

Key Points
Autoreactive B cells can be activated effectively in vitro by endogenous DNA or RNA, as well as by DNA- or RNA-binding proteins, through a mechanism that depends on the B-cell receptor and Toll-like receptor 7 (TLR7) and/or TLR9. B-cell responses to RNA-associated autoantigens are markedly increased by type I interferons (IFNs).
Similarly, it seems that TLR7 and TLR9 have important roles in the activation of plasmacytoid dendritic cells, through a mechanism that depends on the low-affinity receptor for IgG (FcγRIIa; also known as CD32), leading to the production of large amounts of type I IFNs.
Mice that inherit the Y-chromosome-linked autoimmune accelerator (Yaa) mutation express twice the normal amount of TLR7 and are hyper-responsive to TLR7 ligands. The increased amount of TLR7 might account for the role of TLR7 in the acceleration of autoimmune disease in these mice.
TLR9-deficient autoimmune-prone mice have a decreased capacity to produce antibodies specific for double-stranded DNA or nucleosomes, but they produce normal or increased amounts of antibodies specific for RNA-associated autoantigens. Nevertheless, TLR9 deficiency can lead to exacerbation of clinical disease in some animal models of systemic lupus erythematosus.
TLR7-deficient autoimmune-prone mice have a decreased capacity to produce antibodies specific for RNA-associated autoantigens. In contrast to TLR9 deficiency, TLR7 deficiency does not lead to exacerbated clinical disease.
In addition to DNA and RNA, other endogenous TLR ligands that are released from dead or dying cells or are released as a result of tissue injury have been found either to promote or to downregulate chronic inflammatory conditions.
Toll-like receptors are well known as sensors of microorganisms, but they can also sense endogenous molecules. This article describes when this might occur and how it might activate autoreactive B cells and plasmacytoid dendritic cells in systemic autoimmune disease.
Abstract
Toll-like receptors (TLRs) have a crucial role in the early detection of pathogen-associated molecular patterns and the subsequent activation of the adaptive immune response. Whether TLRs also have an important role in the recognition of endogenous ligands has been more controversial. Numerous in vitro studies have documented activation of both autoreactive B cells and plasmacytoid dendritic cells by mammalian TLR ligands. The issue of whether these in vitro observations translate to an in vivo role for TLRs in either the initiation or the progression of systemic autoimmune disease is a subject of intense research; data are beginning to emerge showing that this is the case.
Main
A remarkably high proportion of the autoantibodies that are commonly associated with systemic autoimmune diseases — such as systemic lupus erythematosus (SLE), scleroderma and Sjögren's syndrome (Box 1) — bind DNA, RNA or macromolecular complexes that contain DNA or RNA. Why these epitopes have such an important role in autoimmunity is a question that has been debated for many years. One possibility is that these intracellular autoantigens become 'visible' to the immune system when they accumulate on the plasma membrane during apoptotic cell death1; subsequent uptake and processing by activated antigen-presenting cells (APCs) then leads to a loss of tolerance. Another possibility is that apoptosis results in the cleavage of these molecules (by granzyme B, other proteases or nucleases2), thereby creating neo-epitopes that can be recognized as foreign by cells of the adaptive immune system. Neo-epitopes could also be generated by other forms of post-translational modification or by structural modifications induced by environmental factors3,4. However, data continue to accumulate in support of the idea that autoantigens are autoantigens because they are autoadjuvants: that is, they have the capacity to activate the innate immune system directly and therefore promote self-directed immune responses.
Many adjuvants stimulate the immune system through engagement of pattern-recognition receptors (PRRs). Although PRRs were initially identified as receptors that distinguish microbial molecular structures from host tissues5, it is becoming increasingly evident that many of the same PRRs are also involved in the response to injury, the clearance of apoptotic-cell debris and the repair of damaged tissues. The Toll-like receptor (TLR) family constitutes an important group of PRRs. Microbial TLR ligands include a wide range of molecules with strong adjuvant activity (such as lipopolysaccharide, lipopeptides and bacterial DNA), and these ligands can activate dendritic cells (DCs), macrophages and other APCs and allow the effective presentation of microbial antigens to cells of the adaptive immune system. However, endogenous ligands have also been identified for a substantial proportion of TLRs (Table 1). Although the data on endogenous ligands might, in some cases, be confounded by low-level contamination with microbial products6, it is clear that this is not always the case. The identification and further characterization of endogenous ligands for TLRs (as well as other PRRs) provides a novel perspective for examining the aetiology of autoimmune disease. Instead of viewing autoimmunity as an overzealous response to host antigens by the adaptive immune system, we must also consider the possibility that, at least in some cases, autoimmunity results from an overzealous response to exogenous or endogenous ligands by the innate immune system. This Review summarizes recent in vitro and in vivo studies that point to an important connection between DNA- and RNA-containing immune complexes, the production of type I interferons (IFNs; that is, IFNα and IFNβ), the activation of TLRs and subsequent events in the development and/or the progression of systemic autoimmune diseases.
Table 1.
Examples of microbial and endogenous ligands of Toll-like receptors
| TLR | Microbial ligand* | Endogenous ligand | ||||
|---|---|---|---|---|---|---|
| Autoantigen | Natural source of autoantigen | Experimental source of autoantigen | Disease association | Refs | ||
| TLR2 and/or TLR4 |
• Lipoteichoic acid (Gram-positive bacteria) • Lipoarabinomannan (Mycobacterium spp.) • Glycosylphos-phatidylinositol (Trypanosoma cruzi) • Glycolipids (Treponema pallidum) • Porins (Neisseria meningitidis) • Zymosan (fungi) • Lipopeptides, LPS and lipid A (Gramnegative bacteria) • Paclitaxel (Taxus brevifolia)‡ • F protein (RSV) • Hyphae(Aspergillus fumigatus) • HSP60 (Chlamydia trachomatis) • Envelope proteins (MMTV) • Triacyl lipopeptides (bacteria) (with TLR1)§ • Diacyl lipopeptides (Mycobacterium spp.) (with TLR6)§ |
• Necrotic cells | • Cellular injury | • Necrotic cells |
• Inflammation • Tissue repair |
111 |
| • Renal ischaemia–reperfusion injury |
• Pro-inflammatory cytokine production • Renal injury |
103 | ||||
|
• HSP60 • HSP70 • gp96 |
• Cellular injury |
• Recombinant proteins • Transgenic cell-surface proteins |
• Arthritis | 112,113 | ||
| • HMGB1 |
• Passive release from dead or damaged cells • Secretion by activated macrophages |
• Pig thymus |
• Arthritis • Chronic myositis |
114 | ||
| • Hyaluronate |
• Degradation of extracellular matrix • Synovial fluid |
• Clinical-grade sodium hyaluronate • Human umbilical cord |
• Inflammation • Arthritis |
115,116 | ||
|
• Streptococcus equi • Sera from patients with ARDS |
• Pro-inflammatory cytokine production | 106 | ||||
| • High-molecular-weight extracellular matrix | • Transgenic hyaluronate synthase 2 | • Protection from acute non-infectious lung injury | 106 | |||
| • Fibronectin (extra domain A) | • Release from cells in response to tissue damage | • Recombinant protein | • Arthritis | 117 | ||
| • Minimally modified LDL | • Atherosclerotic lesion | • Human plasma LDL exposed to 15-lipoxygenase | • Atherosclerosis | 118 | ||
| • Heparan sulphate | • Release from plasma membrane or extracellular matrix during injury or inflammation | • Bovine kidneys | • Systemic inflammatory-response syndrome | 119 | ||
| • Fibrinogen | • Leakage from vasculature to extravascular space during inflammation | • Clinical-grade fibrinogen |
• Inflammation • Arthritis |
120 | ||
|
• HSPB8 • α crystallin |
• Synovial tissue | • Recombinant proteins | • Arthritis | 121 | ||
| TLR3 | • Double-stranded RNA (viruses) | • Double-stranded RNA | • Necrotic cells | • Synovial fluid | • Arthritis | 122 |
| TLR5 | • Flagellin (bacteria) | ND | – | – | – | – |
| TLR7 and TLR8 (TLR8 in humans only) | • Single-stranded RNA (viruses) | • Single-stranded RNA |
• Dead or dying cells • Purified snRNPs |
• Apoptotic- or necrotic-cell debris • Purified snRNPs • RNA-based oligonucleotides |
• SLE • Scleroderma • Sjögren's syndrome |
17,30,32,46 |
| TLR9 | • DNA (bacteria or HSV) | • DNA | • Dead or dying cells | • Apoptotic- or necrotic-cell debris | • SLE | 27,28,45 |
| TLR11 (mice only) |
• Uropathogenic Escherichia coli • Profilin (Toxoplasma gondii) |
ND | – | – | – | – |
| *Summarized from Refs 123–126. The ligands (both microbial and endogenous) for Toll-like receptor 10 (TLR10; present in humans only), TLR12 (present in mice only) and TLR13 (present in mice only) are not known. | ||||||
| ‡Paclitaxel (Taxol; Bristol-Myer Squibb). | ||||||
| §TLR2 can form heterodimers with TLR1 or TLR6. ARDS, adult respiratory-distress syndrome; F protein, fusion protein; gp96, glycoprotein 96; HMGB1, high-mobility group box 1 protein; HSP, heat-shock protein; HSV, herpes simplex virus; LDL, low-density lipoprotein; LPS, lipopolysaccharide; MMTV, mouse mammary tumour virus; ND, not determined; RSV, respiratory syncytial virus; SLE, systemic lupus erythematosus; snRNP, small nuclear ribonucleoprotein. | ||||||
Role of IFNα in SLE and the link to TLR activation
IFNα promotes many of the clinical features of SLE. There are 13 subtypes of IFNα, and these constitute a highly pleiotropic cytokine family with diverse biological functions that are usually associated with immune responses to viral infection (reviewed in Ref. 7). However, early clinical studies revealed a correlation between aberrant expression of IFNα and SLE8. Direct evidence of a causal relationship subsequently came from the observation that repeated administration of recombinant IFNα to patients with various malignancies or chronic viral infections could lead to the production of antinuclear antibodies (ANAs) and, occasionally, to the development of clinical symptoms associated with SLE or other autoimmune diseases9,10. More recent studies that examined gene expression have identified an 'IFN signature' in most patients with active SLE11,12. IFNα can contribute to disease pathogenesis through various mechanisms, including direct and indirect effects on APCs, T cells and B cells (reviewed in Ref. 13) (Box 2).
pDCs are the main source of IFNα. It is now known that large amounts of IFNα are produced by a relatively rare, but highly efficient, class of immature DCs that were originally known as natural IFN-producing cells but are now more commonly known as plasmacytoid DCS (pDCs)14. In response to viral infection, pDCs produce IFNα and therefore have a crucial role in activating the innate immune system. Most importantly, as shown by insightful studies carried out by Lars Rönnblom and colleagues15,16, immune complexes (isolated from the sera of patients with SLE) can be an unexpectedly potent stimulus for IFNα production, especially for pDC populations that have previously been exposed to type I IFNs or other cytokines such as granulocyte/macrophage colony-stimulating factor. In addition, pDCs can be activated by purified patient-derived IgG mixed with either apoptotic- or necrotic-cell debris. Activation of pDCs by these immune complexes is sensitive to both DNase and RNase17. Because the apoptotic- and necrotic-cell debris are from mammalian cells, these experiments clearly show that endogenous nucleic acids have a key role in the activation of pDCs. Similar to other APCs, pDCs express receptors for the Fc portion of IgG (FcγRs), which can bind and internalize IgG-containing immune complexes and function as activating receptors (reviewed in Ref. 18). Immune complexes from patients with SLE cannot stimulate pDCs that have been pretreated with blocking antibodies directed against FcγRIIa (a low-affinity FcγR; also known as CD32). Moreover, in this study, heat-aggregated IgG, another ligand of FcγRIIa, fails to induce IFNα production19. Together, these data show that only certain immune complexes have IFNα-inducing activity and that FcγRIIa is required for the detection of these complexes.
pDCs express TLR7 and TLR9, which detect nucleic-acid-containing immune complexes. One reason for the efficient response of pDCs to both microbial infection and SLE-associated immune complexes is that, in addition to other PRRs, pDCs constitutively express two members of the TLR family, TLR7 and TLR9 (Ref. 20). TLR9 was originally identified as a receptor that could distinguish between bacterial (or viral) DNA and mammalian (host) DNA, on the basis of the high frequency of hypomethylated CpG motifs in non-mammalian DNA21,22 (Box 3). TLR7 was identified as a receptor for viral single-stranded RNA (ssRNA)23,24,25. In contrast to most other TLR-family members, TLR7 and TLR9 are not expressed at the plasma membrane but, instead, are sequestered in cytoplasmic compartments of the endoplasmic-reticulum–endosome–lysosome lineage26. The link between DNA-containing immune complexes, FcγRs and TLR9 has been formally established by using TLR9-deficient primary cell populations and TLR9-transfected cell lines. Chromatin-containing immune complexes (but not endotoxin-free protein-containing immune complexes) induce mouse bone-marrow-derived DCs to produce large amounts of tumour-necrosis factor, and this response was found to depend on FcγR expression (in this case, mouse FcγRIII), to be blocked by inhibitors of TLR9 and to be markedly reduced in cells from TLR9-deficient mice27. Moreover, DNA-containing immune complexes from the sera of patients with SLE were shown to stimulate the production of cytokine mRNA by HEK293 (human embryonic kidney 293) cells that had been transfected with TLR9 and FcγRIIa, as well as to colocalize with TLR9 and FcγRIIa in acidic lysosomes28. The exact compartment in which the immune-complex–FcγR and TLR pathways intersect is likely to depend on the cell type, and endosomes might be the more relevant compartment in pDCs29.
Similar to the way that DNA-containing immune complexes activate cells through TLR9, RNA-containing immune complexes activate cells through TLR7. Sera from patients with SLE or Sjögren's syndrome often have high titres of antibodies specific for small nuclear ribonucleoproteins (snRNPs), which are macromolecular complexes that consist of small nuclear RNA (snRNA) and associated proteins. Although it has been difficult to show that these sera activate pDCs directly, IgG purified from the sera of patients with SLE induces IFNa production by pDCs if the IgG is mixed with necrotic-cell debris or purified snRNPs. This IFNa-inducing activity is inhibited by chloroquine or bafilomycin, agents that interfere with the acidification of endosomes and block the activation of TLR7 and TLR9. Cytokine production is also blocked by oligodeoxynucleotide (ODN) sequences that are known to inhibit the activation of TLR7 and TLR9 (Refs 30–32) (Box 3). In addition to the immune complexes formed from cell debris and IgG from the sera of patients with SLE, better-defined RNA-containing immune complexes can be formed by combining purified snRNPs (such as U1 snRNA and its associated proteins) with monoclonal antibodies specific for one of the proteins in the particles (for example, the protein SmD). These immune complexes stimulate pDCs from wild-type (TLR7-sufficient) mice but not TLR7-deficient mice33. Together, these in vitro data strongly implicate FcγR-mediated delivery of nucleic-acid-containing immune complexes to a TLR7- or TLR9-containing compartment of pDCs as a key event in the pathogenesis of SLE (Fig. 1a). These immune complexes also activate monocytes and neutrophils28, thereby further contributing to the inflammatory processes and/or regulatory pathways that operate in systemic autoimmune disease.
Figure 1. Receptor-mediated delivery of autoantigens to Toll-like receptor 7 or Toll-like receptor 9.
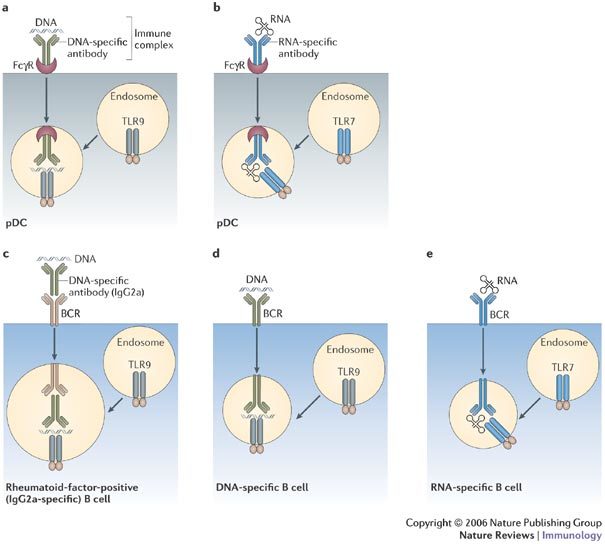
Receptors for the Fc portion of IgG (FcγRs) that are present at the surface of plasmacytoid dendritic cells (pDCs) bind immune complexes and transport both the autoantibody and the autoantigen (such as DNA or RNA) to the cytoplasmic compartment that contains Toll-like receptor 9 (TLR9) (a) or TLR7 (b). The B-cell receptor (BCR) at the surface of AM14 B cells (which are specific for IgG2a) has the same function and can transport both DNA- and RNA-containing immune complexes (c). BCRs that directly bind autoantigen, either DNA (d) or RNA (e), provide the same delivery system.
Origin of IFNα-inducing immune complexes
Microbial ligands potentially contribute to autoantibody production. For the formation of immune complexes, it is necessary to have both autoantigens and autoantibodies. As mentioned in the previous section, many of the common autoantigens are released from dead and dying cells, and might become available to the immune system as a result of excessive cell damage or an inability to appropriately clear apoptotic-cell debris (reviewed in Ref. 34). Autoantibodies are produced by self-reactive B cells, which most probably constitute a reasonably high percentage (5–20%) of the naive B-cell repertoire in most individuals35. Defects in early B-cell tolerance might lead to an even greater percentage of self-reactive B cells in patients who are prone to the development of autoimmune disease36. Several mechanisms, which are not necessarily exclusive of one another, probably account for the activation and the differentiation of these cells that are normally quiescent.
Viral or bacterial infections are frequently associated with the onset of systemic autoimmune disease, as well as with subsequent clinical flares13,37, and these infections are likely to have a pivotal role in the loss of tolerance and the ensuing production of autoantibodies. According to one model that has been proposed16, DCs and macrophages (activated through their various PRRs) upregulate co-stimulatory molecules and start to produce type I IFNs and pro-inflammatory cytokines. Autoantigens that are presented by these APCs then stimulate potentially autoreactive T cells, which in turn promote the activation, clonal expansion and differentiation of the reasonably abundant autoreactive B cells. Under the appropriate conditions, these B cells undergo somatic hypermutation of their B-cell receptor (BCR) and switch to expression of pathogenic immunoglobulin classes. The higher-affinity autoreactive B-cell clones are then selected by autoantigens that become increasingly available during the course of the infection, owing partly to the increased rate of cell death, which is caused by inflammatory cells and cytotoxic effector cells. B-cell differentiation might also be driven by the recognition of virus-infected B cells by T cells38. Excessive amounts of cell debris and autoantibodies subsequently form immune complexes, which then induce a second phase of pDC activation (which is TLR dependent) and establish a feedback loop that further exacerbates the disease process.
A second possible model links infection to the production of antibodies that react with molecular mimics of autoantigens: that is, viral epitopes that crossreact with autoantigens. One of the main examples of this is the crossreactivity between peptides derived from Epstein–Barr virus nuclear antigen 1 (EBNA1) and peptides derived from the autoantigens Ro (also known as SSA) and Sm, which are proteins that are associated with RNA-containing macromolecules39. In this model, B cells that respond to EBNA1 epitopes also recognize the corresponding autoantigens. Macromolecules (or apoptotic- and/or necrotic-cell debris) that contain these autoantigens are taken up by either activated B cells (through the BCR) or APCs (through FcγR-mediated binding of Ro- or Sm-containing immune complexes). This results in the presentation of a more extensive range of autoantigen epitopes in a milieu that can activate a broader range of autoreactive T cells, eventually leading to the phenomenon of epitope spreading and the activation of B cells that produce a more extensive autoantibody repertoire. However, only 40% of patients with SLE produce Ro- or Sm-specific antibodies, and systemic autoimmune diseases such as scleroderma are associated with autoantibodies specific for other sets of autoantigens that are associated with different subcellular macromolecular particles or organelles (such as nucleoli). So either molecular mimics that have yet to be identified account for these reactivities, or additional mechanisms are involved.
A third possibility is that a wide range of infections might activate autoreactive B cells directly through PRRs. Signals that are delivered by a PRR engaged by a microbial ligand plus a BCR engaged by an autoantigen (which separately might be sub-threshold) could combine to promote the activation of autoreactive B cells. There is a precedent for BCR and TLR synergy in this context40.
Endogenous TLR ligands have a role in autoantibody production. Despite the many potential mechanisms for the triggering of autoantibody production by microorganisms, it is also possible that certain autoantigens, independent of infection, are endogenous ligands for PRRs and have a proactive role in the loss of tolerance. Similar to pDCs, B cells express both TLR7 and TLR9. Importantly, IFNα markedly upregulates the expression of TLR7 and the TLR adaptor protein MyD88 (myeloid differentiation primary-response gene 88) in both human B cells41 and mouse B cells (A.M.R. and T. Behrens, unpublished observations) and can also increase the response of B cells to TLR9 ligands42. However, because TLR7 and TLR9 are located in cytoplasmic compartments and not at the cell surface, this route of activation is limited to molecules that can gain access to the appropriate TLR-containing compartment. One such access route is BCR-mediated endocytosis, and this access route is available not only to microorganisms but also to endogenous ligands that are recognized by an autoreactive BCR. Therefore, self antigens that, when available in excessive quantities, can effectively engage both the BCR and either TLR7 or TLR9 might stimulate autoreactive B cells that are normally quiescent, through inherent adjuvant activity. Similarly, aberrant expression or regulation of the relevant TLRs might render B cells hyper-responsive to endogenous ligands and therefore predispose an individual to the development of systemic autoimmune disease.
Supporting evidence for the idea that an endogenous ligand can be a trigger of autoimmune disease initially came from the in vitro analysis of mice transgenic for the AM14 BCR. AM14 B cells express a receptor specific for autologous IgG2a, a specificity that is commonly found in the B-cell repertoire of autoimmune-prone mice. This receptor binds monomeric IgG2a with relatively low affinity43. As a result, in mice that are not prone to autoimmunity, AM14 B cells manage to evade the common mechanisms of tolerance induction and develop into relatively normal mature B cells44. In vitro, AM14 B cells proliferate in response to immune complexes that contain IgG2a bound to DNA or DNA-associated proteins or RNA or RNA-associated proteins, but these B cells fail to respond to immune complexes of IgG2a bound to proteins. These B-cell-stimulatory immune complexes are remarkably similar to the IFNα-inducing immune complexes described earlier. Responses to both RNA- and DNA-containing immune complexes are blocked by inhibitors of TLR7 or TLR9, and B cells that are deficient in TLR7 or TLR9 respond poorly to RNA- and DNA-containing immune complexes, respectively45,46,47. The same BCR and TLR paradigm applies to BCR-transgenic B cells that react directly with DNA48. Together, these experiments strongly indicate that B cells that express low-affinity autoreactive BCRs can be activated effectively by autoantigenic ligands for TLR7 or TLR9 (Fig. 1c, d). As might be predicted by the type-I-IFN-induced upregulation of TLR7, the response to RNA-containing immune complexes was found to be markedly increased by co-culture of the B cells with IFNα or IFNβ. It follows that, during the course of an infection, in the presence of large amounts of type I IFNs, autoreactive B cells are likely to become more responsive to RNA-containing autoantigens, to produce larger amounts of the corresponding autoantibodies and therefore to further activate pDCs (Fig. 2).
Figure 2. Virus-induced interferon-α initiates a self-perpetuating feedback loop to drive autoantibody production.

a | Viral infection induces plasmacytoid dendritic cells (pDCs) to produce interferon-α (IFNα). b | IFNα upregulates expression of Toll-like receptor 7 (TLR7) by B cells, promotes cell death and increased release of certain RNA autoantigens, and primes pDCs to respond more effectively to immune complexes. c | Autoreactive B cells that have upregulated TLR7 expression bind the autoantigen that is released from apoptotic cells. d | The B-cell receptors (BCRs) of these cells deliver RNA autoantigen to TLR7, and engagement of TLR7 leads to proliferation and differentiation of these B cells. e | These B cells produce autoantibody, which combines with autoantigen to form immune complexes. f | Immune complexes bind the receptors for the Fc portion of IgG (FcγRs) at the surface of pDCs that are derived from circulating monocytes. g | The internalized autoantigen engages TLR7, and more IFNα is then produced by the pDCs. IFNαR, IFNα receptor.
Autoantigens can be autoadjuvants. Numerous studies have documented the adjuvant activity of bacterial and viral DNA, as well as DNA from other sources that is rich in hypomethylated CpG motifs21,49,50. By contrast, purified mammalian genomic DNA has remarkably poor adjuvant activity, most probably reflecting the lack of hypomethylated CpG motifs, as well as the presence of sequences that inhibit effective activation of TLR9 by agonist motifs51,52. How then do the DNA-containing immune complexes that are present in the sera of patients with systemic autoimmune disease activate pDCs effectively? Furthermore, mammalian DNA is a very weak immunogen in normal mice, so why are DNA- and chromatin-specific antibodies so common in patients and mice with autoimmune disease? One possibility is that mammalian DNA can activate TLR9 but does not usually have access to the appropriate TLR9-containing cytoplasmic compartment53. FcγR-mediated uptake could, in theory, promote delivery to the TLR9-containing compartment of pDCs, and DNA-binding B cells might simply be more prevalent in patients with SLE. It is also possible that synergistic interactions between the BCR (or FcγR) and TLR9 increase the weakly stimulatory effects of mammalian DNA40. However, the relative inability of immune complexes that consist of hapten-specific antibody and haptenated mammalian genomic DNA to activate B cells effectively compared with immune complexes that consist of hapten-specific antibody bound to haptenated bacterial DNA indicates that neither of those possibilities is likely to occur48.
Alternatively, the DNA that is present in IFNa-inducing or B-cell-stimulatory immune complexes could inherently differ from total mammalian DNA, either because patients with SLE have defects in DNA methylation54 or because segments of the mammalian genome that are enriched for hypomethylated CpG motifs (such as CpG islands or mitochondrial DNA) are preferentially released by apoptotic or necrotic cells. This possibility is consistent with a report that DNA isolated from immune complexes is enriched for CG content55. Notably, therapeutics that are associated with drug-induced lupus are often inhibitors of DNA-methyltransferase56. DNA modifications that result from inflammation and/or cell death might also increase the adjuvant activity of mammalian DNA. For example, reactive oxygen species (ROS) can lead to the oxidation of cellular components, including DNA and RNA. Higher than normal amounts of 8-hydroxy-2′-deoxyguanosine (a marker for ROS-damaged DNA) have been isolated from circulating immune complexes that are present in the sera of patients with SLE57. Moreover, the treatment of DNA with ROS can increase the immunogenicity of DNA58.
Particular features of mammalian RNA subtypes are also likely to confer adjuvant activity. Agonists for TLR7 include ssRNA that is rich in U or a combination of U and G23,24. It therefore follows that many of the snRNPs that are frequently targeted by autoantibodies consist of proteins that are bound to small UG-rich RNAs. Furthermore, RNA-based oligonucleotides that incorporate the UG-rich binding sites of these proteins can activate TLR7 and TLR8 (Ref. 30). The adjuvant activity of RNA also probably depends on the extent of post-translational modification. Mammalian ribosomal RNA is subject to much more extensive modification than bacterial ribosomal RNA, and modifications that are frequently found in mammalian RNA (such as pseudouridine and 5-methylcytidine) can interfere with the capacity of RNA-based oligonucleotides to activate TLR7 (Ref. 59). By this criterion, synthetic (unmodified) RNA that corresponds to mammalian sequences would be expected to activate TLR7 more effectively than native RNA. Therefore, it is possible that mammalian mitochondrial RNA or snRNP-associated RNAs will be found to activate TLR7 (or TLR8) preferentially, based on selective or reduced levels of modification.
One final possibility is that environmental modifications, such as oxidation, might also (in some cases) increase the adjuvant properties of mammalian RNA47. For example, ultraviolet (UV) light (such as sunlight) can trigger flares of SLE and is often associated with cutaneous SLE. UV light can also cause oxidative damage to DNA and RNA60, can generate covalent RNA–protein complexes61 and can induce apoptotic and necrotic cell death62, all of which are factors that could promote the release of potent endogenous TLR7 and TLR9 ligands.
TLRs in in vivo models of SLE
TLR deficiency affects mouse models of SLE. Evaluating the overall impact of TLR activity on the production of autoantibodies and on the development of systemic autoimmune disease requires in vivo verification of the in vitro analyses. A limited number of studies have now provided data consistent with the idea that TLR7 and TLR9 have key roles in the production of pathogenic autoantibodies and/or in the development of clinical features of autoimmunity in experimental animals (Table 2).
Table 2.
Summary of in vivo outcome of aberrant Toll-like-receptor expression on mouse models of systemic lupus erythematosus
| Mouse model* | Genetic background | Effect of deficiency or increase in Toll-like-receptor expression on autoantibody titres‡ | Renal disease | Survival | Refs |
|---|---|---|---|---|---|
| Myd88 −/− lpr/lpr | Mixed |
• HEp-2: no ANAs • Western blotting: no Sm-specific antibodies |
ND | ND | 46 |
| Tlr9 −/− lpr/lpr | Mixed or MRL |
• HEp-2: ↓ dsDNA-specific antibodies • Crithidia: ↓ dsDNA-specific antibodies • Western blotting and ELISA: ↑ Sm-specific antibodies |
Exacerbated | Decreased | 63,64 |
| MRL | • ELISA: ↑ DNA-specific antibodies | Exacerbated | Decreased | 65 | |
| C57BL/6 |
• HEp-2: ↓ dsDNA-specific antibodies; ↑ nucleolus-specific antibodies • ELISA: ↑ DNA-specific antibodies; ↓ nucleosome-specific antibodies |
Exacerbated | Decreased | 67 | |
| Tlr9 −/− Ali5 | C57BL/6 |
• HEp-2: ↓ dsDNA-specific antibodies; ↑ nucleolus-specific antibodies • ELISA: ↔ DNA-specific antibodies; ↓ nucleosome-specific antibodies |
Exacerbated | ND | 66 |
| Tlr3 −/− lpr/lpr | Mixed | • No effect | Unchanged | ND | 63 |
| Tlr7 −/− lpr/lpr | MRL (3–6 backcrosses) | • HEp-2: ↓ speckled nuclear staining by ANAs | Reduced | ND | 64 |
| Tlr7 −/− 564Igi | C57BL/6 | • ELISA: spontaneous secretion of 564 autoantibodies | ND | ND | 70 |
| Fcgr2b −/− Myd88 −/− | C57BL/6 | • ELISA: ↓ DNA-, GBM- and cardiolipin-specific IgG2a and IgG2b | Reduced | Prolonged | 68 |
| Fcgr2b −/− Myd88 −/− 56R | C57BL/6 | • ELISA: ↓ DNA-, GBM- and cardiolipin-specific IgG2a and IgG2b | ND | ND | 68 |
| Fcgr2b −/− Tlr9 −/− 56R | C57BL/6 | • ELISA: ↓ DNA-, GBM- and cardiolipin-specific IgG2a and IgG2b | ND | ND | 68 |
| Fcgr2b −/− Yaa | C57BL/6 | • HEp-2: ↑ speckled nuclear staining by ANAs; ↑ nucleolus-specific antibodies | Exacerbated | Decreased | 71 |
| sle1 Yaa | C57BL/6 | • Protein array: ↑ snRNP-specific IgG | Exacerbated | Decreased | 72 |
| *564 (564Igi mice) and 56R refer to B-cell-receptor-targeted genes that encode antibodies that react with RNA-associated or DNA-associated autoantigens, respectively. Ali5, lpr, sle1 and Yaa (Y-chromosome-linked autoimmune accelerator) are all genetic mutations. Mice with the Ali5 mutation have a gain-of-function mutation in phospholipase Cγ2. Mice that are homozygous for the lpr mutation do not express a functional form of the death receptor CD95. The Yaa mutation results from duplication and translocation of a region of the X chromosome to the Y chromosome; this region includes Toll-like receptor 7 (TLR7). | |||||
| ‡Autoantibody titres were determined by several methods: immunofluorescence (by assays using the human epithelial cell line HEp-2 or the trypanosome Crithidia luciliae), western-blotting analysis, enzyme-linked immunosorbent assay (ELISA) or protein array. ANA, antinuclear antibody; dsDNA, double-stranded DNA; Fcgr2b, low-affinity Fc receptor for IgG; GBM, glomerular basement membrane; Myd88, myeloid differentiation primary-response gene 88; ND, not determined; snRNP, small nuclear ribonucleoprotein. | |||||
Mice that are homozygous for the lpr mutation do not express a functional form of the death receptor CD95 (also known as FAS), and they develop a lymphoproliferative disease that is similar to Canale-Smith syndrome in humans. The disease is associated with the production of antibodies specific for double-stranded DNA (dsDNA), chromatin and snRNPs, as determined by the strong and homogeneous nuclear-staining pattern of HEp-2 cells. By contrast, lpr/lpr mice deficient in the TLR adaptor protein MyD88 do not produce ANAs (even as they age), and these mice develop marked lymphoproliferative disease. Sera from about one-third of lpr/lpr mice also have antibodies specific for SmD or SmB, whereas sera from MyD88-deficient lpr/lprmice show only negligible activity against these RNA-associated proteins46.
Mice with the lpr mutation have also been bred with mice that lack TLR9. These TLR9-deficient lpr/lpr mice have a much more selective defect in autoantibody production than do the MyD88-deficient lpr/lpr mice. Although they still produce ANAs, sera from TLR9-deficient lpr/lpr mice result in a speckled, rather than a homogeneous, nuclear-staining pattern of HEp-2 cells. In addition, these sera fail to stain the mitotic plates of HEp-2 cells or the kinetoplasts of trypanosomes (such as Crithidia luciliae), criteria that are used to identify antibodies that react with dsDNA. It should be noted that serum samples from about one-half of TLR9-deficient lpr/lpr mice show strong cytoplasmic reactivity, but this reactivity is relatively rare among lpr/lpr mice that are sufficient in TLR9 (Ref. 63). Overall, these staining data are best interpreted as a failure to make dsDNA- or chromatin-specific antibodies while continuing to produce antibodies specific for cytoplasmic and nuclear RNPs. These results are completely consistent with the in vitro data (summarized earlier) that predicted that the response to DNA and DNA-associated proteins would be TLR9 dependent, whereas the response to RNA and RNA-associated proteins would be TLR7 dependent. However, despite the absence of dsDNA-specific antibodies, TLR9-deficient lpr/lpr mice and TLR9-deficient mice with the Ali5 mutation (which have a gain-of-function mutation in phospholipase-Cγ2 and are another mouse model of SLE) still make antibodies that react with plate-bound DNA (probably single-stranded DNA) in enzyme-linked immunosorbent assays. Remarkably, renal disease in TLR9-deficient autoimmune-prone mice was significantly worse than in their TLR9-sufficient litter-mates64,65,66,67. It remains to be determined whether this reflects a role for TLR9 in the clearance of cell debris, an increase in the pathogenicity of RNA-containing immune complexes, differential expression of TLR7 or TLR9 by a regulatory-cell population or another mechanism.
The effect of TLR9 deficiency has also been examined in the context of another mouse model of SLE, in which C57BL/6 mice fail to express the inhibitory receptor FcγRIIb. In this case, TLR9-deficient autoreactive B cells do not undergo class switching to the pathogenic immunoglobulin isotypes IgG2a and IgG2b68. This is presumably a result of the absence of TLR9-mediated induction of expression of the transcription factor T-bet, which directly promotes switching to IgG2a69. As a consequence, these mice have significantly smaller deposits of IgG in the glomeruli and survive considerably longer than their TLR9-sufficient litter-mates. Together, these studies confirm that B-cell expression of TLR9 has an important role in promoting the antibody response to DNA and DNA-binding proteins, such as histones. Moreover, in at least one mouse model of SLE, the absence of functional TLR9 has a marked effect on disease outcome.
TLR7 deficiency can also influence autoantibody production. C57BL/6 mice that inherit site-directed insertions of the immunoglobulin heavy- and light-chain variable regions encoding the antibody 564 (denoted 564Igi mice) spontaneously produce autoantibodies that react with ssRNA and stain the cytoplasm and nucleoli of HEp-2 cells. These RNA-reactive antibodies are not produced by TLR7-deficient 564Igi mice70. Moreover, TLR7-deficient lpr/lpr mice do not make antibodies that react with RNA-associated autoantigens, and they develop less severe clinical disease than do their TLR7-sufficient litter-mates64. Therefore, TLR7 can have a key role in the activation of autoreactive B cells specific for RNA-associated autoantigens.
The Yaa mutation and TLR7. The Yaa (Y-chromosome-linked autoimmune accelerator) mutation occurred during the generation of the BXSB strain of autoimmune-prone mice. Male BXSB mice develop a severe form of SLE with a much higher incidence than their female counterparts. Recent studies have shown that the phenotype of mice with the Yaa mutation results from translocation of a 4-megabase portion of the X chromosome to the Y chromosome, leading to a twofold increase in the expression of several genes that are normally X linked, including TLR7 (Refs 71,72). B6.SB-Yaa mice (that is, C57BL/6 mice that have been crossed onto BXSB mice, which have the Yaa mutation) do not develop autoimmune disease and make only very small amounts of IgM ANAs. However, FcγRIIb-deficient B6.SB-Yaa mice have a markedly different phenotype from either of the parent strains (that is, B6.SB-Yaa mice and FcγRIIb-deficient C57BL/6 mice). The cumulative mortality of the mice at 6 months increased from 0% for B6.SB-Yaa mice and 15–20% for C57BL/6 mice deficient in FcγRIIb to more than 80% for the FcγRIIb-deficient B6.SB-Yaa mice. This markedly decreased survival rate is associated with more severe proteinuria and renal disease. Importantly, the mainly DNA- and chromatin-specific antibody repertoire of C57BL/6 mice deficient in FcγRIIb (as evidenced by homogeneous nuclear staining of HEp-2 cells) shifts to a mainly nucleolus-specific antibody repertoire (as evidenced by speckled nuclear staining) in the FcγRIIb-deficient B6.SB-Yaa mice71,73.
Similar results were obtained when B6.SB-Yaa mice were crossed onto the moderately autoimmune-prone strain of mice B6.NZM-sle1 (C57BL/6 mice that have been crossed onto NZM2410 mice, which have a mutation in the sle1 locus). B6.NZM-sle1 mice that are 4–6 months of age make reasonably large amounts of IgG specific for DNA and chromatin, but these mice do not develop clinical nephritis. By contrast, both B6.SB-Yaa mice and B6.SB-Yaa mice with the sle1 mutation begin to produce large amounts of IgM autoantibodies by 6–8 weeks of age, and most of these antibodies are specific for nuclear and cytoplasmic RNA-associated autoantigens. The B6.SB-Yaa mice with the sle1 mutation subsequently develop high titres of IgG specific for RNPs, and this shift is associated with the onset of renal disease and with decreased rates of survival. (Cumulative mortality at 10 months is 0% and 60% for B6.SB-Yaa mice and B6.SB-Yaa mice with the sle1 mutation, respectively72,73.)
Considering the specificity of TLR7 for ssRNA, it is highly probable that the RNA-focused antibody response results from autoantigen engagement of TLR7. In fact, in vivo administration of a TLR7 ligand was sufficient to divert the autoantibody repertoire of C57BL/6 mice deficient in FcγRIIb from a homogeneous to a speckled nuclear-staining pattern independently of the Yaa mutation71. Whether the more severe clinical features of the mice that also have the Yaa mutation are the direct outcome of increased TLR7 expression remains to be determined. Other genes in the translocated interval might prove to be important factors in disease pathogenesis. However, it is intriguing that recent clinical data indicate a strong correlation between IFN-activated genes, SLE disease activity and the presence of antibodies specific for autoantigen-associated RNPs74.
TLR7 and TLR9 inhibitors affect mouse models of SLE. Another approach for evaluating the importance of TLR7 and TLR9 ligands in the development of systemic autoimmune disease is to assess the efficacy of therapeutically administered inhibitors. At present, potential TLR7 and TLR9 inhibitors fall into two main categories: antimalarial agents and inhibitory oligonucleotides75. Antimalarial agents have been used to treat SLE for more than a century, and hydroxychloroquine (Plaquenil) is still considered to be an effective treatment for cutaneous SLE and for SLE-associated polyarthralgia, pleuritis and pericarditis (reviewed in Ref. 76). A particularly efficacious formulation (containing chloroquine, hydroxychloroquine and quinacrine) was described in 1959 (Ref. 77) but was withdrawn from the market in 1972 because of general concerns regarding combination therapies. The proposed mechanisms of action of the antimalarial agents are wide ranging and include absorption of UV light, general anti-inflammatory and antiproliferative effects, dissolution of immune complexes and inhibition of various activities that are associated with immune function (such as cytokine release). Of particular relevance here, chloroquine and related compounds can block TLR9 (and TLR7) activation effectively, presumably by preventing the acidification and subsequent maturation of endosomes78,79,80, and these compounds can inhibit the activation of both B cells and myeloid and plasmacytoid DCs that is mediated by DNA- or RNA-containing immune complexes27,32,45,46. Whether the standard clinical regimen of administration of hydroxychloroquine maintains the drug in an amount that is sufficient to block TLR7 and TLR9 signalling in patients remains to be determined.
ODNs that incorporate hypomethylated CpG motifs (such as GACGTT), which are stimulatory, are potent experimental ligands for TLR9 (Ref. 81 (Box 3). Certain other ODN sequences not only fail to activate either B cells or pDCs but inhibit the agonist activity of such stimulatory motifs. Inhibitory sequences were originally discovered in adenoviral vectors82, and specific inhibitory motifs were subsequently identified by functional analyses of sequences related to a stimulatory motif83. Sequences that incorporate either CCTGGCGGGG or CCTGGAGGGG are usually potent inhibitors. These inhibitory ODNs (also known as immunoregulatory sequences) not only block stimulatory ODN-mediated activation of B cells and pDCs but also block the agonist activity of both DNA- and RNA-containing immune complexes31,46. In the case of RNA-containing immune complexes, the activity of inhibitory ODNs was found to be independent of TLR9: inhibitory ODNs blocked the activation of both wild-type and TLR9-deficient B cells by RNA-containing immune complexes46. These data indicate that inhibitory ODNs can antagonize both TLR7 and TLR9 directly, even though ODNs have a DNA backbone. Notably, administration of these ODNs significantly reduces the severity of clinical features of SLE in the autoimmune-prone mouse strains MRL-lpr and (NZB × NZW)F1 (Ref. 84 and F. Barrat, unpublished observations).
Another class of inhibitory ODNs consists of multimers of a repetitive element, TTAGGG, that are present at high frequency in mammalian telomeres52. These inhibitors directly block the engagement of TLR9 by stimulatory ODNs in vitro and in vivo, but they do not inhibit in vitro responses to lipopolysaccharide, which are mediated though TLR4. However, these inhibitors can also bind signal transducer and activator of transcription 1 (STAT1) and STAT4 and therefore block signalling cascades that are triggered by cytokines such as IFNγ or interleukin-12. In addition, bimonthly administration of telomere-derived ODNs to (NZB × NZW)F1 mice, starting at 6 weeks of age, both delayed the onset of renal disease and prolonged survival85. Whether these effects are mainly due to inhibition of TLR9 or to inhibition of STAT1 and/or STAT4 remains to be determined. Overall, however, there is clear evidence that blockade of TLR7 and/or TLR9 might be a useful therapeutic strategy for the treatment of SLE and related systemic autoimmune diseases.
Genetic studies of humans provide evidence. SLE stems from a complex genetic trait: that is, several genetically determined factors contribute to disease susceptibility. Because genes that predispose individuals to autoimmunity are likely to promote key events in pathogenesis, several groups have recently focused on single-nucleotide polymorphisms (SNPs) that are associated with the type-I-IFN-signalling pathway. IFN-regulatory factor 5 (IRF5) is constitutively expressed by B cells and pDCs and has a crucial role in the TLR-induced transcription of pro-inflammatory cytokines86. In human cells, IRF5 regulates the expression of type I IFNs downstream of TLR7 but not TLR3 (Ref. 87). Remarkably, studies of five separate patient cohorts have now identified at least two SNPs associated with IRF5 as being high risk factors for SLE88,89. Transcriptional regulation of IRF5 is a complex process that involves 3 promoters and at least 11 mRNA isoforms. One of these SLE-associated SNPs allows the expression of IRF5 mRNA isoforms initiated by the promoter for exon 1B. The other SNP promotes increased expression of IRF5. Whether the exon-1B-promoter-driven isoforms have unique functional properties or increased stability remains to be determined. Despite the lack of a clear-cut mechanistic explanation, the correlation between increased IRF5 expression and SLE susceptibility further connects TLR-signalling events to SLE.
Role of TLRs in other autoimmune diseases
If endogenous DNA and RNA contribute to the development of systemic autoimmune disease, then are there endogenous ligands for other TLR-family members? And, if these ligands exist, what is their role in health and disease? It is reasonable to assume that the association between infection and autoimmunity is often caused by TLR-mediated induction of pro-inflammatory cytokine and chemokine expression and upregulation of co-stimulatory molecule expression by APCs. Numerous experimental systems have now documented the ability of microbial TLR ligands to trigger disease onset in experimental models of arthritis90,91, multiple sclerosis (experimental allergic encephalomyelitis (EAE), in mice)92,93,94, myocarditis95, diabetes96 and atherosclerosis97. Whether endogenous TLR ligands contribute markedly to the onset or perpetuation of these diseases is less clear-cut. The following endogenous molecules have all been reported to stimulate TLR2 and/or TLR4: breakdown products of the extracellular matrix, such as hyaluronate and heparan sulphate; molecules that have been released from dead or damaged cells, such as high-mobility group box 1 protein (HMGB1), fibronectin and heat-shock proteins; fibrinogen; and modified low-density lipoprotein (Table 1). Many of these molecules accumulate in the joints of patients with rheumatoid arthritis and at other sites of inflammation. The precise mechanism by which TLR2 and TLR4 recognize such a wide range of molecular structures, and the extent of redundancy between TLR2 and TLR4 in the detection of these molecules at sites of cell injury or inflammation, is unresolved.
Linkage studies in patients have not yet found significant correlations between TLR2 or TLR4 activity and human autoimmune disease. A variant of TLR4 with reduced function has been shown to confer reduced risk of developing atherosclerosis98 but not arthritis99; however, in this case, risk outcome could reflect activity of either exogenous or endogenous ligands.
Adoptive-transfer studies carried out in mouse models of arthritis and mice with EAE indicate that endogenous TLR ligands might contribute to the pathogenesis of related autoimmune diseases. K/B×N mice develop spontaneous arthritis that is associated with the production of large amounts of antibody specific for glucose-6-phosphate isomerase. Serum from these mice is sufficient to transfer arthritis to naive recipients. Transfer of serum to TLR4-deficient mice induces joint swelling that resolves more quickly than in TLR4-sufficient (control) mice, indicating that endogenous TLR4 ligands have a role in the perpetuation of disease100. Similarly, injection of mice with the antigen myelin oligodendrocyte glycoprotein in complete Freund's adjuvant results in the development of EAE, and T cells from these mice can transfer disease to naive recipients. Unexpectedly, the transfer of these T cells to MyD88-deficient recipients led to only minimal disease, and TLR9-deficient recipients had a much-attenuated clinical score compared with TLR-sufficient (control) mice101. These data could be explained, in part, by in vitro studies showing that microglia express TLR9 and respond to CpG motifs by the production of pro-inflammatory mediators102; presumably, the ligand in the central nervous system is derived from cells that are damaged by the pathogenic effector T cells.
Perhaps the most convincing in vivo data for physiologically relevant endogenous ligands of TLR2 and TLR4 come from experimental models of tissue injury. In an in vivo model of ischaemia and reperfusion damage to the kidney, TLR2-deficient mice that were subjected to occlusion of the renal arteries produced significantly less pro-inflammatory cytokines and chemokines, showed less leukocyte infiltration and developed less severe renal injury than did TLR-sufficient (control) groups of mice103. A similar role for TLR2 and TLR4 was found in studies of mouse models of myocardial ischaemia and reperfusion104,105; in these studies, mice deficient in TLR2 or TLR4 produced less pro-inflammatory cytokines and developed less severe pathology. Possible TLR ligands include products released from necrotic cells, such as heat-shock proteins and HMGB1. However, TLR-mediated cytokine production is not always detrimental to the host. In a bleomycin-induced lung-injury model, alveolar cells from mice deficient in both TLR2 and TLR4 produced less pro-inflammatory cytokines than did TLR-sufficient (control) cells, but the TLR-deficient mice had a lower survival rate than the TLR-sufficient mice106. The mice deficient in both TLR2 and TLR4 were also more sensitive to hypoxia-induced lung injury and had a much lower survival rate than did wild-type mice. It seems that low-molecular-weight fragments of hyaluronate, which are released from the extracellular matrix as a result of injury-associated degradation, can provoke inflammatory responses. By contrast, intact (high molecular weight) hyaluronate has a protective effect, which is also mediated through engagement of TLR2 and TLR4 (Ref. 106). It will be interesting to see whether other endogenous ligands have the same capacity to tilt the immunological balance from tissue injury to tissue repair.
Concluding remarks
Experimental evidence continues to support a role for TLR7 and TLR9 in the development of systemic autoimmune diseases; however, many questions remain unanswered. In addition to the different forms of SLE, diseases such as scleroderma and Sjögren's syndrome are associated with a characteristic set of ANAs and RNP-specific antibodies107. So how does autoantibody specificity relate to disease mechanisms? And does activation of TLRs contribute to the pathogenesis of systemic autoimmune diseases that are IFNα dependent to different extents? TLR9 deficiency has a large impact on the clinical phenotype of B6.NZM-sle1 mice but only exacerbates the clinical parameters of CD95-deficient mice. Given the many genetic and environmental factors that are likely to contribute to SLE in humans, will the contribution of TLR9 (or TLR7) to disease activity in humans be equally variable? Most likely, it will be necessary to evaluate several models of spontaneous SLE to appreciate the overall impact of deficiency in TLR7, TLR9 or both TLR7 and TLR9 on autoantibody production, as well as on various other clinical parameters. Will blockade of TLR7 and TLR9 prevent and/or ameliorate the clinical features of SLE and related diseases? And, if so, to what extent would such therapeutics compromise the protective capacity of the immune system?
Although it is beyond the scope of this Review, non-TLR sensors of nucleic acids such as PKR (IFN-inducible double-stranded-RNA-dependent protein kinase) and RIG-I (retinoic-acid-inducible gene I) might also recognize endogenous ligands and trigger the production of pro-inflammatory cytokines. From a broader perspective, endogenous ligands for other TLRs (and other PRRs) probably have a crucial role in sensing and repairing various forms of tissue injury. Although we are just beginning to understand how, where and when TLRs recognize self components, appropriate manipulation of these components of the innate immune system might eventually provide the means to treat a broad range of chronic inflammatory conditions.
Box 1 | Common systemic autoimmune diseases: clinical symptoms and immunological features.
Systemic lupus erythematosus (SLE) is a complex chronic inflammatory disease that arises spontaneously and can affect the skin, joints, kidneys, lungs, nervous system, vasculature, serous membranes and other organs. The effector mechanisms of inflammation depend on lymphocytes, neutrophils, monocytes, platelets and mast cells and can involve the complement, kinin and coagulation cascades. In individuals who are genetically predisposed to develop SLE, factors that have been associated with the initiation of symptoms and the subsequent disease flares include infectious agents, stress, toxins and physical agents (such as sunlight). Patients present with a wide range of clinical manifestations. Cutaneous lesions are found in 80–90% of patients and include the following: a characteristic rash across the cheeks (also known as a butterfly rash), which recurs; and discoid lesions, which are more chronic. Other symptoms can include one or more of the following: fatigue, painful joints, nephritis, neuropsychiatric abnormalities, pericarditis and pleuritis. Morbidity and mortality are associated with severe renal disease and accelerated atherosclerosis. SLE is considered to be a systemic autoimmune disease because of the prevalence of antibodies that react with self components in the nucleus and cytoplasm, often macromolecular complexes of proteins and nucleic acids. The autoantibodies form immune complexes that can accumulate in the kidneys and other tissues and induce inflammation. Some autoantibodies might also bind directly to epitopes in the kidneys or the central nervous system, with pathological consequences.
Other examples of chronic inflammatory diseases with an autoimmune aetiology are scleroderma (systemic sclerosis) and Sjögren's syndrome. Clinical features of scleroderma include the following: progressive thickening and hardening of the skin; vascular disease, which manifests particularly in the hands (Raynaud's syndrome); and possibly also inflammation and fibrosis of internal organs such as the heart, lungs and kidneys. Sjögren's syndrome is most commonly associated with lymphocyte infiltration of the lacrimal and salivary glands, resulting in dry eyes and a dry mouth; however, lymphocyte infiltration might extend to the skin, lungs, heart, kidneys and nervous system. Both scleroderma and Sjögren's disease are frequently associated with the production of antinuclear antibodies. Intriguingly, SLE, scleroderma and Sjögren's syndrome are each associated with particular autoantibody reactivities: SLE, with antibodies that react with double-stranded DNA, nucleosomes and the protein Sm; scleroderma, with antibodies that react with topoisomerase I, fibrillarin and centromeres; and Sjögren's syndrome, with antibodies that react with the RNA-binding proteins Ro (also known as SSA) and La (also known as SSB).
Box 2 | Mechanisms by which interferon-α promotes SLE.
There are 13 subtypes of interferon-α (IFNα), and these are encoded in a cluster on chromosome 9 and signal through the type I IFN receptor. IFNα has a broad set of functions. Many of its effects probably contribute to the initiation and the perpetuation of systemic autoimmune diseases such as systemic lupus erythematosus (SLE), Sjögren's syndrome and scleroderma. For example, IFNα promotes the differentiation of monocytes into myeloid dendritic cells (DCs) that are highly efficient at antigen presentation, that express large amounts of co-stimulatory molecules, MHC class I and class II molecules, and transporter associated with antigen processing (TAP) proteins, and that produce T- and B-cell survival factors. Mature DCs can then activate autoreactive T helper cells and cytotoxic effector (CD8+) T cells. IFNα can also contribute to the availability of autoantigen in the following ways: by increasing the sensitivity of target cells to cytotoxic effector mechanisms; by increasing the cytotoxic activity of CD8+ T cells and natural killer cells, through upregulation of expression of granzyme B, CD95 ligand and TRAIL (tumour-necrosis-factor-related apoptosis-inducing ligand); and by increasing the expression of common autoantigens, such as Ro and lamin 1b. Direct effects of IFNα on B cells include promoting plasmablast and/or plasma-cell differentiation, promoting switching to the pathogenic immunoglobulin isotype IgG2a, and upregulating expression of Toll-like receptor 7 (TLR7) and the TLR adaptor protein MyD88 (myeloid differentiation primary-response gene 88). Importantly, IFNα also influences the functional properties of plasmacytoid DCs directly, increasing the expression of IFN-regulatory factor 7 and thereby increasing the responsiveness of plasmacytoid DCs to RNA- and/or DNA-containing immune complexes. The expression of many of the genes that are induced by IFNα is higher in patients with SLE, and this gene-expression pattern, as determined by DNA-microarray analysis, is referred to as the IFN signature.
Box 3 | Stimulatory versus inhibitory oligodeoxynucleotides.
Synthetic oligodeoxynucleotides (ODNs) stabilized by a phosphorothioate backbone (which is nuclease resistant) have been used experimentally as adjuvant mimics of bacterial DNA.
Stimulatory ODNs incorporate a hypomethylated CpG motif, or an unmethylated CG dinucleotide, in a particular base context that recapitulates short sequences that are commonly found in bacterial DNA but not mammalian DNA. The optimal motif for stimulation of mouse cells is GACGTT, and GTCGTT is the optimal motif for stimulation of human cells108. ODNs in which the entire backbone is phosphorothioate linked are sometimes referred to as type B ODNs (also known as type K ODNs) and mainly stimulate B cells and monocytes. ODNs with a mixed phosphodiester–phosphorothioate backbone can also be synthesized; in this case, the CpG motif is flanked by self-complementary bases to form a stem–loop structure that is capped at the 3′ end by a poly(G) tail. These ODNs can be referred to as type A ODNs (also known as type D ODNs) and are particularly effective inducers of interferon-α production by plasmacytoid dendritic cells (pDCs)109. Type C ODNs contain structured elements of both type A ODNs amd type B ODNs, and stimulate both B cells and pDCs effectively110.
ODNs that have inhibitory activity have also been synthesized, and these ODNs can block the activation of Toll-like receptor 7 (TLR7) and/or TLR9 specifically, with minimal effects on other TLR-family members. One type of inhibitory ODN is constructed by replacing the 3′ bases downstream of the CG dinucleotide with a GGG trinucleotide and by incorporating a CCT trinucleotide upstream83. The CCTNNNGGGG motif (where N denotes an unspecified base) is also shared by other inhibitory sequences. Another type of inhibitory ODN consists of repeats of the TTAGGG hexanucleotide, which is commonly found in mammalian telomeres52. Some examples of commonly used ODNs are shown in the table.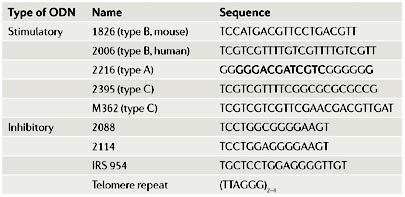
Acknowledgements
The author thanks past and present members of her laboratory for scientific contributions that helped to develop the concepts outlined in this Review. Studies from the author's laboratory have been supported by the National Institutes of Health (United States).
Glossary
- Tolerance
Non-responsiveness of lymphocytes to antigen. This is an active process not a passive lack of response.
- Plasmacytoid DC
(pDC). A dendritic cell (DC) that lacks myeloid markers such as CD11c and CD33 but expresses large amounts of the cell-surface markers HLA-DR and CD123. After activation — for example, when stimulated through Toll-like receptors — these cells produce large amounts of interferon-α.
- Hypomethylated CpG motif
An unmethylated CG dinucleotide in a particular base context. These motifs have been found to induce innate immune responses through interaction with Toll-like receptor 9.
- Drug-induced lupus
The development of systemic lupus erythematosus (SLE)-like symptoms following exposure to drugs such as procainamide or hydralazine. Patients with drug-induced lupus commonly develop high titres of antinuclear antibodies, and this form of SLE is also associated with malaise and polyarthralgia, and sometimes with arthritis, pleuritis or pericarditis. Clinical features tend to resolve after treatment with the drug ceases.
- HEp-2 cells
A human epithelial cell line that is commonly used as a target for immunofluorescent detection of a wide range of nuclear- and cytoplasmic-staining antibodies. Distinct staining patterns are associated with particular antibody specificities. For example, a homogeneous nuclear-staining pattern is indicative of antibodies that react with double-stranded DNA or chromatin, whereas a speckled nuclear-staining pattern is indicative of antibodies that react with small nuclear ribonucleoproteins.
- Mitotic plates
The aligned metaphase chromosomes before cell division.
- Kinetoplast
A DNA-containing organelle of trypanosomes, usually found in an elongated mitochondrion that is located adjacent to the basal body.
- sle1
The systemic lupus erythematosus 1 (sle1) locus encompasses several well-studied SLE-susceptibility genes, including members of the SLAM (signalling lymphocytic activation molecule) family of co-stimulatory molecules.
- (NZB × NZW)F1
The F1 offspring of a cross between NZB and NZW mice develop many of the symptoms of human systemic lupus erythematosus (SLE) and are commonly used as an animal model of SLE.
- Single-nucleotide polymorphism
(SNP). A SNP indicates a particular site in the genome where different bases could be present. In general, alleles of any polymorphism are present at a frequency of 1% or greater in the human genome.
- K/B×N mice
A mouse strain that was formed by crossing non-obese diabetic (NOD)/Lt mice with KRN T-cell-receptor-transgenic mice on a C57BL/6 background. In these mice, T cells recognize a peptide from the autoantigen glucose-6-phosphate isomerase (GPI). The mice develop arthritis that is mediated, and transferable, by circulating antibody specific for GPI.
- Microglial cell
A macrophage-lineage cell that is derived from bone marrow and is present in the central nervous system.
Biography
Ann Marshak-Rothstein received a Ph.D. from the University of Pennsylvania (Philadelphia, Pennsylvania, USA) and did postdoctoral research at Massachusetts Institute of Technology (Cambridge, Massachusetts, USA), before becoming a faculty member at Boston University School of Medicine (Boston, Massachusetts). Her main research interests in recent years have been factors that regulate the activation, function, survival and apoptosis of T cells and B cells, especially in animal models of systemic autoimmune disease. Ongoing studies include evaluation of the pro-apoptotic and pro-inflammatory activities of CD95 ligand in models of chronic inflammation, as well as assessment of the role of endogenous ligands of Toll-like receptors in the activation of autoreactive B cells.
Related links
FURTHER INFORMATION
Boston University School of Medicine Immunology Training Program
Competing interests
US patent application 10/487,885 (entitled Method and Composition for Treating Immune Complex Associated Disorders) and corresponding foreign applications have been licensed and provide royalty income to Ann Marshak-Rothstein.
References
- 1.Casciola-Rosen LA, Anhalt G, Rosen A. Autoantigens targeted in systemic lupus erythematosus are clustered in two populations of surface structures on apoptotic keratinocytes. J. Exp. Med. 1994;179:1317–1330. doi: 10.1084/jem.179.4.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rosen A, Casciola-Rosen L. Autoantigens as substrates for apoptotic proteases: implications for the pathogenesis of systemic autoimmune disease. Cell Death Differ. 1999;6:6–12. doi: 10.1038/sj.cdd.4400460. [DOI] [PubMed] [Google Scholar]
- 3.Utz PJ, Gensler TJ, Anderson P. Death, autoantigen modifications, and tolerance. Arthritis Res. 2000;2:101–114. doi: 10.1186/ar75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Plotz PH. The autoantibody repertoire: searching for order. Nature Rev. Immunol. 2003;3:73–78. doi: 10.1038/nri976. [DOI] [PubMed] [Google Scholar]
- 5.Medzhitov R, Janeway CA., Jr Innate immune recognition: mechanisms and pathways. Immunol. Rev. 2000;173:89–97. doi: 10.1034/j.1600-065X.2000.917309.x. [DOI] [PubMed] [Google Scholar]
- 6.Tsan M-F, Gao B. Endogenous ligands of Toll-like receptors. J. Leukoc. Biol. 2004;76:514–519. doi: 10.1189/jlb.0304127. [DOI] [PubMed] [Google Scholar]
- 7.Theofilopoulos AN, Baccala R, Beutler B, Kono DH. Type I interferons (α/β) in immunity and autoimmunity. Annu. Rev. Immunol. 2005;23:307–336. doi: 10.1146/annurev.immunol.23.021704.115843. [DOI] [PubMed] [Google Scholar]
- 8.Preble OT, Black RJ, Friedman RM, Klippel JH, Vilcek J. Systemic lupus erythematosus: presence in human serum of an unusual acid-labile leukocyte interferon. Science. 1982;216:429–431. doi: 10.1126/science.6176024. [DOI] [PubMed] [Google Scholar]
- 9.Rönnblom LE, Alm GV, Oberg KE. Possible induction of systemic lupus erythematosus by interferon-α treatment in a patient with a malignant carcinoid tumour. J. Intern. Med. 1990;227:207–210. doi: 10.1111/j.1365-2796.1990.tb00144.x. [DOI] [PubMed] [Google Scholar]
- 10.Gota C, Calabrese L. Induction of clinical autoimmune disease by therapeutic interferon-α. Autoimmunity. 2003;36:511–518. doi: 10.1080/08916930310001605873. [DOI] [PubMed] [Google Scholar]
- 11.Baechler EC, et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc. Natl Acad. Sci. USA. 2003;100:2610–2615. doi: 10.1073/pnas.0337679100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bennett L, et al. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J. Exp. Med. 2003;197:711–723. doi: 10.1084/jem.20021553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Banchereau J, Pascual V, Palucka AK. Autoimmunity through cytokine-induced dendritic cell activation. Immunity. 2004;20:539–550. doi: 10.1016/S1074-7613(04)00108-6. [DOI] [PubMed] [Google Scholar]
- 14.Rönnblom L, Alm GV. A pivotal role for the natural interferon α-producing cells (plasmacytoid dendritic cells) in the pathogenesis of lupus. J. Exp. Med. 2001;194:F59–F63. doi: 10.1084/jem.194.12.f59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vallin H, Blomberg S, Alm GV, Cederblad B, Rönnblom L. Patients with systemic lupus erythematosus (SLE) have a circulating inducer of interferon-α (IFN-α) production acting on leucocytes resembling immature dendritic cells. Clin. Exp. Immunol. 1999;115:196–202. doi: 10.1046/j.1365-2249.1999.00772.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rönnblom L, Alm GV. An etiopathogenic role for the type I IFN system in SLE. Trends Immunol. 2001;22:427–431. doi: 10.1016/S1471-4906(01)01955-X. [DOI] [PubMed] [Google Scholar]
- 17.Lovgren T, Eloranta ML, Bave U, Alm GV, Rönnblom L. Induction of interferon-α production in plasmacytoid dendritic cells by immune complexes containing nucleic acid released by necrotic or late apoptotic cells and lupus IgG. Arthritis Rheum. 2004;50:1861–1872. doi: 10.1002/art.20254. [DOI] [PubMed] [Google Scholar]
- 18.Amigorena S, Bonnerot C. Role of B-cell and Fc receptors in the selection of T cell epitopes. Curr. Opin. Immunol. 1998;10:88–92. doi: 10.1016/S0952-7915(98)80037-X. [DOI] [PubMed] [Google Scholar]
- 19.Bave U, et al. FcγRIIa is expressed on natural IFN-α-producing cells (plasmacytoid dendritic cells) and is required for the IFN-α production induced by apoptotic cells combined with lupus IgG. J. Immunol. 2003;171:3296–3302. doi: 10.4049/jimmunol.171.6.3296. [DOI] [PubMed] [Google Scholar]
- 20.Kadowaki N, et al. Subsets of human dendritic cell precursors express different Toll-like receptors and respond to different microbial antigens. J. Exp. Med. 2001;194:863–869. doi: 10.1084/jem.194.6.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Krieg AM, et al. CpG motifs in bacterial DNA trigger direct B-cell activation. Nature. 1995;374:546–549. doi: 10.1038/374546a0. [DOI] [PubMed] [Google Scholar]
- 22.Hemmi H, et al. A Toll-like receptor recognizes bacterial DNA. Nature. 2000;408:740–745. doi: 10.1038/35047123. [DOI] [PubMed] [Google Scholar]
- 23.Diebold SS, Kaisho T, Hemmi H, Akira S, Reis e Sousa C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science. 2004;303:1529–1531. doi: 10.1126/science.1093616. [DOI] [PubMed] [Google Scholar]
- 24.Heil F, et al. Species-specific recognition of single-stranded RNA via Toll-like receptor 7 and 8. Science. 2004;303:1526–1529. doi: 10.1126/science.1093620. [DOI] [PubMed] [Google Scholar]
- 25.Lund JM, et al. TLR7: a new sensor of viral infection. Proc. Natl Acad. Sci. USA. 2004;101:6835–6836. doi: 10.1073/pnas.0400937101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ahmad-Nejad P, et al. Bacterial CpG-DNA and lipopolysaccharides activate Toll-like receptors at distinct cellular compartments. Eur. J. Immunol. 2002;32:1958–1968. doi: 10.1002/1521-4141(200207)32:7<1958::AID-IMMU1958>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 27.Boule MW, et al. Toll-like receptor 9-dependent and -independent dendritic cell activation by chromatin–immunoglobulin G complexes. J. Exp. Med. 2004;199:1631–1640. doi: 10.1084/jem.20031942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Means TK, et al. Human lupus autoantibody–DNA complexes activate DCs through cooperation of CD32 and TLR9. J. Clin. Invest. 2005;115:407–417. doi: 10.1172/JCI23025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Honda K, et al. Spatiotemporal regulation of MyD88–IRF-7 signalling for robust type-I interferon induction. Nature. 2005;434:1035–1040. doi: 10.1038/nature03547. [DOI] [PubMed] [Google Scholar]
- 30.Vollmer J, et al. Autoantigen binding sites within small nuclear RNAs induce innate immunity through Toll-like receptors 7 and 8. J. Exp. Med. 2005;202:1575–1585. doi: 10.1084/jem.20051696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barrat FJ, et al. Nucleic acids of mammalian origin can act as endogenous ligands for Toll-like receptors and may promote systemic lupus erythematosus. J. Exp. Med. 2005;202:1131–1139. doi: 10.1084/jem.20050914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lovgren T, et al. Induction of interferon-α by immune complexes or liposomes containing systemic lupus erythematosus and Sjögren's syndrome autoantigen-associated RNA. Arthritis Rheum. 2006;54:1917–1927. doi: 10.1002/art.21893. [DOI] [PubMed] [Google Scholar]
- 33.Savarese E, et al. U1 small nuclear ribonucleoprotein immune complexes induce type I interferon in plasmacytoid dendritic cells through TLR7. Blood. 2006;107:3229–3234. doi: 10.1182/blood-2005-07-2650. [DOI] [PubMed] [Google Scholar]
- 34.Rifkin IR, Leadbetter EA, Busconi L, Viglianti G, Marshak-Rothstein A. Toll-like receptors, endogenous ligands, and systemic autoimmune disease. Immunol. Rev. 2005;204:27–42. doi: 10.1111/j.0105-2896.2005.00239.x. [DOI] [PubMed] [Google Scholar]
- 35.Wardemann H, et al. Predominant autoantibody production by early human B cell precursors. Science. 2003;301:1374–1377. doi: 10.1126/science.1086907. [DOI] [PubMed] [Google Scholar]
- 36.Yurasov S, et al. Defective B cell tolerance checkpoints in systemic lupus erythematosus. J. Exp. Med. 2005;201:703–711. doi: 10.1084/jem.20042251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen CJ, et al. High prevalence of immunoglobulin A antibody against Epstein–Barr virus capsid antigen in adult patients with lupus with disease flare: case control studies. J. Rheumatol. 2005;32:44–47. [PubMed] [Google Scholar]
- 38.Hunziker L, et al. Hypergammaglobulinemia and autoantibody induction mechanisms in viral infections. Nature Immunol. 2003;4:343–349. doi: 10.1038/ni911. [DOI] [PubMed] [Google Scholar]
- 39.McClain MT, et al. Early events in lupus humoral autoimmunity suggest initiation through molecular mimicry. Nature Med. 2005;11:85–89. doi: 10.1038/nm1167. [DOI] [PubMed] [Google Scholar]
- 40.Wang Y, Krieg AM. Synergy between CpG- or non-CpG DNA and specific antigen for B cell activation. Int. Immunol. 2003;15:223–231. doi: 10.1093/intimm/dxg020. [DOI] [PubMed] [Google Scholar]
- 41.Bekeredjian-Ding IB, et al. Plasmacytoid dendritic cells control TLR7 sensitivity of naive B cells via type I IFN. J. Immunol. 2005;174:4043–4050. doi: 10.4049/jimmunol.174.7.4043. [DOI] [PubMed] [Google Scholar]
- 42.Brummel R, Roberts TL, Stacey KJ, Lenert P. Higher-order CpG-DNA stimulation reveals distinct activation requirements for marginal zone and follicular B cells in lupus mice. Eur. J. Immunol. 2006;36:1951–1962. doi: 10.1002/eji.200535734. [DOI] [PubMed] [Google Scholar]
- 43.Jacobson BA, et al. An isotype switched and somatically mutated rheumatoid factor clone isolated from a MRL-lpr/lpr mouse exhibits limited intraclonal affinity maturation. J. Immunol. 1994;152:4489–4499. [PubMed] [Google Scholar]
- 44.Hannum LG, Ni D, Haberman AM, Weigert MG, Shlomchik MJ. A disease-related rheumatoid factor autoantibody is not tolerized in a normal mouse: implications for the origins of autoantibodies in autoimmune disease. J. Exp. Med. 1996;184:1269–1278. doi: 10.1084/jem.184.4.1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Leadbetter EA, et al. Chromatin–IgG complexes activate autoreactive B cells by dual engagement of sIgM and Toll-like receptors. Nature. 2002;416:603–607. doi: 10.1038/416603a. [DOI] [PubMed] [Google Scholar]
- 46.Lau CM, et al. RNA-associated autoantigens activate B cells by combined BCR/Toll-like receptor 7 engagement. J. Exp. Med. 2005;202:1171–1177. doi: 10.1084/jem.20050630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Busconi, L. et al. DNA and RNA autoantigens as autoadjuvants. J. Endotoxin Res. (in the press). [DOI] [PubMed]
- 48.Viglianti GA, et al. Activation of autoreactive B cells by CpG dsDNA. Immunity. 2003;19:837–847. doi: 10.1016/S1074-7613(03)00323-6. [DOI] [PubMed] [Google Scholar]
- 49.Messina JP, Gilkeson GS, Pisetsky DS. Stimulation of in vitro murine lymphocyte proliferation by bacterial DNA. J. Immunol. 1991;147:1759–1764. [PubMed] [Google Scholar]
- 50.Sun S, Beard C, Jaenisch R, Jones P, Sprent J. Mitogenicity of DNA from different organisms for murine B cells. J. Immunol. 1997;159:3119–3125. [PubMed] [Google Scholar]
- 51.Stacey KJ, et al. The molecular basis for the lack of immunostimulatory activity of vertebrate DNA. J. Immunol. 2003;170:3614–3620. doi: 10.4049/jimmunol.170.7.3614. [DOI] [PubMed] [Google Scholar]
- 52.Gursel I, et al. Repetitive elements in mammalian telomeres suppress bacterial DNA-induced immune activation. J. Immunol. 2003;171:1393–1400. doi: 10.4049/jimmunol.171.3.1393. [DOI] [PubMed] [Google Scholar]
- 53.Barton GM, Kagan JC, Medzhitov R. Intracellular localization of Toll-like receptor 9 prevents recognition of self DNA but facilitates access to viral DNA. Nature Immunol. 2006;7:49–56. doi: 10.1038/ni1280. [DOI] [PubMed] [Google Scholar]
- 54.Richardson B, et al. Evidence for impaired T cell DNA methylation in systemic lupus erythematosus and rheumatoid arthritis. Arthritis Rheum. 1990;33:1665–1673. doi: 10.1002/art.1780331109. [DOI] [PubMed] [Google Scholar]
- 55.Sano H, Morimoto C. DNA isolated from DNA/anti-DNA antibody immune complexes in systemic lupus erythematosus is rich in guanine–cytosine content. J. Immunol. 1982;128:1341–1345. [PubMed] [Google Scholar]
- 56.Cornacchia E, et al. Hydralazine and procainamide inhibit T cell DNA methylation and induce autoreactivity. J. Immunol. 1988;140:2197–2200. [PubMed] [Google Scholar]
- 57.Lunec J, Herbert K, Blount S, Griffiths HR, Emery P. 8-Hydroxydeoxyguanosine: a marker of oxidative DNA damage in systemic lupus erythematosus. FEBS Lett. 1994;348:131–138. doi: 10.1016/0014-5793(94)00583-4. [DOI] [PubMed] [Google Scholar]
- 58.Cooke MS, Mistry N, Wood C, Herbert KE, Lunec J. Immunogenicity of DNA damaged by reactive oxygen species — implications for anti-DNA antibodies in lupus. Free Radic. Biol. Med. 1997;22:151–159. doi: 10.1016/S0891-5849(96)00283-3. [DOI] [PubMed] [Google Scholar]
- 59.Kariko K, Buckstein M, Ni H, Weissman D. Suppression of RNA recognition by Toll-like receptors: the impact of nucleoside modification and the evolutionary origin of RNA. Immunity. 2005;23:165–175. doi: 10.1016/j.immuni.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 60.Emerit I, Michelson AM. Mechanism of photosensitivity in systemic lupus erythematosus patients. Proc. Natl Acad. Sci. USA. 1981;78:2537–2540. doi: 10.1073/pnas.78.4.2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Andrade F, Casciola-Rosen LA, Rosen A. Generation of novel covalent RNA–protein complexes in cells by ultraviolet B irradiation: implications for autoimmunity. Arthritis Rheum. 2005;52:1160–1170. doi: 10.1002/art.20992. [DOI] [PubMed] [Google Scholar]
- 62.Caricchio R, McPhie L, Cohen PL. Ultraviolet B radiation-induced cell death: critical role of ultraviolet dose in inflammation and lupus autoantigen redistribution. J. Immunol. 2003;171:5778–5786. doi: 10.4049/jimmunol.171.11.5778. [DOI] [PubMed] [Google Scholar]
- 63.Christensen SR, et al. Toll-like receptor 9 controls anti-DNA autoantibody production in murine lupus. J. Exp. Med. 2005;202:321–331. doi: 10.1084/jem.20050338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Christensen SR, et al. TLR7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity. 2006;25:417–428. doi: 10.1016/j.immuni.2006.07.013. [DOI] [PubMed] [Google Scholar]
- 65.Wu X, Peng SL. Toll-like receptor 9 signaling protects against murine lupus. Arthritis Rheum. 2006;54:336–342. doi: 10.1002/art.21553. [DOI] [PubMed] [Google Scholar]
- 66.Yu P, et al. Toll-like receptor 9-independent aggravation of glomerulonephritis in a novel model of SLE. Int. Immunol. 2006;18:1211–1219. doi: 10.1093/intimm/dxl067. [DOI] [PubMed] [Google Scholar]
- 67.Lartigue A, et al. Role of TLR9 in anti-nucleosome and anti-DNA antibody production in lpr mutation-induced murine lupus. J. Immunol. 2006;177:1349–1354. doi: 10.4049/jimmunol.177.2.1349. [DOI] [PubMed] [Google Scholar]
- 68.Ehlers M, Fukuyama H, McGaha TL, Aderem A, Ravetch JV. TLR9/MyD88 signaling is required for class switching to pathogenic IgG2a and 2b autoantibodies in SLE. J. Exp. Med. 2006;203:553–561. doi: 10.1084/jem.20052438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Peng SL, Szabo SJ, Glimcher LH. T-bet regulates IgG class switching and pathogenic autoantibody production. Proc. Natl Acad. Sci. USA. 2002;99:5545–5550. doi: 10.1073/pnas.082114899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Berland R, et al. Toll-like receptor 7-dependent loss of B cell tolerance in pathogenic autoantibody knockin mice. Immunity. 2006;25:429–440. doi: 10.1016/j.immuni.2006.07.014. [DOI] [PubMed] [Google Scholar]
- 71.Pisitkun P, et al. Autoreactive B cell responses to RNA-related antigens due to TLR7 gene duplication. Science. 2006;312:1669–1672. doi: 10.1126/science.1124978. [DOI] [PubMed] [Google Scholar]
- 72.Subramanian S, et al. A Tlr7 translocation accelerates systemic autoimmunity in murine lupus. Proc. Natl Acad. Sci. USA. 2006;103:9970–9975. doi: 10.1073/pnas.0603912103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bolland S, Yim YS, Tus K, Wakeland EK, Ravetch JV. Genetic modifiers of systemic lupus erythematosus in FcγRIIB−/− mice. J. Exp. Med. 2002;195:1167–1174. doi: 10.1084/jem.20020165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kirou KA, et al. Activation of the interferon-α pathway identifies a subgroup of systemic lupus erythematosus patients with distinct serologic features and active disease. Arthritis Rheum. 2005;52:1491–1503. doi: 10.1002/art.21031. [DOI] [PubMed] [Google Scholar]
- 75.Lenert PS. Targeting Toll-like receptor signaling in plasmacytoid dendritic cells and autoreactive B cells as a therapy for lupus. Arthritis Res. Ther. 2006;8:203–214. doi: 10.1186/ar1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wallace DJ. Dubois' Lupus Erythematosus. 1987. pp. 563–573. [Google Scholar]
- 77.Tye MJ, White H, Appel B, Ansell HB. Lupus erythematosus treated with a combination of quinacrine, hydroxychloroquine, and chloroquine. N. Engl. J. Med. 1959;260:63–66. doi: 10.1056/NEJM195901082600203. [DOI] [PubMed] [Google Scholar]
- 78.Macfarlane DE, Manzel L. Antagonism of immunostimulatory CpG-oligodeoxynucleotides by quinacrine, chloroquine and structurally related compounds. J. Immunol. 1998;160:1122–1131. [PubMed] [Google Scholar]
- 79.Hacker H, et al. CpG-DNA-specific activation of antigen-presenting cells requires stress kinase activity and is preceded by non-specific endocytosis and endosomal maturation. EMBO J. 1998;17:6230–6240. doi: 10.1093/emboj/17.21.6230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yi A-K, et al. CpG motifs in bacterial DNA activate leukocytes through the pH-dependent generation of reactive oxygen species. J. Immunol. 1998;160:4755–4761. [PubMed] [Google Scholar]
- 81.Krieg AM. CpG motifs: the active ingredient in bacterial extracts? Nature Med. 2003;9:831–835. doi: 10.1038/nm0703-831. [DOI] [PubMed] [Google Scholar]
- 82.Krieg AM, et al. Sequence motifs in adenoviral DNA block immune activation by stimulatory CpG motifs. Proc. Natl Acad. Sci. USA. 1998;95:12631–12636. doi: 10.1073/pnas.95.21.12631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ashman RF, Goeken JA, Drahos J, Lenert P. Sequence requirements for oligodeoxyribonucleotide inhibitory activity. Int. Immunol. 2005;17:411–420. doi: 10.1093/intimm/dxh222. [DOI] [PubMed] [Google Scholar]
- 84.Patole PS, et al. G-rich DNA suppresses systemic lupus. J. Am. Soc. Nephrol. 2005;16:3273–3280. doi: 10.1681/ASN.2005060658. [DOI] [PubMed] [Google Scholar]
- 85.Dong L, Ito S, Ishii KJ, Klinman DM. Suppressive oligodeoxynucleotides delay the onset of glomerulonephritis and prolong survival in lupus-prone NZB × NZW mice. Arthritis Rheum. 2005;52:651–658. doi: 10.1002/art.20810. [DOI] [PubMed] [Google Scholar]
- 86.Takaoka A, et al. Integral role of IRF-5 in the gene induction programme activated by Toll-like receptors. Nature. 2005;434:243–249. doi: 10.1038/nature03308. [DOI] [PubMed] [Google Scholar]
- 87.Schoenemeyer A, et al. The interferon regulatory factor, IRF5, is a central mediator of Toll-like receptor 7 signaling. J. Biol. Chem. 2005;280:17005–17012. doi: 10.1074/jbc.M412584200. [DOI] [PubMed] [Google Scholar]
- 88.Sigurdsson S, et al. Polymorphisms in the tyrosine kinase 2 and interferon regulatory factor 5 genes are associated with systemic lupus erythematosus. Am. J. Hum. Genet. 2005;76:528–537. doi: 10.1086/428480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Graham RR, et al. A common haplotype of interferon regulatory factor 5 (IRF5) regulates splicing and expression and is associated with increased risk of systemic lupus erythematosus. Nature Genet. 2006;38:550–555. doi: 10.1038/ng1782. [DOI] [PubMed] [Google Scholar]
- 90.Joosten LAB, et al. Toll-like receptor 2 pathway drives streptococcal cell wall-induced joint inflammation: critical role of myeloid differentiation factor 88. J. Immunol. 2003;171:6145–6153. doi: 10.4049/jimmunol.171.11.6145. [DOI] [PubMed] [Google Scholar]
- 91.Deng GM, Nilsson IM, Verdrengh M, Collins LV, Tarkowski A. Intra-articularly localized bacterial DNA containing CpG motifs induces arthritis. Nature Med. 1999;5:702–705. doi: 10.1038/9554. [DOI] [PubMed] [Google Scholar]
- 92.Waldner H, Collins M, Kuchroo VK. Activation of antigen-presenting cells by microbial products breaks self tolerance and induces autoimmune disease. J. Clin. Invest. 2004;113:990–997. doi: 10.1172/JCI19388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kerfoot SM, et al. TLR4 contributes to disease-inducing mechanisms resulting in central nervous system autoimmune disease. J. Immunol. 2004;173:7070–7077. doi: 10.4049/jimmunol.173.11.7070. [DOI] [PubMed] [Google Scholar]
- 94.Ichikawa HT, Williams LP, Segal BM. Activation of APCs through CD40 or Toll-like receptor 9 overcomes tolerance and precipitates autoimmune disease. J. Immunol. 2002;169:2781–2787. doi: 10.4049/jimmunol.169.5.2781. [DOI] [PubMed] [Google Scholar]
- 95.Eriksson U, et al. Dendritic cell-induced autoimmune heart failure requires cooperation between adaptive and innate immunity. Nature Med. 2003;9:1484–1490. doi: 10.1038/nm960. [DOI] [PubMed] [Google Scholar]
- 96.Lang KS, et al. Toll-like receptor engagement converts T-cell autoreactivity into overt autoimmune disease. Nature Med. 2005;11:138–145. doi: 10.1038/nm1176. [DOI] [PubMed] [Google Scholar]
- 97.Yumoto H, et al. Sensitization of human aortic endothelial cells to lipopolysaccharide via regulation of Toll-like receptor 4 by bacterial fimbria-dependent invasion. Infect. Immun. 2005;73:8050–8059. doi: 10.1128/IAI.73.12.8050-8059.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kiechl S, et al. Toll-like receptor 4 polymorphisms and atherogenesis. N. Engl. J. Med. 2002;347:185–192. doi: 10.1056/NEJMoa012673. [DOI] [PubMed] [Google Scholar]
- 99.Kilding R, et al. A biologically important single nucleotide polymorphism within the Toll-like receptor-4 gene is not associated with rheumatoid arthritis. Clin. Exp. Rheumatol. 2003;21:340–342. [PubMed] [Google Scholar]
- 100.Choe J-Y, Crain B, Wu SR, Corr M. Interleukin 1 receptor dependence of serum transferred arthritis can be circumvented by Toll-like receptor 4 signaling. J. Exp. Med. 2003;197:537–542. doi: 10.1084/jem.20021850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Prinz M, et al. Innate immunity mediated by TLR9 modulates pathogenicity in an animal model of multiple sclerosis. J. Clin. Invest. 2006;116:456–464. doi: 10.1172/JCI26078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Iliev AI, Stringaris AK, Nau R, Neumann H. Neuronal injury mediated via stimulation of microglial Toll-like receptor-9 (TLR9) FASEB J. 2004;18:412–414. doi: 10.1096/fj.03-0670fje. [DOI] [PubMed] [Google Scholar]
- 103.Leemans JC, et al. Renal-associated TLR2 mediates ischemia/reperfusion injury in the kidney. J. Clin. Invest. 2005;115:2894–2903. doi: 10.1172/JCI22832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Shishido T, et al. Toll-like receptor 2 modulates ventricular remodeling after myocardial infarction. Circulation. 2003;108:2905–2910. doi: 10.1161/01.CIR.0000101921.93016.1C. [DOI] [PubMed] [Google Scholar]
- 105.Oyama J, et al. Reduced myocardial ischemia–reperfusion injury in Toll-like receptor 4-deficient mice. Circulation. 2004;109:784–789. doi: 10.1161/01.CIR.0000112575.66565.84. [DOI] [PubMed] [Google Scholar]
- 106.Jiang D, et al. Regulation of lung injury and repair by Toll-like receptors and hyaluronan. Nature Med. 2005;11:1173–1179. doi: 10.1038/nm1315. [DOI] [PubMed] [Google Scholar]
- 107.Tan E. Antinuclear antibodies: diagnostic markers for autoimmune diseases and probes for cell biology. Adv. Immunol. 1989;44:93–151. doi: 10.1016/S0065-2776(08)60641-0. [DOI] [PubMed] [Google Scholar]
- 108.Krieg AM. CpG motifs in bacterial DNA and their immune effects. Annu. Rev. Immunol. 2002;20:709–760. doi: 10.1146/annurev.immunol.20.100301.064842. [DOI] [PubMed] [Google Scholar]
- 109.Verthelyi D, Zeuner RA. Differential signaling by CpG DNA in DCs and B cells: not just TLR9. Trends Immunol. 2003;24:519–522. doi: 10.1016/S1471-4906(03)00243-6. [DOI] [PubMed] [Google Scholar]
- 110.Hartmann G, et al. Rational design of new CpG oligonucleotides that combine B cell activation with high IFN-α induction in plasmacytoid dendritic cells. Eur. J. Immunol. 2003;33:1633–1641. doi: 10.1002/eji.200323813. [DOI] [PubMed] [Google Scholar]
- 111.Li M, et al. An essential role of the NF-κB/Toll-like receptor pathway in induction of inflammatory and tissue-repair gene expression by necrotic cells. J. Immunol. 2001;166:7128–7135. doi: 10.4049/jimmunol.166.12.7128. [DOI] [PubMed] [Google Scholar]
- 112.Vabulas RM, Wagner H, Schild H. Heat shock proteins as ligands of Toll-like receptors. Curr. Top. Microbiol. Immunol. 2002;270:169–184. doi: 10.1007/978-3-642-59430-4_11. [DOI] [PubMed] [Google Scholar]
- 113.Liu B, et al. Cell surface expression of an endoplasmic reticulum resident heat shock protein gp96 triggers MyD88-dependent systemic autoimmune diseases. Proc. Natl Acad. Sci. USA. 2003;100:15824–15829. doi: 10.1073/pnas.2635458100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Park JS, et al. Involvement of Toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J. Biol. Chem. 2004;279:7370–7377. doi: 10.1074/jbc.M306793200. [DOI] [PubMed] [Google Scholar]
- 115.Termeer C, et al. Oligosaccharides of hyaluronan activate dendritic cells via Toll-like receptor 4. J. Exp. Med. 2002;195:99–111. doi: 10.1084/jem.20001858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Taylor KR, et al. Hyaluronan fragments stimulate endothelial recognition of injury through TLR4. J. Biol. Chem. 2004;279:17079–17084. doi: 10.1074/jbc.M310859200. [DOI] [PubMed] [Google Scholar]
- 117.Okamura Y, et al. The extra domain A of fibronectin activates Toll-like receptor 4. J. Biol. Chem. 2001;276:10229–10233. doi: 10.1074/jbc.M100099200. [DOI] [PubMed] [Google Scholar]
- 118.Miller YI, et al. Minimally modified LDL binds to CD14, induces macrophage spreading via TLR4/MD-2, and inhibits phagocytosis of apoptotic cells. J. Biol. Chem. 2003;278:1561–1568. doi: 10.1074/jbc.M209634200. [DOI] [PubMed] [Google Scholar]
- 119.Johnson GB, Brunn GJ, Platt JL. An endogenous pathway to systemic inflammatory response syndrome (SIRS)-like reactions through Toll-like receptor 4. J. Immunol. 2004;172:20–24. doi: 10.4049/jimmunol.172.1.20. [DOI] [PubMed] [Google Scholar]
- 120.Smiley ST, King JA, Hancock WW. Fibrinogen stimulates macrophage chemokine secretion through Toll-like receptor 4. J. Immunol. 2001;167:2887–2894. doi: 10.4049/jimmunol.167.5.2887. [DOI] [PubMed] [Google Scholar]
- 121.Roelofs MF, et al. Identification of small heat shock protein B8 (HSP22) as a novel TLR4 ligand and potential involvement in the pathogenesis of rheumatoid arthritis. J. Immunol. 2006;176:7021–7027. doi: 10.4049/jimmunol.176.11.7021. [DOI] [PubMed] [Google Scholar]
- 122.Brentano F, Schorr O, Gay RE, Gay S, Kyburz D. RNA released from necrotic synovial fluid cells activates rheumatoid arthritis synovial fibroblasts via Toll-like receptor 3. Arthritis Rheum. 2005;52:2656–2665. doi: 10.1002/art.21273. [DOI] [PubMed] [Google Scholar]
- 123.Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu. Rev. Immunol. 2003;21:335–376. doi: 10.1146/annurev.immunol.21.120601.141126. [DOI] [PubMed] [Google Scholar]
- 124.Lien E, Ingalis RR. Toll-like receptors. Crit. Care Med. 2002;30:S1–S11. doi: 10.1097/00003246-200201001-00001. [DOI] [PubMed] [Google Scholar]
- 125.Zhang D, et al. A Toll-like receptor that prevents infection by uropathogenic bacteria. Science. 2004;303:1522–1526. doi: 10.1126/science.1094351. [DOI] [PubMed] [Google Scholar]
- 126.Yarovinsky F, et al. TLR11 activation of dendritic cells by a protozoan profilin-like protein. Science. 2005;308:1626–1629. doi: 10.1126/science.1109893. [DOI] [PubMed] [Google Scholar]