

Liver transplantation is often used to treat patients with end-stage liver disease as a result of infection with HCV. However, the optimal choice of immunosuppressants in these patients is still undecided. In this article Luc van der Laan and colleagues explore the effect of different immunosuppressants on the complex cellular events involved in HCV infection and interferon signalling.
Subject terms: Cell signalling, Hepatitis C, Immunosuppression, Transplantation
Abstract
Liver transplantation is an effective treatment for end-stage liver disease that is attributable to chronic HCV infection. However, long-term outcomes are compromised by universal virological recurrence in the graft. Reinfection that occurs after transplantation has increased resistance to current interferon-based antiviral therapy and often leads to accelerated development of cirrhosis. Important risk factors for severe HCV recurrence are linked to immunosuppression. Owing to the lack of good randomized, controlled trials, the optimal choice of immunosuppressants is still debated. By contrast, much progress has been made in the understanding of HCV biology and the antiviral action of interferons. These new insights have greatly expanded our knowledge of the molecular interplay between HCV and immunosuppressive drugs. In this article, we explore the effect of different immunosuppressants on the complex cellular events involved in HCV infection and interferon signalling. Potential implications for clinical practice and future drug development are discussed.
Introduction
End-stage liver diseases that are caused by chronic HCV infection are currently the leading indications for liver transplantation.1 However, HCV reinfection of the graft occurs universally and often results in accelerated recurrence of liver fibrosis and early development of cirrhosis.2 HCV infection is also the most frequent cause of liver disease after kidney transplantation and is an independent risk factor for graft loss.3 In general, the current standard therapy for patients with HCV (PEG-IFN-α in combination with ribavirin) is much less effective in patients who have had a liver transplant than in those who have not had a transplant.4 An aggravated course of infection and increased resistance to antiviral therapy have been attributed to several host and viral factors, in particular the application of specific immunosuppressive medication.5 Over the past five decades, the immunosuppressive regimen has evolved from being mainly based on glucocorticosteroids, azathioprine and ciclosporin A to steroid-sparing regimens involving anti-IL-2 receptor antibody induction therapy, calcineurin inhibition by tacrolimus and the use of new-generation immunosuppressants—mycophenolate mofetil and rapamycin. The changes in drug types and drug combinations together with increases in the age of donors have probably contributed to the reported worsening of HCV recurrence since the early 1990s.6 Although enormous efforts have been devoted to clinical research,7 the choice of immunosuppressants in HCV-positive recipients is still a subject of debate.
The lack of good model systems hampered HCV research in its early stages. However, this deficiency has improved dramatically in the past decade with the development of various in vitro systems of cell culture (Box 1). The HCV pseudoparticles, the HCV replicons and the JFH-1-derived infectious HCV cell culture systems have all demonstrated their importance in studies on different aspects of the viral life cycle and pathophysiology, including viral entry, trafficking in the cell and assembly and release of de novo virus particles.8 These models have enabled basic research to make considerable contributions to an improved understanding of how immunosuppressive compounds can actually influence HCV infection and the antiviral action of interferons.
In this article, we explore the potential molecular interaction of different immunosuppressants with HCV and interferon signalling. We also discuss how to extrapolate these laboratory findings into clinical practice and drug development.
Box 1 | HCV culture models.
The first full-length HCV clones could infect chimpanzees but replication in vitro remained ineffective. By creating antibiotic-resistant HCV genomes, selection of replication-competent viral clones became possible. This advance led to the development of subgenomic or bicistronic (two cistrons) replicons, which mimic HCV replication. HCV replicons are most efficient in a human hepatoma cell line (Huh7), and can be linked to fluorescent or luminescent reporter genes
Early studies of HCV viral entry used pseudoparticles incorporating HCV envelope proteins E1 and E2 onto a retrovirus or lentivirus
The highly effective HCV infectious model was based on the first genotype 2a clone called JFH-1, isolated from a Japanese patient with fulminant hepatitis C. JFH-1-derived HCV models can be used to study the complete viral life cycle and chimeric genomes have been constructed of all seven known genotypes
Calcineurin inhibitors
The primary cellular targets of the calcineurin inhibitors are immunophilins. Ciclosporin A binds to cyclophilins, and tacrolimus (also known as FK506) binds to FK binding proteins (FKBPs); both events result in a profound inhibition of the phosphatase activity of calcineurin. Immunophilin-dependent signal transduction via calcineurin represents a key event in the activation of T-cell proliferation by regulating expression of the gene that encodes IL-2 (Figure 1a). Apart from their role in calcineurin signalling, immunophilins are catalysts of protein folding and as such have a supportive role in viral infection. Viruses, including HIV, herpes simplex virus, vaccinia virus, vesicular stomatitis virus and coronaviruses, take advantage of immunophilins for their replication, and this virus–host interaction is inhibited by ciclosporin A.9,10 Moreover, cyclophilin A can actually incorporate itself into these viral particles.9,11 For HCV, it is well established that cyclophilins have an essential role in viral replication and de novo virus production. Early studies suggest that HCV replication is dependent on the interaction between cyclophilin B and nonstructural protein 5B (NS5B, HCV RNA polymerase) to stimulate its RNA binding activity and thereby promote the de novo synthesis of positive and negative stranded RNA.12,13 Ciclosporin A blocks the binding of cyclophilin B to NS5B RNA and thereby inhibits viral replication.12,13,14 Cyclophilin A also directly interacts with NS5B15 and is involved in modulating the polyprotein cleavage activity of NS2.16 In addition, cyclophilin A interacts with NS5A and stimulates its binding with HCV RNA.15,17
Figure 1. Mechanisms of action of GCs and CaN inhibitors.

Mechanisms are shown in a | leukocytes and b | hepatocytes. GCs diffuse into cells and bind to the GR. The GC–GR complex translocates to the nucleus to regulate gene expression. The GC–GR complex interacts with GRE promoter elements and suppresses the expression of inflammatory genes and cytokines, such as IL-2, thereby inhibiting T-cell proliferation. Steroids facilitate HCV entry in hepatocytes by upregulating gene expression of two essential HCV entry receptors—occludin and scavenger receptor class B type I—conceivably via positive GRE promoter elements. GR and type I interferon signalling share the coregulator GRIP1. Ciclosporin A binds to cyclophilin, while tacrolimus binds to FK506-binding protein, forming a complex to block calcineurin. Suppression of the serine/threonine phosphatase activity of calcineurin results in inhibition of TCR/CD3 induced T-cell proliferation by blockage of IL-2 production. Ciclosporin A binds to cyclophilin A and cyclophilin B to block the function of NS2, NS5A or NS5B to affect viral replication in hepatocytes. Protein phosphorylation induced by signal transduction is indicated by P in orange. Abbreviations: AP1, activator protein 1; CaN, calcineurin; CsA, ciclosporin A; CyP, cyclophilin; FKPB, FK506-binding protein; GC, glucocorticosteroid; GR, cytoplasmic receptor; GRE, glucocorticoid response element; GRIP1, glucocorticoid receptor-interacting protein 1, IRF9, interferon regulatory factor 9; ISG, interferon-stimulated gene; ISRE, interferon stimulated response element; JAK1, Janus kinase 1; LD, lipid droplet; NF-ATc, nuclear factor of activated T cells; NFκB, nuclear factor κB; NS, nonstructural protein; SR-BI, scavenger receptor class B type I; STAT, signal transducers and activators of transcription; Tac, tacrolimus; TCR, T-cell receptor; TYK2, nonreceptor tyrosine-protein kinase.
As ciclosporin A interacts with both cyclophilin A and cyclophilin B, it is conceivable that it affects multiple steps in the virus life cycle (Figure 1b). The anti-HCV activity of ciclosporin A is not mediated by an interferon response and is independent of calcineurin signalling.14 Although studies showed a role for FKBP8 in HCV replication18 through interaction with NS5A,19 FKBP8 lacks several amino acid residues thought to be important for tacrolimus binding.18,20 No evidence exists that tacrolimus inhibits HCV replication.14
Several in vitro studies have shown that ciclosporin A can act in synergy with IFN-α,21,22,23,24 but clinical evidence to support these findings is still limited.25 The results of a prospective, randomized, controlled pilot study published in 2010 demonstrated that a switch from tacrolimus to ciclosporin A resulted in a modest drop in serum levels of HCV RNA and an enhancement of the response to antiviral interferon therapy in patients who had received a liver transplant.26 This finding supports those of a previous study that observed a benefit of converting from tacrolimus to ciclosporin A during pre-emptive IFN-α and ribavirin antiviral therapy in patients who were HCV positive and had undergone liver transplantation.27 The mechanism by which ciclosporin A can promote the response to interferon is an intriguing issue. Hirano et al.28 suggested that tacrolimus, but not ciclosporin A, interferes with interferon signal transduction. However, a later study did not confirm this suggestion, instead demonstrating in two state-of-the-art cell culture models that tacrolimus does not interfere with either interferon signalling or the antiviral activity of interferon.29 Therefore, no further mechanistic understanding has been unveiled on this issue.
In fact, the first in vitro evidence that ciclosporin A but not tacrolimus can inhibit HCV replication14 sparked the clinical debate on the possible differential effect of these two drugs on HCV recurrence.30 As HCV only infects humans and chimpanzees, small animal models for HCV infection are still difficult to create. One of the few established experimental animal models to study HCV infection is the SCID/uPA (severe combined immunodeficient, urokinase plasminogen activator transgenic) mouse engrafted with primary human hepatocytes. A study published in 2011 by the group that pioneered the development of this mouse model has attempted to define the effect of ciclosporin A and tacrolimus on serum HCV titres and antiviral interferon therapy in vivo.31 Although no statistically significant differences were observed between ciclosporin A and tacrolimus either on HCV viral titres or the efficacy of interferon therapy, effects could have been masked by some technical limitations. Firstly, the baseline HCV titres differed between the tested groups, with a trend toward higher baseline HCV RNA levels in the ciclosporin A arms than in the tacrolimus arms. Secondly, the antiviral response to interferon in this model was much less robust than that observed in patients. Moreover, models in immunodeficient mice cannot evaluate indirect effects of immunosuppressive drugs on antiviral immune responses unless one of the latest immunocompetent mouse models is used.32
The development of ciclosporin A derivatives strongly indicates that ciclosporin A would have anti-HCV activity both in vitro and in patients. Three cyclophilin inhibitors are currently being evaluated in preclinical and clinical studies: Debio-025, NIM811 and SCY-635.33 These compounds are developed by structural modifications of ciclosporin A to enhance the antiviral activity and reduce the immunosuppression capacity by increasing cyclophilin binding and abolishing calcineurin affinity, respectively. Debio-025 has been the first to enter clinical trials and the first results are encouraging. In a cohort infected with HCV and HIV, 2 weeks of treatment with Debio-025 decreased HCV viral load by more than 3-log, including in patients infected with HCV genotypes 1, 3 and 4. By contrast, the anti-HIV effects were much less pronounced (1-log reduction of viral load).34 In treatment-naive patients infected with just HCV, treatment with Debio-025 alone resulted in a more than 2-log reduction of viral load in those with genotypes 1 and 4, and a more than 4-log reduction in those with genotypes 2 and 3.35 When combined with PEG-IFN-α and ribavirin, additive antiviral effects were demonstrated.35
Glucocorticosteroids
Glucocorticosteroids have been used since the early years of organ transplantation. Prednisolone and its close analogue dexamethasone are potent suppressors of the immune system, as they modulate cellular and inflammatory responses via stimulation or inhibition of gene transcription (Figure 1).36 Although evidence indicates that the oestrogen receptor is functionally involved in HCV replication by promoting NS5B association with the viral replication complex,37 the role of other glucocorticosteroid receptors is largely unknown. A study using the HCV subgenomic replicon model38 found that both prednisolone and dexamethasone have no stimulatory effects on RNA levels, but rather have minor inhibitory effects.39 Using an infectious HCV model (the JFH1-derived chimera Jc1 in Huh-7.5 cells), similar results were obtained showing ∼50% inhibition of viral replication at high doses of prednisolone.40 However, in this infectious model, prednisolone stimulated HCV infection by enhancing virus entry.40 This specific enhancement was mechanistically linked to the upregulation of the two essential HCV entry factors—occludin and scavenger receptor class B type I—at both mRNA and protein levels.40 On the basis of their observations, the authors speculated that treatment with a steroid bolus (>250 mg prednisolone per day) in patients with HCV might foster virus dissemination through facilitation of virus entry into hepatocytes, thus aggravating HCV recurrence. Moreover, treatment with prednisolone probably suppresses the patient's immune control of the HCV-infected cells, by effects on T cells41 and plasmacytoid dendritic cells,42 and thereby might indirectly accentuate infection.39 Plasmacytoid dendritic cells are of particular interest in HCV infection, as they produce large amounts of IFN-α; however, the capability of plasmacytoid dendritic cells to produce IFN-α seems to be reduced in patients with chronic HCV.43 Experimental data from our group have shown that treatment with prednisolone abolishes the anti-HCV activity of plasmacytoid dendritic cells in vitro (P. E. de Ruiter, personal communication).
In liver transplantation, glucocorticosteroids are often given as an induction protocol during surgery and low doses in combination with other immunosuppressants are used as maintenance immune suppression after transplantation. In cases of acute cellular rejection episodes, patients receive several boluses of methylprednisolone to reverse the rejection. The most compelling evidence that steroids affect HCV viral load actually came from studies in patients with HCV who did not undergo a liver transplant.44,45,46 In a transplant setting, the general consensus is that steroid avoidance47 or slow tapering of the dose48 is associated with reduced disease recurrence, whereas boluses for treating acute rejection can increase the viral load.49,50 However, a randomized multicentre study comparing steroid-free therapy and standard immunosuppression showed no clear advantage of not using steroids in liver transplant recipients who had HCV.51
Two systematic Cochrane reviews with meta-analyses of randomized, controlled trials have been performed in the settings of chronic HCV52 and HBV53 infection to comprehensively assess the effects of glucocorticosteroids on interferon antiviral therapy. No statistically significant effect of steroids on the virological response at the end of treatment was found in patients with HCV.52 By contrast, pretreatment with steroids was associated with a considerably higher frequency of loss of hepatitis B e antigen and HBV DNA, although no statistically significant effect on clinical outcomes was found.53 However, mechanistic studies have shown that glucocorticosteroids can interfere with signal transduction of the interferon receptor. The glucocorticosteroid receptor and type I interferon receptor signalling pathways share the cofactor glucocorticoid receptor-interacting protein 1 (GRIP1).54,55 Glucocorticosteroids suppress interferon responses by antagonizing the heterotrimeric STAT1–STAT2–IRF9 (ISGF3) activity via GRIP156 or attenuation of STAT1 activation through induction of SOCS1, which is a suppressor of cytokine signalling 1 (Figure 1).57 However, these events probably differ between cell types and no direct link between these molecular interactions and viral infection has been well established. Therefore, it is of particular relevancy to further investigate the actual effects of steroids on interferon antiviral response in HCV models.
MMF and MPA
Mycophenolic acid (MPA) is the activated form of the prodrug mycophenolate mofetil (MMF). MMF and MPA are part of the class of antimetabolite immunosuppressive agents that also includes azathioprine. The effect of azathioprine on HCV has not been extensively studied, despite one early study suggesting that this agent has antiviral effects on two Flaviviridae viruses.58
MMF is commonly used in kidney transplantation, but it is also approved by the FDA for the prophylaxis of allograft rejection after heart or liver transplantation.59 In addition to its potent immunosuppressive capacity, MPA also has a broad spectrum of antiviral activity in vitro against numerous DNA and RNA viruses, including dengue virus,60 West Nile virus,23 yellow fever virus,61 Chikungunya virus,62 HBV63 and HCV.23 MPA is an uncompetitive inhibitor of inosine monophosphate dehydrogenase (IMPDH), in particular the isoform IMPDH2. Inhibition of IMPDH decreases intracellular levels of guanosine nucleotide pools resulting in inadequate quantities for nominal DNA duplication, which was thought to be the antiproliferative and immunosuppressive mechanism of MPA (Figure 2a).64 This mechanism is probably also responsible for its antiviral effects against at least the West Nile,23 yellow fever61 and Chikungunya viruses,62 as supplementation by exogenous guanosine almost completely overcomes these inhibitory effects. By contrast, supplementation of guanosine had little effect on the inhibition of HCV replication.23,65 Consistently, ectopic expression of IMPDH2 mutants lacking the binding site for MPA largely restored its antiproliferative effect, but with only minor effects on HCV replication, suggesting other mechanisms are involved in its anti-HCV action.66 Unexpectedly, MPA was found to induce the expression of important antiviral interferon-stimulated genes in HCV cell culture models, including interferon regulatory factor (IRF) 1, IRF9 and interferon-induced transmembrane protein 3 (IFITM3).66 Using an RNAi-based loss-of-function approach, IRF1 was demonstrated to be directly involved in the anti-HCV activity of MPA.66
Figure 2. Mechanisms of action of mycophenolic acid and rapamycin.
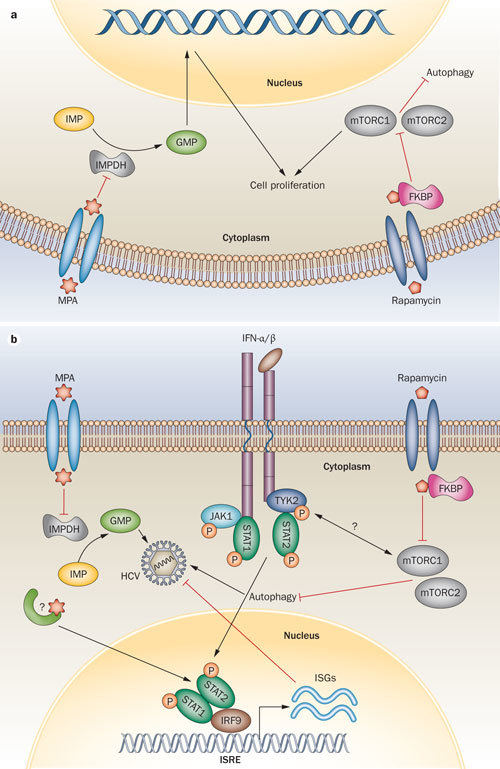
Mechanisms shown in a | leukocytes and b | hepatocytes. The uptake of MPA by cells involves organic anion transporting polypeptides and probably other transporters as well. MPA is an uncompetitive inhibitor of IMPDH and results in the inhibition of de novo nucleotide biosynthesis. Insufficient intracellular levels of guanosine nucleotide pools result in a defect of nominal DNA duplication that is thought to be the immunosuppressive mechanism of MPA. In hepatocytes, persistent attenuation of nucleotide biosynthesis by MPA partially contributes to the inhibition of HCV replication. In addition to this IMPDH-dependent antiviral pathway, MPA also conveys a rapid antiviral effect mediated by induction of the expression of interferon-stimulated genes. Through an unknown mechanism, MPA directly potentiates the activity of the ISRE promoter element both in the presence or absence of IFN-α stimulation. Rapamycin engages the cytosolic protein FKBP12 to form a complex. This complex inhibits the mTOR pathway by directly binding to the mTORC1, resulting in blockage of cell cycle progression at the G1 to S phase and thereby causing inhibition of T-cell proliferation. Rapamycin is a potent autophagy inducer, which facilitates HCV infection. The mTOR pathway directly interacts with interferon-activated JAK–STAT signalling. Protein phosphorylation induced by signal transduction is indicated by P in orange. Abbreviations: FKBP12, FK506 binding protein 12; GMP, guanosine monophosphate; IMP, inosine monophosphate; IMPDH, inosine monophosphate dehydrogenase; IRF9, interferon regulatory factor 9; ISG, interferon-stimulated gene; ISRE, interferon stimulated response element; JAK1, Janus kinase 1; MPA, mycophenolic acid; mTOR, mammalian target of rapamycin; mTORC, mammalian target of rapamycin complex; STAT, signal transducers and activators of transcription; TYK2, nonreceptor tyrosine-protein kinase.
The expression of a panel of interferon-stimulated genes was also considerably higher in peripheral blood mononuclear cells of patients who had received a kidney transplant and were maintained on MMF immunosuppression, compared with peripheral blood mononuclear cells of healthy control individuals.66 Similar results have been reported for ribavirin, which also induces expression of a broad spectrum of interferon-stimulated genes.67,68 Interestingly, like MPA, ribavirin is a known inhibitor of IMPDH.69 Inhibiting IMPDH and inducing the expression of interferon-stimulated genes probably simultaneously contribute to the anti-HCV action of MPA (Figure 2b). Whether IMPDH by itself is involved in the induction of the expression of interferon-stimulated genes is not fully clear. However, supplementation with exogenous guanosine did not interfere with inducing the expression of these genes.66
Although the safety and efficacy of MMF as an immunosuppressive medication in patients with HCV who received a transplant has been demonstrated, the exact effects on HCV recurrence have not been clearly studied in prospective, randomized and double-blinded trials. Results from prospective nonrandomized or retrospective studies remain controversial at this point of time. However, the in vivo efficacy of many antiviral compounds (such as ribavirin) can be observed only when combined with other antivirals. Ribavirin has shown substantial anti-HCV activity in vitro but is generally considered to have little or no detectable antiviral activity as a monotherapy in patients.70 However, when combined with IFN-α, ribavirin increases the sustained virologic response by threefold.71 Similarly, a previous clinical study showed that combined treatment with MMF and PEG-IFN-α in patients with chronic HCV resulted in a considerably improved end-of-therapy response rate in a difficult-to-treat population of previous nonresponders to standard therapy.72 The sustained virologic response of the IFN-α and MMF combination group, however, was lower than that of the group treated with IFN-α and ribavirin.72 A possible explanation for these limited sustained virologic responses is that MMF interferes with a robust antiviral immune response required to eliminate HCV.23 Notably, MPA works in synergy with IFN-α on HCV replication in vitro.23 When combined with IFN-α, MPA augments the transcription of multiple interferon-stimulated genes that are mediated by interferon-stimulated response elements.66 These latest findings have shed some light on the molecular basis of how MPA works in synergy with IFN-α.
Rapamycin
Rapamycin, a new-generation immunosuppressant, has been gaining increasing favour in the transplantation context, mainly attributable to its low nephrotoxicity and potential anticancer properties.73 Rapamycin engages the cytosolic protein FKBP12 to form a complex. This complex inhibits the mammalian target of rapamycin (mTOR) pathway by directly binding to the mTOR complex 1, resulting in blockage of cell cycle progression at the G1 to S phase and thereby causing inhibition of T-cell proliferation (Figure 2).
One particular interesting feature of rapamycin is that it induces autophagy by inhibition of mTOR. Autophagy is a process for catabolising organelles and other cytoplasmic components to balance cellular metabolism and to promote cell survival during stressful conditions. Autophagy is also an important event in regulation of the cellular response against viral infections.74 The role of autophagy is complex and can have both antiviral and proviral effects in different virus infections. During infection, autophagy can be a surveillance mechanism that delivers viral antigens to endosomal compartments to stimulate innate immune signalling and to provide processed antigens for presentation in the major histocompatibility complex. Virus infection can trigger the induction of autophagy, but can also block the function of the autophagy pathway or even subvert autophagy. By contrast, viruses can directly exploit the autophagy machinery to facilitate replication or to promote their maturation and the dissemination of virions.75
Several studies have shown that HCV infection induces autophagy in hepatocytes via the unfolded protein response.76,77 However, the induced autophagy appears incomplete, as the maturation of autophagosomes to autolysosomes is blocked. Therefore, autophagosomes are not degraded but instead support viral replication.77 HCV viral proteins, such as NS5B, directly interact with the host proteins that are required for the induction of autophagy.78 In addition, induction of autophagy by HCV seems to facilitate infection by reducing the innate immunity of host cells, including interferon responses79,80 Indeed, accumulating evidence exists for a direct link between the mTOR signalling pathway and interferon-activated JAK-STAT signalling.81,82,83 Type I interferon induces phosphorylation and activation of mTOR in a PI3-kinase-dependent manner.83 In addition, mTOR complex 1 was found to stimulate the production of type I interferons through activation of IRF5 and IRF7.82 On the basis of these mechanistic insights, it is conceivable that rapamycin could affect HCV recurrence and antiviral interferon therapy, though clinical evidence to support this theory is still limited.84,85
Novel immunosuppressants
Despite the success of current immunosuppressive agents in reducing the incidence of acute cellular rejection, the toxicity associated with these regimens has now become a major obstacle for positive long-term outcomes of liver transplantation. Currently, novel agents, including small molecules, are at various stages of clinical development.86 These novel small molecules include the new generation ciclosporin A analogue, ISA247, the protein kinase C (PKC) inhibitor, AEB071, and the selective Janus kinase inhibitor, CP-690550.
Given the evidence for an important role of cyclophilins in the life cycle of HCV,12,15,16 investigation of the effects of ISA247 on HCV infection would be of particular interest. ISA247 only differs from ciclosporin A by a single amino acid residue. The structural change induced by this substitution is considered to result in a greater calcineurin inhibitory potency and a more predictable pharmacokinetic profile than ciclosporin A. An X-ray crystal structure study has revealed the basis for the cyclophilin A binding affinity and immunosuppressive potency of ISA247.87 As cyclophilin A is a host factor that modulates infection of a number of viruses, it will be important to study the potential antiviral activity of ISA247 compared with ciclosporin A.
AEB071 is a small-molecule inhibitor that targets multiple members of the PKC family. PKC inhibition results in decreased T-cell activation. Several members of the PKC family have been linked to both HCV infection and interferon signalling.88,89 Using cell culture models, however, no stimulatory effect of AEB071 on HCV infection was observed; instead, at high drug concentrations a reduction in viral replication was found.90 This reduction might be related to inhibition of cell proliferation by AEB071. Therefore, it was concluded in this study that AEB071 had no direct effect on HCV infection.90 Further investigation is required to draw firm conclusions.
CP-690550 was developed as a selective inhibitor of JAK3, which is predominantly expressed in immune cells and is only bound by γ-chain-bearing cytokine receptors involved in the JAK–STAT signalling pathway.91 Some of the immunosuppressive mechanism of CP-690550 can be explained as the result of IL-2 inhibition, as JAK3 functions in the receptor signalling pathway for this cytokine.92 However, CP-690550 has also been shown to inhibit other members of the JAK family, including JAK1, JAK2 and TYK2.93 These pathways have considerable implications in both defence of virus infection and interferon responses. In addition, expression and activation of JAK3, which is involved in defence against dengue virus infection, have been reported in hepatic cells.94 Therefore, studying the effects and mechanisms of CP-690550 on both HCV infection and interferon signalling would be highly relevant.
Conclusions
The development of subgenomic HCV cell culture models has greatly contributed to the discovery of the antiviral effects of ciclosporin A and MPA on viral replication. The use of a recently developed infectious culture model has revealed the negative effects of steroids, specifically on HCV entry.40 However, a common question from transplant clinicians is why these clear in vitro effects of HCV are not univocally observed in clinical practice. For instance, the clear antiviral activity of ciclosporin A and MPA in vitro is yet to be confirmed in proper prospective clinical studies, and retrospective data sketch a murky picture. One probable answer to this in vivo and in vitro discrepancy is that tampering of the host immune system against the virus masks the direct antiviral effects of these immunosuppressive agents. The antiviral potency of particular immunosuppressants might not be strong enough to result in a dramatic effect on viral load, but could potentially be reflected in other parameters, such as slowing down fibrosis progression.95,96,97,98 Perhaps the potent effects of the derivates of ciclosporin A, such as Debio-025, in patients were achieved by both deprivation of immunosuppressive properties and enhancement of cyclophilin binding affinity.34,35
A major new milestone for HCV treatment is the approval of two protease inhibitors by the FDA in 2011. Although boceprevir and telaprevir are only approved in combination with PEG-IFN-α and ribavirin for treating adults with chronic HCV genotype 1 infection who have compensated liver disease, trials are expected to be designed to treat patients both before and after transplantation. As a caution, a phase I study found that telaprevir interferes with the metabolism of both ciclosporin A and tacrolimus by inhibition of cytochrome P450 3A enzymatic activity.99 Telaprevir caused a tremendous increase in blood concentrations of both these calcineurin inhibitors and could potentially lead to serious or even life-threatening adverse effects. Therefore, the interaction of new antiviral compounds and immunosuppressants requires extensive studies of drug–drug interactions before designing any antiviral therapy trial in the post-transplantation setting.
In summary, progress in understanding HCV biology is important in evaluating the complex effects of immunosuppressive medication on HCV recurrence. Despite clear effects of ciclosporin A and MPA in HCV culture models, reliable clinical evidence for their effect on HCV recurrence is still lacking. Inhibition of the immune response against HCV by immunosuppressants probably counteracts the direct antiviral effects. The effectiveness of interferon-based antiviral therapy is reduced after transplantation, but, to date, no concrete evidence from HCV culture models that immunosuppressive drugs interfere with interferon signal transduction exists. Immunosuppressive medication for transplant recipients who have HCV should be further improved. The application of newly registered anti-HCV compounds in transplant recipients should be carefully tested because of potential concerns regarding adverse drug–drug interactions with immunosuppressants.
Although basic science alone will never resolve the clinical debate, it already provides mechanistic insight into how particular immunosuppressants can affect the course of infection and outcomes of antiviral therapy. The knowledge gained in a liver-transplant setting will also definitely be a valuable reference for the management of patients who are HCV positive and receive a kidney transplant. Conceivably, these mechanistic understandings will promote future investment in the initiation of randomized, controlled studies and development of new-generation immunosuppressants for HCV-positive transplantation.
Acknowledgements
The authors would like to thank Petra de Ruiter for critically reading the manuscript and sharing unpublished data. The authors received financial support from the Erasmus MC Translational Research Fund and the Liver Research Foundation Rotterdam (SLO).
Biographies
Qiuwei Pan is a postdoctorial research associate at the Laboratory of Gastroenterology and Hepatology, Erasmus MC-University Medical Center, Rotterdam, The Netherlands. He obtained BSc and MSc degrees in China. In 2007, he moved to Rotterdam to carry out his PhD research and aimed to improve the outcome of HCV-positive liver transplantation. He focused on the effect of immunosuppressants and the development of novel small RNA and stem cell-based antiviral strategies. In 2009, he was elected as Rising Star by the International Liver Transplantation Society. Currently, he is recruiting PhD students and building a novel translational research line on hepatitis virus–host interaction at the same lab.
Hugo Tilanus is Professor of Surgery and Vice-Chairman of the Department of Surgery, at Erasmus MC-University Medical Center, Rotterdam, The Netherlands. He graduated in Medicine at Leiden University, The Netherlands, in 1976. He finished his surgical training in Rotterdam in 1984. A year before he defended his PhD thesis regarding the femoropopliteal bypass. Over the years his scientific interests, starting with vascular surgery, were devoted to clinical and basic research regarding oesophageal carcinoma and liver transplantation.
Herold Metselaar is Professor of Liver Failure & Liver Transplantation at Erasmus MC-University Medical Center, Rotterdam, The Netherlands. He studied medicine from 1973 to 1979 and received a PhD in Medicine in 1990 in Rotterdam. In 1995, he was appointed as Medical Director of the Rotterdam liver transplant program and started a translational research program in liver transplantation with an emphasis on immunosuppressive therapy and viral recurrence. Recent achievements of his team include the identification and characterization of the antiviral effects of immunosuppressive agents. His group is heavily involved in the development of tolerogenic strategies.
Harry Janssen is Professor of Hepatology and Chief of Liver Diseases & Transplantation, at Erasmus MC-University Medical Center, Rotterdam, The Netherlands. He graduated from medical school at the Radboud University of Nijmegen, The Netherlands, and obtained his PhD in Rotterdam. Following his registration as a Gastroenterologist he went to the Mayo Clinic, USA, for a research fellowship in Hepatology at the Center of Basic Research in Digestive Diseases. In addition to his longstanding expertise in antiviral therapy of chronic viral hepatitis, he is also a leading scientist in the field of vascular disorders of the liver.
Luc van der Laan is Associate Professor of Regenerative Transplantation Research and Head of the Laboratory of Experimental Transplantation and Intestinal Surgery, LETIS, at the Department of Surgery, Erasmus MC-University Medical Center, Rotterdam, The Netherlands. He studied Biology at the University of Amsterdam, The Netherlands, and received his PhD degree at the Vrije University in Amsterdam (1998). As a fellow he worked at the Scripps Research Institute, USA, on the xeno-infectious risk of porcine pancreatic islet transplantation. His current research line focuses on immunological and virological aspects of liver transplantation. The research is translational in character and aims to provide basic and applied knowledge to improve the outcome of liver transplantation.
Author Contributions
Q. Pan and L. J. W. van der Laan contributed equally to all aspects of the article. H. W. Tilanus, H. J. Metselaar and H. L. A. Janssen contributed equally to discussion of the content and review/editing of the manuscript before submission.
Competing interests
H. J. Metselaar has been a consultant and speaker for Astrellas and Novarits. He has also received grant/research support from Astrellas. The other authors declare no competing interests.
Contributor Information
Harry L. A. Janssen, Email: l.vanderlaan@erasmusmc.nl
Luc J. W. van der Laan, Email: l.vanderlaan@erasmusmc.nl
References
- 1.Brown RS. Hepatitis C and liver transplantation. Nature. 2005;436:973–978. doi: 10.1038/nature04083. [DOI] [PubMed] [Google Scholar]
- 2.Yilmaz N. A prospective evaluation of fibrosis progression in patients with recurrent hepatitis C virus following liver transplantation. Liver Transpl. 2007;13:975–983. doi: 10.1002/lt.21117. [DOI] [PubMed] [Google Scholar]
- 3.Huskey J, Wiseman AC. Chronic viral hepatitis in kidney transplantation. Nat. Rev. Nephrol. 2011;7:156–165. doi: 10.1038/nrneph.2010.192. [DOI] [PubMed] [Google Scholar]
- 4.Samuel D. Interferon-alpha 2b plus ribavirin in patients with chronic hepatitis C after liver transplantation: a randomized study. Gastroenterology. 2003;124:642–650. doi: 10.1053/gast.2003.50095. [DOI] [PubMed] [Google Scholar]
- 5.McCaughan GW, Shackel NA, Bertolino P, Bowen DG. Molecular and cellular aspects of hepatitis C virus reinfection after liver transplantation: how the early phase impacts on outcomes. Transplantation. 2009;87:1105–1111. doi: 10.1097/TP.0b013e31819dfa83. [DOI] [PubMed] [Google Scholar]
- 6.Roche B, Samuel D. Risk factors for hepatitis C recurrence after liver transplantation. J. Viral Hepat. 2007;14(Suppl. 1):89–96. doi: 10.1111/j.1365-2893.2007.00920.x. [DOI] [PubMed] [Google Scholar]
- 7.Watt K, Veldt B, Charlton M. A practical guide to the management of HCV infection following liver transplantation. Am. J. Transplant. 2009;9:1707–1713. doi: 10.1111/j.1600-6143.2009.02702.x. [DOI] [PubMed] [Google Scholar]
- 8.Boonstra A, van der Laan LJ, Vanwolleghem T, Janssen HL. Experimental models for hepatitis C viral infection. Hepatology. 2009;50:1646–1655. doi: 10.1002/hep.23138. [DOI] [PubMed] [Google Scholar]
- 9.Towers GJ. The control of viral infection by tripartite motif proteins and cyclophilin A. Retrovirology. 2007;4:40. doi: 10.1186/1742-4690-4-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de Wilde AH. Cyclosporin A inhibits the replication of diverse coronaviruses. J. Gen. Virol. 2011;92:2542–2548. doi: 10.1099/vir.0.034983-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Franke EK, Yuan HE, Luban J. Specific incorporation of cyclophilin A into HIV-1 virions. Nature. 1994;372:359–362. doi: 10.1038/372359a0. [DOI] [PubMed] [Google Scholar]
- 12.Watashi K. Cyclophilin B is a functional regulator of hepatitis C virus RNA polymerase. Mol. Cell. 2005;19:111–122. doi: 10.1016/j.molcel.2005.05.014. [DOI] [PubMed] [Google Scholar]
- 13.Nakagawa M. Suppression of hepatitis C virus replication by cyclosporin A is mediated by blockade of cyclophilins. Gastroenterology. 2005;129:1031–1041. doi: 10.1053/j.gastro.2005.06.031. [DOI] [PubMed] [Google Scholar]
- 14.Watashi K, Hijikata M, Hosaka M, Yamaji M, Shimotohno K. Cyclosporin A suppresses replication of hepatitis C virus genome in cultured hepatocytes. Hepatology. 2003;38:1282–1288. doi: 10.1053/jhep.2003.50449. [DOI] [PubMed] [Google Scholar]
- 15.Fernandes F. Sensitivity of hepatitis C virus to cyclosporine A depends on nonstructural proteins NS5A and NS5B. Hepatology. 2007;46:1026–1033. doi: 10.1002/hep.21809. [DOI] [PubMed] [Google Scholar]
- 16.Ciesek S. Cyclosporine A inhibits hepatitis C virus nonstructural protein 2 through cyclophilin A. Hepatology. 2009;50:1638–1645. doi: 10.1002/hep.23281. [DOI] [PubMed] [Google Scholar]
- 17.Foster TL, Gallay P, Stonehouse NJ, Harris M. Cyclophilin A interacts with domain II of hepatitis C virus NS5A and stimulates RNA binding in an isomerase-dependent manner. J. Virol. 2011;85:7460–7464. doi: 10.1128/JVI.00393-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Okamoto T. Hepatitis C virus RNA replication is regulated by FKBP8 and Hsp90. EMBO J. 2006;25:5015–5025. doi: 10.1038/sj.emboj.7601367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Okamoto T. A single-amino-acid mutation in hepatitis C virus NS5A disrupting FKBP8 interaction impairs viral replication. J. Virol. 2008;82:3480–3489. doi: 10.1128/JVI.02253-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lam E, Martin M, Wiederrecht G. Isolation of a cDNA encoding a novel human FK506-binding protein homolog containing leucine zipper and tetratricopeptide repeat motifs. Gene. 1995;160:297–302. doi: 10.1016/0378-1119(95)00216-S. [DOI] [PubMed] [Google Scholar]
- 21.Goto K. Evaluation of the anti-hepatitis C virus effects of cyclophilin inhibitors, cyclosporin A, and NIM811. Biochem. Biophys. Res. Commun. 2006;343:879–884. doi: 10.1016/j.bbrc.2006.03.059. [DOI] [PubMed] [Google Scholar]
- 22.Firpi RJ. Cyclosporine suppresses hepatitis C virus in vitro and increases the chance of a sustained virological response after liver transplantation. Liver Transpl. 2006;12:51–57. doi: 10.1002/lt.20532. [DOI] [PubMed] [Google Scholar]
- 23.Henry SD. Mycophenolic acid inhibits hepatitis C virus replication and acts in synergy with cyclosporin A and interferon-alpha. Gastroenterology. 2006;131:1452–1462. doi: 10.1053/j.gastro.2006.08.027. [DOI] [PubMed] [Google Scholar]
- 24.Paeshuyse J. The non-immunosuppressive cyclosporin DEBIO-025 is a potent inhibitor of hepatitis C virus replication in vitro. Hepatology. 2006;43:761–770. doi: 10.1002/hep.21102. [DOI] [PubMed] [Google Scholar]
- 25.Roche B. Hepatitis C virus therapy in liver transplant recipients: response predictors, effect on fibrosis progression, and importance of the initial stage of fibrosis. Liver Transpl. 2008;14:1766–1777. doi: 10.1002/lt.21635. [DOI] [PubMed] [Google Scholar]
- 26.Firpi RJ. The use of cyclosporine for recurrent hepatitis C after liver transplant: a randomized pilot study. Dig. Dis. Sci. 2010;55:196–203. doi: 10.1007/s10620-009-0981-3. [DOI] [PubMed] [Google Scholar]
- 27.Sugawara Y, Kaneko J, Makuuchi M. Cyclosporin a for treatment of hepatitis C virus after liver transplantation. Transplantation. 2006;82:579–580. doi: 10.1097/01.tp.0000229397.81425.51. [DOI] [PubMed] [Google Scholar]
- 28.Hirano K. Differential effects of calcineurin inhibitors, tacrolimus and cyclosporin a, on interferon-induced antiviral protein in human hepatocyte cells. Liver Transpl. 2008;14:292–298. doi: 10.1002/lt.21358. [DOI] [PubMed] [Google Scholar]
- 29.Pan Q. Calcineurin inhibitor tacrolimus does not interfere with the suppression of hepatitis C virus infection by interferon-alpha. Liver Transpl. 2010;16:520–526. doi: 10.1002/lt.22032. [DOI] [PubMed] [Google Scholar]
- 30.Berenguer M. Viral hepatitis: Ciclosporin versus tacrolimus for HCV transplant recipients. Nat. Rev. Gastroenterol. Hepatol. 2011;8:422–424. doi: 10.1038/nrgastro.2011.124. [DOI] [PubMed] [Google Scholar]
- 31.Kneteman NM. Impact of calcineurin inhibitors with and without interferon on HCV titers in a chimeric mouse model of HCV infection. Liver Transpl. 2011;18:38–44. doi: 10.1002/lt.22400. [DOI] [PubMed] [Google Scholar]
- 32.Dorner M. A genetically humanized mouse model for hepatitis C virus infection. Nature. 2011;474:208–211. doi: 10.1038/nature10168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gallay PA. Cyclophilin inhibitors. Clin. Liver Dis. 2009;13:403–417. doi: 10.1016/j.cld.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 34.Flisiak R. The cyclophilin inhibitor Debio-025 shows potent anti-hepatitis C effect in patients coinfected with hepatitis C and human immunodeficiency virus. Hepatology. 2008;47:817–826. doi: 10.1002/hep.22131. [DOI] [PubMed] [Google Scholar]
- 35.Flisiak R. The cyclophilin inhibitor Debio 025 combined with PEG IFNalpha2a significantly reduces viral load in treatment-naive hepatitis C patients. Hepatology. 2009;49:1460–1468. doi: 10.1002/hep.22835. [DOI] [PubMed] [Google Scholar]
- 36.Lowenberg M, Verhaar AP, van den Brink GR, Hommes DW. Glucocorticoid signaling: a nongenomic mechanism for T-cell immunosuppression. Trends Mol. Med. 2007;13:158–163. doi: 10.1016/j.molmed.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 37.Watashi K. Anti-hepatitis C virus activity of tamoxifen reveals the functional association of estrogen receptor with viral RNA polymerase NS5B. J. Biol. Chem. 2007;282:32765–32772. doi: 10.1074/jbc.M704418200. [DOI] [PubMed] [Google Scholar]
- 38.Lohmann V. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science. 1999;285:110–113. doi: 10.1126/science.285.5424.110. [DOI] [PubMed] [Google Scholar]
- 39.Henry SD, Metselaar HJ, Van Dijck J, Tilanus HW, van der Laan LJ. Impact of steroids on hepatitis C virus replication in vivo and in vitro. Ann. NY Acad. Sci. 2007;1110:439–447. doi: 10.1196/annals.1423.046. [DOI] [PubMed] [Google Scholar]
- 40.Ciesek S. Glucocorticosteroids increase cell entry by hepatitis C virus. Gastroenterology. 2010;138:1875–1884. doi: 10.1053/j.gastro.2010.02.004. [DOI] [PubMed] [Google Scholar]
- 41.Rosen HR. Association of multispecific CD4(+) response to hepatitis C and severity of recurrence after liver transplantation. Gastroenterology. 1999;117:926–932. doi: 10.1016/S0016-5085(99)70352-5. [DOI] [PubMed] [Google Scholar]
- 42.Boor PP. Prednisolone suppresses the function and promotes apoptosis of plasmacytoid dendritic cells. Am. J. Transplant. 2006;6:2332–2341. doi: 10.1111/j.1600-6143.2006.01476.x. [DOI] [PubMed] [Google Scholar]
- 43.Dolganiuc A, Szabo G. Dendritic cells in hepatitis C infection: can they (help) win the battle? J. Gastroenterol. 2011;46:432–447. doi: 10.1007/s00535-011-0377-y. [DOI] [PubMed] [Google Scholar]
- 44.Magrin S. Hepatitis C viremia in chronic liver disease: relationship to interferon-alpha or corticosteroid treatment. Hepatology. 1994;19:273–279. doi: 10.1002/hep.1840190203. [DOI] [PubMed] [Google Scholar]
- 45.Fong TL. Short-term prednisone therapy affects aminotransferase activity and hepatitis C virus RNA levels in chronic hepatitis C. Gastroenterology. 1994;107:196–199. doi: 10.1016/0016-5085(94)90077-9. [DOI] [PubMed] [Google Scholar]
- 46.Calleja JL. Interferon and prednisone therapy in chronic hepatitis C with non-organ-specific antibodies. J. Hepatol. 1996;24:308–312. doi: 10.1016/S0168-8278(96)80009-2. [DOI] [PubMed] [Google Scholar]
- 47.Segev DL. Steroid avoidance in liver transplantation: meta-analysis and meta-regression of randomized trials. Liver Transpl. 2008;14:512–525. doi: 10.1002/lt.21396. [DOI] [PubMed] [Google Scholar]
- 48.Vivarelli M. Influence of steroids on HCV recurrence after liver transplantation: a prospective study. J. Hepatol. 2007;47:793–798. doi: 10.1016/j.jhep.2007.07.023. [DOI] [PubMed] [Google Scholar]
- 49.Gane EJ. A longitudinal analysis of hepatitis C virus replication following liver transplantation. Gastroenterology. 1996;110:167–177. doi: 10.1053/gast.1996.v110.pm8536853. [DOI] [PubMed] [Google Scholar]
- 50.Gane EJ. Long-term outcome of hepatitis C infection after liver transplantation. N. Engl. J. Med. 1996;334:815–820. doi: 10.1056/NEJM199603283341302. [DOI] [PubMed] [Google Scholar]
- 51.Klintmalm GB. A randomized multicenter study comparing steroid-free and standard immunosuppression for liver transplant recipients with chronic hepatitis C. Liver Transpl. 2011;17:1394–1403. doi: 10.1002/lt.22417. [DOI] [PubMed] [Google Scholar]
- 52.Brok, J., Mellerup, M. T., Krogsgaard, K. & Gluud, C. Glucocorticosteroids for viral hepatitis C. Cochrane Database of Systematic Reviews, Issue 2. Art. No.: CD002904. 10.1002/14651858.CD002904.pub2. [DOI] [PMC free article] [PubMed]
- 53.Mellerup, M. T., Krogsgaard, K., Mathurin, P., Gluud, C. & Poynard, T. Sequential combination of glucocorticosteroids and alfa interferon versus alfa interferon alone for HBeAg-positive chronic hepatitis B. Cochrane Database of Systematic Reviews, Issue 3. Art. No.: CD000345 10.1002/14651858.CD000345.pub2.
- 54.Reily MM, Pantoja C, Hu X, Chinenov Y, Rogatsky I. The GRIP1:IRF3 interaction as a target for glucocorticoid receptor-mediated immunosuppression. EMBO J. 2006;25:108–117. doi: 10.1038/sj.emboj.7600919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Flammer JR, Rogatsky I. Minireview: glucocorticoids in autoimmunity: unexpected targets and mechanisms. Mol. Endocrinol. 2011;25:1075–1086. doi: 10.1210/me.2011-0068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Flammer JR. The type I interferon signaling pathway is a target for glucocorticoid inhibition. Mol. Cell. Biol. 2010;30:4564–4574. doi: 10.1128/MCB.00146-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bhattacharyya S, Zhao Y, Kay TW, Muglia LJ. Glucocorticoids target suppressor of cytokine signaling 1 (SOCS1) and type 1 interferons to regulate Toll-like receptor-induced STAT1 activation. Proc. Natl Acad. Sci. USA. 2011;108:9554–9559. doi: 10.1073/pnas.1017296108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stangl JR, Carroll KL, Illichmann M, Striker R. Effect of antimetabolite immunosuppressants on Flaviviridae, including hepatitis C virus. Transplantation. 2004;77:562–567. doi: 10.1097/01.TP.0000114610.40412.C6. [DOI] [PubMed] [Google Scholar]
- 59.Shipkova M, Armstrong VW, Oellerich M, Wieland E. Mycophenolate mofetil in organ transplantation: focus on metabolism, safety and tolerability. Expert Opin. Drug Metab. Toxicol. 2005;1:505–526. doi: 10.1517/17425255.1.3.505. [DOI] [PubMed] [Google Scholar]
- 60.Diamond MS, Zachariah M, Harris E. Mycophenolic acid inhibits dengue virus infection by preventing replication of viral RNA. Virology. 2002;304:211–221. doi: 10.1006/viro.2002.1685. [DOI] [PubMed] [Google Scholar]
- 61.Leyssen P, Balzarini J, De Clercq E, Neyts J. The predominant mechanism by which ribavirin exerts its antiviral activity in vitro against flaviviruses and paramyxoviruses is mediated by inhibition of IMP dehydrogenase. J. Virol. 2005;79:1943–1947. doi: 10.1128/JVI.79.3.1943-1947.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Khan M, Dhanwani R, Patro IK, Rao PV, Parida MM. Cellular IMPDH enzyme activity is a potential target for the inhibition of Chikungunya virus replication and virus induced apoptosis in cultured mammalian cells. Antiviral Res. 2011;89:1–8. doi: 10.1016/j.antiviral.2010.10.009. [DOI] [PubMed] [Google Scholar]
- 63.Gong ZJ. Mycophenolic acid, an immunosuppressive agent, inhibits HBV replication in vitro. J. Viral Hepat. 1999;6:229–236. doi: 10.1046/j.1365-2893.1999.00163.x. [DOI] [PubMed] [Google Scholar]
- 64.Allison AC, Eugui EM. Mycophenolate mofetil and its mechanisms of action. Immunopharmacology. 2000;47:85–118. doi: 10.1016/S0162-3109(00)00188-0. [DOI] [PubMed] [Google Scholar]
- 65.Mori K. Mechanism of action of ribavirin in a novel hepatitis C virus replication cell system. Virus Res. 2011;157:61–70. doi: 10.1016/j.virusres.2011.02.005. [DOI] [PubMed] [Google Scholar]
- 66.Pan, Q. et al. Mycophenolic acid augments interferon-stimulated gene expression and inhibits hepatitis C virus infection in vitro and in vivo. Hepatology10.1002/hep.25562. [DOI] [PubMed]
- 67.Thomas E. Ribavirin potentiates interferon action by augmenting interferon-stimulated gene induction in hepatitis C virus cell culture models. Hepatology. 2011;53:32–41. doi: 10.1002/hep.23985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pan Q, Tilanus HW, Janssen HL, van der Laan LJ. Ribavirin enhances interferon-stimulated gene transcription by activation of the interferon-stimulated response element. Hepatology. 2011;53:1400–1401. doi: 10.1002/hep.24202. [DOI] [PubMed] [Google Scholar]
- 69.Feld JJ, Hoofnagle JH. Mechanism of action of interferon and ribavirin in treatment of hepatitis C. Nature. 2005;436:967–972. doi: 10.1038/nature04082. [DOI] [PubMed] [Google Scholar]
- 70.Lee JH. Effect of ribavirin on virus load and quasispecies distribution in patients infected with hepatitis C virus. J. Hepatol. 1998;29:29–35. doi: 10.1016/S0168-8278(98)80175-X. [DOI] [PubMed] [Google Scholar]
- 71.Poynard T. Randomised trial of interferon alpha2b plus ribavirin for 48 weeks or for 24 weeks versus interferon alpha2b plus placebo for 48 weeks for treatment of chronic infection with hepatitis C virus. International Hepatitis Interventional Therapy Group (IHIT) Lancet. 1998;352:1426–1432. doi: 10.1016/S0140-6736(98)07124-4. [DOI] [PubMed] [Google Scholar]
- 72.Herrine SK. Peginterferon alpha-2a combination therapies in chronic hepatitis C patients who relapsed after or had a viral breakthrough on therapy with standard interferon alpha-2b plus ribavirin: a pilot study of efficacy and safety. Dig. Dis. Sci. 2005;50:719–726. doi: 10.1007/s10620-005-2563-3. [DOI] [PubMed] [Google Scholar]
- 73.Rostaing L, Kamar N. mTOR inhibitor/proliferation signal inhibitors: entering or leaving the field? J. Nephrol. 2010;23:133–142. [PubMed] [Google Scholar]
- 74.Dreux M, Chisari FV. Viruses and the autophagy machinery. Cell Cycle. 2010;9:1295–1307. doi: 10.4161/cc.9.7.11109. [DOI] [PubMed] [Google Scholar]
- 75.Jordan TX, Randall G. Manipulation or capitulation: virus interactions with autophagy. Microbes Infect. 2011;4:126–139. doi: 10.1016/j.micinf.2011.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ait-Goughoulte M. Hepatitis C virus genotype 1a growth and induction of autophagy. J. Virol. 2008;82:2241–2249. doi: 10.1128/JVI.02093-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sir D. Induction of incomplete autophagic response by hepatitis C virus via the unfolded protein response. Hepatology. 2008;48:1054–1061. doi: 10.1002/hep.22464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Guévin C. Autophagy protein ATG5 interacts transiently with the hepatitis C virus RNA polymerase (NS5B) early during infection. Virology. 2010;405:1–7. doi: 10.1016/j.virol.2010.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ke PY, Chen SS. Activation of the unfolded protein response and autophagy after hepatitis C virus infection suppresses innate antiviral immunity in vitro. J. Clin. Invest. 2011;121:37–56. doi: 10.1172/JCI41474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Shrivastava S, Raychoudhuri A, Steele R, Ray R, Ray RB. Knockdown of autophagy enhances the innate immune response in hepatitis C virus-infected hepatocytes. Hepatology. 2011;53:406–414. doi: 10.1002/hep.24073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kaur S. Regulatory effects of mammalian target of rapamycin-activated pathways in type I and II interferon signaling. J. Biol. Chem. 2007;282:1757–1768. doi: 10.1074/jbc.M607365200. [DOI] [PubMed] [Google Scholar]
- 82.Cao W. Toll-like receptor-mediated induction of type I interferon in plasmacytoid dendritic cells requires the rapamycin-sensitive PI(3)K-mTOR-p70S76K pathway. Nat. Immunol. 2008;9:1157–1164. doi: 10.1038/ni.1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lekmine F. Activation of the p70 S6 kinase and phosphorylation of the 4E-BP1 repressor of mRNA translation by type I interferons. J. Biol. Chem. 2003;278:27772–27780. doi: 10.1074/jbc.M301364200. [DOI] [PubMed] [Google Scholar]
- 84.Asthana S. The impact of sirolimus on hepatitis C recurrence after liver transplantation. Can. J. Gastroenterol. 2011;25:28–34. doi: 10.1155/2011/201019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wagner D. Sirolimus has a potential to influent viral recurrence in HCV positive liver transplant candidates. Int. Immunopharmacol. 2010;10:990–993. doi: 10.1016/j.intimp.2010.05.006. [DOI] [PubMed] [Google Scholar]
- 86.Selzner N, Grant DR, Shalev I, Levy GA. The immunosuppressive pipeline: meeting unmet needs in liver transplantation. Liver Transpl. 2010;16:1359–1372. doi: 10.1002/lt.22193. [DOI] [PubMed] [Google Scholar]
- 87.Kuglstatter A. Structural basis for the cyclophilin A binding affinity and immunosuppressive potency of E-ISA247 (voclosporin) Acta Crystallogr. D Biol. Crystallogr. 2011;67:119–123. doi: 10.1107/S0907444910051905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Murakami Y. Identification of bisindolylmaleimides and indolocarbazoles as inhibitors of HCV replication by tube-capture-RT-PCR. Antiviral Res. 2009;83:112–117. doi: 10.1016/j.antiviral.2009.03.008. [DOI] [PubMed] [Google Scholar]
- 89.Redig AJ. Activation of protein kinase C{eta} by type I interferons. J. Biol. Chem. 2009;284:10301–10314. doi: 10.1074/jbc.M807254200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.von Hahn T. The novel immunosuppressive protein kinase C inhibitor sotrastaurin has no pro-viral effects on the replication cycle of hepatitis B or C virus. PLoS ONE. 2011;6:e24142. doi: 10.1371/journal.pone.0024142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Quaedackers ME. Monitoring of the immunomodulatory effect of CP-690,550 by analysis of the JAK/STAT pathway in kidney transplant patients. Transplantation. 2009;88:1002–1009. doi: 10.1097/TP.0b013e3181b9ced7. [DOI] [PubMed] [Google Scholar]
- 92.Changelian PS. Prevention of organ allograft rejection by a specific Janus kinase 3 inhibitor. Science. 2003;302:875–878. doi: 10.1126/science.1087061. [DOI] [PubMed] [Google Scholar]
- 93.Williams NK. Dissecting specificity in the Janus kinases: the structures of JAK-specific inhibitors complexed to the JAK1 and JAK2 protein tyrosine kinase domains. J. Mol. Biol. 2009;387:219–232. doi: 10.1016/j.jmb.2009.01.041. [DOI] [PubMed] [Google Scholar]
- 94.Tsai YT, Chen YH, Chang DM, Chen PC, Lai JH. Janus kinase/signal transducer and activator of transcription 3 signaling pathway is crucial in chemokine production from hepatocytes infected by dengue virus. Exp. Biol. Med. (Maywood) 2011;236:1156–1165. doi: 10.1258/ebm.2011.011060. [DOI] [PubMed] [Google Scholar]
- 95.Wiesner R. A randomized double-blind comparative study of mycophenolate mofetil and azathioprine in combination with cyclosporine and corticosteroids in primary liver transplant recipients. Liver Transpl. 2001;7:442–450. doi: 10.1053/jlts.2001.23356. [DOI] [PubMed] [Google Scholar]
- 96.Jain A. A prospective randomized trial of mycophenolate mofetil in liver transplant recipients with hepatitis C. Liver Transpl. 2002;8:40–46. doi: 10.1053/jlts.2002.29763. [DOI] [PubMed] [Google Scholar]
- 97.van der Laan LJ. Results of a two-center study comparing hepatic fibrosis progression in HCV-positive liver transplant patients receiving cyclosporine or tacrolimus. Transplant. Proc. 2010;42:4573–4577. doi: 10.1016/j.transproceed.2010.10.013. [DOI] [PubMed] [Google Scholar]
- 98.Manzia TM. Long-term, maintenance MMF monotherapy improves the fibrosis progression in liver transplant recipients with recurrent hepatitis C. Transpl. Int. 2011;24:461–468. doi: 10.1111/j.1432-2277.2011.01228.x. [DOI] [PubMed] [Google Scholar]
- 99.Garg V. Effect of telaprevir on the pharmacokinetics of cyclosporine and tacrolimus. Hepatology. 2011;54:20–27. doi: 10.1002/hep.24443. [DOI] [PubMed] [Google Scholar]