

Key Points
On the 50th anniversary of the discovery of interferon (IFN), we offer a perspective from more than 100,000 published papers, highlighting initial pivotal discoveries and more recent findings of conceptual importance. This covers the mechanisms of IFN induction, the cellular actions of IFN and IFN-stimulated genes (ISGs), and human therapeutic applications.
The synthesis of IFNs requires stimulation by viruses or microbial products binding to Toll-like receptors, or chemical inducers. The development of small-molecule modulators is still in its infancy, but the delineation of the responsible signalling pathways has identified many target proteins.
IFNs constitute a large protein family that can be subdivided into three types, binding to different receptors. These receptors initiate signalling by activating a complex signalling cascade regulated at many levels, resulting in a diverse pattern of ISG induction.
ISGs are a diverse group of more than 300 genes, which can have direct antiviral and antitumour functions. These are attractive targets for high-throughput screening for the identification of new modulators of the IFN system.
IFNs were initially investigated for their potential as antivirals, and are now commonly used in anti-HBV (hepatitis B virus) and anti-HCV (hepatitis C virus) therapy. They might also have prophylactic or therapeutic effectiveness in SARS (severe acute respiratory syndrome), influenza or another virus pandemic.
The first FDA approval of an IFN was, however, not for virus infection but for cancer. The mechanisms of antitumour action are incompletely understood. Aberrations of the IFN system are also emerging as important contributors to cancer development.
IFNs also proved effectiveness in relapsing, remitting multiple sclerosis. It is now common practice to initiate IFN-β treatment at the time of diagnosis.
Because of the effectiveness of IFNs in limiting virus replication, reducing tumour cell mass, controlling disease symptoms and prolonging survival, market sales of IFNs approach US$4 billion. As all the effects of IFNs are mediated through ISGs, understanding of the function of these genes might lead to more efficacious antiviral and anti-cancer drugs.
Interferons (IFNs) provide fundamental cellular defence mechanisms against viral infections and cancer. On the 50th anniversary of the discovery of IFNs, the authors provide a comprehensive overview of IFN biology, human therapeutic applications and potential drug targets within the IFN system.
Abstract
The family of interferon (IFN) proteins has now more than reached the potential envisioned by early discovering virologists: IFNs are not only antivirals with a spectrum of clinical effectiveness against both RNA and DNA viruses, but are also the prototypic biological response modifiers for oncology, and show effectiveness in suppressing manifestations of multiple sclerosis. Studies of IFNs have resulted in fundamental insights into cellular signalling mechanisms, gene transcription and innate and acquired immunity. Further elucidation of the multitude of IFN-induced genes, as well as drug development strategies targeting IFN production via the activation of the Toll-like receptors (TLRs), will almost certainly lead to newer and more efficacious therapeutics. Our goal is to offer a molecular and clinical perspective that will enable IFNs or their TLR agonist inducers to reach their full clinical potential.
Main
The discovery and molecular understanding of the cellular mechanisms and clinical use of interferons (IFNs) have been a major advance in biomedicine over the past 50 years. This family of secreted autocrine and paracrine proteins stimulates intracellular and intercellular networks that regulate resistance to viral infections, enhance innate and acquired immune responses, and modulate normal and tumour cell survival and death. After their discovery in 1957 (Ref. 1), it was soon appreciated that IFNs were critically important to the health of animals and humans and that the IFN system had potential as therapies for infections for both RNA and DNA viruses (Timeline).
However, advances in molecular biology a decade later and into the 1970s were required before the promise could be realized. The 1980s saw their introduction into the clinic as the first pharmaceutical products of the budding biotechnology industry,and, importantly, as a demonstration of the effectiveness of IFNs not only for viral diseases and cancer but also for multiple sclerosis (MS). The 1990s were marked by an expansion in their clinical applications with regulatory approvals worldwide and a further understanding of molecular events influencing biological actions. Current studies have led to new insights into IFNs as a fundamental component of the innate immune system. Additionally, studies have revealed how IFN production is induced through Toll-like receptors (TLRs), the actions of IFN-stimulated genes (ISGs), the identification of viral mechanisms that resist actions of this potent protein, and how mutation and suppression of gene products of the IFN system in and by malignant cells may affect the initiation and progression of cancer.
After binding to high-affinity receptors, IFNs initiate a signalling cascade through signalling proteins that can also be activated by other cytokines, which were first identified through studies of IFNs. Cellular actions are mediated through specific ISGs, which underlie the antiviral effects, as well as immunoregulatory and antitumour effects. Future drugs that could act as molecular activators for ISGs, many of which exist in a latent state or as agonists for TLRs, might be expected to have potent antiviral, antitumour and/or immunomodulatory effects.
From more than 100,000 published papers, we offer a perspective with a focus on human IFNs to stimulate the future investigation of important questions. Although IFNs function as an integrated system, conceptually it helps to consider their production, which is mediated through TLR activation, and their action, which is mediated through JAK/STAT (Janus kinase/Signal Transducers and Activators of Transcription) and other signalling pathways. The production of IFNs is important for understanding the role of IFNs in innate immunity, while their effects relate to dissecting underlying and future mechanisms of action and application. Highlighting initial pivotal discoveries and more recent findings of conceptual importance, we review how IFNs are induced, the cellular actions of IFNs and ISGs, human therapeutic applications, and summarize important questions for biomedicine and drug and clinical development initiatives.
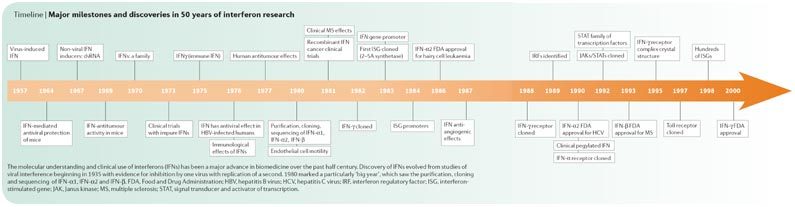
Major milestones and discoveries in 50 years of interferon research
How synthesis is induced
Production of IFNs, both in vitro and in vivo, is transient and requires stimulation by viruses, microbial products or chemical inducers. In the course of the discovery of IFNs, either live or heat-inactivated influenza viruses were initially identified as inducers1. Subsequently other microbial products, including those of bacteria, protozoa, and RNA and DNA viruses, were also recognized to induce IFN2. It was also shown that microbial nucleic acids, lipids, polysaccharides or proteins trigger induction of IFNs through activation of TLRs (Fig. 1). An early pivotal discovery identified double-stranded (ds) RNAs, both natural and synthetic, as potent inducers3, leading to the simplistic paradigm that viruses induce IFNs by producing dsRNA; in reality, it is only one of the viral gene products responsible for induction. Nonetheless, dissection of cellular responses that lead to induction were spearheaded by the analysis of dsRNA-mediated signalling pathways.
Figure 1. Mammalian Toll-like receptors (TLRs) and their ligands.

TLR1, TLR2, TLR4, TLR5 and TLR6 are located on the cell surface. Their extracellular domains (depicted as rods) bind specific microbial products that act as ligands and the intracellular domains (depicted as spheres) signal via specific cytoplasmic signalling proteins. TLRs function as homodimers or heterodimers.The ligands specific for several such dimers are listed at the top of the figure. Several other TLRs, such as TLR3, TLR7/8 and TLR9, recognize specific nucleic acids that are often produced by viruses. They span the endosomal membrane with the ligand-binding domains inside the lumen and the signalling domains in the cytoplasm. They also function as dimers and recognize double-stranded (ds) RNA, single-stranded (ss) RNA or dsDNA containing CpG sequences. GPI, glycosylphosphatidylinisotol; LPS, lipopolysaccharide.
dsRNA is recognized by TLR3, which is present mostly in endosomal membranes4, and also by two cytoplasmic RNA helicases, retinoic acid-inducible gene I (RIG-I) and melanoma differentiation associated protein 5 (MDA5)5 (Fig. 2). The cytoplasmic proteins can also recognize single-stranded (ss) RNAs but only if they have 5′-triphosphates. Mice in which the TLR3 (Ref. 6), the RIG- I7 or the MDA5 (Ref. 8) gene has been disrupted are more susceptible to virus infection; however the relative importance of the three proteins for inhibitory activity varies for the immune defence against different viruses. Surprisingly, the presence of TLR3 can enhance pathogenesis in mice infected with influenza A virus9 or West Nile virus10. An adaptor protein for TLR3 signalling is TLR adapter molecule 1 (TRIF), whereas the mitochondrial protein IFN-β-promoter stimulator 1 (IPS1; also known as VISA) is an adaptor for RIG-I and MDA5; both TRIF and IPS1 recruit inhibitor of nuclear factor-κB (NFκB) kinase (IKK) and TANK-binding kinase (TBK1), the common activator kinases11. Other nucleic acids such as ssRNA, acting through TLR7 and TLR8, and bacterial oligodeoxyribonucleotides, acting through TLR9, are also potent inducers12. Bacterial lipopolysaccharides induce IFNs through TLR4 and also recruit TRIF; viral glycoproteins bind and activate different TLRs. An additional cytoplasmic receptor exists for recognizing viral DNA13.
Figure 2. Different interferon (IFN) signalling pathways activated by dsRNA and viruses.
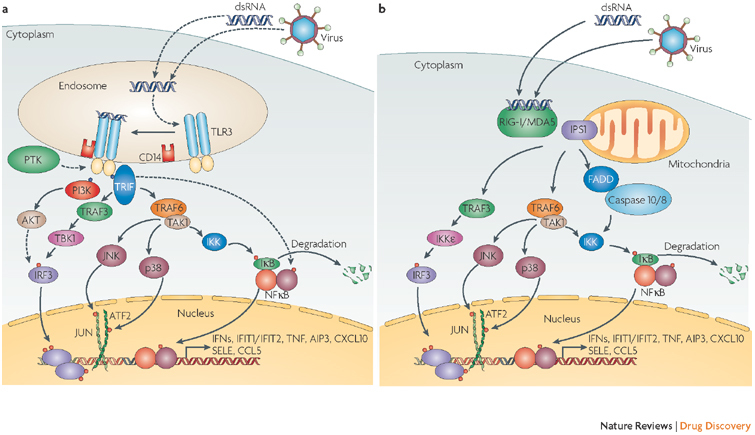
Extracellular double-stranded (ds) RNA or intracellular dsRNA produced during viral replication can activate different signalling pathways triggered by either membrane-bound Toll-like receptor 3 (TLR3) or cytoplasmic retinoic acid-inducible gene I (RIG-I; also known as DDX58) or melanoma differentiation associated protein 5 (MDA5; also known as IFIH1). a | TLR3 recognizes dsRNA in the lumen of the endosome, which causes phosphorylation of specific tyrosine residues in TLR3 by an unidentified protein tyrosine kinase (PTK). TLR3 dimerizes, binds to CD14 and activates the signalling complex assembled by TLR adaptor molecule 1 (TRIF). Two major pathways bifurcate from TRIF. One, composed of tumour necrosis factor (TNF) receptor-associated factor 3 (TRAF3) and TANK-binding kinase (TBK1/IKKE), leads to phosphorylation of the transcription factor IFN regulatory factor 3 (IRF3). IRF3 requires further phosphorylation by the phosphatidylinositol 3-kinase (P13K)/AKT pathway for its full activation, which is initiated by binding PI3K to phosphorylated TLR3. The other branch acts through TRAF6 and transforming growth factor-β-activated kinase 1 (TAK1; also known as MAP3K7) leading to the activation of nuclear factor-κB (NFκB), JUN and activating transcription factor 2 (ATF2) transcription factors. The activated transcription factors translocate from the cytoplasm to the nucleus, bind to the cognate sites in the promoters of the target genes and singly or in combinations induce their transcription. b | The cytoplasmic RNA helicases RIG-I and MDA5 recognize dsRNA or 5′ triphosphorylated single-stranded (ss) RNA and use the mitochondrial membrane-bound protein IFN-β-promoter stimulator 1 (IPS1; also known as VISA) as the specific adaptor. IPS1 functions like TRIF and activates the same transcription factors leading to the induction of similar genes. In addition, they cause apoptosis by activating caspases 8 and 10 through the interaction of FADD with IPS1. Solid arrows denote steps that have been fully delineated, stippled arrows show steps that contain as yet unknown intermediaries. AIP3, atrophin-1 interacting protein 3; CCL5, chemokine (C-C motif) ligand 5; CXCL10, chemokine (C-X-C motif) ligand 10; IFIT1/2, interferon-induced protein with tetratricopeptide repeats 1/2; IKK, inhibitor of NFκB kinase; SELE, selectin E (endothelial adhesion molecule 1).
IFN genes, which are normally transcriptionally silent, are induced by the binding of TLR-activated transcription factors to their promoters. Transcriptional induction of IFN-β has been a model experimental system for defining interactions of transcription factors as an enhanceosome multiprotein complex with DNA14. The most important transcription factors for induction are proteins of the IFN regulatory factor (IRF), specifically IRF3 and IRF7, and NFκB families15,16. IRFs are activated by the kinases TBK1 or IKKɛ; activated IRFs then dimerize and translocate to the nucleus15. The IKK protein kinase complex phosphorylates IκB and releases it from NFκB; NFκB is then further activated by phosphorylation by other kinases17.
To evade the IFN system, viruses have evolved many mechanisms to block IFN synthesis and actions — acting at almost every step of the signalling pathway18,19. For example, a hepatitis C virus (HCV)-encoded protease can cleave IPS1 off the mitochondrial membrane and block RIG-I/MDA5-mediated signalling20. Hepatitis B virus (HBV) ORF-C and terminal proteins can also block induction. Influenza virus, Ebola virus, papilloma viruses and the human herpes Kaposi's sarcoma-associated virus (KSHV) encode proteins that interfere with IRF activation or induction and actions of IFNs21. For example, KSHV can downregulate one of the receptor chains for IFN-γ, and the NS1 protein of influenza viruses prevents establishment of an antiviral state through the interaction with RIG-I22,23. Conversely, patients with defects in the production of type I IFNs, due to mutation of the UNC93B gene, are highly susceptible to herpes simplex virus 1 (HSV-1) encephalitis24.
Development of small-molecule activators of induction are only beginning; however, delineation of the responsible signalling pathway has identified many target proteins. CpG oligonucleotides are activators of TLR9 (Ref. 25); the quinolinamine imiquimod26 and its analogues activate TLR7 (Refs 25, 26); and DMXAA induces IFN synthesis through a TLR-independent pathway27. Development of chemical modulators that selectively activate IFN synthesis or block the synthesis of inflammatory cytokines could have a broad therapeutic potential28,29.
IFN proteins and their receptors
The realization that IFNs constitute a protein family arose initially from the definition of antigenic differences between human fibroblast (IFN-β) and leukocyte IFNs (IFN-α)30. Although protein purification studies suggested the potential multigenic nature of IFN-α, this was only firmly established by the cloning of IFN-α1, IFN-α2 and IFN-β31,32,33, which, when accomplished, helped solidify the financial future of the nascent biotechnology industry. The number of functional genes identified that encode type I IFNs has grown subsequently: 17 non-allelic genes have now been described in humans. All lack introns and cluster on chromosome 9 (Refs 34, 35). Of the type I IFNs, there are 13 IFN-αs (plus an additional synthetic concensus sequence and also additional minor allelic variants), whereas there is only one type of IFN-β, IFN-ω, IFN-ɛ or IFN-κ (Fig. 3). Among mammals, the number of type I IFN genes is variable; some have unique types (for example, IFN-δ occurs only in pigs and IFN-τ only in ruminants) and others are devoid of a particular type (for example, IFN-ω in mice). Consistent with the universal biological definition of IFNs (that is, proteins inducing relatively species-specific antiviral effects), all type I IFNs, which are mostly non-glycosylated proteins of 165–200-plus amino acids, share homologies that range from 30–85% within a species. Essentially all have relatively high specific potencies (1 × 107 to 1 × 109 antiviral units per mg protein).
Figure 3. Receptor activation or ligand–receptor complex assembled by type I, type II or type III interferons.
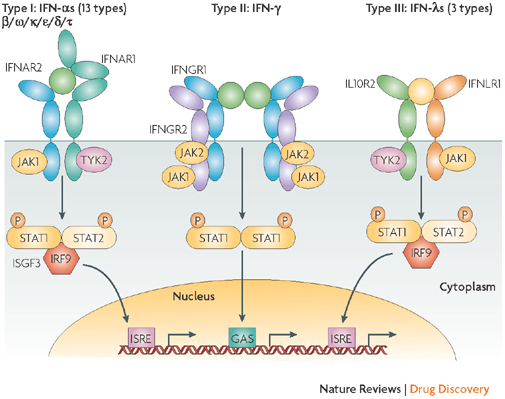
Type I interferons (IFNs) (α, β ω, κ, ɛ, δ (pigs), τ (ruminants)) interact with IFN (α, β and ω) receptor 1 (IFNAR1) and IFNAR2; type II IFN-γ with IFN-γ receptor 1 (IFNGR1) and IFNGR2; and type III IFN-λs with IFN-λ receptor 1 (IFNLR1; also known as IL28RA) and interleukin 10 receptor 2 (IL10R2; also known as IL10RB). Type II IFN-γ is an antiparallel homodimer exhibiting a two-fold axis of symmetry. It binds two IFNGR1 receptor chains, assembling a complex that is stabilized by two IFNGR2 chains. These receptors are associated with two kinases from the JAK family: JAK1 and TYK2 for type I and III IFNs; JAK1 and JAK2 for type II IFN. All IFN receptor chains belong to the class 2 helical cytokine receptor family, which is defined by the structure of the extracellular domains of their members: approximately 200 amino acids structured in two subdomains of 100 amino acids (fibronectin type III modules), themselves structured by seven β-strands arranged in a β-sandwich. The 200 amino-acids domain usually contain the ligand binding site. IFNAR2, IFNLR1, IL10R2, IFNGR1 and IFNGR2 are classical representatives of this family, while IFNAR1 is atypical as its extracellular domain is duplicated. GAS, IFN-γ-activated site; IRF9, IFN regulatory factor 9; ISGF3, IFN-stimulated gene factor 3, refers to the STAT1–STAT2–IRF9 complex; ISRE, IFN-stimulated response element; P, phosphate; STAT1/2, signal transducers and activators of transcription 1/2.
Although type I IFNs have qualitative and quantitative differences in their antiviral and other actions33,34,35,36,37,38,39,40, the reason for origin and maintenance through evolution of these related proteins is unknown. All mammalian species have retained, however, at least one IFN-α and one IFN-β41. In humans, expression of IFN-κ and IFN-ɛ seems tissue specific35,42, but all cells are able to produce other IFNs. In monocyte-derived dendritic cells, in which viral infection induces expression of the 15 IFN-α/IFN-β/IFN-ω subtypes, stimulation of TLR3 or TLR4 induces mostly IFN-β and IFN-α1, which emphasizes the differences in the promoter sequences for the IFN-αs, IFN-ω, and IFN-β genes that govern the response to different inducers43.
Type I IFNs belong to the helical cytokine family with secondary structures of a five α-helix bundle held in position by two disulphide bonds44. They act through a cell-surface receptor composed of two ubiquitously expressed transmembrane proteins, IFN (α, β and ω) receptor 1 (IFNAR1) and IFNAR2 (the genes for which are clustered on chromosome 21), and are associated with two cytoplasmic tyrosine kinases, TYK2 and JAK1 (Ref. 39) (Fig. 3). Formation of the IFN–receptor complex involves one side of the IFN protein interacting with IFNAR2 in a region forming the hinge between the two fibronectin type III (FnIII) domains (Fig. 3); binding affinity is in the nanomolar range39. IFNAR1 binds IFNs with an affinity 1,000-fold weaker than that of IFNAR2, with a binding site located opposite to the IFNAR2 binding site. Binding studies are consistent with the ternary complex between IFNAR1, IFN and IFNAR2 having a 1:1:1 stoichiometry, and a similar if not identical architecture for all type I IFNs. Ternary complex assembly is a two-step process; the ligand binds first to one IFNAR and then recruits the second with no identified interaction between the two IFNARs39.
As affinities for IFNAR2 are generally much higher than for IFNAR1, a binding pathway in which IFNs bind first to IFNAR2 and then IFNAR1 should have a higher probability. However, with IFN-α1 having a low affinity for IFNAR2, the relevance of the reverse-binding pathway, which could lead to differing cellular effects, has been confirmed45. If differences in the structures of the IFN–receptor complexes cannot account for the differential activities of type I IFNs, then a body of argument — which includes studies on the activities of engineered IFNs — suggests that differential affinities for IFNARs and thus, ternary complex stability, govern differential biological activities39,46,47,48. The cell-surface concentration of IFNARs and their lateral organization into microdomains could also be important cellular parameters that shape responsiveness to individual IFNs49. Similar or other changes in receptor organization may also account for the increased susceptibility to HBV infection that occurs with polymorphisms in class II cytokine receptor genes50.
IFN-γ is a single glycosylated protein of 140 amino acids that is designated as a type II IFN because of its distant amino-acid sequence homology with type I IFNs, and its production by natural killer (NK) or activated T cells. Like type I IFNs, it binds to two class II cytokine receptor proteins; when ligand bound it forms a complex of two of each of the receptor proteins linked to an antiparallel homodimer of IFN-γ51 (Fig. 3). IFN-γ receptor 1 (IFNGR1) maps to chromosome 6 and has a JAK1 binding domain and a STAT1 docking site. IFNGR2 contains a JAK2 binding domain and maps to chromosome 21q22.1 in a cluster that also contains IFNAR1, IFNAR2 and interleukin 10 receptor 2 (IL10R2; also known as IL10RB)52. Although IFNGR1 is constitutively present on all cells, IFNGR2 is tightly regulated and less widely expressed. Promoter polymorphisms and/or mutations within both chains have been associated with increased susceptibility to malaria and mycobacteria — in a few patients these defects have been reconstituted by marrow transplantation53. The type III IFN family with three subtypes of IFN-λ, which are co-produced with IFN-β, activate the same main signalling pathway as type I IFNs but have evolved a completely different receptor structure54 (Fig. 3).
How cells respond to IFNs
Initial studies identified several genes that were induced by type I IFNs and analysis of their promoters identified conserved DNA elements55,56,57,58. Proteins bound to these elements after treatment with type I IFNs were purified and identified as STAT1, STAT2 and IRF9 (Refs 59–62). To identify other components of IFN-dependent signalling cascades, the promoter element of the 6–16 gene was used to drive IFN-dependent expression of guanine phosphoribosyl transferase; cells that did not respond to IFNs were then selected with 6-thioguanine. Following chemical mutagenesis, several mutant cell lines were obtained, each lacking a protein essential for signalling. For example, mutant U1A was shown by complementation to lack the tyrosine kinase TYK2 (Refs 60, 62). Subsequently seven STAT and four JAK family members were identified; these transcription factors and tyrosine kinases have been shown to be essential for responses not only to IFNs but also to other cytokines and growth factors as well61.
Minimum requirements for a response to type I IFNs are the heterodimeric IFN receptor; the tyrosine kinases TYK2 and JAK1, which reciprocally transphosphorylate the receptor chains when activated; STAT1 and STAT2, which are phosphorylated in response to signalling; and the unphosphorylated IRF9 (Fig. 3). Transcription in response to IFN-dependent signalling is initiated by high-affinity binding to specific palindromic promoter sequences of the trimeric complex of STAT1, STAT2 and IRF9. The response to IFN-γ requires only the two receptor proteins, the kinases JAK1 and JAK2, and STAT1. STAT1 and STAT3 bind competitively to the same phosphotyrosine residue of IFNGR1, with the binding of STAT1 greatly favoured63. Although initial work was carried out primarily in human fibroblasts, recent studies have identified additional complexity that allows individual cell types to respond by activating different STATs in response to the same IFN (reviewed in Refs 64, 65). Analysis of defects in the IFN system has identified germline mutations in humans that result in deficiencies of STAT1 or TYK2, with enhanced susceptibility to infection by viruses61,66. Although the mouse has been a useful model, the human defects are not always identical to the effects that result from the targeted deletions of these genes in mice61.
Upon activation of receptors, the JAKs undergo autophosphorylation and transphosphorylation to increase their activity, and then phosphorylate the IFN receptors and finally STATs. However, the kinase activities of the JAKs are not sufficient to explain all nuances of signalling. Tissue-specific differences in activating additional protein kinases probably contribute to the differential responses of various cells to a single type of IFN. In at least some cell types, the p85 subunit of phosphatidylinositol 3-kinase (PI3K) is associated with IFNAR1. The activation of p85 by IFN leads to AKT phosphorylation and expression of the chemokine (C-X-C motif) ligand 11 (CXCL11) gene, encoding an important chemokine67. Type I IFNs also activate p38, and inactivation of p38 blocks induction by IFN-β of CXCL11 and TNFSF10 (encoding tumour necrosis factor-related apoptosis-inducing ligand, APO2L/TRAIL)68 and of CXCL10 (encoding the chemokine IP-10; also known as IFN-γ-induced peptide, 10 kDa) in primary leukocytes69.
An important function of the activation of protein serine kinases such as p38 and protein kinase C (PKC) in response to IFN-dependent signalling is phosphorylation, directly or indirectly, of transcription factors70. Serine 727 of STAT1 is phosphorylated in response to IFN-γ by the kinase cascade PI3K—AKT—PKC—MKK4—p38 (MKK4, mitogen-activated protein kinase kinase 4; also known as MAP2K4), with some variation in the activation of different PKC or MKK proteins in different cells65. IFN-dependent activation of PI3K, extracellular response kinases (ERKs) and p38 stimulates the phosphorylation of NFκB (but not IκB), AP-1 and possibly PU.1, respectively. These activated transcription factors may then either drive gene expression independently of activated STATs or cooperate with activated STATs on certain promoters (Fig. 4). Conversely, transcription initiated by phosphorylated STATs does not proceed indefinitely; homeostasis and balance result from the actions of phosphatases such as SHP1 and SHP2 and a family of ISGs, the suppressor of cytokine signalling (SOCS) proteins71,72. SOCS inhibit receptor signalling both by directly inhibiting JAKs and by targeting the receptor complex for proteasomal degradation71.
Figure 4. Complexity of the signalling response.

Different types of cells respond differentially to a single type of interferon (IFN) by varying the activation of specific signal transducers and activators of transcription (STATs), additional transcription factors (TFs) and kinases in addition to the Janus kinases (JAKs). Priming of cells by pre-treatment with another cytokine modulates the response further by increasing the amounts of negative regulators and by modulating other processes. Most genes require STATs, with or without additional TFs, and several genes respond only to activated TFs and not to STATs. The STATs bind to IFN-γ-activated site (GAS) elements or, together with IFN regulatory factor (IRF) proteins, to IFN-stimulated response elements (ISREs), and the TFs bind to specific binding elements (TFBE). CIS, cytokine inducible SH2-containing protein; PTP, protein tyrosine phosphatase; SOCS1, suppressor of cytokine signalling 1.
Prior exposure to other cytokines conditions how a cell will respond and, conversely, IFNs condition responses to other cytokines64,65,73. An excellent example of such an effect is prior exposure of human macrophages to IFN-γ, which changes the response to IL10 from activation of STAT3 to activation of STAT1 (Ref. 74). Although, as reductionist scientists, we tend to study the responses of cells in culture to treatment with IFNs alone, the situation in vivo is obviously much more complex.
Both STAT1 and STAT3 have activities in addition to their roles as cytokine-activated transcription factors. STAT1 is activated in response to both type I and type II IFNs and STAT3 is activated in response to gp130 cytokines such as IL6. As STAT1 and STAT3 drive essentially opposite biological responses, large signal-dependent changes in their concentrations will affect their relative activation by a further signal. Indeed, an increase in the ratio of STAT1–STAT3 after IFN-α2 treatment of patients with melanoma correlated with survival75. Another consequence of cytokine-dependent increases in STAT expression is that unphosphorylated STAT1 and STAT3 have important functions that are quite distinct from those of the phosphorylated proteins76,77. For example, unphosphorylated STAT3 activates a subset of κB-dependent genes by forming a complex with NFκB78. Thus the role of STATs in signalling after receptor binding has expanded from kinase-activated transcription factors to proteins that, even in the absence of ligand activation, activate transcription and participate in cell-type specificity, resulting in diverse patterns of ISG induction in different cell types in response to a single IFN.
ISGs: molecular mechanisms of antiviral action
ISGs are a diverse group of more than 300 genes (which and how many are a function of cell-type signalling variations as discussed above) that mediate the biological and therapeutic effects of IFN stimulation79,80 (Table 1). Studies of their mode of action have resulted in fundamental discoveries concerning translational control, regulation of RNA stability and editing, and protein transport and turnover18. Furthermore, proteins that are induced upon IFN stimulation, especially those that can be activated or inhibited in vivo, are targets for high-throughput screening for identification of new modulators of the IFN system.
Table 1.
ISGs in the antiviral and anticancer effects of IFNs
| Gene | Protein function | Mechanism of action | Refs |
|---|---|---|---|
| OAS, RNASEL | RNA cleavage | Degrades viral and cellular RNA, induces IFN-α/β | 81–91 |
| PKR | EIF2α phosphorylation | Blocks protein synthesis, transcriptional signalling | 94–98 |
| p56 | Binds EIF3 | Blocks protein synthesis | 99,100 |
| Mx1 | Wraps around viral nucleocapsids | Interferes with intracellular trafficking | 101–104 |
| ISG15 | ISGylation | Cytokine-like, protein modification | 105–107 |
| PLSCR1 | Phospholipid migration, DNA binding | Enhances expression of some ISGs | 110–112 |
| TRAIL/APO2L | Ligand of death receptor | Apoptosis | 113–115 |
| XAF1 | Blocks inhibitor of apoptosis (XIAP) | Apoptosis | 247 |
| G1P3 (6–16) | Inhibits caspase 3 | Anti-apoptotic | 116,117 |
| ISG12 | Not known | Antiviral | 119,120 |
| GBP1 | GTPase | Antiviral, angiogenesis inhibitor | 121,268 |
| ISG20 | 3′-exonuclease for RNA and DNA | Antiviral | 122 |
| PML | Not known | Antiviral, antitumor | 123 |
| ADAR1 | Adenosine deaminase for dsRNA | RNA editing, altered translation | 124 |
| Viperin (cig5) | Not known | Antiviral | 125 |
| iNOS | Nitric oxide synthase | Antiviral | 126 |
| Nup98/Nup96 | Nucleoporin, RNA and protein transporters | Antiviral | 127,270 |
| IRF7, RIG-I, MDA5, STAT1 | Signalling to IFN-α/β genes or to ISGs | Induction of type I IFNs | 79,80 |
| ADAR1, adenosine deaminase, RNA-specific; dsRNA, double-stranded RNA; EIF2α/3, eukaryotic initiation factor 2α/3; G1P3, interferon, α-inducible protein; GPB1, guanylate binding protein 1, interferon-inducible, 67kDa; iNOS, inducible nitric oxide synthase; IRF7, interferon regulatory factor 7; ISGs, interferon-stimulated genes; MDA5, melanoma differentiation associated protein 5 (also known as IFIH1); Mx1, myxovirus (influenza virus) resistance 1, interferon-inducible protein p78; Nup98/Nup96, nucleoporin 98/96 kDa; OAS, 2′-5′-oligoadenylate synthetase; PKR, protein kinase R; PLSCR1, phospholipid scramblase 1; PML, promyelocytic leukaemia; RIG-I, retinoic acid-inducible gene I (also known as DDX58); RNASEL, ribonuclease L; STAT1, signal transducer and activator of transcription 1, 91 kDa; TRAIL/APO2L, tumour necrosis factor-related apoptosis-inducing ligand (also known as TNFSF10); Viperin (cig5), also known as RSAD2; XAF1, X-linked inhibitor of apoptosis-associated factor 1; XIAP, X-linked inhibitor of apoptosis protein (also known as BIRC4). | |||
Examples for this are 2′,5′-oligoadenylate synthetases (OASs) and ribonuclease L (RNASEL), which inhibit a broad range of RNA viruses81. Viral dsRNA can directly activate one of several human OAS proteins to produce a unique 2′-to-5′ linked oligoadenylate of 3–6 bases (2–5A) from ATP82. The only well-established function of 2–5A is activation of the ubiquitous, latent enzyme, RNASEL83. 2–5A binding to RNASEL induces monomeric, inactive RNASEL to dimerize into a potent endoribonuclease that cleaves single-stranded regions of RNA on the 3′ side of UpUp and UpAp dinucleotides84,85,86. The OAS–RNASEL pathway can inhibit the replication of encephalomyocarditis virus, Coxsackie virus B4, West Nile virus, some retroviruses and HCV81. Furthermore, degradation of cellular mRNA and rRNA by RNASEL damages the host cell machinery that is required for viral replication and can result in apoptosis, contributing to both antiviral and antitumour actions87,88,89. RNASEL also cleaves self-RNA into small degradation products that activate the recognition receptors, RIG-I and MDA5, to induce IFN-β, similar to that of non-self viral RNA90, thus perpetuating and amplifying the production of IFN-β. A high-throughput screen has resulted in the identification of small molecules that can activate RNASEL and produce broad-spectrum antiviral effects91.
The dsRNA-activated protein kinase (PKR) and OAS were the first enzymes identified that uniquely respond to IFNs92,93. PKR is a serine/threonine kinase that mediates translational and transcriptional control in response to dsRNA and other signals92,93,94,95,96. In addition, the cellular protein PACT (also known as PRKRA) activates PKR in the absence of dsRNA97. PKR mediates translational control by phosphorylating the protein synthesis initiation factor EIF2α, resulting in an inactive complex between EIF2–GDP and the recycling factor, EIF2B. These events produce global inhibition of protein synthesis that blocks further viral replication and full amplification of the viral-induced cellular stress response. Many viruses, however, evade PKR through a range of strategies such as binding and sequestering dsRNA, thus depriving PKR of its activator or inhibition of its kinase activity98.
Another ISG family that influences translation is the strongly induced p56-related proteins (IFIT gene products). Two of these, p56 and p54, inhibit protein synthesis by blocking the action of the translation initiation factor EIF3 (Ref. 99). p56 and p54 bind to different subunits of EIF3 and block some of its diverse functions. HCV mRNA translation is inhibited more strongly by p56 than by cellular mRNAs, because its initiation is internal ribosome entry site (IRES)-mediated, and not CAP-mediated; thus it selectively inhibits viral protein synthesis100.
The ISG-encoded antiviral protein Mx was identified because of the resistance of mouse strain A2G to influenza A viruses (Mx1, orthomyxovirus resistance gene 1)101,102,103. Mx proteins are large (∼80 kDa) GTPases in the dynamin superfamily that self-assemble and bind viral nucleocapsids. This interferes with intracellular trafficking and activity of viral polymerases, thus inhibiting replication of many RNA viruses including influenza and measles viruses104. The human homologue, MXA, is a cytoplasmic protein that associates with intracellular membranes.
ISG15 encodes a ubiquitin-like, 15 kDa protein that modifies more than 100 proteins through a process known as ISGylation105,106,107. ISG15 inhibits HIV-1 release from cells, mimicking the effect of IFN108 and infections by influenza, herpes and Sindbis viruses109. The antiviral mechanism of ISG15 in vivo is unknown but could relate to its cytokine-like properties or to its ability to conjugate and modify the function of cellular or viral proteins. ISG15 is also a target gene for dysregulation of the p53 and ISG pathways that occurs in many types of cancer, suggesting an additional role in tumorigenesis107.
Phospholipid scramblase 1 (PLSCR1), a protein implicated in Ca2+-dependent reorganization of plasma membrane phospholipids110, either inserts into the plasma membrane or binds DNA in the nucleus depending on its palmyitoylation111. PLSCR1 has antiviral activity that possibly results from the enhanced transcription of a subset of ISGs112. TRAIL/APO2L is an ISG that contributes to apoptosis and therefore probably to both the antiviral and antitumour effects of IFNs113,114,115. Although many ISGs promote apoptosis, some promote cell survival. For example, the ISG G1P3 (or 6–16)116,117 localizes to mitochondria and has anti-apoptotic actions, including inhibition of caspase-3 (Ref. 118). A related ISG, encoded by IFI27 (ISG12)119, promotes an age-dependent resistance to alphavirus encephalitis in mice without affecting either levels of apoptosis or viral yields120. Additional ISGs with probable or confirmed antiviral activities include the guanylate-binding protein 1 (GBP1); a 3′,5′-exonuclease encoded by ISG20; the promyelocytic leukaemia protein (PML); adenosine deaminase (ADAR1); the endoplasmic reticulum-associated protein Viperin (cig5) that can inhibit human cytomegalovirus; inducible nitric oxide synthase (iNOS); and the nucleoporins Nup98 and Nup96 (Refs 121–128).
Finally, many IFN-pathway signalling proteins are themselves ISGs, thus providing an autocrine loop that amplifies IFN responses; examples are IRF7, RIG-I, MDA5 and STAT1. As ISGs with high levels of transcriptional induction are still poorly characterized functionally79,80,128, some could prove to be critical mediators of antiviral and other actions. Because all biological effects of IFNs are mediated through the action of ISGs (Table 1), further research into understanding the functions of the protein products of these may lead to more efficacious antiviral and other therapeutics.
Innate and adaptive immunity
In addition to direct inhibition of viral replication by ISGs, a second level of IFN action augments adaptive and acquired immune responses. Early warning of pathogen presence is delivered by tissue-associated and circulating dendritic cells, one type of which, the plasmacytoid dendritic cell, is the circulating type I IFN-producing cell129. In addition to TLR activation on cells at the sites of pathogen invasion or replication, this response culminates with dendritic cell-mediated presentation to CD4+ T cells of pathogen-derived peptide fragments that are bound to surface major histocompatibility complex (MHC) class II molecules. MHC class II proteins are selectively upregulated by IFN-γ, whereas type I IFNs fail to do so owing to the STAT2-dependent induction of SOCS1 (Ref. 130). Infected cells that display peptide fragments associated with MHC class I molecules on the surface are recognized and subsequently eliminated by CD8+ T cells, thereby clearing the virus. Either type I IFNs or IFN-γ can markedly upregulate MHC class I-dependent antigen presentation. In addition to MHC molecules, other ISGs involved in antigen processing include the lysosomal membrane permeabilization (LMP) components of proteasomes and transporters for antigen processing (TAPs), which shuttle peptides into the endoplasmic reticulum for loading onto nascent MHC class I proteins131,132,133,134,135.
IFNs also promote accumulation of leukocytes at sites of pathogen invasion; specifically, IFNs (along with cytokines such as TNFα and IL1β), strongly promote the expression of vascular adhesion molecules including intracellular adhesion molecule 1 (ICAM1). Furthermore, IFNs induce the production of chemotactic cytokines (chemokines), which participate in leukocyte recruitment. As examples, three closely related chemokines involved in accumulation of activated T cells and macrophages are the ISGs CXCL9 (also known as MIG, monokine induced by IFN-γ); CXCL10 (also known as IP-10, IFN-γ 10 kD inducible protein); and CXCL11 (also known as I-TAC, interferon-inducible T-cell α-chemoattractant)136,137,138,139. True to their names, these chemokines are not expressed in the absence of IFN signalling.
In the development of an adaptive immune response, IFN-γ is produced by an early warning NK cell, or by activated T cells140. IFN-γ governs expression of class II transactivator (CIITA), a master regulator of transcription of the MHC class II molecules themselves, as well as the associated invariant chain, which helps stabilize MHC class II heterodimers. HLA-DM catalyses the displacement of the invariant chain from the MHC class II peptide binding site as the mature MHC class II-peptide conjugate is finalized for insertion into the plasma membrane141,142,143,144,145,146,147,148,149. Finally, of substantial importance in the host response to the virus is the IFN-mediated activation of cytotoxic effector function among cells of innate and adaptive immunity including NK cells, dendritic cells, macrophages and T cells. Indeed, the property of stimulating macrophages contributed substantially to the recognition of IFN-γ as a biologically important lymphokine and as a 'different' IFN150.
Human therapeutic applications
Based on preclinical studies of broad spectrum inhibition of virus replication, IFNs were initially investigated as antivirals with activity against RNA and DNA viruses. Clinical effectiveness for both has now been established. But development of relatively specific, low molecular mass antivirals has largely supplanted broad application except for HBV and HCV chronic infections. The first US Food and Drug Administration (FDA) approval for IFN-α2, however, was not for virus infection but for cancer, which was driven by interest created by publicity resulting from its effectiveness in American Cancer Society trials. Subsequently, placebo-controlled randomized trials established the effectiveness of IFN-β for relapsing, remitting MS — an apparent paradox in terms of the mechanistic understanding of IFN actions, as IFNs, as discussed above, are generally immune augmenting rather than immunosuppressive. Presently, a number of drugs are being or have been designed to target different components of the IFN system for different therapeutic indications (Fig. 5).
Figure 5. Potential drug targets in the interferon (IFN) system.
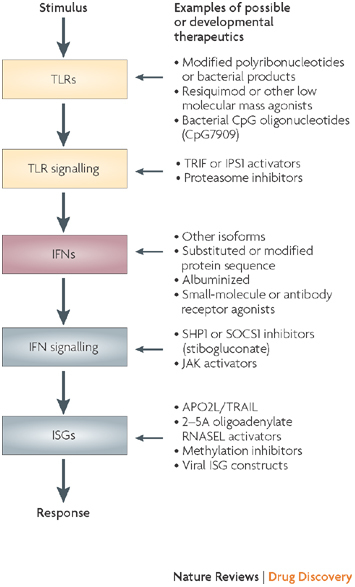
Examples of potential or developmental drugs targeted at different steps in the pathways are presented. IPS1, IFN-β promoter stimulator 1 (also known as VISA); ISG, IFN-stimulated gene; JAK, Janus kinase; RNASEL, ribonulcease L; SOCS1, suppressor of cytokine signalling 1; TRAIL, tumour necrosis factor-related apoptosis-inducing ligand (also known as APO2L); TRIF, TLR adapter molecule 1.
Viruses. The recognition that HBV often caused a chronic infection leading to cirrhosis and hepatocellular carcinoma suggested that infected patients might benefit from IFNs151. Initial clinical trials of impure IFN-α152 suggested benefit but studies with impure IFN-β were less promising153. These low-dose studies were followed, however, by higher doses of recombinant IFNs, when they became available, which then confirmed beneficial effects154.
HBV chronic infection evolves with hepatitis e antigen (HBeAg)-positive quiescent viruses escaping inhibition during conversion of an immunotolerant to an immunoactive phase, with enhanced immune elimination of infected hepatocytes155,156. In many, this immune response causes suppression of viral replication and HBeAg loss. A quiescent phase or 'healthy carriage' may ensue but disease reactivation is common (HBeAg-negative disease157). In HBeAg-positive early HBV infection, IFNs have not been particularly effective. However, in the immunoactive chronic phase, HBV is sensitive to IFN-α2 and the ongoing immune response is augmented, leading to quiescent HBeAg-negative disease in up to 40% of patients. IFN-α2 (Roferon-A, Hoffmann-LaRoche; Intron-A, Schering–Plough), now usually in the form of a long-acting pegylated version, has been widely used to treat HBeAg-positive HBV infections158.
IFN-α2 has also been used in the HBeAg-negative disease that develops when viral mutations permit viral reactivation following HBeAg loss. IFNs reduce viraemia (usually by over 90%) and induce host responses, but drug withdrawal often leads to disease recurrence; however, a proportion of patients (approximately 10–15%) have a prolonged period of viral suppression159. Therapy for chronic HBV infection illustrates the two complementary activities of IFNs: in HBeAg-positive disease IFN increases an immune response, whereas in HBeAg-negative disease IFNs act as direct antivirals.
In the late 1980s 'non-A, non-B hepatitis' or 'post-transfusion hepatitis' was effectively treated with IFNs160. Subsequent studies identified the causative agent as HCV161. Initial clinical studies of IFN-α2 resulted in sustained and curative virological responses in up to 20% of patients162. Response rates to monotherapy with IFN-α2 for chronic HCV infection were transformed in the mid-1990s by combined use with the weak antiviral ribavirin — over 40% of patients responded163. These results mimic studies for HSV keratitis in which therapy with a combination of IFNs and a weak antiviral agent (acyclovir) were synergistic164. Therapy for chronic HCV infection has now evolved and current regimes commonly use a long-acting pegylated IFN-α2 plus ribavirin with cure of up to 60% of patients. Possibly as a result of selection against RNASEL cleavage sites in its genome, or inhibition of PKR, effectiveness is much less for genotype 1 than for genotypes 2 and 3 of HCV162,271,272.
In addition to HBV and HCV infections, other chronic viral infections have been effectively treated. Both systemic and topical IFN-αs and IFN-β have reduced virus titres and decreased clinical manifestations of herpes zoster, HSV and cytomegalovirus infections165,166,167,168,169. Almost simultaneous introduction of acyclovir and its analogues, however, which proved to have greater clinical efficacy and reduced side effects, ended clinical development of IFNs for these indications. Papilloma virus infections of skin, larynx and genitals were found to respond with regression of warts upon either intralesional or systemic administration of IFN-αs and IFN-β170,171,172,173,174,175. When compared with placebo, useful therapeutic effects resulted for patients with extensive or refractory disease, but permanent eradication was infrequent. These studies did, however, establish a basis for the use of the TLR7 IFN-inducing agonist imiquimod topically for genital warts with decreases in HPV DNA and with complete response frequencies of approximately 50% compared with 5% in placebo controls176,177,178. In studies of HIV, virus recovery and clinical manifestations of both early and late stages were reduced179. However, effectiveness of azidothymidine or protease inhibitors was not enhanced when IFNs were used in combination180.
Once adequate quantities of human IFNs became available through recombinant DNA production, prophylaxis and treatment of acute respiratory virus infections were assessed. Reduction in virus yield, infection frequency and symptom scores resulted from administration of intranasal IFNs in experimental challenge infections with rhinoviruses, influenza viruses and coronaviruses181,182,183,184. Prophylactic efficacy for natural rhinovirus colds in family and work settings was also identified185,186,187. However, as a result of the nasal erosions and bleeding resulting from mucosal irritation by IFN-α2 and IFN-β, symptom scores under field conditions were significantly higher in treated patients when compared with placebo. Case-based use in the setting of the severe acute respiratory syndrome (SARS) epidemic suggested clinical effectiveness of the consensus sequence type I IFN against the coronavirus aetiological agent188. These findings and the preclinical and clinical antiviral effectiveness with topical and/or high-dose administration suggest that IFNs might have prophylactic or therapeutic effectiveness in SARS or in an influenza or other virus pandemic.
Multiple sclerosis (MS). In about 85% of patients with MS, an inflammatory demyelinating disorder of the central nervous system, disease begins with approximately annual episodes of transient neurological dysfunction (relapsing–remitting MS or RR-MS). Initial studies of IFNs in the 1970s followed tissue-culture studies suggesting that cells from MS patients secreted less IFN-like activity following viral induction than did controls. These findings, combined with a notion that a slow or chronic viral infection might be causative, resulted in the evaluation of using an intrathecal, impure IFN-β as therapy that identified a reduction in relapses189; however, subsequent clinical trials were either inconclusive (IFN-a2) or detrimental (IFN-γ)190,191,192. But in 1993, recombinant IFN-β given subcutaneously in a randomized placebo-controlled trial for RR-MS reduced relapses by about a third and resulted in marked reductions in subclinical disease, as assessed by magnetic resonance imaging (MRI)193,194. This report ushered in the modern age of MS therapeutics: by showing that the natural history of MS could be modified; by documenting that IFN-β was clinically beneficial; and by the demonstration of MRI lesions as a useful surrogate of clinical effectiveness, now widely used in MS drug development. It is now common clinical practice to initiate IFN-β (Betaferon/Betaserom, Bayer Schering/Chiron; Avonex, Biogen Idec; Rebif, Merck Serono (or medications of comparable efficacy)) at the time of diagnosis195. Attacks decrease by about 30%, numbers of new and active MRI lesions (which reflect inflammation) are often reduced as soon as 1-month after initiation, and long-term clinical benefits are now considered plausible196. Although it has unequivocally represented a breakthrough, improvements on IFN-β are needed as it is only partially effective and is expensive for the life-long, non-curative use195,197.
Pathogenesis of MS remains unknown but evidence implicates genetic–environmental interactions with critical timing of exposures to initiating factors. Epidemiological studies highlight Epstein–Barr virus and low plasma levels of vitamin D, and genetic studies implicate several polymorphic variants of immune-response genes198,199,200,201. The most obvious clinical outcome from IFN-β is a reduction in MRI lesions202,203,204, and protein products of ISGs probably mediate these effects.
As one example, an IFN-β ISG product, CD69, forms an inhibitory association with a sphingosine 1-phosphate receptor (S1PR). The consequence in vivo is suppressed lymphocyte exit from lymph nodes and restriction in numbers of circulating lymphocytes available to cross the blood–brain barrier205. Reduction in expression of matrix metalloproteinase 9 (MMP9) in activated lymphocytes and increased soluble vascular cell adhesion molecule (sVCAM) levels in plasma have also been identified and assigned putative roles in the beneficial effects of IFN-β for patients with MS202,206,207. Expression-array studies and candidate gene evaluations have been applied, without success, in attempts to identify molecular biomarkers of the therapeutic effect of IFN-β in MS208. Development of validated outcome measures for treatment success (and failure) will aid in this process209.
Cancer. Based on the reduction in disease morbidities210,211, initial regulatory approvals for the marketing of IFN-α2 for a chronic B (hairy) cell leukaemia occurred within 5 years from clinical introduction as a result of close collaboration between academic institutions, government and industry. In hairy cell leukaemia and chronic myelogenous leukemia (CML), IFN-α2 decreased marrow infiltration with malignant cells and normalized peripheral haematological parameters210,212,213,214. In CML, in addition to reductions in leukaemic cell mass, a decrease resulted in cells with the abnormal, activated BCR–ABL kinase212,213,214. Over 90% of patients with CML with complete cytogenetic response were in remission at 10 years213. However, the survival advantage for IFN-α2, when compared with chemotherapy for CML, has now been exceeded by the even greater effectiveness of the targeted inhibitor of the activated BCR–ABL kinase, such as imatinib (Gleevec; Novartis) and now other newer tyrosine kinase inhibitors.
In addition to hairy cell leukaemia and CML, therapeutic effectiveness of IFN-α2 in causing at least partial disease regression has been identified in more than a dozen other malignancies including myeloma, lymphomas, melanoma, renal cell and bladder carcinoma, and Kaposi's sarcoma215. For example, in lymphomas of various histologies and of both B-cell and T-cell phenotypes, IFN-α2 has been effective in inducing tumour regressions in almost half of the patients involved in the study, and even in patients previously treated with chemotherapy215,216. Prolonged disease-free and overall survival in intermediate prognosis lymphomas has resulted from IFN-α2 in combination with chemotherapy, even given for limited periods, in randomized multicenter trials215,216. International Phase III trials have been conducted with survival impact confirmed in metastatic renal carcinoma, but like in CML, the orally active, targeted tyrosine kinase inhibitors have changed the natural history of renal carcinoma, extending survival in metastatic disease more than the injectable IFN-α2.
Cure of metastatic malignancies can result when micrometastases are eliminated in patients at highest risk for recurrence after surgical removal of a primary tumour. Effectiveness as a surgical adjuvant for murine tumours provided the rationale leading to pioneering clinical studies that suggested benefit of impure IFN-α when given after surgery for osteosarcoma217,218. This surgical adjuvant approach was the basis for evaluation of IFN-α2 in patients at high risk for recurrence of melanoma. Initial beneficial effects of significant prolongation of disease-free survival have now largely been validated by combined analyses of multi-institutional trials, by subsequent studies that have included evaluation of pegylated IFN-α2 and by meta-analyses219,220,221.
Like other potent physiological mediators such as glucocorticoids, IFNs have toxicities when administered with pharmacological intent222,223. These have been dose related and particularly difficult at the high dose used for melanoma. With the initial dose, malaise, fever and chills, which last for a few hours, dominate but tachyphylaxis occurs with subsequent injections. Fatigue and anorexia, the aetiology of which is not understood, are often dose-limiting with chronic administration for cancer or MS; at higher doses weight loss occurs and may be significant (>10%). Reversible elevation of hepatic transaminases may occur, as may haematological effects, most markedly granulocytopaenia.
Like in MS, failure to fully understand the mechanism(s) of antitumour action has slowed further development. Suppression, mutation and polymorphisms of IFNs and their signalling mechanisms in and by malignant cells are emerging as important contributors to cancer development224,225,226,227,228,229,230,231,232,233,234. Mutations in RNASEL have been associated with prostate carcinoma and with presence of the retrovirus XMRV235,273,274,275. Epigenetic and genetic silencing of IFN-signalling or ISG expression may also influence tumour development236. Reversal of these effects are likely to be the basis for effectiveness of IFNs and/or inducers in murine carcinogen-induced tumours, and may contribute to effectiveness in advanced disease, and provides a rational for developing TLR agonists for chemoprevention237,238,239. Indeed, TLR agonists appear to be effective and are already establishing a role in treatment of malignancy with the proven effectiveness of the TLR7 agonist imiquimod used topically for basal cell carcinomas as an example. Furthermore, relative clinical safety has been established for phosphorthioate oligoribonucelotide agonists for TLR9.
Induction of apoptosis by the ISG products APO2L/TRAIL and Fas has been identified in many malignant cell types, as has induction of APO2L/TRAIL on immune effector cell surfaces, thus sensitizing tumour cells to T-cell, NK cell and macrophage-mediated cytotoxicity240,241,242,243 (Table 1). Intralesional administration of IFN-α into basal cell carcinomas increased Fas expression and correlated with regression244. IFN-γ has increased susceptibility to apoptosis by Fas activators and cytotoxic chemotherapies in many cell types including melanoma and colorectal carcinoma245,246. Through interactions with p53 and the inhibitor of apoptosis, XIAP, the ISG product XAF1 may allow APO2L/TRAIL to fully activate downstream caspases247,248. In addition, the ISG product IRF1 can suppress another anti-apoptotic protein, Survivin249.
Antitumour activity in vivo may also be mediated by augmented lytic activity of immune effector cells and by enhanced immunogenicity of tumour cells. Both T-cell and NK-cell trafficking, expansion and lytic activity can be promoted by IFNs and ISGs; furthermore, IFN-γ is secreted from these activated cells into the tumour microenvironment250,251,252,253,254,255. In addition to stimulating immune effector cells, IFNs have critical roles in antigen processing and presentation, as discussed above, both by T cells and dendritic cells256. In addition, IFN-γ can upregulate the tumour-associated antigens, carcinoembryonic antigen and TAG72, both in vitro and in vivo257.
IFNs can also inhibit angiogenesis by altering the stimuli from tumour cells and by directly inhibiting endothelial cells — indeed, they were the first angiogenic inhibitor identified258. Endothelial cells are inhibited in motility259, undergo coagulation necrosis in vitro and inhibition of angiogenesis occurs in vivo within 24 hours of tumour cell inoculation260,261. Suppression of basic fibroblast growth factor (bFGF; also known as FGF2) correlated with reduced vascularization and tumour growth262,263. IFNs also inhibit vascular endothelial growth factor (VEGF) mRNA and protein expression by regulating its promoter264. IL8, a mediator of angiogenesis, was inhibited in vitro and in vivo by IFN-α2b and IFN-β; other angiogenesis inhibitory members of the chemokine family, CXCL9, CXCL10 and CXCL11, are ISGs265,266,267. In endothelial cells, the ISG product guanylate binding protein 1, interferon-inducible, 67 kDa (GBP1), functioned as an inflammatory response factor inhibiting endothelial cell proliferation and angiogenesis in part through MMPs268. Clinically, IFN-α2 has proved effective in the treatment of infantile haemangiomas, haemangioblastomas, giant cell tumour of the mandible and Kaposi's sarcoma215,269. Thus, induction of ISGs that function as angiostatic inhibitors, coupled with secondary downregulation of angiogenic factors, may contribute to antitumour effects269.
Perspective
IFNs provide fundamental cellular defence mechanisms against viral infections and cancer and are thus critically important to the health of animals and humans. Because of their clinical effectiveness in limiting virus replication, reducing tumour cell mass, controlling disease symptoms and prolonging survival, IFNs are now licensed worldwide for the treatment of various viral, malignant and immune disorders; market sales approach US$4 billion. As part of the innate immune response, IFNs are not only a principal cytokine that blocks viral replication through the action of specific ISGs, but also (particularly IFN-g) mediate critical elements of the cellular immune response for recurring bacterial infections in chronic granulomatous disease and for mycobacteria. Because all biological effects of IFNs are mediated through the action of ISGs, understanding the functions of these genes may lead to more efficacious anticancer and antiviral therapeutics. For example, certain IFN-regulated proteins, such as OAS, RNASEL and PKR, exist in either latent inactive or active states, which could be targeted for potent antitumour and/or antiviral effects (Fig. 5).
IFNs have therefore more than reached the effectiveness anticipated by early virologists: they are not only an antiviral with a spectrum of clinical effectiveness against both RNA and DNA viruses, but have been the prototypical biological response modifiers for oncology, and have proved to have effectiveness in suppressing manifestations of MS. The study of IFNs has resulted in fundamental insights into cellular signalling mechanisms and innate and acquired immunity. In addition, their therapeutic use has improved the quality and quantity of life for millions of patients worldwide. However, to fully realize their potential, many questions remain unanswered (Box 1). As exemplified by recent publications276,277,278, further investigations will only enable IFNs to have even greater impacts in biomedicine.
Box 1 | Important research questions for the future.
What specific roles do the multiple isoforms of interferons (IFNs) play in host defences?
What new roles will be identified for the IFN system and their protein products in the crosstalk between innate and cellular immunity?
How do cellular gene products avoid activating Toll-like receptor (TLR) pathways while microbial gene products activate them very efficiently?
What are cell type differences in other alternative signalling pathways interacting with JAK/STAT?
What are cell and tissue specific differences in molecular responses?
How do responses differ in cells of different maturities (for example, stem cells, senescent cells)?
What are the functions of the still many uncharacterized IFN-stimulated genes (ISGs)?
Which of the ISGs are most important for the specific cellular and clinical effects?
Will ways of activating the IFN system prove therapeutically useful for the newly human papilloma virus (HPV)-associated squamous neoplasms of upper airways?
What mechanisms cause the difficult spectrum of clinical side effects of fatigue and anorexia?
Do IFNs play a bystander or pathogenic role in lupus erythematosis, rheumatoid arthritis or aplastic anaemia?
Do low or high-affinity antibodies to IFNs or their protein products influence innate immunity?
What are the molecular causes of resistance to IFNs in viral diseases? Multiple sclerosis? Cancer?
Will helicases or proteases augment activity of IFNs for hepatitis B virus infections?
Will anti-angiogenic or promoter methylation inhibitors increase antitumour activity?
Can novel receptor agonists and antagonists be engineered for specific functions?
Can a physicochemical rather than biological standard be developed for scientific and regulatory comparison?
Will small-molecule activators of IFN-inducible proteins have antiviral and/or antitumour effects?
How effective will components of the IFN system prove as immunological adjuvants?
Can effective oral systemic inducers be identified?
Acknowledgements
The authors are indebted to Sandya Rani and Kristin Kraus for careful reading of parts of the manuscript. This work was supported in part by grants to the authors from NIH R01 CA90914, CA115,494, CA044059, CA103943, CA089132, CA62220, CA68782, M01 RR018390, NS32151, NS38667, ARC 3158.
Glossary
- CpG oligonucleotides
Bacterial DNA oligodeoxynucleotide sequences that include a cytosine–guanosine sequence and certain flanking nucleotides that have been found to induce innate immune responses through interaction with Toll-like receptor 9.
- DMXAA
5,6-dimethylxanthenone-4-acetic acid. An experimental anticancer drug currently in clinical trials for lung and prostate cancer. It is classified as a vascular disruption agent, causing apoptosis (death) of vessel endothelial cells and the release of vasoactive molecules, which inhibit the formation of new tumour blood vessels.
- Antiviral unit
An antiviral unit is the concentration of an interferon required to inhibit virus replication in vitro by 50%; an international WHO standard provides a reference base for each major interferon type.
- Caspase
A group of enzymes that have a role in promoting apoptosis (that is, programmed cell death). Inhibition of such enzymes might be useful for combating cell and tissue damage in conditions such as myocardial infarction, stroke, inflammatory diseases and neurodegenerative diseases. Augmentation of such enzymes, through the production of pro-apoptotic proteins, might be useful for combating proliferative conditions, such as cancer.
- Inducible nitric oxide synthase
(iNOS). An inducible haem-containing enzyme that produces nitric oxide in response to inflammatory signals.
- Nucleoporins
Proteins that function in the nuclear transport of protein and RNA.
- MRI lesions
Regions of abnormal signals in the brain or spinal cord, detected by magnetic resonance imaging (MRI), and indicative of tissue changes related to the multiple sclerosis pathogenic process. Depending on the imaging technique, these MRI changes can reflect inflammation, demyelination, axonal destruction or scarring.
- Glucocorticoids
A group of compounds that belongs to the corticosteroid family. These compounds can either be naturally produced (hormones) or synthetic. They affect metabolism and have anti-inflammatory and immunosuppressive effects. Many synthetic glucocorticoids (for example, dexamethasone) are used in clinical medicine as anti-inflammatory drugs.
Biographies
Ernest C. Borden obtained his undergraduate degree from Harvard College, Cambridge, Massachusetts, USA, and his medical degree from Duke University, Durham, North Carolina. After appointments at the University of Wisconsin Comprehensive Cancer Center and then as Director of the Cancer Centers of the Medical College of Wisconsin and the University of Maryland, he joined Cleveland Clinic, Ohio, in 1998 to direct the Center for Cancer Drug Discovery and Development. In 2005, he was named Director of Center for Hematology and Oncology Molecular Therapeutics (CHOMT). He is also a staff member in the Departments of Solid Tumor Oncology and Cancer Biology and is a Professor of Molecular Medicine in Cleveland Clinic Lerner College of Medicine, Case Western Reserve University. In the 1980s, he was amongst the first doing clinical trials of interferons for cancer. In addition to developing improved approaches to clinically assess interferons and their inducers, Dr Borden's laboratory has focused on the function and action of genes that are stimulated by interferons and on the anti-tumour effects of other protein therapeutics. He has published over 200 articles and book chapters on interferons. Borden also has an international reputation for research and treatment of melanomas and sarcomas. He received the Milstein Award from the International Society of Interferon and Cytokine Research (ISICR) in 2004, and an American Cancer Society Distinguished Service Award in 1984. He also serves on the Scientific Advisory Boards of Cleveland BioLabs and Alios Bio Pharma.
Ganes C. Sen received his Ph.D. in biochemistry from McMaster University, Canada. During his postdoctoral training with Peter Lengyel at Yale University, USA, he began investigating the interferon (IFN) system. He has continued and expanded his activities in this area of research in his own laboratory, first at the Memorial Sloan-Kettering Cancer Center, USA, and then at the Lerner Research Institute of The Cleveland Clinic, USA, where he nucleated the formation of a strong cytokine research group. He is currently the Interim Chair and professor of molecular genetics at The Cleveland Clinic and Vice-Chair of the Lerner Research Institute. Sen has published extensively on the mechanism of actions of IFN-induced proteins and the mode of induction and actions of double-stranded RNA-stimulated genes. For his contributions to IFN research, he received the Milstein Award in 2002. In another line of research, Sen studies the physiological roles of angiotensin-converting enzyme in blood pressure regulation, male fertility and kidney functions. Sen is a consultant for the National Institutes of Health (NIH) and the American Foundation for AIDS Research. Currently, he is a Senior Editor of the Journal of Virology and Editor-in-Chief of the Journal of Interferon and Cytokine Research.
Gilles Uzé (uze@univ-montp2.fr) was trained in the laboratory of Ion Gresser (Villejuif, France) and received his Ph.D. in 1986 from the University of Paris VII. In 1993, he moved to Montpellier (France) where he is currently Research Director at the Centre National de la Recherche Scientifique (CNRS). He has a long-term experience in the study of structure–function relationship of IFN and receptor components, and he has contributed to the understanding of the biological significance of the multiple type I IFN subtypes.
Robert H. Silverman holds the Mal and Lea Bank Chair in the Department of Cancer Biology at the Lerner Research Institute at the Cleveland Clinic. He is professor of biochemistry and molecular biology and microbiology at Case Western Reserve University, Ohio, USA, and professor of chemistry at Cleveland State University, Ohio. Silverman received his B.Sc. from Michigan State University, USA, and his Ph.D. in molecular, cellular and developmental biology from Iowa State University, USA. His postdoctoral training was at the Roche Institute for Molecular Biology, New Jersey, USA, and at the National Institute for Medical Research and Imperial Cancer Research Fund Laboratories, London, UK. He was professor in the Department of Pathology at the Uniformed Services University of the Health Sciences in Bethesda, USA, prior to joining the Cleveland Clinic in 1991. Among his honours and awards are the Milstein Award (1993), Harold L. Stewart Lecture in Experimental Oncology (2006), Sam and Maria Miller Scientific Achievement Award in Basic Research (2006), and the Standing Tall Tribute from the American Cancer Society (2007). He has published 180 papers, which are principally focused on the molecular mechanisms of IFN action against viruses and cancer cells.
Richard M. Ransohoff is Director of the Neuroinflammation Research Center in the Department of Neurosciences of the Lerner Research Institute, professor of molecular medicine at The Cleveland Clinic Lerner College of Medicine, and Staff Neurologist in the Mellen Center for Multiple Sclerosis Treatment and Research, both at the Cleveland Clinic Foundation. Ransohoff received several honours and awards, including The Cleveland Clinic Lerner Research Institute's Award for Excellence in Science in 2006. He has been cited from 1996 through to 2007 in the “Best Doctors in America” for his expertise in the clinical care of patients with multiple sclerosis. Ransohoff was elected to the American Association of Physicians in 2006. For the past decade, Ransohoff's research has focused on the functions of chemokines and chemokine receptors in the development and pathology of the nervous system. He also has a long-standing and continuing interest in the mechanisms of action of IFN-β. Ransohoff has received research support from the NIH and the National Multiple Sclerosis Society. He has published more than 150 scientific reports, more than 50 reviews and book chapters, and three edited books.
Graham R. Foster is the Professor of Hepatology at Queen Mary's School of Medicine, London, UK, and a consultant hepatologist at Barts and The London NHS Trust in East London. He trained in Medicine at Oxford and London Universities in the 1980s and completed a Ph.D. in molecular biology in 1992. Foster has a long-standing interest in the management of chronic viral hepatitis and runs a clinical research programme studying the natural history of viral hepatitis, its effect on patients and their communities, and novel therapies for this disease. He supervises a laboratory research programme investigating the mode of action of the different type I interferons. He is the sub-editor of The Journal of Viral Hepatitis and has published widely in the field of viral liver disease. He is a member of a number of patient advocacy groups and is a member of the UK Department of Health Advisory Group on Hepatitis.
George R. Stark received his Ph.D. degree in chemistry from Columbia University in 1959. After a postdoctoral fellowship with William Stein and Stanford Moore at the Rockefeller University, USA, he joined the Department of Biochemistry at Stanford University, USA, in 1963, becoming professor in 1971. In 1983, he moved to the Imperial Cancer Research Fund in London, UK, as Associate Director of Research. In July 1992, he became the Chair of the Lerner Research Institute of The Cleveland Clinic Foundation, a position he held until August 2002. He is currently the distinguished scientist of The Cleveland Clinic Foundation, with a laboratory in the Department of Molecular Genetics, and a professor of genetics at Case Western Reserve University, USA. Stark was elected to the National Academy of Sciences in 1986, to the Fellowship of the Royal Society in 1990 and to the Institute of Medicine in 2002. He has also received the Sober, Milstein and Coley Awards. Stark's work with IFN, together with that of James Darnell, led to the discovery of the family of JAK–STAT (Janus kinase–signal transducers and activators of transcription) signalling pathways.
Related links
DATABASES
OMIM
Competing interests
The authors declare that they have no competing interests that constitute a conflict of interest for content and interpretations contained herein with the following exceptions: E.C.B. is a member of the Scientific Advisory Boards of Cleveland BioLabs and Alios BioPharma and in the past 3 years that of Coley Pharma together with honoaria for scientfic or educational presentations underwritten by Schering Plough, Maxygen, and Novartis; R.H.S. is a member of the Scientific Advisory Board of Alios BioPharma, Inc.; G.R.F. has lectured and consulted for Roche, Maxygen, Novartis, and Human Genome Sciences. Within the past 5 years, R.M.R. has received occasional honouraria for scientific presentations from (in alphabetical order) Berlex, Biogen and Serono. These presentations did not serve marketing interests.
Contributor Information
Ernest C. Borden, Email: bordene@ccf.org
Gilles Uze, Email: uze@univ-montp2.fr.
References
- 1.Isaacs A, Lindenmann J. Virus interference. I. The interferon. Proc. R. Soc. Lond., B, Biol. Sci. 1957;147:258–267. [PubMed] [Google Scholar]
- 2.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 3.Field AK, Tytell AA, Lampson GP, Hilleman MR. Inducers of interferon and host resistance. II. Multistranded synthetic polynucleotide complexes. Proc. Natl Acad. Sci. USA. 1967;58:1004–1010. doi: 10.1073/pnas.58.3.1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sen GC, Sarkar SN. Transcriptional signaling by double-stranded RNA: role of TLR3. Cytokine Growth Factor Rev. 2005;16:1–14. doi: 10.1016/j.cytogfr.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 5.Yoneyama M, et al. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nature Immunol. 2004;5:730–737. doi: 10.1038/ni1087. [DOI] [PubMed] [Google Scholar]
- 6.Tabeta K, et al. Toll-like receptors 9 and 3 as essential components of innate immune defense against mouse cytomegalovirus infection. Proc. Natl Acad. Sci. USA. 2004;101:3516–3521. doi: 10.1073/pnas.0400525101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kato H, et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. 2006;441:101–105. doi: 10.1038/nature04734. [DOI] [PubMed] [Google Scholar]
- 8.Gitlin L, et al. Essential role of mda-5 in type I IFN responses to polyriboinosinic:polyribocytidylic acid and encephalomyocarditis picornavirus. Proc. Natl Acad. Sci. USA. 2006;103:8459–8464. doi: 10.1073/pnas.0603082103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Le Goffic R, et al. Detrimental contribution of the Toll-like receptor (TLR)3 to influenza A virus-induced acute pneumonia. PLoS Pathog. 2006;2:e53. doi: 10.1371/journal.ppat.0020053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang T, et al. Toll-like receptor 3 mediates West Nile virus entry into the brain causing lethal encephalitis. Nature Med. 2004;10:1366–1373. doi: 10.1038/nm1140. [DOI] [PubMed] [Google Scholar]
- 11.Meylan E, Tschopp J. Toll-like receptors and RNA helicases: two parallel ways to trigger antiviral responses. Mol. Cell. 2006;22:561–569. doi: 10.1016/j.molcel.2006.05.012. [DOI] [PubMed] [Google Scholar]
- 12.Kawai T, Akira S. Antiviral signaling through pattern recognition receptors. J. Biochem. 2007;141:137–145. doi: 10.1093/jb/mvm032. [DOI] [PubMed] [Google Scholar]
- 13.Yoneyama M, Fujita T. Cytoplasmic double-stranded DNA sensor. Nature Immunol. 2007;8:907–908. doi: 10.1038/ni0907-907. [DOI] [PubMed] [Google Scholar]
- 14.Panne D, Maniatis T, Harrison SC. An atomic model of the interferon-β enhanceosome. Cell. 2007;129:1111–1123. doi: 10.1016/j.cell.2007.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Honda K, Takaoka A, Taniguchi T. Type I interferon gene induction by the interferon regulatory factor family of transcription factors. Immunity. 2006;25:349–360. doi: 10.1016/j.immuni.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 16.Tenoever BR, et al. Multiple functions of the IKK-related kinase IKKɛ in interferon-mediated antiviral immunity. Science. 2007;315:1274–1278. doi: 10.1126/science.1136567. [DOI] [PubMed] [Google Scholar]
- 17.Fitzgerald KA, et al. IKKɛ and TBK1 are essential components of the IRF3 signaling pathway. Nature Immunol. 2003;4:491–496. doi: 10.1038/ni921. [DOI] [PubMed] [Google Scholar]
- 18.Biron CA, Sen GC, et al. Fields Virology. 2006. pp. 249–278. [Google Scholar]
- 19.Hiscott J, Nguyen T-LA, Arguello M, Nakhaei P, Paz S. Manipulation of the nuclear factor-κB pathway and the innate immune response by viruses. Oncogene. 2006;25:6844–6867. doi: 10.1038/sj.onc.1209941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Foy E, et al. Control of antiviral defenses through hepatitis C virus disruption of retinoic acid-inducible gene-I signaling. Proc. Natl Acad. Sci. USA. 2005;102:2986–2991. doi: 10.1073/pnas.0408707102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Garcia-Sastre A, Biron CA. Type 1 interferons and the virus–host relationship: a lesson in detente. Science. 2006;312:879–882. doi: 10.1126/science.1125676. [DOI] [PubMed] [Google Scholar]
- 22.Finlay BB, McFadden G. Anti-immunology: evasion of the host immune system by bacterial and viral pathogens. Cell. 2006;124:767–782. doi: 10.1016/j.cell.2006.01.034. [DOI] [PubMed] [Google Scholar]
- 23.Pichlmair A, et al. RIG-I mediated antiviral responses to single-stranded RNA bearing 5′ phosphates. Science. 2006;314:997–1001. doi: 10.1126/science.1132998. [DOI] [PubMed] [Google Scholar]
- 24.Casrouge A, et al. Herpes simplex virus encephalitis in human UNC-93B deficiency. Science. 2006;314:308–312. doi: 10.1126/science.1128346. [DOI] [PubMed] [Google Scholar]
- 25.Hemmi H, et al. A Toll-like receptor recognizes bacterial DNA. Nature. 2000;408:740–745. doi: 10.1038/35047123. [DOI] [PubMed] [Google Scholar]
- 26.Hemmi H, et al. Small anti-viral compounds activate immune cells via the TLR7 MyD88-dependent signaling pathway. Nature Immunol. 2002;3:196–200. doi: 10.1038/ni758. [DOI] [PubMed] [Google Scholar]
- 27.Roberts ZJ, et al. The chemotherapeutic agent DMXAA potently and specifically activates the TBK1-IRF-3 signaling axis. J. Exp. Med. 2007;204:1559–1569. doi: 10.1084/jem.20061845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.O'Neill LA. Targeting signal transduction as a strategy to treat inflammatory diseases. Nature Rev. Drug Discov. 2006;5:549–563. doi: 10.1038/nrd2070. [DOI] [PubMed] [Google Scholar]
- 29.Krieg AM. Therapeutic potential of Toll-like receptor 9 activation. Nature Rev. Drug Discov. 2006;5:471–484. doi: 10.1038/nrd2059. [DOI] [PubMed] [Google Scholar]
- 30.Havell EA, et al. Two antigenically distinct species of human interferon. Proc. Natl Acad. Sci. USA. 1975;72:2185–2187. doi: 10.1073/pnas.72.6.2185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Streuli M, Nagata S, Weissmann C. At least three human type α-interferons: structure of α 2. Science. 1980;209:1343–1347. doi: 10.1126/science.6158094. [DOI] [PubMed] [Google Scholar]
- 32.Taniguchi T, et al. Human leukocyte and fibroblast interferons are structurally related. Nature. 1980;285:547–549. doi: 10.1038/285547a0. [DOI] [PubMed] [Google Scholar]
- 33.Goeddel DV, et al. The structure of eight distinct cloned human leukocyte interferon cDNAs. Nature. 1981;290:20–26. doi: 10.1038/290020a0. [DOI] [PubMed] [Google Scholar]
- 34.Pestka S, Krause CD, Walter R. Interferons, interferon-like cytokines, and their receptors. Immunol. Rev. 1998;202:8–32. doi: 10.1111/j.0105-2896.2004.00204.x. [DOI] [PubMed] [Google Scholar]
- 35.Hardy MP, Owczarek CM, Jermiin LS, Ejdeback M, Hertzog PJ. Characterization of the type I interferon locus and identification of novel genes. Genomics. 2004;84:331–345. doi: 10.1016/j.ygeno.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 36.Coelho LF, de Freitas Almeida GM, Mennechet FJ, Blangy A, Uze G. Interferon-α and -β differentially regulate osteoclastogenesis: role of differential induction of chemokine CXCL11 expression. Proc. Natl Acad. Sci. USA. 2005;102:11917–11922. doi: 10.1073/pnas.0502188102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Foster GR, et al. IFN-α subtypes differentially affect human T cell motility. J. Immunol. 2004;173:1663–1670. doi: 10.4049/jimmunol.173.3.1663. [DOI] [PubMed] [Google Scholar]
- 38.Hilkens CM, Schlaak JF, Kerr IM. Differential responses to IFN-α subtypes in human T cells and dendritic cells. J. Immunol. 2003;171:5255–5263. doi: 10.4049/jimmunol.171.10.5255. [DOI] [PubMed] [Google Scholar]
- 39.Uze G, Schreiber G, Piehler J, Pellegrini S. The receptor of the type I interferon family. Cur. Topics Microbiol. Immunol. 2007;316:71–95. doi: 10.1007/978-3-540-71329-6_5. [DOI] [PubMed] [Google Scholar]
- 40.Walker J, Tough DF. Modification of TLR-induced activation of human dendritic cells by type I IFN: synergistic interaction with TLR4 but not TLR3 agonists. Eur. J. Immunol. 2006;36:1827–1836. doi: 10.1002/eji.200635854. [DOI] [PubMed] [Google Scholar]
- 41.Roberts RM, Liu L, Alexenko A. New and atypical families of type I interferons in mammals: comparative functions, structures, and evolutionary relationships. Prog. Nucleic Acid Res. Mol. Biol. 1997;56:287–325. doi: 10.1016/s0079-6603(08)61008-9. [DOI] [PubMed] [Google Scholar]
- 42.LaFleur DW, et al. Interferon-κ, a novel type I interferon expressed in human keratinocytes. J. Biol. Chem. 2001;276:39765–39771. doi: 10.1074/jbc.M102502200. [DOI] [PubMed] [Google Scholar]
- 43.Coccia EM, et al. Viral infection and Toll-like receptor agonists induce a differential expression of type I and λ interferons in human plasmacytoid and monocyte-derived dendritic cells. Eur. J. Immunol. 2004;34:796–805. doi: 10.1002/eji.200324610. [DOI] [PubMed] [Google Scholar]
- 44.Mitsui Y, Senda T. Elucidation of the basic three-dimensional structure of type I interferons and its functional and evolutionary implications. J. Interferon Cytokine Res. 1997;17:319–326. doi: 10.1089/jir.1997.17.319. [DOI] [PubMed] [Google Scholar]
- 45.Gavutis M, Jaks E, Lamken P, Piehler J. Determination of the two-dimensional interaction rate constants of a cytokine receptor complex. Biophys. J. 2006;90:3345–3355. doi: 10.1529/biophysj.105.072546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jaitin DA, et al. Inquiring into the differential action of interferons (IFNs): an IFN-α2 mutant with enhanced affinity to IFNAR1 is functionally similar to IFN-β. Mol. Cell Biol. 2006;26:1888–1897. doi: 10.1128/MCB.26.5.1888-1897.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jaks E, Gavutis M, Uze G, Martal J, Piehler J. Differential receptor subunit affinities of type I interferons govern differential signal activation. J. Mol. Biol. 2007;366:525–539. doi: 10.1016/j.jmb.2006.11.053. [DOI] [PubMed] [Google Scholar]
- 48.Kalie E, Jaitin DA, Abramovich R, Schreiber G. An interferon α2 mutant optimized by phage display for IFNAR1 binding confers specifically enhanced antitumor activities. J. Biol. Chem. 2007;282:11602–11611. doi: 10.1074/jbc.M610115200. [DOI] [PubMed] [Google Scholar]
- 49.Severa M, et al. Differential responsiveness to IFN-α and IFN-β of human mature DC through modulation of IFNAR expression. J. Leukoc. Biol. 2006;79:1286–1294. doi: 10.1189/jlb.1205742. [DOI] [PubMed] [Google Scholar]
- 50.Frodsham AJ, et al. Class II cytokine receptor gene cluster is a major locus for hepatitis B persistence. Proc. Natl Acad. Sci. USA. 2006;103:9148–9153. doi: 10.1073/pnas.0602800103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bach EA, Aguet M, Schreiber RD. The IFN γ receptor: a paradigm for cytokine receptor signaling. Annu. Rev. Immunol. 1997;15:563–591. doi: 10.1146/annurev.immunol.15.1.563. [DOI] [PubMed] [Google Scholar]
- 52.Kotenko SV, Langer JA. Full house: 12 receptors for 27 cytokines. Int. Immunopharmacol. 2004;4:593–608. doi: 10.1016/j.intimp.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 53.Roesler J, et al. Listeria monocytogenes and recurrent mycobacterial infections in a child with complete interferon-γ-receptor (IFN-γR1) deficiency: mutational analysis and evaluation of therapeutic options. Exp. Hematol. 1999;27:1368–1374. doi: 10.1016/s0301-472x(99)00077-6. [DOI] [PubMed] [Google Scholar]
- 54.Uze G, Monneron D. IL-28 and IL-29: newcomers to the interferon family. Biochimie. 2007;89:729–734. doi: 10.1016/j.biochi.2007.01.008. [DOI] [PubMed] [Google Scholar]
- 55.Zinn K, DiMaio D, Maniatis T. Identification of two distinct regulatory regions adjacent to the human β-interferon gene. Cell. 1983;34:865–879. doi: 10.1016/0092-8674(83)90544-5. [DOI] [PubMed] [Google Scholar]
- 56.Merlin G, Chebath J, Benech P, Metz R, Revel M. Molecular cloning and sequence of partial cDNA for interferon-induced (2′-5′)oligo(A) synthetase mRNA from human cells. Proc. Natl Acad. Sci. USA. 1983;80:4904–4908. doi: 10.1073/pnas.80.16.4904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Friedman RL, Manly SP, McMahon M, Kerr IM, Stark GR. Transcriptional and posttranscriptional regulation of interferon-induced gene expression in human cells. Cell. 1984;38:745–755. doi: 10.1016/0092-8674(84)90270-8. [DOI] [PubMed] [Google Scholar]
- 58.Friedman RL, Stark GR. α-Interferon-induced transcription of HLA and metallothionein genes containing homologous upstream sequences. Nature. 1985;314:637–639. doi: 10.1038/314637a0. [DOI] [PubMed] [Google Scholar]
- 59.Fu XY, Schindler C, Improta T, Aebersold R, Darnell JE., Jr. The proteins of ISGF-3, the interferon α-induced transcriptional activator, define a gene family involved in signal transduction. Proc. Natl Acad. Sci. USA. 1992;89:7840–7843. doi: 10.1073/pnas.89.16.7840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stark GR, Kerr IM, Williams BR, Silverman RH, Schreiber RD. How cells respond to interferons. Annu. Rev. Biochem. 1998;67:227–264. doi: 10.1146/annurev.biochem.67.1.227. [DOI] [PubMed] [Google Scholar]
- 61.Schindler C, Levy DE, Decker T. JAK–STAT signaling: from interferons to cytokines. J. Biol. Chem. 2007;282:20059–20063. doi: 10.1074/jbc.R700016200. [DOI] [PubMed] [Google Scholar]
- 62.Velazquez L, Fellous M, Stark GR, Pellegrini S. A protein tyrosine kinase in the interferon α/β signaling pathway. Cell. 1992;70:313–322. doi: 10.1016/0092-8674(92)90105-l. [DOI] [PubMed] [Google Scholar]
- 63.Qing Y, Stark GR. Alternative activation of STAT1 and STAT3 in response to interferon-γ. J. Biol. Chem. 2004;279:41679–41685. doi: 10.1074/jbc.M406413200. [DOI] [PubMed] [Google Scholar]
- 64.van Boxel-Dezaire AH, Rani MR, Stark GR. Complex modulation of cell type-specific signaling in response to type I interferons. Immunity. 2006;25:361–372. doi: 10.1016/j.immuni.2006.08.014. [DOI] [PubMed] [Google Scholar]
- 65.van Boxel-Dezaire AH, Stark GR. Cell type-specific signaling in response to interferon-γ. Curr. Top. Microbiol. Immunol. 2007;316:119–154. doi: 10.1007/978-3-540-71329-6_7. [DOI] [PubMed] [Google Scholar]
- 66.Jouanguy E, et al. Human primary immunodeficiencies of type I interferons. Biochimie. 2007;89:878–883. doi: 10.1016/j.biochi.2007.04.016. [DOI] [PubMed] [Google Scholar]
- 67.Rani MR, Hibbert L, Sizemore N, Stark GR, Ransohoff RM. Requirement of phosphoinositide 3-kinase and Akt for interferon-β-mediated induction of the β-R1 (SCYB11) gene. J. Biol. Chem. 2002;277:38456–38461. doi: 10.1074/jbc.M203204200. [DOI] [PubMed] [Google Scholar]
- 68.Rani MR, Ransohoff RM. Alternative and accessory pathways in the regulation of IFN-β-mediated gene expression. J. Interferon Cytokine Res. 2005;25:788–798. doi: 10.1089/jir.2005.25.788. [DOI] [PubMed] [Google Scholar]
- 69.Hilkens CM, Schlaak JF, Kerr IM. Differential responses to IFN-α subtypes in human T cells and dendritic cells. J. Immunol. 2003;171:5255–5263. doi: 10.4049/jimmunol.171.10.5255. [DOI] [PubMed] [Google Scholar]
- 70.Platanias LC. Mechanisms of type-I- and type-II- interferon-mediated signaling. Nature Rev. Immunol. 2005;5:375–386. doi: 10.1038/nri1604. [DOI] [PubMed] [Google Scholar]
- 71.Alexander WS, Hilton DJ. The role of suppressors of cytokine signaling (SOCS) proteins in regulation of the immune response. Annu. Rev. Immunol. 2004;22:503–529. doi: 10.1146/annurev.immunol.22.091003.090312. [DOI] [PubMed] [Google Scholar]
- 72.Yi T, et al. Anticancer activity of sodium stibogluconate in synergy with IFNs. J. Immunol. 2002;169:5978–5985. doi: 10.4049/jimmunol.169.10.5978. [DOI] [PubMed] [Google Scholar]
- 73.Ho HH, Ivashkiv LB. Role of STAT3 in type I interferon responses. Negative regulation of STAT1-dependent inflammatory gene activation. J. Biol. Chem. 2006;281:14111–14118. doi: 10.1074/jbc.M511797200. [DOI] [PubMed] [Google Scholar]
- 74.Hu X, et al. Sensitization of IFN-γ Jak–STAT signaling during macrophage activation. Nature Immunol. 2002;3:859–866. doi: 10.1038/ni828. [DOI] [PubMed] [Google Scholar]
- 75.Kirkwood J, et al. Modulation of STAT1 and STAT3 signaling in melanoma by high-dose IFN-α2b. Clin. Cancer Res. 2007;13:1523–1531. doi: 10.1158/1078-0432.CCR-06-1387. [DOI] [PubMed] [Google Scholar]
- 76.Kumar A, Commane M, Flickinger TW, Horvath CM, Stark GR. Defective TNF-α-induced apoptosis in STAT1-null cells due to low constitutive levels of caspases. Science. 1997;278:1630–1632. doi: 10.1126/science.278.5343.1630. [DOI] [PubMed] [Google Scholar]
- 77.Yang J, et al. Novel roles of unphosphorylated STAT3 in oncogenesis and transcriptional regulation. Cancer Res. 2005;65:939–947. [PubMed] [Google Scholar]
- 78.Yang J, et al. Unphosphorylated STAT3 accumulates in response to IL-6 and activates transcription by binding to NFκB. Genes Dev. 2007;21:1396–1408. doi: 10.1101/gad.1553707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Der SD, Zhou A, Williams BR, Silverman RH. Identification of genes differentially regulated by interferon α, β, or γ using oligonucleotide arrays. Proc. Natl Acad. Sci. USA. 1998;95:15623–15628. doi: 10.1073/pnas.95.26.15623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.de Veer MJ, et al. Functional classification of interferon-stimulated genes identified using microarrays. J. Leukoc. Biol. 2001;69:912–920. [PubMed] [Google Scholar]
- 81.Silverman R. H. Viral Encounters with 2',5'-Oligoadenylate Synthetase and RNase L during the Interferon Antiviral Response. Journal of Virology. 2007;81(23):12720–12729. doi: 10.1128/JVI.01471-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kerr IM, Brown RE. pppA2′p5′A2′p5′A: an inhibitor of protein synthesis synthesized with an enzyme fraction from interferon-treated cells. Proc. Natl Acad. Sci. USA. 1978;75:256–260. doi: 10.1073/pnas.75.1.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhou A, Hassel BA, Silverman RH. Expression cloning of 2–5A-dependent RNAase: a uniquely regulated mediator of interferon action. Cell. 1993;72:753–765. doi: 10.1016/0092-8674(93)90403-d. [DOI] [PubMed] [Google Scholar]
- 84.Dong B, Silverman RH. 2–5A-dependent RNase molecules dimerize during activation by 2-5A. J. Biol. Chem. 1995;270:4133–4137. doi: 10.1074/jbc.270.8.4133. [DOI] [PubMed] [Google Scholar]
- 85.Wreschner DH, McCauley JW, Skehel JJ, Kerr IM. Interferon action — sequence specificity of the ppp(A2′p)nA-dependent ribonuclease. Nature. 1981;289:414–417. doi: 10.1038/289414a0. [DOI] [PubMed] [Google Scholar]
- 86.Floyd-Smith G, Slattery E, Lengyel P. Interferon action: RNA cleavage pattern of a (2′-5′)oligoadenylate-dependent endonuclease. Science. 1981;212:1030–1032. doi: 10.1126/science.6165080. [DOI] [PubMed] [Google Scholar]
- 87.Zhou A, et al. Interferon action and apoptosis are defective in mice devoid of 2′, 5′-oligoadenylate-dependent RNase, L. EMBO J. 1997;16:6355–6363. doi: 10.1093/emboj/16.21.6355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Castelli JC, et al. The role of 2′-5′ oligoadenylate-activated ribonuclease L in apoptosis. Cell Death Differ. 1998;5:313–320. doi: 10.1038/sj.cdd.4400352. [DOI] [PubMed] [Google Scholar]
- 89.Castelli JC, et al. A study of the interferon antiviral mechanism: apoptosis activation by the 2–5A system. J. Exp. Med. 1997;186:967–972. doi: 10.1084/jem.186.6.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Malathi K, Dong B, Gale M, Jr, Silverman RH. Small self RNA generates by RNase L amplifies antiviral innate immunity. Nature. 2007;448:816–819. doi: 10.1038/nature06042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Thakur CS, et al. Small-molecule activators of RNase L with broad-spectrum antiviral activity. Proc. Natl Acad. Sci. USA. 2007;104:9585–9590. doi: 10.1073/pnas.0700590104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Roberts WK, Hovanessian A, Brown RE, Clemens MJ, Kerr IM. Interferon-mediated protein kinase and low-molecular-weight inhibitor of protein synthesis. Nature. 1976;264:477–480. doi: 10.1038/264477a0. [DOI] [PubMed] [Google Scholar]
- 93.Zilberstein A, Kimchi A, Schmidt A, Revel M. Isolation of two interferon-induced translational inhibitors: a protein kinase and an oligo-isoadenylate synthetase. Proc. Natl Acad. Sci. USA. 1978;75:4734–4738. doi: 10.1073/pnas.75.10.4734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Meurs E, et al. Molecular cloning and characterization of the human double-stranded RNA-activated protein kinase induced by interferon. Cell. 1990;62:379–390. doi: 10.1016/0092-8674(90)90374-n. [DOI] [PubMed] [Google Scholar]
- 95.Williams BR. Signal integration via PKR. Sci STKE. 2001;89:RE2. doi: 10.1126/stke.2001.89.re2. [DOI] [PubMed] [Google Scholar]
- 96.Williams BR. PKR; a sentinel kinase for cellular stress. Oncogene. 1999;18:6112–6120. doi: 10.1038/sj.onc.1203127. [DOI] [PubMed] [Google Scholar]
- 97.Patel RC, Sen GC. PACT, a protein activator of the interferon-induced protein kinase, PKR. EMBO J. 1998;17:4379–4390. doi: 10.1093/emboj/17.15.4379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Gale M, Jr., Katze MG. Molecular mechanisms of interferon resistance mediated by viral-directed inhibition of PKR, the interferon-induced protein kinase. Pharmacol. Ther. 1998;78:29–46. doi: 10.1016/s0163-7258(97)00165-4. [DOI] [PubMed] [Google Scholar]
- 99.Sarkar SN, Sen GC. Novel functions of proteins encoded by viral stress-inducible genes. Pharmacol. Ther. 2004;103:245–259. doi: 10.1016/j.pharmthera.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 100.Wang C, et al. α Interferon induces distinct translational control programs to suppress hepatitis C virus RNA replication. J. Virol. 2003;77:3898–3912. doi: 10.1128/JVI.77.7.3898-3912.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lindenmann J. Inheritance of resistance to influenza virus in mice. Proc. Soc. Exp. Biol. Med. 1964;116:506–509. doi: 10.3181/00379727-116-29292. [DOI] [PubMed] [Google Scholar]
- 102.Horisberger MA, Staeheli P, Haller O. Interferon induces a unique protein in mouse cells bearing a gene for resistance to influenza virus. Proc. Natl Acad. Sci. USA. 1983;80:1910–1914. doi: 10.1073/pnas.80.7.1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Staeheli P, Haller O, Boll W, Lindenmann J, Weissmann C. Mx protein: constitutive expression in 3T3 cells transformed with cloned Mx cDNA confers selective resistance to influenza virus. Cell. 1986;44:147–158. doi: 10.1016/0092-8674(86)90493-9. [DOI] [PubMed] [Google Scholar]
- 104.Haller O, Staeheli P, Kochs G. Interferon-induced Mx proteins in antiviral host defense. Biochimie. 2007;89:812–818. doi: 10.1016/j.biochi.2007.04.015. [DOI] [PubMed] [Google Scholar]
- 105.Recht M, Borden EC, Knight E., Jr. A human 15-kDa IFN-induced protein induces the secretion of IFN-γ. J. Immunol. 1991;147:2617–2623. [PubMed] [Google Scholar]
- 106.D'Cunha J, et al. In vitro and in vivo secretion of human ISG15, an IFN-induced immunomodulatory cytokine. J. Immunol. 1996;157:4100–4108. [PubMed] [Google Scholar]
- 107.Andersen JB, Hassel BA. The interferon regulated ubiquitin-like protein, ISG15, in tumorigenesis: friend or foe? Cytokine Growth Factor Rev. 2006;17:411–421. doi: 10.1016/j.cytogfr.2006.10.001. [DOI] [PubMed] [Google Scholar]
- 108.Okumura A, Lu G, Pitha-Rowe I, Pitha PM. Innate antiviral response targets HIV-1 release by the induction of ubiquitin-like protein ISG15. Proc. Natl Acad. Sci. USA. 2006;103:1440–1445. doi: 10.1073/pnas.0510518103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lenschow DJ, et al. IFN-stimulated gene 15 functions as a critical antiviral molecule against influenza, herpes, and Sindbis viruses. Proc. Natl Acad. Sci. USA. 2007;104:1371–1376. doi: 10.1073/pnas.0607038104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zhou Q, et al. Molecular cloning of human plasma membrane phospholipid scramblase. A protein mediating transbilayer movement of plasma membrane phospholipids. J. Biol. Chem. 1997;272:18240–18244. doi: 10.1074/jbc.272.29.18240. [DOI] [PubMed] [Google Scholar]
- 111.Ben-Efraim I, Zhou Q, Wiedmer T, Gerace L, Sims PJ. Phospholipid scramblase 1 is imported into the nucleus by a receptor-mediated pathway and interacts with DNA. Biochemistry. 2004;43:3518–3526. doi: 10.1021/bi0356911. [DOI] [PubMed] [Google Scholar]
- 112.Dong B, et al. Phospholipid scramblase 1 potentiates the antiviral activity of interferon. J. Virol. 2004;78:8983–8993. doi: 10.1128/JVI.78.17.8983-8993.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kayagaki N, et al. Type I interferons (IFNs) regulate tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) expression on human T cells: a novel mechanism for the antitumor effects of type I IFNs. J. Exp. Med. 1999;189:1451–1460. doi: 10.1084/jem.189.9.1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Chawla-Sarkar M, Leaman DW, Borden EC. Preferential induction of apoptosis by interferon (IFN)-β compared with IFN-α2: correlation with TRAIL/Apo2L induction in melanoma cell lines. Clin. Cancer. Res. 2001;7:1821–1831. [PubMed] [Google Scholar]
- 115.Chen Q, et al. Apo2L/TRAIL and Bcl-2-related proteins regulate type I interferon-induced apoptosis in multiple myeloma. Blood. 2001;98:2183–2192. doi: 10.1182/blood.v98.7.2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kelly JM, et al. Characterization of a human gene inducible by α- and β-interferons and its expression in mouse cells. EMBO J. 1986;5:1601–1606. doi: 10.1002/j.1460-2075.1986.tb04402.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Martensen PM, Justesen J. Small ISGs coming forward. J. Interferon Cytokine Res. 2004;24:1–19. doi: 10.1089/107999004772719864. [DOI] [PubMed] [Google Scholar]
- 118.Tahara E, Jr., et al. G1P3, an interferon inducible gene 6–16, is expressed in gastric cancers and inhibits mitochondrial-mediated apoptosis in gastric cancer cell line TMK-1 cell. Cancer Immunol. Immunother. 2005;54:729–740. doi: 10.1007/s00262-004-0645-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Martensen PM, et al. The interferon α induced protein ISG12 is localized to the nuclear membrane. Eur. J. Biochem. 2001;268:5947–5954. doi: 10.1046/j.0014-2956.2001.02545.x. [DOI] [PubMed] [Google Scholar]
- 120.Labrada L, Liang XH, Zheng W, Johnston C, Levine B. Age-dependent resistance to lethal alphavirus encephalitis in mice: analysis of gene expression in the central nervous system and identification of a novel interferon-inducible protective gene, mouse ISG12. J. Virol. 2002;76:11688–11703. doi: 10.1128/JVI.76.22.11688-11703.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Anderson SL, Carton JM, Lou J, Xing L, Rubin BY. Interferon-induced guanylate binding protein-1 (GBP-1) mediates an antiviral effect against vesicular stomatitis virus and encephalomyocarditis virus. Virology. 1999;256:8–14. doi: 10.1006/viro.1999.9614. [DOI] [PubMed] [Google Scholar]
- 122.Espert L, et al. ISG20, a new interferon-induced RNase specific for single-stranded RNA, defines an alternative antiviral pathway against RNA genomic viruses. J. Biol. Chem. 2003;278:16151–16158. doi: 10.1074/jbc.M209628200. [DOI] [PubMed] [Google Scholar]
- 123.Regad T, et al. PML mediates the interferon-induced antiviral state against a complex retrovirus via its association with the viral transactivator. EMBO J. 2001;20:3495–3505. doi: 10.1093/emboj/20.13.3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Liu Y, George CX, Patterson JB, Samuel CE. Functionally distinct double-stranded RNA-binding domains associated with alternative splice site variants of the interferon-inducible double-stranded RNA-specific adenosine deaminase. J. Biol. Chem. 1997;272:4419–4428. doi: 10.1074/jbc.272.7.4419. [DOI] [PubMed] [Google Scholar]
- 125.Chin KC, Cresswell P. Viperin (cig5), an IFN-inducible antiviral protein directly induced by human cytomegalovirus. Proc. Natl Acad. Sci. USA. 2001;98:15125–15130. doi: 10.1073/pnas.011593298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Melkova Z, Esteban M. Inhibition of vaccinia virus DNA replication by inducible expression of nitric oxide synthase. J. Immunol. 1995;155:5711–5718. [PubMed] [Google Scholar]
- 127.Enninga J, Levy DE, Blobel G, Fontoura BM. Role of nucleoporin induction in releasing an mRNA nuclear export block. Science. 2002;295:1523–1525. doi: 10.1126/science.1067861. [DOI] [PubMed] [Google Scholar]
- 128.Samuel CE. Antiviral actions of interferons. Clin. Microbiol. Rev. 2001;14:778–809. doi: 10.1128/CMR.14.4.778-809.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Siegal FP, et al. The nature of the principal type 1 interferon-producing cells in human blood. Science. 1999;284:1835–1837. doi: 10.1126/science.284.5421.1835. [DOI] [PubMed] [Google Scholar]
- 130.Zhao W, Cha EN, Lee C, Park CY, Schindler C. Stat2-dependent regulation of MHC class II expression. J. Immunol. 2007;179:463–471. doi: 10.4049/jimmunol.179.1.463. [DOI] [PubMed] [Google Scholar]
- 131.Cresswell P. Intracellular surveillance: controlling the assembly of MHC class I-peptide complexes. Traffic. 2000;1:301–305. doi: 10.1034/j.1600-0854.2000.010402.x. [DOI] [PubMed] [Google Scholar]
- 132.Drozina G, Kohoutek J, Jabrane-Ferrat N, Peterlin BM. Expression of MHC II genes. Curr. Top. Microbiol. Immunol. 2005;290:147–170. doi: 10.1007/3-540-26363-2_7. [DOI] [PubMed] [Google Scholar]
- 133.Fruh K, Yang Y. Antigen presentation by MHC class I and its regulation by interferon γ. Curr. Opin. Immunol. 1999;11:76–81. doi: 10.1016/s0952-7915(99)80014-4. [DOI] [PubMed] [Google Scholar]
- 134.Gaczynska M, Rock KL, Goldberg AL. γ-Interferon and expression of MHC genes regulate peptide hydrolysis by proteasomes. Nature. 1993;365:264–267. doi: 10.1038/365264a0. [DOI] [PubMed] [Google Scholar]
- 135.Groothuis T, Neefjes J. The ins and outs of intracellular peptides and antigen presentation by MHC class I molecules. Curr. Top. Microbiol. Immunol. 2005;300:127–148. doi: 10.1007/3-540-28007-3_6. [DOI] [PubMed] [Google Scholar]
- 136.Cole KE, et al. Interferon-inducible T cell alpha chemoattractant (I-TAC): a novel non-ELR CXC chemokine with potent activity on activated T cells through selective high affinity binding to CXCR3. J. Exp. Med. 1998;187:2009–2021. doi: 10.1084/jem.187.12.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Farber JM. A macrophage mRNA selectively induced by γ-interferon encodes a member of the platelet factor 4 family of cytokines. Proc. Natl Acad. Sci. USA. 1990;87:5238–5242. doi: 10.1073/pnas.87.14.5238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Rani MR, et al. Characterization of β-R1, a gene that is selectively induced by interferon β (IFN-β) compared with IFN-α. J. Biol. Chem. 1996;271:22878–22884. doi: 10.1074/jbc.271.37.22878. [DOI] [PubMed] [Google Scholar]
- 139.Luster AD, Unkeless JC, Ravetch JV. γ-Interferon transcriptionally regulates an early-response gene containing homology to platelet proteins. Nature. 1985;315:672–676. doi: 10.1038/315672a0. [DOI] [PubMed] [Google Scholar]
- 140.Shi FD, Van KL. Reciprocal regulation between natural killer cells and autoreactive T cells. Nature Rev. Immunol. 2006;6:751–760. doi: 10.1038/nri1935. [DOI] [PubMed] [Google Scholar]
- 141.Fontes JD, Kanazawa S, Nekrep N, Peterlin BM. The class II transactivator CIITA is a transcriptional integrator. Microbes Infect. 1999;1:863–869. doi: 10.1016/s1286-4579(99)00232-4. [DOI] [PubMed] [Google Scholar]
- 142.LeibundGut-Landmann S, et al. Specificity and expression of CIITA, the master regulator of MHC class II genes. Eur. J. Immunol. 2004;34:1513–1525. doi: 10.1002/eji.200424964. [DOI] [PubMed] [Google Scholar]
- 143.Wright KL, Ting JP. Epigenetic regulation of MHC-II and CIITA genes. Trends Immunol. 2006;27:405–412. doi: 10.1016/j.it.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 144.Denzin LK, Hammond C, Cresswell P. HLA-DM interactions with intermediates in HLA-DR maturation and a role for HLA-DM in stabilizing empty HLA-DR molecules. J. Exp. Med. 1996;184:2153–2165. doi: 10.1084/jem.184.6.2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Denzin LK, Robbins NF, Carboy-Newcomb C, Cresswell P. Assembly and intracellular transport of HLA-DM and correction of the class II antigen-processing defect in T2 cells. Immunity. 1994;1:595–606. doi: 10.1016/1074-7613(94)90049-3. [DOI] [PubMed] [Google Scholar]
- 146.Gruneberg U, et al. The structure and function of the novel MHC class II molecule, HLA-DM. Biochem. 1997;25:208S. doi: 10.1042/bst025208s. [DOI] [PubMed] [Google Scholar]
- 147.Kropshofer H, Arndt SO, Moldenhauer G, Hammerling GJ, Vogt AB. HLA-DM acts as a molecular chaperone and rescues empty HLA-DR molecules at lysosomal pH. Immunity. 1997;6:293–302. doi: 10.1016/s1074-7613(00)80332-5. [DOI] [PubMed] [Google Scholar]
- 148.Morris P, et al. An essential role for HLA-DM in antigen presentation by class II major histocompatibility molecules. Nature. 1994;368:551–554. doi: 10.1038/368551a0. [DOI] [PubMed] [Google Scholar]
- 149.Vogt AB, Kropshofer H. HLA-DM — an endosomal and lysosomal chaperone for the immune system. Trends Biochem. Sci. 1999;24:150–154. doi: 10.1016/s0968-0004(99)01364-x. [DOI] [PubMed] [Google Scholar]
- 150.Kleinschmidt WJ, Schultz RM. Similarities of murine γ interferon and the lymphokine that renders macrophages cytotoxic. J. Interferon Res. 1982;2:291–299. doi: 10.1089/jir.1982.2.291. [DOI] [PubMed] [Google Scholar]
- 151.Blumberg BS. Australia antigen and the biology of hepatitis B. Science. 1977;197:17–25. doi: 10.1126/science.325649. [DOI] [PubMed] [Google Scholar]
- 152.Greenberg HB, et al. Effect of human leukocyte interferon on hepatitis B virus infection in patients with chronic active hepatitis. N. Engl. J. Med. 1976;295:517–522. doi: 10.1056/NEJM197609022951001. [DOI] [PubMed] [Google Scholar]
- 153.Weimar W, et al. Fibroblast interferon in HBsAg-positive chronic active hepatitis. Lancet. 1977;310:1282. doi: 10.1016/s0140-6736(77)92682-4. [DOI] [PubMed] [Google Scholar]
- 154.Hoofnagle JH, et al. Randomized, controlled trial of recombinant human α-interferon in patients with chronic hepatitis B. Gastroenterology. 1988;95:1318–1325. doi: 10.1016/0016-5085(88)90367-8. [DOI] [PubMed] [Google Scholar]
- 155.Lok AS, McMahon BJ. Chronic hepatitis B. Hepatology. 2007;45:507–539. doi: 10.1002/hep.21513. [DOI] [PubMed] [Google Scholar]
- 156.Bertoletti A, Maini M, Williams R. Role of hepatitis B virus specific cytotoxic T cells in liver damage and viral control. Antiviral Res. 2003;60:61–66. doi: 10.1016/j.antiviral.2003.08.012. [DOI] [PubMed] [Google Scholar]
- 157.Carmen W, et al. Mutation preventing formation of hepatitis B e antigen in patients with chronic HBV infection. Lancet. 1989;2:588–591. doi: 10.1016/s0140-6736(89)90713-7. [DOI] [PubMed] [Google Scholar]
- 158.Lau GK, et al. Peginterferon Alfa-2a, lamivudine, and the combination for HBeAg-positive chronic hepatitis B. N. Engl. J. Med. 2005;352:2682–2695. doi: 10.1056/NEJMoa043470. [DOI] [PubMed] [Google Scholar]
- 159.Marcellin P, et al. Peginterferon alfa-2a alone, lamivudine alone, and the two in combination in patients with HBeAg-negative chronic hepatitis B. N. Engl. J. Med. 2004;351:1206–1217. doi: 10.1056/NEJMoa040431. [DOI] [PubMed] [Google Scholar]
- 160.Hoofnagle J, Mullen K, Jones D. Treatment of chronic nonA nonB hepatitis with recombinant human α interferon: a preliminary report. N. Engl. J. Med. 1986;328:465–470. doi: 10.1056/NEJM198612183152503. [DOI] [PubMed] [Google Scholar]
- 161.Choo Q-L, et al. Isolation of a cDNA clone derived from a blood borne non-A, non-B viral hepatitis clone. Science. 1989;244:359–361. doi: 10.1126/science.2523562. [DOI] [PubMed] [Google Scholar]
- 162.Heathcote J, Main J. Treatment of hepatitis C. J. Viral Hepat. 2005;12:223–235. doi: 10.1111/j.1365-2893.2005.00600.x. [DOI] [PubMed] [Google Scholar]
- 163.Foster GR. Past, present, and future hepatitis C treatments. Semin. Liver Dis. 2004;24(Suppl. 2):97–104. doi: 10.1055/s-2004-832934. [DOI] [PubMed] [Google Scholar]
- 164.de Koning EW, van Bijsterveld OP, Cantell K. Combination therapy for dendritic keratitis with acyclovir and α-interferon. Arch. Ophthalmol. 1983;101:1866–1868. doi: 10.1001/archopht.1983.01040020868006. [DOI] [PubMed] [Google Scholar]
- 165.Jones BR, Coster DJ, Falcon MG, Cantell K. Topical therapy of ulcerative herpetic keratitis with human interferon. Lancet. 1976;17:128. doi: 10.1016/s0140-6736(76)92850-6. [DOI] [PubMed] [Google Scholar]
- 166.Merigan TC, et al. Human leukocyte interferon for the treatment of herpes zoster in patients with cancer. N. Engl. J. Med. 1978;298:981–987. doi: 10.1056/NEJM197805042981801. [DOI] [PubMed] [Google Scholar]
- 167.Pazin GJ, et al. Prevention of reactivated herpes simplex infection by human leukocyte interferon after operation on the trigeminal root. N. Engl. J. Med. 1979;301:225–230. doi: 10.1056/NEJM197908023010501. [DOI] [PubMed] [Google Scholar]
- 168.Winston DJ, et al. Recombinant interferon α-2a for treatment of herpes zoster in immunosuppressed patients with cancer. Am. J. Med. 1988;85:147–151. doi: 10.1016/s0002-9343(88)80333-4. [DOI] [PubMed] [Google Scholar]
- 169.Lui SF, et al. Double-blind, placebo-controlled trial of human lymphoblastoid interferon prophylaxis of cytomegalovirus infection in renal transplant recipients. Nephrol. Dial. Transplant. 1992;7:1230–1237. doi: 10.1093/ndt/7.12.1230. [DOI] [PubMed] [Google Scholar]
- 170.Haglund S, Lundquist PG, Cantell K, Strander H. Interferon therapy in juvenile laryngeal papillomatosis. Arch. Otolaryngol. 1981;107:327–332. doi: 10.1001/archotol.1981.00790420001001. [DOI] [PubMed] [Google Scholar]
- 171.Pazin GJ, et al. Effects of interferon-α on human warts. J. Interferon Res. 1982;2:235–243. doi: 10.1089/jir.1982.2.235. [DOI] [PubMed] [Google Scholar]
- 172.Eron LJ, et al. Interferon therapy for condylomata acuminata. N. Engl. J. Med. 1986;315:1059–1064. doi: 10.1056/NEJM198610233151704. [DOI] [PubMed] [Google Scholar]
- 173.Healy GB, et al. Treatment of recurrent respiratory papillomatosis with human leukocyte interferon. Results of a multicenter randomized clinical trial. N. Engl. J. Med. 1988;319:401–407. doi: 10.1056/NEJM198808183190704. [DOI] [PubMed] [Google Scholar]
- 174.Weck PK, Buddin DA, Whisnant JK. Interferons in the treatment of genital human papillomavirus infections. Am. J. Med. 1988;85:159–164. [PubMed] [Google Scholar]
- 175.Deuñas L, et al. Use of interferon-α in laryngeal papillomatosis: eight years of the Cuban national programme. J. Laryngol. Otol. 1997;111:134–140. doi: 10.1017/s0022215100136667. [DOI] [PubMed] [Google Scholar]
- 176.Beutner KR, et al. Imiquimod, a patient-applied immune-response modifier for treatment of external genital warts. Antimicrob. Agents Chemother. 1998;42:789–794. doi: 10.1128/aac.42.4.789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Tyring SK, et al. A randomized, controlled, molecular study of condylomata acuminata clearance during treatment with imiquimod. J. Infect. Dis. 1998;178:551–555. doi: 10.1086/517472. [DOI] [PubMed] [Google Scholar]
- 178.Moore RA, Edwards JE, Hopwood J, Hicks D. Imiquimod for the treatment of genital warts: a quantitative systematic review. BMC Infect. Dis. 2001;1:e3. doi: 10.1186/1471-2334-1-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Lane HC, et al. Interferon-α in patients with asymptomatic human immunodeficiency virus (HIV) infection. A randomized, placebo-controlled trial. Ann. Intern. Med. 1990;112:805–811. doi: 10.7326/0003-4819-112-11-805. [DOI] [PubMed] [Google Scholar]
- 180.Krown SE, Aeppli D, Balfour HH., Jr. Phase II, randomized, open-label, community-based trial to compare the safety and activity of combination therapy with recombinant interferon-α2b and zidovudine versus zidovudine alone in patients with asymptomatic to mildly symptomatic HIV infection. HIV Protocol C91–253 Study Team. J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 1999;20:245–254. doi: 10.1097/00042560-199903010-00005. [DOI] [PubMed] [Google Scholar]
- 181.Merigan TC, Reed SE, Hall TS, Tyrrell DA. Inhibition of respiratory virus infection by locally applied interferon. Lancet. 1973;301:563–567. doi: 10.1016/s0140-6736(73)90714-9. [DOI] [PubMed] [Google Scholar]
- 182.Scott GM, et al. Prevention of rhinovirus colds by human interferon α-2 from Escherichia coli. Lancet. 1982;320:186–188. doi: 10.1016/s0140-6736(82)91031-5. [DOI] [PubMed] [Google Scholar]
- 183.Higgins PG, et al. Intranasal interferon as protection against experimental respiratory coronavirus infection in volunteers. Antimicrob. Agents Chemother. 1983;24:713–715. doi: 10.1128/aac.24.5.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Treanor JJ, Betts RF, Erb SM, Roth FK, Dolin R. Intranasally administered interferon as prophylaxis against experimentally induced influenza A virus infection in humans. J. Infect. Dis. 1987;156:379–383. doi: 10.1093/infdis/156.2.379. [DOI] [PubMed] [Google Scholar]
- 185.Hayden FG, Kaiser DL, Albrecht JK. Intranasal recombinant α-2b interferon treatment of naturally occurring common colds. Antimicrob. Agents Chemother. 1988;32:224–230. doi: 10.1128/aac.32.2.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Douglas RM, et al. Prophylactic efficacy of intranasal α 2-interferon against rhinovirus infections in the family setting. N. Engl. J. Med. 1986;314:65–70. doi: 10.1056/NEJM198601093140201. [DOI] [PubMed] [Google Scholar]
- 187.Farr BM, Gwaltney JM, Jr., Adams KF, Hayden FG. Intranasal interferon-α 2 for prevention of natural rhinovirus colds. Antimicrob. Agents Chemother. 1984;6:31–34. doi: 10.1128/aac.26.1.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Loutfy MR, et al. Interferon alfacon-1 plus corticosteroids in severe acute respiratory syndrome: a preliminary study. JAMA. 2003;290:3222–3228. doi: 10.1001/jama.290.24.3222. [DOI] [PubMed] [Google Scholar]
- 189.Jacobs L, O'Malley J, Freeman A, Ekes R. Intrathecal interferon reduces exacerbations of multiple sclerosis. Science. 1981;214:1026–1028. doi: 10.1126/science.6171035. [DOI] [PubMed] [Google Scholar]
- 190.Knobler RL, et al. Systemic α-interferon therapy of multiple sclerosis. Neurology. 1984;34:1273–1279. doi: 10.1212/wnl.34.10.1273. [DOI] [PubMed] [Google Scholar]
- 191.Panitch HS, Hirsch RL, Haley AS, Johnson KP. Exacerbations of multiple sclerosis in patients treated with γ interferon. Lancet. 1987;329:893–895. doi: 10.1016/s0140-6736(87)92863-7. [DOI] [PubMed] [Google Scholar]
- 192.Panitch HS, Hirsch RL, Schindler J, Johnson KP. Treatment of multiple sclerosis with γ interferon: exacerbations associated with activation of the immune system. Neurology. 1987;37:1097–1102. doi: 10.1212/wnl.37.7.1097. [DOI] [PubMed] [Google Scholar]
- 193.IFNB MS Study Group. Interferon β-1b is effective in relapsing-remitting multiple sclerosis. I. Clinical results of a multicenter, randomized, double-blind, placebo-controlled trial. Neurology43, 655–661 (1993). [DOI] [PubMed]
- 194.Paty DW, Li DK. Interferon β-1b is effective in relapsing-remitting multiple sclerosis. II. MRI analysis results of a multicenter, randomized, double-blind, placebo-controlled trial. UBC MS/MRI Study Group and the IFNB Multiple Sclerosis Study Group. Neurology. 1993;43:662–667. doi: 10.1212/wnl.43.4.662. [DOI] [PubMed] [Google Scholar]
- 195.Bermel RA, Rudick RA. Interferon-β treatment for multiple sclerosis. Neurotherapeutics. 2007;4:633–646. doi: 10.1016/j.nurt.2007.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Rudick RA, Cutter G. Interferon-β for multiple sclerosis: long-term benefits? Ann. Neurol. 2007;61:283–285. doi: 10.1002/ana.21119. [DOI] [PubMed] [Google Scholar]
- 197.Hauser SL, Johnston SC. Recombinant therapeutics: from bench to bedside (if your health plan concurs) Ann. Neurol. 2006;60:10A–11A. doi: 10.1002/ana.21041. [DOI] [PubMed] [Google Scholar]
- 198.Ascherio A, Munger KL. Environmental risk factors for multiple sclerosis. Part II: noninfectious factors. Ann. Neurol. 2007;61:504–513. doi: 10.1002/ana.21141. [DOI] [PubMed] [Google Scholar]
- 199.Ascherio A, Munger KL. Environmental risk factors for multiple sclerosis. Part I: the role of infection. Ann. Neurol. 2007;61:288–299. doi: 10.1002/ana.21117. [DOI] [PubMed] [Google Scholar]
- 200.Lincoln MR, et al. A predominant role for the HLA class II region in the association of the MHC region with multiple sclerosis. Nature Genet. 2005;37:1108–1112. doi: 10.1038/ng1647. [DOI] [PubMed] [Google Scholar]
- 201.Peltonen L. Old suspects found guilty — the first genome profile of multiple sclerosis. N. Engl. J. Med. 2007;357:927–929. doi: 10.1056/NEJMe078147. [DOI] [PubMed] [Google Scholar]
- 202.Calabresi PA, et al. Increases in soluble VCAM-1 correlate with a decrease in MRI lesions in multiple sclerosis treated with interferon β-1b. Ann. Neurol. 1997;41:669–674. doi: 10.1002/ana.410410517. [DOI] [PubMed] [Google Scholar]
- 203.Frank JA, et al. Serial contrast-enhanced magnetic resonance imaging in patients with early relapsing-remitting multiple sclerosis: implications for treatment trials. Ann. Neurol. 1994;36:S86–S90. doi: 10.1002/ana.410360719. [DOI] [PubMed] [Google Scholar]
- 204.Stone LA, et al. The effect of interferon-β on blood–brain barrier disruptions demonstrated by contrast-enhanced magnetic resonance imaging in relapsing-remitting multiple sclerosis. Ann. Neurol. 1995;37:611–619. doi: 10.1002/ana.410370511. [DOI] [PubMed] [Google Scholar]
- 205.Shiow LR, et al. CD69 acts downstream of interferon-α/β to inhibit S1P1 and lymphocyte egress from lymphoid organs. Nature. 2006;440:540–544. doi: 10.1038/nature04606. [DOI] [PubMed] [Google Scholar]
- 206.Stuve O, et al. Interferon β-1b decreases the migration of T lymphocytes in vitro: effects on matrix metalloproteinase-9. Ann. Neurol. 1996;40:853–863. doi: 10.1002/ana.410400607. [DOI] [PubMed] [Google Scholar]
- 207.Leppert D, Waubant E, Burk MR, Oksenberg JR, Hauser SL. Interferon β-1b inhibits gelatinase secretion and in vitro migration of human T cells: a possible mechanism for treatment efficacy in multiple sclerosis. Ann. Neurol. 1996;40:846–852. doi: 10.1002/ana.410400606. [DOI] [PubMed] [Google Scholar]
- 208.Fernald GH, et al. Genome-wide network analysis reveals the global properties of IFN-β immediate transcriptional effects in humans. J. Immunol. 2007;178:5076–5085. doi: 10.4049/jimmunol.178.8.5076. [DOI] [PubMed] [Google Scholar]
- 209.Rudick RA, Lee JC, Simon J, Ransohoff RM, Fisher E. Defining interferon β response status in multiple sclerosis patients. Ann. Neurol. 2004;56:548–555. doi: 10.1002/ana.20224. [DOI] [PubMed] [Google Scholar]
- 210.Gutterman JU. Cytokine therapeutics: lessons from interferon α. Proc. Natl Acad. Sci. USA. 1994;91:1198–1205. doi: 10.1073/pnas.91.4.1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Kurzrock R, Talpaz M, Gutterman JU. Hairy cell leukemia: review of treatment. Br. J. Haematol. 1991;79(Suppl. 1):17–20. doi: 10.1111/j.1365-2141.1991.tb08112.x. [DOI] [PubMed] [Google Scholar]
- 212.The Italian Cooperative Study Group on Chronic Myeloid Leukemia. Long-term follow-up of the Italian trial of interferon-α versus conventional chemotherapy in chronic myeloid leukemia. Blood92, 1541–1548 (1998). [PubMed]
- 213.Talpaz M. Interferon-α-based treatment of chronic myeloid leukemia and implications of signal transduction inhibition. Semin. Hematol. 2001;38:22–27. doi: 10.1016/s0037-1963(01)90114-3. [DOI] [PubMed] [Google Scholar]
- 214.Bonifazi F, et al. Chronic myeloid leukemia and interferon-α: a study of complete cytogenetic responders. Blood. 2001;98:3074–3081. doi: 10.1182/blood.v98.10.3074. [DOI] [PubMed] [Google Scholar]
- 215.Borden EC, et al. Cancer Medicine. 2006. pp. 733–743. [Google Scholar]
- 216.Rohatiner AZ, et al. Meta-analysis to evaluate the role of interferon in follicular lymphoma. J. Clin. Oncol. 2005;23:2215–2223. doi: 10.1200/JCO.2005.06.146. [DOI] [PubMed] [Google Scholar]
- 217.Gresser I, Maury C, Belardelli F. Anti-tumor effects of interferon in mice injected with interferon-sensitive and interferon-resistant Friend leukemia cells. VI. Adjuvant therapy after surgery in the inhibition of liver and spleen metastases. Int. J. Cancer. 1987;39:789–792. doi: 10.1002/ijc.2910390623. [DOI] [PubMed] [Google Scholar]
- 218.Müller CR, Smeland S, Bauer HC, Saeter G, Strander H. Interferon-α as the only adjuvant treatment in high-grade osteosarcoma: long term results of the Karolinska Hospital series. Acta Oncol. 2005;44:475–480. doi: 10.1080/02841860510029978. [DOI] [PubMed] [Google Scholar]
- 219.Kirkwood JM, et al. A pooled analysis of Eastern Cooperative Oncology Group and intergroup trials of adjuvant high-dose interferon for melanoma. Clin. Cancer Res. 2004;10:1670–1677. doi: 10.1158/1078-0432.ccr-1103-3. [DOI] [PubMed] [Google Scholar]
- 220.Wheatley K, et al. Does adjuvant interferon-α for high-risk melanoma provide a worthwhile benefit? A meta-analysis of the randomised trials. Cancer Treat. Rev. 2003;29:241–252. doi: 10.1016/s0305-7372(03)00074-4. [DOI] [PubMed] [Google Scholar]
- 221.Eggermont AMM, Gore M. Randomized adjuvant therapy trials in melanoma: surgical and systemic trials. Semin. Oncol. 2007;34:507–513. doi: 10.1053/j.seminoncol.2007.09.003. [DOI] [PubMed] [Google Scholar]
- 222.Weiss K. Safety profile of interferon-α therapy. Semin. Oncol. 1998;25:S9–S13. [PubMed] [Google Scholar]
- 223.Kirkwood JM, et al. Mechanisms and management of toxicities associated with high-dose interferon α-2b therapy. J. Clin. Oncol. 2002;20:3703–3718. doi: 10.1200/JCO.2002.03.052. [DOI] [PubMed] [Google Scholar]
- 224.Diaz MO, et al. Deletions of interferon genes in acute lymphoblastic leukemia. N. Engl. J. Med. 1990;322:77–82. doi: 10.1056/NEJM199001113220202. [DOI] [PubMed] [Google Scholar]
- 225.Fountain JW, et al. Homozygous deletions within human chromosome band 9p21 in melanoma. Proc. Natl Acad. Sci. USA. 1992;89:10557–10561. doi: 10.1073/pnas.89.21.10557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Olopade OI, et al. Homozygous loss of the interferon genes defines the critical region on 9p that is deleted in lung cancers. Cancer Res. 1993;53(Suppl. 10):2410–2415. [PubMed] [Google Scholar]
- 227.DeYoung KL, et al. Cloning a novel member of the human interferon-inducible gene family associated with control of tumorigenicity in a model of human melanoma. Oncogene. 1997;15:453–457. doi: 10.1038/sj.onc.1201206. [DOI] [PubMed] [Google Scholar]
- 228.Ding Y, Lee JF, Lu H, Lee MH, Yan DH. Interferon-inducible protein IFIXα1 functions as a negative regulator of HDM2. Mol. Cell Biol. 2006;26:1979–1996. doi: 10.1128/MCB.26.5.1979-1996.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Johnsen A, France J, Sy MS, Harding CV. Down-regulation of the transporter for antigen presentation, proteasome subunits, and class I major histocompatibility complex in tumor cell lines. Cancer Res. 1998;58:3660–3667. [PubMed] [Google Scholar]
- 230.Shou J, et al. Expression profiling of a human cell line model of prostatic cancer reveals a direct involvement of interferon signaling in prostate tumor progression. Proc. Natl Acad. Sci. USA. 2002;99:2830–2835. doi: 10.1073/pnas.052705299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Seliger B, et al. Characterization of human lymphocyte antigen class I antigen-processing machinery defects in renal cell carcinoma lesions with special emphasis on transporter-associated with antigen-processing down-regulation. Clin. Cancer Res. 2003;9:1721–1727. [PubMed] [Google Scholar]
- 232.Hoek K, et al. Expression profiling reveals novel pathways in the transformation of melanocytes to melanomas. Cancer Res. 2004;64:5270–5282. doi: 10.1158/0008-5472.CAN-04-0731. [DOI] [PubMed] [Google Scholar]
- 233.Pitha-Rowe I, et al. Microarray analyses uncover UBE1L as a candidate target gene for lung cancer chemoprevention. Cancer Res. 2004;64:8109–8115. doi: 10.1158/0008-5472.CAN-03-3938. [DOI] [PubMed] [Google Scholar]
- 234.Ito N, et al. STAT3 polymorphism predicts interferon-α response in patients with metastatic renal cell carcinoma. J. Clin. Oncol. 2007;25:2785–2791. doi: 10.1200/JCO.2006.09.8897. [DOI] [PubMed] [Google Scholar]
- 235.Casey G, et al. RNASEL Arg462Gln variant is implicated in up to 13% of prostate cancer cases. Nature Genet. 2002;32:581–583. doi: 10.1038/ng1021. [DOI] [PubMed] [Google Scholar]
- 236.Borden EC. Augmentation of effects of interferon-stimulated genes by reversal of epigenetic silencing: potential application to melanoma. Cytokine Growth Factor Rev. 2007;18:491–501. doi: 10.1016/j.cytogfr.2007.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Nakaii M, et al. IFN-α prevents the growth of pre-neoplastic lesions and inhibits the development of hepatocellular carcinoma in the rat. Carcinogenesis. 2004;25:389–397. doi: 10.1093/carcin/bgh028. [DOI] [PubMed] [Google Scholar]
- 238.Borden EC, Sidky YA, Ertürk E, Wierenga W, Bryan GT. Protection from carcinogen-induced murine bladder carcinoma by interferons and an oral interferon-inducing pyrimidinone, bropirimine. Cancer Res. 1990;50:1071–1074. [PubMed] [Google Scholar]
- 239.Kulik LM. Can therapy of hepatitis C affect the development of hepatocellular carcinoma? J. Natl Comp. Cancer Netw. 2006;4:751–757. doi: 10.6004/jnccn.2006.0065. [DOI] [PubMed] [Google Scholar]
- 240.Chawla-Sarkar M, et al. Apoptosis and interferons: role of interferon-stimulated genes as mediators of apoptosis. Apoptosis. 2003;8:237–249. doi: 10.1023/a:1023668705040. [DOI] [PubMed] [Google Scholar]
- 241.Ballestrero A, et al. Tumor necrosis factor-related apoptosis-inducing ligand cooperates with anticancer drugs to overcome chemoresistance in antiapoptotic Bcl-2 family members expressing jurkat cells. Clin. Cancer Res. 2004;10:1463–1470. doi: 10.1158/1078-0432.ccr-1365-02. [DOI] [PubMed] [Google Scholar]
- 242.Sato K, et al. Antiviral response by natural killer cells through TRAIL gene induction by IFN-α/β. Eur. J. Immunol. 2001;31:3138–3146. doi: 10.1002/1521-4141(200111)31:11<3138::aid-immu3138>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 243.Chaperot L, et al. Virus or TLR agonists induce TRAIL-mediated cytotoxic activity of plasmacytoid dendritic cells. J. Immunol. 2006;176:248–255. doi: 10.4049/jimmunol.176.1.248. [DOI] [PubMed] [Google Scholar]
- 244.Buechner SA, et al. Regression of basal cell carcinoma by intralesional interferon-α treatment is mediated by CD95 (Apo-1/Fas)-CD95ligand-induced suicide. J. Clin. Invest. 1997;100:2691–2696. doi: 10.1172/JCI119814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Ugurel S, et al. Heterogenous susceptibility to CD95-induced apoptosis in melanoma cells correlates with Bcl-2 and Bcl-x expression and is sensitive to modulation by interferon-γ. Int. J. Cancer. 1999;82:727–736. doi: 10.1002/(sici)1097-0215(19990827)82:5<727::aid-ijc17>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 246.Schwartzberg LS, et al. Modulation of the Fas signaling pathway by IFN-γ in therapy of colon cancer: Phase I trial and correlative studies of IFN-γ, 5-fluorouracil, and leukovorin. Clin. Cancer. Res. 2002;8:2488–2498. [PubMed] [Google Scholar]
- 247.Leaman DW, et al. Identification of X-linked inhibitor of apoptosis-associated factor-1 (XAF1) as an interferon-stimulated gene that augments TRAIL/Apo2L-induced apoptosis. J. Biol. Chem. 2002;277:28504–28511. doi: 10.1074/jbc.M204851200. [DOI] [PubMed] [Google Scholar]
- 248.Lee MG, et al. Promoter CpG hypermethylation and downregulation of XAF1 expression in human urogenital malignancies: implication for attenuated p53 response to apoptotic stresses. Oncogene. 2006;25:5807–5822. doi: 10.1038/sj.onc.1209867. [DOI] [PubMed] [Google Scholar]
- 249.Pizzoferrato E, et al. Ectopic expression of interferon regulatory factor-1 promotes human breast cancer cell death and results in reduced expression of survivin. Cancer Res. 2004;64:8381–8388. doi: 10.1158/0008-5472.CAN-04-2223. [DOI] [PubMed] [Google Scholar]
- 250.Pfeffer LM, et al. Biological properties of recombinant α-interferons: 40th anniversary of the discovery of interferons. Cancer Res. 1998;58:2489–2499. [PubMed] [Google Scholar]
- 251.Tannenbaum CS, Hamilton TA. Immune-inflammatory mechanisms in IFN-γ-mediated anti-tumor activity. Semin. Cancer Biol. 2000;10:113–123. doi: 10.1006/scbi.2000.0314. [DOI] [PubMed] [Google Scholar]
- 252.Glimcher LH, Townsend MJ, Sullivan BM, Lord GM. Recent developments in the transcriptional regulation of cytolytic effector cells. Nature Rev. Immunol. 2004;4:900–911. doi: 10.1038/nri1490. [DOI] [PubMed] [Google Scholar]
- 253.Mikhak Z, et al. STAT1 in peripheral tissue differentially regulates homing of antigen-specific Th1 and Th2 cells. J. Immunol. 2006;176:4959–4967. doi: 10.4049/jimmunol.176.8.4959. [DOI] [PubMed] [Google Scholar]
- 254.Dunn GP, Koebel CM, Schreiber RD. Interferons, immunity and cancer immunoediting. Nature Rev. Immunol. 2006;6:836–848. doi: 10.1038/nri1961. [DOI] [PubMed] [Google Scholar]
- 255.Swann JB, et al. Type I IFN contributes to NK cell homeostasis, activation, and antitumor functions. J. Immunol. 2007;178:7540–7549. doi: 10.4049/jimmunol.178.12.7540. [DOI] [PubMed] [Google Scholar]
- 256.Strehl B, et al. Interferon-γ, the functional plasticity of the ubiquitin-proteasome system, and MHC class I antigen processing. Immunol. Rev. 2005;207:19–30. doi: 10.1111/j.0105-2896.2005.00308.x. [DOI] [PubMed] [Google Scholar]
- 257.Greiner JW, et al. Intraperitoneal administration of interferon-γ to carcinoma patients enhances expression of tumor-associated glycoprotein-72 and carcinoembryonic antigen on malignant ascites cells. J. Clin. Oncol. 1992;10:735–746. doi: 10.1200/JCO.1992.10.5.735. [DOI] [PubMed] [Google Scholar]
- 258.Folkman J. Angiogenesis: an organizing principle for drug discovery. Nature Rev. Drug Discovery. 2007;6:273–286. doi: 10.1038/nrd2115. [DOI] [PubMed] [Google Scholar]
- 259.Brouty-Boye D, Zetter BR. Inhibition of cell motility by interferons. Science. 1980;208:516–518. doi: 10.1126/science.6154315. [DOI] [PubMed] [Google Scholar]
- 260.Dvorak HF, Gresser I. Microvascular injury in pathogenesis of interferon-induced necrosis of subcutaneous tumors in mice. J. Natl Cancer Inst. 1989;81:497–502. doi: 10.1093/jnci/81.7.497. [DOI] [PubMed] [Google Scholar]
- 261.Sidky YA, Borden EC. Inhibition of angiogenesis by interferons: effects on tumor- and lymphocyte-induced vascular responses. Cancer Res. 1987;47:5155–5161. [PubMed] [Google Scholar]
- 262.Dinney CP, et al. Inhibition of basic fibroblast growth factor expression, angiogenesis, and growth of human bladder carcinoma in mice by systemic interferon-α administration. Cancer Res. 1998;58:808–814. [PubMed] [Google Scholar]
- 263.McCarty MF, Bielenberg D, Donawho C, Bucana CD, Fidler IJ. Evidence for the causal role of endogenous interferon-α/β in the regulation of angiogenesis, tumorigenicity, and metastasis of cutaneous neoplasms. Clin. Exp. Metastasis. 2002;19:609–615. doi: 10.1023/a:1020923326441. [DOI] [PubMed] [Google Scholar]
- 264.von Marschall Z, et al. Effects of interferon α on vascular endothelial growth factor gene transcription and tumor angiogenesis. J. Natl Cancer Inst. 2003;95:437–448. doi: 10.1093/jnci/95.6.437. [DOI] [PubMed] [Google Scholar]
- 265.Reznikov LL, et al. Spontaneous and inducible cytokine responses in healthy humans receiving a single dose of IFN-α2b: increased production of interleukin-1 receptor antagonist and suppression of IL-1-induced IL-8. J. Interferon Cytokine Res. 1998;18:897–903. doi: 10.1089/jir.1998.18.897. [DOI] [PubMed] [Google Scholar]
- 266.Yang J, Richmond A. The angiostatic activity of interferon-inducible protein-10/CXCL10 in human melanoma depends on binding to CXCR3 but not to glycosaminoglycan. Mol. Ther. 2004;9:846–855. doi: 10.1016/j.ymthe.2004.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Feldman ED, et al. Interferon γ-inducible protein 10 selectively inhibits proliferation and induces apoptosis in endothelial cells. Ann. Surg. Oncol. 2006;13:125–133. doi: 10.1245/ASO.2006.03.038. [DOI] [PubMed] [Google Scholar]
- 268.Guenzi E, et al. The guanylate binding protein-1 GTPase controls the invasive and angiogenic capability of endothelial cells through inhibition of MMP-1 expression. EMBO J. 2003;22:3772–3782. doi: 10.1093/emboj/cdg382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Lindner DJ. Interferons as antiangiogenic agents. Curr. Oncol. Rep. 2002;4:510–514. doi: 10.1007/s11912-002-0065-4. [DOI] [PubMed] [Google Scholar]
- 270.Faria AM, et al. The nucleoporin Nup96 is required for proper expression of interferon-regulated proteins and functions. Immunity. 2006;24:295–304. doi: 10.1016/j.immuni.2006.01.014. [DOI] [PubMed] [Google Scholar]
- 271.Han JQ, Barton DJ. Activation and evasion of the antiviral 2′-5′ oligoadenylate synthetase/ribonuclease L pathway by hepatitis C virus mRNA. RNA. 2002;8:512–525. doi: 10.1017/s1355838202020617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Gale MJ, Jr, et al. Evidence that hepatitis C virus resistance to interferon is mediated through repression of the PKR protein kinase by the nonstructural 5A protein. Virology. 1997;230:217–227. doi: 10.1006/viro.1997.8493. [DOI] [PubMed] [Google Scholar]
- 273.Carpten J, et al. Germline mutations in the ribonuclease L gene in families showing linkage with HPC1. Nature Genet. 2002;30:181–184. doi: 10.1038/ng823. [DOI] [PubMed] [Google Scholar]
- 274.Urisman A, et al. Identification of a novel Gammaretrovirus in prostate tumors of patients homozygous for R462Q RNASEL variant. PLoS Pathog. 2006;2:e25. doi: 10.1371/journal.ppat.0020025. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 275.Dong B, et al. An infectious retrovirus susceptible to an IFN antiviral pathway from human prostate tumors. Proc. Natl Acad. Sci. USA. 2007;104:1655–1660. doi: 10.1073/pnas.0610291104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Pedersen IM, et al. Interferon modulation of cellular microRNAs as an antiviral mechanism. Nature. 2007;449:919–922. doi: 10.1038/nature06205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Brideau-Andersen AD, et al. Directed evolution of gene-shuffled IFN-α molecules with activity profiles tailored for treatment of chronic viral diseases. Proc. Natl Acad. Sci. USA. 2007;104:8269–8274. doi: 10.1073/pnas.0609001104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Walsh RJ, et al. Type I interferon-inducible gene expression in blood is present and reflects disease activity in dermatomyositis and polymyositis. Arthritis Rheum. 2007;56:3784–3792. doi: 10.1002/art.22928. [DOI] [PMC free article] [PubMed] [Google Scholar]