

Key Points
Several drugs are available that could be used, either alone or in combination, for the treatment (prophylaxis or therapy) of an influenza pandemic. These include adamantan(amin)e derivatives (amantadine), neuraminidase inhibitors (zanamivir and oseltamivir), ribavirin and interferon.
Amantadine was the first synthetic compound shown to inhibit influenza-virus replication. It blocks the migration of H+ ions into the interior of the virus particles within endosomes, a process that is needed for uncoating to occur.
Neuraminidase inhibitors such as oseltamivir and zanamivir interfere with the release of progeny influenza virions from the surface of infected host cells. In doing so, the neuraminidase inhibitors prevent virus infection of new host cells and thereby halt the spread of infection in the respiratory tract.
Ribavirin targets a cellular enzyme — inosine 5′-monophosphate dehydrogenase, which has a key role in the biosynthesis of GTP and viral RNA synthesis — and is active against both human and avian influenza viruses.
In addition to the available drugs, attempts to further design and develop new antivirals should be intensified, whether based on known molecular targets, such as the neuraminidase or viral uncoating process, or on as-yet relatively unexplored targets such as viral RNA polymerase.
There are currently serious concerns about the control measures that should be taken if a pandemic of influenza A were to strike. De Clercq discusses the therapeutic potential of agents that have been shown to be active against influenza A viruses, and describes emerging strategies for targeting these viruses.
Abstract
The recent outbreaks of avian influenza A (H5N1) virus, its expanding geographic distribution and its ability to transfer to humans and cause severe infection have raised serious concerns about the measures available to control an avian or human pandemic of influenza A. In anticipation of such a pandemic, several preventive and therapeutic strategies have been proposed, including the stockpiling of antiviral drugs, in particular the neuraminidase inhibitors oseltamivir (Tamiflu; Roche) and zanamivir (Relenza; GlaxoSmithKline). This article reviews agents that have been shown to have activity against influenza A viruses and discusses their therapeutic potential, and also describes emerging strategies for targeting these viruses.
Main
In the face of the persistent threat of human influenza A (H3N2, H1N1) and B infections, the outbreaks of avian influenza (H5N1) in Southeast Asia, and the potential of a new human or avian influenza A variant to unleash a pandemic, there is much concern about the shortage in both the number and supply of effective anti-influenza-virus agents1,2,3,4. There are, in principle, two mechanisms by which pandemic influenza could originate: first, by direct transmission (of a mutated virus perhaps) from animal (bird) to humans, as happened in 1918 with the 'Spanish influenza' (H1N1)5; or second, through reassortment of an avian influenza virus with a human influenza virus, as occurred in 1957 with the 'Asian influenza' (H2N2) and, again, in 1968 with the 'Hong Kong influenza' (H3N2)6,7 (Fig. 1).
Figure 1. The two mechanisms by which pandemic influenza originates.
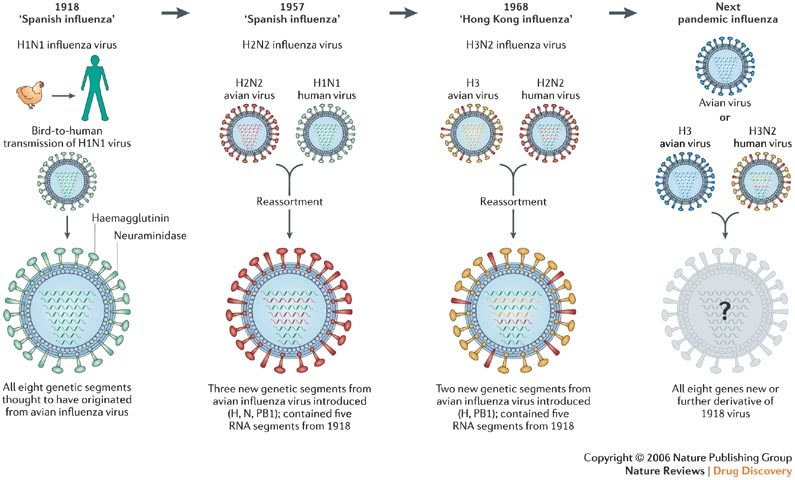
In 1918, the 'Spanish influenza' H1N1 virus, closely related to an avian virus, adapted to replicate efficiently in humans. In 1957 and 1968, reassortment events led to, respectively, the 'Asian influenza' H2N2 virus and the 'Hong Kong influenza' H3N2 virus. The 'Asian influenza' H2N2 virus acquired three genetic segments from an avian species (a haemagglutinin (H), a neuraminidase (N) and a polymerase (PB1) gene). The 'Hong Kong influenza' H3N2 virus acquired two genetic segments from an avian species (H and PB1). Future pandemic strains could arise through either mechanism. Figure adapted, with permission, from Ref. 7 © (2005) Massachusetts Medical Society.
Whether a new influenza pandemic could arise through antigenic 'drift' from an avian influenza virus or antigenic 'shift' through recombination of an avian and human influenza virus can only be speculated on. However, although this question is of crucial importance for future vaccine development, it has much less bearing on antiviral-drug design, as the antiviral drug targets shown in Fig. 2, and others which will be discussed here, should be relevant to all variants of influenza A virus8. In this article, I focus on agents that have been shown to have activity against influenza A viruses, and consider their therapeutic potential.
Figure 2. Inhibition of the influenza-virus replication cycle by antiviral agents.
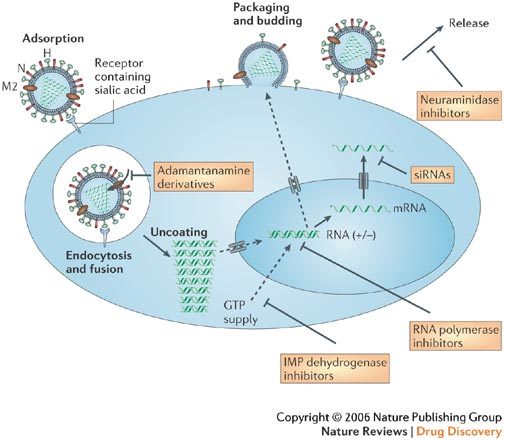
After binding to sialic-acid receptors, influenza virions are internalized by receptor-mediated endocytosis. The low pH in the endosome triggers the fusion of viral and endosomal membranes, and the influx of H+ ions through the M2 channel releases the viral RNA genes in the cytoplasm. Adamantan(amin)e derivatives block this uncoating step. RNA replication and transcription occur in the nucleus. This process can be blocked by inhibitors of inosine 5′-monophosphate (IMP) dehydrogenase (a cellular enzyme) or viral RNA polymerase. The stability of the viral mRNA and its translation to viral protein might be prevented by small interfering RNAs (siRNAs). Packaging and budding of virions occur at the cytoplasmic membrane. Neuraminidase (N) inhibitors block the release of the newly formed virions from the infected cells. Figure adapted with permission from Ref. 8 © (2004) Macmillan Magazines Ltd. H, haemagglutinin.
Adamantan(amin)e derivatives
The first synthetic compound shown to inhibit influenza-virus replication was amantadine9. As indicated in Fig. 2, amantadine blocks the migration of H+ ions into the interior of the virus particles (virions) within endosomes, a process that is needed for the uncoating to occur. The H+ ions are imported through the M2 (matrix 2) channels10; the transmembrane domain of the M2 protein, with the amino-acid residues facing the ion-conducting pore, is shown in Fig. 3a (Ref. 11). Amantadine has been postulated to block the interior channel within the tetrameric M2 helix bundle12.
Figure 3. Adamantan(amin)e derivatives as antiviral drugs.
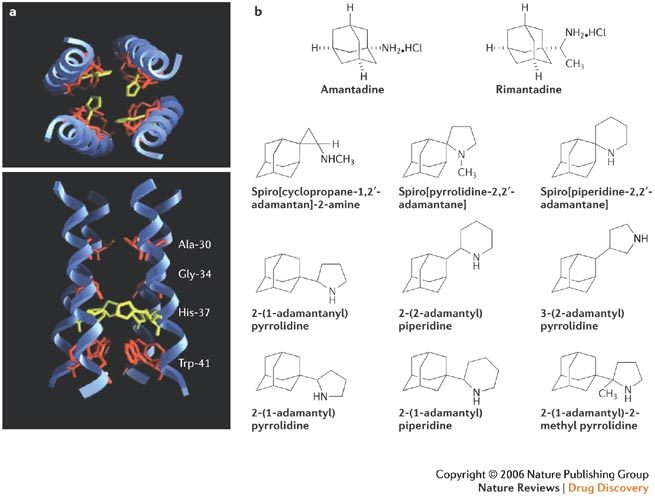
a | Amantadine, rimantadine and adamantanamine derivatives share several common structural features which relate to their mode of action: blockade of the M2 channel, which is responsible for transporting H+ ions (protons) into the interior of the virions and initiating the viral uncoating process (Fig. 2). The figure shows a model of the proposed transmembrane domain of the M2 protein with a top view as seen from the extracellular side and a cross-section in the plane of the lipid layer. Residues which were identified as facing the ion-conducting aqueous pore are indicated. b | Structures of the adamantan(amin)e derivatives amantadine and rimantadine, and various new adamantanamine derivatives: spiro[cyclopropane-1,2′-adamantan]-2-amine16, spiro[pyrrolidine-2,2′-adamantane]16, spiro[piperidine-2,2′-adamantane]17, 2-(2-adamantyl)piperidine18, 3-(2-adamantyl)pyrrolidine19, rimantadine 2-isomers20, 2-(1-adamantyl)piperidine21, 2-(1-adamantyl)pyrrolidine21 and 2-(1-adamantyl)-2-methyl-pyrrolidine22. Panel a reproduced with permission from Ref. 11 © (2000) American Society for Microbiology.
The adamantan(amin)e derivatives amantadine and rimantadine (Fig. 3b) have long been available for both the prophylaxis and therapy of influenza A virus infections, but their use has been limited because of the rapid emergence of drug resistance, the ready transmissibility of drug-resistant viruses, and, particularly for amantadine, the occurrence of central nervous system (CNS) side effects.These drawbacks compromise the potential usefulness of amantadine or rimantadine if used as single agents in the treatment of avian or human influenza A virus infections.
In particular, the incidence of adamantane resistance among influenza A (H3N2) viruses isolated in the United States13 and worldwide14 has been a cause for concern. More than 98% of the adamantane-resistant isolates identified worldwide between 1995 and 2005 contain the same S31N substitution14. The global circulation of adamantane-resistant H3N2 viruses is unprecedented and does not seem to be mediated by continued selective drug pressure. It could be argued that if resistance exists in a relatively homogeneous strain of H3N2, and if antiviral use would be curtailed, susceptible strains might (re-)emerge and adamantanes might regain their utility against both epidemic and pandemic influenza15.
In addition to amantadine and rimantadine, various new adamantan(amin)e derivatives have shown marked activity against influenza A (H2N2 and/or H3N2)16,17,18,19,20,21,22 (Fig. 3b). Whether any of these new derivatives might offer any advantage in terms of potency, selectivity, safety or resistance profile over the parent compounds amantadine and rimantadine needs to be further explored. Also, further investigation of the antiviral potential of other 'cage-like' compounds structurally related to the adamantyl entity, such as bananin23, might be worthwhile. At present, it can only be speculated whether any of the new adamantan(amin)e derivatives might be active against amantadine-resistant variants and efficacious in vivo in humans or relevant animal models.
Neuraminidase inhibitors
Viral haemagglutinin (H) is needed for the virus to interact with the receptor bearing N-acetylneuraminic acid (NANA, sialic acid). The viral neuraminidase (N) then cleaves off NANA from the cell-surface glycoprotein at a specific bond: SAα2,3Gal (sialic acid linked to galactose by an α-2,3 linkage) or SAα2,6Gal (sialic acid linked to galactose by an α-2,6 linkage (Fig. 4)). This enables the progeny virions to leave the infected cells and to spread to other host cells. So, by blocking the release of these newly formed virus particles, neuraminidase inhibitors should prevent further spread of the virus24,25 (Fig. 4). The neuraminidase might also have a role early in influenza infection of the human airway epithelium26.
Figure 4. Viral neuraminidase inhibition.
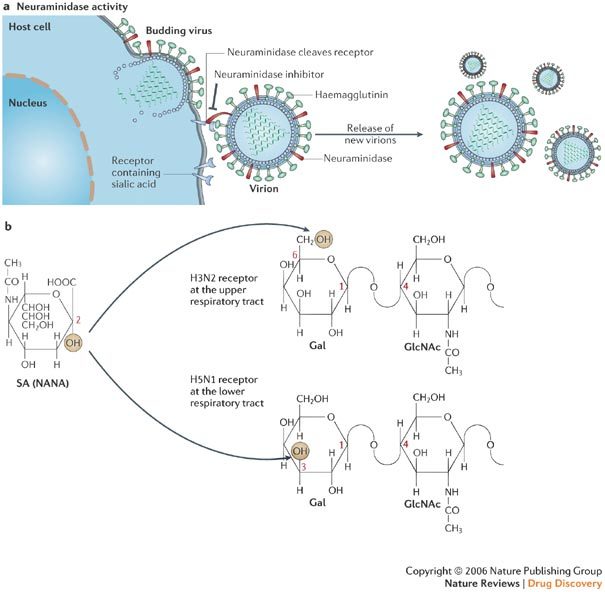
a | The neuraminidase cleaves off sialic acid (SA, also known as N-acetylneuraminic acid or NANA) from the cell receptor for influenza virus (b), so that the newly formed virus particles can be released from the cells. Neuraminidase inhibitors, such as zanamivir and oseltamivir (Fig. 5), interfere with the release of progeny influenza virions from the surface of infected host cells. In doing so, the neuraminidase inhibitors prevent virus infection of new host cells and thereby halt the spread of infection in the respiratory tract. b | SA linked to galactose (Gal) by an α2–3 linkage (SAα2–3Gal) or α2–6 linkage (SAα2–6Gal). Galactose is linked to N-acetylglucosamine (GlcNAc) through a β1–4 linkage. Panel a adapted with permission from Ref. 25 © (2005) Massachusetts Medical Society.
Avian (H5N1) influenza viruses and human (H3N2, H1N1) influenza viruses seem to target different receptors of the human respiratory tract: whereas human-derived viruses preferentially recognize SAα2,6Gal located on epithelial cells of the nasal mucosa, paranasal sinuses, pharynx, trachea and bronchi, avian viruses seem to preferentially recognize SAα2,3Gal located more deeply in the respiratory tract, at the alveolar cell wall and junction between the respiratory bronchiole and alveolus27. The avian influenza (H5N1) virus might cause severe lower respiratory tract (LRT) disease in humans because it attaches predominantly to type II pneumocytes, alveolar macrophages and non-ciliated bronchiolar cells of the human LRT28. In terms of the effectiveness of neuraminidase inhibitors, it would not, in theory, matter whether NANA is bound through an α-2,3 or α-2,6 linkage, as the neuraminidase inhibitors act as transition state analogues29 of NANA, irrespective of how it is bound to the penultimate galactose unit.
Oseltamivir and zanamivir. The first neuraminidase inhibitors designed according to the 'transition state analogue' principle29 were DANA (2-deoxy-2,3-didehydro-N-acetylneuraminic acid) and FANA (2-deoxy-2,3-dehydro-N-trifluoroacetylneuraminic acid). They served as the lead compounds for the development of the neuraminidase inhibitors that are marketed at present for the treatment (and prophylaxis) of influenza A and B virus infections: zanamivir (Relenza, 4-guanidino-Neu5Ac2en, GG167)30 and oseltamivir (Tamiflu, GS4071 ethyl ester, GS4104, Ro64-0796)31 (Fig. 5a). Both compounds have been found to be highly potent inhibitors (IC50 ≤ 1 ng ml−1) of the influenza neuraminidase, to inhibit influenza A and B virus replication in vitro and in vivo (mice, ferrets), to be well tolerated, and to be both prophylactically (significant reduction in number of ill subjects) and therapeutically (significant reduction in duration of illness) effective against influenza A and B virus infection in humans. A crucial difference between zanamivir and oseltamivir, however, is that zanamivir has to be administered by inhalation (10 mg twice daily), whereas oseltamivir can be administered orally (75 or 150 mg twice daily).
Figure 5. Neuraminidase inhibitors.
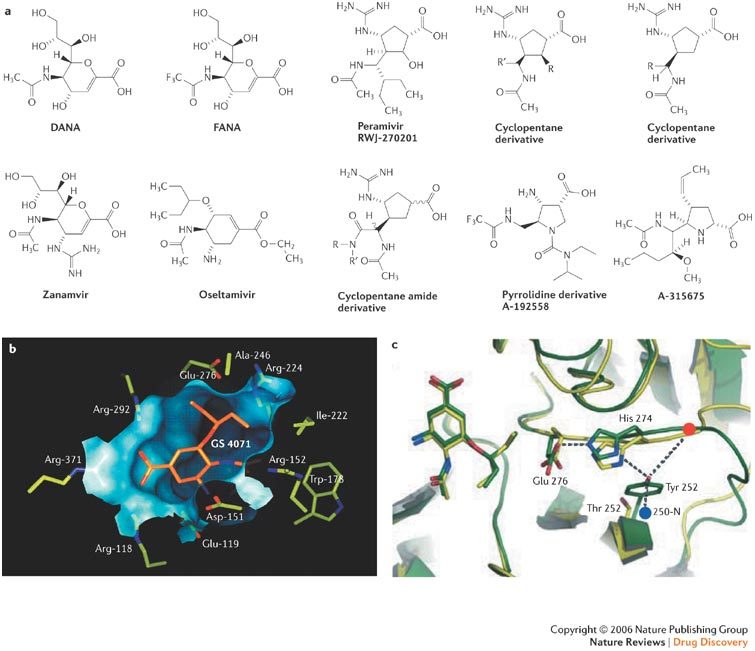
a | Structures of DANA, FANA, zanamivir (4-guanidino-Neu5Ac2en, GG167), oseltamivir (GS4071 ethyl ester, GS4104, Ro64-0796), peramivir (RWJ-270201), cyclopentane derivatives, a cyclopentane amide derivative and pyrrolidine derivatives (A-192558 and A-315675). b | GS4071 within the active site of the influenza A viral neuraminidase. c | Locations of oseltamivir-resistance mutations (H274Y) showing that the tyrosine at position 252 is involved in a network of hydrogen bonds in group-1 (H5N1 and H1N1) neuraminidases38. Panel b reproduced with permission from Ref. 31 © (1997) American Chemical Society and Ref. 37 © (2002) Macmillan Magazines Ltd. Panel c reproduced with permission from Ref. 38 © (2006) Macmillan Magazines Ltd.
Neuraminidase inhibitors are anticipated to reduce illness duration by 1–3 days, to reduce the risk of virus transmission to household or healthcare contacts, to reduce the number and severity of complications (sinusitis, bronchitis), to reduce the use of antibiotics and to prevent seasonal influenza-virus infection. As shown in particular for oseltamivir, the earlier the administration, the shorter the duration of fever, the greater the alleviation of symptoms and the faster the return to baseline activity and health scores32. Oseltamivir treatment of influenza illness reduces lower respiratory tract complications, particularly bronchitis and pneumonia, concomitantly with a reduction in antibiotic use and need for hospitalization33. Also, post-exposure prophylaxis with oseltamivir, 75 mg once daily for seven days, was found to protect close contacts of influenza-infected persons against influenza illness and to prevent spread within households34. Post-exposure prophylaxis with oseltamivir can be considered an effective option to prevent the transmission of influenza within households35. It should be recognized, however, that oseltamivir is less effective against influenza B than against influenza A with regard to duration of fever and virus persistence36.
The neuraminidase inhibitor GS4071 has been found to be positioned in the active centre of the neuraminidase31,37 (Fig. 5b). Two structures of the influenza A virus neuraminidase have now been solved: the first containing the N1 neuraminidase of H5N1, and the second containing the N2 neuraminidase as in H3N238 (Fig. 5c). The neuraminidase inhibitors make contact with arginine in position 292 through their carboxylic acid group, and with glutamic acid in position 119 through their basic amine (in the case of oseltamivir) or guanidinium (in the case of zanamivir) group. So, it is not surprising that at these positions mutations (R292K, E119G) can arise that engender resistance to both zanamivir and oseltamivir39. The R292K mutation causes high-level resistance to oseltamivir but only low-level (5–30-fold) resistance to zanamivir. In a comprehensive study of more than 1,000 clinical influenza isolates recovered from 1996 to 1999, there was no evidence of naturally occurring resistance to either oseltamivir or zanamivir in any of the isolates40. However, in children treated for influenza with oseltamivir, Kiso et al.41 found neuraminidase mutations in viruses from nine patients (18%), six of whom had mutations at position 292 (R292K) and two at position 119 (E119V). Zanamivir-resistant influenza H3N2 viruses might not readily arise in vivo owing to their poor viability (reduced fitness)42.
Recombinant viruses containing either the wild-type neuraminidase or a single amino-acid change at residue 119 (E119V) or 292 (R292K) were generated in the influenza A (H3N2) virus by reverse genetics: both mutants showed decreased sensitivity to oseltamivir, and the R292K virus showed cross-resistance to zanamivir. The R292K mutation was associated with compromised viral growth and transmissibility (in accordance with earlier studies43,44), whereas the growth and transmissibility of the E119V virus were comparable to those of wild-type virus45.
Of note, influenza A (H3N2) virus carrying the R292K mutation in the neuraminidase gene did not transmit to ferrets under conditions in which the wild-type virus was readily transmitted43. However, other mutant viruses of influenza A (H3N2) — that is, E119V and H274Y, both engendering resistance to oseltamivir — were found to be readily transmissible in ferrets, although the H274Y mutant required a 100-fold higher dose for infection and was transmitted more slowly than the wild type44.
The use of neuraminidase inhibitors in the therapy and prophylaxis of influenza A and B virus infections has been considered a new 'millennium conundrum', and the stockpiling of zanamivir and/or oseltamivir has been proposed in preparation of an influenza pandemic46. It was also hypothesized that these compounds could, in theory, also inhibit the virus that caused the 1918 pandemic46. In fact, recombinant viruses possessing the 1918 neuraminidase or both the 1918 neuraminidase and 1918 haemagglutinin were shown to be effectively inhibited, both in vitro and in vivo (mice), by zanamivir and oseltamivir, and a recombinant virus possessing the 1918 M2 ion channel could be effectively inhibited by amantadine and rimantadine. This indicates that current antiviral strategies could be effective in curbing a re-emerging 1918 or 1918-like influenza (H1N1) virus47.
Recently, resistance of influenza A (H5N1) virus to oseltamivir owing to the H274Y mutation in the neuraminidase gene was described48: the patient from whom the oseltamivir-resistant H5N1 strain was isolated recovered from the disease, and the virus was found to be less pathogenic in ferrets than the parent strain and did not show cross-resistance to zanamivir. Although the H274Y mutation in influenza A (H1N1) neuraminidase had been previously reported49,50, its occurrence in influenza A (H5N1) infection raised concern as it was associated with death in two of the eight influenza A (H5N1)-infected patients51. However, whether there was a causal relationship between the emergence of the H274Y mutation and the lethal outcome was not established51.
The efficacy of oseltamivir in the treatment of H5N1 infection in humans could, because of its anecdotal use, not be unequivocally shown. However, it should be noted that oseltamivir has been shown to protect ferrets against lethal influenza H5N1 infection: treatment with oseltamivir at 5 mg kg−1 per day for 5 days twice daily (orally) resulted in complete inhibition of virus replication in the lungs and small intestine on day 5 post-infection and, consequently, prevented mortality52. Similarly, oseltamivir has proven efficacious in the treatment of mice infected with the highly pathogenic H5N1 A/Vietnam/1203/04 influenza strain, although prolonged and higher-dose oseltamivir regimens were required for achieving the most beneficial antiviral effect53.
Peramivir and other cyclopentane or pyrrolidine derivatives. Whereas oseltamivir can be described as a cyclohexenyl derivative, there are several cyclopentane and pyrrolidine derivatives that have been described as neuraminidase inhibitors: peramivir (RWJ-270201, BCX-1182)54,55,56 and other cyclopentane56,57 and cyclopentane amide58 derivatives, as well as various pyrrolidine derivatives, including A-192558 (Ref. 59), A-315675 (Refs 60–62) and other pyrrolidines63 (Fig. 5a). In addition, 2,3-disubstituted tetrahydrofuran-5-carboxylic acid derivatives have been described as influenza-neuraminidase inhibitors, albeit with reduced inhibitory potency compared with the corresponding pyrrolidine analogues64.
Peramivir and A-315675 retain activity against various zanamivir- and oseltamivir-resistant influenza A and B viruses65. Specifically, a new oseltamivir-resistant influenza B variant that carries the D198N substitution in the viral neuraminidase was found to retain susceptibility to peramivir and A-315675. In addition, the neuraminidase E119V mutant, which shows 6,000-fold lower susceptibility to oseltamivir and 175-fold lower susceptibility to zanamivir than wild-type virus, retained full susceptibility to A-315675 (Ref. 66). Taken together, the studies performed with influenza A (H3N2) virus mutants41,42,43,44,45,65,66 indicate that neuraminidase inhibitors might select for mutations at several positions (E119V, R152K, D198N, H274Y and R292K) that only partially overlap67 — that is, only partially engender cross-resistance.
In vivo, peramivir was found to strongly suppress influenza A (H1N1) infection in mice following a single intramuscular injection (10 mg kg−1). This was ascribed to tight binding of peramivir to neuraminidase68. Similarly, a single intramuscular/intravenous injection of peramivir one hour before virus exposure offered protection against influenza A (H5N1) in mice69. When given orally to humans, however, peramivir did not offer robust protection against human influenza A infection, which was attributed to the very low oral bioavailability (<5%) of peramivir70; further studies with parenteral formulations of peramivir are therefore warranted.
Dimeric neuraminidase inhibitors. Starting from zanamivir, 7-alkyl ether derivatives and bicyclic ether derivatives were synthesized which showed improved influenza A virus plaque-reduction activity in vitro71, and improved oral efficacy in vivo (mice)72, respectively, compared with the parent compound, zanamivir. Dimeric derivatives of zanamivir with linking groups of 14–18 atoms in length were found to be 100-fold more potent inhibitors of influenza-virus replication in vitro and in vivo than zanamivir73,74. These compounds showed long-lasting antiviral activity owing to extremely long persistence times in the lungs, allowing a once-weekly dosing regimen. This raises the prospect of a new type of anti-influenza drug that could be administered as a single dose in the treatment of influenza, or just once a week in the prevention of infection74.
Ribavirin and viramidine
Ribavirin has long been recognized as a broad-spectrum antiviral agent with particularly distinct activity against orthomyxoviruses (that is, influenza) and paramyxoviruses (that is, measles and respiratory syncytial virus (RSV))75. RSV infection is the only (−)RNA virus infection for which aerosolized ribavirin has been formally approved. Oral ribavirin is also used, in combination with parenteral PEGylated interferon-α, in the treatment of chronic hepatitis C virus (HCV) infection. The intravenous form of ribavirin has been registered for the treatment of haemorrhagic fever with renal syndrome (HFRS). In addition to ribavirin, viramidine — which can be considered to be the amidine prodrug of ribavirin(Fig. 6) — has been accredited with marked potential as anti-influenza drug76. Ribavirin is notable for not generating drug resistance, and resistance of influenza-virus replication to ribavirin has, to the best of my knowledge, not been reported so far. Obviously, the lack of drug-resistance is due to the fact that ribavirin's main target of antiviral action is a cellular enzyme: inosine 5′-monophosphate (IMP) dehydrogenase (responsible for the conversion of IMP to xanthosine 5′-monophosphate), a key enzyme involved in the biosynthesis of GTP and viral RNA synthesis (Fig. 6).
Figure 6. IMP dehydrogenase inhibition.
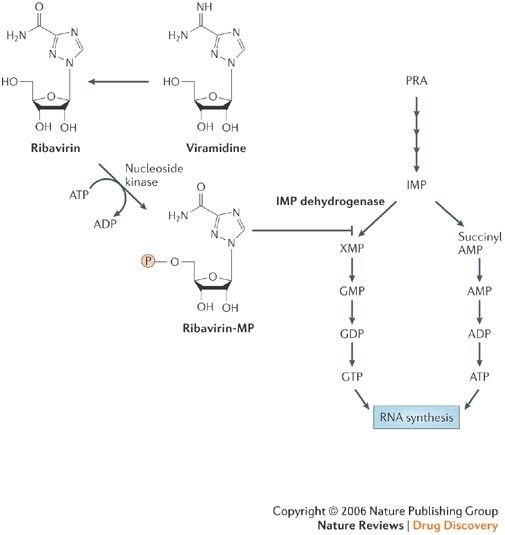
Viramidine acts as a prodrug (precursor) of ribavirin, which is converted intracellularly to its 5′-monophosphate derivative, ribavirin-MP. The latter inhibits inosine 5′-monophosphate (IMP) dehydrogenase, a crucial enzyme in the biosynthesis of RNA, including viral RNA. IMP dehydrogenase is responsible for the conversion of IMP into xanthosine 5′-monophosphate (XMP) which, in turn, is further converted to GMP (guanosine 5′-monophosphate), GDP (guanosine 5′-diphosphate) and GTP (guanosine 5′-triphosphate). The latter serves as substrate, together with ATP, UTP and CTP, in the synthesis of RNA.
Ribavirin is active against both human and avian (H5N1) influenza viruses within the 50% effective concentration (EC50) range of 6–22 μM76. Of the three routes (oral, aerosolized and intravenous) by which ribavirin could be administered in the treatment of influenza, the intravenous route is preferred for therapy of acute influenza-virus infection. Oral ribavirin did not offer the expected clinical or virological efficacy in earlier studies with influenza A (H1N1)77. Ribavirin aerosol has been used successfully (based on reduction of virus shedding and clinical symptoms) in the treatment of influenza-virus infections in college students78. Intravenous ribavirin (producing a mean plasma concentration of 20–60 μM) was associated with symptomatic improvements and elimination of influenza virus from nasopharyngeal swabbings and tracheal aspirates79.
Intravenous ribavirin has been further investigated, with success, in the treatment of Lassa fever80 and HFRS81. Both studies showed significant benefits of ribavirin in terms of survival and reduction of disease severity. The dosing regimen for intravenous ribavirin consists of a loading dose of 2 grams of ribavirin followed by 1 gram every 6 hours for 4 days. During the following 5 days, administering a maintenance dose of 0.5 gram every 8 hours should generate the concentrations needed to achieve suppression of (human and avian) influenza-virus replication. The dose-limiting toxicity would be haemolytic anaemia, which should be reversible on cessation of therapy.
Sialidase fusion protein and sialylglycopolymers
Recently, a recombinant fusion protein composed of the sialidase (neuraminidase) catalytic domain derived from Actinomyces viscosus fused with a cell-surface-anchoring sequence was reported as a novel broad-spectrum inhibitor of influenza-virus infection82. The sialidase fusion protein is to be applied topically as an inhalant to remove the influenza-virus receptors — sialic acids — from the airway epithelium. A sialidase fusion construct, DAS181 (Fludase), was shown to effectively cleave sialic-acid receptors used by both human and avian influenza viruses. DAS181 showed potent antiviral and cell-protective efficacies against a panel of laboratory strains and clinical isolates of influenza A and influenza B, with virus-replication inhibition EC50 values in the range of 0.04–0.9 nM. Significant in vivo efficacy of the sialidase fusion construct was noted in both prophylactic and therapeutic approaches82.
The sialic-acid receptors could also be targeted by sialic-acid polyacrylamide conjugates, also termed sialylglycopolymers83,84. Sialylglycopolymers inhibit influenza-virus attachment to the cells. In vivo, when administered in aerosol form within 24–110 hours of infection, they were found to completely prevent mortality in mice infected with mouse-adapted influenza A strains (H3N2, H1N1). These sialylglycopolymers target the receptor determinant SAα2-6Galβ1-4GlcNAc, recognized by human influenza A and B viruses. This would make them potentially valuable for protection against any newly emerging (human) influenza virus strains.
siRNAs and phosphorothioate oligonucleotides
Small interfering RNAs (siRNAs) specific for conserved regions of influenza-virus genes were found to reduce virus production in the lungs of infected mice when the siRNAs were given intravenously in complexes with a polycation carrier either before or after initiating virus infection85. Delivery of siRNAs specific for highly conserved regions of the nucleoprotein or acidic polymerase significantly reduced lung virus titres in mice infected with influenza A virus and protected the animals from lethal challenge. This protection was specific and not mediated by an antiviral interferon response. The influenza-specific siRNA treatment was broadly effective and protected animals against lethal challenge with highly pathogenic avian influenza A viruses of the H5 and H7 subtypes86. It could be predicted that specific siRNAs would be effective against influenza from equally effective results obtained with other specific siRNAs against the SARS (severe acute respiratory syndrome) coronavirus, in comparable situations87.
Phosphorothioate oligonucleotides (PS-ONs) (that is, REP 9, a 40-mer PS-ON) offer potential, when administered as aerosol in the prophylaxis and therapy of influenza infection88. Similarly, antisense phosphorodiamidate morpholino oligomers (ARP-PMOs) could be further pursued for their potential in the treatment of H5N1 influenza A virus infections89.
Influenza-virus RNA-polymerase inhibitors
The influenza-virus RNA polymerase consists of a complex of three virus-encoded polypeptides (PB1, PB2 and PA) which, in addition to the RNA replicative activity, also contains an endonuclease activity so as to ensure 'cap snatching' to initiate the transcription and subsequent translation process90. The polymerase-complex genes contribute to the high virulence of the human H5N1 influenza-virus isolate A/Vietnam/1203/04 (Ref. 91). This observation highlights the importance of novel antivirals that target the polymerase for further development of therapy and prophylaxis of human and avian influenza-virus infections.
Few compounds have been reported to be operating at either the RNA replicase (RNA polymerase) or endonuclease level. Like the inhibitors that have been found to inhibit the reverse transcriptase (RNA-dependent DNA polymerase) of HIV or RNA replicase (RNA-dependent RNA polymerase) of HCV, influenza RNA-replicase inhibitors can be divided into two classes: nucleosides and non-nucleosides. Examples of the nucleoside type of inhibitors are 2′-deoxy-2′-fluoroguanosine (FdG)92,93 and T-70594,95,96 (Fig. 7).
Figure 7. Influenza-virus RNA-polymerase inhibitors.
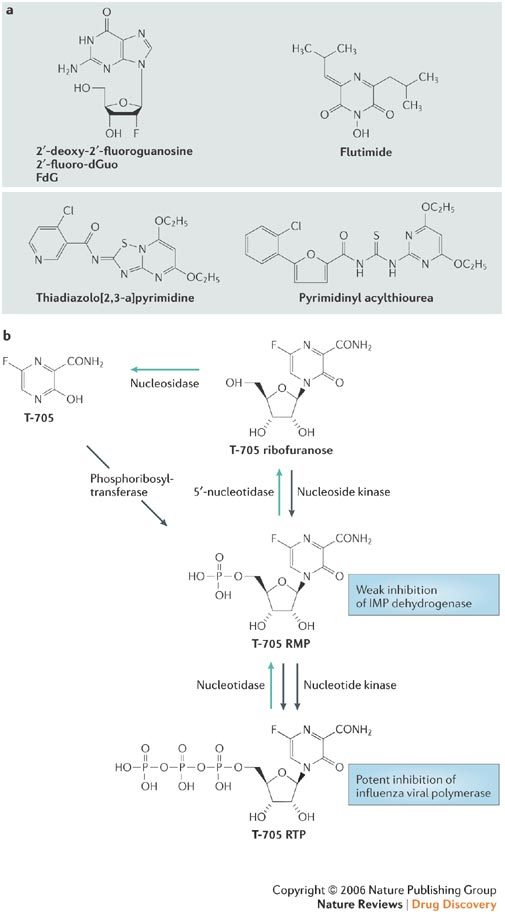
a | FdG, Flutimide, thiadiazolo [2,3-a]pyrimidine and pyrimidinyl acylthiourea. b | The postulated mode of action of T-705, according to Furuta and colleagues96.
T-705 is a substituted pyrazine with potent anti-influenza-virus activity in vitro and in vivo. According to a comparative study, T-705 would even be more potent than oseltamivir when increasing the multiplicity of infection (in vitro) or using a higher virus-challenge dose (in vivo)95. It has been postulated that T-705 is converted intracellularly to the ribonucleotide T-705-4-ribofuranosyl-5′-monophosphate (T-705 RMP) by a phosphoribosyl transferase and, on further phosphorylation to its 5′-triphosphate (Fig. 7), T-705 RTP would then inhibit influenza-virus RNA polymerase in a GTP-competitive manner96. Unlike ribavirin 5′-monophosphate, T-705 RMP did not significantly inhibit IMP dehydrogenase, indicating that it owes its anti-influenza virus activity mainly, if not exclusively, to inhibition of the influenza-virus RNA polymerase.
In addition to the RNA polymerase, the 'cap snatching' or 'cap scavenging' endonuclease activity associated with the PB1–PB2–PA complex could be an attractive target for influenza-virus inhibitors: it can be inhibited by 4-substituted 2,4-dioxobutanoic acid derivatives97 and N-hydroxamic acid/N-hydroxy-imide derivatives98. Likewise, flutimide, a 2,6-diketopiperazine (Fig. 7) identified in extracts of the fungal species Delitschia confertaspora, has been shown to specifically inhibit the cap-dependent endonuclease activity associated with influenza viral RNA polymerase and to inhibit the replication of influenza A and B virus in cell culture99. Both the viral RNA polymerase and endonuclease should be further explored as targets for the development of anti-influenza agents.
Recently, a new class of potent influenza-virus inhibitors (EC50 for virus replication: 0.08–0.09 μM) has been reported100, represented by thiadiazolo[2,3-a]pyrimidine and pyrimidinyl acylthiourea (Fig. 7). Although the mechanism of action of this class of highly potent and selective inhibitors of influenza virus remains to be established, they represent a highly interesting lead worth pursuing. A series of novel bisheterocycle tandem derivatives consisting of methyloxazole and thiazole might also serve as leads for further optimization, although the lead compounds showed only modest activity against influenza A virus101.
Interferon (inducers)
Interferon was originally discovered almost 50 years ago, with influenza virus as its inducer102. In fact, Baron and Isaacs alluded to the absence of interferon in lungs from fatal cases of influenza103. In some earlier studies, interferon instilled by the intranasal route did not offer significant protection in the prophylaxis of influenza-A-virus infections104,105.
Since then, PEGylated interferon-α (injected parenterally), in combination with (oral) ribavirin has become the standard therapy for chronic HCV infections. So, extensive experience with this combination has been accumulated106, which could be readily implemented in the prophylaxis and therapy of human as well as avian influenza-virus infections in humans. In the prophylaxis and therapy of influenza-virus infections, the duration of treatment would be much shorter than in the treatment of hepatitis C, which would obviously affect the convenience (cost/benefit) and side effects that are inherently linked to the use of interferon and ribavirin.
In addition to interferon, interferon inducers such as poly(I)·poly(C), discovered ∼40 years ago107 might also have a role in the control of influenza-virus infections. Prophylaxis using liposome-encapsulated double-stranded RNA (poly(I)·poly(C)) provided complete and long-lasting protection against influenza A infection108. Furthermore, when combined with (intranasal) vaccination, poly(I)·poly(C) conferred complete protection against influenza-virus infection, which might have been mediated by an upregulated expression of Toll-like receptor 3 and interferon-α/β as well as TH1- and TH2-related cytokines109. It is unclear whether the use of exogenous interferon or the induction of endogenous interferon by poly(I).poly(C) or other double-stranded RNAs might help in the prophylaxis or therapy of avian or human influenza-virus infections, but in view of the 'renaissance' of interferon in the treatment of HCV infection, the potential of interferon for influenza might well deserve to be revisited.
Signal-transduction inhibitors
Signal-transduction inhibitors, such as those targeted at either ErbB tyrosine kinase110 or the Abl kinase family (that is, imatimib111), have recently been shown to suppress the in vitro replication and in vivo dissemination of poxviruses. Likewise, the signalling cascade could be considered an attractive opportunity for future strategies to block influenza-virus production. The export of the influenza-virus ribonucleoprotein (RNP) from the nucleus depends on the cellular Raf/MEK/ERK kinase (MAPK) signalling cascade112, and this PKCα-mediated activation of ERK signalling is specifically triggered by the accumulation of influenza-A-virus haemagglutinin at the cell membrane113. The PKCα-mediated ERK activation could therefore be considered a potential target for intervention with influenza-virus propagation.
Other potential targets
Recently, the CPSF30-binding site on the NS1A protein of influenza A virus was proposed as a potential target for the development of antivirals directed against influenza A virus114. The NS1A protein inhibits the 3′-end processing of cellular pre-mRNAs by binding to the 30-kDa subunit of cleavage and polyadenylation specificity factor (CPSF30). This binding site is also required for efficient virus replication. A fragment of CPSF30, termed F2F3 because it spans the second and third zinc-finger domains, was found to specifically bind to the CPSF30 binding site: it inhibited influenza-A-virus replication but did not inhibit the 3′-end processing of cellular pre-mRNAs. This might indicate that the CPSF30-binding site of NS1A could possibly be targeted by low-molecular-mass inhibitors of HIV replication114.
Combination therapy
Drug-combination regimens used in the treatment of Mycobacterium tuberculosis and HIV infections achieve greater benefit than each compound given individually, reduce the likelihood of drug-resistance development and might allow the individual drug doses to be lowered, thereby diminishing adverse effects. In the therapy (or prophylaxis) of influenza-virus infections, the combination of (PEGylated) interferon and ribavirin could be further complemented with amantadine (or rimantadine). This triple-drug combination has shown efficacy in the treatment of chronic HCV infection115. Against influenza, such a triple-drug regimen might theoretically be expected to yield a beneficial outcome. The three drugs are, individually, all active against influenza-virus replication in vitro and act through different mechanisms, which implies that, when combined, they might achieve an additive or even synergistic action, while reducing the risk of emergence of drug-resistant virus variants. As early as 1984, Hayden et al.116 pointed to the additive synergistic action between interferon-α2 and rimantadine or ribavirin. Similarly, combinations of (PEGylated) interferon with neuraminidase inhibitors (zanamivir, oseltamivir or peramivir) could also be considered, and so might combinations of ribavirin (or viramidine) with the neuraminidase inhibitors.
Combinations of the adamantan(amin)es (amantadine or rimantadine) with the neuraminidase inhibitors (zanamivir or oseltamivir) should also receive attention. In vitro, rimantadine was found to act synergistically with zanamivir, oseltamivir or peramivir in reducing the extracellular yield of influenza A (H3N2) virus117. In vivo, oseltamivir at 10 mg kg−1 per day and amantadine at 15 mg kg−1 per day provided similar protection against influenza A (H5N1)-associated death risk in mice, but when both were combined they provided an incremental protection against lethality compared with both compounds given as single-agent chemotherapy118. The only controlled study in humans was a comparison of rimantadine alone versus rimantadine plus inhaled zanamivir in hospitalized (adult) patients with serious influenza119. Although preliminary, this study pointed to a higher clinical benefit for the combination of zanamivir with rimantadine.
Conclusions
Several drugs are available that could be used, either alone or in combination, for the treatment (prophylaxis or therapy) of a pandemic influenza-virus infection, whether avian or human. These include adamantan(amin)e derivatives (amantadine), neuraminidase inhibitors (zanamivir and oseltamivir), ribavirin and interferon. In the meantime, attempts should be intensified to further design and develop new antivirals, whether based on known molecular targets, such as the neuraminidase or viral uncoating process, or on as-yet relatively unexplored targets such as the viral RNA polymerase. The latter could, in principle, be targeted by both nucleoside and non-nucleoside inhibitor types, an approach which has proven most successful in the cases of the HIV reverse transcriptase and HCV RNA polymerase.
Acknowledgements
I thank C. Callebaut for her invaluable editorial assistance.
Glossary
- HXNY
In the naming system for virus strains, H refers to haemagglutinin and N to neuraminidase.
- Alveoli
Lung structures responsible for gas (O2 ↔ CO2) exchange.
- Transition state analogue
A structural mimic of the intermediary state between reactant(s) and product(s) in a given reaction.
- PEGylation
Addition of poly(ethylene glycol) (PEG) groups to proteins can increase their resistance to proteolytic degradation, improve their water solubility and reduce their antigenicity.
- Prodrug
A pharmacologically inactive compound that is converted to the active form of the drug by endogenous enzymes or metabolism. It is generally designed to overcome problems associated with stability, toxicity, lack of specificity or limited (oral) bioavailability.
- Small interfering RNAs
(siRNAs). Small (∼20 nucleotide) RNA constructs that interfere with RNA translation.
- Phosphorothioate oligonucleotides
Antisense oligonucleotides in which phosphate groups are replaced by thiophosphate groups.
Biography
Erik De Clercq has been Chairman of the Department of Microbiology and Immunology at the Katholieke Universiteit (KU) Leuven (Belgium) and Chairman of the Directory Board of the Rega Institute for Medical Research. He is President of the Rega Foundation (Belgium). As a full professor since 1977, he has been teaching the courses of cell biology, biochemistry, microbiology and virology at the KU Leuven Medical School (Belgium). He is a director of the Belgian Royal Academy of Medicine and a member of the Academia Europaea. De Clercq's scientific interests are in the antiviral chemotherapy field, and in particular, the development of new antiviral agents for various viral infections, including herpes simplex virus (HSV), varicella-zoster virus, cytomegalovirus (CMV) and human immunodeficiency virus. He has co-discovered several drugs that are used for the treatment of HSV (valaciclovir), herpes zoster (brivudin), CMV (cidofovir), hepatitis B (adefovir dipivoxil) and AIDS (tenofovir disoproxil fumarate, marketed as Viread, and, in combination with emtricitabine, as Truvada, and in combination with emtricitabine and efavirenz, as Atripla).
Related links
FURTHER INFORMATION
Competing interests
The author declares no competing financial interests.
References
- 1.Ferguson NM, et al. Strategies for containing an emerging influenza pandemic in Southeast Asia. Nature. 2005;437:209–214. doi: 10.1038/nature04017. [DOI] [PubMed] [Google Scholar]
- 2.Ferguson NM, et al. Strategies for mitigating an influenza pandemic. Nature. 2006;442:448–452. doi: 10.1038/nature04795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kaye D, Pringle CR. Avian influenza viruses and their implication for human health. Clin. Infect. Dis. 2005;40:108–112. doi: 10.1086/427236. [DOI] [PubMed] [Google Scholar]
- 4.Beigel JH, et al. Avian influenza A (H5N1) infection in humans. N. Engl. J. Med. 2005;353:1374–1385. doi: 10.1056/NEJMra052211. [DOI] [PubMed] [Google Scholar]
- 5.Taubenberger JK, Morens DM. 1918 influenza: the mother of all pandemics. Emerging Infect. Dis. 2006;12:15–22. doi: 10.3201/eid1201.050979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kilbourne ED. Influenza pandemics of the 20th century. Emerging Infect. Dis. 2006;12:9–14. doi: 10.3201/eid1201.051254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Belshe RB. The origins of pandemic influenza — lessons from the 1918 virus. N. Engl. J. Med. 2005;353:2209–2211. doi: 10.1056/NEJMp058281. [DOI] [PubMed] [Google Scholar]
- 8.Palese P. Influenza: old and new threats. Nature Med. 2004;10:S82–S87. doi: 10.1038/nm1141. [DOI] [PubMed] [Google Scholar]
- 9.Davies WL, et al. Antiviral activity of 1-adamantanamine (amantadine) Science. 1964;144:862–863. doi: 10.1126/science.144.3620.862. [DOI] [PubMed] [Google Scholar]
- 10.Horimoto T, Kawaoka Y. Influenza: lessons from past pandemics, warnings from current incidents. Nature Rev. Microbiol. 2005;3:591–600. doi: 10.1038/nrmicro1208. [DOI] [PubMed] [Google Scholar]
- 11.Shuck K, Lamb RA, Pinto LH. Analysis of the pore structure of the influenza A virus M2 ion channel by the substituted-cysteine accessibility method. J. Virol. 2000;74:7755–7761. doi: 10.1128/jvi.74.17.7755-7761.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sansom MS, Kerr ID. Influenza virus M2 protein: a molecular modelling study of the ion channel. Protein Eng. 1993;6:65–74. doi: 10.1093/protein/6.1.65. [DOI] [PubMed] [Google Scholar]
- 13.Bright RA, et al. Adamantane resistance among influenza A viruses isolated early during the 2005–2006 influenza season in the United States. JAMA. 2006;295:891–894. doi: 10.1001/jama.295.8.joc60020. [DOI] [PubMed] [Google Scholar]
- 14.Bright RA, et al. Incidence of adamantane resistance among influenza A (H3N2) viruses isolated worldwide from 1994 to 2005: a cause for concern. Lancet. 2005;366:1175–1181. doi: 10.1016/S0140-6736(05)67338-2. [DOI] [PubMed] [Google Scholar]
- 15.Weinstock DM, Zuccotti G. Adamantane resistance in influenza A. JAMA. 2006;295:934–936. doi: 10.1001/jama.295.8.934. [DOI] [PubMed] [Google Scholar]
- 16.Kolocouris N, et al. Synthesis and antiviral activity evaluation of some aminoadamantane derivatives. J. Med. Chem. 1994;37:2896–2902. doi: 10.1021/jm00044a010. [DOI] [PubMed] [Google Scholar]
- 17.Kolocouris N, et al. Synthesis and antiviral activity evaluation of some new aminoadamantane derivatives. 2. J. Med. Chem. 1996;39:3307–3318. doi: 10.1021/jm950891z. [DOI] [PubMed] [Google Scholar]
- 18.Kolocouris N, et al. Synthesis of 2-(2-adamantyl)piperidines and structure anti-influenza virus A activity relationship study using a combination of NMR spectroscopy and molecular modeling. Bioorg. Med. Chem. Lett. 1999;9:3465–3470. doi: 10.1016/s0960-894x(99)00631-9. [DOI] [PubMed] [Google Scholar]
- 19.Stamatiou G, et al. Novel 3-(2-adamantyl)pyrrolidines with potent activity against influenza A virus — identification of aminoadamantane derivatives bearing two pharmacophoric amine groups. Bioorg. Med. Chem. Lett. 2001;11:2137–2142. doi: 10.1016/s0960-894x(01)00388-2. [DOI] [PubMed] [Google Scholar]
- 20.Zoidis G, et al. Are the 2-isomers of the drug rimantadine active anti-influenza A agents? Antiviral Chem. Chemother. 2003;14:153–164. doi: 10.1177/095632020301400305. [DOI] [PubMed] [Google Scholar]
- 21.Stamatiou G, et al. Heterocyclic rimantadine analogues with antiviral activity. Bioorg. Med. Chem. 2003;11:5485–5492. doi: 10.1016/j.bmc.2003.09.024. [DOI] [PubMed] [Google Scholar]
- 22.Zoidis G, et al. Heterocyclic rimantadine analogues with antiviral activity. Bioorg. Med. Chem. 2006;14:3341–3348. doi: 10.1016/j.bmc.2005.12.056. [DOI] [PubMed] [Google Scholar]
- 23.Tanner JA, et al. The adamantane-derived bananins are potent inhibitors of the helicase activities and replication of SARS coronavirus. Chem. Biol. 2005;12:303–311. doi: 10.1016/j.chembiol.2005.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.De Clercq E. Antivirals and antiviral strategies. Nature Rev. Microbiol. 2004;2:704–720. doi: 10.1038/nrmicro975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moscona A. Neuraminidase inhibitors for influenza. N. Engl. J. Med. 2005;353:1363–1373. doi: 10.1056/NEJMra050740. [DOI] [PubMed] [Google Scholar]
- 26.Matrosovich M, Matrosovich TY, Gray T, Roberts NA, Klenk H-D. Neuraminidase is important for the initiation of influenza virus infection in human airway epithelium. J. Virol. 2004;78:12665–12667. doi: 10.1128/JVI.78.22.12665-12667.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shinya K, et al. Avian flu: influenza virus receptors in the human airway. Nature. 2006;440:435–436. doi: 10.1038/440435a. [DOI] [PubMed] [Google Scholar]
- 28.van Riel D, et al. H5N1 virus attachment to lower respiratory tract. Science. 2006;312:399. doi: 10.1126/science.1125548. [DOI] [PubMed] [Google Scholar]
- 29.Abdel-Magid AF, Marvanoff CA, Mehrman SJ. Synthesis of influenza neuraminidase inhibitors. Curr. Opin. Drug Discov. Devel. 2001;4:776–791. [PubMed] [Google Scholar]
- 30.von Itzstein M, et al. Rational design of potent sialidase-based inhibitors of influenza virus replication. Nature. 1993;363:418–423. doi: 10.1038/363418a0. [DOI] [PubMed] [Google Scholar]
- 31.Kim CU, et al. Influenza neuraminidase inhibitors possessing a novel hydrophobic interaction in the enzyme active site: design, synthesis, and structural analysis of carbocyclic sialic acid analogues with potent anti-influenza activity. J. Am. Chem. Soc. 1997;119:681–690. doi: 10.1021/ja963036t. [DOI] [PubMed] [Google Scholar]
- 32.Aoki FY, et al. Early administration of oral oseltamivir increases the benefits of influenza treatment. J. Antimicrob. Chemother. 2003;51:123–129. doi: 10.1093/jac/dkg007. [DOI] [PubMed] [Google Scholar]
- 33.Kaiser L, et al. Impact of oseltamivir treatment on influenza-related lower respiratory tract complications and hospitalizations. Arch. Intern. Med. 2003;163:1667–1672. doi: 10.1001/archinte.163.14.1667. [DOI] [PubMed] [Google Scholar]
- 34.Welliver R, et al. Effectiveness of oseltamivir in preventing influenza in household contacts. JAMA. 2001;285:748–754. doi: 10.1001/jama.285.6.748. [DOI] [PubMed] [Google Scholar]
- 35.Hayden FG, et al. Management of influenza in households: a prospective, randomized comparison of oseltamivir treatment with or without postexposure prophylaxis. J. Infect. Dis. 2004;189:440–449. doi: 10.1086/381128. [DOI] [PubMed] [Google Scholar]
- 36.Kawai N, et al. A comparison of the effectiveness of oseltamivir for the treatment of influenza A and influenza B: a Japanese multicenter study of the 2003–2004 and 2004–2005 influenza seasons. Clin. Infect. Dis. 2006;43:439–444. doi: 10.1086/505868. [DOI] [PubMed] [Google Scholar]
- 37.De Clercq E. Strategies in the design of antiviral drugs. Nature Rev. Drug Discov. 2002;1:13–25. doi: 10.1038/nrd703. [DOI] [PubMed] [Google Scholar]
- 38.Russell RJ, et al. The structure of H5N1 avian influenza neuraminidase suggests new opportunities for drug design. Nature. 2006;443:45–49. doi: 10.1038/nature05114. [DOI] [PubMed] [Google Scholar]
- 39.McKimm-Breschkin JL. Resistance of influenza viruses to neuraminidase inhibitors — a review. Antiviral Res. 2000;47:1–17. doi: 10.1016/s0166-3542(00)00103-0. [DOI] [PubMed] [Google Scholar]
- 40.McKimm-Breschkin J, et al. Neuraminidase sequence analysis and susceptibilities of influenza virus clinical isolates to zanamivir and oseltamivir. Antimicrob. Agents Chemother. 2003;47:2264–2272. doi: 10.1128/AAC.47.7.2264-2272.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kiso M, et al. Resistant influenza A viruses in children treated with oseltamivir: descriptive study. Lancet. 2004;364:759–765. doi: 10.1016/S0140-6736(04)16934-1. [DOI] [PubMed] [Google Scholar]
- 42.Zürcher T, et al. Mutations conferring zanamivir resistance in human influenza virus N2 neuraminidases compromise virus fitness and are not stably maintained in vitro. J. Antimicrob. Chemother. 2006;58:723–732. doi: 10.1093/jac/dkl321. [DOI] [PubMed] [Google Scholar]
- 43.Herlocher ML, et al. Influenza virus carrying an R292K mutation in the neuraminidase gene is not transmitted in ferrets. Antiviral Res. 2002;54:99–111. doi: 10.1016/s0166-3542(01)00214-5. [DOI] [PubMed] [Google Scholar]
- 44.Herlocher ML, et al. Influenza viruses resistant to the antiviral drug oseltamivir: transmission studies in ferrets. J. Infect. Dis. 2004;190:1627–1630. doi: 10.1086/424572. [DOI] [PubMed] [Google Scholar]
- 45.Yen H-L, et al. Neuraminidase inhibitor-resistant influenza viruses may differ substantially in fitness and transmissibility. Antimicrob. Agents Chemother. 2005;49:4075–4084. doi: 10.1128/AAC.49.10.4075-4084.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Oxford J, Balasingam S, Lambkin R. A new millennium conundrum: how to use a powerful class of influenza anti-neuraminidase drugs (NAIs) in the community. J. Antimicrob. Chemother. 2004;53:133–136. doi: 10.1093/jac/dkh037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tumpey TM, et al. Existing antivirals are effective against influenza viruses with genes from the 1918 pandemic virus. Proc. Natl Acad. Sci. USA. 2002;99:13849–13854. doi: 10.1073/pnas.212519699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Le QM, et al. Avian flu: isolation of drug-resistant H5N1 virus. Nature. 2005;437:1108. doi: 10.1038/4371108a. [DOI] [PubMed] [Google Scholar]
- 49.Gubareva LV, Kaiser L, Matrosovich MN, Soo-Hoo Y, Hayden FG. Selection of influenza virus mutants in experimentally infected volunteers treated with oseltamivir. J. Infect. Dis. 2001;183:523–531. doi: 10.1086/318537. [DOI] [PubMed] [Google Scholar]
- 50.Ives JA, et al. The H274Y mutation in the influenza A/H1N1 neuraminidase active site following oseltamivir phosphate treatment leave virus severely compromised both in vitro and in vivo. Antiviral Res. 2002;55:307–317. doi: 10.1016/s0166-3542(02)00053-0. [DOI] [PubMed] [Google Scholar]
- 51.de Jong MD, et al. Oseltamivir resistance during treatment of influenza A (H5N1) infection. N. Engl. J. Med. 2005;353:2667–2672. doi: 10.1056/NEJMoa054512. [DOI] [PubMed] [Google Scholar]
- 52.Govorkova EA, Ilyushina NA, Smith J, Webster RG. Oseltamivir protects ferrets against lethal H5N1 influenza virus infection. Antiviral Res. 2006;70:A29. [Google Scholar]
- 53.Yen H-L, Monto AS, Webster RG, Govorkova EA. Virulence may determine the necessary duration and dosage of oseltamivir treatment for highly pathogenic A/Vietnam/1203/04 influenza virus in mice. J. Infect. Dis. 2005;192:665–672. doi: 10.1086/432008. [DOI] [PubMed] [Google Scholar]
- 54.Smee DF, Huffman JH, Morrison AC, Barnard DL, Sidwell RW. Cyclopentane neuraminidase inhibitors with potent in vitro anti-influenza virus activities. Antimicrob. Agents Chemother. 2001;45:743–748. doi: 10.1128/AAC.45.3.743-748.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sidwell RW, et al. In vivo influenza virus-inhibitory effects of the cyclopentane neuraminidase inhibitor RJW-270201. Antimicrob. Agents Chemother. 2001;45:749–757. doi: 10.1128/AAC.45.3.749-757.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chand P, et al. Comparison of the anti-influenza virus activity of cyclopentane derivatives with oseltamivir and zanamivir in vivo. Bioorg. Med. Chem. 2005;13:4071–4077. doi: 10.1016/j.bmc.2005.03.048. [DOI] [PubMed] [Google Scholar]
- 57.Chand P, et al. Systematic structure-based design and stereoselective synthesis of novel multisubstituted cyclopentane derivatives with potent antiinfluenza activity. J. Med. Chem. 2001;44:4379–4392. doi: 10.1021/jm010277p. [DOI] [PubMed] [Google Scholar]
- 58.Chand P, et al. Syntheses and neuraminidase inhibitory activity of multisubstituted cyclopentane amide derivatives. J. Med. Chem. 2004;47:1919–1929. doi: 10.1021/jm0303406. [DOI] [PubMed] [Google Scholar]
- 59.Wang GT, et al. Design, synthesis, and structural analysis of influenza neuraminidase inhibitors containing pyrrolidine cores. J. Med. Chem. 2001;44:1192–1201. doi: 10.1021/jm000468c. [DOI] [PubMed] [Google Scholar]
- 60.DeGoey DA, et al. Enantioselective synthesis of antiinfluenza compound A-315675. J. Org. Chem. 2002;67:5445–5453. doi: 10.1021/jo0162890. [DOI] [PubMed] [Google Scholar]
- 61.Hanessian S, Bayrakdarian M, Luo X. Total synthesis of A-315675: a potent inhibitor of influenza neuraminidase. J. Am. Chem. Soc. 2002;124:4716–4721. doi: 10.1021/ja0126226. [DOI] [PubMed] [Google Scholar]
- 62.Kati WM, et al. In vitro characterization of A-315675, a highly potent inhibitor of A and B strain influenza virus neuraminidases and influenza virus replication. Antimicrob. Agents Chemother. 2002;46:1014–1021. doi: 10.1128/AAC.46.4.1014-1021.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Maring CJ, et al. Structure-based characterization and optimization of novel hydrophobic binding interactions in a series of pyrrolidine influenza neuraminidase inhibitors. J. Med. Chem. 2005;48:3980–3990. doi: 10.1021/jm049276y. [DOI] [PubMed] [Google Scholar]
- 64.Wang GT, et al. Design, synthesis, and structural analysis of inhibitors of influenza neuraminidase containing a 2,3-disubstituted tetrahydrofuran-5-carboxylic acid core. Bioorg. Med. Chem. Lett. 2005;15:125–128. doi: 10.1016/j.bmcl.2004.10.022. [DOI] [PubMed] [Google Scholar]
- 65.Mishin V, Hayden FG, Gubareva LV. Susceptibilities of antiviral-resistant influenza viruses to novel neuraminidase inhibitors. Antimicrob. Agents Chemother. 2005;49:4515–4520. doi: 10.1128/AAC.49.11.4515-4520.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Molla A, et al. In vitro selection and characterization of influenza A (A/N9) virus variants resistant to a novel neuraminidase inhibitor, A-315675. J. Virol. 2002;76:5380–5386. doi: 10.1128/JVI.76.11.5380-5386.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gubareva LV. Molecular mechanisms of influenza virus resistance to neuraminidase inhibitors. Virus Res. 2004;103:199–203. doi: 10.1016/j.virusres.2004.02.034. [DOI] [PubMed] [Google Scholar]
- 68.Bantia S, Arnold CS, Parker CD, Upshaw R, Chand P. Anti-influenza virus activity of peramivir in mice with single intramuscular injection. Antiviral Res. 2006;69:39–45. doi: 10.1016/j.antiviral.2005.10.002. [DOI] [PubMed] [Google Scholar]
- 69.Sidwell RW, et al. Effect of single i. m. or i. v. injection of peramivir on an influenza A (H5N1) virus infection in mice. Antiviral Res. 2006;70:A50. [Google Scholar]
- 70.Barroso L, Treanor J, Gubareva L, Hayden FG. Efficacy and tolerability of the oral neuraminidase inhibitor peramivir in experimental human influenza: randomized, controlled trials for prophylaxis and treatment. Antiviral Ther. 2005;10:901–910. [PubMed] [Google Scholar]
- 71.Honda T, et al. Synthesis and anti-influenza virus activity of 7-O-alkylated derivatives related to zanamivir. Bioorg. Med. Chem. Lett. 2002;12:1925–1928. doi: 10.1016/s0960-894x(02)00329-3. [DOI] [PubMed] [Google Scholar]
- 72.Masuda T, et al. Synthesis and anti-influenza evaluation of orally active bicyclic ether derivatives related to zanamivir. Bioorg. Med. Chem. Lett. 2003;13:669–673. doi: 10.1016/s0960-894x(02)01039-9. [DOI] [PubMed] [Google Scholar]
- 73.Macdonald SJ, et al. Dimeric zanamivir conjugates with various linking groups are potent, long-lasting inhibitors of influenza neuraminidase including H5N1 avian influenza. J. Med. Chem. 2005;48:2964–2971. doi: 10.1021/jm040891b. [DOI] [PubMed] [Google Scholar]
- 74.Macdonald SJF, et al. Potent and long-acting dimeric inhibitors of influenza virus neuraminidase are effective at a once-weekly dosing regimen. Antimicrob. Agents Chemother. 2004;48:4542–4549. doi: 10.1128/AAC.48.12.4542-4549.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sidwell RW, et al. Broad-spectrum antiviral activity of virazole: 1-β-D-ribofuranosyl-1,2,4-triazole-3-carboxamide. Science. 1972;177:705–706. doi: 10.1126/science.177.4050.705. [DOI] [PubMed] [Google Scholar]
- 76.Sidwell RW, Bailey KW, Wong M-H, Barnard DL, Smee DF. In vitro and in vivo influenza virus-inhibitory effects of viramidine. Antiviral Res. 2005;68:10–17. doi: 10.1016/j.antiviral.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 77.Smith CB, Charette RP, Fox JP, Cooney MK, Hall CE. Lack of effect of oral ribavirin in naturally occurring influenza A virus (H1N1) infection. J. Infect. Dis. 1980;141:548–554. doi: 10.1093/infdis/141.5.548. [DOI] [PubMed] [Google Scholar]
- 78.Knight V, Gilbert BE. Ribavirin aerosol treatment of influenza. Infect. Dis. Clin. North Am. 1987;1:441–457. [PubMed] [Google Scholar]
- 79.Hayden FG, Sable CA, Connor JD, Lane J. Intravenous ribavirin by constant infusion for serious influenza and parainfluenzavirus infection. Antiviral Ther. 1996;1:51–56. [PubMed] [Google Scholar]
- 80.McCormick JB, et al. Lassa fever. Effective therapy with ribavirin. N. Engl. J. Med. 1986;314:20–26. doi: 10.1056/NEJM198601023140104. [DOI] [PubMed] [Google Scholar]
- 81.Huggins JW, et al. Prospective, double-blind, concurrent, placebo-controlled clinical trial of intravenous ribavirin therapy of hemorrhagic fever with renal syndrome. J. Infect. Dis. 1991;164:1119–1127. doi: 10.1093/infdis/164.6.1119. [DOI] [PubMed] [Google Scholar]
- 82.Malakhov MP, et al. Sialidase fusion protein as a novel broad-spectrum inhibitor of influenza virus infection. Antimicrob. Agents Chemother. 2006;50:1470–1479. doi: 10.1128/AAC.50.4.1470-1479.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gambaryan AS, et al. Polymeric inhibitor of influenza virus attachment protects mice from experimental influenza infection. Antiviral Res. 2002;55:201–205. doi: 10.1016/s0166-3542(02)00020-7. [DOI] [PubMed] [Google Scholar]
- 84.Gambaryan AS, et al. Polymer-bound 6′ sialyl-N-acetyllactosamine protects mice infected by influenza virus. Antiviral Res. 2005;68:116–123. doi: 10.1016/j.antiviral.2005.07.008. [DOI] [PubMed] [Google Scholar]
- 85.Ge Q, et al. Inhibition of influenza virus production in virus-infected mice by RNA interference. Proc. Natl Acad. Sci. USA. 2004;101:8676–8681. doi: 10.1073/pnas.0402486101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tompkins SM, Lo CY, Tumpey TM, Epstein SL. Protection against lethal influenza virus challenge by RNA interference in vivo. Proc. Natl Acad. Sci. USA. 2004;101:8682–8686. doi: 10.1073/pnas.0402630101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Li BJ, et al. Using siRNA in prophylactic and therapeutic regimens against SARS coronavirus in Rhesus macaque. Nature Med. 2005;11:944–951. doi: 10.1038/nm1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Vaillant A, et al. Rep 9: a potent broad spectrum aerosol prophylaxis and therapy against influenza infection in vivo. Antiviral Res. 2006;70:A52. [Google Scholar]
- 89.Iversen P, et al. Inhibition of multiple influenza A subtypes in cell culture with antisense phosphorodiamidate morpholino oligomers. Antiviral Res. 2006;70:A49. [Google Scholar]
- 90.Deng T, Sharps J, Fodor E, Brownlee GG. In vitro assembly of PB2 with a PB1–PA dimer supports a new model of assembly of influenza A virus polymerase subunits into a functional trimeric complex. J. Virol. 2005;79:8669–8674. doi: 10.1128/JVI.79.13.8669-8674.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Salomon R, et al. The polymerase complex genes contribute to the high virulence of the human H5N1 influenza virus isolate A/Vietnam/1203/04. J. Exp. Med. 2006;203:689–697. doi: 10.1084/jem.20051938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Tuttle JV, Tisdale M, Krenitsky TA. Purine 2′-deoxy-2′-fluororibosides as antiinfluenza virus agents. J. Med. Chem. 1993;36:119–125. doi: 10.1021/jm00053a015. [DOI] [PubMed] [Google Scholar]
- 93.Tisdale M, Ellis M, Klumpp K, Court S, Ford M. Inhibition of influenza virus transcription by 2′-deoxy-2′-fluoroguanosine. Antimicrob. Agents Chemother. 1995;39:2454–2458. doi: 10.1128/aac.39.11.2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Furuta Y, et al. In vitro and in vivo activities of anti-influenza virus compound T-705. Antimicrob. Agents Chemother. 2002;46:977–981. doi: 10.1128/AAC.46.4.977-981.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Takahashi K, et al. In vitro and in vivo activities of T-705 and oseltamivir against influenza virus. Antiviral Chem. Chemother. 2003;14:235–241. doi: 10.1177/095632020301400502. [DOI] [PubMed] [Google Scholar]
- 96.Furuta Y, et al. Mechanism of action of T-705 against influenza virus. Antimicrob. Agents Chemother. 2005;49:981–986. doi: 10.1128/AAC.49.3.981-986.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Tomassini J, et al. Inhibition of cap (m7GpppXm)-dependent endonuclease of influenza virus by 4-substituted 2,4-dioxobutanoic acid compounds. Antimicrob. Agents Chemother. 1994;38:2827–2837. doi: 10.1128/aac.38.12.2827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Cianci C, et al. Identification of N-hydroxamic acid and N-hydroxy-imide compounds that inhibit the influenza virus polymerase. Antiviral Chem. Chemother. 1996;7:353–360. [Google Scholar]
- 99.Tomassini J, et al. A novel antiviral agent which inhibits the endonuclease of influenza viruses. Antimicrob. Agents Chemother. 1996;40:1189–1193. doi: 10.1128/aac.40.5.1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sun C, et al. A novel class of potent influenza virus inhibitors: polysubstituted acylthiourea and its fused heterocycle derivatives. Bioorg. Med. Chem. Lett. 2006;16:162–166. doi: 10.1016/j.bmcl.2005.09.033. [DOI] [PubMed] [Google Scholar]
- 101.Wang W-L, et al. Synthesis and biological evaluation of novel bisheterocycle-containing compounds as potential anti-influenza virus agents. Bioorg. Med. Chem. Lett. 2005;15:5284–5287. doi: 10.1016/j.bmcl.2005.08.046. [DOI] [PubMed] [Google Scholar]
- 102.Isaacs A, Lindenmann J. Virus interference. I. The interferon. Proc. R. Soc. Lond. B Biol. Sci. 1957;147:258–267. [PubMed] [Google Scholar]
- 103.Baron S, Isaacs A. Absence of interferon in lungs from fatal cases of influenza. Br. Med. J. 1962;5270:18–20. doi: 10.1136/bmj.1.5270.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Isomura S, et al. The preventive effect of human interferon-α on influenza infection; modification of clinical manifestations of influenza in children in a closed community. Biken J. 1982;25:131–137. [PubMed] [Google Scholar]
- 105.Phillpotts RJ, et al. Intranasal lymphoblastoid interferon (“Wellferon”) prophylaxis against rhinovirus and influenza virus in volunteers. J. Interferon Res. 1984;4:535–541. doi: 10.1089/jir.1984.4.535. [DOI] [PubMed] [Google Scholar]
- 106.Fried MW, et al. Peginterferon α-2a plus ribavirin for chronic hepatitis C virus infection. N. Engl. J. Med. 2002;347:975–982. doi: 10.1056/NEJMoa020047. [DOI] [PubMed] [Google Scholar]
- 107.Field AK, Tytell AA, Lampson GP, Hilleman MR. Inducers of interferon and host resistance. II. Multistranded synthetic polynucleotide complexes. Proc. Natl Acad. Sci. USA. 1967;58:1004–1010. doi: 10.1073/pnas.58.3.1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Saravolac EG, et al. Immunoprophylactic strategies against respiratory influenza virus infection. Vaccine. 2001;19:2227–2232. doi: 10.1016/s0264-410x(00)00450-3. [DOI] [PubMed] [Google Scholar]
- 109.Ichinohe T, et al. Synthetic double-stranded RNA poly(I:C) combined with mucosal vaccine protects against influenza virus infection. J. Virol. 2005;79:2910–2919. doi: 10.1128/JVI.79.5.2910-2919.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Yang H, et al. Antiviral chemotherapy facilitates control of poxvirus infections through inhibition of cellular signal transduction. J. Clin. Invest. 2005;115:379–387. doi: 10.1172/JCI23220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Reeves PM, et al. Disabling poxvirus pathogenesis by inhibition of Abl-family tyrosine kinases. Nature Med. 2005;11:731–739. doi: 10.1038/nm1265. [DOI] [PubMed] [Google Scholar]
- 112.Pleschka S, et al. Influenza virus propagation is impaired by inhibition of the Raf/MEK/ERK signalling cascade. Nature Cell Biol. 2001;3:301–305. doi: 10.1038/35060098. [DOI] [PubMed] [Google Scholar]
- 113.Marjuki H, et al. Membrane accumulation of influenza A virus hemagglutinin triggers nuclear export of the viral genome via protein kinase Cα-mediated activation of ERK signaling. J. Biol. Chem. 2006;281:16707–16715. doi: 10.1074/jbc.M510233200. [DOI] [PubMed] [Google Scholar]
- 114.Twu KY, Noah DL, Rao P, Kuo R-L, Krug RM. The CPSF30 binding site on the NS1A protein of influenza A virus is a potential antiviral target. J. Virol. 2006;80:3957–3965. doi: 10.1128/JVI.80.8.3957-3965.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Bizollon T, et al. Triple antiviral therapy with amantadine for IFN-ribavirin nonresponders with recurrent posttransplantation hepatitis C. Transplantation. 2005;79:325–329. doi: 10.1097/01.tp.0000149499.78996.b3. [DOI] [PubMed] [Google Scholar]
- 116.Hayden FG, Schlepushkin AN, Pushkarskaya NL. Combined interferon-α2, rimantadine hydrochloride, and ribavirin inhibition of influenza virus replication in vitro. Antimicrob. Agents Chemother. 1984;25:53–57. doi: 10.1128/aac.25.1.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Govorkova EA, Fang H-B, Tan M, Webster RG. Neuraminidase inhibitor-rimantadine combinations exert additive and synergistic anti-influenza virus effects in MDCK cells. Antimicrob. Agents Chemother. 2004;48:4855–4863. doi: 10.1128/AAC.48.12.4855-4863.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ilyushina N, Hoffmann E, Salomon R, Webster R, Govorkova E. Advantages of combination chemotherapy for highly pathogenic A/Vietnam/1203/04 (H5N1) influenza virus in mice. Antiviral Res. 2006;70:A29. [Google Scholar]
- 119.Ison MG, et al. Safety and efficacy of nebulized zanamivir in hospitalized patients with serious influenza. Antiviral Ther. 2003;8:183–190. [PubMed] [Google Scholar]