

Key Points
Viruses are detected by different classes of pattern-recognition receptors (PRRs), including Toll-like receptors (TLRs), RIG-I-like receptors (RLRs) and cytoplasmic DNA receptors. This leads to the activation of downstream signalling pathways that culminate in the activation of transcription factors and the production of type I interferons (IFNs).
Investigating how viruses interact with PRR signalling pathways provides insights into how the pathways function, which host proteins are involved and how the pathways might be manipulated therapeutically.
TLRs and RLRs are inhibited in various ways by viruses: the main viral mechanism for inhibiting TLR signalling to be identified so far is the disabling of the TIR (TLR/interleukin-1 receptor)-domain-containing adaptor proteins, which are required for the activation of transcription factors. Although described only recently, RLRs have already been shown to be crucial for sensing viruses. Consequently, many viral strategies that are targeted against them have already been identified.
All PRR signalling pathways converge with the activation of IκB kinase (IKK) family members. So, direct inhibition of IKKs is an efficient viral strategy to disable multiple PRR pathways and it is used by many viruses.
The transcription factors that control IFN induction, IFN-regulatory factor 3 (IRF3), IRF7 and nuclear factor-κB, are also directly antagonized by viruses. IRF3 in particular is subject to virus-induced degradation.
Viruses also actively subvert TLRs to manipulate the host cytokine environment for their own benefit.
This Review highlights how understanding the mechanisms by which viruses evade and subvert host signalling by pattern-recognition receptors has provided insights into the function of these signalling pathways, the host proteins that are involved and ways in which the pathways might be manipulated therapeutically.
Abstract
The expression of pattern-recognition receptors (PRRs) by immune and tissue cells provides the host with the ability to detect and respond to infection by viruses and other microorganisms. Significant progress has been made from studying this area, including the identification of PRRs, such as Toll-like receptors and RIG-I-like receptors, and the description of the molecular basis of their signalling pathways, which lead to the production of interferons and other cytokines. In parallel, common mechanisms used by viruses to evade PRR-mediated responses or to actively subvert these pathways for their own benefit are emerging. Accumulating evidence on how viral infection and PRR signalling pathways intersect is providing further insights into the function of the pathways involved, their constituent proteins and ways in which they could be manipulated therapeutically.
Main
Viruses are the most abundant, diverse and rapidly evolving pathogens that the host immune system is challenged by and they therefore represent a serious threat to human health. Paradoxically, viruses are obligate parasites that depend on host cells for survival and, throughout evolution, they have developed strategies to evade and subvert the immune response, and to use host proteins for their own life cycle. Viral infection of host cells leads to the initiation of antiviral innate immune responses, which results in the induction of expression of the type I interferons (IFNs) IFNα and IFNβ, and of pro-inflammatory cytokines. The importance of type I IFNs in directing the antiviral response has been characterized in detail1 and validated in vivo. For example, mice that are deficient in the type I IFN receptor have increased susceptibility to many viruses, and humans with genetic defects in IFN signalling components die of viral infections1. Some of the important IFN antiviral effector pathways, which comprise host mechanisms to directly target the viral life cycle and to amplify host detection of viruses and IFN production, are shown in Fig. 1. In addition to influencing innate immune responses, type I IFNs direct the adaptive immune response by priming T helper cells and cytotoxic T cells (CTLs), thereby resulting in the induction of antigen-specific responses.
Figure 1. Activation of the interferon response triggered by the detection of viral pathogen-associated molecular patterns.
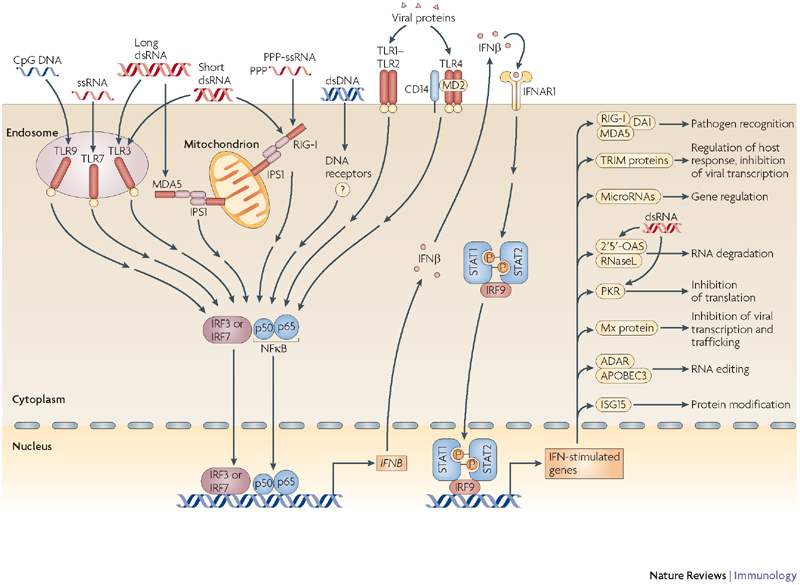
All of the pattern-recognition receptors (PRRs) initiate signalling pathways that converge at the activation of the transcription factors interferon (IFN)-regulatory factor 3 (IRF3), IRF7 and/or nuclear factor-κB (NFκB); this leads to the expression of IFNβ. IFNβ then initiates an antiviral effector programme in the infected cell and neighbouring cells, which involves the expression of numerous IFN-stimulated genes (ISGs). Some of the ISGs shown here, such as RIG-I (retinoic-acid-inducible gene I), MDA5 (melanoma differentiation-associated gene 5), DAI (DNA-dependent activator of IRFs), some microRNAs and the TRIM (tripartite motif-containing) family of proteins, are involved in the amplification and regulation of the IFN response. Other ISGs shown here, such as 2′5′-oligoadenylate synthetase (OAS) and ribonuclease L (RNaseL), IFN-inducible dsRNA-dependent protein kinase (PKR), myxovirus resistance (Mx) protein, adenosine deaminase RNA-specific (ADAR) and apolipoprotein B mRNA-editing enzyme, catalytic polypeptide 3 (APOBEC3), are involved in antiviral mechanisms that interfere with the life cycle of individual viruses . OAS and PKR are further activated by double-stranded RNA (dsRNA). IFNAR1, interferon-α receptor; IPS1, IFNB-promoter stimulator 1; ISG15, IFN-stimulated protein of 15 kDa; MD2, myeloid differentiation protein 2; PPP, 5′ triphosphate; ssRNA, single-stranded RNA; STAT, signal transducer and activator of transcription; TLR, Toll-like receptor.
Eight years ago, almost nothing was known about the cellular mechanisms that are used to detect viruses and to subsequently produce IFNs and pro-inflammatory cytokines. It is now known that viruses, similar to bacteria and fungi, are initially recognized by host pattern-recognition receptors (PRRs), as was predicted by Janeway almost 20 years ago2. In a short time, a large body of research has accumulated and now provides a detailed understanding of the molecular basis of PRR-mediated recognition of viral nucleic acids, and of the subsequent activation of signalling pathways that lead to the transcription of genes that encode pro-inflammatory cytokines and IFNs. Two families of PRRs that recognize viral nucleic acid have been characterized in detail. The first is a subfamily of Toll-like receptors (TLRs) that is made up of TLR3, TLR7, TLR8 and TLR9, which are mainly expressed in the endosomes of some cell types. The second family of receptors comprises the retinoic-acid-inducible gene I (RIG-I)-like receptors (RLRs), encompassing RIG-I, melanoma differentiation-associated gene 5 (MDA5; also known as IFIH1) and laboratory of genetics and physiology 2 (LGP2), which are ubiquitously expressed in the cytoplasm (Fig. 1). An additional family of intracellular PRRs that is probably important in the sensing of viral DNA in the cytoplasm is also emerging3,4,5. Viral nucleic acid (both RNA and DNA) is the most important pathogen-associated molecular pattern (PAMP) that is recognized by the host (Box 1), and there is now an excellent understanding of how viral nucleic acids bind to and activate PRRs at the molecular level (reviewed in Refs 6,7). In addition, the fundamental aspects of the PRR signalling pathways, and the components that are involved, have also been well characterized8,9.
There has long been a substantial amount of literature on how the IFN effector phase of the antiviral response is antagonized by viruses, but with the more recent discoveries identifying how virus triggering of PRRs leads to IFN induction, the mechanisms by which viruses evade early events in PRR signalling pathways have become apparent. It is now clear that, following infection, viruses can induce complex intracellular events that affect many components of host signalling pathways. The effects of viruses on host defence not only inhibit viral detection, but also allow the virus to manipulate host signalling pathways for their own benefit. So, the activation of a certain PRR by a virus can be either part of the host's protective immune response or a mechanism that is used by the virus to aid its replication and spread. Owing to the fine-tuning of immune signalling by both virus and host, different viruses may activate the same PRR with either positive or negative outcomes on disease progression and immune response.
Elucidating the strategies that are used by viruses to inhibit and to manipulate host responses is important for two reasons. First, this knowledge will contribute to our understanding of viral pathogenesis. As viruses (especially RNA viruses) mutate very rapidly, the identification of mutations that increase or decrease the activity of a viral protein that targets the immune response may help to define the virulence factors of different strains or species of viruses that infect humans. For example, Ebola virus, which is usually highly virulent in humans and triggers lethal haemorrhagic fever, induces a minimal adaptive immune response (which requires innate immune detection of the virus by PRRs) in patients who succumb to infection, whereas those who survive infection mount an effective adaptive immune response10. Furthermore, a mutant virus with a single amino-acid change in the Ebola virus protein (VP35) that inhibits PRR signalling (Table 1) induces a markedly different innate immune response in human liver cells than the wild-type virus11.
Table 1.
Representative viral proteins that interfere with PPR signalling pathways at multiple points
| Protein | Virus | Assigned functions in evading or subverting PRR signalling pathways | Refs |
|---|---|---|---|
| VP35 | Ebola virus | Sequesters viral dsRNA | 112 |
| Inhibits IRF3 activation downstream of IPS1 | 112 | ||
| NS3–4A | Hepatitis C virus | Degrades TRIF to inhibit TLR3 signalling | 35 |
| Cleaves IPS1 from its mitochondrial tether to disable RLR signalling pathways | 48,69,70 | ||
| Inhibits IRF3 phosphorylation by disrupting the TBK1–IRF3 interaction | 75 | ||
| NS5A | Hepatitis C virus | Inhibits PKR through direct binding | 113 |
| Inhibits OAS through direct binding | 114 | ||
| Inhibits TLR signalling by binding MyD88 | 36 | ||
| E3L | Vaccinia virus | Sequesters viral dsRNA | 115 |
| Inhibits PKR through direct binding | 116 | ||
| Prevents DAI from interacting with DNA | 5 | ||
| Binds to and disables ISG15 | 93 | ||
| A52R | Vaccinia virus | Inhibits TLR-induced NFκB activation by binding IRAK2 | 38 |
| Enhances TLR-induced IL-10 production by binding TRAF6 | 106 | ||
| NS1 | Influenza A virus | Sequesters viral dsRNA | 117 |
| Binds to RIG-I and suppresses RIG-I signalling | 51 | ||
| DAI, DNA-dependent activator of IRFs; ds, double stranded; IFN, interferon; IL-10, interleukin-10; IL-1R, IL-1 receptor; IPS1, IFNB-promoter stimulator 1; IRAK2, IL-1R-associated kinase 2; IRF3, IFN-regulatory factor 3; ISG15, IFN-stimulated protein of 15 kDa; MyD88, myeloid differentiation primary-response gene 88; NFκB, nuclear factor-κB; NS1, nonstructural protein 1; OAS, 2′, 5′-oligoadenylate synthetase; PKR, IFN-inducible dsRNA-dependent protein kinase; PPR, pattern-recognition receptor; RLR, retinoic-acid-inducible gene I (RIG-I)-like receptor; TANK, TRAF-family-member-associated NFκB activator; TBK1, TANK-binding kinase 1; TLR, Toll-like receptor; TRAF6, TNFR-associated factor 6; TRIF, TIR-domain-containing adaptor protein inducing IFNβ. | |||
Second, understanding how viruses manipulate and inhibit PRR detection pathways has led to further insights into how these host pathways function (Box 2). This information will be important for our understanding of the mechanism by which human PRRs detect viruses and can complement forward genetics and reverse genetics, which are used to investigate the mechanisms of mouse PRR detection and signalling12.
In this Review, we describe recent advances and emerging themes in our understanding of how viruses evade detection by host PRRs and discuss how viruses inhibit and manipulate PRR signalling to gain an early advantage over the host immune response. We describe viral strategies that function proximal to distinct PRRs (such as TLRs or RLRs), and also those that act further downstream on shared components of the PRR signalling cascades (such as IFN-regulatory factors (IRFs) or nuclear factor-κB (NFκB)).
TLRs and viruses
Role of TLRs in detecting viruses. The identification of TLR4 as the signalling receptor for bacterial lipopolysaccharide was a major breakthrough in the field of innate immune research. The first evidence that TLRs might also respond to viruses came with the finding that the fusion protein of respiratory syncytial virus (RSV) stimulated the secretion of interleukin-6 (IL-6) from mouse macrophages, and this depended on TLR4 signalling13. In addition, it was discovered that proteins from vaccinia virus (VACV) antagonized TLR signalling in cultured cells14, further supporting a role for TLRs in sensing viruses15. In 2001, TLR3 was identified as a PRR for viral double-stranded RNA (dsRNA) based on the observation that mice deficient in IFN-inducible dsRNA-dependent protein kinase (PKR; also known as EIF2AK2 and previously the only known dsRNA receptor) still responded to the synthetic dsRNA mimic polyinosinic–polycytidylic acid (polyI:C)16. As stimulation of cells with polyI:C was known to mimic many aspects of viral infection, TLR3 was expected to have a broad and crucial role in antiviral immunity. However, it soon became clear that this was not the case, as viral infection of hosts that were deficient in TLR3 did not alter viral pathogenesis or impair the host immune response to several viruses17. It now seems that the absence of TLR3 is actually protective in the case of some viral infections (see later). Nonetheless, important and specific roles for TLR3 in the antiviral response have been shown, including host defence against certain viruses (reviewed in Refs 18–20).
TLR3 is part of a subset of TLRs that are present in endosomes and sense nucleic acids. Others include TLR7, TLR8 and TLR9. Human TLR8 and mouse TLR7 (TLR8 is non-functional in mice) detect single-stranded RNA (ssRNA) from RNA viruses, including HIV-1, vesicular stomatitis virus (VSV) and influenza A virus, and this leads to the induction of IFNα expression21. TLR9 senses unmethylated CpG DNA in the genomes of DNA viruses, such as herpes simplex virus (HSV), and this induces the production of type I IFNs by plasmacytoid dendritic cells (pDCs)22,23. TLR7 and TLR9 seem to have a more important role than TLR3 in antiviral immunity, being responsible for the sensing of viruses by pDCs and their subsequent production of type I IFNs. Indeed, Akira and colleagues recently showed that TLR-mediated recognition of the ssRNA lymphocytic choriomeningitis virus (LCMV) in pDCs leads to the production of type I IFNs and the subsequent CTL response in vivo24.
Viral evasion of TLR signalling pathways. Engagement of TLRs by viral PAMPs causes receptor dimerization followed by the initiation of intracellular signalling pathways that culminate in the activation of the transcription factors NFκB, IRF3 and IRF7 (for recent reviews, see Refs 9,25). The cytoplasmic region of TLRs contains the conserved Toll/IL-1 receptor (IL-1R) (TIR) domain, which is common to the IL-1R family and is essential for signalling that is mediated by TLRs and IL-1Rs25. Receptor dimerization triggers homotypic interactions between the receptor TIR domain and TIR-domain-containing adaptor proteins. There are five TIR-domain-containing adaptor proteins that control signalling from activated TLRs, namely myeloid differentiation primary-response gene 88 (MyD88), MyD88-adaptor-like (MAL; also known as TIRAP), TIR-domain-containing adaptor protein inducing IFNβ (TRIF), TRIF-related adaptor molecule (TRAM) and sterile-α- and armadillo-motif-containing protein (SARM)25. MyD88 is required for signalling from all mouse TLRs except TLR3, as well as signalling from the IL-1R25, although different TLRs signal through different repertoires of TIR-domain-containing adaptors. For example, TLR2 signalling requires MyD88 and MAL, TLR3 signalling needs TRIF, and TLR7 and TLR9 signalling requires only MyD88 (Ref. 25). The TLR-induced signalling pathways that lead to IRF3, IRF7 and NFκB activation are outlined in Fig. 2. Some of the factors that are involved in TLR signalling are shared with other signalling pathways. More specifically, it has recently been shown that TRADD (tumour-necrosis factor receptor (TNFR)-associated via death domain) is required for the signalling pathways that are downstream of the TNFR, for TRIF-dependent TLR signalling and for both RIG-I and MDA5 signalling pathways26,27,28. This might provide the opportunity for crosstalk between the different innate immune signalling events.
Figure 2. Viral evasion of Toll-like receptor signalling.
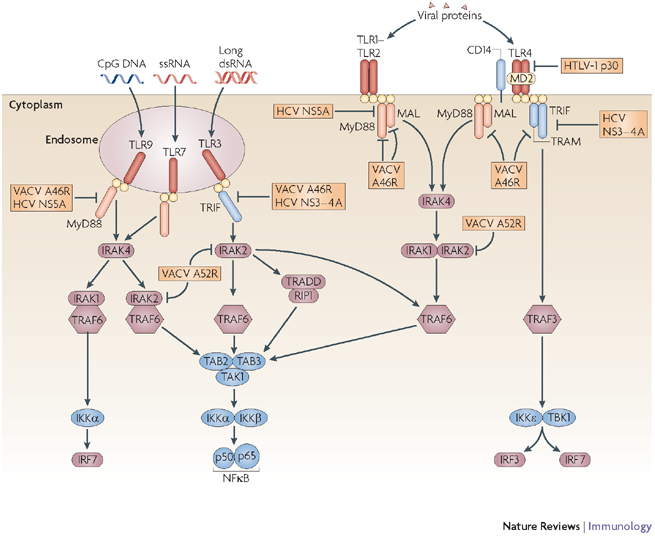
Following activation, Toll-like receptors (TLRs) recruit the adaptor proteins myeloid differentiation primary-response gene 88 (MyD88), TIR-domain-containing adaptor protein inducing IFNβ (TRIF), MyD88-adaptor-like (MAL) and TRIF-related adaptor protein (TRAM), as indicated. These then initiate signalling cascades involving IL-1R-associated kinase (IRAK) and TNFR-associated factor (TRAF) proteins, which finally converge at the activation of the IκB kinase (IKK) family members IKKα, IKKβ, IKKɛ and TBK1 (TANK-binding kinase 1). The vaccinia virus (VACV) protein A46R sequesters all these adaptor proteins, whereas the hepatitis C virus (HCV) protein NS5A selectively binds MyD88 and the HCV NS3–4A protease cleaves TRIF. Human T-cell leukaemia virus type 1 (HTLV-1) protein p30 acts even further upstream by reducing the expression of TLR4. VACV A52R binds to and inhibits IRAK2, thereby affecting several TLR pathways that lead to nuclear factor-κB (NFκB) activation. dsRNA, double-stranded RNA; IFN, interferon; IL-1R, interleukin-1 receptor; IRF, IFN-regulatory factor; IκB, inhibitor of NFκB; MD2, myeloid differentiation protein 2; ssRNA, single-stranded RNA; RIP1, receptor-interacting protein 1; TAB, TAK1-binding protein; TAK1, transforming-growth-factor-β-activated kinase 1; TANK, TRAF-family-member-associated NFκB activator; TIR, TLR/IL-1R; TNFR, tumour-necrosis factor receptor; TRADD, TNFR-associated via death domain.
Many viral antagonists of TLR signalling have been identified (Fig. 2). One of the first characterized viral inhibitors of a TLR signalling pathway was the VACV protein A46R, which was found to directly target specific TIR-domain-containing adaptor proteins. VACV, a large dsDNA virus of the Poxviridae family, is now known to encode many inhibitors of innate immune signalling pathways, many of which also have a non-redundant, independent role in virulence29,30. A46R contains a TIR domain, which allows it to interact with TIR-domain-containing complexes and to bind directly to the TIR-domain-containing adaptors MyD88, MAL, TRIF and TRAM; it can therefore inhibit the activation of both NFκB and IRFs that is normally triggered by multiple TLR pathways31. The affinity of the interaction between A46R and TIR-domain-containing adaptors in an infected cell may be sufficient to prevent the recruitment of adaptors to TLRs and thereby inhibit host defence, although this theory has not yet been supported experimentally. When the gene that encodes A46R was deleted from VACV, the virus was attenuated31. In keeping with this observation several TLRs have been shown to participate in the host cytokine and IFN response to poxvirus infection30. It is worth noting that A46R does not interact with SARM, which is a negative regulator of TLR signalling32 and is therefore favourable in the context of VACV infection.
Although A46R is still the only known TIR-domain-containing viral protein, bacterial proteins containing TIR domains have been identified and have been found to have a role in bacterial virulence33,34, possibly by using mechanisms that are similar to those used by A46R. However, another non-TIR-domain-containing viral protein, namely the NS3–4A heterodimer from the ssRNA flavivirus hepatitis C virus (HCV), has also been shown to use a similar strategy to disable TIR adaptor proteins. NS3–4A has serine protease activity, is essential for the survival of the virus and has many functions in evading or subverting PRR signalling pathways (Table 1). One function is to cleave an HCV precursor polypeptide to generate mature structural proteins during viral replication. Further work showed that TRIF was also specifically cleaved by NS3–4A into two polypeptides that could no longer activate TLR-dependent transcription from the IFNB promoter35. NS3–4A expression potently decreased the polyI:C-induced activity of the IFNB promoter, the polyI:C-mediated activation of NFκB and IRF3, and the induction of IRF3-dependent genes35. Therefore, by targeting TRIF, HCV inhibits TLR3-mediated antiviral signalling. Although the importance of TLR3 in the host immune response to HCV infection is currently unclear, the targeting of TRIF by HCV indicated that TLR3 has an important role in the antiviral response to HCV. Furthermore, it has recently been shown that when four different HCV proteins (one of which was NS3–4A) were expressed individually in a mouse macrophage cell line, they inhibited TLR-induced cytokine production. The target of the HCV protein NS5A was found to be MyD88 (Ref. 36).
Another VACV protein, A52R, specifically targets IL-1R and TLR signalling and contributes to virulence37. A52R interacts with TNFR-associated factor 6 (TRAF6) and IL-1R-associated kinase 2 (IRAK2), two signalling molecules that are downstream of the TIR-domain-containing adaptors, and is an effective inhibitor of NFκB activation37. Although TRAF6 was known to have a central role in TLR signalling that leads to NFκB activation, the role of the pseudokinase IRAK2 was unknown. Surprisingly, it was shown that a mutant version of A52R, which could interact with IRAK2 but not TRAF6, induced maximal inhibition of NFκB38. This key observation led to a study which showed that IRAK2 is more important for NFκB activation than was previously appreciated, and certainly more important than IRAK1 for some TLR signalling pathways38. Because A52R, unlike A46R, had no effect on TLR-induced IRF activation, this suggested that IRAK2 was not involved in the TLR–IRF axis. Indeed, it is now appreciated that IRAK1 has a crucial role in this signalling pathway (Fig. 2), whereas IRAK2 is involved in NFκB activation, as confirmed by the study of IRAK2-deficient mice39. So, the study of A52R is a clear example of what we can learn about PRR signalling pathways by investigating viral evasion strategies (Box 2). Similar to NS3–4A, A52R also has many functions in evading or subverting PRR signalling (Table 1), and its ability to interact with TRAF6 might represent subversion (rather than evasion) of the TLR pathways by VACV (see later).
In addition to VACV and HCV, other viruses that target TLR signalling pathways include human T-cell leukaemia virus type 1 (HTLV-1) and West Nile virus (WNV). HTLV-I downregulates the host cell-surface expression of TLR4 through the viral protein p30, which binds to and disables the transcription factor PU.1 (which is normally required for TLR4 expression)40. By contrast, the WNV protein non-structural protein 1 (NS1) inhibits TLR3-mediated induction of IFNβ expression by preventing the translocation of NFκB and IRF3 to the nucleus41. However, it is currently unclear whether inhibition that is mediated by NS1 is specific to the TLR3 signalling pathway or whether it also affects the induction of IFNβ expression downstream of other PRRs.
Important principles of viral evasion are illustrated by VACV and HCV targeting of TLR signalling pathways. Viral antagonists of innate immunity often have a similar sequence to the host proteins they target (as in the case of A46R), which can provide clues about their function, or they can be completely unrelated in sequence to host proteins (as in the case of NS3–4A). Furthermore, viral proteins with known functions (such as NS5A42) may have additional roles, which illustrates the high efficiency with which viruses use their limited protein-coding resources (Table 1).
RLRs and viruses
Recognition of viral RNA by RIG-I and MDA5. TLR7 and TLR9 are important for the recognition of viral nucleic acids in the endosomes of pDCs, but most other cell types recognize viral RNA through the RLRs RIG-I and MDA5 (Refs 43,44,45). RIG-I and MDA5 are closely related proteins that contain two amino (N)-terminal caspase-recruitment domains (CARDs), a central ATPase and helicase domain and a carboxy (C)-terminal regulatory domain. The CARDs of RIG-I and MDA5 recruit the signalling adaptor protein IFNB-promoter stimulator 1 (IPS1; also known as CARDIF, VISA or MAVS)46,47,48,49. IPS1 resides at the outer mitochondrial membrane and interacts with RIG-I and MDA5 through its CARD. This interaction provides the link between the RLRs and the downstream kinases TANK-binding kinase 1 (TBK1) and IκB kinase-ɛ (IKKɛ), which phosphorylate and activate IRF3 and IRF7 (Fig. 3). Recently, a protein known as stimulator of IFN genes (STING) has been identified as an additional factor in the RIG-I–IPS1 signalling complex, but not in the complex containing MDA5 (Ref. 50). STING is located in the endoplasmic reticulum, which suggests that this intracellular compartment is also involved in RIG-I-mediated signalling.
Figure 3. Viral evasion of retinoic-acid-inducible-gene-I-like receptor signalling.
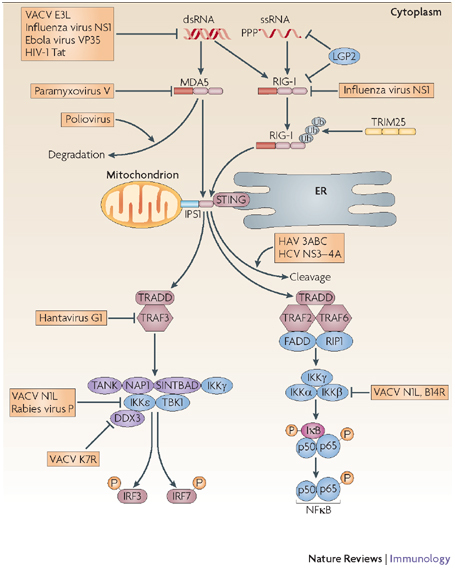
RIG-I (retinoic-acid-inducible gene I) and MDA5 (melanoma differentiation-associated gene 5), termed RIG-I-like receptors (RLRs), are activated by cytoplasmic RNA during viral infection. Both signal using IFNB-promoter stimulator 1 (IPS1), which is tethered to the mitochondrial membrane. When IPS1 is engaged by RLRs, it recruits downstream signalling complexes that lead to the activation of the IFN-regulatory factors (IRFs) and nuclear factor-κB (NFκB). In addition, signalling through RIG-I requires the adaptor STING (stimulator of IFN genes), which resides in the endoplasmic reticulum (ER). RLR signalling is inhibited by viral proteins that either bind RIG-I, MDA5 or IPS1 directly or cause their degradation. The IκB kinase (IKK) family members are also a common target for viral proteins. DDX3, DEAD-box protein 3; dsRNA, double-stranded RNA; FADD, FAS-associated via death domain; HAV, hepatitis A virus; HCV, hepatitis C virus; IFN, interferon; IκB, inhibitor of NFκB; LGP2, laboratory of genetics and physiology 2; NAP1, NFκB-activating kinase-associated protein 1; NS1, nonstructural protein 1; PPP, 5′ triphosphate; RIP1, receptor-interacting protein 1; SINTBAD, similar to NAP1 TBK1 adaptor; ssRNA, single-stranded RNA; TANK, TRAF-family-member-associated NFκB activator; TBK1, TANK-binding kinase 1; TNFR, tumour-necrosis factor receptor; TRADD, TNFR-associated via death domain; TRAF, TNFR-associated factor; TRIM25, tripartite motif-containing 25; ub, ubiquitin; VACV, vaccinia virus.
Both RIG-I and MDA5 were originally thought to recognize similar types of viral dsRNA. Recently, however, the exact nature of their nucleic acid ligands has been defined and shown to be distinct. Characterization of the mechanism by which the influenza virus A nonstructural protein 1 (NS1) inhibited RIG-I-mediated detection of viral RNA revealed that the main ligand for RIG-I is ssRNA that has a 5′ triphosphate51,52,53, such as that generated by many viral RNA polymerases. Although the 5′ triphosphate is necessary, it is not sufficient for RIG-I activation54; the composition of the RNA sequence also has a role, as viral RNAs that contain a 5′ triphosphate and a sequence of poly-uridine or riboadenine are particularly potent activators of RIG-I54. RIG-I can also be activated by short fragments of dsRNA55, whereas MDA5 is the main cytoplasmic receptor for longer molecules of viral dsRNA and for polyI:C7. On the one hand, viruses that produce large amounts of dsRNA during their life cycle, such as picornaviruses, are sensed mainly by MDA5 (Refs 56,57). On the other hand, negative-stranded RNA viruses, such as influenza virus, which produce barely detectable levels of dsRNA, are sensed by RIG-I, as confirmed by studies using mice deficient in RIG-I and MDA5 (Ref. 57). Endogenous host RNAs do not activate RIG-I or MDA5, as they are generally ssRNAs and have 5′ ends that either are protected by a methylguanosine cap (as in the case of mRNAs and small nuclear RNAs) or are monophosphates (as in the case of ribosomal RNAs, tRNAs and microRNAs). In addition, abundant cellular RNAs, such as tRNAs and rRNAs, are extensively modified with unusual bases, such as pseudouridine, which prevent the activation of RIG-I and MDA5 (Ref. 52).
Modification of viral RNAs and viral RNA-binding proteins. Viruses with an RNA genome mimic some of the strategies that are used by host cells to prevent the recognition of their RNA by RIG-I and MDA5. Many viruses either use the host mRNA processing machinery to cap their newly synthesized viral mRNAs, encode their own capping enzymes (such as poxviruses and rotaviruses) or even 'snatch' capped 5′ fragments from cellular mRNAs (a strategy used by influenza A virus58). Some RNA viruses, such as Borna disease virus, express a phosphatase that converts the triphosphate into a monophosphate59, and picornaviruses protect the 5′ end of their genomic RNA with the covalently linked protein VPg60,61. By contrast, genomic RNA from many ssRNA viruses, such as influenza A and rabies viruses, contains a 5′ triphosphate moiety and consequently activates RIG-I52.
Many viruses must produce dsRNA at some stage during their life cycle, which is why it is a useful target of host PRRs. For example, viruses with a dsRNA genome are susceptible to recognition by MDA5, as are positive-sense RNA viruses that use a dsRNA intermediate during their replication cycle. Even DNA viruses can produce considerable amounts of dsRNA owing to convergent transcription (that is, when two adjacent open reading frames are transcribed at opposite directions, resulting in a partial 3′ overlap between their transcripts62. To avoid the antiviral responses that are initiated by RLRs (and by the dsRNA-activated effector proteins PKR and 2′5′-oligoadenylate synthase; Fig. 1), some viruses encode cytoplasmic dsRNA-binding proteins, including VACV E3L, Ebola virus VP35 (Ref. 63 and HIV-1 Tat64, that shield the dsRNA species from recognition by PRRs. In addition to sequestering viral RNA, some of these proteins also bind to PKR and/or RIG-I directly, thereby inhibiting the antiviral response at many levels65. Furthermore, it is possible that these cytoplasmic proteins may also prevent the redistribution of dsRNA to endosomes, where it would be recognized by TLR3.
Interference with RIG-I and MDA5 signalling. Although the RLRs were discovered less than 5 years ago, many studies have already revealed that viruses can target these receptors and their adaptor IPS1 directly and inhibit their function; this validates the importance of these PRRs for the recognition of viral infections. Influenza A virus NS1 protein binds to the RIG-I–IPS1 complex and blocks downstream signalling66. Analogously, the V proteins of many paramyxoviruses interact with MDA5 and inhibit its function45, and poliovirus induces the cleavage of MDA5 by caspases67.
Cleavage of the downstream adaptor IPS1 is also a common event that has been observed during infection with several viruses. More specifically, the protease precursor protein 3ABC of hepatitis A virus triggers the degradation of IPS1 at the mitochondrial membrane, which disrupts RIG-I- and MDA5-mediated activation of IRF3 (Ref. 68). In addition, as well as targeting TRIF (as discussed earlier), HCV uses the protease NS3–4A to cleave the short C-terminal transmembrane domain of IPS1, causing it to dissociate from the mitochondrial membrane and rendering it incapable of RLR signal transduction48,69,70. These studies confirmed the hypothesis that IPS1 must be tethered to the mitochondrial membrane to mediate its antiviral function47.
Intracellular DNA receptors
The most recent addition to the expanding family of viral PRRs is the intracellular DNA receptor DNA-dependent activator of IRFs (DAI; previously known as Z-DNA binding protein). When dsDNA is introduced into the cytoplasm by transfection, by bacterial pore-forming proteins or by viral infection, it can elicit a TLR-independent innate immune response that involves the activation of IRF3 by TBK1 and STING3,50,71,72,73. DAI is one such cytoplasmic DNA receptor4, but its role in the in vivo recognition of cytoplasmic DNA has been controversial; additional cytoplasmic DNA receptors also probably have an important role5,74. Interestingly, the VACV dsRNA-binding protein E3L also has a Z-DNA-binding domain, which can inhibit the DNA-induced expression of IFNβ and prevent the interaction of DAI with DNA5. So, E3L may protect the VACV DNA genome from recognition by DNA receptors. As the DNA-responsive pathways are currently being investigated in more detail, additional virus-encoded inhibitors will probably be identified. Elucidating the mechanisms by which viruses disrupt DNA receptor signalling in host cells will probably be instrumental in defining the signalling mechanisms of these receptors.
Viral evasion of downstream PRR signalling
Many PRR signalling pathways converge at the level of IKKs. All of the PRRs described previously, namely the TLRs, RLRs and cytoplasmic DNA receptor (or receptors), activate signalling cascades that converge at the level of the IKK family of proteins (Figs 2,3). IKKα and IKKβ, which form a complex with the regulatory subunit IKKγ (also known as NEMO), phosphorylate inhibitor of NFκB (IκB), which leads to its degradation and consequently the release and nuclear translocation of active NFκB. A second IKK complex, which is especially important for the induction of type I IFN expression, consists of IKKɛ, TBK1 and associated proteins. IKKɛ and TBK1 phosphorylate and activate IRF3 and IRF7, which homodimerize and translocate to the nucleus (Fig. 3). In addition to activating the IKKs, PRR signalling also activates mitogen-activated protein kinase (MAPK) signalling cascades, thereby leading to the activation of activator protein 1 (AP1) family members. Together, NFκB, IRF3, IRF7 and AP1 form an active complex on the IFNB promoter that leads to transcriptional activation of this promoter. In the case of TLR7, TLR8 and TLR9, induction of IFNα expression is largely mediated through IRF7 activation, which involves IKKα. Thus, the IKK family and the transcription factors they activate are an attractive target for inhibition by viral proteins, allowing viruses to disable the pathways that are triggered by many PRRs at the same time.
Viral inhibition of IRF3 and IRF7 activation at the level of IKKs. The interaction of the multifunctional HCV protein NS3 (Ref. 75), or of the VACV protein N1L76, with TBK1 inhibits the activation of downstream transcription factors. TBK1 is also inhibited by the phosphoprotein of many negative-sense RNA viruses, such as rabies virus and Borna disease virus77,78. Furthermore, the G1 protein of pathogenic hantaviruses can inhibit TBK1 function by disrupting the TRAF3–TBK1 interaction that is required for signalling79 (Fig. 3). In cells in which the activity of TBK1 is inhibited, IRF3 is not phosphorylated and cannot dimerize or translocate to the nucleus77,79. The dimerization and nuclear translocation of IRF3 is also inhibited by papain-like protease (PLpro) of severe acute respiratory syndrome (SARS)-associated coronavirus, which directly interacts with it80.
A compelling example of how the study of viral evasion can reveal new host signalling mechanisms is a recent study of the VACV protein K7R81. K7R was found to inhibit the PRR-mediated induction of IFNβ by preventing the TBK1–IKKɛ-mediated activation of IRFs owing to its ability to target human DEAD-box protein 3 (DDX3)81. Further work confirmed that DDX3 is part of the TBK1–IKKɛ complex downstream of both RLRs and cytoplasmic DNA receptors, showing for the first time that a DEAD-box helicase is involved in TBK1–IKKɛ-mediated activation of IRFs81. The role of DDX3 in the activation of the IFNB promoter downstream of PRR signalling was confirmed by another recent study82.
Direct targeting of IRFs. Several viruses have viral IRF mimics that inhibit IRF3 and IRF7 signalling through various mechanisms (Fig. 4). For example, the V proteins of paramyxoviruses, such as mumps virus and parainfluenza virus 5, act as IRF3 mimics that compete with cellular IRF3 for phosphorylation by the TBK1–IKKɛ complex83. Human herpesvirus 8 also expresses several IRF homologues that act as dominant-negative inhibitors by preventing the association of IRF3 with its co-activators CREB-binding protein (CBP) and p300 (Ref. 84). Similarly, a viral IRF from Kaposi's sarcoma-associated herpesvirus (KSHV) dimerizes with host IRF7 and inhibits its DNA-binding activity85. Another KSHV protein, the transcription factor K-bZIP, competes with host IRF3 for binding sites in the IFNB promoter, thereby blocking promoter activation86. Alternatively, HSV infected cell protein 0 (ICP0) binds to host IRF3 and sequesters it together with CBP and p300 in nuclear bodies away from its normal binding sites on host genes87.
Figure 4. Inhibition of interferon-regulatory factor 3 (IRF3) and IRF7 by viral proteins.
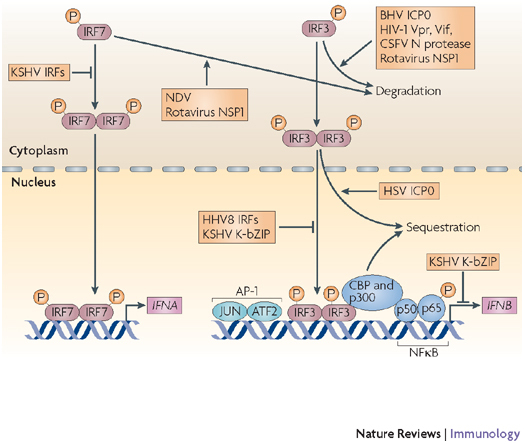
IRF3 and IRF7 are activated by phosphorylation, they then homodimerize and translocate to the nucleus, where they interact with their co-activators CREB-binding protein (CBP) and p300 and induce the expression of genes such as interferon-α (IFNA) and IFNB. Viruses inhibit IRFs by inducing their degradation, sequestering them or competing with them for binding to promoter sequences. AP1, activator protein 1; ATF2, activating transcription factor 2; BHV, bovine herpesvirus; CREB, cyclic-AMP-responsive-element-binding protein; CSFV, classical swine fever virus; HHV, human herpesvirus; HSV, herpes simplex virus; ICP0, infected cell protein 0; NDV, Newcastle disease virus; NFκB, nuclear factor-κB; NSP1, non-structural protein 1; KSHV, Kaposi's sarcoma-associated herpesvirus.
The related ICP0 protein from bovine herpesvirus also targets host IRF3, but it causes its degradation instead of temporarily sequestering it88. Virus-induced IRF3 degradation is a commonly used mechanism of viral evasion — other IRF3-interacting viral proteins that mediate this effect include the HIV-1 proteins Vpr and Vif89, and the Flaviviridae family protease N90. The rotavirus non-structural protein 1 (NSP1) targets IRF5 and IRF7, in addition to IRF3, for degradation91. Therefore, during viral infection the expression levels of the IRFs are dynamically regulated by viral factors. In fact, it has been suggested that one crucial function of the IFN effector protein ISG15 (IFN-stimulated protein of 15 kDa) (Fig. 1) is to counteract virus-mediated proteolysis of IRF3, as ISG15 covalently binds to and stabilizes IRF3, and can prevent its ubiquitin-mediated degradation by Newcastle disease virus92. Interestingly, VACV E3L protein has recently been shown to directly target and disable ISG15 function, which adds to the impressive repertoire of inhibitory strategies used by this viral protein93 (Table 1).
Viral modulation of NF κ B activity. The role of NFκB during viral infection is more complex than that of IRF3 and IRF7. Although NFκB is involved in inducing the production of IFNβ and chemokines, both of which have antiviral effects, NFκB also inhibits apoptosis and promotes proliferation of host cells, which is beneficial to the virus as apoptosis is an important host defence strategy for the deletion of virus-infected cells. Therefore, some viruses activate NFκB to prevent the cell from undergoing apoptosis94. However, at early stages of infection viruses can temporarily block NFκB activity to delay the innate immune response until an infection is established and the virus is less vulnerable to elimination. An example of this biphasic regulation of NFκB activity is observed during infection with African swine fever virus (ASFV). The ASFV protein A238L is an early expressed degradation-resistant IκBα homologue that inhibits NFκB activity at early stages of infection by sequestering host NFκB in the cytoplasm95. However, once infection progresses, the late-stage protein A224L is expressed, which activates NFκB and inhibits caspases96.
As in the case of IRFs, NFκB can also be regulated by viral proteins through the disruption of IKK function. Similarly to VACV N1L (described earlier), which targets multiple IKKs76, VACV B14R specifically interacts with IKKβ and prevents its activation by inhibiting the phosphorylation of its activation loop97. Conversely, the KSHV protein K13 interacts with the IKKα–IKKβ complex such that NFκB is activated98.
The many roles of NFκB illustrate the dynamic nature of transcription factor activation, which is continually adjusted to respond to the precise needs of the cell or, as it may be, the virus.
Beyond inhibition: viral subversion of PRRs
In contrast to viral evasion of host detection, it is now clear that viruses can subvert aspects of PRR-mediated signalling in addition to NFκB activation for their own benefit. There are now examples of viruses that use and manipulate TLR signalling pathways to create a host cytokine environment that favours the viral life cycle. Interestingly, RLRs seem less amenable to subversion, probably because they have a more important role in viral detection than TLRs.
Studies of TLR3-deficient mice that had been infected with WNV, influenza A virus and VACV showed that the absence of TLR3 actually favoured the host, suggesting that these viruses might use TLR3-mediated inflammation to disseminate and establish an infection99–101. For example, infection with WNV led to the TLR3-dependent production of pro-inflammatory cytokines and the subsequent disruption of the blood–brain barrier, which facilitated entry of the virus into the brain. By contrast, infection in the absence of TLR3 led to a reduced viral load and dampened inflammatory responses in the brain99. However, this may have been a consequence of mouse experimental models of viral infection in which the high viral titres that are used may lead to an overpronounced inflammatory response compared with the more protective TLR3-mediated host response that occurs during natural infections102. Evidence for a protective role for TLR3 in natural infections comes from recent studies showing that human patients that bear mutations in TLR3 are predisposed to HSV-associated encephalitis103.
Another potential effect of viral manipulation of TLRs involves the selective stimulation of TLR pathways that lead to the production of IL-10, a cytokine that is required for viral persistence104,105. For example, the VACV protein A52R inhibits the activation of NFκB by interacting with IRAK2 (as discussed earlier), but it also interacts with TRAF6 in a way that leads to the activation of the MAPK p38 and the subsequent activation of the IL10 promoter106. Thus, under conditions of TLR stimulation (as may occur during VACV infection), A52R inhibits the expression of NFκB-dependent genes, such as IL8, and instead enhances the production of IL-10 (Ref. 106), which might contribute to the role of A52R in VACV virulence37. The HTLV-I protein p30 achieves the same effect as A52R, but using a different mechanism40. Furthermore, mouse mammary tumour virus was shown to induce IL-10 production through stimulation of TLR4 in B cells, which was necessary for viral persistence in the host107. In mice lacking functional TLR4, the virus was eliminated by the cytotoxic immune response107.
Viruses can also use signalling proteins to support their life cycle. Although VACV K7R binds to DDX3 to inhibit its ability to enhance IFNβ expression81, HIV-1 and HCV go a step further as they manipulate host DDX3 for different purposes. Both viruses have proteins that are known to interact with DDX3: HCV requires DDX3 for replication108 and HIV-1 uses DDX3 to shuttle viral mRNAs out of the host-cell nucleus109. Therefore, for these viruses, targeting DDX3 may provide the dual benefit of suppressing the host type I IFN response in parallel to facilitating the viral life cycle.
Conclusions and future perspectives
In this Review, we have described how advances in the past 5 years of research have increased our understanding of how host cells sense viruses through PRRs and how viruses evade and subvert their detection. For some aspects of PRR-mediated signalling there is now unprecedented molecular detail, provided in part by 'seeing' the host immune response from the perspective of the virus. In the near future, we can expect a more complete description of a new family of PRRs, the cytoplasmic DNA receptors, and it will be of interest to decipher how large DNA viruses, such as poxviruses and herpesviruses, interact with these receptors.
Stepping back from what we now know about virus–host interactions, some common themes are becoming apparent. Viruses adopt many different strategies that inhibit PRR signalling: viral RNA can be 'hidden' from PRRs, RLRS can be directly inhibited, TIR-domain-containing adaptors can be targeted by both DNA (VACV) and RNA (HCV) viruses, TLR signalling pathways can be actively subverted to manipulate the cytokine environment and benefit viruses, IKK family members can be disabled to prevent the activation of transcription factors at common points of convergence of PRR pathways and IRFs can be degraded.
A key challenge in future studies will be to harness the information from viral evasion studies for the benefit of human health. It will be important to identify the in vitro and mouse in vivo studies that are directly relevant to humans, which is a difficult task given the host-range specificities of most viruses. However, there are already some clues, as the effect of polymorphisms in genes that encode PRRs and signalling components on the susceptibility to viral disease in humans is being uncovered20,110. In addition, it is now clear that the molecular basis of viral pathogenesis in humans can be defined by understanding how viral PRR inhibitors act10,11.
Further studies that aim to understand PRR evasion and subversion will have several implications in human health. First, the molecular mechanisms that are revealed by such studies are relevant to other pathogens and diseases, as the signalling components are shared among PRRs that sense other types of pathogen. For example, in addition to its recently described role in RLR-mediated IRF3 and IRF7 activation81, DDX3 was also found to be important for the induction of IFNβ expression by Listeria monocytogene s82. Second, PRR signalling discoveries are also relevant to non-pathogen-induced disease, as many PRR pathways can contribute to autoimmunity and inflammatory disease if they inappropriately sense host molecules. So, some of the identified targets of viral proteins (such as IRAK2 and DDX3) might turn out to be new drug targets for treating a range of diseases. Third, the viral proteins themselves, or derivatives of them, may have therapeutic uses in suppressing inappropriate PRR signalling; viruses have been 'examining' the host immune machinery for millions of years, so their proteins are optimized to specifically and maximally inhibit their targets, which is analogous to a naturally occurring drug-development programme. Finally, targeting viral inhibitors therapeutically may be beneficial for restoring innate immune defences during chronic viral infections, as has been shown in in vitro studies of HCV in which NS3–4A protease inhibitors restored RIG-I signalling during infection111.
Box 1 | Sensing of different classes of viruses.
Viruses with a double-stranded DNA (dsDNA) genome are detected by Toll-like receptor 9 (TLR9) and presumably other cytoplasmic DNA receptors. They can also produce dsRNA by convergent transcription and thus activate TLR3, IFN-inducible dsRNA-dependent protein kinase (PKR), and possibly melanoma differentiation-associated gene 5 (MDA5) and/or retinoic-acid-inducible gene I (RIG-I). dsDNA viruses include the poxviruses (such as vaccinia and variola virus), the herpesviruses (such as herpes simplex virus, human herpesvirus 8, bovine herpesviruses and Kaposi's sarcoma-associated herpesvirus) and African swine fever virus.
Viruses with a dsRNA genome may be detected by TLR3 (for example, rotaviruses) or by MDA5. Single-stranded RNA (ssRNA) viruses with a positive-sense RNA genome (one that can be directly used as mRNA) produce a dsRNA intermediate during replication, which can be recognized by MDA5 in the cytoplasm. This is the case for the picornaviruses (such as poliovirus and hepatitis A virus). The positive-sense genome of flaviviruses (for example, hepatitis C virus and West Nile virus) can also be recognized by RIG-I. Severe acute respiratory syndrome (SARS)-associated coronavirus may also be sensed by either RIG-I or MDA5.
Retroviruses (such as HIV-1 and human T-lymphotrophic virus) are a special group of positive-sense ssRNA viruses that reverse transcribe their RNA into DNA, which is then integrated into the host genome. HIV-1 ssRNA is sensed by TLR7 and TLR8. A role for RLRs or possibly cytoplasmic DNA receptors has not yet been investigated.
Negative-sense ssRNA viruses include the orthomyxoviruses (such as influenza A virus), the paramyxoviruses (such as mumps virus, Newcastle disease virus, respiratory syncytial virus and parainfluenza virus 5), the rhabdoviruses (rabies virus and vesicular stomatitis virus), and the hantaviruses, as well as lymphocytic choriomeningitis virus, Borna disease virus and Ebola virus. These viruses produce very limited amounts of dsRNA during their life cycle, but their ssRNA genome can activate RIG-I.
Box 2 | Recent lessons about PRR signalling pathways learnt from viruses.
As viruses target host proteins for the specific purpose of gaining an advantage over the host, newly discovered or ill-defined host proteins that interact with viral proteins can be implicated in the innate immune response. Recent examples show how understanding these virus–host interactions has clarified aspects of the pattern-recognition receptor (PRR) response. Vaccinia virus (VACV) protein A52R was found to be an inhibitor of Toll-like receptor (TLR)-mediated activation of nuclear factor-κB (NFκB), through an interaction with interleukin-1 receptor-associated kinase 2 (IRAK2)38. Prior to this it was unclear whether IRAK2 had a non-redundant role in PRR signalling, but further study of A52R showed that IRAK2 has a crucial role in human TLR signalling pathways38, and the study of IRAK2-deficient mice recently confirmed that it also has a role in mouse TLR signalling39. Studies using another VACV PRR signalling inhibitor, K7R, showed that human DEAD-box protein 3 (DDX3) is involved in PRR signalling that leads to the expression of interferon-β (IFNβ). More specifically, DDX3 was found to be the host target of K7R, which explained the ability of K7R to inhibit PRR-induced activation of IFN-regulatory factors (IRFs)81. It is now clear that DDX3 is a component of the IRF-activating complex that contains TANK-binding kinase 1 (TBK1) and inhibitor of NFκB kinase-ɛ (IKKɛ), which is essential for signalling by many PRRs. Additional aspects about PRR signalling were revealed by studies of the influenza A virus nonstructural protein 1 (NS1), this time clarifying the nature of the ligand for the PRR retinoic-acid-inducible gene I (RIG-I), which at that time was thought to be double-stranded RNA (dsRNA). NS1 was known to suppress the induction of type I IFN expression, and this was thought to be mediated through dsRNA binding. However, studies that aimed to elucidate the mechanism of action of NS1 revealed that influenza A virus infection produces very little dsRNA, and instead NS1 binds to and inhibits RIG-I directly51. This led the authors to conclude that RIG-I actually recognized influenza A virus genomic single-stranded RNA containing a 5′ triphosphate51.
Acknowledgements
The work in our laboratory is supported by Science Foundation Ireland and the Health Research Board.
Glossary
- Pattern-recognition receptor
A host receptor (such as Toll-like receptors) that can sense pathogen-associated molecular patterns and initiate signalling cascades that lead to an innate immune response.
- Endosome
An acidified intracellular vesicle that is derived from the plasma membrane. Endosomes are involved in sorting endocytosed material before it is transported to lysosomes, and they are also the site of action for certain Toll-like receptors that sense viral nucleic acid.
- Pathogen-associated molecular pattern
(PAMP). Molecular components of pathogens that are not normally found in mammals and are often common to whole classes of microorganisms. PAMPs are sensed by host pattern-recognition receptors. Double-stranded RNA is an important viral PAMP.
- Forward genetics
A classical genetic analysis approach that proceeds from phenotype to genotype by positional cloning or candidate-gene analysis.
- Reverse genetics
A genetic analysis that proceeds from genotype to phenotype by gene-manipulation techniques, such as homologous recombination in embryonic stem cells.
- Plasmacytoid dendritic cell
(pDC). An immature DC that belongs to a DC subset with a morphology that resembles that of a plasmablast. Plasmacytoid DCs produce large amounts of type I interferons in response to viral infection.
- TIR domain
An intracellular domain that is common to all members of the Toll-like receptor and interleukin-1 receptor family that can transduce a signal. This domain is essential for intracellular signalling and acts to recruit TIR-domain-containing adaptor proteins.
- Caspase-recruitment domain
A protein domain that is found in certain initiator caspases (for example, mammalian caspase-9) and their adaptor proteins (for example, apoptotic-protease-activating factor 1). This domain mediates protein–protein interactions.
Biographies
Andrew G. Bowie is an associate professor in immunology in the School of Biochemistry and Immunology at Trinity College Dublin, Ireland. His Ph.D. studies focused on the mechanisms by which nuclear factor-κB is regulated by oxidative stress, and his subsequent postdoctoral research in Trinity College focused on Toll-like receptor signal transduction. This led to his current interest in viral evasion and subversion of innate immune signalling, with a view to defining the role of proteins such as sterile α- and armadillo-motif-containing protein (SARM), interleukin-1-receptor-associated kinase 2 (IRAK2) and DEAD-box protein 3 (DDX3).
Leonie Unterholzner received her Ph.D. from the Gene Expression Programme at the European Molecular Biology Laboratory in Heidelberg, Germany, and the University of Dundee, UK. She is currently a postdoctoral fellow in the School of Biochemistry and Immunology at Trinity College Dublin, where she is investigating the function of new vaccinia-virus proteins that inhibit the interferon response in human cells.
Related links
FURTHER INFORMATION
References
- 1.Sadler AJ, Williams BR. Interferon-inducible antiviral effectors. Nature Rev. Immunol. 2008;8:559–568. doi: 10.1038/nri2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Janeway CA., Jr. Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb. Symp. Quant. Biol. 1989;54:1–13. doi: 10.1101/sqb.1989.054.01.003. [DOI] [PubMed] [Google Scholar]
- 3.Ishii KJ, et al. A Toll-like receptor-independent antiviral response induced by double-stranded B-form DNA. Nature Immunol. 2006;7:40–48. doi: 10.1038/ni1282. [DOI] [PubMed] [Google Scholar]
- 4.Takaoka A, et al. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature. 2007;448:501–505. doi: 10.1038/nature06013. [DOI] [PubMed] [Google Scholar]
- 5.Wang Z, et al. Regulation of innate immune responses by DAI (DLM-1/ZBP1) and other DNA-sensing molecules. Proc. Natl Acad. Sci. USA. 2008;105:5477–5482. doi: 10.1073/pnas.0801295105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jin MS, Lee JO. Structures of TLR-ligand complexes. Curr. Opin. Immunol. 2008;29:182–191. doi: 10.1016/j.coi.2008.06.002. [DOI] [PubMed] [Google Scholar]
- 7.Saito T, Gale M., Jr. Differential recognition of double-stranded RNA by RIG-I-like receptors in antiviral immunity. J. Exp. Med. 2008;205:1523–1527. doi: 10.1084/jem.20081210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Takeuchi O, Akira S. MDA5/RIG-I and virus recognition. Curr. Opin. Immunol. 2008;20:17–22. doi: 10.1016/j.coi.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 9.O'Neill LA. When signaling pathways collide: positive and negative regulation of toll-like receptor signal transduction. Immunity. 2008;29:12–20. doi: 10.1016/j.immuni.2008.06.004. [DOI] [PubMed] [Google Scholar]
- 10.Zampieri CA, Sullivan NJ, Nabel GJ. Immunopathology of highly virulent pathogens: insights from Ebola virus. Nature Immunol. 2007;8:1159–1164. doi: 10.1038/ni1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hartman AL, Ling L, Nichol ST, Hibberd ML. Whole-genome expression profiling reveals that inhibition of host innate immune response pathways by Ebola virus can be reversed by a single amino acid change in the VP35 protein. J. Virol. 2008;82:5348–5358. doi: 10.1128/JVI.00215-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Beutler B, et al. Genetic analysis of resistance to viral infection. Nature Rev. Immunol. 2007;7:753–766. doi: 10.1038/nri2174. [DOI] [PubMed] [Google Scholar]
- 13.Kurt-Jones EA, et al. Pattern recognition receptors TLR4 and CD14 mediate response to respiratory syncytial virus. Nature Immunol. 2000;1:398–401. doi: 10.1038/80833. [DOI] [PubMed] [Google Scholar]
- 14.Bowie A, et al. A46R and A52R from vaccinia virus are antagonists of host IL-1 and toll-like receptor signaling. Proc. Natl Acad. Sci. USA. 2000;97:10162–10167. doi: 10.1073/pnas.160027697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Janeway C, Jr., Medzhitov R. Viral interference with IL-1 and toll signaling. Proc. Natl Acad. Sci. USA. 2000;97:10682–10683. doi: 10.1073/pnas.97.20.10682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-κB by Toll-like receptor 3. Nature. 2001;413:732–738. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- 17.Edelmann KH, et al. Does Toll-like receptor 3 play a biological role in virus infections? Virology. 2004;322:231–238. doi: 10.1016/j.virol.2004.01.033. [DOI] [PubMed] [Google Scholar]
- 18.Schroder M, Bowie AG. TLR3 in antiviral immunity: key player or bystander? Trends Immunol. 2005;26:462–468. doi: 10.1016/j.it.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 19.Vercammen E, Staal J, Beyaert R. Sensing of viral infection and activation of innate immunity by Toll-like receptor 3. Clin. Microbiol Rev. 2008;21:13–25. doi: 10.1128/CMR.00022-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Quintana-Murci L, Alcais A, Abel L, Casanova J-L. Immunology in natura: clinical, epidemiological and evolutionary genetics of infectious diseases. Nature Immunol. 2007;8:1165–1171. doi: 10.1038/ni1535. [DOI] [PubMed] [Google Scholar]
- 21.Bowie AG. Translational mini-review series on Toll-like receptors: recent advances in understanding the role of Toll-like receptors in anti-viral immunity. Clin. Exp. Immunol. 2007;147:217–226. doi: 10.1111/j.1365-2249.2006.03301.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lund J, Sato A, Akira S, Medzhitov R, Iwasaki A. Toll-like receptor 9-mediated recognition of herpes simplex virus-2 by plasmacytoid dendritic cells. J. Exp. Med. 2003;198:513–520. doi: 10.1084/jem.20030162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krug A, et al. Herpes simplex virus type 1 activates murine natural interferon-producing cells through toll-like receptor 9. Blood. 2004;103:1433–1437. doi: 10.1182/blood-2003-08-2674. [DOI] [PubMed] [Google Scholar]
- 24.Jung A, et al. Lymphocytoid choriomeningitis virus activates plasmacytoid dendritic cells and induces a cytotoxic T-cell response via MyD88. J. Virol. 2008;82:196–206. doi: 10.1128/JVI.01640-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.O'Neill LA, Bowie AG. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nature Rev. Immunol. 2007;7:353–364. doi: 10.1038/nri2079. [DOI] [PubMed] [Google Scholar]
- 26.Pobezinskaya YL, et al. The function of TRADD in signaling through tumor necrosis factor receptor 1 and TRIF-dependent Toll-like receptors. Nature Immunol. 2008;9:1047–1054. doi: 10.1038/ni.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Michallet MC, et al. TRADD protein is an essential component of the RIG-like helicase antiviral pathway. Immunity. 2008;28:651–661. doi: 10.1016/j.immuni.2008.03.013. [DOI] [PubMed] [Google Scholar]
- 28.Ermolaeva MA, et al. Function of TRADD in tumor necrosis factor receptor 1 signaling and in TRIF-dependent inflammatory responses. Nature Immunol. 2008;9:1037–1046. doi: 10.1038/ni.1638. [DOI] [PubMed] [Google Scholar]
- 29.Haga IR, Bowie AG. Evasion of innate immunity by vaccinia virus. Parasitology. 2005;130:S11–S25. doi: 10.1017/S0031182005008127. [DOI] [PubMed] [Google Scholar]
- 30.Hurst T, Bowie AG. Innate immune signaling pathways: lessons from vaccinia virus. Future Virol. 2008;3:147–156. [Google Scholar]
- 31.Stack Julianne, Haga Ismar R., Schröder Martina, Bartlett Nathan W., Maloney Geraldine, Reading Patrick C., Fitzgerald Katherine A., Smith Geoffrey L., Bowie Andrew G. Vaccinia virus protein A46R targets multiple Toll-like–interleukin-1 receptor adaptors and contributes to virulence. The Journal of Experimental Medicine. 2005;201(6):1007–1018. doi: 10.1084/jem.20041442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carty M, et al. The human adaptor SARM negatively regulates adaptor protein TRIF-dependent Toll-like receptor signaling. Nature Immunol. 2006;7:1074–1081. doi: 10.1038/ni1382. [DOI] [PubMed] [Google Scholar]
- 33.Newman RM, Salunkhe P, Godzik A, Reed JC. Identification and characterization of a novel bacterial virulence factor that shares homology with mammalian Toll/interleukin-1 receptor family proteins. Infect. Immun. 2006;74:594–601. doi: 10.1128/IAI.74.1.594-601.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cirl C, et al. Subversion of Toll-like receptor signaling by a unique family of bacterial Toll/interleukin-1 receptor domain-containing proteins. Nature Med. 2008;14:399–406. doi: 10.1038/nm1734. [DOI] [PubMed] [Google Scholar]
- 35.Li, K. et al. Immune evasion by hepatitis C virus NS3/4A protease-mediated cleavage of the Toll-like receptor 3 adaptor protein TRIF. Proc. Natl Acad. Sci. USA102, 2992–2997 (2005). This is the first demonstration that HCV can target TLR3 by using the viral protease NS3–4A to cleave and disable TRIF. [DOI] [PMC free article] [PubMed]
- 36.Abe T, et al. Hepatitis C virus nonstructural protein 5A modulates the Toll-like receptor-MyD88-dependent signaling pathway in macrophage cell lines. J. Virol. 2007;81:8953–8966. doi: 10.1128/JVI.00649-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Harte MT, et al. The poxvirus protein A52R targets Toll-like receptor signaling complexes to suppress host defense. J. Exp. Med. 2003;197:343–351. doi: 10.1084/jem.20021652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Keating SE, Maloney GM, Moran EM, Bowie AG. IRAK-2 participates in multiple toll-like receptor signaling pathways to NFκB via activation of TRAF6 ubiquitination. J. Biol. Chem. 2007;282:33435–33443. doi: 10.1074/jbc.M705266200. [DOI] [PubMed] [Google Scholar]
- 39.Kawagoe T, et al. Sequential control of Toll-like receptor-dependent responses by IRAK1 and IRAK2. Nature Immunol. 2008;9:684–691. doi: 10.1038/ni.1606. [DOI] [PubMed] [Google Scholar]
- 40.Datta A, Sinha-Datta U, Dhillon NK, Buch S, Nicot C. The HTLV-I p30 interferes with TLR4 signaling and modulates the release of pro- and anti-inflammatory cytokines from human macrophages. J. Biol. Chem. 2006;281:23414–23424. doi: 10.1074/jbc.M600684200. [DOI] [PubMed] [Google Scholar]
- 41.Wilson JR, de Sessions PF, Leon MA, Scholle F. West Nile Virus nonstructural protein 1 inhibits TLR-3 signal transduction. J. Virol. 2008;82:8262–8271. doi: 10.1128/JVI.00226-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Macdonald A, Harris M. Hepatitis C virus NS5A: tales of a promiscuous protein. J. Gen. Virol. 2004;85:2485–2502. doi: 10.1099/vir.0.80204-0. [DOI] [PubMed] [Google Scholar]
- 43.Yoneyama M, et al. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nature Immunol. 2004;5:730–737. doi: 10.1038/ni1087. [DOI] [PubMed] [Google Scholar]
- 44.Yoneyama M, et al. Shared and unique functions of the DExD/H-Box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J. Immunol. 2005;175:2851–2858. doi: 10.4049/jimmunol.175.5.2851. [DOI] [PubMed] [Google Scholar]
- 45.Andrejeva J, et al. The V proteins of paramyxoviruses bind the IFN-inducible RNA helicase, MDA-5, and inhibit its activation of the IFN-β promoter. Proc. Natl Acad. Sci. USA. 2004;101:17264–17269. doi: 10.1073/pnas.0407639101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kawai T, et al. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nature Immunol. 2005;6:981–988. doi: 10.1038/ni1243. [DOI] [PubMed] [Google Scholar]
- 47.Seth RB, Sun L, Ea C-K, Chen ZJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-κB and IRF3. Cell. 2005;122:669–682. doi: 10.1016/j.cell.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 48.Meylan E, et al. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature. 2005;437:1167–1172. doi: 10.1038/nature04193. [DOI] [PubMed] [Google Scholar]
- 49.Xu L-G, et al. VISA is an adapter protein required for virus-triggered IFN-β signaling. Mol. Cell. 2005;19:727–740. doi: 10.1016/j.molcel.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 50.Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. 2008;455:674–678. doi: 10.1038/nature07317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pichlmair A, et al. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science. 2006;314:997–1001. doi: 10.1126/science.1132998. [DOI] [PubMed] [Google Scholar]
- 52.Hornung V, et al. 5′-triphosphate RNA is the ligand for RIG-I. Science. 2006;314:994–997. doi: 10.1126/science.1132505. [DOI] [PubMed] [Google Scholar]
- 53.Cui S, et al. The C-terminal regulatory domain is the RNA 5′-triphosphate sensor of RIG-I. Mol. Cell. 2008;29:169–179. doi: 10.1016/j.molcel.2007.10.032. [DOI] [PubMed] [Google Scholar]
- 54.Saito T, Owen DM, Jiang F, Marcotrigiano J, Gale M. Innate immunity induced by composition-dependent RIG-I recognition of hepatitis C virus RNA. Nature. 2008;454:523–527. doi: 10.1038/nature07106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kato H, et al. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J. Exp. Med. 2008;205:1601–1610. doi: 10.1084/jem.20080091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gitlin L, et al. Essential role of MDA-5 in type I IFN responses to polyriboinosinic:polyribocytidylic acid and encephalomyocarditis picornavirus. Proc. Natl Acad. Sci. USA. 2006;103:8459–8464. doi: 10.1073/pnas.0603082103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kato H, et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. 2006;441:101–105. doi: 10.1038/nature04734. [DOI] [PubMed] [Google Scholar]
- 58.Plotch SJ, Bouloy M, Ulmanen I, Krug RM. A unique cap(m7GpppXm)-dependent influenza virion endonuclease cleaves capped RNAs to generate the primers that initiate viral RNA transcription. Cell. 1981;23:847–858. doi: 10.1016/0092-8674(81)90449-9. [DOI] [PubMed] [Google Scholar]
- 59.Habjan M, et al. Processing of genome 5′ termini as a strategy of negative-strand RNA viruses to avoid RIG-I-dependent interferon induction. PLoS ONE. 2008;3:e2032. doi: 10.1371/journal.pone.0002032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Flanegan JB, Petterson RF, Ambros V, Hewlett NJ, Baltimore D. Covalent linkage of a protein to a defined nucleotide sequence at the 5′-terminus of virion and replicative intermediate RNAs of poliovirus. Proc. Natl Acad. Sci. USA. 1977;74:961–965. doi: 10.1073/pnas.74.3.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lee YF, Nomoto A, Detjen BM, Wimmer E. A protein covalently linked to poliovirus genome RNA. Proc. Natl Acad. Sci. USA. 1977;74:59–63. doi: 10.1073/pnas.74.1.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Weber F, Wagner V, Rasmussen SB, Hartmann R, Paludan SR. Double-stranded RNA is produced by positive-strand RNA viruses and DNA viruses but not in detectable amounts by negative-strand RNA viruses. J. Virol. 2006;80:5059–5064. doi: 10.1128/JVI.80.10.5059-5064.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Haasnoot J, et al. The Ebola virus VP35 protein is a suppressor of RNA silencing. PLoS Pathog. 2007;3:e86. doi: 10.1371/journal.ppat.0030086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Weeks KM, Ampe C, Schultz SC, Steitz TA, Crothers DM. Fragments of the HIV-1 Tat protein specifically bind TAR RNA. Science. 1990;249:1281–1285. doi: 10.1126/science.2205002. [DOI] [PubMed] [Google Scholar]
- 65.Brand SR, Kobayashi R, Mathews MB. The Tat protein of human immunodeficiency virus type 1 is a substrate and inhibitor of the interferon-induced, virally activated protein kinase, PKR. J. Biol. Chem. 1997;272:8388–8395. doi: 10.1074/jbc.272.13.8388. [DOI] [PubMed] [Google Scholar]
- 66.Mibayashi M, et al. inhibition of retinoic acid-inducible gene I-mediated induction of β interferon by the NS1 protein of influenza A virus. J. Virol. 2007;81:514–524. doi: 10.1128/JVI.01265-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Barral PM, et al. MDA-5 is cleaved in poliovirus-infected cells. J. Virol. 2007;81:3677–3684. doi: 10.1128/JVI.01360-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yang Y, et al. Disruption of innate immunity due to mitochondrial targeting of a picornaviral protease precursor. Proc. Natl Acad. Sci. USA. 2007;104:7253–7258. doi: 10.1073/pnas.0611506104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Loo YM, et al. Viral and therapeutic control of IFN-β promoter stimulator 1 during hepatitis C virus infection. Proc. Natl Acad. Sci. USA. 2006;103:6001–6006. doi: 10.1073/pnas.0601523103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lin R, et al. Dissociation of a MAVS/IPS-1/VISA/Cardif-IKKɛ molecular complex from the mitochondrial outer membrane by hepatitis C virus NS3–4A proteolytic cleavage. J. Virol. 2006;80:6072–6083. doi: 10.1128/JVI.02495-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Stetson DB, Medzhitov R. Recognition of cytosolic DNA activates an IRF3-dependent innate immune response. Immunity. 2006;24:93–103. doi: 10.1016/j.immuni.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 72.Okabe Y, Kawane K, Akira S, Taniguchi T, Nagata S. Toll-like receptor-independent gene induction program activated by mammalian DNA escaped from apoptotic DNA degradation. J. Exp. Med. 2005;202:1333–1339. doi: 10.1084/jem.20051654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nociari M, Ocheretina O, Schoggins JW, Falck-Pedersen E. Sensing infection by adenovirus: Toll-like receptor-independent viral DNA recognition signals activation of the interferon regulatory factor 3 master regulator. J. Virol. 2007;81:4145–4157. doi: 10.1128/JVI.02685-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ishii KJ, et al. TANK-binding kinase-1 delineates innate and adaptive immune responses to DNA vaccines. Nature. 2008;451:725–729. doi: 10.1038/nature06537. [DOI] [PubMed] [Google Scholar]
- 75.Otsuka M, et al. Interaction between the HCV NS3 protein and the host TBK1 protein leads to inhibition of cellular antiviral responses. Hepatology. 2005;41:1004–1012. doi: 10.1002/hep.20666. [DOI] [PubMed] [Google Scholar]
- 76.DiPerna G, et al. Poxvirus protein N1L targets the I-κB kinase complex, inhibits signaling to NF-κB by the tumor necrosis factor superfamily of receptors, and inhibits NF-κB and IRF3 signaling by Toll-like receptors. J. Biol. Chem. 2004;279:36570–36578. doi: 10.1074/jbc.M400567200. [DOI] [PubMed] [Google Scholar]
- 77.Unterstab G, et al. Viral targeting of the interferon-β-inducing Traf family member-associated NF-κB activator (TANK)-binding kinase-1. Proc. Natl Acad. Sci. USA. 2005;102:13640–13645. doi: 10.1073/pnas.0502883102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Brzozka K, Finke S, Conzelmann K-K. Identification of the rabies virus α/β interferon antagonist: phosphoprotein P interferes with phosphorylation of interferon regulatory factor 3. J. Virol. 2005;79:7673–7681. doi: 10.1128/JVI.79.12.7673-7681.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Alff PJ, Sen N, Gorbunova E, Gavrilovskaya IN, Mackow ER. The NY-1 hantavirus Gn cytoplasmic tail co-precipitates TRAF3 and inhibits cellular interferon responses by disrupting TBK–TRAF3 complex formation. J. Virol. 2008;80:9676–9686. doi: 10.1128/JVI.00290-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Devaraj SG, et al. Regulation of IRF-3-dependent innate immunity by the papain-like protease domain of the severe acute respiratory syndrome coronavirus. J. Biol. Chem. 2007;282:32208–32221. doi: 10.1074/jbc.M704870200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Schroder M, Baran M, Bowie AG. Viral targeting of DEAD box protein 3 reveals its role in TBK1/IKKvarɛ-mediated IRF activation. Embo J. 2008;27:2147–2157. doi: 10.1038/emboj.2008.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Soulat D, et al. The DEAD-box helicase DDX3X is a critical component of the TANK-binding kinase 1-dependent innate immune response. Embo J. 2008;27:2135–2146. doi: 10.1038/emboj.2008.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lu LL, Puri M, Horvath CM, Sen GC. Select paramyxoviral V proteins inhibit IRF3 activation by acting as alternative substrates for inhibitor of κB kinase ɛ (IKKɛ)/TBK1. J. Biol. Chem. 2008;283:14269–14276. doi: 10.1074/jbc.M710089200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lin R, et al. HHV-8 encoded vIRF-1 represses the interferon antiviral response by blocking IRF-3 recruitment of the CBP/p300 coactivators. Oncogene. 2001;20:800–811. doi: 10.1038/sj.onc.1204163. [DOI] [PubMed] [Google Scholar]
- 85.Joo CH, et al. Inhibition of interferon regulatory factor 7 (IRF7)-mediated interferon signal transduction by the Kaposi's sarcoma-associated herpesvirus viral IRF homolog vIRF3. J. Virol. 2007;81:8282–8292. doi: 10.1128/JVI.00235-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lefort S, Soucy-Faulkner A, Grandvaux N, Flamand L. Binding of Kaposi's sarcoma-associated herpesvirus K-bZIP to interferon-responsive factor 3 elements modulates antiviral gene expression. J. Virol. 2007;81:10950–10960. doi: 10.1128/JVI.00183-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Melroe GT, Silva L, Schaffer PA, Knipe DM. Recruitment of activated IRF-3 and CBP/p300 to herpes simplex virus ICP0 nuclear foci: potential role in blocking IFN-β induction. Virology. 2007;360:305–321. doi: 10.1016/j.virol.2006.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Saira K, Zhou Y, Jones C. The infected cell protein 0 encoded by bovine herpesvirus 1 (bICP0) induces degradation of interferon response factor 3 and, consequently, inhibits β interferon promoter activity. J. Virol. 2007;81:3077–3086. doi: 10.1128/JVI.02064-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Okumura A, et al. HIV-1 accessory proteins VPR and Vif modulate antiviral response by targeting IRF-3 for degradation. Virology. 2008;373:85–97. doi: 10.1016/j.virol.2007.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bauhofer O, et al. Classical swine fever virus Npro interacts with interferon regulatory factor 3 and induces its proteasomal degradation. J. Virol. 2007;81:3087–3096. doi: 10.1128/JVI.02032-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Barro M, Patton JT. Rotavirus NSP1 inhibits expression of type I interferon by antagonizing the function of interferon regulatory factors IRF3, IRF5, and IRF7. J. Virol. 2007;81:4473–4481. doi: 10.1128/JVI.02498-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lu G, et al. ISG15 enhances the innate antiviral response by inhibition of IRF-3 degradation. Cell. Mol. Biol. (Noisy-le-grand) 2006;52:29–41. [PubMed] [Google Scholar]
- 93.Guerra S, Caceres A, Knobeloch KP, Horak I, Esteban M. Vaccinia virus E3 protein prevents the antiviral action of ISG15. PLoS Pathog. 2008;4:e1000096. doi: 10.1371/journal.ppat.1000096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hiscott J, Nguyen TL, Arguello M, Nakhaei P, Paz S. Manipulation of the nuclear factor-κB pathway and the innate immune response by viruses. Oncogene. 2006;25:6844–6867. doi: 10.1038/sj.onc.1209941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Tait SW, Reid EB, Greaves DR, Wileman TE, Powell PP. Mechanism of inactivation of NF-κB by a viral homologue of IκBα. Signal-induced release of IκBα results in binding of the viral homologue to NF-κB. J. Biol. Chem. 2000;275:34656–34664. doi: 10.1074/jbc.M000320200. [DOI] [PubMed] [Google Scholar]
- 96.Rodriguez CI, et al. African swine fever virus IAP-like protein induces the activation of nuclear factor κB. J. Virol. 2002;76:3936–3942. doi: 10.1128/JVI.76.8.3936-3942.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Chen RAJ, Ryzhakov G, Cooray S, Randow F, Smith GL. Inhibition of IκB kinase by vaccinia virus virulence factor B14. PLoS Pathog. 2008;4:e22. doi: 10.1371/journal.ppat.0040022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Matta H, et al. Kaposi's sarcoma-associated herpesvirus (KSHV) oncoprotein K13 bypasses TRAFs and directly interacts with the IκB kinase complex to selectively activate NF-κB without JNK activation. J. Biol. Chem. 2007;282:24858–24865. doi: 10.1074/jbc.M700118200. [DOI] [PubMed] [Google Scholar]
- 99.Wang T, et al. Toll-like receptor 3 mediates West Nile virus entry into the brain causing lethal encephalitis. Nature Med. 2004;10:1366–1373. doi: 10.1038/nm1140. [DOI] [PubMed] [Google Scholar]
- 100.Le Goffic R, et al. Detrimental contribution of the Toll-like receptor (TLR)3 to influenza A virus-induced acute pneumonia. PLoS Pathog. 2006;2:e53. doi: 10.1371/journal.ppat.0020053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hutchens M., Luker K. E., Sottile P., Sonstein J., Lukacs N. W., Nunez G., Curtis J. L., Luker G. D. TLR3 Increases Disease Morbidity and Mortality from Vaccinia Infection. The Journal of Immunology. 2007;180(1):483–491. doi: 10.4049/jimmunol.180.1.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Gowen BB, et al. TLR3 deletion limits mortality and disease severity due to Phlebovirus infection. J. Immunol. 2006;177:6301–6307. doi: 10.4049/jimmunol.177.9.6301. [DOI] [PubMed] [Google Scholar]
- 103.Zhang SY, et al. TLR3 deficiency in patients with herpes simplex encephalitis. Science. 2007;317:1522–1527. doi: 10.1126/science.1139522. [DOI] [PubMed] [Google Scholar]
- 104.Brooks DG, et al. Interleukin-10 determines viral clearance or persistence in vivo. Nature Med. 2006;12:1301–1309. doi: 10.1038/nm1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ejrnaes M, et al. Resolution of a chronic viral infection after interleukin-10 receptor blockade. J. Exp. Med. 2006;203:2461–2472. doi: 10.1084/jem.20061462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Maloney G, Schroder M, Bowie AG. Vaccinia virus protein A52R activates p38 mitogen-activated protein kinase and potentiates lipopolysaccharide-induced interleukin-10. J. Biol. Chem. 2005;280:30838–30844. doi: 10.1074/jbc.M501917200. [DOI] [PubMed] [Google Scholar]
- 107.Jude BA, et al. Subversion of the innate immune system by a retrovirus. Nature Immunol. 2003;4:573–578. doi: 10.1038/ni926. [DOI] [PubMed] [Google Scholar]
- 108.Ariumi Y, et al. DDX3 DEAD-box RNA helicase is required for hepatitis C virus RNA replication. J. Virol. 2007;81:13922–13926. doi: 10.1128/JVI.01517-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Yedavalli VSRK, Neuveut C, Chi Y-H, Kleiman L, Jeang K-T. Requirement of DDX3 DEAD Box RNA helicase for HIV-1 Rev-RRE export function. Cell. 2004;119:381–392. doi: 10.1016/j.cell.2004.09.029. [DOI] [PubMed] [Google Scholar]
- 110.Awomoyi AA, et al. Association of TLR4 polymorphisms with symptomatic respiratory syncytial virus infection in high-risk infants and young children. J. Immunol. 2007;179:3171–3177. doi: 10.4049/jimmunol.179.5.3171. [DOI] [PubMed] [Google Scholar]
- 111.Johnson CL, Owen DM, Gale M., Jr. Functional and therapeutic analysis of hepatitis c virus NS3.4a protease control of antiviral immune defense. J. Biol. Chem. 2007;282:10792–10803. doi: 10.1074/jbc.M610361200. [DOI] [PubMed] [Google Scholar]
- 112.Cardenas WB, et al. Ebola virus VP35 protein binds double-stranded RNA and inhibits α/β interferon production induced by RIG-I signaling. J. Virol. 2006;80:5168–5178. doi: 10.1128/JVI.02199-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Gale MJ, Jr, et al. Evidence that hepatitis C virus resistance to interferon is mediated through repression of the PKR protein kinase by the nonstructural 5A protein. Virology. 1997;230:217–227. doi: 10.1006/viro.1997.8493. [DOI] [PubMed] [Google Scholar]
- 114.Taguchi T, et al. Hepatitis C virus NS5A protein interacts with 2′, 5′-oligoadenylate synthetase and inhibits antiviral activity of IFN in an IFN sensitivity-determining region-independent manner. J. Gen. Virol. 2004;85:959–969. doi: 10.1099/vir.0.19513-0. [DOI] [PubMed] [Google Scholar]
- 115.Chang HW, Watson JC, Jacobs BL. The E3L gene of vaccinia virus encodes an inhibitor of the interferon-induced, double-stranded RNA-dependent protein kinase. Proc. Natl Acad. Sci. USA. 1992;89:4825–4829. doi: 10.1073/pnas.89.11.4825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Romano PR, et al. Inhibition of double-stranded RNA-dependent protein kinase PKR by vaccinia virus E3: role of complex formation and the E3 N-terminal domain. Mol. Cell. Biol. 1998;18:7304–7316. doi: 10.1128/mcb.18.12.7304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Hatada E, Fukuda R. Binding of influenza A virus NS1 protein to dsRNA in vitro. J. Gen. Virol. 1992;73:3325–3329. doi: 10.1099/0022-1317-73-12-3325. [DOI] [PubMed] [Google Scholar]