

Abstract
Will our increasing understanding of virus-host interactions translate into a new generation of antiviral therapeutics or steer us toward an expensive journey to nowhere?
Systems biology... is about putting together rather than taking apart, integration rather than reduction. It requires that we develop ways of thinking about integration that are as rigorous as our reductionist programmes, but different.... It means changing our philosophy, in the full sense of the term1.
Although traditional antiviral drug-discovery approaches have yielded notable successes in recent years, the propensity of viruses to develop drug resistance that results in suboptimal treatment outcomes owing to poor therapy compliance by patients is continuing to provide commercial and medical incentive for developing more innovative and effective therapeutics. With the increasing adoption of high-throughput technologies, the possibility of getting a more detailed view of virus-host interactions is coming into focus. But as information from 'omics experiments begins populating databases on a grand scale, our ability to stitch together such vast amounts of data and thereby investigate, model and understand biological processes at a systems level will be crucial to the pursuit of new therapeutic targets. In this article, we provide a snapshot of realized and potential applications of systems biology in antiviral research using specific case studies and discuss the associated shortcomings that will need to be addressed to advance these technologies to their fullest potential.
The current environment
Over the past few years, several major pharmaceutical companies have either deprioritized or discontinued their antiviral vaccine programs, while smaller and less risk-averse biotech comp-anies have entered the antiviral arena. The payoffs can be lucrative, with the antiviral and vaccine markets each grossing global sales of $10 billion in 2005, and several factors have contributed to the apparent resurgence of the antiviral and vaccine marketplace, including the emergence and spread of viral diseases worldwide, particularly HIV/AIDS, recent severe acute respiratory syndrome (SARS) and avian influenza outbreaks; the growing perceived and actual threats of bioterrorism; and recent approvals for vaccines against certain strains of human papilloma virus (HPV) associated with high rates of cervical cancer. This, in turn, is creating a demand for technological advances in molecular biology for the development of affordable and rapid viral assays, which will spur the growth of the molecular diagnostic industry.
As much as the global public health demands for antiviral and vaccine products grow, the fact is that antiviral therapeutics remain an extremely challenging goal. Inevitably, the industry will need to implement fundamental and innovative changes in its antiviral drug-discovery processes to be successful. The opportunities should be more ample than ever, with technological advances in whole-genome sequencing and profiling ushering in an era of large-scale, high-throughput biology.
In the field of virology specifically, the US National Center for Biotechnology Information's (NCBI; Bethesda, MD) GenBank contains an ever-growing list of over 2,000 completely sequenced viral genomes. At the same time, although there are approximately 60 US Food and Drug Administration (FDA)-approved antiviral drugs on the market, many either target the same viral proteins or are variations of the same drug. Furthermore, a large part of the human genome remains poorly annotated or understood, highlighting the limitations in our knowledge of host biology. The key question remains: how do viruses, which encode relatively few genes, constantly outmaneuver hosts like us, which encode >20,000 genes? We believe that integrated system-wide approaches (Fig. 1), including transcriptomics, metabolomics, proteomics and high-throughput techniques, combined with mathematical modeling and computational biology, hold great potential for understanding the biological complexity of virus-host interactions and translating it into preventive and personalized medicine to combat viral infections.
Figure 1.

Conceptual illustration of how various systems approaches can be applied to study virus-host interactions as a means to understand disease biology through model systems and chemical or genetic perturbations that would provide insights into a repertoire of novel targets for therapeutic intervention strategies, host factors for biomarker discovery and viral determinants for diagnostics.
A host-oriented antiviral discovery paradigm
Many of the current antiviral drugs are directed against virally encoded enzymes that are essential for viral replication. Despite the remarkable success of these viral-inhibitor drugs in controlling viral infections and disease progression (for example, combinations of HIV-1 reverse transcriptase and protease inhibitors in AIDS), they suffer from serious drawbacks and, in particular, have had limited success in eradicating chronic viral infections.
One drawback of this approach is many of these drugs can only be used to treat a specific viral species, or even subtype, because they are designed to target and inhibit specific viral enzymes. This narrow spectrum of action, while reducing the potential for toxicity associated with targeting of nonspecific enzymes, also greatly limits the usage and market potential of each of these drugs. Furthermore, many medically important viruses, such as HIV-1, influenza viruses and hepatitis C virus (HCV), have the inherent capacity to rapidly mutate and become resistant to even the most potent of drugs because of their low-fidelity replication mode. For this reason, single antiretroviral drugs targeting HIV-1 are no longer recommended for clinical use, necessitating the use of combination therapy.
Although combination therapy is more effective at suppressing viral replication, such regimens are expensive and are associated with greater adverse side effects, resulting in poor patient adherence. The alternative, to search for new generations of antiviral drugs, is made difficult by the fact that many clinically important viruses are small RNA viruses with genomes usually encoding no more than a dozen genes, with less than a handful of the products of those genes being enzymes with known binding pockets for small molecules and thus considered 'druggable' by the pharmaceutical industry.
For the reasons mentioned above, it is not unreasonable to argue that treating viral infection by stimulating, manipulating or subverting the host antiviral response or by targeting cellular enzymes or cofactors required for the viral life cycle could provide advantages over the traditional viral-inhibitor approach. Host cells offer a wealth of proven, druggable targets, such as cell surface receptors, protein kinases, nuclear receptors and proteasomes. Moreover, antiviral drugs targeting cellular pathways may be less susceptible to the emergence of drug resistance because the human genes encoding targeted cellular proteins are less likely to mutate in response to therapy. Indeed, experimental evidence supporting this notion is provided by the utility of pharmacological inhibitors of cyclin-dependent kinases (CDKs; Fig. 2) in blocking the replication of herpes simplex virus (HSV)-1 and HIV-1 and by the fact that attempts to isolate resistant strains of these viruses have been unsuccessful in cell culture systems2,3. Importantly, several CDK inhibitors are being developed as potential anticancer drugs, and thus far no major toxicity has been observed in human clinical trials. Thus, by combining drugs targeting common cellular pathways that are required for the life cycle of different viruses with antivirals and/or immunomodulators, we may be able to develop regimens capable of treating multiple viral diseases.
Figure 2. Example highlighting the distinct mechanisms of antiviral actions by pharmacological inhibitors of cellular cyclin-dependent kinases (CDKs) compared to conventional antiviral drugs.
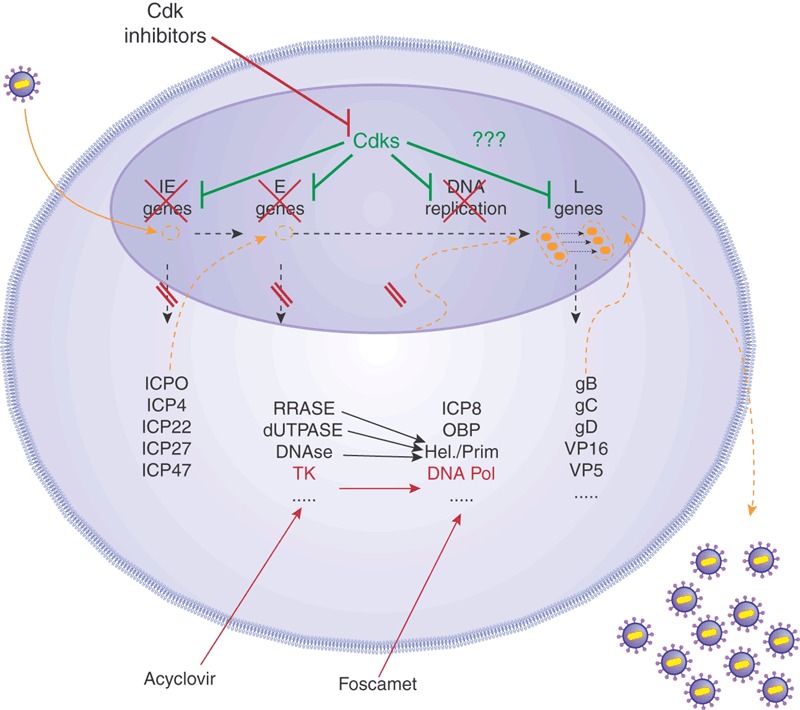
The illustration depicts the viral functions that are directly repressed by CDK inhibitors or by two typical antiviral drugs, acyclovir and foscarnet. Because CDKs are required for transcription of HSV immediate-early (IE), early (E) and perhaps late (L) genes, and for viral DNA replication, CDK inhibitors can target several stages of the viral replication cycle as opposed to blocking a specific viral target(s), such as the viral thymidine kinase (TK) or DNA polymerase (DNA pol). Thus, a single drug targeting a single cellular protein that is required for several viral functions is akin to combination antiviral therapy, in which each drug targets a different viral enzymes. Hel./prim, helicase-primase. Reproduced with permission from Luis Schang.
Targeting host cell factors for antiviral therapy should not be viewed as a major risk in the industry, as there is sound genetic evidence that specific genomic changes are well tolerated by mammals yet offer strong protection against viral infections. For example, naturally occurring homozygous mutation in the human CCR5 (C-C-motif receptor 5) protein confers resistance to HIV infection4, and Pfizer's (New York, NY) Selzentry/Celsentri (maraviroc) is an example of a recently approved drug targeting CCR5 for anti-HIV therapy5.
It is likely that other potential resistance mechanisms can also be exploited in mammals. Furthermore, the recent initiation by Romark Laboratories (Tampa, FL) of a phase 2 clinical trial of nitazoxanide for treating chronic hepatitis C in the United States represents the first of a new class of small-molecule drugs called the thiazolides that target cell signaling pathways used in viral replication. It is also noteworthy that the current standard treatment for HCV is a combination of a PEGylated interferon (IFN)-α and ribavirin, both of which are, ironically, nonspecific antiviral drugs targeting cellular factors6. IFNs activate multiple intracellular pathways to establish intracellular antiviral immunity, and ribavirin acts, at least in part, by depleting intracellular pools of guanine nucleotides through inhibition of the cellular inosine monophosphate dehydrogenase. The Toll-like receptor (TLR)-7 agonist imiquimod, an immune modifier that rapidly induces IFNs and other antiviral effector molecules, is approved for the treatment of genital warts caused by HSV. Indeed, there is considerable development of host response modulation in antiviral treatment. For example, systemic administration of ANA-245 (7-thia-8 oxoguanosine), a selective TLR7 agonist, in patients resulted in dose-dependent induction of immunological biomarkers and a statistically significant antiviral effect with relatively few and mild side effects7. In addition, TLR7 ligand SM360320 (9-benzyl-8-hydroxy-2-(2-methoxyethoxy)adenine) reduces HCV RNA levels in Huh-7 cells carrying the HCV replicon, at least in part by induction of type I IFN through TLR7 stimulation8. Besides TLR7 agonists, CPG10101—a TLR9 agonist and a CpG-containing oligonucleotide—in combination with PEGylated IFN and/or ribavirin also induced an early response in chronically HCV-infected patients with prior relapse response.
Critics will contend that such host-oriented pharmacological approaches will cause more side effects than those targeting viral targets, although antiviral drugs of other types are not immune to safety concerns—as demonstrated by the recent halting of several trials of promising antiviral candidates9,10. For those pathways that are vital to fundamental cellular processes, it will be necessary to modulate rather than ablate the enzymes involved so as to minimize the potential for severe side effects. If we have been successful in identifying small molecules to treat various chronic human disorders with manageable adverse effects, why can't we do the same for viral infections? As Paracelsus (1493–1541) so aptly noted: “All things are poison and nothing (is) without poison; only the dose makes that a thing is no poison.”
Hope and hype in systems biology
Although it appears that the majority of large pharmaceutical companies have yet to visibly embrace the potential of systems biology approaches11,12, a growing number of virologists in academia have embarked on a quest to refine and apply the tools of systems biology to understand more fully the interactions between virus and host (Box 1). Deciphering the mechanisms of any disease requires a deep knowledge of how multiple and concurrent signal-transduction pathways operate and are deregulated. Inescapably, the intricacies of signaling pathways, which are often highly interconnected and temporally and spatially regulated, can be dissected only by system-level approaches. Coupled with compendium analysis, systems biology has the potential to discover novel pro-host therapeutic targets. By providing a more robust overview of the host cellular machinery and its response and interaction with a virus, these kinds of analyses offer inroads toward the development of innovative therapeutics that can act in concert with the host defense mechanism. Several notable case studies support the use of full systems-scale analyses in rational antiviral drug development. These can be categorized into three general areas: viral pathogenesis, functional identification of novel viral and host genes as drug targets, and prediction of antiviral drug response and patient stratification.
Viral pathogenesis—a 90-year-old mystery solved
There is increasing evidence from functional genomics experiments that the patterns of cellular response to a variety of viral infections may reflect the pathogenic properties of the viruses. We contend that dissection of the critical, and often subtly different, cellular pathways will eventually unveil opportunities for manipulating the host immune response to fight off viral infection, control pathogenesis or both. A primary objective of the research in the laboratory of M.G.K. is to determine the viral and cellular factors responsible for the increased virulence of pandemic viral strains, particularly the 1918 influenza virus. We found that mice infected with a virus containing all eight genes from that pandemic virus showed dramatic activation of proinflammatory and cell-death pathways by 24 hours after infection—a phenomenon that continued unabated until the animals' death on day 5 (ref. 12). This contrasted with the situation with influenza viruses containing only subsets of the 1918 genes, which induced less marked host immune responses (as measured at the gene expression level using microarrays), produced less severe disease pathology and killed infected mice more slowly. This study12 represents the first comprehensive analysis of the global host response induced by the 1918 influenza virus in an animal model. As a specific illustration of where genomics approaches can lead us, in Figure 3 a biological network of selected genes induced twofold or more (P < 0.01) in the lungs of mice infected with the recombinant 1918 influenza virus (r1918), as compared with uninfected controls, is used to depict the activation of cell-death responses during r1918 infection.
Figure 3. Functional relationships of activated of cell death responses during r1918 influenza virus infection.
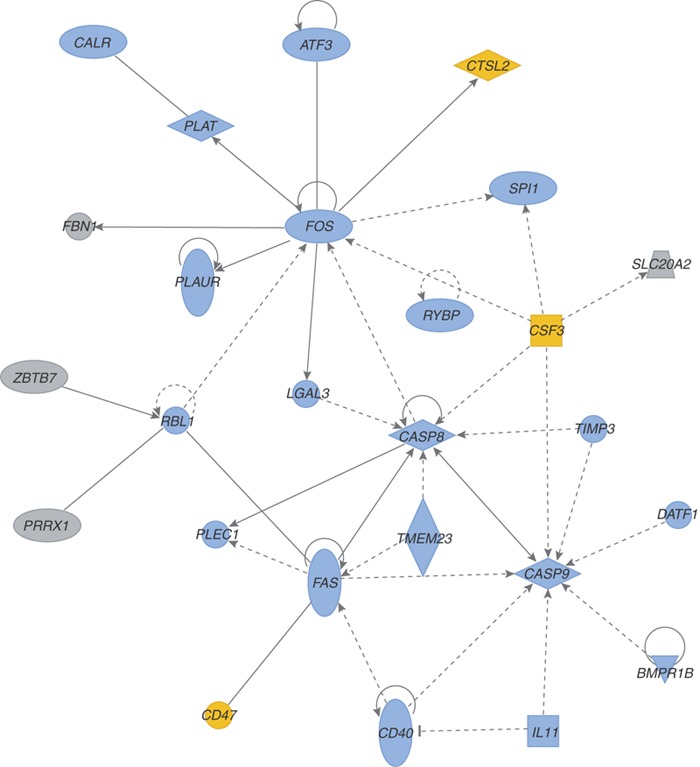
Biological network of selected genes that were induced at least twofold (P < 0.01) in r1918-infected mouse lung as compared with uninfected controls. This diagram shows the direct (solid lines) and indirect (dashed lines) interactions reported for these cell death– (blue shading) and immune response–related genes (yellow shading); gray denotes genes with multiple and/or undefined biological function. Biological network analysis was performed using the Ingenuity Systems (Redwood City, CA) Ingenuity Pathway Analysis program and showed statistical significance when assessed using Fisher's exact test, which was used to calculate a P value determining the probability that each biological function and/or disease assigned to that dataset was due to chance alone.
Because macaque monkeys have been used extensively as models for a wide variety of human diseases (and in AIDS research in particular), we have also taken on the challenge of developing genomic resources focused on macaque species. This effort has resulted in the sequencing of the rhesus macaque genome13 and the development of an oligonucleotide microarray containing over 17,000 unique macaque sequences14 (additional information is available through http://www.macaque.org). Infections of macaques with the reconstructed 1918 virus15 have also been performed by our collaborators, and the results of the genomic analysis of these infections have recently been published16. Our studies suggest that the lethality associated with the 1918 virus is a culmination of a cooperative interactions between the 1918 viral genes that results in a one-two blow to the host: an attenuation of the expression of specific innate immune response genes, including certain genes associated with the type I IFN response, followed by severe and persistent inflammatory and cell-death responses that may contribute to severe immunopathology. Indeed, these data suggest that the best treatment for such a virulent virus would be to combine an antiviral compound with existing anti-inflammatory drugs.
Work in the laboratory of M.G.K. has also addressed a wide range of other viral pathogens, including HCV17, Ebola virus18, West Nile virus19, SARS-associated coronavirus20, HSV21 and HIV22. The long-term goal is to assemble massive databases describing the host responses to multiple viral agents in multiple model systems. We believe that a careful dissection of these databases will reveal the secrets necessary to devise effective antiviral strategies against host cell processes. As microarray analysis measures gene expression only at the transcriptional level, we have also begun to incorporate other complementary approaches, such as proteomics and bioinformatics, to apply systems biology to the study of viral infections and virus-associated diseases23,24. By collecting 'clinical' data on humans and animals, we hope to integrate genomic, proteomic and clinical datasets to enhance our ability to understand the underlying mechanisms of host defense and response to viral infection. This will be important in dissecting the impact of viral infection on host pathways and vice versa.
It also is becoming apparent that functional 'virogenomics' is rapidly evolving, as exemplified by ongoing work in other laboratories. Ideker and colleagues25 have combined gene expression profiles with known protein-protein interactions to unravel transcriptional regulatory networks governing the latency and early reactivation phases of HIV-1. In another study, Uetz et al.26 have recently used a docking algorithm to infer virus-host protein interaction maps for two herpesviruses—Kaposi sarcoma–associated herpesvirus (KSHV) and varicella zoster virus (VZV)—on the basis of experimental data generated from yeast two-hybrid screening and comparative proteomics. These studies suggest not only that viral and host 'interactomes' possess distinct network topologies, but also that their interplay may lead to emergent new system properties that represent specific features of the viral pathogenesis.
In time, we should be able to combine experimental and computational studies to model viral infection processes. Chan et al.27 demonstrated an important step toward this goal by redesigning the genome of bacteriophage T7 and its host, Escherichia coli, incorporating parameters describing the host and phage nucleic acid polymerases and the host protein synthesis machinery, as well as the temporal expression of the phage genes. Their results suggest the intriguing notion that the primary limitation on T7 growth is the number of ribosomes, whereas too much polymerase actually causes excessive transcription of the phage early genes and diversion of the ribosomes away from making capsid proteins from late-gene transcripts.
Identification of new antiviral and pro-host drug targets
The availability of protein interaction networks and large-scale virus-host interaction data will boost our knowledge of the function of many still poorly characterized viral proteins as well as the large number of remaining 'unknown' genes in host pathways. This will lead to a more detailed understanding of viral pathogenesis and provide potential new targets for interfering with either the virus or host at key points in the infection. For instance, a recent unbiased global proteome analysis has led to the identification of previously unknown immunomodulatory functions for the MIR2 pathogenic protein of KSHV28, and a bioinformatics gene detection system capable of detecting a group of novel HIV regulatory genes, which repress expression of host genes by binding to their untranslated regions, has also been reported29. Indeed, similar viral regulatory genes, known as microRNAs, have been identified in several DNA viruses, including herpesviruses and polyomavirus30,31,32,33,34,35,36.
The laboratory of M.G.K has recently published the first host proteome analysis of HIV-1–infected cells37. This study analyzed the expression levels of 3,200 proteins in the CD4 CEMx174 cell line after infection with the Lai strain of HIV-1; the proteins were assessed using liquid chromatography–mass spectrometry coupled with stable isotope labeling and the accurate mass and time tag approach. The analysis revealed that 687 proteins changed in abundance at the peak of virus production at 36 hours after infection. Pathway analysis revealed that the differential expression of proteins was concentrated in select biological pathways, such as ubiquitin-conjugating enzymes, carrier proteins in nucleocytoplasmic transport, CDKs in cell cycle progression and pyruvate dehydrogenase of the citrate cycle pathways. Moreover, changes were observed in the abundance of proteins that are known to interact with HIV-1 viral proteins. This proteomic analysis captured changes in the host protein milieu at the time of robust virus production, depicting changes in cellular processes that may contribute to virus replication. These host proteins may indeed prove to be future targets for antiviral therapies.
Medically important RNA viruses such as HIV tend to encode 'Swiss army knife' proteins that are capable of performing more than one function at different stages during the viral life cycle. They often accomplish this by hijacking host enzymes to carry out post-translational modifications, such as protein phosphorylation, producing versions of the same viral proteins with different biological functions. Understanding the process and timing of viral gene expression and protein post-translational modifications and their role in the viral life cycle not only will reveal mechanisms of virus replication and/or pathogenesis, as discussed above, but may also identify host cellular targets, such as protein kinases, that are suitable for therapeutic intervention3. For example, microarray studies38 have revealed that elevated levels of prostaglandin E2 are required for efficient replication of human cytomegalovirus (HCMV) in fibroblasts, leading to the experimental demonstration that cyclooxygenase-2 inhibitors can block the accumulation of immediate-early viral mRNA and protein in cell culture. This suggests that drugs targeting this cellular pathway could be used to treat HCMV infection. Similarly, functional genomics studies have revealed that c-Kit is one of the most consistently KSHV-induced genes39. Inhibition of c-Kit activity with the pharmacological inhibitor of c-Kit signaling STI571 reverses the KSHV-induced morphological transformation in cell culture. Lck and heterogeneous nuclear ribonucleoproteins (hnRNPs) are upregulated in latently HIV-infected CD4+ T cells40. Interestingly, Lck signaling events can be linked to RNA processing and translation via the Lck kinase–mediated phosphorylation of hnRNP K, which may regulate HIV transcription, translation and/or RNA processing, suggesting that Lck could be a promising drug target for HIV latency.
Systems biology approaches using cell-based screens can identify drug candidates against host targets with unsuspected roles. A recent proof of concept for this strategy was provided by Sakamoto et al.41. Using an HCV replicon system, the researchers screened and identified a small-molecule HCV replication inhibitor, NA255, that worked by preventing the de novo synthesis of sphingolipids by inhibiting host serine palmitoyltransferase, disrupting assembly of HCV nonstructural proteins on lipid rafts. Coupled with genome-wide RNA interference screen, such systems cell biology screening could be a powerful approach for identifying host factors required for viral replication and pathogenesis. Indeed, Pelkmans et al.42 have demonstrated the power of using kinome-wide RNA interference (RNAi) screens to identify host factors that facilitate the infectious entry of vesicular stomatitis virus and simian virus 40. Interestingly, they found limited overlap between the kinases regulating the entry of the two viruses, suggesting that the viruses harness different host endocytic machinery to gain entry. This may offer a selective approach to target various classes of viruses. Taken together, these studies suggest that systems biology has the potential to uncover a common viral theme based on shared molecular signatures or networks that may be vulnerable to disruption and that it could lead to the development of broadly applicable control strategies for viruses.
Genetic variation and antiviral drug responses
A highly attractive niche for the application of high-throughput genomics to antiviral drug and vaccine development lies in surveying host and viral variation to identify sources of virulence and susceptibility (Box 2). The advent of high-throughput genotyping platforms has also made it possible to carry out unbiased genome-wide association studies to uncover medium-penetrance risk alleles. An illustrative example is the recent report of the first whole-genome association study to identify host determinants of HIV-1 infection43. Researchers found three polymorphisms that explain nearly 15% of the variation in HIV-1 viral load among individuals, which could lead to improved therapies and new targets for vaccine development. Pharmacogenomics initiatives will be enhanced by higher-density platforms, such as the '6.0 SNP array' that detects ∼900,000 single-nucleotide polymorphisms (SNPs) in a single experiment. Detailing the association between genotype and individual response to infection or treatment will allow physicians to limit the use of drugs that may be dangerous for subsets of the population to the groups of individuals that can benefit from these treatments. These kinds of studies are already underway with therapies for HIV44,45,46, HCV47,48, HPV49 and hepatitis B virus50.
Concluding remarks
We have entered into an unprecedented time of information production via high-throughput technologies and computational advances. But this is just the tip of the iceberg. The prospects for systems biology in studies of virus-host interactions are more exciting than ever with recent advances in noninvasive technologies to image fundamental processes (for example, transcriptional regulation, signal-transduction cascades, protein-protein interactions and cell trafficking) in living cells51,52,53 and animal models54. For example, a neglected area of investigation is the replication sites for positive-strand RNA viruses (such as HCV and the SARS coronavirus), which replicate their genomes on intracellular membrane compartments while simultaneously avoiding, minimizing or delaying double-stranded RNA–induced host antiviral responses, such as activation of protein kinase, RNA-activated (PKR) and RNase L or RNA interference55. Many of these intracellular compartments also harbor host defense sensors, such as TLR3, TLR7 and TLR9 on endosomal vesicles, and antiviral effectors, such as RIG-I (retinoic-acid-inducible gene I) and PERK (protein kinase (PKR)-like ER kinase) on mitochondria and endoplasmic reticulum, respectively. Despite being a major battleground in the host-virus conflict, the exact sites of viral RNA synthesis, the components of host and viral factors, and the organization and functioning of the intracellular membrane compartments are poorly understood. By integrating ultrastructural and live-animal imaging technologies with functional genomics and computational analysis, it should now be possible to characterize the features of membrane-associated RNA replication complexes in other viruses and generate a 'systems-based model' that will facilitate our understanding of the general principles and mechanisms of positive-strand RNA virus replication as well as of the viruses' evasion of the immune system.
In the field of antiviral therapy, where combination therapies are becoming the norm, a systems biology approach that allows one to formulate multicomponent drugs to achieve optimal efficacy with reduced mechanism-based toxicity will undoubtedly have a significant impact in antiviral drug discovery and development. In vaccine development, another promising approach is the development of newly emerging peptide microarrays that use antigen peptides as fixed probes and serum antibodies as targets to identify antibody reactivity patterns (and potentially valuable clinical biomarkers) involved in virus infections. This concept is highlighted by studies examining antibody responses to simian immunodeficiency virus and HIV using an array displaying peptides derived from viral amino acid sequences; these studies found that a reduction in the repertoire of the antibody response is associated with the development of AIDS56. By correlating the efficacy of vaccine candidates with host immune responses, we may gain a better molecular understanding of, and thus become better able to predict efficacy of, vaccine candidates55.
So why hasn't the promise of large-scale biology provided us yet with a raft of new antiviral therapeutics? Currently, systems biology approaches suffer from several experimental and computational drawbacks. High-throughput approaches are sensitive to the way in which samples are collected and handled, and a variety of factors, such as RNA and protein degradation and the presence of contaminating tissue, can influence gene expression and proteome analysis. Although a lot of time and effort has gone into ameliorating these kinds of problems to generate more reproducible results in genomics experiments, variability across platforms and between laboratories remains an issue for those attempting to integrate datasets from different sources. The challenge now is the need to integrate not only multiple levels of biological data from an individual experiment but also data from different groups for the same assay, and to translate high-throughput data into digested results that can be easily interpreted by a broader audience, including clinicians, governmental regulators and other scientists. This will require close collaborations among virologists, pathologists, clinicians, biologists, statisticians and bioinformaticians, and collaborative consortia and large-scale networked science with partnerships between industry and academia to ensure high quality of the samples, data generation, processing and analysis, as well as ease of data accessibility and interpretation.
Box 1: Virus-host interactions as a paradigm for systems analysis.
Researchers at the Institute for Systems Biology (ISB, Seattle, WA) have nicely summarized the properties of biological systems that make them attractive for systems-level exploration—emergent properties, robustness and modularity57. Emergence is a trait in which the whole is greater than the sum of the parts; robustness is characteristic of resilience to fluctuations in the immediate environment resulting from redundancy and control mechanisms; modularity is a phenomenon that explains the 'clustering' of parts into a functional or structural entity. That said, certain features of virus-host interactions make them particularly interesting for global investigations. A case in point for emergent properties is the discovery of aberrant innate immune responses in macaques lethally infected by the 1918 influenza virus16. Robustness of HIV through latency and rapid low-fidelity turnover is widely recognized as a factor for continued infection and resistance. Modularity in predicted varicella zoster virus–human and KSHV-human networks was seen in Uetz's work26, although the intraviral interactomes lacked clustering and were tightly connected. Taken together, these examples demonstrate that virus-host interplay provides a rich opportunity for large-scale analyses (see main text for a variety of illustrative studies).
Box 2: Systems virology—'viromics'.
Innovations in sequencing technologies, particularly the rapid, high-throughput pyrosequencing platforms, continue to transform large-scale biology. To illustrate this point, NCBI's GenBank contains an overwhelming ∼2,000 completely sequenced unique viral genomes, and ∼2,500 complete viral genomes of influenza A virus have been sequenced to date58. This, along with the '$1,000 genome' goals of the Personalized Human Genome project, necessitate advances in bioinformatics and computational biology to carry out whole-genome comparisons for intrahost and interhost strains as well as for host-host variation. A stimulating commentary by Holmes58 asks thought-provoking questions about the use of genomics data to understand evolutionary interactions between concurrently circulating pathogens in a host and their influence on disease etiology (for example, the rise of opportunistic pathogen-related infections with the HIV pandemic) and how host immunity shapes pathogen diversity and response to infection (for example, interactions among the four dengue serotypes). Furthermore, intrahost and interhost genetic variability is important in understanding cross-species transmission and emergence, with the avian H5N1 flu virus being a prime example. The fields of viral metagenomics and viral diagnostics are therefore poised for rapid expansion. Beyond the burgeoning expression profiling datasets in databases, such as NCBI's GEO, M.G.K.'s laboratory has generated over 5,000 array datasets over the past seven years using commercial and internally developed microarray platforms. Such datasets will provide clues to host determinants of infections and usher in an era of 'personalized viromics'. Along the same lines, Braga-Neto and colleagues59 provide an illuminating discussion on the overwhelming scale and potential of immunomics, which aims to uncover host immunological responses to antigens. These developments set the stage for systems-level analyses to develop better and safer personalized treatment and preventive modalities.
References
- 1.Noble D. The Music of Life: Biology Beyond the Genome. 2006. [Google Scholar]
- 2.Salerno D, et al. Direct inhibition of CDK9 blocks HIV-1 replication without preventing T-cell activation in primary human peripheral blood lymphocytes. Gene. 2007;405:65–78. doi: 10.1016/j.gene.2007.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schang LM. First demonstration of the effectiveness of inhibitors of cellular protein kinases in antiviral therapy. Expert Rev. Anti Infect. Ther. 2006;4:953–956. doi: 10.1586/14787210.4.6.953. [DOI] [PubMed] [Google Scholar]
- 4.de Angelis DS, Freire WS, Pannuti CS, Succi RC, Machado DM. CCR5 genotypes and progression to HIV disease in perinatally infected children. Braz. J. Infect. Dis. 2007;11:196–198. doi: 10.1590/S1413-86702007000200004. [DOI] [PubMed] [Google Scholar]
- 5.Wheeler J, McHale M, Jackson V, Penny M. Assessing theoretical risk and benefit suggested by genetic association studies of CCR5: experience in a drug development programme for maraviroc. Antivir. Ther. 2007;12:233–245. [PubMed] [Google Scholar]
- 6.Tan SL, He Y, Huang Y, Gale M., Jr. Strategies for hepatitis C therapeutic intervention: now and next. Curr. Opin. Pharmacol. 2004;4:465–470. doi: 10.1016/j.coph.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 7.Horsmans Y, et al. Isatoribine, an agonist of TLR7, reduces plasma virus concentration in chronic hepatitis C infection. Hepatology. 2005;42:724–731. doi: 10.1002/hep.20839. [DOI] [PubMed] [Google Scholar]
- 8.Lee J, et al. Activation of anti-hepatitis C virus responses via Toll-like receptor 7. Proc. Natl. Acad. Sci. USA. 2006;103:1828–1833. doi: 10.1073/pnas.0510801103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vanwolleghem T, et al. Ultra-rapid cardiotoxicity of the hepatitis C virus protease inhibitor BILN 2061 in the urokinase-type plasminogen activator mouse. Gastroenterology. 2007;133:1144–1155. doi: 10.1053/j.gastro.2007.07.007. [DOI] [PubMed] [Google Scholar]
- 10.ViroPharma Inc. Potential Safety Issue Identified in Ongoing Phase 2 Clinical Study of HCV-796 (press release) (ViroPharma Inc., Exton, Pennsylvania, USA, 2007).
- 11.Davidov E, Holland J, Marple E, Naylor S. Advancing drug discovery through systems biology. Drug Discov. Today. 2003;8:175–183. doi: 10.1016/S1359-6446(03)02600-X. [DOI] [PubMed] [Google Scholar]
- 12.Butcher EC, Berg EL, Kunkel EJ. Systems biology in drug discovery. Nat. Biotechnol. 2004;22:1253–1259. doi: 10.1038/nbt1017. [DOI] [PubMed] [Google Scholar]
- 13.Gibbs RA, et al. Evolutionary and biomedical insights from the rhesus macaque genome. Science. 2007;316:222–234. doi: 10.1126/science.1139247. [DOI] [PubMed] [Google Scholar]
- 14.Wallace JC, et al. High-density rhesus macaque oligonucleotide microarray design using early-stage rhesus genome sequence information and human genome annotations. BMC Genomics. 2007;8:28. doi: 10.1186/1471-2164-8-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tumpey TM, et al. Characterization of the reconstructed 1918 Spanish influenza pandemic virus. Science. 2005;310:77–80. doi: 10.1126/science.1119392. [DOI] [PubMed] [Google Scholar]
- 16.Kobasa D, et al. Aberrant innate immune response in lethal infection of macaques with the 1918 influenza virus. Nature. 2007;445:319–323. doi: 10.1038/nature05495. [DOI] [PubMed] [Google Scholar]
- 17.Walters KA, et al. Application of functional genomics to the chimeric mouse model of HCV infection: optimization of microarray protocols and genomics analysis. Virol. J. 2006;3:37–44. doi: 10.1186/1743-422X-3-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kash JC, et al. Global suppression of the host antiviral response by Ebola- and Marburgviruses: increased antagonism of the type 1 interferon response is associated with enhanced virulence. J. Virol. 2006;80:3009–3020. doi: 10.1128/JVI.80.6.3009-3020.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fredericksen BL, Smith M, Katze MG, Shi PY, Gale M., Jr. The host response to West Nile Virus infection limits viral spread through the activation of the interferon regulatory factor 3 pathway. J. Virol. 2004;78:7737–7747. doi: 10.1128/JVI.78.14.7737-7747.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baas T, Taubenberger J, Chong PY, Chui P, Katze MG. SARS-CoV virus-host interactions and comparative etiologies of acute respiratory distress syndrome as determined by transcriptional and cytokine profiling of formalin-fixed paraffin-embedded tissues. J. Interferon Cytokine Res. 2006;26:309–317. doi: 10.1089/jir.2006.26.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pasieka TJ, et al. Functional genomics analysis of herpes simplex virus type 1 counteraction of the host innate response. J. Virol. 2006;80:7600–7612. doi: 10.1128/JVI.00333-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li Y, Chan EY, Katze MG. Functional genomics analyses of differential macaque peripheral blood mononuclear cell infections by human immunodeficiency virus-1 and simian immunodeficiency virus. Virology. 2007;366:137–149. doi: 10.1016/j.virol.2007.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wallace JC, Korth MJ, Diamond DL, Proll SC, Katze MG. Virology in the 21st century: finding function with functional genomics. Future Virol. 2006;1:47–53. doi: 10.2217/17460794.1.1.47. [DOI] [Google Scholar]
- 24.Diamond DL, et al. Proteomic profiling of human liver biopsies: hepatitis C virus-induced fibrosis and mitochondrial dysfunction. Hepatology. 2007;46:649–657. doi: 10.1002/hep.21751. [DOI] [PubMed] [Google Scholar]
- 25.Bandyopadhyay S, Kelley R, Ideker T. Discovering regulated networks during HIV-1 latency and reactivation. Pac. Symp. Biocomput. 2006;11:354–366. [PMC free article] [PubMed] [Google Scholar]
- 26.Uetz P, et al. Herpesviral protein networks and their interaction with the human proteome. Science. 2006;311:239–242. doi: 10.1126/science.1116804. [DOI] [PubMed] [Google Scholar]
- 27.Chan LY, Kosuri S, Endy D. Refactoring bacteriophage T7. Mol. Syst. Biol. 2005;1:2005.0018. doi: 10.1038/msb4100025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bartee E, McCormack A, Fruh K. Quantitative membrane proteomics reveals new cellular targets of viral immune modulators. PLoS Pathog. 2006;2:e107. doi: 10.1371/journal.ppat.0020107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bentwich, I. Bioinformatically detectable group of novel HIV regulatory genes and uses thereof. US Patent 7,217,807 (2007).
- 30.Burnside J, et al. Marek's disease virus encodes MicroRNAs that map to meq and the latency-associated transcript. J. Virol. 2006;80:8778–8786. doi: 10.1128/JVI.00831-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cai X, et al. Kaposi's sarcoma-associated herpes-virus expresses an array of viral microRNAs in latently infected cells. Proc. Natl. Acad. Sci. USA. 2005;102:5570–5575. doi: 10.1073/pnas.0408192102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cui C, et al. Prediction and identification of herpes simplex virus 1-encoded microRNAs. J. Virol. 2006;80:5499–5508. doi: 10.1128/JVI.00200-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Grey F, et al. Identification and characterization of human cytomegalovirus-encoded microRNAs. J. Virol. 2005;79:12095–12099. doi: 10.1128/JVI.79.18.12095-12099.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pfeffer S. Identification of virally encoded microRNAs. Methods Enzymol. 2007;427:51–63. doi: 10.1016/S0076-6879(07)27003-X. [DOI] [PubMed] [Google Scholar]
- 35.Sullivan CS, Grundhoff AT, Tevethia S, Pipas JM, Ganem D. SV40-encoded microRNAs regulate viral gene expression and reduce susceptibility to cytotoxic T cells. Nature. 2005;435:682–686. doi: 10.1038/nature03576. [DOI] [PubMed] [Google Scholar]
- 36.Grundhoff A, Sullivan CS, Ganem D. A combined computational and microarray-based approach identifies novel microRNAs encoded by human gamma-herpes-viruses. RNA. 2006;12:733–750. doi: 10.1261/rna.2326106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chan EY, et al. Quantitative analysis of human immunodeficiency virus type 1-infected CD4+ cell proteome: dysregulated cell cycle progression and nuclear transport coincide with robust virus production. J. Virol. 2007;81:7571–7583. doi: 10.1128/JVI.00288-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhu H, Cong JP, Mamtora G, Gingeras T, Shenk T. Cellular gene expression altered by human cytomegalovirus: global monitoring with oligonucleotide arrays. Proc. Natl. Acad. Sci. USA. 1998;95:14470–14475. doi: 10.1073/pnas.95.24.14470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moses AV, et al. A functional genomics approach to Kaposi's sarcoma. Ann. NY Acad. Sci. 2002;975:180–191. doi: 10.1111/j.1749-6632.2002.tb05951.x. [DOI] [PubMed] [Google Scholar]
- 40.Chun TW, et al. Gene expression and viral prodution in latently infected, resting CD4+ T cells in viremic versus aviremic HIV-infected individuals. Proc. Natl. Acad. Sci. USA. 2003;100:1908–1913. doi: 10.1073/pnas.0437640100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sakamoto H, et al. Host sphingolipid biosynthesis as a target for hepatitis C virus therapy. Nat. Chem. Biol. 2005;1:333–337. doi: 10.1038/nchembio742. [DOI] [PubMed] [Google Scholar]
- 42.Pelkmans L, et al. Genome-wide analysis of human kinases in clathrin- and caveolae/raft-mediated endocytosis. Nature. 2005;436:78–86. doi: 10.1038/nature03571. [DOI] [PubMed] [Google Scholar]
- 43.Fellay J, et al. A whole-genome association study of major determinants for host control of HIV-1. Science. 2007;317:944–947. doi: 10.1126/science.1143767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Foulkes AS, et al. Associations among race/ethnicity, ApoC-III genotypes, and lipids in HIV-1-infected individuals on antiretroviral therapy. PLoS Med. 2006;3:e52. doi: 10.1371/journal.pmed.0030052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Haas DW. Human genetic variability and HIV treatment response. Curr. HIV/AIDS Rep. 2006;3:53–58. doi: 10.1007/s11904-006-0018-x. [DOI] [PubMed] [Google Scholar]
- 46.Dahri K, Ensom MH. Efavirenz and nevirapine in HIV-1 infection: is there a role for clinical pharmaco-kinetic monitoring? Clin. Pharmacokinet. 2007;46:109–132. doi: 10.2165/00003088-200746020-00002. [DOI] [PubMed] [Google Scholar]
- 47.Wright M, Grieve R, Roberts J, Main J, Thomas HC. Health benefits of antiviral therapy for mild chronic hepatitis C: randomised controlled trial and economic evaluation. Health Technol. Assess. 2006;10:1–113. doi: 10.3310/hta10210. [DOI] [PubMed] [Google Scholar]
- 48.Xie Y, et al. Predictive factors for sustained response to interferon treatment in patients with chronic hepatitis C: a randomized, open, and multi-center controlled trial. Hepatobiliary Pancreat. Dis. Int. 2005;4:213–219. [PubMed] [Google Scholar]
- 49.Angulo M, Carvajal-Rodriguez A. Evidence of recombination within human alpha-papillomavirus. Virol. J. 2007;4:33–40. doi: 10.1186/1743-422X-4-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ding X R, Yang J, Sun D C, Lou S K, Wang S Q. Whole genome expression profiling of hepatitis B virus-transfected cell line reveals the potential targets of anti-HBV drugs. The Pharmacogenomics Journal. 2007;8(1):61–70. doi: 10.1038/sj.tpj.6500459. [DOI] [PubMed] [Google Scholar]
- 51.Damm EM, Pelkmans L. Systems biology of virus entry in mammalian cells. Cell. Microbiol. 2006;8:1219–1227. doi: 10.1111/j.1462-5822.2006.00745.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Silver PA, Way JC. Molecular systems biology in drug development. Clin. Pharmacol. Ther. 2007;82:586–590. doi: 10.1038/sj.clpt.6100293. [DOI] [PubMed] [Google Scholar]
- 53.Giuliano KA, Johnston PA, Gough A, Taylor DL. Systems cell biology based on high-content screening. Methods Enzymol. 2006;414:601–619. doi: 10.1016/S0076-6879(06)14031-8. [DOI] [PubMed] [Google Scholar]
- 54.Piwnica-Worms D, Schuster DP, Garbow JR. Molecular imaging of host-pathogen interactions in intact small animals. Cell. Microbiol. 2004;6:319–331. doi: 10.1111/j.1462-5822.2004.00379.x. [DOI] [PubMed] [Google Scholar]
- 55.He ML, et al. Inhibition of SARS-associated coronavirus infection and replication by RNA interference. JAMA. 2003;290:2665–2666. doi: 10.1001/jama.290.20.2665. [DOI] [PubMed] [Google Scholar]
- 56.Neuman de Vegvar HE, et al. Microarray profiling of antibody responses against simian-human immunodeficiency virus: postchallenge convergence of reactivities independent of host histocompatibility type and vaccine regimen. J. Virol. 2003;77:11125–11138. doi: 10.1128/JVI.77.20.11125-11138.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Aderem A. Systems biology: its practice and challenges. Cell. 2005;121:511–513. doi: 10.1016/j.cell.2005.04.020. [DOI] [PubMed] [Google Scholar]
- 58.Holmes EC. Viral evolution in the genomic age. PLoS Biol. 2007;5:e278. doi: 10.1371/journal.pbio.0050278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Braga-Neto UM, Marques ET. Jr. From functional genomics to functional immunomics: new challenges, old problems, big rewards. PLoS Comput. Biol. 2006;2:e81. doi: 10.1371/journal.pcbi.0020081. [DOI] [PMC free article] [PubMed] [Google Scholar]