

Key Points
Oxidative stress results in the oxidative damage of membrane lipids, leading to the formation of neo-self epitopes, which are known as oxidation-specific epitopes (OSEs).
Major carriers of OSEs are dying cells, microvesicles and damaged proteins and lipoproteins, such as oxidized low-density lipoproteins.
OSEs are recognized by multiple arcs of the innate immune system, such as soluble pattern recognition receptors (natural antibodies and complement components) and cellular pattern recognition receptors (Toll-like receptors and scavenger receptors).
OSEs have an important role in physiological processes by serving as markers of oxidatively modified endogenous structures, allowing the immune system to mediate their clearance and to maintain homeostasis.
The accumulation of OSEs can trigger sterile inflammation.
OSEs have been suggested to contribute, as drivers of disease development, to various chronic and acute inflammatory diseases, including atherosclerosis, non-alcoholic steatohepatitis and age-related macular degeneration.
Supplementary information
The online version of this article (doi:10.1038/nri.2016.63) contains supplementary material, which is available to authorized users.
Subject terms: Innate immunity, Inflammation, Lipid peroxides, Atherosclerosis
Oxidation-specific epitopes (OSEs) function as markers of oxidative damage of membrane lipids. This Review discusses the immune recognition of OSEs, as well as their role in the maintenance of tissue homeostasis and their contribution to the development of inflammatory diseases.
Supplementary information
The online version of this article (doi:10.1038/nri.2016.63) contains supplementary material, which is available to authorized users.
Abstract
Ageing, infections and inflammation result in oxidative stress that can irreversibly damage cellular structures. The oxidative damage of lipids in membranes or lipoproteins is one of these deleterious consequences that not only alters lipid function but also leads to the formation of neo-self epitopes — oxidation-specific epitopes (OSEs) — which are present on dying cells and damaged proteins. OSEs represent endogenous damage-associated molecular patterns that are recognized by pattern recognition receptors and the proteins of the innate immune system, and thereby enable the host to sense and remove dangerous biological waste and to maintain homeostasis. If this system is dysfunctional or overwhelmed, the accumulation of OSEs can trigger chronic inflammation and the development of diseases, such as atherosclerosis and age-related macular degeneration. Understanding the molecular components and mechanisms that are involved in this process will help to identify individuals with an increased risk of developing chronic inflammation, and will also help to indicate novel modes of therapeutic intervention.
Supplementary information
The online version of this article (doi:10.1038/nri.2016.63) contains supplementary material, which is available to authorized users.
Main
In the course of evolution, most organisms have developed mechanisms to utilize oxygen for various biological processes, such as the synthesis of biomolecules, energy production and phagocytosis. Cellular usage of oxygen leads to the constant generation of reactive oxygen species (ROS), which are mainly generated by the mitochondria. In addition, ROS are generated in peroxisomes, the endoplasmic reticulum and the plasma membrane. Within eukaryotic organisms, superoxide, peroxide and hydroxyl radicals represent the main types of ROS that have high reactivity and low stability and that actively drive many cellular reactions. The biological effects of ROS greatly depend on the amounts of ROS present, which supports the idea that cellular ROS generation has characteristics of hormesis1. Indeed, at low concentrations, ROS participate in physiological processes, such as cell renewal, cellular metabolism, proliferation and differentiation1,2,3,4. However, high ROS production during an oxidative burst is crucial for host defence to ensure microbial killing5 and the formation of neutrophil extracellular traps (NETs)6. These cellular responses and the modulation of immune responses by ROS have been recently reviewed1,3,4,7. However, both exogenous triggers, such as ultraviolet (UV) irradiation, chemical toxins and hyperthermia, and endogenous conditions, including senescence, metabolic changes and inflammation, can impair the balance between the generation and the elimination of intracellular ROS, which can lead to ROS accumulation and oxidative stress. The harmful effects that are caused by oxidative stress are typically limited by antioxidant responses, which include the induction of enzymes and proteins that eliminate ROS. However, if unresolved, oxidative stress generates cellular and extracellular damage by irreversibly modifying DNA, RNA, proteins and lipids, which ultimately leads to molecular modifications that disrupt normal functions and often results in cell0 death. Oxidative modifications of biomolecules also lead to the generation of secondary free radicals, which further propagate oxidative damage1,3,4,7.
Lipids, particularly phospholipids, are prime targets for oxidation owing to their abundance as important building blocks of cells, extracellular vesicles and lipoproteins. The oxidation of lipids not only alters their biological function but also results in the generation of various degradation products through a process that is known as lipid peroxidation8 (Box 1). The newly generated products propagate the effect of oxidative stress by directly modulating cellular functions and responses such as metabolism and inflammation9,10,11,12,13,14,15,16,17,18. Furthermore, as the degradation products of oxidized lipids are often highly reactive, they modify self-molecules and thereby generate structural neo-epitopes that are recognized by receptors of the immune system. These neo-epitopes have been termed oxidation-specific epitopes (OSEs) and represent a common set of epitopes present on various oxidatively modified self-proteins and lipids14,19. OSEs, including oxidized phospholipids (OxPLs) and malondialdehyde (MDA)-modified amino groups, have been documented on the surface of apoptotic cells, microvesicles and damaged structures such as oxidized low-density lipoproteins (OxLDLs)14,17,20. They are sensed by humoral and cellular components of the innate immune system, which mediate their removal and prevent inflammatory effects. However, if not efficiently cleared, they can also act as damage-associated molecular patterns (DAMPs) and can trigger sterile inflammation21,22 (Box 2).
In this Review, we discuss the role of OSEs in health and disease, and the innate immune responses that specifically target OSEs as markers of oxidative stress.
Box 1: Lipid peroxidation.
Lipid peroxidation is the oxidative damage of lipids that is initiated by the removal of a hydrogen atom from the CH2 group within the double bonds of polyunsaturated fatty acids (PUFAs). The subsequent addition of oxygen radicals and the formation of lipid-peroxyl radicals, which are transformed into lipid-hydroperoxide molecules can further propagate this reaction. Both enzymatic and non-enzymatic mechanisms can lead to lipid peroxidation. The enzymatic mechanisms involve lipoxygenases, cyclooxygenases and cytochrome P450, and result in the generation of products with high stereo-specificity23,144. Depending on the PUFA substrate, major products of lipoxygenase oxidation are hydroxyperoxyeicosatetraenoic acids (HpETEs) hydroxyeicosatetraenoic acids (HETEs), leukotrienes, lipoxins, hydroxyoctadecadienoic acids (HODEs), hepoxylins and resolvins. Oxygenation by cyclooxygenases results in the generation of prostaglandins, prostacyclin and thromboxanes, whereas cytochrome P450 generates epoxyeicosatrienoic acids (EETs), 20-HETE, thromboxanes and prostacyclins27,145. Non-enzymatic mechanisms are mediated by free radicals that are generated by NADPH oxidases and nitric oxide synthases in the presence of transition metal ions (Fe2+ and Cu2+) and result in a mixture of nonspecific stereoisomers23,27. Moreover, the enzyme myeloperoxidase can also initiate non-enzymatic lipid peroxidation through the generation of reactive oxygen species (ROS), such as HOCl, HOBr and ·NO145,146. In contrast to products of enzymatic lipid oxidation, the products of non-enzymatic processes are considered to be more toxic and damaging to the host147. Continuous degradation of lipid-hydroxyperoxides by cyclization and fragmentation in the presence of reducing metal ions (Fe2+ and Cu2+) results in the generation of highly reactive terminal degradation products such as reactive aldehydes 4-hydroxynonenal (4-HNE), malondialdehyde (MDA) and 2-(ω carboxyethyl) pyrrole (CEP)8,24. Lipid peroxidation products that are generated by both mechanisms modulate a multitude of physiological processes such as cell signalling, wound healing, immune tolerance, skin barrier function, coagulation, vasodilation, modulation and the resolution of inflammation12,13,16,23,145,148,149. They are rapidly removed in healthy tissues but accumulate in many pathological conditions in which they can cause adverse effects.
Box 2: Sterile inflammation.
Sterile inflammation is an inflammatory process that is elicited in response to damage-associated molecular patterns (DAMPs), which are released locally in response to tissue damage22. DAMPs are intracellular and extracellular host-derived molecules that are not usually sensed by the immune system but that are released or become modified into altered self-molecules upon tissue damage21. In analogy to pathogen- induced inflammation, sterile inflammation is triggered by the activation of the innate immune response through the recognition of DAMPs by pattern recognition receptors (PRRs), resulting in the enhanced secretion of cytokines and chemokines. Membrane- bound PRRs, such as Toll-like receptors (TLRs), and intracellular PRRs, such as the inflammasome, are key mediators of sterile inflammation. Cytokines belonging to the interleukin-1 (IL-1) family have been proposed to be important drivers of sterile inflammation150. Increased cytokine and chemokine secretion at the site of initial damage ultimately results in an enhanced recruitment of immune cells, such as neutrophils and macrophages. The resolution of sterile inflammation should lead to tissue repair and the re-establishment of homeostasis. Unresolved sterile inflammation is implicated in the development of several medical conditions, such as gout, Alzheimer disease, rheumatoid arthritis and atherosclerosis.
The generation of OSEs
Oxidized lipids, and their degradation products, can interfere with the normal function of proteins and lipids. However, a unique aspect of some of these lipid derivatives is their ability to modify proteins and lipids and to form OSEs, which are then recognized by the immune system in a hapten-specific manner; for example, the same OSE can be present on different proteins and can be recognized by a common set of innate immune receptors. Several mechanisms can lead to the oxidation of lipids, particularly of polyunsaturated fatty acids (PUFAs)8,23,24, and important insights into the generation of OSEs have been obtained from studies on the biological activities of OxLDL that are central to the pathogenesis of atherosclerosis25,26. Lipid peroxidation involves both enzymatic mechanisms, including lipoxygenases, cyclooxygenases and cytochrome p450, and non-enzymatic mechanisms, which are mediated by free radicals and which require catalysis by transition metals or hemin23,26,27,28 (Box 1). Moreover, there is now ample evidence that during apoptosis membrane lipids undergo oxidation, which results in the generation of OSEs. The exact mechanisms that lead to the oxidation of cellular membrane lipids are still being elucidated17,29.
PUFA-containing phospholipids, such as the major membrane phospholipid phosphatidylcholine, are particularly prone to oxidative damage, and their peroxidation results in the generation of a complex mixture of OxPLs and terminal degradation products8,23,24 (Box 1). These, in turn, form adducts with, for example, the amino group of lysine residues or aminophospholipids to generate OSEs. In particular, the oxidation of the PUFA chain at the sn-2 position of phosphatidylcholine results in its fragmentation and the generation of reactive PUFA breakdown products such as MDA and 4-hydroxynonenal (4-HNE), which can modify self-molecules and form OSEs (Fig. 1). OSEs can also be generated by the modification of proteins with truncated phospholipids, such as oxidized phosphatidylcholine, oxidized cardiolipin (OxCL), oxidized phosphatidylserine (OxPS) and oxidized phosphatidylethanolamine (OxPE)8,30,31,32. Furthermore, 2-(ω-carboxyethyl)pyrrole (CEP) represents an adduct between (E)-4-hydroxy-7-oxohept-5-enoic acid — an oxidative fragment of docosahexaenoic acid — and the amino groups of lysines or aminophospholipids24,33,34. Notably, some of these breakdown products, such as MDA and 4-HNE, have been studied as prototypical markers of oxidative stress and can be measured by the frequently used 2-thiobarbituric acid reaction (TBAR) method8. As a vast diversity of OxPLs can be generated, many more OSEs derived from these may exist23,28. Moreover, the oxidation of other lipids, such as cholesterol and cholesteryl esters, can lead to structural changes and altered biological activities, but their ability to be recognized by innate immune receptors is still being characterized10,25,35. It will be important to elucidate the exact structures of the relevant OSEs to better understand their recognition by the immune system. Owing to their biological activities, these products of lipid peroxidation have been implicated in a wide variety of physiological and pathological processes, and immune responses that target these products can modulate many of these effects14,30,36.
Figure 1. The generation of OSEs.

Tissue damage, cellular stress and cell death result in increased oxidative stress, which promotes lipid peroxidation. Lipid peroxidation can occur through non-enzymatic mechanisms, such as reactive oxygen species (ROS), and through enzymatic mechanisms, including myeloperoxidases, 12/15-lipoxygenases, cyclooxygenases and cytochrome P450. The oxidation of sn-2 polyunsaturated fatty acids (PUFAs) of membrane phospholipids leads to fragmentation and the generation of highly reactive breakdown products, such as malondialdehyde (MDA) and 4-hydroxynonenal (4-HNE)8,30,31,32. In addition, different types of oxidized phospholipids (OxPLs) can be generated from different phospholipid backbones, including oxidized phosphatidylcholine, oxidized phosphatidylethanolamine (OxPE), oxidized phosphatidylserine (OxPS) and oxidized cardiolipin (OxCL). The newly generated breakdown products and the oxidized and truncated residual core OxPLs can in turn react with free amino groups of protein side chains or lipids that are localized in their vicinity, forming stable covalent adducts and creating oxidation-specific epitopes (OSEs). Because phosphocholine (PC) as an OSE is only presented as an epitope in the context of OxPL, these epitopes are termed PC-OxPLs for clarity. The PC moiety can also be a component of the capsular polysaccharide of bacteria, where it is not part of a phospholipid and is constitutively presented as an epitope. In addition, an adduct between an oxidative fragment of docosahexaenoic acid, (E)-4-hydroxy-7-oxohept-5-enoic acid and lysines of proteins (or aminophospholipids) can lead to the formation of 2-(ω-carboxyethyl) pyrrole (CEP). OSE-modified proteins or lipids are sensed by innate immune responses and represent a unique class of damage-associated molecular patterns (DAMPs).
Immune recognition of OSEs
OSEs have important roles in physiological processes, as they mark oxidatively modified endogenous molecules, such as proteins and/or lipoproteins and apoptotic cells, as being damaged by increased oxidative stress. This facilitates their removal by the housekeeping functions of the immune system to maintain homeostasis. In pathological situations — in which oxidative events are greatly increased and the homeostatic functions of innate immunity are overwhelmed and/or impaired — the accumulation of OSEs leads to sterile inflammation, which is a mechanism that should ultimately restore normal tissue integrity22 (Box 2). If this fails as a result of persistent tissue damage and/or impaired resolution, the accumulation of OSEs can trigger chronic inflammation. Therefore, OSEs have important roles in both the regulation of tissue homeostasis and inflammatory responses (Fig. 2). The detailed characterization of OSEs and the immune responses against these OSEs have shown that they are recognized by both cellular and soluble pattern recognition receptors (PRRs), including natural IgM antibodies14,31 (Table 1).
Figure 2. Role of innate immune responses targeting OSEs.
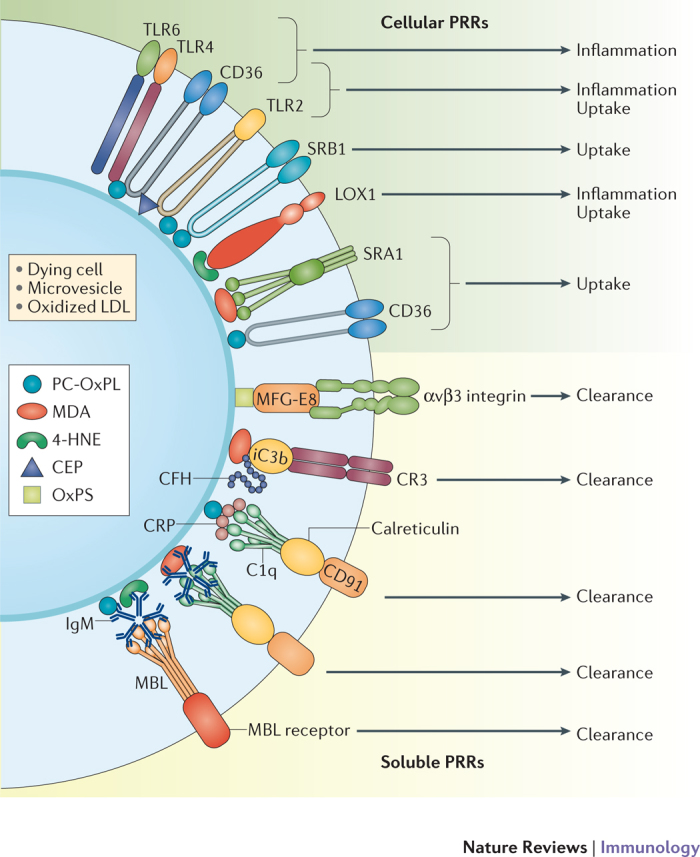
Oxidized low-density lipoproteins (OxLDL), apoptotic cells, microvesicles and cellular debris display various types of oxidation-specific epitopes (OSEs)17,20,67,81,88. Components of cellular and humoral immune responses sense OSEs and mediate sterile inflammation or clearance and neutralization depending on the availability and context. Macrophages sense OSEs through various pattern recognition receptors (PRRs) that are expressed on their surface. Scavenger receptor CD36 preferentially binds and mediates the uptake of the phosphocholine (PC) of OxPL (PC-OxPL), oxidized phosphatidylserine (OxPS) and 2-(ω-carboxyethyl)pyrrole (CEP). Scavenger receptor SRB1 recognizes PC-OxPL; SRA1 andlectin-like oxidized LDL receptor 1 (LOX1) bind malondialdehyde (MDA); and LOX1 recognizes 4-hydroxynonenal (4-HNE)14,37. Toll-like receptors (TLRs) mediate pro-inflammatory signals. PC-OxPL transmit inflammatory signals through a heterotrimer of TLR4–TLR6–CD36, as well as through TLR2, and CEP signals through TLR2 in cooperation with CD36 (Refs 33,46). Humoral immune responses to OSEs include natural IgM antibodies, C-reactive protein (CRP), members of complement and other soluble PRRs that are involved in the clearance of apoptotic cells. Soluble PRRs inhibit the recognition of OSEs by cellular PRRs and mediate uptake via alternative pathways. Natural IgM bound to PC-OxPL, MDA and 4-HNE epitopes are taken up by macrophages by C1q–calreticulin–CD91-dependent or mannose-binding lectin (MBL)- and MBL receptor-dependent mechanisms. CRP bound to PC-OxPL is taken up by C1q–calreticulin–CD91-dependent mechanisms103,151. Alternative recognition of C1q is mediated by CD93 (not shown). Complement factor H (CFH) bound to MDA provides cofactor activity for the cleavage of C3b into iC3b opsonins67 that mediate anti-inflammatory clearance via complement receptor 3 (CR3). Milk fat globule-epidermal growth factor 8 (MFG-E8) bound to OxPS or OxPE mediate clearance via αvβ3 integrins72,73.
Table 1. Cellular and humoral immune responses targeting OSEs.
| Innate immune response | OSE bound | Effect of OSE binding | Refs | |
|---|---|---|---|---|
| Class | PRR | |||
| Scavenger receptors | SRA1 and SRA2 | MDA | Uptake | 47,48 |
| LOX1 | MDA | Nitric oxide production by the endothelium | 49 | |
| CD36 |
• PC-OxPL • OxPS • CEP |
• Uptake • Enhanced efferocytosis • Inflammation |
33,43,44,46 | |
| SRB1 | PC-OxPL | Uptake | 45 | |
| LOX1 | 4-HNE |
• Uptake • Inflammation |
50 | |
| TLRs | TLR4–TLR6 | PC-OxPL | Inflammation | 51,53 |
| TLR2 and TLR2–TLR6 |
• CEP • OxPL |
• Inflammation • Angiogenesis • ER stress |
33,46,54 | |
| Annexin A5 | OxCL | Neutralization | 74 | |
| Complement | CFH | MDA |
• Neutralization • Opsonization (iC3b) |
67 |
| C3a | MDA | Enhanced clearance of C3a | 71 | |
| CRP | PC-OxPL | Enhanced efferocytosis | 65 | |
| Other PRRs | MFG-E8 |
• OxPS • OxPE |
• Enhanced efferocytosis • Modulation of efferocytosis |
73,97 |
| Annexin A5 | OxCL | Neutralization | 74 | |
| CRP | PC-OxPL | Enhanced efferocytosis | 65 | |
| Natural IgM antibodies | LR01 | OxCL | Unknown | 77 |
| LR04 | MDA | Neutralization | 20 | |
| NA17 | MDA | Enhanced efferocytosis | 75 | |
| T15/E06 | PC-OxPL |
• Neutralization • Enhanced efferocytosis • Inhibition of foam cell formation |
18,51,77,78,88 | |
| E014 | MDA | Neutralization (?) | 78,89 | |
| IgM | 4-HNE | Unknown | 75 | |
4-HNE, 4-hydroxynonenal; CEP, 2-(ω-carboxyethyl) pyrrole; CFH, complement factor H; CRP, C-reactive protein; ER, endoplasmic reticulum; LOX1, lectin-like oxidized LDL receptor 1; MDA, malondialdehyde; MFG-E8, milk fat globule-epidermal growth factor 8; OSE, oxidation- specific epitope; OxCL, oxidized cardiolipin; OxPE, oxidized phosphatidylethanolamine; OxPL, oxidized phospholipid; OxPS, oxidized phosphatidylserine; PC-OxPL, phosphocholine (PC)-containing OxPL; PRR, pattern recognition receptor; SR, scavenger receptor; TLR, Toll-like receptor.
OSE sensing by cellular PRRs. OSEs are recognized by a wide variety of cell surface receptors (Table 1), most of which are expressed by macrophages. Macrophages mediate the uptake and phagocytosis of oxidatively altered molecules and cells, and macrophages also sense the accumulation of OSEs in the local microenvironment, which triggers signalling pathways that induce the secretion of inflammatory chemokines and cytokines14,17,31.
Scavenger receptors represent the prototypical class of innate receptors for OSEs37. They are multi-ligand receptors that bind both modified self-structures and non-self molecules. Different types of scavenger receptors have been identified, including CD36, SRA1, SRA2, SRB1, CD68 and lectin-like oxidized LDL receptor 1 (LOX1; also known as OLR1)37. Among these, SRA1, SRA2 and CD36 are considered to be responsible for most of the OxLDL uptake by macrophages in in vitro assays, because macrophages that are deficient in CD36, SRA1 and SRA2 show 75–90% decreased binding and degradation of OxLDL38. Detailed structural studies have shown that OxPL is a high-affinity ligand that mediates OxLDL binding to CD36 (Ref. 39), and, specifically, that this is mediated by the phosphocholine (PC) headgroup of OxPL (PC-OxPL) but the PC of unoxidized phospholipids does not serve as a ligand40. Binding can also be mediated by the truncated sn-2 acyl chain of OxPLs41,42, which indicates that other classes of oxidized phospholipids such as OxPS can bind to CD36 (Refs 43,44). Similarly, PC-OxPL also mediates the uptake of OxLDL by SRB1 on macrophages45. Examples of other OSEs that are recognized by scavenger receptors include CEP-modified proteins, which are recognized by CD36 (Ref. 46) and MDA-modified proteins, which are taken up by SRA1 and SRA247,48. A recent study showed that the endothelial scavenger receptor LOX1 mediates MDA-induced nitric oxide (NO) production49 and this receptor also binds 4-HNE50.
Toll-like receptors (TLRs) also recognize and respond to OSEs. Macrophages stimulated with oxidized 1-palmitoyl-2-arachidonyl-sn-glycero-3-phosphocholine (OxPAPC) secrete interleukin-6 (IL-6), whereas IL-6 production is absent in macrophages that lack TLR4 or TIR-domain-containing adaptor protein inducing interferon-β (TRIF; also known as TICAM1)51. Sensing of PC-OxPL seems to be important for the TLR4-mediated effects, as the PC-specific IgM T15/E06 inhibited IL-6 secretion induced by OxPAPC but did not inhibit IL-6 secretion induced by lipopolysaccharide (LPS)51. OxPLs have also been reported to stimulate macrophages via a TLR2 pathway52. Moreover, chemokine secretion by macrophages stimulated with OxLDL has been shown to require the cooperation of CD36 with a TLR4–TLR6 heterodimer53. Conversely, OxPLs induce apoptosis in macrophages via TLR2–TLR6 in cooperation with a CD36 ligand54. As most of these studies have used mixtures of different OxPL products, individual OxPL moieties might use TLR receptors in varying combinations to generate different signalling pathways. The common role of TLRs as sensors of OSEs is further supported by the findings that CEP adducts are bound by TLR2, but not by TLR4, on both endothelial cells33 and macrophages46. Furthermore, CEP selectively augments TLR2–TLR1 signalling in macrophages55 and has been shown to activate platelets via TLR2–TLR1 (Ref. 56). TLRs have not yet been directly implicated in chemokine secretion that is induced by MDA or 4-HNE, but this process is probably also mediated by the cooperation of scavenger receptors with other signalling PRRs57. Oxidized cholesteryl esters (OxCEs) trigger pro-inflammatory macrophage responses via TLR4 and these involve non-classical signalling via spleen tyrosine kinase (SYK)35,58. Thus, some OSEs represent endogenous TLR ligands that have the capacity to trigger inflammatory responses. Notably, in endotoxin-induced inflammation, soluble OxPLs impair complex formation of TLR4 with CD14, MD2 and LPS-binding protein and thereby dampen LPS-induced responses23,59. Recently, epoxycyclopentenones that are present in OxPAPC were identified as active components responsible for the inhibitory effects of OxPAPC on the activation of specific TLRs by synthetic ligands60. An interesting property of the inflammatory responses to OxPL is the tight cooperation with scavenger receptors, such as CD36, which mediate recognition of OxPLs and probably also facilitate sensing and signalling by TLRs. Although OxLDL uptake via CD36 seems to be necessary for some macrophage signalling pathways, it is unknown to what extent scavenger receptor-mediated uptake of other OSEs is required for their cellular responses.
OSE sensing by soluble PRRs. Soluble PRRs include secreted versions of cellular PRR, pentraxins and proteins of the complement system61. Whereas some of these proteins interact with cellular PRRs to mediate the signals of bound ligands, others participate in the housekeeping functions of the host. Soluble PRRs distinguish between healthy and damaged tissues by sensing metabolic byproducts and OSEs presented on dying cells (Table 1).
C-reactive protein (CRP) was originally identified as the C-reactive component of plasma that could bind the PC present on the capsular polysaccharide of Streptococcus pneumoniae62,63,64. However, this PC moiety is not part of a phospholipid and CRP was later found to also bind PC-OxPL found on OxLDL and on apoptotic cell membranes65. This molecular mimicry between microbial PC and PC-OxPL enables CRP to respond to a common OSE present during both microbial infections and oxidative stress. By analogy, Porphyromonas gingivalis and group A Streptococcus have been suggested to carry MDA-like epitopes, but their ability to be recognized by MDA-specific PRRs has not been tested in detail66.
The identification of complement-regulatory protein complement factor H (CFH) as a receptor for MDA-epitopes has shed important light on its function in disease67. CFH consists of 20 short consensus repeats (SCRs)68 and two of its domains — SCR7 and SCR19-20 — mediate the binding of CFH to MDA67. Importantly, these binding sites are hotspots for disease-associated single nucleotide polymorphisms (SNPs)69; for example, SNP rs1061170 results in a Y>H exchange of amino acid 402 in SCR7 and reduces the ability of plasma CFH to bind MDA by more than 65% for homozygous carriers compared with controls67. In addition, the use of recombinant variants of the SCR19-20 domain demonstrated that other SNPs in SCR19-20 could alter binding to MDA70. Thus, genetic variants of both MDA-binding sites of CFH may determine the ability of CFH to bind MDA. Considering the conserved structure of the regulators of complement activation, MDA-epitopes could also serve as binding sites for other members of this family68. Interestingly, the C3 cleavage product C3a, which acts as a pro-inflammatory anaphylatoxin, was also shown to bind MDA-epitopes71.
Other soluble PRRs may recognize oxidized rather than native lipid structures on cellular surfaces, but the exact nature of these interactions is less well understood. For example, milk fat globule-epidermal growth factor 8 (MFG-E8) has recently been shown to specifically recognize OxPS72 and OxPE73. Moreover, another phosphatidylserine-binding protein, annexin A5, has been suggested to bind to OxCL and to neutralize its biological effects74.
Therefore, OSEs represent targets for several soluble PRRs of the innate immune system. As different OSEs are often present on the same surfaces, different soluble PRRs binding to them may synergize and cooperate in their effector functions. For example, apoptotic cells and microvesicles carry both MDA and PC-OxPL epitopes20,75, each of which recruits different soluble PRRs to the same surface.
OSEs are major targets of natural IgM antibodies. OSEs have been identified as prominent antigens of natural IgM antibodies (Table 1). IgM antibodies in human umbilical cord blood, which represent naive natural antibodies of fetal origin, have specificity for OSEs75. In fact, titres of IgM with specificity for OxLDL and MDA-LDL were higher in cord blood samples compared with matched maternal blood samples. The specific binding properties of natural IgM antibodies have been investigated by characterizing IgM antibodies derived both from in vitro stimulated B-1 cells and from the plasma of mice lacking recombination-activating gene 1 (RAG1) that were selectively reconstituted with B-1 cells75. These studies revealed that several OSEs, including PC-OxPL, MDA and 4-HNE adducts are bound by up to 30% of all natural IgM found in the plasma of both wild-type and gnotobiotic mice, and by a similar percentage of IgM-secreting cells from the spleens of wild-type mice75. Furthermore, several monoclonal natural IgM antibodies with specificity for different OSEs have been characterized. For example, the well-studied PC-binding T15/E06 IgM — which contains a variable region that is composed of a canonical rearrangement of germline genes in both the VL and VH chains — was originally cloned from the spleens of atherosclerosis-prone apolipoprotein E (Apoe−/−) mice as an antibody with specificity for OxLDL, which was later found to be specific for PC-OxPL76,77,78. The same antibody idiotype, as an IgA, was originally described due to its ability to bind PC present in the capsular polysaccharide of S. pneumoniae76,79,80. In addition, several other IgMs that bind different OSEs have been identified, such as the OxCL-specific LR01 and a number of others with specificity for MDA-type adducts, including E014, NA17 and LR04. Similar to T15/E06, all of the OSE-specific natural IgM antibodies that have been described exhibit germline or near-germline usage in their complementary-determining region 3 (CDR3), identifying them as natural IgM75,78,81,82. Thus, natural IgM antibodies prominently participate in the response to OSEs.
Role of OSEs in the maintenance of homeostasis
Physiological carriers of OSEs. Tissue homeostasis depends on the efficient removal of dying cells, which is achieved by molecular tags that mark cellular debris at different stages of cellular death. The quiescent removal of apoptotic cells is mainly mediated by phagocytes that sense several 'find-me' and 'eat-me' signals on apoptotic cells83,84,85. However, in situations of increased cell death and/or inefficient clearance, apoptotic cells can release DAMPs that trigger sterile inflammation22 and render these cells immunogenic17. OSEs on late-stage apoptotic cells have been identified as important ligands for the clearance of apoptotic cells under these conditions17,18,75,81,86,87. In fact, apoptotic mammalian cells of every cellular origin tested display OSEs on their surface independently of the apoptosis trigger, and these OSEs are identical to the ones on OxLDL18 (Fig. 2). Notably, only a small proportion of early apoptotic cells display OSEs, which is in contrast to late apoptotic cells as >50% of this cell population carries OSEs18,81. A subset of apoptotic cells is bound by natural IgM antibodies with specificity for PC-OxPL and different types of MDA epitopes, and some are bound by monoclonal antibodies with specificity for OxCL17,18,75,81,82. Furthermore, mass spectrometry has demonstrated that lipid extracts of thymocytes, in which apoptosis was induced by several triggers, were enriched in different OxPLs17. Similarly, microvesicles generated from monocytes that were loaded with unesterified cholesterol, as well as t-BuOOH-oxidized microvesicles that were derived from endothelial cells, carried both MDA and PC-OxPL epitopes in their membranes88,89.
More recently, we also identified a subset of circulating microvesicles as carriers of various OSEs20. Microvesicles have been implicated in several diseases owing to their pro-inflammatory and pro-coagulant activities90. Interestingly, MDA adducts consisting of simple MDA-lysine adducts, as well as more complex adducts made up of a lysine residue and two or more MDA moieties, were the most prominent OSE found on ∼50% of circulating microvesicles, whereas PC-OxPLs were found on fewer microvesicles but were found to a greater extent on the surface of the same microvesicles. The presence of OSEs was generally independent of cellular origin, supporting their universal presence20. Moreover, a portion of the microvesicles also carried IgM bound on their surface, which was primarily directed against MDA, consistent with their prominent presence. Interestingly, a subset of circulating microvesicles also carry CRP on their surface, which is consistent with the presence of PC-OxPL91.
Thus, different mechanisms of cell death and generation of microvesicles result in the surface presentation of various types of OSEs. An interesting area for future investigations includes understanding whether the presence of OSEs is associated with other types of cell death such as pyroptosis, necroptosis and ferroptosis85,92.
Physiological role of OSEs. In situations that are associated with increased oxidative stress, which favour OSE generation, a large amount of cellular waste is generated. For example, exposure to high light intensities in an oxygen-rich environment results in enhanced lipid peroxidation. Consequently, the retina is one of the tissues with the highest cell turnover rate, and accordingly requires high phagocytic activity for the clearance of aged photoreceptors, which are enriched in long-chain PUFAs. Both retinal pigment epithelium (RPE) and microvascular endothelial cells express several scavenger receptors that mediate the clearance of apoptotic cells, cellular debris and oxidized lipids93. Indeed, rats with a deletion variant of CD36 exhibit impaired phagocytosis of photoreceptor rod outer segments94. Whereas anionic phospholipids on apoptotic cells have been suggested to mediate this binding95, mass spectrometry studies have shown that the exposure of rat retinal cells to light results in elevated levels of OxPLs96. Importantly, OxPL inhibited the uptake of rod outer segments by wild-type cells but not by Cd36−/− RPE cells. OSEs may also be involved in efferocytosis via MFG-E8, which binds OxPS exposed on the surface of apoptotic cells72. MFG-E8 facilitates engulfment by phagocytes via the αvβ3 and αvβ5 integrin receptors, and has been shown to have an important role in the phagocytosis of photoreceptors of the outer segment97. Recent work has identified the 12/15-lipoxygenase product OxPE as a ligand for MFG-E8 with unique functional consequences in the context of apoptotic cell clearance during peritonitis73. In this study, OxPE that is exposed on the surface of alternatively activated macrophages led to the sequestration of MFG-E8, which favours the engulfment of apoptotic cells via phosphatidylserine receptors, such as T cell immunoglobulin and mucin domain 4 (TIM4), but blocks MFG-E8-dependent uptake of apoptotic cells by pro-inflammatory LY6Chi monocytes. Thus, in environments with high oxidative stress, the OSE-dependent clearance of cellular debris represents a physiological housekeeping function that directs the clearance of dying cells to specific pathways.
Accumulating evidence indicates that the recognition of OSEs may also be crucial for complement-mediated clearance mechanisms, particularly of late apoptotic and necrotic cells, including the activity of several complement proteins and receptors, such as CRP and natural IgM, as well as pentraxin83,98,99. Natural IgM antibodies are important in apoptotic cell clearance, as mice deficient in secreted IgM are more prone to developing autoimmunity when crossed with the lupus-prone MRL/lpr background100. To date, all characterized OSE-specific natural IgM antibodies have also been found to bind apoptotic cells, and the PC-specific T15/E06 IgM and the MDA-specific NA17 antibodies enhance the uptake of apoptotic cells in mice in vivo75,101. Interestingly, MDA and 4-HNE accumulate in the serum of aged MRL/lpr lupus-prone mice102. MDA and PC-OxPL have also been suggested to be dominant drivers in IgM-C1q-mediated efferocytosis by dendritic cells87. Moreover, the administration of T15/E06 protected arthritis-prone DBA1 mice from collagen-induced arthritis86, suggesting an anti-inflammatory potential of neutralizing PC-OxPL. As dying cells display different OSEs at the same time, IgM with different OSE specificity may have functionally additive effects. Similarly, CRP, which also binds apoptotic cells via PC-OxPL, enhances the clearance of dying cells in a complement-dependent manner103. Although CRP and IgM are potent activators of complement, their binding on apoptotic cell surfaces does not lead to terminal complement activation because this is limited by the recruitment of the soluble regulators CFH and C4 binding protein (C4BP)104. Importantly, recruitment of CFH to apoptotic blebs and to necrotic RPE cells can be mediated by MDA67 and other previously described ligands105. This might allow CFH to provide cofactor activity for factor I on dying cells to inactivate C3b into iC3b. The phagocytosis of dying cells that have been opsonized with iC3b can inhibit nuclear factor-κB (NF-κB) transcription, and can thereby promote anti-inflammatory clearance by phagocytes106. Notably, CFH mediates cofactor activity on MDA-decorated surfaces, and MDA binding does not interfere with this process. Moreover, owing to its reduced MDA-binding capacity, the H402 variant of CFH displayed significantly impaired cofactor activity compared with the Y402 variant, which may result in the less efficient clearance of MDA-decorated structures and increased inflammation67,107. Interestingly, CFH was reported to slow down efferocytosis in vitro104,108, which is beneficial in circumstances that require decreased release of ROS, nitrogen intermediates and lysosomal enzymes by phagocytes108,109. During this process, additional complement-independent neutralizing functions of CFH and OSE-specific IgM may also be crucial to prevent potential pro-inflammatory effects of the OSEs that have not yet been cleared. For example, CFH blocks MDA-induced CXC-chemokine ligand 8 (CXCL8) and CXCL1 production in vitro and in vivo, respectively67.
Similarly, the MDA-specific IgM LR04 and the PC-specific IgM T15/E06 inhibited microvesicle-induced CXCL8 secretion in human monocytes and the activation of endothelial cells, respectively20,88. These findings provide insights into the important physiological function of OSE-specific immune responses in the recognition and neutralization of cellular debris in the circulation.
In summary, OSEs mark dying cells and their cellular debris and distinguish them from viable and healthy tissues. This enables specific humoral immune responses such as natural IgM antibodies to coordinate the safe disposal of dying cells with a regulated activation of the early steps of the complement cascade. In addition, scavenger receptors that bind to OSEs mediate efferocytosis. However, if the enhanced generation of dying cells exceeds the clearance capacity of available phagocytes, the OSEs present on the accumulating apoptotic cells mediate pro-inflammatory signals and may promote autoimmunity.
Innate sensing of OSEs in disease
During acute inflammatory conditions OSEs can trigger sterile inflammation to ultimately restore tissue integrity. However, under certain conditions, the balance between the generation and the clearance of OSEs by innate immune functions is lost owing to the increased generation of OSEs and/or dysfunctional innate immune responses, which leads to increased or chronic inflammation.
OSEs have been documented in diseased tissues of patients with, for example, infectious and sterile acute lung injury, atherosclerosis, hepatitis, age-related macular degeneration (AMD), multiple sclerosis and Alzheimer disease31. The investigation of these diseases has provided important insights into the functional role of OSE-specific immunity in both acute and chronic inflammatory models.
Role of OSEs in acute inflammation. The OSE CEP transiently accumulates during acute inflammation in a mouse model of wound healing, and induces angiogenesis in endothelial cells in a TLR2–MYD88-dependent manner to stimulate wound healing and to protect against hindlimb ischaemia33,46. In this context, F4/80hi and alternatively activated macrophages scavenge and degrade accumulated CEP in a process that requires binding of both TLR2 and CD36 (Ref. 46). Notably, CEP accumulates in ageing tissues, which may lead to adverse consequences.
OSEs also accumulate in a wide variety of acute situations33,59,110. They accumulate in the lungs of mice following acute lung injury induced by sterile acid aspiration, as well as following infection with the avian influenza virus H5N1 (Ref. 51). The pulmonary lavage fluid of injured mice contains OxPL that stimulates the production of IL-6 in macrophages, which can be ameliorated by the natural IgM antibody T15/E06. Administration of OxPL promoted acute lung injury in mice in a TLR4- and TRIF-dependent manner. In addition to PC-OxPL, MDA adducts also formed in the lungs of these mice51, and intranasal administration of MDA-modified bovine serum albumin (BSA) induced CXCL1 secretion and neutrophil recruitment into the lungs111. This is consistent with the capacity of MDA-adducts to trigger chemokine secretion in macrophages, and further demonstrates the pro-inflammatory potential of OSEs in vivo. Notably, both PC-OxPL and MDA have been documented in the lung tissues of patients who have died of H5N1 and SARS infections and in mice infected with lethal pathogens such as H5N1, Bacillus anthracis, SARS coronavirus and Yersinia pestis51.
Role of OSEs in atherosclerosis. Atherosclerosis is the prototypical chronic inflammatory disease in which the innate immune responses to OSEs are involved (Fig. 3). It is characterized by the deposition of LDL in the intima of large and medium-sized arteries, and the LDL subsequently undergoes oxidation to generate OxLDL. OSEs on OxLDL not only mediate uptake by macrophages to generate foam cells but also mediate many of the chronic inflammatory events that lead to the development of plaques. If plaques rupture, they can trigger thrombus formation, resulting in severe clinical manifestations such as myocardial infarction and stroke112.
Figure 3. Sensing of OSEs in atherosclerosis.

Endothelial cells sense oxidation-specific epitopes (OSEs) present on microvesicles and oxidized low-density lipoprotein (OxLDL)20,114. This results in the expression of adhesion molecules and the secretion of chemoattractants leading to monocyte recruitment to the intima of the artery wall. Macrophages internalize OxLDL via scavenger receptors such as scavenger receptor A1 (SRA1), lectin-like oxidized LDL receptor 1 (LOX1), SRB1 and CD36 (Ref. 37), and in cooperation with Toll-like receptor 2 (TLR2) and TLR4–TLR6 receive pro-inflammatory signals from OSEs. The enhanced uptake of OxLDL via scavenger receptors leads to the excess accumulation of intracellular cholesterol and the formation of lipid-laden foam cells, as well as the secretion of cytokines and chemokines. Excessive free cholesterol accumulation induces cholesterol crystal formation that triggers lysosome rupture and activation of the NOD-, LRR- and pyrin domain-containing protein 3 (NLRP3) inflammasome120 primed by pro-inflammatory OSE-induced signalling. Free intracellular cholesterol also induces endoplasmic reticulum (ER) stress, leading to macrophage apoptosis. As a consequence of this, and impaired efferocytosis, late-stage apoptotic cells accumulate, contributing — together with OxLDL and microvesicles — to a growing pool of OSEs inside the plaque. Natural IgM specific for OSEs blocks scavenger receptor-mediated uptake and neutralizes the pro-inflammatory effects of OSEs by promoting their complement-dependent clearance. Complement factor H (CFH) blocks the pro-inflammatory effects of malondialdehyde (MDA) and facilitates the anti-inflammatory clearance of MDA-decorated surfaces through Factor I-dependent iC3b generation67. The milk fat globule-epidermal growth factor 8 (MFG-E8) is also involved in the clearance of OSEs by recognizing oxidized phosphatidylserine (OxPS). Impaired functions of these humoral immune responses as a result of low abundance or decreased binding capacities, as well as excessive accumulation of OxLDL, microvesicles and apoptotic cells, favours the recognition of OSEs by macrophage receptors, leading to sustained inflammation. CRP, C-reactive protein; CXCL8, CXC-chemokine ligand 8; HNE, 4-hydroxynonenal; IL-1β, interleukin-1β; NF-κB, nuclear factor-κB; PC-OxPL, phosphocholine (PC) of oxidized phospholipid.
PC-OxPL, MDA, 4-HNE, OxCE and CEP have all been documented in atherosclerotic lesions in humans and in animal models of disease46,76,113,114,115. Endothelial cells and macrophages — which express different sets of scavenger receptors and TLRs — are major cellular sensors of OSEs in atherosclerosis. OxPLs induce the expression of chemoattractants, such as CC-chemokine ligand 2 (CCL2; also known as MCP1), fibronectin containing connecting segment 1, CXCL8 and P-selectin, and also trigger monocyte binding to endothelial cells23, which is partly mediated by TLR4 (Ref. 116). 4-HNE also induces ERK1/2 and NF-κB activation in endothelial cells via LOX1 (Ref. 50). LOX1 deficiency results in decreased lesion formation in Ldlr−/− mice117. CEP may also trigger a pro-atherogenic endothelial cell response via TLR2 (Ref. 33). The expression of TLR2 is increased in endothelial cells at sites of disturbed blood flow, and TLR2 deficiency in non-haematopoietic tissues reduces atherosclerosis in Ldlr−/− mice118. Thus, OSE sensing by endothelial cells is a key response in the development of atherosclerosis. Moreover, a recent study demonstrated that CEP accelerates platelet activation and thrombus formation in hyperlipidaemic Apoe−/− mice in a TLR2-dependent manner56, indicating a role for OSEs in triggering the clinical events that result from atherosclerosis.
A hallmark of atherosclerotic lesions is the formation of lipid-laden macrophages, known as foam cells, which occurs because of the enhanced uptake of OxLDL mediated by OSE binding to scavenger receptors, such as CD36 and SRA1 (Ref. 119). The enhanced uptake of cholesterol-rich LDL and subsequent inefficient removal of intracellular cholesterol can lead to a situation of supersaturation of cholesterol, leading to the formation of cholesterol crystals that damage lysosomes and that activate the NOD-, LRR- and pyrin domain-containing protein 3 (NLRP3) inflammasome, resulting in IL-1β secretion120. Indeed, cholesterol loading of macrophages with OxLDL induces IL-1β secretion, and Ldlr−/− mice reconstituted with NLRP3-deficient bone marrow develop less atherosclerosis120. Moreover, deficiency of CD36 and/or SRA1 in Apoe−/− and Ldlr−/− mice, respectively, reduces the size and/or complexity of lesions compared with control mice119. In addition, binding of OxLDL to CD36 induces chemokine expression and inflammasome priming through NF-κB activation, which is dependent on heterotrimer formation with TLR4–TLR6 (Refs 53,121). TLR4 signalling is important, as Tlr4−/−Apoe−/− mice exhibit significantly reduced lesion size122, and clinical studies have found an SNP in TLR4, which results in impaired signalling, to be associated with reduced plaque burden and acute coronary events123,124. These studies demonstrate that sensing of OSEs, and in particular PC-OxPL, by CD36 is central to several pro-atherogenic responses in macrophages. OxLDL, as well as excessive foam cell formation, induces macrophage apoptosis, which is observed in advanced atherosclerotic lesions125. As a result of inefficient efferocytosis, apoptotic macrophages accumulate, which results in increased plaque necrosis and long-term atherosclerotic burden. Several studies demonstrate that deficiencies in certain proteins or receptors that mediate efferocytosis enhance lesion formation in atherosclerosis-prone mice125. Furthermore, the accumulation of apoptotic macrophages within lesions, and their released microvesicles, which all bear OSEs, may compete with both OxLDL and apoptotic cells for clearance by macrophages and thereby further impair efferocytosis.
Humoral immune responses are increasingly being recognized as important modulators of atherogenesis126. Although the ability of CRP to bind OSEs and to mediate apoptotic cell clearance suggests a role in atherosclerosis, there is no real support for an active role of CRP itself in the pathogenesis of the disease from animal or human studies127,128. However, CRP is an excellent biomarker of inflammation, and increased levels of CRP independently predict manifestations of atherosclerosis64. By contrast, natural IgM antibodies clearly modulate atherosclerotic lesion formation, as deficiency in secreted IgM promotes atherosclerosis in Ldlr−/− mice126. As many natural IgM antibodies bind OSEs75, their atheroprotective function may be mostly mediated by OSE-specific IgM, which includes housekeeping mechanisms and other protective effects. For example, T15/E06 IgM neutralizes the pro-inflammatory effects on endothelial cells and macrophages of late apoptotic cells and/or blebs carrying PC-OxPL17,51,88. T15/E06 IgM antibodies have also been shown to block the binding and uptake of OxLDL in macrophages, preventing foam cell formation77. Importantly, raising plasma levels of T15/E06 IgM by immunization with PC-containing pneumococcal extracts or passive infusion decreased atherosclerosis in Ldlr−/− mice and vein graft atherosclerosis in Apoe−/− mice, respectively129,130. In analogy to PC-OxPL, MDA-epitopes also trigger tumour necrosis factor (TNF) and CXCL8 in monocytes and macrophages67. In addition, the numbers of MDA-positive microvesicles substantially increased at the culprit lesion sites of patients suffering from an acute coronary event compared with the number found in peripheral circulation, which could further propagate inflammation20. Thus, OSE-specific IgM can protect by neutralizing the pro-inflammatory effects of OSEs in the context of OxLDL, microvesicles and apoptotic cells, and can promote the anti-inflammatory clearance of cellular debris. In line with this, several clinical studies have shown that lower levels of IgM antibodies directed against MDA-LDL and OxLDL are linked to an increased risk of cardiovascular disease (CVD)126.
Our recent identification of CFH as a major MDA-binding protein in plasma also suggests its potential involvement in atherosclerosis67. CFH is present in human atherosclerotic lesions, where it localizes to areas rich in MDA-epitopes67,131. Although several studies have reported an association of the 402H allele — which decreases binding of CFH to MDA — with an increased risk of heart disease and stroke, a meta-analysis of eight different study populations failed to find a significant association of this gene variant with CVD132. However, to fully understand this function of CFH in CVD, the net contribution of all SNPs that potentially affect MDA binding needs to be explored.
These data support an important role for immune responses that target OSEs in atherogenesis. OxLDL and cellular debris contribute to an excessive accumulation of OSEs, which cannot be adequately targeted by beneficial clearance mechanisms and, consequently, pro-inflammatory responses in macrophages and endothelial cells are sustained. Similarly, OSEs may promote non-alcoholic steatohepatitis133, which is increasingly being recognized as a risk factor for CVD. Studies in cholesterol-fed Ldlr−/− mice indicate that the expression of CD36 and SRA1 in Kupffer cells mediates hepatic inflammation134, whereas inducing plasma levels of the T15/E06 IgM is protective135. Consistent with these data, we recently showed that deficiency of sialic acid-binding immunoglobulin-type lectin G (Siglec-G), a negative regulator of B-1 cell function, results in increased levels of OSE-specific IgM and reduced atherosclerosis and hepatic inflammation in cholesterol-fed Ldlr−/− mice136.
OSEs in AMD. OSEs have also been implicated in AMD137, which is characterized by the accumulation of cellular waste (drusen) in the retina. Several OSEs, including PC-OxPLs, and MDA- and CEP-modified proteins, have been identified as drusen components and have been shown to trigger sterile inflammation138. For example, CEP-modified proteins induce pro-inflammatory gene expression and prime the NLRP3 inflammasome in monocytes via TLR2 (Refs 55,139). Drusen extracts isolated from AMD lesions activate the NLRP3 inflammasome, resulting in the secretion of IL-1β and IL-18 (Ref. 139). Similarly, MDA-modified proteins induce the expression of CXCL8 and CXCL1 in RPE cells in in vitro and in vivo models, respectively67. Dysregulation of complement represents another major pathological driver of AMD, and genome-wide association studies have identified SNPs in genes encoding complement proteins as genetic risk factors of AMD69,137. Among these, the SNP rs1061170 in CFH predisposes to increased risk for AMD140,141. Necrotic RPE cells and blebs thereof display MDA-epitopes that are recognized by CFH, which in turn facilitates the generation of iC3b for opsonization and anti-inflammatory removal. Compared with the Y402 CFH variant, the cofactor activity on MDA-decorated surfaces is severely impaired when the 402H CFH variant is used, demonstrating a functional consequence. As a result, MDA may accumulate and promote inflammation67. Indeed, transgenic mice that express the human 402H variant have increased susceptibility to oxidative stress-mediated injury in the retina as a result of MDA accumulation and increased inflammation142.
Conclusions and future directions
The recognition of OSEs by cellular and humoral immune responses enables the immune system to mediate important housekeeping functions, such as the removal of dying cells, cellular debris and damaged molecules. In situations of increased oxidative stress, the generation and burden of OSEs is greatly increased. As a consequence, OSEs are sensed by cellular signalling receptors, leading to the secretion of chemokines and pro-inflammatory cytokines. Thus, OSEs represent a unique class of DAMPs, which both mediate the recognition of cellular debris for housekeeping functions and, under certain circumstances, act as pro-inflammatory danger signals themselves. When this fine balance between the presence of OSEs and the capacities of the immune system to survey and respond to them is lost owing to the excessive generation of OSEs and/or their impaired removal, the development and propagation of chronic inflammatory diseases become manifest.
Although there have been a few attempts to treat patients with antioxidants to reduce the progression of CVD, these have not been successful143. These very limited studies have been criticized as premature as we currently lack sufficient knowledge to adequately design an effective therapeutic trial to test the role of oxidation in atherogenesis26. As discussed in this Review, oxidative events and the associated immune responses have beneficial effects as well as adverse consequences and we currently lack insight into the relative importance of these in vivo, which could affect the proper design of antioxidant trials.
A better understanding of the role of OSEs and OSE-reactive immune responses in the maintenance of homeostasis and in controlling inflammation are important areas of future investigations. Moreover, it is important to elucidate how different components of OSE-specific immune responses cooperate at a functional level. The identification of the genetic variants that affect some of these responses, and mechanistic studies characterizing these, may provide exciting insights into different individual predispositions to responding to oxidative stress and to developing chronic inflammation.
Acknowledgements
The work carried out in the authors' laboratories related to this Review is supported by the Austrian Science Fund (SFB-30 and SFB-54 to C.J.B.) and the US National Institutes of Health (grants HL086559, HL119828 and HL055798 to J.L.W.). The authors thank V. Krajina for help with the illustrations.
Glossary
- Hormesis
A dose–response phenomenon that is characterized by beneficial effects at low doses and toxicity at high doses.
- Oxidation-specific epitopes
(OSEs). Novel epitopes that form as a result of lipid peroxidation in response to various physiological and pathological situations. These epitopes are enriched on oxidized low-density lipoprotein and on membranes of microvesicles and apoptotic cells. They are recognized by humoral and cellular immune responses.
- Microvesicles
Small (0.1–1 μm) membrane vesicles shed from activated or dying cells. They expose phosphatidylserine and carry surface markers of their parental cells. They mediate the transfer of proteins, nucleic acids and lipids between cells. Elevated levels of circulating microvesicles have been reported in acute and chronic inflammatory diseases.
- Oxidized low-density lipoproteins
(OxLDLs). Low-density lipoprotein (LDL) particles (20–25 nm) are the major transporters of cholesterol in the circulation. LDL particles consist of 45% cholesterol, 20% phospholipids, 10% triglycerides and 25% protein. LDL can be oxidatively modified by enzymatic and non-enzymatic mechanisms that result in many different changes to the lipid and protein moiety of LDL. Unlike LDL, OxLDL is immunogenic and pro-inflammatory, and can be recognized by macrophage scavenger receptors.
- Damage-associated molecular patterns
(DAMPs). Intracellular and extracellular components that are typically hidden from recognition by the immune system until cellular damage occurs. Once released into the extracellular environment they trigger sterile inflammation. Prototypical examples include high mobility group box 1 protein (HMGB1), heat shock proteins (HSPs), uric acid, mitochondrial proteins, DNA, RNA and cholesterol crystals.
- 2-thiobarbituric acid reaction
(TBAR). The TBAR method is used to measure lipid peroxidation products, such as malondialdehyde (MDA) or other reactive aldehydes, as these products generate thiobarbituric acid-reactive substances that can be detected by colorimetric or fluorometric measurements.
- Natural IgM antibodies
Found in individuals that have not had any previous known exposure to the antigen recognized by the antibodies. They are mainly of the IgM isotype, have not undergone somatic mutations and have reactivity for many microbial pathogens and self antigens.
- Scavenger receptors
Cell-membrane proteins that take up oxidatively or otherwise modified low-density lipoproteins and mediate their clearance by macrophages and other cell types. They have subsequently been shown to bind a large variety of different modified proteins as well as pathogens.
- C-reactive protein
(CRP). A highly conserved pentraxin that is induced and rapidly secreted by the liver in response to bacterial infections. Increased plasma CRP levels are also found in a wide variety of other non-infectious chronic inflammatory states.
- Complement factor H
(CFH). A soluble glycoprotein (155 kDa) that is made up of 20 short consensus repeats (SCRs) organized in a 'bead on the string' manner. It is the major inhibitor of the alternative complement pathway and is highly abundant in plasma at steady-state levels of 400–700 μg/ml.
- Milk fat globule-epidermal growth factor 8
(MFG-E8). A ∼53 kDa secreted glycoprotein with versatile functions, including binding to exposed phosphatidylserine to mediate the clearance of apoptotic cells via the αvβ3 and αvβ5 integrin receptors of phagocytic cells.
- Efferocytosis
The multi-step process by which dying cells are cleared by phagocytic cells, which is initiated by recognition signals exposed on apoptotic cells recognized by soluble receptors or membrane-bound receptors expressed on phagocytes, resulting in their engulfment and removal.
- Opsonized
The deposition of opsonins on the surface of dying cells or invading pathogens to enhance their clearance by specific receptors expressed on the surface of leukocytes. Specific opsonins not only enhance uptake but also determine whether the uptake has pro- or anti-inflammatory consequences.
- Age-related macular degeneration
(AMD). A condition predominantly found in the elderly that is caused by the accumulation of cellular debris (drusen) between the Bruch's membrane and the retinal pigment epithelium and by the loss of photoreceptors. AMD is divided in to a dry form resulting from atrophy and a wet form resulting from abnormal vascularization of the retina.
- Foam cells
Cholesterol-laden macrophages with a foamy appearance that typify the early atherosclerotic lesion. The excess cholesterol is believed to occur because of unregulated and excessive uptake of modified low-density lipoprotein (LDL), such as oxidized LDL via scavenger receptors. They also secrete pro-inflammatory cytokines and promote atherosclerotic lesion formation and are hallmark cells of atherosclerotic plaques.
- Non-alcoholic steatohepatitis
Characterized by lipid accumulation in the liver and inflammation, it represents a critical stage in the progression of non-alcoholic fatty liver disease to more severe potentially life-threatening conditions that are associated with liver cirrhosis and end-stage liver disease.
Biographies
Christoph J. Binder received his M.D. degree from the University of Vienna, Austria, and his Ph.D. degree from the University of California San Diego, USA. Following additional postdoctoral training with Dr Joseph Witztum in San Diego, he returned to the Medical University of Vienna to establish his research group and to specialize in laboratory medicine. He is currently Professor of Atherosclerosis Research at the Medical University of Vienna and Principal Investigator at the Research Center for Molecular Medicine of the Austrian Academy of Sciences. His research focuses on immune mechanisms of atherosclerosis and the role of natural IgM antibodies and humoral immunity targeting oxidation-specific epitopes in health and disease. Christoph J. Binder's homepage.
Nikolina Papac-Milicevic received her Ph.D. from the University of Vienna, Austria, under the supervision of Bernd R. Binder. Her graduate research focused on the role of nuclear receptor NRA41 in vascular biology and vascular inflammation. She is currently a staff scientist at the Medical University of Vienna in Christoph J. Binder's laboratory with a focus on homeostatic functions of the complement system and humoral immunity in health and chronic inflammation.
Joseph L. Witztum received an M.D. from Washington University, St. Louis, USA, followed by a medicine residency at Mt. Sinai Hospital, New York City, USA, and an endocrinology/metabolism fellowship at Washington University. He subsequently joined the Endocrine/Metabolism Division in the Department of Medicine at the University California San Diego, USA, in 1979, where he is currently a Distinguished Professor of Medicine. Together with Dr Daniel Steinberg, he developed the concept that oxidation of low-density lipoprotein (LDL) was a central mechanism in atherogenesis and that innate and adaptive immune responses to oxidation-specific epitopes were important mediators in converting atherosclerosis into a chronic inflammatory disease.
PowerPoint slides
Competing interests
J.L.W. is an inventor and receives royalties from patents or patent applications owned by the University of California San Diego, USA, on oxidation-specific antibodies.
References
- 1.Schieber M, Chandel NS. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014;24:R453–R462. doi: 10.1016/j.cub.2014.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Savina A, et al. NOX2 controls phagosomal pH to regulate antigen processing during crosspresentation by dendritic cells. Cell. 2006;126:205–218. doi: 10.1016/j.cell.2006.05.035. [DOI] [PubMed] [Google Scholar]
- 3.Nathan C, Cunningham-Bussel A. Beyond oxidative stress: an immunologist's guide to reactive oxygen species. Nat. Rev. Immunol. 2013;13:349–361. doi: 10.1038/nri3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature. 2000;408:239–247. doi: 10.1038/35041687. [DOI] [PubMed] [Google Scholar]
- 5.Klebanoff SJ. Myeloperoxidase: friend and foe. J. Leukoc. Biol. 2005;77:598–625. doi: 10.1189/jlb.1204697. [DOI] [PubMed] [Google Scholar]
- 6.Almyroudis NG, et al. NETosis and NADPH oxidase: at the intersection of host defense, inflammation, and injury. Front. Immunol. 2013;4:45. doi: 10.3389/fimmu.2013.00045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bae YS, Oh H, Rhee SG, Yoo YD. Regulation of reactive oxygen species generation in cell signaling. Mol. Cells. 2011;32:491–509. doi: 10.1007/s10059-011-0276-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Esterbauer H, Schaur RJ, Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic. Biol. Med. 1991;11:81–128. doi: 10.1016/0891-5849(91)90192-6. [DOI] [PubMed] [Google Scholar]
- 9.Cyster JG, Dang EV, Reboldi A, Yi T. 25-Hydroxycholesterols in innate and adaptive immunity. Nat. Rev. Immunol. 2014;14:731–743. doi: 10.1038/nri3755. [DOI] [PubMed] [Google Scholar]
- 10.Spann NJ, Glass CK. Sterols and oxysterols in immune cell function. Nat. Immunol. 2013;14:893–900. doi: 10.1038/ni.2681. [DOI] [PubMed] [Google Scholar]
- 11.Bauer J, et al. Pathophysiology of isoprostanes in the cardiovascular system: implications of isoprostane-mediated thromboxane A2 receptor activation. Br. J. Pharmacol. 2014;171:3115–3131. doi: 10.1111/bph.12677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Serhan CN. Pro-resolving lipid mediators are leads for resolution physiology. Nature. 2014;510:92–101. doi: 10.1038/nature13479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rothe T, et al. 12/15-Lipoxygenase-mediated enzymatic lipid oxidation regulates DC maturation and function. J. Clin. Invest. 2015;125:1944–1954. doi: 10.1172/JCI78490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miller YI, et al. Oxidation-specific epitopes are danger-associated molecular patterns recognized by pattern recognition receptors of innate immunity. Circ. Res. 2011;108:235–248. doi: 10.1161/CIRCRESAHA.110.223875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kawamoto Y, et al. Cyclopentenone prostaglandins as potential inducers of phase II detoxification enzymes. 15-deoxy-Δ(12,14)-prostaglandin j2-induced expression of glutathione S-transferases. J. Biol. Chem. 2000;275:11291–11299. doi: 10.1074/jbc.275.15.11291. [DOI] [PubMed] [Google Scholar]
- 16.Clark SR, et al. Characterization of platelet aminophospholipid externalization reveals fatty acids as molecular determinants that regulate coagulation. Proc. Natl Acad. Sci. USA. 2013;110:5875–5880. doi: 10.1073/pnas.1222419110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chang MK, et al. Apoptotic cells with oxidation-specific epitopes are immunogenic and proinflammatory. J. Exp. Med. 2004;200:1359–1370. doi: 10.1084/jem.20031763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chang MK, et al. Monoclonal antibodies against oxidized low-density lipoprotein bind to apoptotic cells and inhibit their phagocytosis by elicited macrophages: evidence that oxidation-specific epitopes mediate macrophage recognition. Proc. Natl Acad. Sci. USA. 1999;96:6353–6358. doi: 10.1073/pnas.96.11.6353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chou MY, et al. Oxidation-specific epitopes are important targets of innate immunity. J. Intern. Med. 2008;263:479–488. doi: 10.1111/j.1365-2796.2008.01968.x. [DOI] [PubMed] [Google Scholar]
- 20.Tsiantoulas D, et al. Circulating microparticles carry oxidation-specific epitopes and are recognized by natural IgM antibodies. J. Lipid. Res. 2015;56:440–448. doi: 10.1194/jlr.P054569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gallucci S, Matzinger P. Danger signals: SOS to the immune system. Curr. Opin. Immunol. 2001;13:114–119. doi: 10.1016/S0952-7915(00)00191-6. [DOI] [PubMed] [Google Scholar]
- 22.Chen GY, Nunez G. Sterile inflammation: sensing and reacting to damage. Nat. Rev. Immunol. 2010;10:826–837. doi: 10.1038/nri2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bochkov VN, et al. Generation and biological activities of oxidized phospholipids. Antioxid. Redox Signal. 2010;12:1009–1059. doi: 10.1089/ars.2009.2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Salomon RG. Structural identification and cardiovascular activities of oxidized phospholipids. Circ. Res. 2012;111:930–946. doi: 10.1161/CIRCRESAHA.112.275388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Harkewicz R, et al. Cholesteryl ester hydroperoxides are biologically active components of minimally oxidized low density lipoprotein. J. Biol. Chem. 2008;283:10241–10251. doi: 10.1074/jbc.M709006200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Steinberg D, Witztum JL. Oxidized low-density lipoprotein and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2010;30:2311–2316. doi: 10.1161/ATVBAHA.108.179697. [DOI] [PubMed] [Google Scholar]
- 27.O'Donnell VB, Murphy RC. New families of bioactive oxidized phospholipids generated by immune cells: identification and signaling actions. Blood. 2012;120:1985–1992. doi: 10.1182/blood-2012-04-402826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee S, et al. Role of phospholipid oxidation products in atherosclerosis. Circ. Res. 2012;111:778–799. doi: 10.1161/CIRCRESAHA.111.256859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fadeel B, Xue D, Kagan V. Programmed cell clearance: molecular regulation of the elimination of apoptotic cell corpses and its role in the resolution of inflammation. Biochem. Biophys. Res. Commun. 2010;396:7–10. doi: 10.1016/j.bbrc.2010.02.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Salomon RG, Hong L, Hollyfield JG. Discovery of carboxyethylpyrroles (CEPs): critical insights into AMD, autism, cancer, and wound healing from basic research on the chemistry of oxidized phospholipids. Chem. Res. Toxicol. 2011;24:1803–1816. doi: 10.1021/tx200206v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Weismann D, Binder CJ. The innate immune response to products of phospholipid peroxidation. Biochim. Biophys. Acta. 2012;1818:2465–2475. doi: 10.1016/j.bbamem.2012.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Harnett W, Harnett MM. Phosphorylcholine: friend or foe of the immune system? Immunol. Today. 1999;20:125–129. doi: 10.1016/S0167-5699(98)01419-4. [DOI] [PubMed] [Google Scholar]
- 33.West XZ, et al. Oxidative stress induces angiogenesis by activating TLR2 with novel endogenous ligands. Nature. 2010;467:972–976. doi: 10.1038/nature09421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang H, et al. 4-Hydroxy-7-oxo-5-heptenoic acid (HOHA) lactone is a biologically active precursor for the generation of 2-(ωa-Carboxyethyl)pyrrole (CEP) derivatives of proteins and ethanolamine phospholipids. Chem. Res. Toxicol. 2015;28:967–977. doi: 10.1021/acs.chemrestox.5b00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Choi SH, et al. Polyoxygenated cholesterol ester hydroperoxide activates TLR4 and SYK dependent signaling in macrophages. PLoS ONE. 2013;8:e83145. doi: 10.1371/journal.pone.0083145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Witztum JL, Lichtman AH. The influence of innate and adaptive immune responses on atherosclerosis. Annu. Rev. Pathol. 2014;9:73–102. doi: 10.1146/annurev-pathol-020712-163936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Greaves DR, Gordon S. The macrophage scavenger receptor at 30 years of age: current knowledge and future challenges. J. Lipid Res. 2009;50:S282–S286. doi: 10.1194/jlr.R800066-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kunjathoor VV, et al. Scavenger receptors class A-I/II and CD36 are the principal receptors responsible for the uptake of modified low density lipoprotein leading to lipid loading in macrophages. J. Biol. Chem. 2002;277:49982–49988. doi: 10.1074/jbc.M209649200. [DOI] [PubMed] [Google Scholar]
- 39.Boullier A, et al. The binding of oxidized low density lipoprotein to mouse CD36 is mediated in part by oxidized phospholipids that are associated with both the lipid and protein moieties of the lipoprotein. J. Biol. Chem. 2000;275:9163–9169. doi: 10.1074/jbc.275.13.9163. [DOI] [PubMed] [Google Scholar]
- 40.Boullier A, et al. Phosphocholine as a pattern recognition ligand for CD36. J. Lipid Res. 2005;46:969–976. doi: 10.1194/jlr.M400496-JLR200. [DOI] [PubMed] [Google Scholar]
- 41.Li XM, Salomon RG, Qin J, Hazen SL. Conformation of an endogenous ligand in a membrane bilayer for the macrophage scavenger receptor CD36. Biochemistry. 2007;46:5009–5017. doi: 10.1021/bi700163y. [DOI] [PubMed] [Google Scholar]
- 42.Greenberg ME, et al. The lipid whisker model of the structure of oxidized cell membranes. J. Biol. Chem. 2008;283:2385–2396. doi: 10.1074/jbc.M707348200. [DOI] [PubMed] [Google Scholar]
- 43.Fadok VA, Warner ML, Bratton DL, Henson PM. CD36 is required for phagocytosis of apoptotic cells by human macrophages that use either a phosphatidylserine receptor or the vitronectin receptor (αvβ3) J. Immunol. 1998;161:6250–6257. [PubMed] [Google Scholar]
- 44.Greenberg ME, et al. Oxidized phosphatidylserine–CD36 interactions play an essential role in macrophage-dependent phagocytosis of apoptotic cells. J. Exp. Med. 2006;203:2613–2625. doi: 10.1084/jem.20060370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gillotte-Taylor K, Boullier A, Witztum JL, Steinberg D, Quehenberger O. Scavenger receptor class B type I as a receptor for oxidized low density lipoprotein. J. Lipid Res. 2001;42:1474–1482. [PubMed] [Google Scholar]
- 46.Kim YW, et al. Receptor-mediated mechanism controlling tissue levels of bioactive lipid oxidation products. Circ. Res. 2015;117:321–332. doi: 10.1161/CIRCRESAHA.117.305925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Duryee MJ, et al. Scavenger receptors on sinusoidal liver endothelial cells are involved in the uptake of aldehyde-modified proteins. Mol. Pharmacol. 2005;68:1423–1430. doi: 10.1124/mol.105.016121. [DOI] [PubMed] [Google Scholar]
- 48.Shechter I, et al. The metabolism of native and malondialdehyde-altered low density lipoproteins by human monocyte-macrophages. J. Lipid Res. 1981;22:63–71. [PubMed] [Google Scholar]
- 49.Besler C, et al. Mechanisms underlying adverse effects of HDL on eNOS-activating pathways in patients with coronary artery disease. J. Clin. Invest. 2011;121:2693–2708. doi: 10.1172/JCI42946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kumano-Kuramochi M, et al. Identification of 4-hydroxy-2-nonenal-histidine adducts that serve as ligands for human lectin-like oxidized LDL receptor-1. Biochem. J. 2012;442:171–180. doi: 10.1042/BJ20111029. [DOI] [PubMed] [Google Scholar]
- 51.Imai Y, et al. Identification of oxidative stress and Toll-like receptor 4 signaling as a key pathway of acute lung injury. Cell. 2008;133:235–249. doi: 10.1016/j.cell.2008.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kadl A, et al. Oxidized phospholipid-induced inflammation is mediated by Toll-like receptor 2. Free Radic. Biol. Med. 2011;51:1903–1909. doi: 10.1016/j.freeradbiomed.2011.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stewart CR, et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat. Immunol. 2010;11:155–161. doi: 10.1038/ni.1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Seimon TA, et al. Atherogenic lipids and lipoproteins trigger CD36–TLR2-dependent apoptosis in macrophages undergoing endoplasmic reticulum stress. Cell. Metab. 2010;12:467–482. doi: 10.1016/j.cmet.2010.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Saeed AM, et al. The oxidative stress product carboxyethylpyrrole potentiates TLR2/TLR1 inflammatory signaling in macrophages. PLoS ONE. 2014;9:e106421. doi: 10.1371/journal.pone.0106421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Biswas, S. et al. Novel phosphatidylethanolamine derivatives accumulate in circulation in hyperlipidemic ApoE−/− mice and activate platelets via TLR2. Blood10.1182/blood-2015-08-664300 (2016). [DOI] [PMC free article] [PubMed]
- 57.Gargiulo S, et al. Relation between TLR4/NF-κB signaling pathway activation by 27-hydroxycholesterol and 4-hydroxynonenal, and atherosclerotic plaque instability. Aging Cell. 2015;14:569–581. doi: 10.1111/acel.12322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Choi SH, et al. Lipoprotein accumulation in macrophages via Toll-like receptor-4-dependent fluid phase uptake. Circ. Res. 2009;104:1355–1363. doi: 10.1161/CIRCRESAHA.108.192880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bochkov VN, et al. Protective role of phospholipid oxidation products in endotoxin-induced tissue damage. Nature. 2002;419:77–81. doi: 10.1038/nature01023. [DOI] [PubMed] [Google Scholar]
- 60.Bretscher P, et al. Phospholipid oxidation generates potent anti-inflammatory lipid mediators that mimic structurally related pro-resolving eicosanoids by activating Nrf2. EMBO Mol. Med. 2015;7:593–607. doi: 10.15252/emmm.201404702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Litvack ML, Palaniyar N. Review: soluble innate immune pattern-recognition proteins for clearing dying cells and cellular components: implications on exacerbating or resolving inflammation. Innate Immun. 2010;16:191–200. doi: 10.1177/1753425910369271. [DOI] [PubMed] [Google Scholar]
- 62.Volanakis JE, Kaplan MH. Specificity of C-reactive protein for choline phosphate residues of pneumococcal C-polysaccharide. Proc. Soc. Exp. Biol. Med. 1971;136:612–614. doi: 10.3181/00379727-136-35323. [DOI] [PubMed] [Google Scholar]
- 63.Volanakis JE. Human C-reactive protein: expression, structure, and function. Mol. Immunol. 2001;38:189–197. doi: 10.1016/S0161-5890(01)00042-6. [DOI] [PubMed] [Google Scholar]
- 64.Verma S, Szmitko PE, Ridker PM. C-reactive protein comes of age. Nat. Clin. Pract. Cardiovasc. Med. 2005;2:29–36. doi: 10.1038/ncpcardio0074. [DOI] [PubMed] [Google Scholar]
- 65.Chang MK, Binder CJ, Torzewski M, Witztum JL. C-reactive protein binds to both oxidized LDL and apoptotic cells through recognition of a common ligand: phosphorylcholine of oxidized phospholipids. Proc. Natl Acad. Sci. USA. 2002;99:13043–13048. doi: 10.1073/pnas.192399699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Turunen SP, et al. Recognition of Porphyromonas gingivalis gingipain epitopes by natural IgM binding to malondialdehyde modified low-density lipoprotein. PLoS ONE. 2012;7:e34910. doi: 10.1371/journal.pone.0034910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Weismann D, et al. Complement factor H binds malondialdehyde epitopes and protects from oxidative stress. Nature. 2011;478:76–81. doi: 10.1038/nature10449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zipfel PF, Skerka C. Complement regulators and inhibitory proteins. Nat. Rev. Immunol. 2009;9:729–740. doi: 10.1038/nri2620. [DOI] [PubMed] [Google Scholar]
- 69.Zipfel PF, Heinen S, Jozsi M, Skerka C. Complement and diseases: defective alternative pathway control results in kidney and eye diseases. Mol. Immunol. 2006;43:97–106. doi: 10.1016/j.molimm.2005.06.015. [DOI] [PubMed] [Google Scholar]
- 70.Hyvarinen S, Uchida K, Varjosalo M, Jokela R, Jokiranta TS. Recognition of malondialdehyde-modified proteins by the C terminus of complement factor H is mediated via the polyanion binding site and impaired by mutations found in atypical hemolytic uremic syndrome. J. Biol. Chem. 2014;289:4295–4306. doi: 10.1074/jbc.M113.527416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Veneskoski M, et al. Specific recognition of malondialdehyde and malondialdehyde acetaldehyde adducts on oxidized LDL and apoptotic cells by complement anaphylatoxin C3a. Free Radic. Biol. Med. 2011;51:834–843. doi: 10.1016/j.freeradbiomed.2011.05.029. [DOI] [PubMed] [Google Scholar]
- 72.Borisenko GG, Iverson SL, Ahlberg S, Kagan VE, Fadeel B. Milk fat globule epidermal growth factor 8 (MFG-E8) binds to oxidized phosphatidylserine: implications for macrophage clearance of apoptotic cells. Cell Death Differ. 2004;11:943–945. doi: 10.1038/sj.cdd.4401421. [DOI] [PubMed] [Google Scholar]
- 73.Uderhardt S, et al. 12/15-lipoxygenase orchestrates the clearance of apoptotic cells and maintains immunologic tolerance. Immunity. 2012;36:834–846. doi: 10.1016/j.immuni.2012.03.010. [DOI] [PubMed] [Google Scholar]
- 74.Wan M, et al. Oxidized but not native cardiolipin has pro-inflammatory effects, which are inhibited by Annexin A5. Atherosclerosis. 2014;235:592–598. doi: 10.1016/j.atherosclerosis.2014.05.913. [DOI] [PubMed] [Google Scholar]
- 75.Chou MY, et al. Oxidation-specific epitopes are dominant targets of innate natural antibodies in mice and humans. J. Clin. Invest. 2009;119:1335–1349. doi: 10.1172/JCI36800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Shaw PX, et al. Natural antibodies with the T15 idiotype may act in atherosclerosis, apoptotic clearance, and protective immunity. J. Clin. Invest. 2000;105:1731–1740. doi: 10.1172/JCI8472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Horkko S, et al. Monoclonal autoantibodies specific for oxidized phospholipids or oxidized phospholipid-protein adducts inhibit macrophage uptake of oxidized low-density lipoproteins. J. Clin. Invest. 1999;103:117–128. doi: 10.1172/JCI4533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Palinski W, et al. Cloning of monoclonal autoantibodies to epitopes of oxidized lipoproteins from apolipoprotein E-deficient mice. Demonstration of epitopes of oxidized low density lipoprotein in human plasma. J. Clin. Invest. 1996;98:800–814. doi: 10.1172/JCI118853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Potter M, Lieberman R. Common individual antigenic determinants in five of eight BALB-c IgA myeloma proteins that bind phosphoryl choline. J. Exp. Med. 1970;132:737–751. doi: 10.1084/jem.132.4.737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Briles DE, Forman C, Hudak S, Claflin JL. Anti-phosphorylcholine antibodies of the T15 idiotype are optimally protective against Streptococcus pneumoniae. J. Exp. Med. 1982;156:1177–1185. doi: 10.1084/jem.156.4.1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Amir S, et al. Peptide mimotopes of malondialdehyde epitopes for clinical applications in cardiovascular disease. J. Lipid Res. 2012;53:1316–1326. doi: 10.1194/jlr.M025445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tuominen A, et al. A natural antibody to oxidized cardiolipin binds to oxidized low-density lipoprotein, apoptotic cells, and atherosclerotic lesions. Arterioscler. Thromb. Vasc. Biol. 2006;26:2096–2102. doi: 10.1161/01.ATV.0000233333.07991.4a. [DOI] [PubMed] [Google Scholar]
- 83.Arandjelovic S, Ravichandran KS. Phagocytosis of apoptotic cells in homeostasis. Nat. Immunol. 2015;16:907–917. doi: 10.1038/ni.3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Poon IK, Lucas CD, Rossi AG, Ravichandran KS. Apoptotic cell clearance: basic biology and therapeutic potential. Nat. Rev. Immunol. 2014;14:166–180. doi: 10.1038/nri3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ravichandran KS. Beginnings of a good apoptotic meal: the find-me and eat-me signaling pathways. Immunity. 2011;35:445–455. doi: 10.1016/j.immuni.2011.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chen Y, et al. Regulation of dendritic cells and macrophages by an anti-apoptotic cell natural antibody that suppresses TLR responses and inhibits inflammatory arthritis. J. Immunol. 2009;183:1346–1359. doi: 10.4049/jimmunol.0900948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chen Y, Park YB, Patel E, Silverman GJ. IgM antibodies to apoptosis-associated determinants recruit C1q and enhance dendritic cell phagocytosis of apoptotic cells. J. Immunol. 2009;182:6031–6043. doi: 10.4049/jimmunol.0804191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Huber J, et al. Oxidized membrane vesicles and blebs from apoptotic cells contain biologically active oxidized phospholipids that induce monocyte-endothelial interactions. Arterioscler. Thromb. Vasc. Biol. 2002;22:101–107. doi: 10.1161/hq0102.101525. [DOI] [PubMed] [Google Scholar]
- 89.Liu ML, Scalia R, Mehta JL, Williams KJ. Cholesterol-induced membrane microvesicles as novel carriers of damage-associated molecular patterns: mechanisms of formation, action, and detoxification. Arterioscler. Thromb. Vasc. Biol. 2012;32:2113–2121. doi: 10.1161/ATVBAHA.112.255471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Rautou PE, et al. Microparticles, vascular function, and atherothrombosis. Circ. Res. 2011;109:593–606. doi: 10.1161/CIRCRESAHA.110.233163. [DOI] [PubMed] [Google Scholar]
- 91.Biro E, et al. Activated complement components and complement activator molecules on the surface of cell-derived microparticles in patients with rheumatoid arthritis and healthy individuals. Ann. Rheum. Dis. 2007;66:1085–1092. doi: 10.1136/ard.2006.061309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Linkermann A, Stockwell BR, Krautwald S, Anders HJ. Regulated cell death and inflammation: an auto-amplification loop causes organ failure. Nat. Rev. Immunol. 2014;14:759–767. doi: 10.1038/nri3743. [DOI] [PubMed] [Google Scholar]
- 93.Duncan KG, Bailey KR, Kane JP, Schwartz DM. Human retinal pigment epithelial cells express scavenger receptors BI and BII. Biochem. Biophys. Res. Commun. 2002;292:1017–1022. doi: 10.1006/bbrc.2002.6756. [DOI] [PubMed] [Google Scholar]
- 94.Sparrow JR, Ryeom SW, Abumrad NA, Ibrahimi A, Silverstein RL. CD36 expression is altered in retinal pigment epithelial cells of the RCS rat. Exp. Eye Res. 1997;64:45–56. doi: 10.1006/exer.1996.0177. [DOI] [PubMed] [Google Scholar]
- 95.Ryeom SW, Silverstein RL, Scotto A, Sparrow JR. Binding of anionic phospholipids to retinal pigment epithelium may be mediated by the scavenger receptor CD36. J. Biol. Chem. 1996;271:20536–20539. doi: 10.1074/jbc.271.34.20536. [DOI] [PubMed] [Google Scholar]
- 96.Sun M, et al. Light-induced oxidation of photoreceptor outer segment phospholipids generates ligands for CD36-mediated phagocytosis by retinal pigment epithelium: a potential mechanism for modulating outer segment phagocytosis under oxidant stress conditions. J. Biol. Chem. 2006;281:4222–4230. doi: 10.1074/jbc.M509769200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Nandrot EF, et al. Essential role for MFG-E8 as ligand for αvβ5 integrin in diurnal retinal phagocytosis. Proc. Natl Acad. Sci. USA. 2007;104:12005–12010. doi: 10.1073/pnas.0704756104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Peng Y, Kowalewski R, Kim S, Elkon KB. The role of IgM antibodies in the recognition and clearance of apoptotic cells. Mol. Immunol. 2005;42:781–787. doi: 10.1016/j.molimm.2004.07.045. [DOI] [PubMed] [Google Scholar]
- 99.Taylor PR, et al. A hierarchical role for classical pathway complement proteins in the clearance of apoptotic cells in vivo. J. Exp. Med. 2000;192:359–366. doi: 10.1084/jem.192.3.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Boes M, et al. Accelerated development of IgG autoantibodies and autoimmune disease in the absence of secreted IgM. Proc. Natl Acad. Sci. USA. 2000;97:1184–1189. doi: 10.1073/pnas.97.3.1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ogden CA, Kowalewski R, Peng Y, Montenegro V, Elkon KB. IGM is required for efficient complement mediated phagocytosis of apoptotic cells in vivo. Autoimmunity. 2005;38:259–264. doi: 10.1080/08916930500124452. [DOI] [PubMed] [Google Scholar]
- 102.Wang G, Li H, Firoze Khan M. Differential oxidative modification of proteins in MRL+/+ and MRL/lpr mice: Increased formation of lipid peroxidation-derived aldehyde-protein adducts may contribute to accelerated onset of autoimmune response. Free Radic. Res. 2012;46:1472–1481. doi: 10.3109/10715762.2012.727209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Gershov D, Kim S, Brot N, Elkon KB. C-Reactive protein binds to apoptotic cells, protects the cells from assembly of the terminal complement components, and sustains an antiinflammatory innate immune response: implications for systemic autoimmunity. J. Exp. Med. 2000;192:1353–1364. doi: 10.1084/jem.192.9.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Trouw LA, et al. C4b-binding protein and factor H compensate for the loss of membrane-bound complement inhibitors to protect apoptotic cells against excessive complement attack. J. Biol. Chem. 2007;282:28540–28548. doi: 10.1074/jbc.M704354200. [DOI] [PubMed] [Google Scholar]
- 105.Trouw LA, Blom AM, Gasque P. Role of complement and complement regulators in the removal of apoptotic cells. Mol. Immunol. 2008;45:1199–1207. doi: 10.1016/j.molimm.2007.09.008. [DOI] [PubMed] [Google Scholar]
- 106.Amarilyo G, et al. iC3b-opsonized apoptotic cells mediate a distinct anti-inflammatory response and transcriptional NF-κB-dependent blockade. Eur. J. Immunol. 2010;40:699–709. doi: 10.1002/eji.200838951. [DOI] [PubMed] [Google Scholar]
- 107.Lauer N, et al. Complement regulation at necrotic cell lesions is impaired by the age-related macular degeneration-associated factor-H His402 risk variant. J. Immunol. 2011;187:4374–4383. doi: 10.4049/jimmunol.1002488. [DOI] [PubMed] [Google Scholar]
- 108.Kang YH, Urban BC, Sim RB, Kishore U. Human complement factor H modulates C1q-mediated phagocytosis of apoptotic cells. Immunobiology. 2012;217:455–464. doi: 10.1016/j.imbio.2011.10.008. [DOI] [PubMed] [Google Scholar]
- 109.Gresham HD, et al. Negative regulation of phagocytosis in murine macrophages by the Src kinase family member, Fgr. J. Exp. Med. 2000;191:515–528. doi: 10.1084/jem.191.3.515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Matt U, et al. WAVE1 mediates suppression of phagocytosis by phospholipid-derived DAMPs. J. Clin. Invest. 2013;123:3014–3024. doi: 10.1172/JCI60681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wyatt TA, et al. Malondialdehyde-acetaldehyde-adducted protein inhalation causes lung injury. Alcohol. 2012;46:51–59. doi: 10.1016/j.alcohol.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Libby P, Lichtman AH, Hansson GK. Immune effector mechanisms implicated in atherosclerosis: from mice to humans. Immunity. 2013;38:1092–1104. doi: 10.1016/j.immuni.2013.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Jurgens G, et al. Immunostaining of human autopsy aortas with antibodies to modified apolipoprotein B and apoprotein(a) Arterioscler. Thromb. 1993;13:1689–1699. doi: 10.1161/01.ATV.13.11.1689. [DOI] [PubMed] [Google Scholar]
- 114.Yla-Herttuala S, et al. Evidence for the presence of oxidatively modified low density lipoprotein in atherosclerotic lesions of rabbit and man. J. Clin. Invest. 1989;84:1086–1095. doi: 10.1172/JCI114271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.van Dijk RA, et al. Differential expression of oxidation-specific epitopes and apolipoprotein(a) in progressing and ruptured human coronary and carotid atherosclerotic lesions. J. Lipid Res. 2012;53:2773–2790. doi: 10.1194/jlr.P030890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Walton KA, et al. Receptors involved in the oxidized 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphorylcholine-mediated synthesis of interleukin-8. A role for Toll-like receptor 4 and a glycosylphosphatidylinositol-anchored protein. J. Biol. Chem. 2003;278:29661–29666. doi: 10.1074/jbc.M300738200. [DOI] [PubMed] [Google Scholar]
- 117.Mehta JL, et al. Deletion of LOX-1 reduces atherogenesis in LDLR knockout mice fed high cholesterol diet. Circ. Res. 2007;100:1634–1642. doi: 10.1161/CIRCRESAHA.107.149724. [DOI] [PubMed] [Google Scholar]
- 118.Mullick AE, et al. Increased endothelial expression of Toll-like receptor 2 at sites of disturbed blood flow exacerbates early atherogenic events. J. Exp. Med. 2008;205:373–383. doi: 10.1084/jem.20071096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Moore KJ, Sheedy FJ, Fisher EA. Macrophages in atherosclerosis: a dynamic balance. Nat. Rev. Immunol. 2013;13:709–721. doi: 10.1038/nri3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Duewell P, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464:1357–1361. doi: 10.1038/nature08938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Sheedy FJ, et al. CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nat. Immunol. 2013;14:812–820. doi: 10.1038/ni.2639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Michelsen KS, et al. Lack of Toll-like receptor 4 or myeloid differentiation factor 88 reduces atherosclerosis and alters plaque phenotype in mice deficient in apolipoprotein E. Proc. Natl Acad. Sci. USA. 2004;101:10679–10684. doi: 10.1073/pnas.0403249101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Ameziane N, et al. Association of the Toll-like receptor 4 gene Asp299Gly polymorphism with acute coronary events. Arterioscler. Thromb. Vasc. Biol. 2003;23:e61–e64. doi: 10.1161/01.ATV.0000101191.92392.1D. [DOI] [PubMed] [Google Scholar]
- 124.Kiechl S, et al. Toll-like receptor 4 polymorphisms and atherogenesis. N. Engl. J. Med. 2002;347:185–192. doi: 10.1056/NEJMoa012673. [DOI] [PubMed] [Google Scholar]
- 125.Tabas I. Macrophage death and defective inflammation resolution in atherosclerosis. Nat. Rev. Immunol. 2010;10:36–46. doi: 10.1038/nri2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Tsiantoulas D, Diehl CJ, Witztum JL, Binder CJ. B cells and humoral immunity in atherosclerosis. Circ. Res. 2014;114:1743–1756. doi: 10.1161/CIRCRESAHA.113.301145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Tanigaki K, et al. Fcγ receptors and ligands and cardiovascular disease. Circ. Res. 2015;116:368–384. doi: 10.1161/CIRCRESAHA.116.302795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Zacho J, et al. Genetically elevated C-reactive protein and ischemic vascular disease. N. Engl. J. Med. 2008;359:1897–1908. doi: 10.1056/NEJMoa0707402. [DOI] [PubMed] [Google Scholar]
- 129.Binder CJ, et al. Pneumococcal vaccination decreases atherosclerotic lesion formation: molecular mimicry between Streptococcus pneumoniae and oxidized LDL. Nat. Med. 2003;9:736–743. doi: 10.1038/nm876. [DOI] [PubMed] [Google Scholar]
- 130.Faria-Neto JR, et al. Passive immunization with monoclonal IgM antibodies against phosphorylcholine reduces accelerated vein graft atherosclerosis in apolipoprotein E-null mice. Atherosclerosis. 2006;189:83–90. doi: 10.1016/j.atherosclerosis.2005.11.033. [DOI] [PubMed] [Google Scholar]
- 131.Oksjoki R, et al. Association between complement factor H and proteoglycans in early human coronary atherosclerotic lesions: implications for local regulation of complement activation. Arterioscler. Thromb. Vasc. Biol. 2003;23:630–636. doi: 10.1161/01.ATV.0000057808.91263.A4. [DOI] [PubMed] [Google Scholar]
- 132.Sofat R, et al. Genetic variation in complement factor H and risk of coronary heart disease: eight new studies and a meta-analysis of around 48,000 individuals. Atherosclerosis. 2010;213:184–190. doi: 10.1016/j.atherosclerosis.2010.07.021. [DOI] [PubMed] [Google Scholar]
- 133.Walenbergh SM, Koek GH, Bieghs V, Shiri-Sverdlov R. Non-alcoholic steatohepatitis: the role of oxidized low-density lipoproteins. J. Hepatol. 2013;58:801–810. doi: 10.1016/j.jhep.2012.11.014. [DOI] [PubMed] [Google Scholar]
- 134.Bieghs V, et al. Internalization of modified lipids by CD36 and SR-A leads to hepatic inflammation and lysosomal cholesterol storage in Kupffer cells. PLoS ONE. 2012;7:e34378. doi: 10.1371/journal.pone.0034378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Bieghs V, et al. Specific immunization strategies against oxidized low-density lipoprotein: a novel way to reduce nonalcoholic steatohepatitis in mice. Hepatology. 2012;56:894–903. doi: 10.1002/hep.25660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Gruber S, et al. Sialic acid-binding immunoglobulin-like lectin G promotes atherosclerosis and liver inflammation by suppressing the protective functions of B-1 cells. Cell Rep. 2016;14:2348–2361. doi: 10.1016/j.celrep.2016.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.McHarg S, Clark SJ, Day AJ, Bishop PN. Age-related macular degeneration and the role of the complement system. Mol. Immunol. 2015;67:43–50. doi: 10.1016/j.molimm.2015.02.032. [DOI] [PubMed] [Google Scholar]
- 138.Handa JT. How does the macula protect itself from oxidative stress? Mol. Aspects Med. 2012;33:418–435. doi: 10.1016/j.mam.2012.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Doyle SL, et al. NLRP3 has a protective role in age-related macular degeneration through the induction of IL-18 by drusen components. Nat. Med. 2012;18:791–798. doi: 10.1038/nm.2717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Haines JL, et al. Complement factor H variant increases the risk of age-related macular degeneration. Science. 2005;308:419–421. doi: 10.1126/science.1110359. [DOI] [PubMed] [Google Scholar]
- 141.Edwards AO, et al. Complement factor H polymorphism and age-related macular degeneration. Science. 2005;308:421–424. doi: 10.1126/science.1110189. [DOI] [PubMed] [Google Scholar]
- 142.Aredo B, et al. A chimeric Cfh transgene leads to increased retinal oxidative stress, inflammation, and accumulation of activated subretinal microglia in mice. Invest. Ophthalmol. Vis. Sci. 2015;56:3427–3440. doi: 10.1167/iovs.14-16089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Kris-Etherton PM, et al. Antioxidant vitamin supplements and cardiovascular disease. Circulation. 2004;110:637–641. doi: 10.1161/01.CIR.0000137822.39831.F1. [DOI] [PubMed] [Google Scholar]
- 144.Niki E, Yoshida Y, Saito Y, Noguchi N. Lipid peroxidation: mechanisms, inhibition, and biological effects. Biochem. Biophys. Res. Commun. 2005;338:668–676. doi: 10.1016/j.bbrc.2005.08.072. [DOI] [PubMed] [Google Scholar]
- 145.Thomas CP, O'Donnell VB. Oxidized phospholipid signaling in immune cells. Curr. Opin. Pharmacol. 2012;12:471–477. doi: 10.1016/j.coph.2012.02.013. [DOI] [PubMed] [Google Scholar]
- 146.Nicholls SJ, Hazen SL. Myeloperoxidase, modified lipoproteins, and atherogenesis. J. Lipid Res. 2009;50:S346–S351. doi: 10.1194/jlr.R800086-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Mauerhofer C, Philippova M, Oskolkova OV, Bochkov VN. Mol. Aspects Med. 2016;49:78–90. doi: 10.1016/j.mam.2016.02.003. [DOI] [PubMed] [Google Scholar]
- 148.Krieg P, et al. Aloxe3 knockout mice reveal a function of epidermal lipoxygenase-3 as hepoxilin synthase and its pivotal role in barrier formation. J. Invest. Dermatol. 2013;133:172–180. doi: 10.1038/jid.2012.250. [DOI] [PubMed] [Google Scholar]
- 149.Chiang N, Arita M, Serhan CN. Anti-inflammatory circuitry: lipoxin, aspirin-triggered lipoxins and their receptor ALX. Prostaglandins Leukot. Essent. Fatty Acids. 2005;73:163–177. doi: 10.1016/j.plefa.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 150.Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 2013;13:397–411. doi: 10.1038/nri3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Marnell L, Mold C, Du Clos TW. C-reactive protein: ligands, receptors and role in inflammation. Clin. Immunol. 2005;117:104–111. doi: 10.1016/j.clim.2005.08.004. [DOI] [PubMed] [Google Scholar]