

Abstract
Annual outbreaks of influenza A infection are an ongoing public health threat and novel influenza strains can periodically emerge to which humans have little immunity, resulting in devastating pandemics. The 1918 pandemic killed at least 40 million people worldwide and pandemics in 1957 and 1968 caused hundreds of thousands of deaths. The influenza A virus is capable of enormous genetic variation, both by continuous, gradual mutation and by reassortment of genome segments between viruses. Both the 1957 and 1968 pandemic strains are thought to have originated as reassortants in which one or both human-adapted viral surface proteins were replaced by proteins from avian influenza strains. Analyses of the genes of the 1918 pandemic virus, however, indicate that this strain might have had a different origin. The haemagglutinin and nucleoprotein genome segments in particular are unlikely to have come directly from an avian source that is similar to those that are currently being sequenced. Determining whether a pandemic influenza virus can emerge by different mechanisms will affect the scope and focus of surveillance and prevention efforts.
Main
Influenza afflicts approximately 10–20% of the population and kills an estimated 36,000 people each year in the United States1. The annual return of the disease is driven by the antigenic variability of the influenza virus; two or three amino acid changes in the haemagglutinin (HA) protein on the virus surface can greatly reduce the effectiveness of existing antibodies, leaving people vulnerable to repeated influenza infections throughout their lives. In addition to this gradual change in the influenza virus, which is known as ANTIGENIC DRIFT, influenza A viruses can acquire novel surface proteins against which existing antibodies are ineffective2. When this kind of ANTIGENIC SHIFT occurs, up to one-third of the population can become ill in an explosive global pandemic3,4.
Two characteristics of influenza A biology contribute to its ability to undergo antigenic shifts. First, its RNA genome comprises eight separate segments. Co-infection of a cell with two different influenza virus strains can result in the generation of hybrid viruses that contain some segments from each progenitor — a process known as REASSORTMENT5. Second, influenza A viruses can infect many animal hosts, including wild waterfowl, poultry, pigs and horses. Under circumstances that have not yet been fully characterized, influenza strains can cross between species. The ability to reassort and the existence of several reservoirs of influenza virus provide many possibilities for the generation of novel influenza virus strains2.
Four pandemics have occurred in recent history — in 1890, 1918, 1957 and 1968 (Refs 2–4). The genetic composition of the viruses responsible for the 1957 and 1968 pandemics is known; these viruses seem to have been generated by reassortment between an avian influenza strain and the strain then circulating in humans. In the 1957 H2N2-SUBTYPE pandemic virus, both influenza surface proteins, HA and neuraminidase (NA), and one internal protein, polymerase B1 (PB1), were closely related to Eurasian wild waterfowl influenza proteins6,7. In 1968, the H3N2 pandemic virus contained novel HA and PB1 proteins, also apparently of Eurasian wild waterfowl origin7,8. Although it is not known exactly how these reassortant viruses were generated, pigs can be infected with both avian and human influenza strains and this species has been suggested as a potential 'mixing vessel' for the generation of pandemic viruses2,9.
In 1918, the most devastating influenza pandemic in history killed at least 40 million people10,11. In addition to a death toll that is several times higher than that of other influenza pandemics, the 1918 H1N1 virus took its greatest toll on young adults instead of the very old and very young who are usually most affected by influenza12,13,14,15,16. Recently, isolation of the genetic material of the 1918 strain from formalin-fixed and frozen case material has made it possible to determine the genetic sequence of this virus. Five of the eight genome segments have been sequenced17,18,19,20,21,22. Analyses of these sequences are underway to try to understand whether the 1918 pandemic strain was generated by the same pattern of reassortment as the 1957 and 1968 strains. It is possible that the unusual characteristics of the 1918 pandemic were, in part, due to an unusual origin. Even if this is not the case, it is important to know whether there is another pathway for the generation of pandemic strains so that surveillance for emerging strains can be optimized.
As data have accumulated, evidence has emerged to indicate that some of the genome segments of the 1918 H1N1 strain might have a novel origin that has not been seen in strains responsible for other pandemics23. The nucleoprotein (NP) gene sequence, in particular, seems to have been acquired directly from a source that is similar to viruses currently found in wild birds at the amino acid level, but very divergent at the nucleotide level, suggesting considerable evolutionary distance between the source of the 1918 NP and the currently sequenced virus strains in wild birds22. In light of this conclusion, it is worth re-examining the data on the other four sequenced 1918 genome segments, to see whether a similarly unusual origin is possible.
Nucleoprotein
PHYLOGENETIC ANALYSES of the NP segment of influenza virus produce trees with two large CLADES — a 'mammalian' clade with human and classical swine subclades, and an 'avian' clade containing equine, gull and waterfowl subclades. The waterfowl subclade is further divided into North American and Eurasian branches. The 1918 NP sequence is placed within the mammalian clade whether total, SYNONYMOUS or NON-SYNONYMOUS substitutions are analysed. However, a BLAST search of the 1918 NP protein reveals that the amino acid sequences to which it is most closely related are avian. It differs from the A/Duck/Bavaria/2/77 (H1N1) strain by only eight amino acids, compared with 11 amino acid differences from the A/Swine/Iowa/15/30 (H1N1) strain and 16 from the A/Wilson-Smith/33 (H1N1) strain (an early human virus). Conversely, the 1918 NP gene differs from the A/Duck/ Bavaria/2/77 NP sequence by 193 nucleotides (87.1% identity), from the A/Swine/Iowa/15/30 sequence by 67 nucleotides (95.5% identity) and from A/Wilson-Smith/33 by 65 nucleotides (95.6% identity). The large number of synonymous differences with the avian sequence would be expected if the NP of the 1918 strain had been retained from the previously circulating human strain. However, the small number of amino acid differences from the avian strain indicates a recent avian origin.
Synonymous/non-synonymous (S/N) ratios vary widely between different lineages in the NP phylogenetic tree. Within the avian clade, where influenza is thought to be endemic, the average S/N ratio is 15.2. This indicates that in avian strains, the NP is so well adapted that most amino acid changes would be deleterious, and silent nucleotide changes therefore predominate. Within the mammalian clade, the average S/N ratio is 3.9, which indicates that NP is an actively evolving protein in which many amino acid changes provide a selective advantage. When the nucleotide sequence of the NP from the 1918 virus is compared with the NP sequence of mammalian isolates, the S/N ratios vary from 2.7 (1918 compared with A/Wilson-Smith/33) to 5.1 (1918 compared with A/Swine/Iowa/15/30). By contrast, when the 1918 sequence is compared with 10 avian species chosen from both the Eurasian and North American avian subclades, the S/N ratio is 10.4–18.0, with an average of 13.9. These data reinforce the results of the BLAST searches — when compared with avian NP sequences, the 1918 sequence has many changes at the nucleotide level, but only a few of these result in amino acid changes.
A subset of synonymous substitutions that are known as FOUR-FOLD DEGENERATE SUBSTITUTIONS because the presence of any of the four bases does not lead to an amino acid replacement have been investigated. When avian sequences are compared with one another, the number of nucleotide differences is usually 20–30%. Comparisons of the 1918 NP with avian NP sequences consistently yields differences of 30–40%, indicating that the 1918 NP is different from known avian NPs. This does not seem to be simply a matter of evolutionary time, as a region of a viral NP gene found in a strain isolated from a bird captured in 1917 (Refs 24,25) shows identical patterns of four-fold degenerate differences — 20–30% differences in comparisons with modern birds and a 44% difference compared with the 1918 pandemic virus sequence.
Given that only a limited number of wild-bird viruses have been sequenced, is it possible that greater sampling would reveal an NP sequence that is more closely related to the 1918 NP sequences? Avian sequences vary from each other at many synonymous sites, but they are all similarly distant from the avian consensus sequence. Therefore, if the 1918 virus acquired its NP from an avian virus that is similar to current avian strains, it should have a similar number of synonymous changes compared with the avian consensus sequence. One would also expect the 1918 strain to be more similar either to the Eurasian or the North American consensus sequences, as is true of the avian strains that have been sequenced so far. Table 1 and Fig. 1 show that this is not the case. The NP sequence of the 1918 virus differs by 173 nucleotides from the overall avian consensus sequence, by 187 nucleotides from the North American avian consensus sequence and by 179 nucleotides from the Eurasian avian consensus sequence. So, it is only slightly more closely related to the consensus sequences than it is to individual strains, and no more closely related to one avian clade than the other. By contrast, individual avian sequences are more closely related to the avian consensus sequences than they are to each other, and much more closely related to the consensus of their own clade than to the consensus of the other clade (Table 1).
Table 1.
Comparison of NPs with avian consensus sequences

Figure 1. Comparison of the nucleoprotein (NP) genome segment from the 1918 pandemic influenza virus and two avian strains with the overall avian consensus sequence.
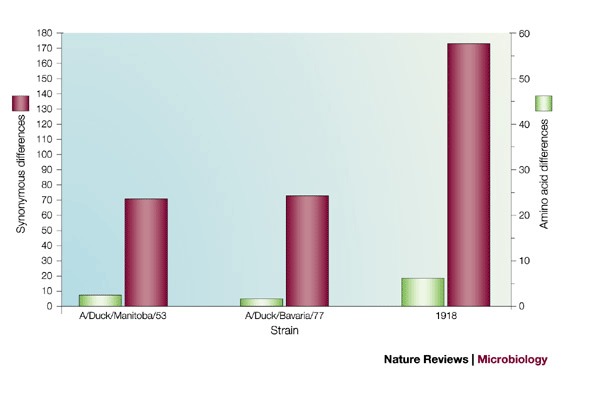
The data show that although the amino acid sequence of the 1918 NP is similar to the overall avian consensus at the amino acid level, it is highly divergent at the nucleotide level.
Haemagglutinin
Phylogenetic analyses performed on the complete HA gene or either of its two domains, HA1 and HA2, consistently place the 1918 sequences within, and near the root of, the mammalian clade17,18,24,26. The 1918 sequence is more similar to avian H1 sequences than is any other mammalian H1, but even when it is only compared with avian sequences, PARSIMONY ANALYSIS places the 1918 sequence in a separate clade18,26. The phylogenetic distance between the 1918 HA sequence and avian H1 HA sequences is much greater than that between either the 1957 H2 and 1968 H3 sequences and avian H2 and H3 sequences, respectively (Table 2; Fig. 2).
Table 2.
Comparison of pandemic HA with avian consensus sequences

Figure 2. Comparison of the haemagglutinin 1 (HA1) domain from the 1918, 1957 and 1968 pandemic viruses and various other avian strains with their avian consensus sequence.
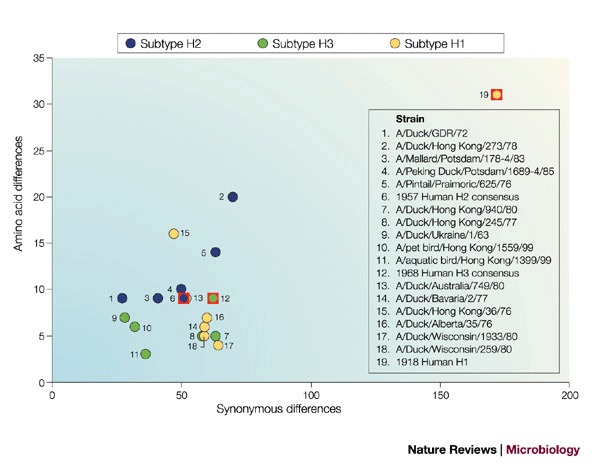
The data show that the HA of the 1918 pandemic virus differs from the H1 avian consensus to a much greater extent than the 1957 human H2 and 1968 human H3 differ from their respective avian consensus sequences.
Within the H2 subtype, North American avian strains differ from Eurasian avian strains by >116 nucleotides and 24 amino acids, on average. Within the Eurasian clade, some strains differ from the avian consensus sequence by as many as 70 nucleotides and 20 amino acids. So, although the HA gene of the 1957 pandemic strain is not a perfect match for any of the avian sequences in the database, with only 60 nucleotide differences from the avian consensus it is clear that it could have come directly from a Eurasian wild waterfowl. Similarly, within the H3 subtype, North American avian strains differ from Eurasian avian strains by >130 nucleotides and 16 amino acids, and one Eurasian strain can differ from another by as many as 60 nucleotides and seven amino acids. Again, the H3 gene of the 1968 pandemic virus is sufficiently similar to the known wild waterfowl sequences to have reassorted directly from this source. By contrast, in the H1 avian clade, strains from North American birds differ from Eurasian bird strains by an average of 92 nucleotides and 13 amino acids, and the differences are even smaller within the two avian subclades. The HA sequence from the 1918 pandemic H1 strain has more than twice as many differences from the avian consensus than any avian sequence. As a result, it seems unlikely that the 1918 pandemic virus acquired its HA directly from an avian source within the avian clades that have been characterized so far.
The possibility that the avian strains circulating around 1918 might have more closely resembled the pandemic strain was tested by sequencing a region of the HA gene from a Brant goose that was captured in 1917 and which was infected with an H1 subtype influenza virus. Phylogenetic analyses placed this 1917 avian sequence within the North American avian clade with a similar number of differences from the 1918 pandemic sequence as modern North American avian HAs24.
An effort to reconcile the phylogenetic distance of the 1918 sequence from the avian clade with the many avian characteristics of the 1918 virus led us to hypothesize that the 1918 HA gene had adapted in a mammalian host for some years before emerging in a pandemic virus. The earliest swine H1 isolate from the outbreak of avian H1N1 influenza in European pigs27,28, the A/Swine/Arnsberg/ 6554/79 (H1N1) isolate, has 40 nucleotide and 19 amino acid differences from the Eurasian avian HA1 consensus sequence — which is comparable to the number of differences between the H2 and H3 pandemic HAs and their avian consensus sequences. Twenty years later, the swine H1 lineage had accumulated 67 nucleotide and 36 amino acid differences (using A/Swine/Belgium/1/98 (H1N1) as an example). So, after 20 years in swine, the avian-derived H1 has acquired as many amino acid differences from the avian consensus as the 1918 strain, but it has fewer than half as many synonymous changes (67 compared with 174 in the 1918 strain). Again, preliminary results indicate that adaptation in an intermediate host such as the pig is unlikely to explain the large number of synonymous differences and the high S/N ratio between the 1918 sequence and avian sequences that have been characterized so far.
Therefore, it seems possible that, like the NP, the 1918 HA might have reassorted directly from a currently unknown host, the HA of which is similar to that in wild birds at the amino acid level, but quite different at the nucleotide level. Again, nucleotide differences at four-fold degenerate sites reinforce this interpretation of the data. When avian HA1 sequences are compared, they differ at four-fold degenerate sites by 20–30%. Comparisons of 1918 viral HA1 with avian sequences give values of 50–60%. As with the NP gene, comparisons with a region of an HA1 gene from a bird captured in 1917 show the same pattern — 20–30% differences with modern birds and a 58% difference compared with the 1918 pandemic virus.
Neuraminidase
Phylogenetic analyses of the N1 NA gene segment produce trees with two clades — a mammalian clade containing human and swine subclades, and an avian clade containing Eurasian and North American subclades. Analyses of the full-length NA coding sequences place the 1918 NA within and near the root of the human/swine clade, as do analyses of synonymous changes alone. However, when non-synonymous substitutions are analysed, the 1918 NA is placed within and near the root of the avian clade. These analyses suggest that the mammalian sequences are characterized by many shared synonymous changes, whereas the 1918 protein is more similar to avian sequences at the amino acid level19. Similar to the NP and HA1 genes, the 1918 viral NA gene has many changes at four-fold degenerate sites. Comparisons of North American and Eurasian avian NA genes show 20–40% differences, whereas comparisons of modern avian sequences with that of the 1918 virus show differences of 30–40%.
Although analyses of the amino acid sequence of the 1918 N1 NA place it in the avian clade, the 1918 sequence shares 13 amino acid changes with strains of the mammalian lineage that are not found in avian strains. Five of these sites are also changed in the avian N1 lineage that has been circulating in European swine for the past 25 years. These sites might be important in the adaptation of avian NA to mammals19,29.
As shown in Table 3, when the sequence of the 1918 N1 is compared with consensus N1 sequences from Eurasian and North American avian strains, it is found to have 136 and 140 synonymous differences from the Eurasian and North American avian lineages, respectively. The 1918 N1 NA protein differs by 30 and 32 amino acids, respectively, from the Eurasian and North American avian consensus amino acid sequences. This is similar to the number of differences that are found between the 1957 N2 NA sequence and consensus sequences derived from avian N2 sequences (Table 3).
Table 3.
Comparison of pandemic NA with avian consensus sequences

When the avian H1N1 strain emerged in European swine in 1979, the N1 of an early swine isolate (A/Swine/Lot/2979/82 (H1N1)) had 49 nucleotide and 9 amino acid differences from the Eurasian avian N1 consensus sequence. By 1997, the swine N1 lineage had accumulated 87 nucleotide and 33 amino acid differences (using A/Swine/Italy/1506-9/97 (H1N1) as an example) from the Eurasian avian consensus sequence. So, 20 years of evolution in pigs resulted in a similar number of amino acid changes as that seen between the 1918 N1 sequence and avian sequences, but only two-thirds as many synonymous changes (87 compared with 136). Again, these results indicate that adaptation in an intermediate host like the pig is unlikely to explain the accumulation of so many synonymous differences between the 1918 sequence and avian sequences obtained so far.
Matrix
The matrix (MA) genome segment encodes two proteins, M1 and M2, both of which are highly conserved. A phylogenetic analysis of M1 nucleotide sequences shows two clades — one comprising human and swine subclades, and a second that includes one subclade containing North American avian and equine sequences and another subclade containing Eurasian birds and sequences from the recent introduction of avian H1N1 into European swine. As with the other 1918 gene segments, the 1918 M1 sequence is positioned within and near the root of the human subclade. The M1 protein is so conserved that phylogenetic analyses yield little information21. If a consensus sequence is made from six Eurasian avian sequences, the individual sequences are found to differ from the consensus by 10–20 nucleotides but their amino acid sequences are virtually identical. The 1918 M1 sequence differs from this Eurasian avian consensus sequence by 51 nucleotides and one amino acid. A consensus sequence derived from four North American avian strains yields similar results; the strains each differ from the consensus by 5–20 nucleotides and the 1918 strain differs from the consensus by 47 nucleotides and, again, one amino acid. The North American and Eurasian avian consensus sequences differ from each other by 47 nucleotides. So, the 1918 M1 is as different from each avian subclade as they are from each other.
Phylogenetic analyses of the M2 gene produce trees in which the mammalian portion of the tree is very similar to the M1 trees. The avian/equine clade, however, has little horizontal depth and does not divide clearly into Eurasian, North American and equine subclades. Comparison of the avian sequences shows that only 18 nucleotide positions display any differences among the avian sequences, with most birds differing from each other by only a few nucleotides. The 1918 M2 sequence differs from the avian consensus by nine nucleotides. At the amino acid level, avian M2 is highly conserved, with only one amino acid difference distinguishing Eurasian and North American avian strains. Strikingly, the 1918 M2 differs from this avian consensus at five amino acid positions. Four of these differences are in the extracellular domain of M2, which is encoded by a reading frame that overlaps with that of the completely conserved M1 carboxyl terminus, indicating that it came from a source that differs markedly from the currently known avian sequences. It is possible that some of these four changes reflect human adaptation. If this is the case, it would indicate that the MA genome segment might have been retained in the 1918 pandemic virus from the previously circulating human influenza virus21.
Non-structural proteins
The non-structural (NS) genome segment is unusual among influenza gene segments in that two distinct alleles, A and B, are found in wild birds. Phylogenetic trees of both NS1 and NS2 position the B allele as an outgroup, and sequences of strains found in gulls and H7N7-subtype equine strains also form distinct clades. The topography of the other large clade is similar to that of M1, with a mammalian clade containing human and swine subclades, and an avian clade containing one subclade of Eurasian birds and another of North American birds and horses. The NS phylogenetic tree differs from the M1 tree in that a small group of old, highly pathogenic avian influenza viruses (previously called fowl plague viruses) is found at the root of the mammalian clade. The 1918 NS sequence, as with all the other 1918 gene segments, is found within and near the root of the mammalian clade20.
NS1 sequences are more diverse than M1 sequences, making it difficult to generate a consensus avian sequence. Within the Eurasian avian clade, sequences differ by as many as 25 nucleotides and six amino acids (comparing A/Chicken/Victoria/1/85 (H7N7) and A/Chicken/Hong Kong/220/97 (H5N1), for example). Within the North American avian clade, sequences differ by as many as 16 nucleotides and two amino acids (comparing A/Pintail/Alberta/119/79 (H4N6) and A/Gull/Delaware/475/86 (H2N2)). Given this diversity, as the 1918 NS1 sequence differs from the most closely related avian sequence, A/Mallard/New York/6750/78 (H2N2), by 24 nucleotides and four amino acids it seems possible that the 1918 NS segment was acquired directly from an avian strain. Eighty years later, the human NS1 lineage (using A/Shiga/25/97 (H3N2) as an example) had accumulated another 39 nucleotide and 33 amino acid differences from the 1918 strain, consistent with the hypothesis that the 1918 NS had not circulated in humans for a long time before the pandemic.
NS2 sequences are more conserved than NS1, with avian sequences varying from each other on average by 10 nucleotides and 1–3 amino acids. The 1918 NS2 sequences differ from most avian sequences by about the same number — for example, from the A/Mallard/New York/6750/78 (H2N2) strain by 11 nucleotides and one amino acid and from the A/Chicken/Victoria/1/85 (H7N7) strain by eight nucleotides and three amino acids. Since 1918, modern human NS2 sequences (again using A/Shiga/25/97 (H3N2) as an example) have accumulated another 26 nucleotide and six amino acid changes. So, the 1918 NS2 gene sequence is also consistent with a recent origin for the NS segment at the time of the pandemic.
Conclusion
The origin of the 1918 pandemic influenza strain remains mysterious. Several of the gene segments — HA, NA and NP — have so many synonymous changes from known sequences of wild-bird strains that it seems unlikely that they could have reassorted directly from an avian strain similar to those that have been sequenced so far. This is especially apparent when one examines differences at four-fold degenerate sites, which should be subject to little selective pressure. At the same time, the 1918 sequences have too few amino acid differences from wild-bird strains to have spent many years adapting in a human or swine intermediate host. One possible explanation is that these gene segments were acquired from a reservoir of influenza virus that has not yet been sampled. Phylogenetic analyses indicate that strains from this unidentified host could form a subclade within the avian clade, similar to the subclades that are formed by the Eurasian and North American waterfowl sequences.
The MA and NS genome segments are much more conserved and therefore sequence analysis alone might not be sufficient to determine whether they were novel in 1918 and, if so, whether they came from avian strains similar to the strains that have been sequenced. The M1 gene, in its pattern of synonymous and non-synonymous differences from avian sequences, resembles the genome segments listed above, consistent with an origin in a novel host. At the same time, the four amino acid changes in the extracellular domain of the M2 protein might indicate that this genome segment was retained from the previously circulating human strain. As the two genes are encoded by a single genome segment, and therefore must have a common origin, more information will be required to determine the origin of the 1918 MA segment.
The genetic composition of the 1957 and 1968 pandemic viruses is consistent with their both having arisen by the same mechanism — reassortment of a Eurasian wild-waterfowl strain with the previously circulating human strain6,7,8. Proof of the hypothesis that the virus responsible for the 1918 pandemic had an origin markedly different from the viruses responsible for the 1957 and 1968 pandemics would require both the discovery of a sample of the human influenza strain that was circulating before 1918 and the discovery of influenza strains in the wild that more closely resemble the 1918 sequences. At present, it is only possible to assert that the sequences of the 1918 pandemic virus do not seem to be consistent with an origin similar to that of the 1957 and 1968 pandemic viruses. Enhanced sampling and surveillance of wild-animal populations to find additional influenza reservoirs could be a prudent course of action30,31,32, given the consequences of missing the emergence of a pandemic strain like that of 1918.
Acknowledgements
The authors are grateful to T. Janczewski and R. Lourens for their valuable contributions to this project.
Glossary
- ANTIGENIC DRIFT
Minor changes in viral antigens due to gradual accumulation of mutations over time.
- ANTIGENIC SHIFT
Sudden change in viral antigens due to acquisition of one or more novel surface-protein-encoding genes by the process of reassortment.
- CLADE
Traits (for example, sequences) that form a distinct group on phylogenetic analysis.
- FOUR-FOLD DEGENERATE SUBSTITUTIONS
A subset of synonymous sites in a codon at which any of the four nucleotides can be present without resulting in an amino acid replacement.
- NON-SYNONYMOUS SUBSTITUTION
Nucleotide substitution in a codon that results in an amino acid replacement.
- PARSIMONY ANALYSIS
A type of phylogenetic analysis in which many possible trees are compared to find the tree requiring the fewest evolutionary changes.
- PHYLOGENETIC ANALYSES
Analysis of the evolutionary connections between traits (for example, sequences). There are several different methods that can be used to analyse phylogenetic connections.
- REASSORTMENT
Due to the segmented nature of the influenza A virus genome (eight individual RNA segments), influenza viruses can undergo a process of genetic reassortment to produce new variant strains of virus. In a cell infected with two different influenza A virus strains, gene segments from each can be packaged into viable hybrid virus strains.
- SUBTYPE
A designation for influenza A viruses describing the antigenic group to which the two dominant surface glycoproteins — haemagglutinin (HA) and neuraminidase (NA) — belong, written in the form HXNX, wherein one of the 15 possible HAs and one of the nine possible NAs is listed, for example, H1N1 or H3N2.
- SYNONYMOUS SUBSTITUTION
Nucleotide substitution in a codon that does not result in an amino acid replacement.
Biographies
Ann Reid has worked at the Armed Forces Institute of Pathology in Washington, DC since 1989 as a research biologist specializing in the adaptation of molecular biology techniques to the analysis of formalin-fixed, paraffin-embedded tissue samples. Since 1996, she has been engaged in the sequencing of the causative virus of the 1918 influenza pandemic from fixed and frozen tissue. Five of the eight gene segments have now been completed. Additional related projects have been the identification of pre-1918 avian influenza strains in alcohol-fixed specimens from the Smithsonian Institution and the examination for influenza RNA of brain samples from victims of Von Economo's encephalitis.
Jeffery K. Taubenberger serves as Chief of the Department of Molecular Pathology at the Armed Forces Institute of Pathology in Washington, DC, a position he has held since 1994. He received his M.D. in 1986 and Ph.D. in 1987 from the Medical College of Virginia, Virginia Commonwealth University, and did a residency in Pathology and postdoctoral research at the National Cancer Institute. His clinical activities involve diagnostic molecular genetics. He holds dual board certifications in Anatomic Pathology and in Molecular Genetic Pathology from the American Board of Pathology and the American Board of Medical Genetics. His clinical interests are chiefly in the development and implementation of molecular diagnostic assays for neoplasia and infectious diseases. His research interests include influenza virus biology; biology of other RNA viruses including marine mammal morbilliviruses and SARS; and gene expression during early lymphocyte differentiation.
Thomas G. Fanning has been a research biologist at the Armed Forces Institute of Pathology in Washington, DC since 1989. He received a Ph.D. in genetics from the University of Wisconsin in 1971. He did post-doctoral research at the University of Cologne, Cologne, Germany, and at the University of California, Davis, California. He spent five years as an investigator at the National Cancer Institute prior to coming to the Armed Forces Institute of Pathology. His research interests include repetitive DNA elements, especially LINE-1 elements in mammalian evolution and neoplasia; and the evolution of influenza A viruses.
Related links
FURTHER INFORMATION
Competing interests
The authors declare no competing financial interests.
References
- 1.Centers for Disease Control and Prevention. Influenza: the disease. [online], <http://www.cdc.gov/flu/about/disease.htm> (2003).
- 2.Webster RG, Bean WJ, Gorman OT, Chambers TM, Kawaoka Y. Evolution and ecology of influenza A viruses. Microbiol. Rev. 1992;56:152–179. doi: 10.1128/mr.56.1.152-179.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Webby RJ, Webster RG. Are we ready for pandemic influenza? Science. 2003;302:1519–1522. doi: 10.1126/science.1090350. [DOI] [PubMed] [Google Scholar]
- 4.Cox NJ, Subbarao K. Global epidemiology of influenza: past and present. Annu. Rev. Med. 2000;51:407–421. doi: 10.1146/annurev.med.51.1.407. [DOI] [PubMed] [Google Scholar]
- 5.Lamb R, Krug R. Fields Virology. 2001. pp. 1487–1531. [Google Scholar]
- 6.Schafer JR, et al. Origin of the pandemic 1957 H2 influenza A virus and the persistence of its possible progenitors in the avian reservoir. Virology. 1993;194:781–788. doi: 10.1006/viro.1993.1319. [DOI] [PubMed] [Google Scholar]
- 7.Kawaoka Y, Krauss S, Webster RG. Avian-to-human transmission of the PB1 gene of influenza A viruses in the 1957 and 1968 pandemics. J. Virol. 1989;63:4603–4608. doi: 10.1128/jvi.63.11.4603-4608.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bean W, et al. Evolution of the H3 influenza virus hemagglutinin from human and nonhuman hosts. J. Virol. 1992;66:1129–1138. doi: 10.1128/jvi.66.2.1129-1138.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Webster RG. Influenza virus: transmission between species and relevance to emergence of the next human pandemic. Arch. Virol. Suppl. 1997;13:105–113. doi: 10.1007/978-3-7091-6534-8_11. [DOI] [PubMed] [Google Scholar]
- 10.Johnson NP, Mueller J. Updating the accounts: global mortality of the 1918–1920 'Spanish' influenza pandemic. Bull. Hist. Med. 2002;76:105–115. doi: 10.1353/bhm.2002.0022. [DOI] [PubMed] [Google Scholar]
- 11.Patterson KD, Pyle GF. The geography and mortality of the 1918 influenza pandemic. Bull. Hist. Med. 1991;65:4–21. [PubMed] [Google Scholar]
- 12.Jordan E. Epidemic Influenza: A Survey. 1927. p. 355. [Google Scholar]
- 13.Linder FE, Grove RD. Vital Statistics Rates in the United States: 1900–1940. 1943. [Google Scholar]
- 14.Marks G, Beatty WK. Epidemics. 1976. [Google Scholar]
- 15.Rosenau MJ, Last JM. Maxcy–Rosenau Preventative Medicine and Public Health. 1980. [Google Scholar]
- 16.Taubenberger JK, Reid AH, Janczewski TA, Fanning TG. Integrating historical, clinical and molecular genetic data in order to explain the origin and virulence of the 1918 Spanish influenza virus. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2001;356:1829–1839. doi: 10.1098/rstb.2001.1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Taubenberger JK, Reid AH, Krafft AE, Bijwaard KE, Fanning TG. Initial genetic characterization of the 1918 'Spanish' influenza virus. Science. 1997;275:1793–1796. doi: 10.1126/science.275.5307.1793. [DOI] [PubMed] [Google Scholar]
- 18.Reid AH, Fanning TG, Hultin JV, Taubenberger JK. Origin and evolution of the 1918 'Spanish' influenza virus hemagglutinin gene. Proc. Natl Acad. Sci. USA. 1999;96:1651–1656. doi: 10.1073/pnas.96.4.1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reid AH, Fanning TG, Janczewski TA, Taubenberger JK. Characterization of the 1918 'Spanish' influenza virus neuraminidase gene. Proc. Natl Acad. Sci. USA. 2000;97:6785–6790. doi: 10.1073/pnas.100140097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Basler CF, et al. Sequence of the 1918 pandemic influenza virus nonstructural gene (NS) segment and characterization of recombinant viruses bearing the 1918 NS genes. Proc. Natl Acad. Sci. USA. 2001;98:2746–2751. doi: 10.1073/pnas.031575198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reid AH, Fanning TG, Janczewski TA, McCall S, Taubenberger JK. Characterization of the 1918 'Spanish' influenza virus matrix gene segment. J. Virol. 2002;76:10717–10723. doi: 10.1128/JVI.76.21.10717-10723.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reid, A. H., Fanning, T. G., Janczewski, T. A., Lourens, R. & Taubenberger, J. K. Novel origin of the 1918 pandemic influenza virus nucleoprotein gene segment. J. Virol. (in the press). [DOI] [PMC free article] [PubMed]
- 23.Reid AH, Taubenberger JK. The origin of the 1918 pandemic influenza virus: a continuing enigma. J. Gen. Virol. 2003;84:2285–2292. doi: 10.1099/vir.0.19302-0. [DOI] [PubMed] [Google Scholar]
- 24.Fanning TG, et al. 1917 avian influenza virus sequences suggest that the 1918 pandemic virus did not acquire its hemagglutinin directly from birds. J. Virol. 2002;76:7860–7862. doi: 10.1128/JVI.76.15.7860-7862.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reid AH, et al. Relationship of pre-1918 avian influenza HA and NP sequences to subsequent avian influenza strains. Avian Dis. 2003;47:921–925. doi: 10.1637/0005-2086-47.s3.921. [DOI] [PubMed] [Google Scholar]
- 26.Fanning TG, Taubenberger JK. Phylogenetically important regions of the influenza A H1 hemagglutinin protein. Virus Res. 1999;65:33–42. doi: 10.1016/S0168-1702(99)00098-2. [DOI] [PubMed] [Google Scholar]
- 27.Scholtissek C, Burger H, Bachmann PA, Hannoun C. Genetic relatedness of hemagglutinins of the H1 subtype of influenza A viruses isolated from swine and birds. Virology. 1983;129:521–523. doi: 10.1016/0042-6822(83)90194-0. [DOI] [PubMed] [Google Scholar]
- 28.Brown IH, et al. Antigenic and genetic analyses of H1N1 influenza A viruses from European pigs. J. Gen. Virol. 1997;78:553–562. doi: 10.1099/0022-1317-78-3-553. [DOI] [PubMed] [Google Scholar]
- 29.Fanning TG, Reid AH, Taubenberger JK. Influenza A virus neuraminidase: regions of the protein potentially involved in virus–host interactions. Virology. 2000;276:417–423. doi: 10.1006/viro.2000.0578. [DOI] [PubMed] [Google Scholar]
- 30.Layne SP, et al. A global lab against influenza. Science. 2001;293:1729. doi: 10.1126/science.293.5536.1729. [DOI] [PubMed] [Google Scholar]
- 31.Alexander DJ, Brown IH. Recent zoonoses caused by influenza A viruses. Rev. Sci. Tech. 2000;19:197–225. doi: 10.20506/rst.19.1.1220. [DOI] [PubMed] [Google Scholar]
- 32.Gensheimer KF, et al. Preparing for pandemic influenza: the need for enhanced surveillance. Vaccine. 2002;20(Suppl.):S63–S65. doi: 10.1016/S0264-410X(02)00135-4. [DOI] [PubMed] [Google Scholar]