

The Ibis T5000 couples nucleic acid amplification to high-performance electrospray mass spectrometry and base-composition analysis and enables the identification and quantification of all known bacteria, all major groups of pathogenic fungi and the major families of viruses that cause disease in humans and animals. Here, Ecker and colleagues describe this new technology.
Abstract
We describe a new technology, the Ibis T5000, for the identification of pathogens in clinical and environmental samples. The Ibis T5000 couples nucleic acid amplification to high-performance electrospray ionization mass spectrometry and base-composition analysis. The system enables the identification and quantification of a broad set of pathogens, including all known bacteria, all major groups of pathogenic fungi and the major families of viruses that cause disease in humans and animals, along with the detection of virulence factors and antibiotic resistance markers.
Main
A walk into a clinical microbiology laboratory today can be like taking a step back in time: culture plates, incubators and microscopes dominate the 'labscape'. It can take days to fully characterize the infectious agents in a patient's sample, beginning with culturing, followed by biochemical tests. Public-health laboratories also deal with the detection and management of infectious diseases, but from the perspective of benefiting society rather than the individual. Epidemiological investigations begin with laborious methods, such as culturing, followed by pulse-field gel electrophoresis (PFGE) or sequencing. Government biodefence and homeland-security agencies have a mandate to detect infectious disease agents to protect against, or respond to, acts of bioterrorism. The US government, for example, has built a large surveillance infrastructure to monitor the environment in major cities for the presence of biological weapons that might be released in an aerosol attack. In these cities, individuals with acute respiratory distress or fevers are treated in hospital emergency departments and doctors' offices. If an individual was infected with a microorganism of bioterrorism significance, it would not be discovered through the government's efforts, as the government's environmental surveillance programme is not integrated into the health-care system. The CDC's PulseNet programme (see Further information) for the comparison of PFGE patterns perhaps presages the future of tracing and following accidental or intentional outbreaks of infectious disease. However, PFGE is time consuming, requires a high level of skill and the pattern results can vary between laboratories. This demonstrates the primary hurdle that prevents rapid and reliable data sharing between clinical microbiology laboratories, public-health laboratories and biodefence agencies: the lack of a reproducible, high-throughput, cost-effective technology that provides digital signatures to detect and identify microorganisms.
The development of this type of technology is a daunting proposition. According to literature reports, more than 1,400 species of microorganism are known to cause disease in humans1. Moreover, each of these species can have hundreds of strain or genotypic variations with different properties of virulence, transmissibility or drug resistance. The ideal diagnostic technology to serve individuals, broad public-health interests and biodefence should provide universal pathogen-identification capability; identify all organisms that are present in a quantitative manner; identify emerging, previously uncharacterized organisms and determine the most closely related species; facilitate outbreak investigations and forensic analysis; facilitate rapid information sharing through a centralized database; allow immediate testing of samples, as well as high-throughput testing; and have low per-sample analysis costs.
The technology that we describe in this article was designed to meet these specifications. We have coupled broad amplification by PCR with electrospray ionization mass spectrometry (ESI–MS) in a system that we call the Ibis T5000. The basic principle of operation is shown in Fig. 1. In brief, multiple pairs of primers are used to amplify carefully selected regions of the genome of the microorganism of interest. Following amplification, a fully automated ESI–MS analysis is performed. The mass spectrometer effectively weighs the amplicons, or mixture of amplicons, with sufficient mass accuracy that the composition of nucleotides (A, G, C and T) can be deduced for each amplicon present. The base composition is compared with a database of calculated base compositions that is derived from the sequences of known organisms to determine the identities of any microorganisms that are present. Thus, analysis using the Ibis T5000 provides detailed information which is analogous to that obtained using a microarray or parallel DNA-sequencing instrument. The Ibis T5000 mass spectrometer analyses each PCR reaction in less than 1 minute, using no consumable products, and is completely automated. We have developed assays for use with the Ibis T5000 that enable the simultaneous identification and quantification of all known bacteria, all major groups of pathogenic fungi and the major families of viruses that cause disease in humans. Here, we describe how this technology can be used for pathogen detection for the benefit of individuals and society.
Figure 1. Flow scheme for Ibis T5000 analysis.
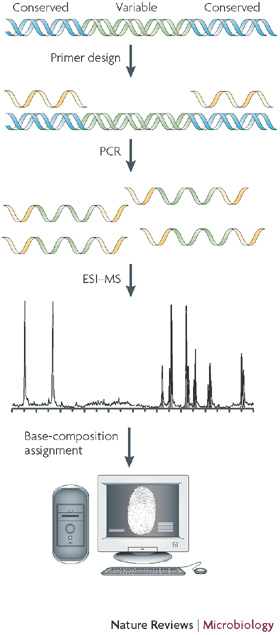
Like any other methodology that uses PCR, the nucleic acids must first be purified from the clinical or environmental sample. Target sites on microbial genomes that are common across broad groups of microorganisms and that flank regions of high information content are selected for primer design. Broad-range PCR is conducted and, following an automated desalting step, amplified nucleic acids are electrosprayed into a time-of-flight (TOF) mass spectrometer and the spectral signal is processed to identify the masses of the amplicons that are present with sufficient mass accuracy to unambiguously calculate the nucleotide base compositions present in each amplicon. The information from all wells is used in a mathematical process that is known as triangulation to convert the base-composition data to a list of the organisms that are present and their relative and absolute quantities. ESI–MS, electrospray ionization mass spectrometry.
The Ibis T5000 universal biosensor
The mass spectrometer component of the Ibis T5000 enables more information to be extracted from PCR reactions than can be obtained with standard individual probes. This enriched information extraction occurs in two dimensions simultaneously. First, a large number of PCR amplicons can be analysed. This enables the use of PCR primers that amplify groups of organisms in mixed populations, rather than single species. For example, primers for viruses can be designed to encompass entire viral families that comprise hundreds of characterized species, and primers for bacteria can be designed that encompass the entire bacterial domain of life. Second, a large amount of information is obtained from each individual amplicon by mass spectrometry. The mass spectrometer weighs each amplicon with sufficient accuracy that the composition of nucleotides (As, Gs, Cs and Ts) can be unambiguously determined. Although not as information rich as the sequence (the linking order is not determined using ESI–MS), for many diagnostic purposes, the nucleotide composition of a nucleic acid can have the same practical value. For example, when a small set of primers is strategically chosen, approximately six PCR reactions can yield sufficient information to identify the bacteria that are present to the species level2. For viruses, primers can be designed to encompass broad genera, such as Alphaviruses3 or Mastadenoviruses4, or even whole virus families, such as the Orthomyxoviridae5 or Coronaviridae6. When primers are designed to amplify all known members within a target group, previously uncharacterized members are also detected. This is a crucial advantage of the Ibis T5000 technology relative to probe-based molecular methods, for which anticipation of the target nucleic acid sequence is required to design the probe.
Bacterial surveillance
Detection and identification of multiple bacterial species in a clinical sample, such as blood, cerebrospinal fluid or sputum, is challenging. The conventional molecular approach is to design specific PCR primers that target specific pathogens. However, this quickly becomes unwieldy because of the large number of possible infecting pathogens. It is not possible to identify rare, emerging or unanticipated pathogens using this conventional method, because the organism's identity must be known in advance to design the test. In the Ibis T5000 system, broad-range primers that can hybridize to virtually all bacterial genomes, rather than specific primers, are used. Broad-range priming is possible because all bacteria share a number of common nucleic acid sequences that are of sufficient length to support the binding of PCR primers. These broad-range primers are used to amplify a product from all bacteria or groups of phylogenetically related bacteria (Fig. 2). The most notable region for broad priming is ribosomal DNA7,8, but there are also broad-priming opportunities in genes that encode other structural RNAs, such as RNase P, and housekeeping proteins9. For this strategy, it is essential that the broad-range primers flank regions of variability such that the base composition of the resulting amplicon is information rich. It would be ideal if an amplicon from a single pair of broad-range primers would provide sufficient information to identify all bacteria and determine the species but, because some bacterial species have the same base composition in conserved primer regions, a number of amplicons from different regions of the microbial genome must be analysed. Primer choices are made based on their ability to provide maximal parsing using base composition as a metric. Microorganisms are distinguished using the aggregate information from the base composition of a number of amplicons; we refer to this as triangulation. The resolving power of base-composition analysis is compared with that of sequencing in Box 1.
Figure 2. The bacterial domain of life depicted using 373 sequenced species and the coverage of 16 primer pairs.
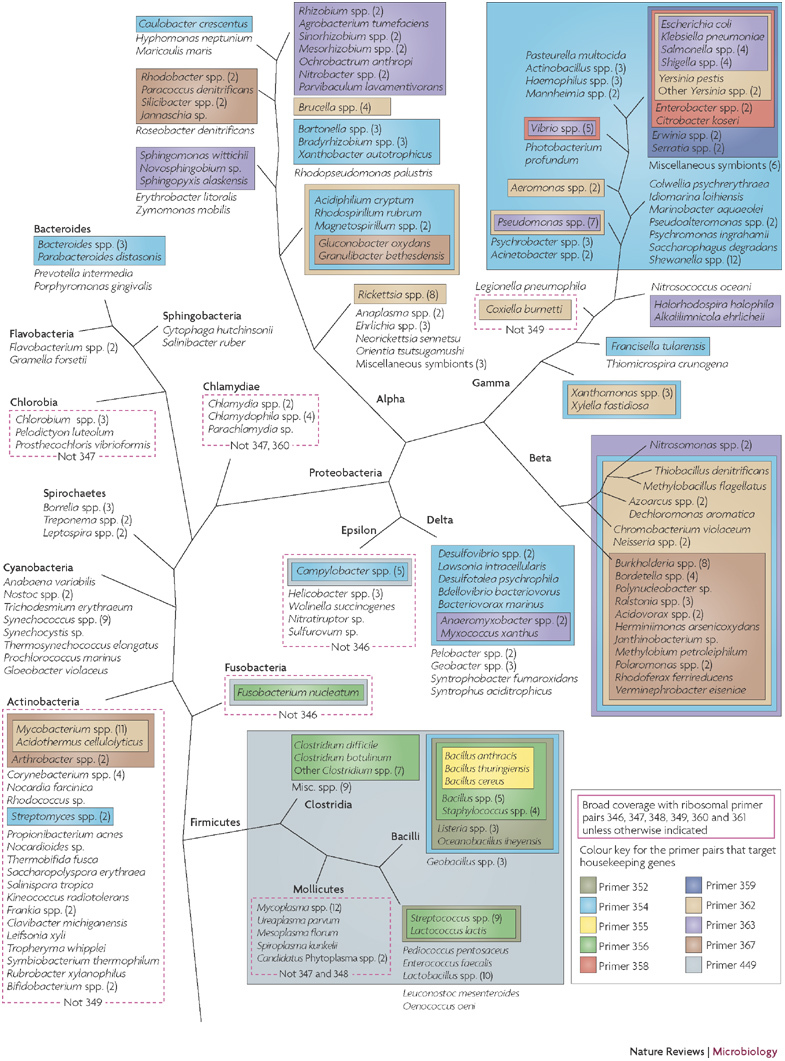
The colour coding depicts the specificity of the broad-range primers that are used in bacterial identification. All the species shown are amplified by the six ribosomal-DNA-targeted primers (346, 347, 348, 349, 360 and 361); exceptions are shown boxed by a red dashed line, along with an indication of the actual subset of primer pairs that is predicted to yield amplicons. Other colours depict coverage of the division- or group-specific primers that target genes which encode conserved housekeeping genes. For clarity, diverse branches that are covered by ribosomal primer pairs only (the Aquificae, Thermotogae and Chloroflexi phyla) are not represented.
Analysis of a sample using multiple broad PCR reactions has several other important benefits. The first relates to coverage of diverse organisms. Broad-range primers designed to amplify DNA from all bacteria, even those targeted to ribosomal DNA, do not match all organisms perfectly and a certain number of mismatches between the PCR primers and some genomes is inevitable. This was considered in the development of PCR conditions for the Ibis T5000, and the conditions are permissive in the first few cycles to ensure hybridization of a partially mismatched oligonucleotide primer to the target. Great care is taken to match the 3′ ends of the primers to the targets; this region is the most sensitive to mispairings during PCR initiation. However, priming failures owing to mismatches between primers and targets will occur for some organisms for any given primer pair. The use of multiple PCR reactions that are targeted to different regions of the microbial genome provides an inherent redundancy that protects against some organisms being missed owing to mismatches from single primer pairs.
Triangulation also increases the dynamic range of detection limits. With extremely broad coverage, such as that provided by the ribosomal-DNA-targeted primers, the microorganisms that are present in a sample compete during the amplification process. When organisms differ in concentration by a ratio of more than 100 to 1, the lower abundance microorganisms might not be detected because they exceed the dynamic range of the competitive PCR reactions. However, when two different microorganisms are individually amplified by non-overlapping division-wide primers, the microorganisms do not compete with each other, because they are amplified in different PCR reactions. The mass spectral information from several reactions is used to triangulate the identities of the organisms that are present. Notably, using the triangulation strategy, none of the primers is designed to be specific for any one microorganism, but instead the primers are designed to cover the bacterial tree of life using a redundant, nested-coverage approach. This enables identification of any bacterial species, even previously unknown organisms, with a single test.
An example of a broad-range primer analysis of a complex mixture of microorganisms is shown in Fig. 3. The spectrum was obtained by using a ribosomal-DNA-targeted primer pair for PCR of a sputum sample from a patient with cystic fibrosis. Spectral signals from six distinct microorganisms were present and microorganism identification was based on triangulation of the signals from eight broad-range primers. Use of integrated information from multiple broad-range primers allows the identification of virtually all organisms that are present in a sample. Crucially, the time to identification is 4–6 hours, and therefore appropriate action can begin promptly.
Figure 3. The spectrum of amplicons obtained from a sputum sample from a patient with cystic fibrosis using a primer pair that targets ribosomal DNA.
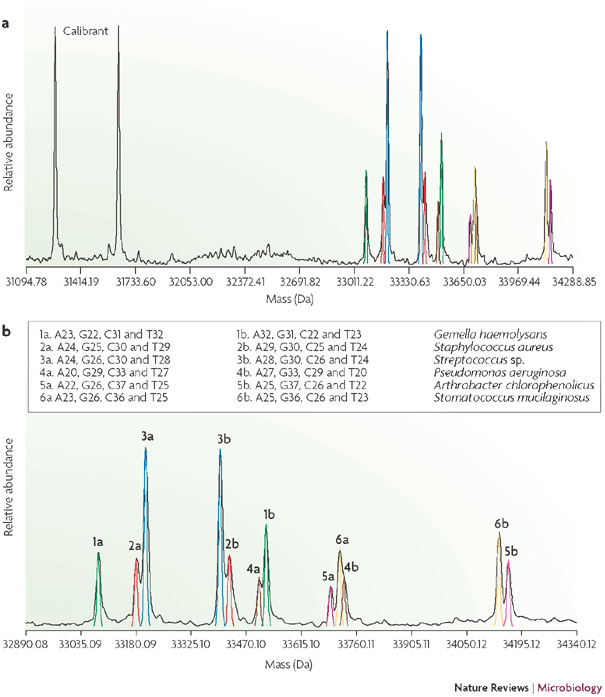
a | Shows a broad mass range that includes each strand of the internal calibration standard. b | Shows an expanded view of the spectral region that excludes the calibration standard. Each amplified product has two peaks that correspond to the two strands of the amplicon. The organisms that are assigned to each peak are based on the collective interpretation from eight broad-range primers.
Bacterial genotyping
The Ibis T5000 method identifies microorganisms by measuring the base composition of selected genomic regions and comparing these digital fingerprints with a database of microorganism signatures that are derived from known sequences. These digital signatures provide a powerful means to identify microorganisms; digitization also enables the technology to be applied to outbreak investigations, forensic analyses, epidemiological tracking and real-time integration of data from various locations. Because regions of high variability in the microbial genomes are amplified, a certain amount of intra-species variability is often observed. For example, primer pairs targeted to variable regions of housekeeping genes that are common to all bacteria will yield a set of base compositions that vary slightly — usually one or two mutations in a few of the target sites — in different strains of the same species. These variations provide distinguishing fingerprints that can be used to track the spread of microorganisms in a hospital setting or follow an epidemiological trail. However, to use the Ibis T5000 technology specifically for epidemiology, a set of genus- or species-specific primer pairs should be designed that target genomic regions that are optimized to distinguish strains within the same species. Genes are selected that provide two levels of evolutionary 'clock speed' for strain genotyping. The first are the housekeeping genes that are used in multilocus sequence typing (MLST)10 and the second are species-specific variable number of tandem repeats (VNTRs)11. MLST and VNTR analyses are conventionally conducted using PCR, followed by sequencing or gel electrophoresis, but both types of targets can be analysed using the high-throughput Ibis T5000. This approach has been used successfully to track a group A Streptococcus outbreak in a military training camp2 and to investigate the source of an outbreak of Acinetobacter spp. in hospital settings12,13.
Virus identification and genotyping
The Ibis T5000 is also useful for identifying and tracking viruses. Despite higher mutation rates and greater sequence variability than bacteria, conserved primer target sites can be identified that enable priming of entire genera or even complete virus families. RNA-dependent RNA polymerase is a housekeeping gene that is common to all RNA viruses and provides several target-site opportunities for developing primers that amplify multiple species within a virus family. This strategy is powerful because a single PCR reaction analysed by mass spectrometry can be used to detect and identify tens to hundreds of related viral species. As shown in Fig. 4, despite the inherent variation of base composition that is found in different flavivirus entries in GenBank, which generates a 'cloud' of base compositions for each species, each species can be distinguished. Because at least two sets of primer pairs targeted to different regions of the viral genome are generally used for each virus group, potential misclassification is avoided, as two regions taken together provide unambiguous species identification. This strategy has been used effectively for broad detection and strain typing of adenoviruses4, influenza viruses5, alphaviruses3, coronaviruses6 and orthopoxviruses9.
Figure 4. Base-composition analysis of flaviviruses.
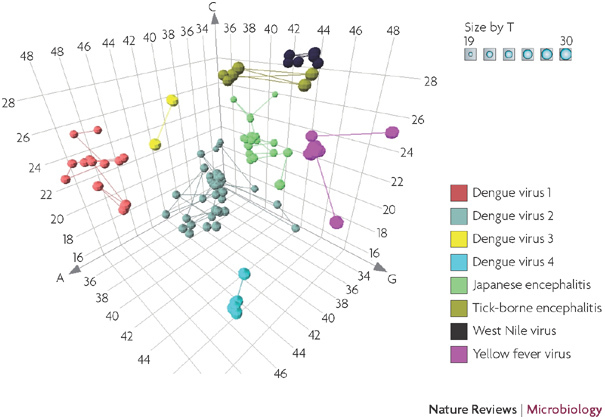
The base composition of a variable fragment of the RNA-dependent RNA polymerase gene flanked by conserved primer target sites is depicted in three dimensions for the flavivirus sequences in GenBank. The region depicted here corresponds to coordinates 9,050–9,150 of Dengue virus 1 (GenBank accession code DQ285559).
Microorganism quantitation
Quantitation in the Ibis T5000 process is achieved by using an internal calibration standard. A nucleic acid with a target sequence that is similar to the primer target sites is added to each PCR reaction at a precisely known concentration. The calibration standard is amplified along with the microbial nucleic acids that are present. The amplicon that is generated from the calibration standard contains a small deletion that unambiguously distinguishes it from the amplified microorganism nucleic acid in the mass spectrum (Fig. 3a). Comparison of spectral peak heights from the calibration standard and the amplified microorganism nucleic acids enables determination of the concentration. This internal calibration method is accurate, within fourfold of the actual concentration, in part because each sample is analysed in multiple PCR reactions that are all independently calibrated and the average over all PCR reactions is reported for each sample9. The calibration standard also serves as an internal positive control. If the calibration standard is not amplified, something in the particular sample might be inhibitory or the PCR might have failed for some other reason. Thus, the calibration standard is valuable as a quality-control check for PCR inhibition and the subsequent steps in mass spectrometry and signal analysis.
High-information-content diagnostics
Key to the use of high-information-content tests in a diagnostic setting are practical considerations, such as the complexity and the skill set that are required to run the assay, costs, throughput and interpretation of complex data. Although several current technologies, including microarrays and parallel sequencing methods, can theoretically provide detailed information about the infectious disease agents that are present in a sample, these methods are currently too complex, slow and expensive for routine use in a diagnostic laboratory. The Ibis T5000 was designed for high-throughput operation in a clinical laboratory setting with consumable costs that are comparable to current PCR-based molecular diagnostic methods. The mass spectrometer uses no consumable products, and therefore the total costs per sample for Ibis T5000 analysis are comparable to the costs of standard molecular diagnostic tests. Samples are analysed at a rate of approximately 30 seconds for each PCR reaction, and the software interprets spectral results, matches signatures with a database and reports the bacterial, viral or fungal identification, along with accessory information, such as high-resolution genotype, virulence factors or antibiotic resistance markers, according to the selected assay configuration. The main drawback to the current Ibis T5000 instrument is its high capital cost, which is driven by the cost of a high-performance mass spectrometer. However, equipment costs can be recovered over time if large numbers of samples are evaluated, which would enable production-scale economy. Moreover, for high-throughput applications in a clinical laboratory, equipment costs are amortized over large numbers of samples, making PCR reagents the dominant cost driver. In addition, mass-spectrometry technology continues to evolve, with yearly performance increases and cost reductions.
Applications and conclusions
Infectious diseases are, by definition, communicable, and yet virtually all infections in individual patients are diagnosed and treated in isolation. What might be learned about the virulence of a microorganism or its response to selected therapies is not easily transferred to aid the treatment of patients who become sequentially infected with the same clonal strain. For example, two paediatricians who work in the same medical complex are likely to see children who have been infected by the same pathogen, but have no way of knowing it. The microorganism in question might be making its way across a country, having been treated successfully or unsuccessfully by other physicians weeks or months earlier in adjacent states, provinces or countries. The technology described here has the potential to integrate infectious disease identification and treatment across time and distance. As the Ibis T5000 provides digital signatures of identified microorganisms, this technology allows the collection and dissemination of epidemiological information in real time. The microbial signatures, along with previous treatment information on that pathogen, can be shared electronically. New signatures can be recorded in a central database and the appearance of those signatures in new locations will enable real-time epidemiological analysis by public-health officials. Likewise, integration of biodefence surveillance with routine clinical diagnostics is facilitated by the use of technology that identifies any organism that infects patients, including bioterrorist agents, rather than only routinely observed infectious agents. We suggest that the infectious disease community should consider the impact of integrating the various communities of stakeholders who are concerned with infectious microorganisms. The Ibis T5000 technology described here is one diagnostic method that has been designed to achieve this objective.
Box 1 | Comparison of the resolving power of base-composition and sequence analysis in bacteria.
We analysed the resolution of 405 unique bacterial species that was provided by the regions amplified by the 16 primer pairs that are used for bacterial surveillance2 (see the figure). Six of these loci are found within the ribosomal subunits and are expected to yield amplicons from virtually all bacteria. By contrast, the ten primer pairs that target essential housekeeping genes typically cover distinct phyla (Fig. 2) and provide additional resolution within the densest parts of the bacterial tree. We first used a computational simulation of PCR to extract the representative sequence and base composition (if any) from each loci for each of the 405 unique, completely sequenced (by September 2007) bacterial species (see Further information for a link to complete microbial genomes from the NCBI database). Thus, we associated each species with a unique 16-loci signature. As the genomes of some species are not amplified by certain primer pairs, on average, there were 7.2 loci with base-composition and sequence signatures. We then determined the number of species that were unambiguously identified by either sequence or base-composition analysis using a variable number of these loci, starting with the six ribosomal loci and adding one locus at a time. Primer pairs were initially sorted by decreasing breadth of coverage, starting with the ribosomal primer pairs (in blue). The resolution of base-composition analysis approximates that of sequence analysis after the addition of the third primer pair, leaving only six extra pairs of closely related organisms that are not separated; this difference disappears after the addition of the ninth primer pair, and neither base-composition nor sequence analysis of the selected genomic regions distinguishes the remaining 19 closely related species.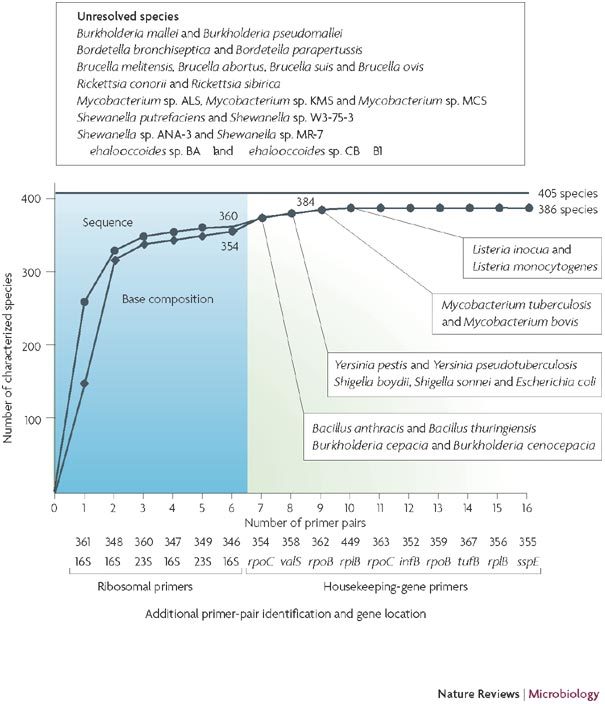
Acknowledgements
The authors acknowledge the government agencies that supported the development of the Ibis T5000 universal biosensor technology, including the Defense Advanced Research Projects Agency (DARPA), the Centers for Disease Control and Prevention, the National Institutes of Health and the Department of Homeland Security.
Biographies
David J. Ecker founded Ibis Biosciences in 1997 and currently serves as its chief scientific officer. He is responsible for the Ibis T5000 universal biosensor programme for the identification and characterization of infectious disease agents and biodefence initiatives, including the science, patents, business development and strategic direction, and the management of government and corporate partnerships. Previously, he was a co-founder of Isis Pharmaceuticals in 1989 and managed several Isis Pharmaceuticals drug-discovery projects in cancer, inflammatory disease, antibacterials and antivirals, using antisense oligonucleotide technology and small molecules that work by RNA-binding mechanisms. From 1984–1989, he was a scientist for Smith Kline and French Laboratories (now GlaxoSmithKline) in drug-discovery programmes in cancer and infectious diseases. From 1982–1984 he was an American Cancer Society fellow in the Department of Chemistry at the University of California, Berkeley, USA, where he studied siderophores and iron metabolism in microrganisms. He completed his Ph.D. from Utah State University, USA, in 1982 in chemistry and biochemistry and his B.A. degree from the College of New Jersey, USA, in 1976.
Rangarajan Sampath is co-founder of Ibis Biosciences and is currently Executive Director of Genomics and Applications Development. He leads Ibis' genomics efforts in microbial detection and diagnosis and is responsible for applications development, validation, data analysis and reporting for the Universal Microbial Detection Platform (Ibis T5000). His research interests include pathogen discovery, epidemiological surveillance and infectious-disease diagnostics. He has been the lead genomics investigator on several programmes that are related to bioinformatics, drug discovery and biological-warfare agent detection and treatment. He holds a Ph.D. in chemical engineering from Rice University, Texas, USA, and a B.S. in chemical engineering from the Indian Institute of Technology, Chennai, India.
Christian Massire joined Ibis Biosciences in 1999. As a member of the Genomics Data Analysis group, he initiates, directs and executes pre-clinical scientific research and/or development strategies for the Ibis T5000. He is responsible for several application- development projects, including health-care-associated infection control, infectious-disease diagnostics and molecular determination of drug resistance. He holds a Ph.D. in bioinformatics and an M.S. in chemical engineering from Louis Pasteur University, Strasbourg, France.
Lawrence B. Blyn is a co-founder of Ibis Biosciences and currently serves as Executive Director of Biology. He has responsibilities in many aspects of research and assay development for the Ibis T5000 Biosensor, including T5000 kit development and sample preparation. He is, and has been, the principle investigator on a number of projects that are involved with biodefence, clinical disease and assay development. From 1991 to 1996, he was a postdoctoral fellow at the University of California, Irvine, USA, and studied the involvement of host cell proteins in the regulation of poliovirus translation and replication. He completed his Ph.D. from the University of Utah, USA, in 1991 in bacterial genetics and has a B.S. degree in biochemistry from Harvey Mudd College, California, USA.
Thomas A. Hall came to Ibis Biosciences in 2000 and has served as an integral part of the biosensor development team since that time. He is responsible for multiple application development projects, including diagnostics and health-care-associated infection characterization. He leads the laboratory development of human forensics applications of the Ibis T5000. He completed his Ph.D. at North Carolina State University, USA, in 2000, studying the information-processing system of the archaeal domain of life. He received his B.S. in biology from the University of the Virgin Islands in 1995.
Mark W. Eshoo is Director of Process Development at Ibis Biosciences. Since 2002, he has led the bioprocess development for the Ibis T5000 biosensor. His research interests include microbial and human forensics analysis and the genetic analysis of clinical samples with low levels of pathogens. He has over 20 years of research experience in the field of genetics and genomics. As Director of Genomics at the Buck Institute for Age Research he supported the genomics research of over 25 researchers. While at the Buck Institute, he helped to develop a number of gene-expression technologies for the isolation, amplification and quantification of RNA from single cells using DNA microarrays. He earned his Ph.D. in genetics from the University of California, USA, and performed his postdoctoral studies at Stanford University, California, USA.
Steven A. Hofstadler is a co-founder of Ibis Biosciences and currently serves as Vice President of Research for Ibis and heads the mass spectrometry (MS) programme. In addition to coordinating the basic research activities of Ibis, he is responsible for the development of advanced MS instrumentation and methods to support clinical-diagnostics development and human forensics efforts. He has coordinated MS efforts for several Defense Advanced Research Projects Agency (DARPA) initiatives, including Drugs for Engineered Biological Warfare Bacteria, Universal Pathogen Detection, Ibis T5000, a recently completed CDC programme, Automated Simultaneous Detection Bioterrorism Agents and several on-going programmes that are sponsored by the Department of Homeland Security and the Federal Bureau of Investigation. He received a Ph.D. in analytical chemistry from The University of Texas, USA.
Related links
DATABASES
Entrez–Gene
FURTHER INFORMATION
Competing interests
The authors are employees of Ibis Biosciences.
References
- 1.Taylor LH, Latham SM, Woolhouse ME. Risk factors for human disease emergence. Philos. Trans. R. Soc. Lond. B. 2001;356:983–989. doi: 10.1098/rstb.2001.0888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ecker DJ, et al. Rapid identification and strain-typing of respiratory pathogens for epidemic surveillance. Proc. Natl Acad. Sci. USA. 2005;102:8012–8017. doi: 10.1073/pnas.0409920102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Eshoo MW, et al. Direct broad-range detection of alphaviruses in mosquito extracts. Virology. 2007;368:286–295. doi: 10.1016/j.virol.2007.06.016. [DOI] [PubMed] [Google Scholar]
- 4.Blyn LB, et al. Rapid detection and molecular serotyping of adenovirus by use of PCR followed by electrospray ionization mass spectrometry. J. Clin. Microbiol. 2008;46:644–651. doi: 10.1128/JCM.00801-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sampath R, et al. Global surveillance of emerging influenza virus genotypes by mass spectrometry. PLoS ONE. 2007;2:e489. doi: 10.1371/journal.pone.0000489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sampath R, et al. Rapid identification of emerging pathogens: coronavirus. Emerg. Infect. Dis. 2005;11:373–379. doi: 10.3201/eid1103.040629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nikkari S, et al. Broad-range bacterial detection and the analysis of unexplained death and critical illness. Emerg. Infect. Dis. 2002;8:188–194. doi: 10.3201/eid0802.010150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kroes I, Lepp PW, Relman DA. Bacterial diversity within the human subgingival crevice. Proc. Natl Acad. Sci. USA. 1999;96:14547–14552. doi: 10.1073/pnas.96.25.14547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hofstadler SA, et al. TIGER: the universal biosensor. Int. J. Mass Spectrom. 2005;242:23–41. doi: 10.1016/j.ijms.2004.09.014. [DOI] [Google Scholar]
- 10.Enright MC, Spratt BG. Multilocus sequence typing. Trends Microbiol. 1999;7:482–487. doi: 10.1016/S0966-842X(99)01609-1. [DOI] [PubMed] [Google Scholar]
- 11.Johansson A, et al. Worldwide genetic relationships among Francisella tularensis isolates determined by multiple-locus variable-number tandem repeat analysis. J. Bacteriol. 2004;186:5808–5818. doi: 10.1128/JB.186.17.5808-5818.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hujer KM, et al. Analysis of antibiotic resistance genes in multidrug-resistant Acinetobacter sp. isolates from military and civilian patients treated at the Walter Reed Army Medical Center. Antimicrob. Agents Chemother. 2006;50:4114–4123. doi: 10.1128/AAC.00778-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ecker JA, et al. Identification of Acinetobacter species and genotyping of Acinetobacter baumannii by multilocus PCR and mass spectrometry. J. Clin. Microbiol. 2006;44:2921–2932. doi: 10.1128/JCM.00619-06. [DOI] [PMC free article] [PubMed] [Google Scholar]