus induces broadly cross-reactive, but weakly neutralizing as well as non-neutralizing antibodies94,95. ADE occurs when the concentration of neutralizing antibody falls below the stoichiometric threshold for neutralization and becomes sub-neutralizing. These preexisting sub-neutralizing (and non-neutralizing) antibodies and the infecting Dengue virus form complexes that bind to Fcγ receptor–bearing cells, leading to increased virus uptake and replication36,37,75.
Box 3: CR57 and CR4098 nAb cocktail against rabies.
Although the epitope of CR57 is highly conserved and mutated in only 1% of natural rabies virus isolates, another human nAb CR4098 was developed to complement CR57. nAbs CR57 and CR4098 recognize distinct epitopes and complementarily neutralize their escape mutants. The global coverage of natural rabies virus isolates by the CR57 and CR4098 cocktail is demonstrated by a spectrum of in vitro neutralization studies. Escape viruses are no longer obtainable in vitro under the selective pressure of a CR57 and CR4098 cocktail. A Syrian hamster animal study showed the CR57 and CR4098 cocktail is a safe and efficacious alternative to hyperimmune rabies immunoglobulin in rabies post exposure prophylaxis57.
Acknowledgements
We would like to acknowledge Caitlyn Silhan for her assistance in compiling the large amount of data that are presented in the tables and to thank the referees for their helpful comments. This work has been supported by the National Institutes of Health grants AI061318, AI074518, AI070343, AI52829 and AI060456.
Contributor Information
Wayne A Marasco, Email: wayne_marasco@dfci.harvard.edu.
Jianhua Sui, Email: jianhua_sui@dfci.harvard.edu.
References
- 1.Casadevall A, Scharff MD. Return to the past: the case for antibody-based therapies in infectious diseases. Clin. Infect. Dis. 1995;21:150–161. doi: 10.1093/clinids/21.1.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Good RA, Lorenz E. Historic aspects of intravenous immunoglobulin therapy. Cancer. 1991;68:1415–1421. doi: 10.1002/1097-0142(19910915)68:6+<1415::aid-cncr2820681402>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 3.Hemming VG. Use of intravenous immunoglobulins for prophylaxis or treatment of infectious diseases. Clin. Diagn. Lab. Immunol. 2001;8:859–863. doi: 10.1128/CDLI.8.5.859-863.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Keller MA, Stiehm ER. Passive immunity in prevention and treatment of infectious diseases. Clin. Microbiol. Rev. 2000;13:602–614. doi: 10.1128/cmr.13.4.602-614.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Casadevall A, Dadachova E, Pirofski LA. Passive antibody therapy for infectious diseases. Nat. Rev. Microbiol. 2004;2:695–703. doi: 10.1038/nrmicro974. [DOI] [PubMed] [Google Scholar]
- 6.Carter PJ. Potent antibody therapeutics by design. Nat. Rev. Immunol. 2006;6:343–357. doi: 10.1038/nri1837. [DOI] [PubMed] [Google Scholar]
- 7.Hoogenboom HR. Selecting and screening recombinant antibody libraries. Nat. Biotechnol. 2005;23:1105–1116. doi: 10.1038/nbt1126. [DOI] [PubMed] [Google Scholar]
- 8.Ho M, Nagata S, Pastan I. Isolation of anti-CD22 Fv with high affinity by Fv display on human cells. Proc. Natl. Acad. Sci. USA. 2006;103:9637–9642. doi: 10.1073/pnas.0603653103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bowley DR, Labrijn AF, Zwick MB, Burton DR. Antigen selection from an HIV-1 immune antibody library displayed on yeast yields many novel antibodies compared to selection from the same library displayed on phage. Protein Eng. Des. Sel. 2007;20:81–90. doi: 10.1093/protein/gzl057. [DOI] [PubMed] [Google Scholar]
- 10.Sloan SE, et al. Identification and characterization of a human monoclonal antibody that potently neutralizes a broad panel of rabies virus isolates. Vaccine. 2007;25:2800–2810. doi: 10.1016/j.vaccine.2006.12.031. [DOI] [PubMed] [Google Scholar]
- 11.Coughlin M, et al. Generation and characterization of human monoclonal neutralizing antibodies with distinct binding and sequence features against SARS coronavirus using XenoMouse. Virology. 2007;361:93–102. doi: 10.1016/j.virol.2006.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Greenough TC, et al. Development and characterization of a severe acute respiratory syndrome-associated coronavirus-neutralizing human monoclonal antibody that provides effective immunoprophylaxis in mice. J. Infect. Dis. 2005;191:507–514. doi: 10.1086/427242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bernasconi NL, Traggiai E, Lanzavecchia A. Maintenance of serological memory by polyclonal activation of human memory B cells. Science. 2002;298:2199–2202. doi: 10.1126/science.1076071. [DOI] [PubMed] [Google Scholar]
- 14.Traggiai E, et al. An efficient method to make human monoclonal antibodies from memory B cells: potent neutralization of SARS coronavirus. Nat. Med. 2004;10:871–875. doi: 10.1038/nm1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Simmons CP, et al. Prophylactic and therapeutic efficacy of human monoclonal antibodies against H5N1 influenza. PLoS Med. 2007;4:e178. doi: 10.1371/journal.pmed.0040178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lanzavecchia A, et al. Understanding and making use of human memory B cells. Immunol. Rev. 2006;211:303–309. doi: 10.1111/j.0105-2896.2006.00403.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kashmiri SV, De Pascalis R, Gonzales NR, Schlom J. SDR grafting—a new approach to antibody humanization. Methods. 2005;36:25–34. doi: 10.1016/j.ymeth.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 18.Reading SA, Dimmock NJ. Neutralization of animal virus infectivity by antibody. Arch. Virol. 2007;152:1047–1059. doi: 10.1007/s00705-006-0923-8. [DOI] [PubMed] [Google Scholar]
- 19.Klasse PJ, Sattentau QJ. Occupancy and mechanism in antibody-mediated neutralization of animal viruses. J. Gen. Virol. 2002;83:2091–2108. doi: 10.1099/0022-1317-83-9-2091. [DOI] [PubMed] [Google Scholar]
- 20.Klasse PJ. Virology. 2007. Modeling how many envelope glycoprotein trimers per virion participate in human immunodeficiency virus infectivity and its neutralization by antibody. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Burton DR, Saphire EO, Parren PW. A model for neutralization of viruses based on antibody coating of the virion surface. Curr. Top. Microbiol. Immunol. 2001;260:109–143. doi: 10.1007/978-3-662-05783-4_7. [DOI] [PubMed] [Google Scholar]
- 22.Parren PW, Burton DR. The antiviral activity of antibodies in vitro and in vivo. Adv. Immunol. 2001;77:195–262. doi: 10.1016/S0065-2776(01)77018-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Burton DR. Antibodies, viruses and vaccines. Nat. Rev. Immunol. 2002;2:706–713. doi: 10.1038/nri891. [DOI] [PubMed] [Google Scholar]
- 24.Dimitrov DS. Virus entry: molecular mechanisms and biomedical applications. Nat. Rev. Microbiol. 2004;2:109–122. doi: 10.1038/nrmicro817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Harrison SC. Mechanism of membrane fusion by viral envelope proteins. Adv. Virus Res. 2005;64:231–261. doi: 10.1016/S0065-3527(05)64007-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Smith AE, Helenius A. How viruses enter animal cells. Science. 2004;304:237–242. doi: 10.1126/science.1094823. [DOI] [PubMed] [Google Scholar]
- 27.Earp LJ, Delos SE, Park HE, White JM. The many mechanisms of viral membrane fusion proteins. Curr. Top. Microbiol. Immunol. 2005;285:25–66. doi: 10.1007/3-540-26764-6_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Skehel JJ, Wiley DC. Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu. Rev. Biochem. 2000;69:531–569. doi: 10.1146/annurev.biochem.69.1.531. [DOI] [PubMed] [Google Scholar]
- 29.Imai M, Sugimoto K, Okazaki K, Kida H. Fusion of influenza virus with the endosomal membrane is inhibited by monoclonal antibodies to defined epitopes on the hemagglutinin. Virus Res. 1998;53:129–139. doi: 10.1016/s0168-1702(97)00143-3. [DOI] [PubMed] [Google Scholar]
- 30.Outlaw MC, Dimmock NJ. IgG neutralization of type A influenza viruses and the inhibition of the endosomal fusion stage of the infectious pathway in BHK cells. Virology. 1993;195:413–421. doi: 10.1006/viro.1993.1391. [DOI] [PubMed] [Google Scholar]
- 31.Burton DR, et al. HIV vaccine design and the neutralizing antibody problem. Nat. Immunol. 2004;5:233–236. doi: 10.1038/ni0304-233. [DOI] [PubMed] [Google Scholar]
- 32.Zwick MB, et al. Anti-human immunodeficiency virus type 1 (HIV-1) antibodies 2F5 and 4E10 require surprisingly few crucial residues in the membrane-proximal external region of glycoprotein gp41 to neutralize HIV-1. J. Virol. 2005;79:1252–1261. doi: 10.1128/JVI.79.2.1252-1261.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zwick MB, et al. Broadly neutralizing antibodies targeted to the membrane-proximal external region of human immunodeficiency virus type 1 glycoprotein gp41. J. Virol. 2001;75:10892–10905. doi: 10.1128/JVI.75.22.10892-10905.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Webster RG, Laver WG. Preparation and properties of antibody directed specifically against the neuraminidase of influenza virus. J. Immunol. 1967;99:49–55. [PubMed] [Google Scholar]
- 35.Bregenholt S, Jensen A, Lantto J, Hyldig S, Haurum JS. Recombinant human polyclonal antibodies: A new class of therapeutic antibodies against viral infections. Curr. Pharm. Des. 2006;12:2007–2015. doi: 10.2174/138161206777442173. [DOI] [PubMed] [Google Scholar]
- 36.Pierson TC, et al. The stoichiometry of antibody-mediated neutralization and enhancement of West Nile virus infection. Cell Host Microbe. 2007;1:135–145. doi: 10.1016/j.chom.2007.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Klasse PJ, Burton DR. Antibodies to West Nile virus: a double-edged sword. Cell Host Microbe. 2007;1:87–89. doi: 10.1016/j.chom.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 38.Hangartner L, Zinkernagel RM, Hengartner H. Antiviral antibody responses: the two extremes of a wide spectrum. Nat. Rev. Immunol. 2006;6:231–243. doi: 10.1038/nri1783. [DOI] [PubMed] [Google Scholar]
- 39.Mehlhop E, Diamond MS. Protective immune responses against West Nile virus are primed by distinct complement activation pathways. J. Exp. Med. 2006;203:1371–1381. doi: 10.1084/jem.20052388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hessell AJ, et al. Fc receptor but not complement binding is important in antibody protection against HIV. Nature. 2007;449:101–104. doi: 10.1038/nature06106. [DOI] [PubMed] [Google Scholar]
- 41.La S, Kim E, Kwon B. In vivo ligation of glucocorticoid-induced TNF receptor enhances the T-cell immunity to herpes simplex virus type 1. Exp. Mol. Med. 2005;37:193–198. doi: 10.1038/emm.2005.26. [DOI] [PubMed] [Google Scholar]
- 42.Mills KH. Regulatory T cells: friend or foe in immunity to infection? Nat. Rev. Immunol. 2004;4:841–855. doi: 10.1038/nri1485. [DOI] [PubMed] [Google Scholar]
- 43.Belkaid Y, Rouse BT. Natural regulatory T cells in infectious disease. Nat. Immunol. 2005;6:353–360. doi: 10.1038/ni1181. [DOI] [PubMed] [Google Scholar]
- 44.Freeman GJ, Wherry EJ, Ahmed R, Sharpe AH. Reinvigorating exhausted HIV-specific T cells via PD-1-PD-1 ligand blockade. J. Exp. Med. 2006;203:2223–2227. doi: 10.1084/jem.20061800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Petrovas C, et al. PD-1 is a regulator of virus-specific CD8+ T cell survival in HIV infection. J. Exp. Med. 2006;203:2281–2292. doi: 10.1084/jem.20061496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Day CL, et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature. 2006;443:350–354. doi: 10.1038/nature05115. [DOI] [PubMed] [Google Scholar]
- 47.Trautmann L, et al. Upregulation of PD-1 expression on HIV-specific CD8+ T cells leads to reversible immune dysfunction. Nat. Med. 2006;12:1198–1202. doi: 10.1038/nm1482. [DOI] [PubMed] [Google Scholar]
- 48.Barber DL, et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439:682–687. doi: 10.1038/nature04444. [DOI] [PubMed] [Google Scholar]
- 49.Kaufmann DE, et al. Upregulation of CTLA-4 by HIV-specific CD4+ T cells correlates with disease progression and defines a reversible immune dysfunction. Nat. Immunol. 2007;8:1246–1254. doi: 10.1038/ni1515. [DOI] [PubMed] [Google Scholar]
- 50.Strand V, Kimberly R, Isaacs JD. Biologic therapies in rheumatology: lessons learned, future directions. Nat. Rev. Drug Discov. 2007;6:75–92. doi: 10.1038/nrd2196. [DOI] [PubMed] [Google Scholar]
- 51.Farzaneh L, Kasahara N, Farzaneh F. The strange case of TGN1412. Cancer Immunol. Immunother. 2007;56:129–134. doi: 10.1007/s00262-006-0189-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Simoes EA, et al. Palivizumab prophylaxis, respiratory syncytial virus, and subsequent recurrent wheezing. J. Pediatr. 2007;151:34–42. doi: 10.1016/j.jpeds.2007.02.032. [DOI] [PubMed] [Google Scholar]
- 53.Wu H, et al. Development of motavizumab, an ultra-potent antibody for the prevention of respiratory syncytial virus infection in the upper and lower respiratory tract. J. Mol. Biol. 2007;368:652–665. doi: 10.1016/j.jmb.2007.02.024. [DOI] [PubMed] [Google Scholar]
- 54.MedImmune. MedImmune's Motavizumab reduced RSV hospitalizations by 83 percent among high-risk Native American, full-term infants in placebo-controlled Phase 3 study. Medimmune Press Release <http://phx.corporate-ir.net/phoenix.zhtml?c=83037&p=irol-investornewsArticle&ID=1043517&highlight=motavizumab> (2007).
- 55.Oswald WB, et al. Neutralizing antibody fails to impact the course of Ebola virus infection in monkeys. PLoS Pathog. 2007;3:e9. doi: 10.1371/journal.ppat.0030009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bakker AB, et al. Novel human monoclonal antibody combination effectively neutralizing natural rabies virus variants and individual in vitro escape mutants. J. Virol. 2005;79:9062–9068. doi: 10.1128/JVI.79.14.9062-9068.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.de Kruif J, et al. A human monoclonal antibody cocktail as a novel component of rabies postexposure prophylaxis. Annu. Rev. Med. 2007;58:359–368. doi: 10.1146/annurev.med.58.061705.145053. [DOI] [PubMed] [Google Scholar]
- 58.Goudsmit J, et al. Comparison of an anti-rabies human monoclonal antibody combination with human polyclonal anti-rabies immune globulin. J. Infect. Dis. 2006;193:796–801. doi: 10.1086/500470. [DOI] [PubMed] [Google Scholar]
- 59.Morrey JD, et al. Humanized monoclonal antibody against West Nile virus envelope protein administered after neuronal infection protects against lethal encephalitis in hamsters. J. Infect. Dis. 2006;194:1300–1308. doi: 10.1086/508293. [DOI] [PubMed] [Google Scholar]
- 60.Zhu Z, et al. Potent cross-reactive neutralization of SARS coronavirus isolates by human monoclonal antibodies. Proc. Natl. Acad. Sci. USA. 2007;104:12123–12128. doi: 10.1073/pnas.0701000104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Engle MJ, Diamond MS. Antibody prophylaxis and therapy against West Nile virus infection in wild-type and immunodeficient mice. J. Virol. 2003;77:12941–12949. doi: 10.1128/JVI.77.24.12941-12949.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sui J, et al. Potent neutralization of severe acute respiratory syndrome (SARS) coronavirus by a human mAb to S1 protein that blocks receptor association. Proc. Natl. Acad. Sci. USA. 2004;101:2536–2541. doi: 10.1073/pnas.0307140101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Parren PW, et al. Antibody protects macaques against vaginal challenge with a pathogenic R5 simian/human immunodeficiency virus at serum levels giving complete neutralization in vitro. J. Virol. 2001;75:8340–8347. doi: 10.1128/JVI.75.17.8340-8347.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sui J, et al. Evaluation of human monoclonal antibody 80R for immunoprophylaxis of severe acute respiratory syndrome by an animal study, epitope mapping, and analysis of spike variants. J. Virol. 2005;79:5900–5906. doi: 10.1128/JVI.79.10.5900-5906.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kretzschmar M, van den Hof S, Wallinga J, van Wijngaarden J. Ring vaccination and smallpox control. Emerg. Infect. Dis. 2004;10:832–841. doi: 10.3201/eid1005.030419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pourbohloul B, et al. Modeling control strategies of respiratory pathogens. Emerg. Infect. Dis. 2005;11:1249–1256. doi: 10.3201/eid1108.040449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.McLean AR, May RM, Pattison J, Weiss RA. SARS A Case Study in Emerging Infections. 2005. [Google Scholar]
- 68.Doblas A, et al. Yellow fever vaccine-associated viscerotropic disease and death in Spain. J. Clin. Virol. 2006;36:156–158. doi: 10.1016/j.jcv.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 69.Haaheim LR. Original antigenic sin. A confounding issue? Dev. Biol. (Basel) 2003;115:49–53. [PubMed] [Google Scholar]
- 70.Li H, et al. Optimization of humanized IgGs in glycoengineered Pichia pastoris. Nat. Biotechnol. 2006;24:210–215. doi: 10.1038/nbt1178. [DOI] [PubMed] [Google Scholar]
- 71.Yamane-Ohnuki N, et al. Establishment of FUT8 knockout Chinese hamster ovary cells: an ideal host cell line for producing completely defucosylated antibodies with enhanced antibody-dependent cellular cytotoxicity. Biotechnol. Bioeng. 2004;87:614–622. doi: 10.1002/bit.20151. [DOI] [PubMed] [Google Scholar]
- 72.Lazar GA, et al. Engineered antibody Fc variants with enhanced effector function. Proc. Natl. Acad. Sci. USA. 2006;103:4005–4010. doi: 10.1073/pnas.0508123103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Shields RL, et al. Lack of fucose on human IgG1 N-linked oligosaccharide improves binding to human Fcgamma RIII and antibody-dependent cellular toxicity. J. Biol. Chem. 2002;277:26733–26740. doi: 10.1074/jbc.M202069200. [DOI] [PubMed] [Google Scholar]
- 74.Umana P, Jean-Mairet J, Moudry R, Amstutz H, Bailey JE. Engineered glycoforms of an antineuroblastoma IgG1 with optimized antibody-dependent cellular cytotoxic activity. Nat. Biotechnol. 1999;17:176–180. doi: 10.1038/6179. [DOI] [PubMed] [Google Scholar]
- 75.Goncalvez AP, Engle RE, St Claire M, Purcell RH, Lai CJ. Monoclonal antibody-mediated enhancement of dengue virus infection in vitro and in vivo and strategies for prevention. Proc. Natl. Acad. Sci. USA. 2007;104:9422–9427. doi: 10.1073/pnas.0703498104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Shultz LD, Ishikawa F, Greiner DL. Humanized mice in translational biomedical research. Nat. Rev. Immunol. 2007;7:118–130. doi: 10.1038/nri2017. [DOI] [PubMed] [Google Scholar]
- 77.Burton DR, Wilson IA. Immunology. Square-dancing antibodies. Science. 2007;317:1507–1508. doi: 10.1126/science.1148905. [DOI] [PubMed] [Google Scholar]
- 78.van der Neut Kolfschoten M, et al. Anti-inflammatory activity of human IgG4 antibodies by dynamic Fab arm exchange. Science. 2007;317:1554–1557. doi: 10.1126/science.1144603. [DOI] [PubMed] [Google Scholar]
- 79.Radoshitzky SR, et al. Transferrin receptor 1 is a cellular receptor for New World haemorrhagic fever arenaviruses. Nature. 2007;446:92–96. doi: 10.1038/nature05539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Evans MJ, et al. Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature. 2007;446:801–805. doi: 10.1038/nature05654. [DOI] [PubMed] [Google Scholar]
- 81.Glass WG, et al. Chemokine receptor CCR5 promotes leukocyte trafficking to the brain and survival in West Nile virus infection. J. Exp. Med. 2005;202:1087–1098. doi: 10.1084/jem.20042530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Glass WG, et al. CCR5 deficiency increases risk of symptomatic West Nile virus infection. J. Exp. Med. 2006;203:35–40. doi: 10.1084/jem.20051970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Brekke OH, Sandlie I. Therapeutic antibodies for human diseases at the dawn of the twenty-first century. Nat. Rev. Drug Discov. 2003;2:52–62. doi: 10.1038/nrd984. [DOI] [PubMed] [Google Scholar]
- 84.Reichert JM. Trends in the development and approval of monoclonal antibodies for viral infections. BioDrugs. 2007;21:1–7. doi: 10.2165/00063030-200721010-00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wurm FM. Production of recombinant protein therapeutics in cultivated mammalian cells. Nat. Biotechnol. 2004;22:1393–1398. doi: 10.1038/nbt1026. [DOI] [PubMed] [Google Scholar]
- 86.Ko K, Koprowski H. Plant biopharming of monoclonal antibodies. Virus Res. 2005;111:93–100. doi: 10.1016/j.virusres.2005.03.016. [DOI] [PubMed] [Google Scholar]
- 87.Sivakumar G. Bioreactor technology: a novel industrial tool for high-tech production of bioactive molecules and biopharmaceuticals from plant roots. Biotechnol. J. 2006;1:1419–1427. doi: 10.1002/biot.200600117. [DOI] [PubMed] [Google Scholar]
- 88.Nimmerjahn F, Ravetch JV. Divergent immunoglobulin g subclass activity through selective Fc receptor binding. Science. 2005;310:1510–1512. doi: 10.1126/science.1118948. [DOI] [PubMed] [Google Scholar]
- 89.Kaneko Y, Nimmerjahn F, Ravetch JV. Anti-inflammatory activity of immunoglobulin G resulting from Fc sialylation. Science. 2006;313:670–673. doi: 10.1126/science.1129594. [DOI] [PubMed] [Google Scholar]
- 90.Burton DR, Dwek RA. Immunology. Sugar determines antibody activity. Science. 2006;313:627–628. doi: 10.1126/science.1131712. [DOI] [PubMed] [Google Scholar]
- 91.Morens DM, Halstead SB, Marchette NJ. Profiles of antibody-dependent enhancement of dengue virus type 2 infection. Microb. Pathog. 1987;3:231–237. doi: 10.1016/0882-4010(87)90056-8. [DOI] [PubMed] [Google Scholar]
- 92.Littaua R, Kurane I, Ennis FA. Human IgG Fc receptor II mediates antibody-dependent enhancement of dengue virus infection. J. Immunol. 1990;144:3183–3186. [PubMed] [Google Scholar]
- 93.Halstead SB. In vivo enhancement of dengue virus infection in rhesus monkeys by passively transferred antibody. J. Infect. Dis. 1979;140:527–533. doi: 10.1093/infdis/140.4.527. [DOI] [PubMed] [Google Scholar]
- 94.Roehrig JT, Bolin RA, Kelly RG. Monoclonal antibody mapping of the envelope glycoprotein of the dengue 2 virus, Jamaica. Virology. 1998;246:317–328. doi: 10.1006/viro.1998.9200. [DOI] [PubMed] [Google Scholar]
- 95.Heinz FX. Epitope mapping of flavivirus glycoproteins. Adv. Virus Res. 1986;31:103–168. doi: 10.1016/s0065-3527(08)60263-8. [DOI] [PubMed] [Google Scholar]
- 96.Richardson D, Conner H. Immunization against measles. J. Am. Med. Assoc. 1919;72:1046–1048. [Google Scholar]
- 97.Luke TC, Kilbane EM, Jackson JL, Hoffman SL. Meta-analysis: convalescent blood products for Spanish influenza pneumonia: a future H5N1 treatment? Ann. Intern. Med. 2006;145:599–609. doi: 10.7326/0003-4819-145-8-200610170-00139. [DOI] [PubMed] [Google Scholar]
- 98.Stoll HF. Value of convalesent blood and serum in treatment of influenzal pneumonia. J. Am. Med. Assoc. 1919;73:478–483. [Google Scholar]
- 99.Weech A. The prophylaxis of varicella with convalescents serum. J. Am. Med. Assoc. 1924;82:1245–1246. [Google Scholar]
- 100.Stinebaugh BJ, et al. Bolivian hemorrhagic fever. A report of four cases. Am. J. Med. 1966;40:217–230. doi: 10.1016/0002-9343(66)90103-3. [DOI] [PubMed] [Google Scholar]
- 101.Ruggiero, H.A. et al. Presse Med. Treatment of Argentine hemorrhagic fever with convalescents plasma. 4433 cases. 15, 2239–2242 (1986). [PubMed]
- 102.Maiztegui JI, Fernandez NJ, de Damilano AJ. Efficacy of immune plasma in treatment of Argentine haemorrhagic fever and association between treatment and a late neurological syndrome. Lancet. 1979;2:1216–1217. doi: 10.1016/s0140-6736(79)92335-3. [DOI] [PubMed] [Google Scholar]
- 103.Leifer E, Gocke DJ, Bourne H. Lassa fever, a new virus disease of man from West Africa. II. Report of a laboratory-acquired infection treated with plasma from a person recently recovered from the disease. Am. J. Trop. Med. Hyg. 1970;19:677–679. doi: 10.4269/ajtmh.1970.19.677. [DOI] [PubMed] [Google Scholar]
- 104.Frame JD, Verbrugge GP, Gill RG, Pinneo L. The use of Lassa fever convalescent plasma in Nigeria. Trans. R. Soc. Trop. Med. Hyg. 1984;78:319–324. doi: 10.1016/0035-9203(84)90107-x. [DOI] [PubMed] [Google Scholar]
- 105.Mupapa K, et al. Treatment of Ebola hemorrhagic fever with blood transfusions from convalescent patients. International Scientific and Technical Committee. J. Infect. Dis. 1999;179(Suppl. 1):S18–S23. doi: 10.1086/514298. [DOI] [PubMed] [Google Scholar]
- 106.Jacobson JM, et al. Passive immunotherapy in the treatment of advanced human immunodeficiency virus infection. J. Infect. Dis. 1993;168:298–305. doi: 10.1093/infdis/168.2.298. [DOI] [PubMed] [Google Scholar]
- 107.Vittecoq D, et al. Passive immunotherapy in AIDS: a double-blind randomized study based on transfusions of plasma rich in anti-human immunodeficiency virus 1 antibodies vs. transfusions of seronegative plasma. Proc. Natl. Acad. Sci. USA. 1995;92:1195–1199. doi: 10.1073/pnas.92.4.1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Guay LA, et al. Phase I/II trial of HIV-1 hyperimmune globulin for the prevention of HIV-1 vertical transmission in Uganda. AIDS. 2002;16:1391–1400. doi: 10.1097/00002030-200207050-00011. [DOI] [PubMed] [Google Scholar]
- 109.Wong VW, Dai D, Wu AK, Sung JJ. Treatment of severe acute respiratory syndrome with convalescent plasma. Hong Kong Med. J. 2003;9:199–201. [PubMed] [Google Scholar]
- 110.Zhou B, Zhong N, Guan Y. Treatment with convalescent plasma for influenza A (H5N1) infection. N. Engl. J. Med. 2007;357:1450–1451. doi: 10.1056/NEJMc070359. [DOI] [PubMed] [Google Scholar]
- 111.Mansbach J, Kunz S, Acholonu U, Clark S, Camargo CA., Jr Evaluation of compliance with palivizumab recommendations in a multicenter study of young children presenting to the emergency department with bronchiolitis. Pediatr. Emerg. Care. 2007;23:362–367. doi: 10.1097/01.pec.0000278406.75815.d3. [DOI] [PubMed] [Google Scholar]
- 112.Mejias A, et al. Comparative effects of two neutralizing anti-respiratory syncytial virus (RSV) monoclonal antibodies in the RSV murine model: time versus potency. Antimicrob. Agents Chemother. 2005;49:4700–4707. doi: 10.1128/AAC.49.11.4700-4707.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Oliphant T, et al. Development of a humanized monoclonal antibody with therapeutic potential against West Nile virus. Nat. Med. 2005;11:522–530. doi: 10.1038/nm1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Throsby M, et al. Isolation and characterization of human monoclonal antibodies from individuals infected with West Nile Virus. J. Virol. 2006;80:6982–6992. doi: 10.1128/JVI.00551-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.van den Brink EN, et al. Molecular and biological characterization of human monoclonal antibodies binding to the spike and nucleocapsid proteins of severe acute respiratory syndrome coronavirus. J. Virol. 2005;79:1635–1644. doi: 10.1128/JVI.79.3.1635-1644.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.ter Meulen J, et al. Human monoclonal antibody combination against SARS coronavirus: synergy and coverage of escape mutants. PLoS Med. 2006;3:e237. doi: 10.1371/journal.pmed.0030237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Arizono H, et al. Pharmacokinetics of a new human monoclonal antibody against cytomegalovirus. Third communication: correspondence of the idiotype activity and virus neutralization activity of the new monoclonal antibody, regavirumab in rat serum and its pharmacokinetics in rats and monkeys. Arzneimittelforschung. 1994;44:909–913. [PubMed] [Google Scholar]
- 118.Tempest PR, White P, Buttle M, Carr FJ, Harris WJ. Identification of framework residues required to restore antigen binding during reshaping of a monoclonal antibody against the glycoprotein gB of human cytomegalovirus. Int. J. Biol. Macromol. 1995;17:37–42. doi: 10.1016/0141-8130(95)93516-z. [DOI] [PubMed] [Google Scholar]
- 119.Kunitomi A, Arima N, Ishikawa T. Epstein-Barr virus-associated post-transplant lymphoproliferative disorders presented as interstitial pneumonia; successful recovery with rituximab. Haematologica. 2007;92:e49–e52. doi: 10.3324/haematol.11142. [DOI] [PubMed] [Google Scholar]
- 120.Yokoyama T, et al. Varicella-zoster virus gH:gL contains a structure reactive with the anti-human gamma chain of IgG near the glycosylation site. J. Gen. Virol. 2001;82:331–334. doi: 10.1099/0022-1317-82-2-331. [DOI] [PubMed] [Google Scholar]
- 121.Kuritzkes DR, et al. Antiretroviral activity of the anti-CD4 monoclonal antibody TNX-355 in patients infected with HIV type 1. J. Infect. Dis. 2004;189:286–291. doi: 10.1086/380802. [DOI] [PubMed] [Google Scholar]
- 122.Borgia G. HepeX-C (XTL Biopharmaceuticals) Curr. Opin. Investig. Drugs. 2004;5:892–897. [PubMed] [Google Scholar]
- 123.Habersetzer F, et al. Characterization of human monoclonal antibodies specific to the hepatitis C virus glycoprotein E2 with in vitro binding neutralization properties. Virology. 1998;249:32–41. doi: 10.1006/viro.1998.9202. [DOI] [PubMed] [Google Scholar]
- 124.Sejvar JJ, et al. Long-term neurological and functional outcome in Nipah virus infection. Ann. Neurol. 2007;62:235–242. doi: 10.1002/ana.21178. [DOI] [PubMed] [Google Scholar]
- 125.Guttieri MC, Bookwalter C, Schmaljohn C. Expression of a human, neutralizing monoclonal antibody specific to puumala virus G2-protein in stably-transformed insect cells. J. Immunol. Methods. 2000;246:97–108. doi: 10.1016/s0022-1759(00)00299-4. [DOI] [PubMed] [Google Scholar]
- 126.de Carvalho Nicacio C, et al. A neutralizing recombinant human antibody Fab fragment against Puumala hantavirus. J. Med. Virol. 2000;60:446–454. doi: 10.1002/(sici)1096-9071(200004)60:4<446::aid-jmv13>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 127.Koch J, Liang M, Queitsch I, Kraus AA, Bautz EK. Human recombinant neutralizing antibodies against hantaan virus G2 protein. Virology. 2003;308:64–73. doi: 10.1016/s0042-6822(02)00094-6. [DOI] [PubMed] [Google Scholar]
- 128.Liang M, et al. Generation of an HFRS patient-derived neutralizing recombinant antibody to Hantaan virus G1 protein and definition of the neutralizing domain. J. Med. Virol. 2003;69:99–107. doi: 10.1002/jmv.10259. [DOI] [PubMed] [Google Scholar]
- 129.Roberts A, et al. Therapy with a severe acute respiratory syndrome-associated coronavirus-neutralizing human monoclonal antibody reduces disease severity and viral burden in golden Syrian hamsters. J. Infect. Dis. 2006;193:685–692. doi: 10.1086/500143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Prabakaran P, et al. Structure of severe acute respiratory syndrome coronavirus receptor-binding domain complexed with neutralizing antibody. J. Biol. Chem. 2006;281:15829–15836. doi: 10.1074/jbc.M600697200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Maruyama T, et al. Ebola virus can be effectively neutralized by antibody produced in natural human infection. J. Virol. 1999;73:6024–6030. doi: 10.1128/jvi.73.7.6024-6030.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Gould LH, et al. Protective and therapeutic capacity of human single-chain Fv-Fc fusion proteins against West Nile virus. J. Virol. 2005;79:14606–14613. doi: 10.1128/JVI.79.23.14606-14613.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Daffis S, et al. Antibody responses against wild-type yellow fever virus and the 17D vaccine strain: characterization with human monoclonal antibody fragments and neutralization escape variants. Virology. 2005;337:262–272. doi: 10.1016/j.virol.2005.04.031. [DOI] [PubMed] [Google Scholar]
- 134.Allander T, et al. Recombinant human monoclonal antibodies against different conformational epitopes of the E2 envelope glycoprotein of hepatitis C virus that inhibit its interaction with CD81. J. Gen. Virol. 2000;81:2451–2459. doi: 10.1099/0022-1317-81-10-2451. [DOI] [PubMed] [Google Scholar]
- 135.Johansson DX, et al. Human combinatorial libraries yield rare antibodies that broadly neutralize hepatitis C virus. Proc. Natl. Acad. Sci. USA. 2007;104:16269–16274. doi: 10.1073/pnas.0705522104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Keck ZY, et al. Human monoclonal antibody to hepatitis C virus E1 glycoprotein that blocks virus attachment and viral infectivity. J. Virol. 2004;78:7257–7263. doi: 10.1128/JVI.78.13.7257-7263.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Kim SH, Park SY. Selection and characterization of human antibodies against hepatitis B virus surface antigen (HBsAg) by phage-display. Hybrid. Hybridomics. 2002;21:385–392. doi: 10.1089/153685902761022742. [DOI] [PubMed] [Google Scholar]
- 138.Park SG, et al. Hepatitis B virus-neutralizing anti-pre-S1 human antibody fragments from large naive antibody phage library. Antiviral Res. 2005;68:109–115. doi: 10.1016/j.antiviral.2005.06.012. [DOI] [PubMed] [Google Scholar]
- 139.Lloyd-Evans P, Gilmour JE. Expression of neutralizing recombinant human antibodies against Varicella Zoster virus for use as a potential prophylactic. Hybridoma. 2000;19:143–149. doi: 10.1089/02724570050031185. [DOI] [PubMed] [Google Scholar]
- 140.Kausmally L, et al. Neutralizing human antibodies to varicella-zoster virus (VZV) derived from a VZV patient recombinant antibody library. J. Gen. Virol. 2004;85:3493–3500. doi: 10.1099/vir.0.80406-0. [DOI] [PubMed] [Google Scholar]
- 141.Hanson BJ, et al. Passive immunoprophylaxis and therapy with humanized monoclonal antibody specific for influenza A H5 hemagglutinin in mice. Respir. Res. 2006;7:126. doi: 10.1186/1465-9921-7-126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Wyde PR, et al. Evaluation of the protective efficacy of reshaped human monoclonal antibody RSHZ19 against respiratory syncytial virus in cotton rats. Pediatr. Res. 1995;38:543–550. doi: 10.1203/00006450-199510000-00012. [DOI] [PubMed] [Google Scholar]
- 143.Zhu Z, et al. Potent neutralization of Hendra and Nipah viruses by human monoclonal antibodies. J. Virol. 2006;80:891–899. doi: 10.1128/JVI.80.2.891-899.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.de Carvalho Nicacio C, et al. Neutralizing human Fab fragments against measles virus recovered by phage display. J. Virol. 2002;76:251–258. doi: 10.1128/JVI.76.1.251-258.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Gigler A, et al. Generation of neutralizing human monoclonal antibodies against parvovirus B19 proteins. J. Virol. 1999;73:1974–1979. doi: 10.1128/jvi.73.3.1974-1979.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Kim SJ, et al. Neutralizing human monoclonal antibodies to hepatitis A virus recovered by phage display. Virology. 2004;318:598–607. doi: 10.1016/j.virol.2003.10.014. [DOI] [PubMed] [Google Scholar]
- 147.Chen Z, et al. Chimpanzee/human mAbs to vaccinia virus B5 protein neutralize vaccinia and smallpox viruses and protect mice against vaccinia virus. Proc. Natl. Acad. Sci. USA. 2006;103:1882–1887. doi: 10.1073/pnas.0510598103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Schmaljohn C, Cui Y, Kerby S, Pennock D, Spik K. Production and characterization of human monoclonal antibody Fab fragments to vaccinia virus from a phage-display combinatorial library. Virology. 1999;258:189–200. doi: 10.1006/viro.1999.9701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Chen Z, et al. Characterization of chimpanzee/human monoclonal antibodies to vaccinia virus A33 glycoprotein and its variola virus homolog in vitro and in a vaccinia virus mouse protection model. J. Virol. 2007;81:8989–8995. doi: 10.1128/JVI.00906-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Higo-Moriguchi K, Akahori Y, Iba Y, Kurosawa Y, Taniguchi K. Isolation of human monoclonal antibodies that neutralize human rotavirus. J. Virol. 2004;78:3325–3332. doi: 10.1128/JVI.78.7.3325-3332.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Burton DR, et al. A large array of human monoclonal antibodies to type 1 human immunodeficiency virus from combinatorial libraries of asymptomatic seropositive individuals. Proc. Natl. Acad. Sci. USA. 1991;88:10134–10137. doi: 10.1073/pnas.88.22.10134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Veazey RS, et al. Prevention of virus transmission to macaque monkeys by a vaginally applied monoclonal antibody to HIV-1 gp120. Nat. Med. 2003;9:343–346. doi: 10.1038/nm833. [DOI] [PubMed] [Google Scholar]
- 153.Buchacher A, et al. Generation of human monoclonal antibodies against HIV-1 proteins; electrofusion and Epstein-Barr virus transformation for peripheral blood lymphocyte immortalization. AIDS Res. Hum. Retroviruses. 1994;10:359–369. doi: 10.1089/aid.1994.10.359. [DOI] [PubMed] [Google Scholar]
- 154.Armbruster C, et al. Passive immunization with the anti-HIV-1 human monoclonal antibody (hMAb) 4E10 and the hMAb combination 4E10/2F5/2G12. J. Antimicrob. Chemother. 2004;54:915–920. doi: 10.1093/jac/dkh428. [DOI] [PubMed] [Google Scholar]
- 155.Trkola A, et al. Delay of HIV-1 rebound after cessation of antiretroviral therapy through passive transfer of human neutralizing antibodies. Nat. Med. 2005;11:615–622. doi: 10.1038/nm1244. [DOI] [PubMed] [Google Scholar]
- 156.Muster T, et al. A conserved neutralizing epitope on gp41 of human immunodeficiency virus type 1. J. Virol. 1993;67:6642–6647. doi: 10.1128/jvi.67.11.6642-6647.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Choudhry V, et al. Cross-reactive HIV-1 neutralizing monoclonal antibodies selected by screening of an immune human phage library against an envelope glycoprotein (gp140) isolated from a patient (R2) with broadly HIV-1 neutralizing antibodies. Virology. 2007;363:79–90. doi: 10.1016/j.virol.2007.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Hadlock KG, et al. Neutralizing human monoclonal antibodies to conformational epitopes of human T-cell lymphotropic virus type 1 and 2 gp46. J. Virol. 1997;71:5828–5840. doi: 10.1128/jvi.71.8.5828-5840.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Champion JM, et al. The development of monoclonal human rabies virus-neutralizing antibodies as a substitute for pooled human immune globulin in the prophylactic treatment of rabies virus exposure. J. Immunol. Methods. 2000;235:81–90. doi: 10.1016/s0022-1759(99)00223-9. [DOI] [PubMed] [Google Scholar]
- 160.Ando T, Yamashiro T, Takita-Sonoda Y, Mannen K, Nishizono A. Construction of human Fab library and isolation of monoclonal Fabs with rabies virus-neutralizing ability. Microbiol. Immunol. 2005;49:311–322. doi: 10.1111/j.1348-0421.2005.tb03735.x. [DOI] [PubMed] [Google Scholar]