

Key Points
The chemokine family is responsible for the specific, specialized trafficking of leukocytes.
Inflammation is characterized by an excessive, inappropriate cellular recruitment.
Chemokine receptors are necessary for HIV infection.
The large number of chemokines and receptors point to a redundant system. In vitro studies support this, but in vivo studies provide evidence for specificity in the chemokine system.
Clinical evidence shows that chemokines and receptors are expressed abundantly in samples and biopsies from patients suffering from inflammatory disorders.
Patients that lack CCR5 are resistant to HIV infection.
Studies using gene-deleted mice, neutralization of ligands or receptors and small molecule inhibitors have all contributed to our understanding of the chemokine system.
Chemokine receptors are valid therapeutic targets for the treatment of inflammation and HIV infection.
Abstract
Chemokines and their receptors are involved in the pathogenesis of diseases ranging from asthma to AIDS. Chemokine receptors are G-protein-coupled serpentine receptors that present attractive tractable targets for the pharmaceutical industry. It is only ten years since the first chemokine receptor was discovered, and the rapidly expanding number of antagonists holds promise for new medicines to combat diseases that are currently incurable. Here, I focus on the rationale for developing antagonists of chemokine receptors for inflammatory disorders and AIDS, and the accumulating evidence that favours this strategy despite the apparent redundancy in the chemokine system.
Main
A characteristic feature of all inflammatory disorders is the excessive recruitment of leukocytes to the site of inflammation. The correct, controlled trafficking of these cells is an essential feature of the immune response to infection, but loss of control results in inflammatory diseases. Leukocyte recruitment is a well-orchestrated process that involves several protein families, including pro-inflammatory cytokines, adhesion molecules, matrix metalloproteinases and the large cytokine subfamily of chemotactic cytokines, the chemokines1,2,3. Anti-inflammatory approaches that target the first three groups of proteins have been studied in vitro and tested in many animal models, and several have been used in the clinic during the past few decades4,5,6. Here, I discuss the rationale for targeting the chemokines and their receptors, and the current status of chemokine therapeutics.
Chemokines
Chemokines are a large family of small proteins that are distinguished from other cytokines by being the only members of the cytokine family that act on the superfamily of G-protein-coupled serpentine receptors. Although chemokines have a relatively low level of sequence identity, their three-dimensional structure shows a remarkable homology in that they all have the same monomeric fold7. This fold, consisting of three β strands, a carboxy (C) terminal helix and a flexible amino (N) terminal region, is conferred on these proteins by a four-cysteine motif that forms two characteristic disulphide bridges. However, as with all rules, there are exceptions in which two cysteines are lacking (as in XCL1 or Lymphotactin) or there is an extra pair (as in CCL21 or the secondary lymphoid-tissue chemokine 6Ckine/SLC). The flexible N-terminal region is believed to be important in receptor activation, because modification of this region has been shown by many laboratories to affect activity8,9,10.
Newcomers to the field of chemokine biology are often daunted by the thought of searching for specific inhibitors of the chemokine system, given that 50 chemokine ligands and 19 functional receptors have been described to date — and how many might be identified now that the sequencing of the human genome is almost complete? The numbers and in vitro studies tell us that the system apparently contains redundancy. There are few receptors that bind a single ligand, and several chemokines can bind to more than one receptor (Fig. 1). However, a closer inspection of the receptors and their ligands shows that they can be broadly categorized into two classes depending on whether they are constitutively produced or are inducible (Fig. 2). Constitutive ligand–receptor pairs usually have a role in basal leukocyte trafficking and development, and it is apparent from the numbering in their systematic names that they have been discovered more recently. This might have been due to their lower level of production (highly likely, because most chemokines were discovered using overexpression or SUBTRACTIVE CLONING techniques). The essential role of chemokines in the establishment of a functional immune system through their properties of basal trafficking and homing is apparent from the phenotypes of mice in which their genes have been deleted. The deletion of CXCR4 or its ligand CXCL12/SDF-1 (stromal-cell-derived factor 1) both result in an embryonic lethal phenotype11,12, whereas deletion of CCR7 or its ligand CCL21/SLC results in mice that, although they are viable, lack the correct architecture of secondary lymphoid tissue13,14. Similarly, in mice deficient in CXCR5 (the receptor for CXCL13/BCA-1, B cell-attracting chemokine-1), the organization of splenic primary follicles is severely impaired15.
Figure 1. Chemokine receptors and their ligands.
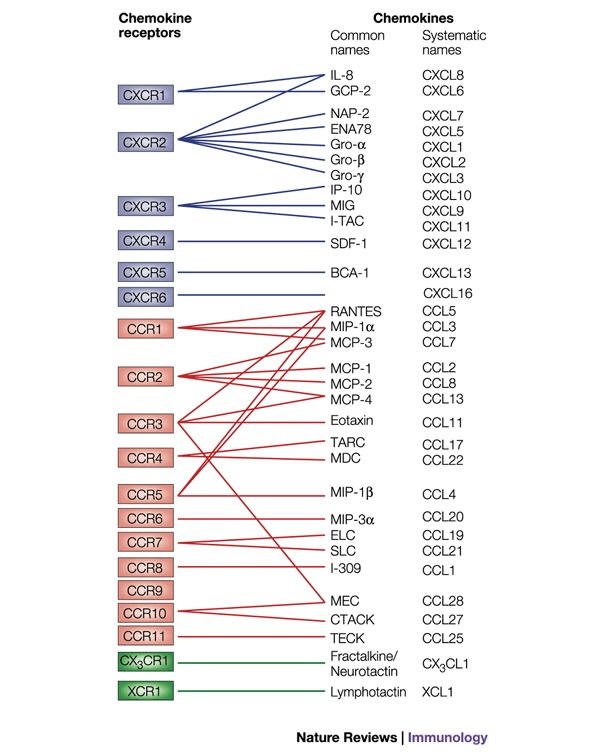
Chemokines are divided into subclasses on the basis of the spacing of the N-terminal cysteine residues. The receptors for the α (or CXC) subclass are shown in blue, the receptors for the β (or CC) subclass in red and the receptors for the minor subclasses (C and CX3C) in green. The pairing of chemokines to their receptors has been carried out mainly by receptor-binding assays. Chemokines were initially named according to their function or from the cell type that produced them, giving rise to names such as monocyte chemoattractant protein 1 (MCP-1), stromal derived factor 1 (SDF-1) and mucosae-associated epithelial chemokine (MEC). The simultaneous identification by different laboratories of a chemokine sequence often resulted in several names, such as MIP-3α, LARC and exodus-1. In order to eliminate the confusion, a systematic nomenclature has recently been adopted3. The abbreviations of the common names are as follows: BCA-1, B-cell-attracting chemokine 1; CTACK, cutaneous T-cell-attracting chemokine; ELC, Epstein–Barr-virus-induced gene 1 ligand chemokine; ENA78, epithelial-cell-derived neutrophil-activating peptide 78; GCP-2, granulocyte chemotactic protein 2; Gro, growth-regulated oncogene; IL-8, interleukin 8; IP-10, interferon-inducible protein 10; I-TAC, interferon-inducible T-cell α chemoattractant; MCP, monocyte chemoattractant protein; MDC, macrophage-derived chemokine; MEC, mucosae-associated epithelial chemokine; MIG, monokine induced by interferon γ; MIP, macrophage inflammatory protein; NAP-2, neutrophil-activating peptide 2; RANTES, regulated on activation, normal T-cell expressed and secreted; SDF-1, stromal-cell-derived factor 1; SLC, secondary lymphoid-tissue chemokine; TARC, thymus and activation-regulated chemokine; TECK, thymus-expressed chemokine.
Figure 2. Chemokine receptors can be expressed constitutively or inducibly.
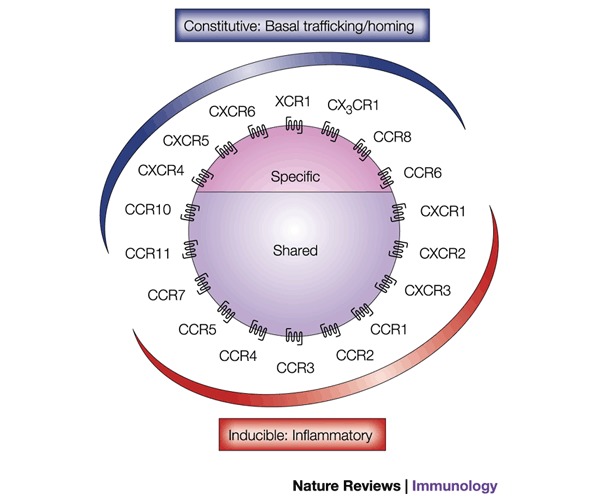
Chemokines can be classified as constitutive (developmentally regulated) or inducible (inflammatory). This division should not be regarded as absolute but rather as a rule with some exceptions. For example, CCR6 is constitutively expressed in immature dendritic cells and in T cells, but it is downregulated as dendritic cells mature. However, it is upregulated in skin lesions. CCR8 is constitutively expressed in the thymus but is upregulated during T-cell activation and is a marker of TH2 cells. Last, CCR7 is upregulated during the antigen recognition process, a requisite for immune surveillance, because it is upregulated as dendritic cells mature. The classification into 'specific' or 'shared' is defined by whether they bind a single or many ligands, respectively.
It is interesting that the inducible chemokine ligands were identified early in the chemokine biology field, probably for the obvious reason that they were produced at high levels in the tissues or tissue cultures in which they were identified. Macrophage inflammatory proteins 1α and β (MIP-1α/CCL3 and MIP-1β/CCL4) and monocyte chemotactic protein 1 (MCP-1/CCL2) were isolated from the culture supernatants of lipopolysaccharide-stimulated macrophages16,17. Similarly, the CXC chemokines interleukin-8 (IL-8/CXCL8)18 and interferon-inducible polypeptide 10 (IP-10/CXCL10) (originally named IP-10 owing to its molecular weight of ∼10 kDa)19 were identified from stimulated monocytes. It was observed from the sequences of these chemokines that they had a highly conserved motif of four cysteines and that the N-terminal cysteine pair was either separated by a single residue, giving rise to the CXC chemokines (α subclass), or adjacent, giving rise to the CC chemokines (β subclass). This observation allowed the rapid cloning of new family members including CCL5 (RANTES, regulated on activation, normal T-cell expressed and secreted) from activated T cells20. These six inducible chemokines are important factors in most inflammatory disorders, as is shown by the involvement of their receptors in the diseases summarized in Fig. 3.
Figure 3. Chemokine receptors and disease.
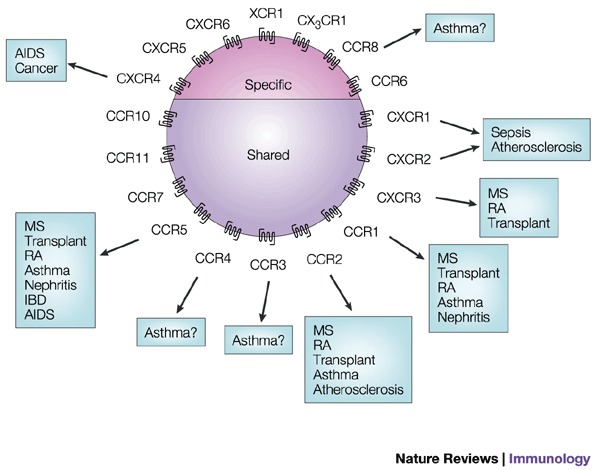
The receptors classified as constitutive in Fig. 2 do not have a great role in inflammatory disorders, with the exception of CCR8. CCR8 is expressed constitutively in the thymus but is upregulated on T-helper type-2 (TH2) cells. The only constitutive receptor involved in disease that has been described to date is the ubiquitous receptor CXCR4, which is one of the main HIV co-receptors and also plays a role in cancer metastasis. IBD, inflammatory bowel disease; MS, multiple sclerosis; RA, rhematoid arthritis.
Excessive recruitment of leukocytes, the hallmark of inflammation, occurs in both acute and chronic inflammatory disorders. For many years, it was thought that CXC chemokines were responsible for acute inflammation, such as acute respiratory distress syndrome (ARDS), in which there is a massive neutrophil infiltrate in the lungs, because CXC chemokines act on these cells. CC chemokines recruit the leukocytes that mediate chronic inflammation, such as eosinophils and T cells in asthma, and monocytes/macrophages and T cells in autoimmune diseases such as multiple sclerosis (MS). The neuronal damage that leads to paralysis in MS patients is thought to be initiated by an inflammatory phase21 in which T cells and monocytes are recruited across the blood–brain barrier (Fig. 4). However, with the discovery that CXCR3 is expressed on activated T cells, this separation of the chemokines into a CXC class that is responsible for acute inflammation and a CC class that is responsible for chronic inflammation, broke down. Current anti-inflammatory therapies mostly act on intracellular targets in leukocytes; that is, they act on the cells that have already been recruited. In fact, certain treatments that are at present successful at treating inflammation have an effect on the recruitment of leukocytes. A good example is the cytokine interferon-β, which, although it is not a cure, is the most effective treatment for MS at present. Although its precise mechanism of action is not fully delineated, one of its properties, at least in vitro, is that it can inhibit the migration of activated leukocytes across an endothelial layer22. A more efficient therapy might be the prevention of excessive recruitment of particular leukocyte populations by antagonizing chemokine receptors, which might act upstream of the current anti-inflammatory agents.
Figure 4. Chemokines are central to the pathogenesis of multiple sclerosis (MS).
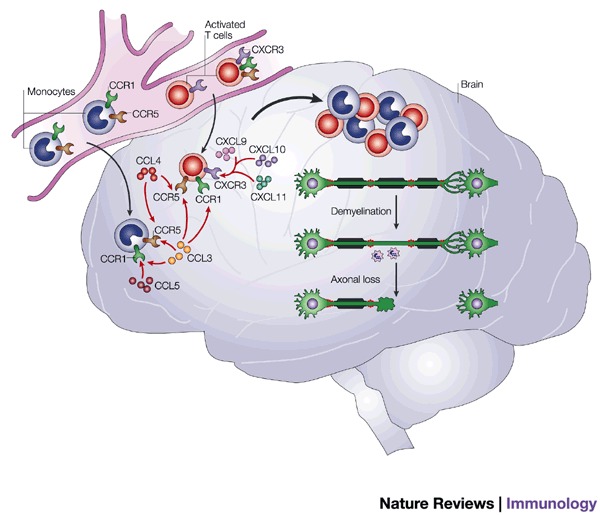
MS is an autoimmune disease associated with a T-helper type 1 phenotype. It is therefore believed that activated T cells have a crucial role. Chemokine receptors have been shown to be highly produced in brain samples from MS patients after autopsy41,42. One of these receptors, CXCR3, is produced by activated T cells, so it might be responsible for the recruitment of auto-aggressive T cells. This hypothesis is further confirmed by the high level of the CXCR3 ligand CXCL10 in these lesion samples. Two CC chemokine receptors, CCR1 and CCR5, have also been observed, and these receptors are both produced on T cells and monocytes/macrophages. The accumulation of these cells is directly correlated with lesions in which demyelination occurs, followed by axonal loss, which ultimately leads to paralysis.
Are chemokines valid targets for therapy?
As already pointed out, receptor–ligand pairing that occurs in vitro in binding and activation studies describes apparent redundancy in the system. Although nature is often described as being wasteful in such redundancy, it is commonly believed that the system has built in several levels of control that lead to specificity in vivo. The mechanisms of specificity are still a long way from being well understood and, although it is beyond the scope of this article to address these issues in detail, I describe a few examples for one of the most promiscuous chemokines, CCL5.
Binding of chemokines to their serpentine receptors activates a complex network of intracellular signalling pathways involving a variety of second messenger systems, such as calcium, cAMP and phospholipids, as well as a concerted interplay of kinase cascades downstream of small GTPases such as Ras and Rac. There is a known link between CCL5-induced leukocyte activation and chemotaxis to second messengers formed by the degradation of plasma-membrane-integral phospholipids by phospholiposes and their synthesis into differently phosphorylated phosphoinositides by phosphoinositide 3-kinase23. However, the roles of the Janus tyrosine kinase (JAK)24 and the signal transducer and activator of transcription (STAT) pathways downstream of CCL5 (Ref. 25) need to be further elucidated. Activation of serpentine receptors induces downmodulation (removal) of the receptors from the cell surface, which is followed either by receptor recycling via endosomes or degradation in the lysosomal compartment. Although CCL5 induces similar downregulation patterns for three of its receptors (CCR1, CCR3 and CCR5), only CCR5 recycles to attain the original cell surface receptor density after exposure to CCL5 (Ref. 26). CCR1 does not recycle to the cell surface after downregulation induced by CCL5, whereas CCR3 is partially, but not completely, restored27,28. Chemokines have a second important interaction aside from their receptors, which is with cell surface glycosaminoglycans (GAGs). This is important for the immobilization of chemokines and could have an important role in governing their local concentration. CCL5 shows a greater range of affinities than CCL2, CCL3 or CXCL8 for different members of the GAG family29. Taken together, these observations support the ideas that the chemokine system does not exhibit total redundancy and that specific points of intervention of the system are attractive targets for therapeutic intervention.
Although the main role of chemokines is leukocyte recruitment, they and their receptors have other biological actions. Their implication in diseases such as cancer should not be neglected30,31. It has been known for over a decade that CCL3 is an inhibitor of haematopoietic stem cell proliferation32, which widens the field of therapeutic interest beyond inflammation to cancer, because haematopoiesis is crucial during chemotherapy. Recently, the role of chemokines in mediating breast cancer metastasis (which requires tumour cell migration) has been elegantly shown33. The discovery five years ago that chemokine receptors were the second element, along with CD4, that is required for human immunodeficiency viruses (HIV) to fuse and enter the host cell34 has again even further broadened their uses as therapeutic targets. This finding has, in fact, had a huge effect on the search for small molecule inhibitors of chemokine receptors.
Target validation from clinical evidence
Given the potential redundancy of the chemokine network, it is crucial to establish the validity of targeting the system for therapeutic intervention. One approach is to examine the levels of expression of chemokines and their receptors in biological fluids, biopsies and tissue specimens from patients suffering from a particular disease. This has the obvious caveat of the feasibility of obtaining such samples. However, certain fluids such as plasma and broncho-alveolar lavage (BAL) fluid are readily accessible, whereas synovial fluid and cerebrospinal fluid require more elaborate surgical intervention, accompanied by more discomfort to the patient.
During sepsis and, in particular, ARDS, there is a massive infiltration of neutrophils to the lungs, and this is accompanied by elevated levels of the principal neutrophil chemoattractant, CXCL8 (Ref. 35). During visits to their physicians, asthmatics are frequently subjected to broncho-alveolar lavages, which contain extensive cellular infiltrates — in this case, of the leukocytes that mediate chronic infiltration, such as T cells and eosinophils (typical of this T-helper type-2 cell (TH2) disorder). It has been known for several decades that eosinophils are strongly associated with allergic lung inflammation and they are thought to be responsible for the tissue damage that occurs in ASTHMA by the release of toxic proteins such as eosinophil cationic protein and major basic protein. A specific chemokine was purified from the BAL of sensitized guinea pigs, CCL11 or eotaxin, which acts only on the main chemokine receptor on eosinophils, CCR3 (Ref. 36), and has been localized to the airway epithelia in humans37. CCL2, CCL3 and CCL5 have also been identified as eosinophilic chemoattractant factors in BALs from allergic ashmatics38. Immunohistochemistry has shown that CCL5 is also upregulated in inflamed tonsils, in certain tumours39 and during cell-mediated rejection of renal transplants40.
Two parallel immunohistochemistry studies on human samples of MS lesions correlated very nicely with the data obtained from an animal model of this disease, experimental autoimmune encephalomyelitis (EAE)41,42. These studies both reported elevated levels of two receptors associated with T-helper type-1 (TH1) cells, CCR5 and CXCR3, and also identified the concomitant high expression of their respective ligands, CCL3 and CXCL10.
Genetic evidence for target validation
Therapeutic strategies have often been inspired from genetic disorders in patients. So far, there is surprisingly little evidence for any involvement of mutations or polymorphisms of a particular chemokine or receptor in a particular disease. This could be attributed to the apparent redundancy in the system — if a certain gene is missing from birth, compensation could occur, as is discussed below for gene deletion experimental animals. However, a striking example of target validation has been reported for the prevention of HIV infection (Box 1). This correlation of CCR5 with HIV infection is perhaps the best target validation evidence so far and has certainly resulted in a huge effort by the pharmaceutical industry to find small molecule inhibitors. Protection from HIV infection and progression to AIDS, although less dramatic than in the homozygous Δ32-CCR5 individuals, is associated with other genetic defects. Polymorphisms in the coding region of CCR2 (Ref. 43) and in the CXCL12 gene (the ligand for the other main HIV receptor, CXCR4) are associated with less severe disease progression44.
However, very little correlation between inflammatory disorders and genetic abnormalities has been reported, with the exception of two mutations in the CCL5 promoter, and one in the CCL2 promoter. A point mutation at position −401 in the CCL5 promoter was found to be associated with atopic dermatitis, but not with asthma45, whereas a mutation at position −403 is associated with both ATOPY and asthma46. The −2518G polymorphism in the CCL2 regulatory region, which results in higher CCL2 levels, has been found to correlate with asthma severity and increased eosinophilia in asthmatic children47.
Target validation in animal models
The data from neutralizing chemokine ligands far outweigh those from neutralizing receptors to date, probably owing to the paucity of neutralizing murine receptor antibodies, although such antibodies are now gradually being produced and characterized42,48. However, anti-human-receptor antibodies can be used in severe combined immunodeficiency (SCID) mice, as was shown by the abrogation of breast cancer metastasis to the lung in mice injected with a human breast carcinoma cell line, thereby confirming the role of CXCR4 in metastasis33. The studies using chemokine-ligand neutralizing antibodies have, however, been more informative.
An excellent example is the delineation of the different roles of various chemokines in the mouse model for MS (EAE) by the use of specific monoclonal antibodies. CCL3 was shown to be essential for the onset of disease symptoms, whereas CCL2 was required for relapses to occur49. In this disease model, neutralization of other ligands such as CXCL10 affected disease progression50, whereas the neutralization of CCL5 had no effect. In the animal model for asthma (allergen-induced airway inflammation in the mouse), the roles of individual chemokines have been extensively studied. Blockade of several ligands that had been identified previously from upregulation of their mRNAs was shown to be effective at reducing symptoms. Surprisingly, an anti-CCL2 antibody had a greater effect than the neutralization of the specific CCR3 ligand CCL11 (Ref. 51). Other chemokines have also been implicated in the pathology of asthmatic inflammation in this model, in which a CCR4 ligand, CCL22, has been shown to be essential in the transit and retention of leukocytes in the lung tissue52.
Target validation using knockout mice
The most convincing evidence for target validation is still that from mice in which the gene coding for the target has been deleted and an appropriate phenotype is observed in a disease model. However, I believe that the absence of a phenotype in a certain disease model using a 'knockout' mouse should be interpreted with caution, owing to the possibility of compensation by other members of the family. Despite this, knockout mice continue to yield valuable information about the chemokine system. Gene deletion of at least five chemokines and eleven receptors has been described to date. Interestingly, as discussed above, the deletion of chemokines and receptors pivotal to basal trafficking and homing have striking phenotypes, whereas those that are inducible (and presumed to have a role in inflammatory disorders) are viable and normal, and phenotypic differences are only evident when the animals are exposed to specific inflammatory situations. Therefore, the deletion of CCL2, CCL3 and CCL11, and their receptors CCR1, CCR2, CCR3 and CCR5, as well as CCR4 and the CXC receptor CXCR3, have no overt phenotype. However, the division into constitutive and inducible chemokines is not absolute, and these knockout mice do show differences in leukocyte compartmentalization in the absence of inflammatory stimuli. Importantly, these mice all had interesting responses in disease models. The data available that are relevant to inducible chemokines and receptors that have a role in inflammation are summarized in Table 1.
Table 1.
Target validation data from knockout mice
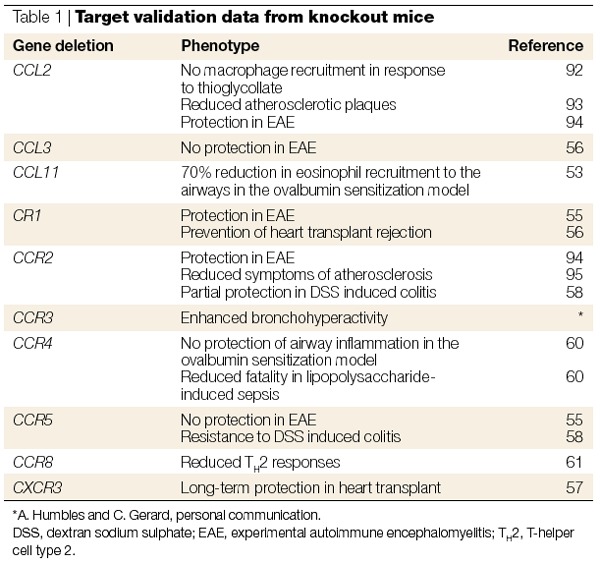
In general, results from the deletion of inflammatory chemokines and their receptors have confirmed the results obtained from antibody studies but have not identified an absolutely essential role for any single ligand. The CCL2 knockout mice have shown the greatest effect of the deletion of a single ligand, which is surprising in that its receptor CCR2 is also activated by four other ligands in mice, CCL7/MCP-3, CCL8/MCP-2, CCL13/MCP-4 and MCP-5 (which, to date, has no human homologue). These results are extremely important because they beautifully show that, in vivo, the redundancy of the chemokine system does not hold. However, certain knockout mice have produced results that contradict those obtained with neutralizing antibody studies, such as the role of CCL3 in disease induction in EAE. The fact that the CCL3 knockout mice developed EAE in the same manner as wild-type mice illustrates the possibility of compensation in knockout mice during development. Surprisingly, there is no report of the effects of the deletion of CCL5 in mice. This might be in accordance with the lack of effects seen in several anti-CCL5-antibody studies but contrasts with the elevated levels of this chemokine in many diseases and in the antagonism of CCL5 receptors with Met-CCL5, described below. When CCR3 was identified as the main receptor on eosinophils and CCL11 was identified as an eosinophil recruitment factor in guinea pigs36, this receptor–ligand pair was immediately believed to be an excellent target for asthma therapy. However, deletion of CCL11 did not completely abolish eosinophil recruitment in the ovalbumin sensitization model of airway inflammation53.
Knockout mice lacking certain chemokine receptors have clarified their relevance in certain diseases but also revealed certain enigmas. CCR1−/− mice are totally resistant to the onset of EAE, as shown by the neutralization of one of its ligands (CCL3; Ref. 54), whereas deletion of CCR5, encoding the other receptor for CCL3, had no phenotype55. The importance of CCR2 and its ligand CCL2 is corroborated by the knockout studies. Organ allograft rejection has clearly shown a role for both CCR1 (Ref. 56) and CXCR3, the receptor expressed on activated T cells and whose ligands are induced by the proinflammatory cytokine interferon γ (Ref. 57). Heart transplants in CXCR3−/− mice are unaffected after more than three months, and this study nicely confirmed the role of this receptor by using a neutralizing antibody to CXCR3 in wild-type mice in a parallel experiment57. A contradictory role for CCR5 has been identified by the study of CCR5−/− mice in experimental colitis, a model of inflammatory bowel disease (IBD), in which very good protection was observed58. However, individuals homogygous for the Δ32-CCR5 allele have not shown a reduced susceptibility to colitis59.
CCR3, CCR4 and CCR8 have been described as markers of TH2 activated lymphocytes and, as discussed above, CCR3 is the main receptor expressed on eosinophils. Therefore, deletion of these receptors would be expected to prevent symptoms in a mouse model of asthma (ovalbumin-induced airway inflammation). However, the complete lack of a phenotype in the CCR4−/− mice was totally unexpected60. Even more surprising was protection from lipopolysaccharide-induced lung inflammation in the CCR4−/− mice, because this is an innate immune response. This protection was presumed to be due to an effect on macrophages rather than on T cells, although further studies are warranted. The effect of the deletion of CCR3 is equally surprising — the CCR3−/− mice have enhanced bronchial hyper-reactivity in the airway-inflammation model (A. Humbles and C. Gerard, personal communication). These results indicate that increased production of a given chemokine and/or receptor might not reflect pathogenicity but in fact be protective or compensatory in a given disease situation. In contrast to the phenotypes observed with the CCR3 and CCR4 deletion mice, CCR8−/− mice showed significant decreases in TH2 responses, resulting in reduced eosinophilia in two models of airway inflammation — as would be predicted if this receptor is a selective TH2 chemokine receptor61.
Antagonism of chemokine receptors
Receptors in disease models can be neutralized using neutralizing antibodies, modified chemokines that act as receptor antagonists and small molecule receptor antagonists. The availability of neutralizing receptor antibodies and small molecule inhibitors that are active in vivo is limited at present, although we expect that this will change rapidly in the near future. There are only two reports so far of the effect of neutralizing a receptor in a disease model. In the first, anti-CXCR3-antibody treatment during heart allograft rejection was shown to produce the same effect as deletion of the CXCR3 gene57; the second involved the prevention of metastasis with an anti-CXCR4 antibody, as described above. The availability of small molecule chemokine receptor antagonists to address the question of redundancy has been eagerly awaited, and the first antagonist is now available. Studies using the Berlex CCR1 inhibitor BX471 confirmed the role of CCR1 in EAE (Ref. 62), in which the data described above were validated.
Many modified chemokines have been described as antagonists in vitro on the basis of several studies involving N-terminal truncation to produce receptor antagonists of the CXCL8 and CCL2 receptors8,9,63,64,65. In the case of the CXCL8 receptor antagonist (6-72)-CXCL8, testing in animal models has been hampered by the fact that mice do not have a CXCL8 homologue. In the case of the truncated CCR2 antagonist (9-68)-CCL2, the efficacy of blockade of CCR2 has been shown in the spontaneous model of arthritis in MRL-lpr mice, in which the administration of the antagonist in a therapeutic protocol significantly reduced the inflammatory symptoms66. We serendipitously produced another CC chemokine receptor antagonist: the initiating methionine was retained at the N-terminus when recombinant CCL5 was produced in Escherichia coli (Met-CCL5)10,67. Another N-terminally extended CCL5 analogue, AOP-CCL5 (a very potent CCR5 agonist), has proved to be very useful in establishing the principle that HIV infection can be prevented through the blockade of CCR5 in vitro68. This modified chemokine can alter CCR5-mediated cell trafficking26 and has properties that would be desirable for a small molecule that would prevent HIV infection of CCR5-using strains69.
The studies with Met-CCL5 in rodent models allow certain conclusions to be drawn. Many of the models studied with Met-CCL5 have been TH1-mediated inflammatory disease models such as collagen-induced arthritis (CIA)70, nephritis71 and colitis72. Typically, these are mediated by activated T cells and monocytes/macrophages — cell types that produce the CCL5 receptors CCR1 and CCR5. However, Met-CCL5 also had an anti-inflammatory effect in a model of TH2 inflammation (ovalbumin-induced airway inflammation)51. CCL5 normally binds to CCR3 but the addition of the Met residue abolishes binding to mouse CCR3, even though high-affinity binding is retained on mouse CCR1 and CCR5 (R. Buser and A. Proudfoot, unpublished observations). Therefore, it is surprising that it was efficient in preventing eosinophil recruitment in the airway-inflammation model; in fact, it was more effective than blocking the specific eosinophil chemokine eotaxin51. This implies that intervention for asthma could be targeted upstream of the CCL11-mediated eosinophil recruitment into the airways via CCR3.
The CIA study highlighted an important feature of the potential use of a receptor antagonist in the guise of a modified chemokine. The protein was administered three times per week intraperitoneally and it still showed a dose-related effect, indicating that the protein is not rapidly eliminated as was previously thought73. I believe that the reason for this is that the chemokine is immobilized on the surface of the endothelium and is released slowly. The interaction of chemokines with cell surface GAGs is well documented29,74,75 and is believed to have a role in increasing local concentrations76. Recently, this long-lasting effect has again been observed in a model in which bacteria induced the recruitment of dendritic cells to the trachea: significant inhibition was observed when Met-CCL5 was administered one and three days before the antigen challenge 77. Furthermore, this study highlights the possibility of specific targeting of the dendritic cells rather than the T cells, monocytes and granulocytes, which are often invoked as the main factors that cause inflammatory diseases. The roles of chemokines in dendritic cell biology and disease should not be neglected78.
Small molecule chemokine receptor antagonists
Chemokine biology is still a very new member of the field of immunology, with the first receptors identified only a decade ago. However, the fact that these receptors are of the serpentine class, which are targeted by approximately half of our current medications, makes them an attractive target for the search for small molecule inhibitors of the immune system. This is in direct contrast to other cytokines, for which the dearth of small molecule receptor inhibitors means that only protein therapeutics are available to date. The results of intense efforts in screening for small molecule chemokine receptor antagonists is beginning to pay off, with the number of patents for small molecule inhibitors growing rapidly.
It is interesting to examine the number of patents published for individual receptors. The large number of patents for small molecule inhibitors of CCR3 certainly reflects the interest of the pharmaceutical industry in a new treatment for asthma, perhaps one of the largest markets in the Western world. The identification of chemokine receptors as HIV co-receptors had a large effect on the screening programmes, which is satisfactorily reflected in the growing number of patented molecules that target CCR5. However, the number of small molecules being patented for the other important HIV co-receptor (CXCR4) is disappointing, particularly in view of its implication in cancer metastasis. It would therefore be interesting to know whether certain receptors are more amenable to the discovery of small molecule inhibitors.
It is now clear that most receptors bind several chemokines, which raises the question of which ligand should be targeted for screening. Ideally, one would like to inhibit the action of all the ligands, which indicates that the inhibitor should be non-competitive. For example, the three ligands of CXCR3 are allotopic, implying that they bind to different receptor activation states and so will not necessarily have the same binding site79. One of the first potent small molecules to be described in the literature was a CCR5 inhibitor, TAK779, which could block the three CCR5 ligands CCL3, CCL4 and CCL5 with nanomolar potency80. Subsequent mapping of its binding site using a panel of CCR5 mutants showed that it was in fact a non-competitive inhibitor because it bound to a cavity near the surface formed between transmembrane helices 1, 2, 3 and 7, and not to the extracellular domains that form the ligand-binding sites81.
The other question that has often been posed is whether an inhibitor should be specific or whether the inhibition of several chemokine receptors would be advantageous. The Berlex CCR1 compound BX471 is reported to be extremely selective82, whereas the CCR5 inhibitor TAK779 was also reasonably potent on CCR2, which is the receptor with the highest homology to CCR5 (Ref. 80). Dual inhibitors have also been reported, such as a molecule that inhibits both CCR1 and CCR3 (Ref. 83). Because both these receptors are expressed on eosinophils, this selectivity could be advantageous for the development of an anti-asthma therapy. A molecule has also been identified that is active in the inhibition of binding to CCR1, CCR3, CCR5 and CXCR4, but does not affect CCR2 or CXCR2 (Ref. 84). Although this molecule is not particularly 'drug-like', because it is large (1,209 Da), it has been shown to have anti-HIV activity in vivo in a SCID mouse model. However, more importantly, it shows that there are molecules that can bind across receptor classes, which is particularly important for anti-HIV therapy in order to inhibit both R5 and X4 strains. In fact, viruses have taught us that this is possible, because the Herpes viral chemokine vMIP-II can also inhibit HIV strains that use CCR3, CCR5 and CXCR4 as co-receptors85.
One of the main hurdles encountered in the development of small molecule receptor antagonists is the loss of species crossreactivity as the affinity for the human receptor increases, which creates problems for testing in animal models, as was found with BX471 (Ref. 86). However, this compound does retain sufficient activity, despite the fact that it has a 2-log drop in affinity for the rat CCR1 receptor, to allow proof of efficacy where it could inhibit EAE symptoms in the rat in a dose-related manner. This result is particularly gratifying because it is a definitive proof of the target validation results obtained with the anti-CCL3-antibody results in EAE, as well as the resistance of CCR1−/− mice to the symptoms of EAE (Ref. 62). Moreover, this molecule is orally bioavailable, which is an enormous advantage for the treatment of a chronic disease such as MS. Another series of inhibitors that encounter species problems are the CXCL8 receptors CXCR1 and CXCR2. Although there is a homologue of CXCR2 in mice, there is no homologue of CXCL8. However, Smithkline Beecham (now GlaxoSmithKline) showed that their small molecule CXCR2 inhibitor could inhibit CXCL8-induced rabbit neutrophil recruitment both in vitro and in vivo. The same company has recently shown activity in a rodent, in which they showed inhibition of neutrophil accumulation in the lungs in newborn rats exposed to hyperoxia87.
Therefore, although the number of small molecule receptor antagonists is rapidly growing, we still lack extensive data in animal disease models. Such data might have to be obtained from primate models, which is a significant expense, but well-conducted toxicity studies in animal species should allow rapid testing in humans. It has been publicly disclosed that certain small molecule programmes for receptors including CXCR2, CXCR4, CCR1 and CCR5 have entered Phase I trials, but no data have yet been published. Certainly, many diseases, including incurable diseases, will profit from rapid clinical trials.
Conclusion
The strategy of selectively blocking leukocyte recruitment to the site of inflammation has been validated by several approaches since the discovery of the chemokine family. The availability of mice with deleted chemokine receptor genes has also answered some, but not all, of the questions, as have studies with neutralizing antibodies and modified chemokine receptor antagonists. The search for new therapeutic targets using any approach depends on the adequacy of animal models to mirror human disease, and this holds equally for antagonists of the chemokine system. However, it is promising that many studies targeting the chemokine receptors have shown that inflammation can be reduced. Although it is still possible that biological therapies could use neutralizing antibodies or modified chemokines as receptor antagonists, the ideal therapeutic is an orally available small molecule. We now await the final proof in humans, because active orally available small molecule inhibitors of the chemokine receptors are now being developed.
One interesting conclusion that is perhaps provocative for a system that shows signs of redundancy can be drawn from knockout mice in organ allograft transplant studies. The deletion of several ligands and receptors has produced remarkable phenotypes in organ transplant models. The in vitro redundancy described from ligand–receptor binding and activation studies certainly seems to be real but, in vivo, the system acts through a coordinated and perhaps sequential chain of events, with temporal and spatial control mechanisms coming into play. Interfering with an essential link in the chain (CCR1 or CXCR3 in the case of organ allograft transplant) might therefore result in a total inhibition of the inflammatory process. We believe that these studies, which showed protection against inflammation by eliminating two unrelated chemokine receptors, provide the strongest validation for targeting the 'redundant' chemokine receptors as therapeutic targets for inflammatory diseases. Validation for AIDS is evident from the Δ32-CCR5 population. A question, however, remains unanswered: what will the long-term effects of such a strategy be? Although the field is still too young to answer this question, and animal model studies have yet to be designed specifically to address this issue, the Δ32-CCR5 individuals again help us by showing that the total inhibition of certain receptors is not harmful to the health of humans. We now look forward to an era of many more studies with small molecule inhibitors that are active in vivo to address and answer these questions.
Box 1 | A human 'knockout' confers resistance to HIV infection.
During the past 5 years, it has become apparent that ∼1% of the Caucasian human population carry a 32-bp deletion in the gene coding for CCR5, resulting in a truncated gene that produces no functional receptor at the cell surface. Individuals homozygous for this Δ32-CCR5 allele are surprisingly resistant to HIV infection, and this mutation was actually discovered in a group of individuals known as the 'exposed uninfected'88. Heterozygous individuals fell into another clinical category, the 'long term nonprogressors'.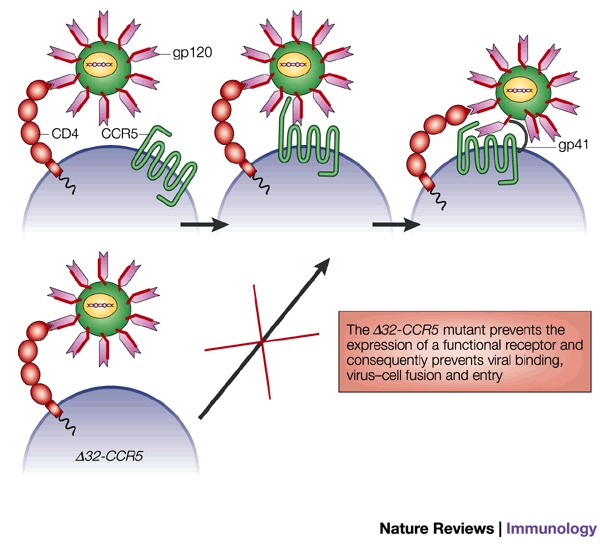
HIV infection requires two interactions with proteins on the host cell surface. First, the viral protein gp120 undergoes a high-affinity interaction with CD4. This induces a conformational change that allows interaction with a chemokine receptor; this receptor is CCR5 during transmission, but CXCR4-using strains emerge as disease progresses to AIDS. The interaction with the chemokine receptor induces a second conformational change in the gp120 protein, exposing the fusion peptide gp41, which fuses with the host cell membrane. The absence of CCR5 in individuals homozygous for the Δ32-CCR5 allele therefore prevents the transmission of HIV.
Evidence from Δ32-CCR5 homozygous individuals has shown a role for CCR5 in allograft acceptance89, but has been fairly disappointing for other inflammatory diseases. Of the 1,227 renal transplant patients screened, 1.7% were homozygous for the Δ32-CCR5 allele and only one of these patients lost transplant function. Homozygous Δ32-CCR5 individuals have been described to develop both rheumatoid arthritis90 and multiple sclerosis (MS). However, these MS patients have not suffered more than a primary attack; in other words, they have not succumbed to the normal relapsing syndrome91. Certainly, the clinical evidence from MS lesions in which CCR5 is seen to be highly expressed41,42 implies that this receptor is involved in pathology.
Acknowledgements
I thank C. Power and T. Wells, and all of our co-workers for the enjoyable and fruitful collaboration over the past few years, without whom my contribution to the chemokine field would not have been possible.
Glossary
- SUBTRACTIVE CLONING
The selective removal of RNA species common to samples from both normal and diseased tissues to identify genes specifically expressed in either state.
- ASTHMA
A chronic inflammatory bronchial disease characterized by increased bronchial hyper-responsiveness to physical, chemical, pharmacological and/or immunological stimuli. Asthma is often associated with atopy.
- ATOPY
A disorder characterized by a sustained, inappropriate IgE response to common environmental antigens (allergens) encountered at the mucosal surface. Interaction of allergens with cells sensitized by binding of surface Fc receptors to IgE is assumed to have a role in the pathogenesis of atopic asthma.
Biography
Amanda E. I. Proudfoot did her undergraduate studies in Biochemistry at the University of Witwatersrand, Johannesburg, followed by a PhD in Biochemistry at the University of Geneva. She then worked as a research assistant in the Medical Biochemistry Department at the University Medical Centre. In 1988, she joined the Glaxo Institute of Molecular Biology and, in 1998, the Serono Pharmaceutical Research Institute. Her principal interests are the structure–function relationships of proteins involved in the immune system. Her research has focused on the chemokine family for the past eight years, with the aim of identifying inhibitors of inflammation and AIDS.
Related links
DATABASES
FURTHER INFORMATION
References
- 1.Rollins BJ. Chemokines. Blood. 1997;90:909–928. [PubMed] [Google Scholar]
- 2.Luster AD. Chemokines — chemotactic cytokines that mediate inflammation. New Engl. J. Med. 1998;338:436–445. doi: 10.1056/NEJM199802123380706. [DOI] [PubMed] [Google Scholar]
- 3.Zlotnik A, Yoshie O. Chemokines: a new classification system and their role in immunity. Immunity. 2000;12:121–127. doi: 10.1016/s1074-7613(00)80165-x. [DOI] [PubMed] [Google Scholar]
- 4.Spencer-Green G. Etanercept (Enbrel): update on therapeutic use. Ann. Rheum. Dis. 2000;59(Suppl. 1):i46–i49. doi: 10.1136/ard.59.suppl_1.i46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lin KC, Castro AC. Very late antigen 4 (VLA4) antagonists as anti-inflammatory agents. Curr. Opin. Chem. Biol. 1998;2:453–457. doi: 10.1016/s1367-5931(98)80120-8. [DOI] [PubMed] [Google Scholar]
- 6.Steward WP, Thomas AL. Marimastat: the clinical development of a matrix metalloproteinase inhibitor. Expert Opin. Invest. Drugs. 2000;9:2913–2922. doi: 10.1517/13543784.9.12.2913. [DOI] [PubMed] [Google Scholar]
- 7.Schwarz MK, Wells TN. Interfering with chemokine networks--the hope for new therapeutics . Curr Opin Chem Biol. 1999;3:407–417. doi: 10.1016/S1367-5931(99)80061-1. [DOI] [PubMed] [Google Scholar]
- 8.Clark-Lewis I, Schumacher C, Baggiolini M, Moser B. Structure–activity relationships of interleukin-8 determined using chemically synthesized analogs. Critical role of NH2-terminal residues and evidence for uncoupling of neutrophil chemotaxis, exocytosis, and receptor binding activities. J. Biol. Chem. 1991;266:23128–23134. [PubMed] [Google Scholar]
- 9.Hebert CA, Vitangcol RV, Baker JB. Scanning mutagenesis of interleukin-8 identifies a cluster of residues required for receptor binding. J. Biol. Chem. 1991;266:18989–18994. [PubMed] [Google Scholar]
- 10.Proudfoot AEI, et al. Extension of recombinant human RANTES by the retention of the initiating methionine produces a potent antagonist. J. Biol. Chem. 1996;271:2599–2603. doi: 10.1074/jbc.271.5.2599. [DOI] [PubMed] [Google Scholar]
- 11.Zou YR, Kottmann AH, Kuroda M, Taniuchi I, Littman DR. Function of the chemokine receptor CXCR4 in haematopoiesis and in cerebellar development. Nature. 1998;393:595–599. doi: 10.1038/31269. [DOI] [PubMed] [Google Scholar]
- 12.Nagasawa T, et al. Defects of B-cell lymphopoiesis and bone-marrow myelopoiesis in mice lacking the CXC chemokine PBSF/SDF-1. Nature. 1996;382:635–638. doi: 10.1038/382635a0. [DOI] [PubMed] [Google Scholar]
- 13.Forster R, et al. CCR7 coordinates the primary immune response by establishing functional microenvironments in secondary lymphoid organs. Cell. 1999;99:23–33. doi: 10.1016/s0092-8674(00)80059-8. [DOI] [PubMed] [Google Scholar]
- 14.Gunn MD, et al. Mice lacking expression of secondary lymphoid organ chemokine have defects in lymphocyte homing and dendritic cell localization. J. Exp. Med. 1999;189:451–460. doi: 10.1084/jem.189.3.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Voigt I, et al. CXCR5-deficient mice develop functional germinal centers in the splenic T cell zone. Eur. J. Immunol. 2000;30:560–567. doi: 10.1002/1521-4141(200002)30:2<560::AID-IMMU560>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 16.Wolpe SD, et al. Macrophages secrete a novel heparin-binding protein with inflammatory and neutrophil chemokinetic properties. J. Exp. Med. 1988;167:570–581. doi: 10.1084/jem.167.2.570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yoshimura T, Robinson EA, Tanaka S, Appella E, Leonard EJ. Purification and amino acid analysis of two human monocyte chemoattractants produced by phytohemagglutinin-stimulated human blood mononuclear leukocytes. J. Immunol. 1989;142:1956–1962. [PubMed] [Google Scholar]
- 18.Walz A, Peveri P, Aschauer H, Baggiolini M. Purification and amino acid sequencing of NAF, a novel neutrophil-activating factor produced by monocytes. Biochem. Biophys. Res. Commun. 1987;149:755–761. doi: 10.1016/0006-291x(87)90432-3. [DOI] [PubMed] [Google Scholar]
- 19.Luster AD, Weinshank RL, Feinman R, Ravetch JV. Molecular and biochemical characterization of a novel γ-interferon-inducible protein. J. Biol. Chem. 1988;263:12036–12043. [PubMed] [Google Scholar]
- 20.Schall TJ, et al. A human T cell-specific molecule is a member of a new gene family. J. Immunol. 1988;141:1018–1025. [PubMed] [Google Scholar]
- 21.Bitsch A, Schuchardt J, Bunkowski S, Kuhlmann T, Bruck W. Acute axonal injury in multiple sclerosis. Correlation with demyelination and inflammation. Brain. 2000;123:1174–1183. doi: 10.1093/brain/123.6.1174. [DOI] [PubMed] [Google Scholar]
- 22.Smeets TJ, et al. The effects of interferon-β treatment of synovial inflammation and expression of metalloproteinases in patients with rheumatoid arthritis. Arthritis Rheum. 2000;43:270–274. doi: 10.1002/1529-0131(200002)43:2<270::AID-ANR5>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 23.Wymann MP, Sozzani S, Altruda F, Mantovani A, Hirsch E. Lipids on the move: phosphoinositide 3-kinases in leukocyte function. Immunol. Today. 2000;21:260–264. doi: 10.1016/s0167-5699(00)01649-2. [DOI] [PubMed] [Google Scholar]
- 24.Ihle JN, et al. Signaling by the cytokine receptor superfamily. Ann. NY Acad. Sci. 1998;865:1–9. doi: 10.1111/j.1749-6632.1998.tb11157.x. [DOI] [PubMed] [Google Scholar]
- 25.Wong M, Fish EN. RANTES and MIP-1-α activate STATS in T cells. J. Biol. Chem. 1998;273:309–314. doi: 10.1074/jbc.273.1.309. [DOI] [PubMed] [Google Scholar]
- 26.Mack M, et al. Aminooxypentane-RANTES induces CCR5 internalization but inhibits recycling: a novel inhibitory mechanism of HIV infectivity. J. Exp. Med. 1998;187:1215–1224. doi: 10.1084/jem.187.8.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Elsner J, et al. Differential activation of CC chemokine receptors by AOP-RANTES. J. Biol. Chem. 2000;275:7787–7794. doi: 10.1074/jbc.275.11.7787. [DOI] [PubMed] [Google Scholar]
- 28.Zimmermann N, Conkright JJ, Rothenberg ME. CC chemokine receptor-3 undergoes prolonged ligand-induced internalization. J. Biol. Chem. 1999;274:12611–12618. doi: 10.1074/jbc.274.18.12611. [DOI] [PubMed] [Google Scholar]
- 29.Kuschert GS, et al. Glycosaminoglycans interact selectively with chemokines and modulate receptor binding and cellular responses. Biochemistry. 1999;38:12959–12968. doi: 10.1021/bi990711d. [DOI] [PubMed] [Google Scholar]
- 30.Chemokines and Cancer (Humana Press, Totowa, New Jersey, 1999).
- 31.Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet. 2001;357:539–545. doi: 10.1016/S0140-6736(00)04046-0. [DOI] [PubMed] [Google Scholar]
- 32.Graham GJ, et al. Identification and characterization of an inhibitor of haemopoietic stem cell proliferation. Nature. 1990;344:442–444. doi: 10.1038/344442a0. [DOI] [PubMed] [Google Scholar]
- 33.Muller A, et al. Involvement of chemokine receptors in breast cancer metastasis. Nature. 2001;410:50–56. doi: 10.1038/35065016. [DOI] [PubMed] [Google Scholar]
- 34.Berger EA. HIV entry and tropism: the chemokine receptor connection. AIDS. 1997;11:S3–S16. [PubMed] [Google Scholar]
- 35.Miller EJ, et al. Elevated levels of NAP-1/interleukin-8 are present in the airspaces of patients with the adult respiratory distress syndrome and are associated with increased mortality. Am. Rev. Respir. Dis. 1992;146:427–432. doi: 10.1164/ajrccm/146.2.427. [DOI] [PubMed] [Google Scholar]
- 36.Jose PJ, et al. Eotaxin: a potent eosinophil chemoattractant cytokine detected in a guinea pig model of allergic airways inflammation. J. Exp. Med. 1994;179:881–887. doi: 10.1084/jem.179.3.881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lilly CM, et al. Expression of eotaxin by human lung epithelial cells: induction by cytokines and inhibition by glucocorticoids. J. Clin. Invest. 1997;99:1767–1773. doi: 10.1172/JCI119341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Holgate ST, et al. Release of RANTES, MIP-1α, and MCP-1 into asthmatic airways following endobronchial allergen challenge. Am. J. Respir. Crit. Care Med. 1997;156:1377–1383. doi: 10.1164/ajrccm.156.5.9610064. [DOI] [PubMed] [Google Scholar]
- 39.von Luettichau I, et al. RANTES chemokine expression in diseased and normal human tissues. Cytokine. 1996;8:89–98. doi: 10.1006/cyto.1996.0012. [DOI] [PubMed] [Google Scholar]
- 40.Pattison J, et al. RANTES chemokine expression in cell-mediated transplant rejection of the kidney. Lancet. 1994;343:209–211. doi: 10.1016/s0140-6736(94)90992-x. [DOI] [PubMed] [Google Scholar]
- 41.Sorensen TL, et al. Expression of specific chemokines and chemokine receptors in the central nervous system of multiple sclerosis patients. J. Clin. Invest. 1999;103:807–815. doi: 10.1172/JCI5150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Balashov KE, Rottman JB, Weiner HL, Hancock WW. CCR5+ and CXCR3+ T cells are increased in multiple sclerosis and their ligands MIP-1α and IP-10 are expressed in demyelinating brain lesions. Proc. Natl Acad. Sci. USA. 1999;96:6873–6878. doi: 10.1073/pnas.96.12.6873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kostrikis LG, et al. A chemokine receptor CCR2 allele delays HIV-1 disease progression and is associated with a CCR5 promoter mutation. Nature Med. 1998;4:350–353. doi: 10.1038/nm0398-350. [DOI] [PubMed] [Google Scholar]
- 44.Winkler C, et al. Genetic restriction of AIDS pathogenesis by an SDF-1 chemokine gene variant. ALIVE Study, Hemophilia Growth and Development Study (HGDS), Multicenter AIDS Cohort Study (MACS), Multicenter Hemophilia Cohort Study (MHCS), San Francisco City Cohort (SFCC) Science. 1998;279:389–393. doi: 10.1126/science.279.5349.389. [DOI] [PubMed] [Google Scholar]
- 45.Nickel RG, et al. Atopic dermatitis is associated with a functional mutation in the promoter of the C-C chemokine RANTES. J. Immunol. 2000;164:1612–1616. doi: 10.4049/jimmunol.164.3.1612. [DOI] [PubMed] [Google Scholar]
- 46.Fryer AA, et al. The −403 G→A promoter polymorphism in the RANTES gene is associated with atopy and asthma. Genes Immun. 2000;1:509–514. doi: 10.1038/sj.gene.6363717. [DOI] [PubMed] [Google Scholar]
- 47.Szalai C, et al. Polymorphism in the gene regulatory region of MCP-1 is associated with asthma susceptibility and severity. J. Allergy Clin. Immunol. 2001;108:375–381. doi: 10.1067/mai.2001.117930. [DOI] [PubMed] [Google Scholar]
- 48.Mack M, et al. Expression and characterization of the chemokine receptors CCR2 and CCR5 in mice. J. Immunol. 2001;166:4697–4704. doi: 10.4049/jimmunol.166.7.4697. [DOI] [PubMed] [Google Scholar]
- 49.Karpus WJ, Kennedy KJ. MIP-1α and MCP-1 differentially regulate acute and relapsing autoimmune encephalomyelitis as well as TH1/TH2 lymphocyte differentiation. J. Leukocyte Biol. 1997;62:681–687. [PubMed] [Google Scholar]
- 50.Fife BT, et al. CXCL10 (IFN-γ-inducible protein-10) control of encephalitogenic CD4+ T cell accumulation in the central nervous system during experimental autoimmune encephalomyelitis. J. Immunol. 2001;166:7617–7624. doi: 10.4049/jimmunol.166.12.7617. [DOI] [PubMed] [Google Scholar]
- 51.Gonzalo JA, et al. The coordinated action of CC chemokines in the lung orchestrates allergic inflammation and airway hyperresponsiveness. J. Exp. Med. 1998;188:157–167. doi: 10.1084/jem.188.1.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gonzalo JA, et al. Mouse monocyte-derived chemokine is involved in airway hyperreactivity and lung inflammation. J. Immunol. 1999;163:403–411. [PubMed] [Google Scholar]
- 53.Rothenberg ME, MacLean JA, Pearlman E, Luster AD, Leder P. Targeted disruption of the chemokine eotaxin partially reduces antigen-induced tissue eosinophilia. J. Exp. Med. 1997;185:785–790. doi: 10.1084/jem.185.4.785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kennedy KJ, Strieter RM, Kunkel SL, Lukacs NW, Karpus WJ. Acute and relapsing experimental autoimmune encephalomyelitis are regulated by differential expression of the CC chemokines macrophage inflammatory protein-1α and monocyte chemotactic protein-1. J. Neuroimmunol. 1998;92:98–108. doi: 10.1016/s0165-5728(98)00187-8. [DOI] [PubMed] [Google Scholar]
- 55.Tran EH, Kuziel WA, Owens T. Induction of experimental autoimmune encephalomyelitis in C57BL/6 mice deficient in either the chemokine macrophage inflammatory protein-1α or its CCR5 receptor. Eur. J. Immunol. 2000;30:1410–1415. doi: 10.1002/(SICI)1521-4141(200005)30:5<1410::AID-IMMU1410>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 56.Gao W, et al. Targeting of the chemokine receptor CCR1 suppresses development of acute and chronic cardiac allograft rejection. J. Clin. Invest. 2000;105:35–44. doi: 10.1172/JCI8126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hancock WW, et al. Requirement of the chemokine receptor CXCR3 for acute allograft rejection. J. Exp. Med. 2000;192:1515–1520. doi: 10.1084/jem.192.10.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Andres PG, et al. Mice with a selective deletion of the CC chemokine receptors 5 or 2 are protected from dextran sodium sulfate-mediated colitis: lack of CC chemokine receptor 5 expression results in a NK1.1+ lymphocyte-associated TH2-type immune response in the intestine. J. Immunol. 2000;164:6303–6312. doi: 10.4049/jimmunol.164.12.6303. [DOI] [PubMed] [Google Scholar]
- 59.Martin K, et al. Δ32 mutation of the chemokine-receptor 5 gene in inflammatory bowel disease. Clin. Immunol. 2001;98:18–22. doi: 10.1006/clim.2000.4943. [DOI] [PubMed] [Google Scholar]
- 60.Chvatchko Y, et al. A key role for CC chemokine receptor 4 in lipopolysaccharide-induced endotoxic shock. J. Exp. Med. 2000;191:1755–1763. doi: 10.1084/jem.191.10.1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chensue SW, et al. Aberrant in vivo T helper type 2 cell response and impaired eosinophil recruitment in CC chemokine receptor 8 knockout mice. J. Exp. Med. 2001;193:573–584. doi: 10.1084/jem.193.5.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liang M, et al. Identification and characterization of a potent, selective, and orally active antagonist of the CC chemokine receptor-1. J. Biol. Chem. 2000;275:19000–19008. doi: 10.1074/jbc.M001222200. [DOI] [PubMed] [Google Scholar]
- 63.Gong JH, Clark-Lewis I. Antagonists of monocyte chemoattractant protein 1 identified by modification of functionally critical NH2-terminal residues. J. Exp. Med. 1995;181:631–640. doi: 10.1084/jem.181.2.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gong JH, Uguccioni M, Dewald B, Baggiolini M, Clark-Lewis I. RANTES and MCP-3 antagonists bind multiple chemokine receptors. J. Biol. Chem. 1996;271:10521–10527. doi: 10.1074/jbc.271.18.10521. [DOI] [PubMed] [Google Scholar]
- 65.Zhang Y, Rollins BJ. A dominant negative inhibitor indicates that monocyte chemoattractant protein 1 functions as a dimer. Mol. Cell. Biol. 1995;15:4851–4855. doi: 10.1128/mcb.15.9.4851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gong JH, Ratkay LG, Waterfield JD, Clark LI. An antagonist of monocyte chemoattractant protein 1 (MCP-1) inhibits arthritis in the MRL-lpr mouse model. J. Exp. Med. 1997;186:131–137. doi: 10.1084/jem.186.1.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Proudfoot AE, et al. Amino-terminally modified RANTES analogues demonstrate differential effects on RANTES receptors. J. Biol. Chem. 1999;274:32478–32485. doi: 10.1074/jbc.274.45.32478. [DOI] [PubMed] [Google Scholar]
- 68.Simmons G, et al. Potent inhibition of HIV-1 infectivity in macrophages and lymphocytes by a novel CCR5 antagonist. Science. 1997;276:276–279. doi: 10.1126/science.276.5310.276. [DOI] [PubMed] [Google Scholar]
- 69.Signoret N, Pelchen-Matthews A, Mack M, Proudfoot AEI, Marsh M. Endocytosis and recycling of the HIV coreceptor CCR5. J. Cell Biol. 2000;151:1281–1293. doi: 10.1083/jcb.151.6.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Plater-Zyberk C, Hoogewerf AJ, Proudfoot AE, Power CA, Wells TN. Effect of a CC chemokine receptor antagonist on collagen induced arthritis in DBA/1 mice. Immunol. Lett. 1997;57:117–120. doi: 10.1016/s0165-2478(97)00075-8. [DOI] [PubMed] [Google Scholar]
- 71.Lloyd CM, et al. RANTES and monocyte chemoattractant protein-1 (MCP-1) play an important role in the inflammatory phase of crescentic nephritis, but only MCP-1 is involved in crescent formation and interstitial fibrosis. J. Exp. Med. 1997;185:1371–1380. doi: 10.1084/jem.185.7.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ajuebor MN, Hogaboam CM, Kunkel SL, Proudfoot AE, Wallace JL. The chemokine RANTES is a crucial mediator of the progression from acute to chronic colitis in the rat. J. Immunol. 2001;166:552–558. doi: 10.4049/jimmunol.166.1.552. [DOI] [PubMed] [Google Scholar]
- 73.Rucinski B, Knight LC, Niewiarowski S. Clearance of human platelet factor 4 by liver and kidney: its alteration by heparin. Am. J. Physiol. 1986;251:H800–H807. doi: 10.1152/ajpheart.1986.251.4.H800. [DOI] [PubMed] [Google Scholar]
- 74.Witt DP, Lander AD. Differential binding of chemokines to glycosaminoglycan subpopulations. Curr. Biol. 1994;4:394–400. doi: 10.1016/s0960-9822(00)00088-9. [DOI] [PubMed] [Google Scholar]
- 75.Rot A. Neutrophil attractant/activation protein-1 (interleukin-8) induces in vitro neutrophil migration by haptotactic mechanism. Eur. J. Immunol. 1993;23:303–306. doi: 10.1002/eji.1830230150. [DOI] [PubMed] [Google Scholar]
- 76.Hoogewerf AJ, et al. Glycosaminoglycans mediate cell surface oligomerization of chemokines. Biochemistry. 1997;36:13570–13578. doi: 10.1021/bi971125s. [DOI] [PubMed] [Google Scholar]
- 77.Stumbles PA, et al. Regulation of dendritic cell recruitment into resting and inflamed airway epithelium: use of alternative chemokine receptors as a function of inducing stimulus. J. Immunol. 2001;167:228–234. doi: 10.4049/jimmunol.167.1.228. [DOI] [PubMed] [Google Scholar]
- 78.Sallusto F, Lanzavecchia A. Understanding dendritic cell and T-lymphocyte traffic through the analysis of chemokine receptor expression. Immunol. Rev. 2000;177:134–140. doi: 10.1034/j.1600-065x.2000.17717.x. [DOI] [PubMed] [Google Scholar]
- 79.Cox MA, et al. Human interferon-inducible 10-kDa protein and human interferon-inducible T cell α chemoattractant are allotopic ligands for human CXCR3: differential binding to receptor states. Mol. Pharmacol. 2001;59:707–715. doi: 10.1124/mol.59.4.707. [DOI] [PubMed] [Google Scholar]
- 80.Baba M, et al. A small-molecule, nonpeptide CCR5 antagonist with highly potent and selective anti-HIV-1 activity. Proc. Natl Acad. Sci. USA. 1999;96:5698–5703. doi: 10.1073/pnas.96.10.5698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Dragic T, et al. A binding pocket for a small molecule inhibitor of HIV-1 entry within the transmembrane helices of CCR5. Proc. Natl Acad. Sci. USA. 2000;97:5639–5644. doi: 10.1073/pnas.090576697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hesselgesser J, et al. Identification and characterization of small molecule functional antagonists of the CCR1 chemokine receptor. J. Biol. Chem. 1998;273:15687–15692. doi: 10.1074/jbc.273.25.15687. [DOI] [PubMed] [Google Scholar]
- 83.Sabroe I, et al. A small molecule antagonist of chemokine receptors CCR1 and CCR3. Potent inhibition of eosinophil function and CCR3-mediated HIV-1 entry. J. Biol. Chem. 2000;275:25985–25992. doi: 10.1074/jbc.M908864199. [DOI] [PubMed] [Google Scholar]
- 84.Howard OMZ, et al. Inhibition of in vitro and in vivo HIV replication by a distamycin analogue that interferes with chemokine receptor function — a candidate for chemotherapeutic and microbicidal application. J. Med. Chem. 1998;41:2184–2193. doi: 10.1021/jm9801253. [DOI] [PubMed] [Google Scholar]
- 85.Kledal TN, et al. A broad-spectrum chemokine antagonist encoded by Kaposi's sarcoma-associated herpesvirus. Science. 1997;277:1656–1659. doi: 10.1126/science.277.5332.1656. [DOI] [PubMed] [Google Scholar]
- 86.Liang M, et al. Species selectivity of a small molecule antagonist for the CCR1 chemokine receptor. Eur. J. Pharmacol. 2000;389:41–49. doi: 10.1016/s0014-2999(99)00863-8. [DOI] [PubMed] [Google Scholar]
- 87.Auten RL, et al. Nonpeptide CXCR2 antagonist prevents neutrophil accumulation in hyperoxia-exposed newborn rats. J. Pharmacol. Exp. Ther. 2001;299:90–95. [PubMed] [Google Scholar]
- 88.Samson M, et al. Resistance to HIV-1 infection in Caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature. 1996;382:722–725. doi: 10.1038/382722a0. [DOI] [PubMed] [Google Scholar]
- 89.Fischereder M, et al. CC chemokine receptor 5 and renal-transplant survival. Lancet. 2001;357:1758–1761. doi: 10.1016/s0140-6736(00)04898-4. [DOI] [PubMed] [Google Scholar]
- 90.Garred P, et al. Dual effect of CCR5 Δ32 gene deletion in HIV-1-infected patients. Copenhagen AIDS Study Group. Lancet. 1997;349:1884. doi: 10.1016/s0140-6736(05)63874-3. [DOI] [PubMed] [Google Scholar]
- 91.Sellebjerg F, Madsen HO, Jensen CV, Jensen J, Garred P. CCR5 Δ32, matrix metalloproteinase-9 and disease activity in multiple sclerosis. J. Neuroimmunol. 2000;102:98–106. doi: 10.1016/s0165-5728(99)00166-6. [DOI] [PubMed] [Google Scholar]
- 92.Gu L, et al. Absence of monocyte chemoattractant protein-1 reduces atherosclerosis in low density lipoprotein receptor-deficient mice. Mol. Cell. 1998;2:275–281. doi: 10.1016/s1097-2765(00)80139-2. [DOI] [PubMed] [Google Scholar]
- 93.Huang D, Wang J, Kivisakk P, Rollins BJ, Ransohoff RM. Absence of monocyte chemoattractant protein 1 in mice leads to decreased local macrophage recruitment and antigen-specific T helper cell type 1 immune response in experimental autoimmune encephalomyelitis. J. Exp. Med. 2001;193:713–726. doi: 10.1084/jem.193.6.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Izikson L, Klein RS, Charo IF, Weiner HL, Luster AD. Resistance to experimental autoimmune encephalomyelitis in mice lacking the CC chemokine receptor (CCR)2. J. Exp. Med. 2000;192:1075–1080. doi: 10.1084/jem.192.7.1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Boring L, Gosling J, Cleary M, Charo IF. Decreased lesion formation in CCR2−/− mice reveals a role for. Nature. 1998;394:894–897. doi: 10.1038/29788. [DOI] [PubMed] [Google Scholar]