

Abstract
Objective:
The cyclin-dependent kinase-like 5 (CDKL5) gene is a known cause of early onset developmental and epileptic encephalopathy, CDKL5 Deficiency Disorder (CDD). We sought to provide 1) a description of seizure types in patients with CDD; 2) an assessment of the frequency of seizure free periods and cortical visual impairment (CVI); 3) correlate these features with genotype and gender; and 4) correlate these features with developmental milestones.
Methods:
This is a cohort study of patients with CDD. Phenotypic features were explored and correlated with gene variant grouping1 and gender. A developmental score was created based on achieving seven primary milestones. Phenotypic variables were correlated with the developmental score to explore markers of better developmental outcomes. Multivariable linear regression was used to account for age at last visit.
Results:
Ninety-two patients with CDD were seen during the enrollment period. Eighteen were male (19%) and with a median age at last visit of 5 years (IQR 2.0, 11.0). Eighty-one percent of patients developed epileptic spasms but only 47% of those also had hypsarrhythmia. Previously described hypertonic-spasm-sequence (HTSS) was seen in only 24% of patients but 56% of patients had seizures with multiple phases (often tonic and spasms). Forty-three percent of patients experienced a seizure free period ranging from 1 to more than 12 months, but only 6% were still seizure free at the last visit. CVI was present in 75% of all CDD patients. None of these features were associated with genotype group or gender. Cortical visual impairment was correlated with reduced milestone achievement after adjusting for age at last visit and a history of hypsarrhythmia.
Significance:
The most common seizure types in CDD are epileptic spasms (often without hypsarrhythmia) and tonic seizures that may cluster together. CVI is a common feature in CDD and is correlated with achieving fewer milestones.
Keywords: CDD, CVI, spasms, hypsarrhythmia, Hypermotor Tonic-spasms sequence
Introduction:
CDKL5 (cyclin-dependent kinase-like 5) Deficiency Disorder (CDD, OMIM 300203, 300672) is a rare neurologic condition characterized by onset of seizures in the first weeks to months of life and significantly reduced developmental milestones2–6. It has also been referred to as CDKL5 encephalopathy, CDKL5 epileptic encephalopathy or CDKL5 related epilepsy. It has an estimated incidence between 1 in 40,000 and 1 in 60,000 live births7; 8. CDD is one of the more common genetic cause of epilepsy in early childhood8. Patients with CDD are reported to have very early onset of epilepsy (median of 6 weeks9) and severe developmental delay with many patients unable to achieve independent walking or speech1; 10. However, CDD also affects several other neurologic domains with evidence of disrupted sleep11 GI problems12, dysautonomia5 and cortical visual impairment5 (CVI). CVI is also seen in rodent models of CDKL513 and has been demonstrated to be an important comorbidity and potentially a marker of clinical improvement in other epileptic encephalopathies14; 15. However, CVI has not been systematically assessed in a large cohort of CDD patients.
Genotype-phenotype correlation has been challenging in CDD due to relatively small cohorts historically and a lack of recurrent pathogenic variants3; 16. To overcome this challenge, a system of classifying variants into four groups has been proposed: Type A - absence of any functional protein; Type B - missense variants in the kinase domain; Type C – truncating variants between aa 172 and 781; and Type D – truncating variants between aa 781 and 9051. This system was originally correlated with achievement of developmental milestones without clear statistical correlations. However, later studies did find associations between variant type and seizure frequency9. This systematic effort to characterize the epilepsy phenotype associated with CDD verified the refractory nature of epilepsy with all but three patients having epilepsy and although many had short periods of seizure freedom this was rarely maintained long-term. Decreased seizure frequency was associated with Type C variants compared to Type A, as well as ability to walk and use of spoken language. However, this study was based on parental questionnaires which limited the assessment of seizure types as a variable9.
Starting in 2014 the International Foundation for CDKL5 Research began establishing CDKL5 Centers of Excellence (Children’s Hospital Colorado, Cleveland Clinic and Boston Children’s Hospital). Centers have been collecting standardized clinical data on CDD patients informed by investigators clinical experience with CDD in the NIH Rett and Rett-related disorders Natural History Study consortium (U54 HD061222; ClinicalTrials.gov: NCT00299312/NCT02738281). Using a combined data set we sought to provide 1) a detailed description of the frequency of seizure free periods, cortical visual impairment, and the seizure types seen in CDD, with a particular focus on the frequency of infantile spasms and hypsarrhythmia; 2) an assessment of whether gene grouping is associated with phenotypic features; and 3) an exploration of whether these specific clinical features are associated with achievement of developmental milestones after adjusting for age.
Methods:
A cross sectional study exploring phenotypic features associated with CDKL5 Deficiency Disorder (CDD) was performed on patients seen at one of the three CDKL5 Centers of Excellence (Children’s Hospital Colorado, Cleveland Clinic Children’s, and Boston Children’s Hospital) between April 2014 and December 2017. These centers frequently received tertiary referrals of patients with CDD. Data was collected at each center using standardized history and exam forms as well as chart review for retrospective data. Data collection forms were developed, modified and expanded based on investigator experience in evaluating patients with CDD in clinical care and in the NIH Rett and Rett-related disorders Natural History Study consortium (U54 HD061222; ClinicalTrials.gov: NCT00299312/NCT02738281) and in collaboration with the International Foundation for CDKL5 research. Specifically, data collected was considered part of clinical care and was collected in the format and time-frame of a routine clinical visit. Each site had its own IRB (CO protocol 13–2020, CC protocol 14–478, BCH protocol P00016602) and consented patients directly. National Institutes of Health National Database for Autism Research (NDAR) Global Unique Identifiers were used to ensure that patient data was not included more than once if subjects were seen at more than one site. De-identified site data were combined for final analysis.
Seizure types were determined by the physician history at each center with an effort to classify according to International League Against Epilepsy classification of seizure types when possible17. Available electroencephalographic (EEG) records were reviewed for the presence of hypsarrhythmia at any time in the first three years of life. Presence of hypsarrhythmia was determined by the clinical report. EEGs were not available for three patients. The standard history assessed for periods of seizure freedom.
Cortical visual impairment (CVI) was determined by both parental report of the patient’s functional abilities and neurologist’s physical exam findings. While there is no specific approach deemed a gold-standard to diagnosing CVI18, we followed recommended practice and a proposed method for in-clinic assessment19. Given variability in exam technique, vision findings were reduced to a global provider impression of whether the patient had CVI. Patients were considered to have CVI if there was at least one of the following exam findings: impaired fix and follow, nystagmus, roving eye movements or abnormal optokinetic nystagmus response without any evidence of structural eye abnormalities.
Developmental outcomes were based on parental report of milestones achieved and provider assessment at the time of the visit. Given the severity of developmental impairment in CDD, a typical developmental quotient would not capture existing variation between CDD patients. Therefore, a novel score for development was calculated on a scale of 0–7 by summing up the number of developmental milestones reached: independent sitting, independent standing, independent walking, raking grasp, pincer grasp, babble and single word as these milestones have been used previously in genotype-phenotype correlation1. This is referred to throughout this manuscript as the CDKL5 Developmental Score (CDS).
Gene variants were categorized according to the Fehr et al gene groupings previously published and described above1. Pathogenicity of all variants were classified according to ACMG criteria20 using the evidence available as of March 201921. Patient were included even if they had variants of uncertain significance if the clinical CDD expert at each COE felt that their phenotype was appropriate. There no patient with “benign” or “likely benign” variants.
Standard frequencies and proportions were used to summarize categorical data. Means (standard deviation) and medians (interquartile range) summarized continuous data. Patient and clinical characteristics were compared across gene types by Chi Square, Kruskal Wallis, and ANOVA tests. ANOVA was used to compare CDS across groupings of age at last visit. For subjects at least 1 year of age, mean CDS was compared across clinical variables (including: epilepsy feature, hypsarrhythmia and presence of CVI) using t-tests. Individual linear models were used to adjust clinical variable associations with CDS for age (at last visit) and test the interaction of age with each clinical variable. Insignificant interaction terms were not considered in further regression modeling. The percentage of variability explained by the specific variable after adjusting for age was calculated (R2) for each individual linear model. Forward stepwise selection was used to build a multivariable linear regression model based on selected clinical variables with a significant (p<0.05) age adjusted association with CDS. Since previous literature suggest that hypsarrhythmia may influence developmental outcomes22, this was kept in the multivariable model regardless of significance. Linear regression assumptions and goodness of fit were assessed with diagnostic plots and adjusted R2. Cohen’s F2 was calculated as an effect size estimate and were evaluated according to Cohen’s (1988) guidelines where f2>=0.02 represents a small effect size, f2>=0.15 represents a medium effect size, and f2 >=0.35 represents a large effect side23. The statistical analysis was done in SAS 9.4. All tests were performed with a 0.05 level of significance. Given the exploratory nature of this study adjustments were not made for multiple comparisons, but this was taken in to account when interpreting results.
Results:
Ninety-two patients with CDD were seen at the three centers during the enrollment period. Eighteen were male (19%) and there was a median age of 5 years (IQR 2.0, 11.0) at last visit. Seven patients had variants of uncertain significance. The remainder hall had pathogenic or likely pathogenic variants based on ACMG classification (Supplemental Table 1). There were 5 patients (4 male and 1 female) who were mosaic for their CDKL5 variant based on clinical laboratory reporting. All patients had epilepsy with a median age of onset of 5 weeks (IQR 3.0, 8.0 – Table 1) though one patient had onset at 9 years of age.
Table 1:
Bivariable Analysis Gene Type
| Gene Type | Total (n=92) n (%) | A (n=23) n (%) | B (n=26) n (%) | C (n=32) n (%) | D (n=11) n (%) | P-value* |
|---|---|---|---|---|---|---|
| Age at Seizure Onset (weeks) Median (IQR) | 5.0 (3.0,8.0) | 4.0 (2.0,24.0) | 5.0 (3.0,8.0) | 6.0 (4.0,10.5) | 5.0 (3.0,8.0) | 0.68 |
| Age at Last Visit (years) Median (IQR) | 5.0 (2.0,11.0) | 4.5 (2.5,9.0) | 5.0 (1.5,13.0) | 4.0 (2.0,9.0) | 9.5 (1.8,17.0) | 0.83 |
| Male (%) | 18 (20) | 5 (22) | 5 (19) | 5 (16) | 3 (27) | 0.85 |
| Seizure Type (Initial) (%) | ||||||
| Epileptic Spasms | 21 (23) | 5 (22) | 5 (19) | 9 (28) | 2 (18) | 0.84 |
| Tonic | 29 (32) | 7 (30) | 12 (46) | 7 (22) | 3 (27) | 0.25 |
| HTSS | 9 (10) | 1 (4) | 2 (8) | 3 (9) | 3 (27) | 0.19 |
| GTC | 20 (22) | 7 (30) | 2 (8) | 8 (25) | 3 (27) | 0.22 |
| Myoclonic | 6 (7) | 1 (4) | 2 (8) | 3 (9) | 0 (0) | 0.70 |
| Clonic | 8 (9) | 0 (0) | 4 (15) | 3 (9) | 1 (9) | 0.30 |
| Focality | 20 (22) | 6 (26) | 8 (31) | 5 (16) | 1 (9) | 0.36 |
| Hypsarrhythmia (%)* | 35 (38) | 9 (39) | 10 (38) | 9 (28) | 7 (64) | 0.25 |
| Seizure Types (Anytime) (%) | ||||||
| Epileptic Spasms | 75 (82) | 17 74) | 23 (88) | 26 (81) | 9 (82) | 0.63 |
| Tonic | 59 (64) | 16 (70) | 15 (58) | 21 (66) | 7 (64) | 0.85 |
| HTSS Combined | 52 (57) | 13 (57) | 17 (65) | 17 (53) | 5 (45) | 0.68 |
| HTSS | 22 (24) | 6 (26) | 8 (31) | 5 (16) | 3 (27) | 0.57 |
| HTSS like | 31 (34) | 8 (35) | 9 (35) | 12 (38) | 2 (18) | 0.70 |
| Generalized Tonic Clonic | 31 (34) | 8 (35) | 7 (27) | 13 (41) | 3 (27) | 0.70 |
| Myoclonic | 36 (39) | 9 (39) | 11 (42) | 12 (38) | 4 (36) | 0.98 |
| Clonic | 10 (11) | 4 (17) | 3 (12) | 2 (6) | 1 (9) | 0.62 |
| Atonic | 8 (9) | 1 (4) | 1 (4) | 3 (9) | 3 (27) | 0.11 |
| Absence | 2 (2) | 0 (0) | 0 (0) | 1 (3) | 1 (9) | 0.30 |
| Focality | 28 (30) | 10 (43) | 9 (35) | 6 (19) | 3 (27) | 0.24 |
| Period of Seizure Freedom (%) | ||||||
| 1–3 months | 8 (9) | 2 (9) | 2 (8) | 2 (6) | 2 (18) | 0.68 |
| 3–6 months | 11 (12) | 3 (13) | 1 (4) | 6 (19) | 1 (9) | 0.37 |
| 6–12 months | 10 (11) | 3 (13) | 4 (15) | 2 (6) | 1 (9) | 0.71 |
| > 12 months | 11 (12) | 3 (13) | 2 (8) | 4 (13) | 2 (18) | 0.83 |
| Combined Seizure Freedom FrffsdfFreeFreedom | 40 (43) | 11 (48) | 9 (35) | 14 (44) | 6 (55) | 0.67 |
| Cortical Visual Impairment (%) | 70 (76) | 18 (78) | 23 (88) | 22 (69) | 7 (64) | 0.25 |
| CDS - Mean (std) | 2.6 (2.1) | 2.7 (1.9) | 2.5 (2.2) | 2.6 (2.2) | 2.8 (2.2) | 0.96 |
Gene type grouping is based on prior publication1 as explained in the introduction and methods. Seizure types are not mutually exclusive. Specifically, focality could represent either a focal seizure or one of the other seizure types with focal features (for example an asymmetric tonic seizure was marked as both focality and tonic seizure). CDS – CDKL5 Developmental Score is a 7-point scale based on achievement of milestones. The p-values shown in the table have not been adjusted for multiple comparisons.
The most common initial seizure type was tonic seizures (32%) followed by epileptic spasms and generalized tonic clonic seizures (23 and 22% respectively). Clonic and myoclonic were less common (9% and 7% respectively). The most common seizure at any point in time was epileptic spasms (82% - which typically started in infancy but often persisted at older ages). However, while epileptic spasms typically started in infancy, only 38% of patients had hypsarrhythmia captured at some point in their course (47% of patient who had epileptic spasms). EEG records were not available for 3 patients. Tonic seizures were the next most common seizure at any time (64%) with myoclonic (39%) and generalized tonic clonic (34%) also being common. Clonic (11%), atonic (9%) and absence (2%) were less common.
Hypermotor tonic spasms sequence seizures (HTSS)24 were described in 24% percent of patients but an additional 34% of patients reported seizures that had multiple phases with clustering of tonic seizures and epileptic spasms (HTSS-like). Often the seizures occurred in a variety of orders even for patients with classically described HTSS (in some cases the hypermotor phase occurred at the end). Overall, 57% of patients had either HTSS or HTSS-like seizures. Thirty percent of all patients had seizures that were either focal or had focality (such as an asymmetric tonic seizure). One patient underwent functional hemispherectomy for persistent hemispheric seizures prior to availability of genetic testing. Patients had an average of 2.8 (STD 1.4) different seizure types at any time. While seizures were typically refractory to treatment 40 patients (43%) did have a seizure free period. Eleven patients (12%) were seizure free for more than a year, but only 6 (6.5%) were still seizure free at the last visit (Table 1).
CVI was diagnosed in 70 patients (76%). Gene groupings failed to show any statistical difference in the proportions of patients with various seizure types, hypsarrhythmia, periods of seizure freedom, CVI or mean CDS (Table 1) (Supplemental Table 1 contains a complete list of all variants analyzed by gene group). Additionally, none of these features were associated with gender except that generalized tonic clonic seizures were less common in males (11% in males compared to 39% in females, p=0.03) (Table 2).
Table 2:
Bivariable Analysis Gender
| Variable | Total (n=92) | Female (n=74) | Male (n=18) | P-value |
|---|---|---|---|---|
| Age at Seizure Onset (weeks) Median (IQR) | 5.0 (3.0,8.0) | 5.0 (3.0,8.0) | 6.0 (4.0,9.0) | 0.35 |
| Age at Last Visit (years) Median (IQR) | 5.0 (2.0,11.0) | 5.0 (2.0,10.0) | 4.8 (2.5,12.0) | 0.73 |
| Seizure Type (Initial) (%) | ||||
| Epileptic Spasms | 21 (23) | 14 (19) | 7 (39) | 0.11 |
| Tonic | 29 (32) | 25 (34) | 4 (22) | 0.34 |
| HTSS | 9 (10) | 8 (11) | 1 (6) | 0.50 |
| GTC | 20 (22) | 18 (24) | 2 (11) | 0.34 |
| Myoclonic | 6 (7) | 4 (5) | 2 (11) | 0.33 |
| Clonic | 8 (9) | 7 (9) | 1 (6) | 0.60 |
| Focality | 20 (22) | 17 (23) | 3 (17) | 0.75 |
| Hypsarrhythmia (%)* | 35 (38) | 25 (34) | 10 (56) | 0.11 |
| Seizure Types (Anytime) (%) | ||||
| Epileptic Spasms | 75 (82) | 61 (82) | 14 (78) | 0.74 |
| Tonic | 59 (64) | 48 (65) | 11 (61) | 0.77 |
| HTSS Combined | 52 (57) | 43 (58) | 9 (50) | 0.53 |
| HTSS | 22 (24) | 19 (26) | 3 (17) | 0.55 |
| HTSS like | 31 (34) | 24 (32) | 7 (39) | 0.60 |
| Generalized Tonic Clonic | 31 (34) | 29 (39) | 2 (11) | 0.03 |
| Myoclonic | 36 (39) | 30 (41) | 6 (33) | 0.57 |
| Clonic | 10 (11) | 7 (9) | 3 (17) | 0.40 |
| Atonic | 8 (9) | 8 (11) | 0 (0) | 0.35 |
| Absence | 2 (2) | 2 (3) | 0 (0) | 0.48 |
| Focality | 28 (30) | 22 (30) | 6 (33) | 0.77 |
| Period of Seizure Freedom (%) | ||||
| 1–3 months | 8 (9) | 5 (7) | 3 (17) | 0.19 |
| 3–6 months | 11 (12) | 8 (11) | 3 (17) | 0.44 |
| 6–12 months | 10 (11) | 8(11) | 2 (11) | 0.97 |
| > 12 months | 11 (12) | 10 (14) | 1 (6) | 0.35 |
| Combined Seizure Freedom | 40 (43) | 31 (42) | 9 (50) | 0.53 |
| Cortical Visual Impairment (%) | 70 (76) | 57 (77) | 13 (72) | 0.67 |
| CDS - Mean (std) | 2.6 (2.1) | 2.6 (2.0) | 2.5 (2.4) | 0.81 |
Table compares the same traits as table one with the same definitions but by gender (P-values are unadjusted).
There was an average developmental score of 2.6 (STD 2.1) out of seven potential developmental milestones. Higher average developmental scores were associated with older patient age at last visit (Table 3). For patients older than 1 year at last visit (n=84), a lack of hypsarrhythmia and absence of cortical visual impairment were significantly associated with higher average developmental scores on bivariable analysis (Table 4). Cortical visual impairment was considered to have a medium effect size according to Cohen’s f2 (Table 4). Individual multivariable regressions determined all interaction terms with age to be insignificant (not reported). A final multivariable linear regression model revealed that on average, after controlling for hypsarrhythmia and age at last visit, the developmental scores for those with cortical visual impairment were 1.58 (2.48,0.67) less than those without cortical visual impairment (p=0.0008, Figure 1). CVI and age at last visit both represented medium effect sizes according to Cohen’s f2.
Table 3:
Developmental Score by Age of Last Visit
| Age | N | Developmental Score Mean (sd) | P-value |
|---|---|---|---|
| <= 2 years | 25 | 1.4 (1.4) | < 0.001 |
| 2–5 years | 23 | 2.0 (2.0) | |
| 6–10 years | 20 | 3.4 (1.9) | |
| > 10 years | 23 | 3.8 (2.1) |
Comparison of mean developmental score across age categories using ANOVA.
Table 4:
Development Score for subjects >=1 years of age at last visit
| Development Score Mean (95% CI) | Adjusted Association* | ||||
|---|---|---|---|---|---|
| Variables | Yes | No | Adjusted Pvalue | R2 | Cohen’s f2 |
| Hypsarrhythmia | 2.2 (1.5,2.8) | 3.2 (2.6,3.8) | 0.0251 | 0.23 | 0.04 |
| Epileptic Spasms | 2.5 (2.1,3.0) | 3.6 (2.5,4.7) | 0.0926 | 0.23 | 0.04 |
| Tonic | 2.6 (2.1,3.2) | 2.9 (2.2,3.7) | 0.7612 | 0.20 | 0.00 |
| HTSS | 2.7 (2.0,3.4) | 2.7 (2.2,3.3) | 0.4590 | 0.21 | 0.01 |
| HTSS like | 2.8 (1.9,3.7) | 2.7 (2.2,3.3) | 0.9062 | 0.20 | 0.00 |
| Any HTSS | 2.7 (2.2,3.3) | 2.8 (2.0,3.5) | 0.0575 | 0.24 | 0.05 |
| GTC | 3.1 (2.2,3.9) | 2.6 (2.0,3.1) | 0.6566 | 0.20 | 0.00 |
| Myoclonic | 2.6 (1.8,3.3) | 2.8 (2.3,3.4) | 0.3130 | 0.21 | 0.01 |
| Clonic | 2.9 (2.0,3.8) | 2.7 (2.2,3.2) | 0.8099 | 0.20 | 0.00 |
| Atonic | 4.0 (2.1,5.9) | 2.6 (2.1,3.1) | 0.2165 | 0.22 | 0.03 |
| Absence | 2.5 (−16.6,21.6) | 2.7 (2.3,3.2) | 0.4113 | 0.21 | 0.01 |
| Focality | 3.1 (2.3,3.9) | 2.6 (2.0,3.1) | 0.2960 | 0.21 | 0.01 |
| Any seizure free period | 3.2 (2.5,3.8) | 2.4 (1.8,3.0) | 0.0279 | 0.25 | 0.07 |
| Cortical Visual Impairment | 2.2 (1.8,2.7) | 4.2 (3.3,5.2) | 0.0001 | 0.34 | 0.21 |
| Age at Last Visit | - | - | <0.0001 | 0.20 | |
Analysis of binary variables association with 7-point CDKL5 Developmental Score adjusted for age at last visit. R2 and Cohen’s f2 effect size are provided as a demonstration of the effect size with f2>=0.02,0.15,0.35 representing small, medium, and large effect sizes respectively23. HTSS – Hypermotor Tonic Spasm Sequence (including HTSS-like as referenced in the text); GTC – Generalized Tonic Clonic; focality could represent either a focal seizure or one of the other seizure types with focal features (for example an asymmetric tonic seizure was marked as both focality and tonic seizure).
Figure 1:
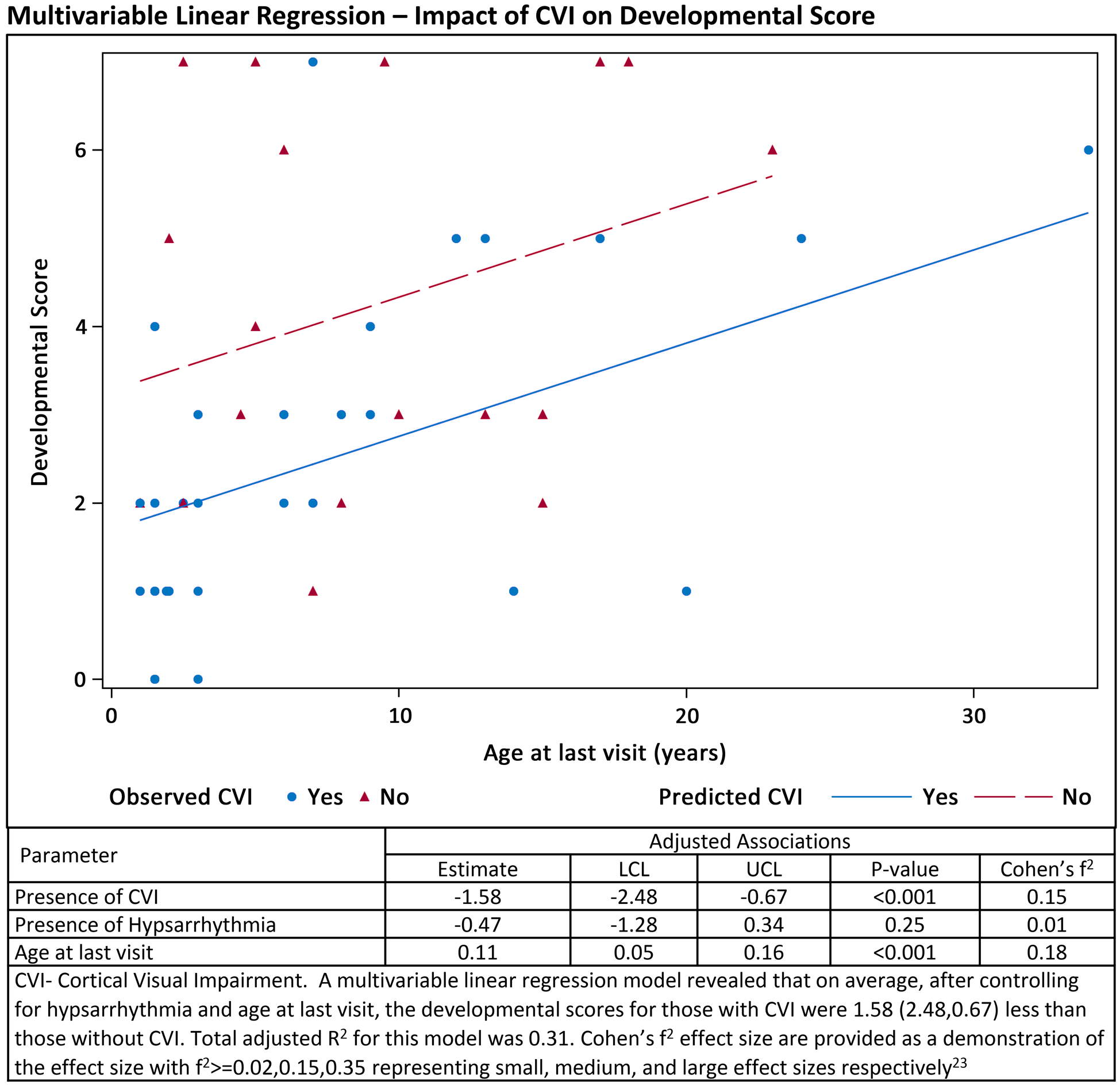
Multivariable Linear Regression Model
Discussion
This combined data from three CDKL5 Centers of Excellence, characterizes seizure types and seizure free periods, demonstrates high prevalence of cortical visual impairment, and correlates presence of CVI with a simple developmental severity scale. Patients with CDD typically have several seizures types with epileptic spasms, tonic seizures, and generalized tonic clonic being the most common both in infancy and as patients get older. The epilepsy associated with CDD is generally thought to be severe and refractory25. As reported previously, patients can have periods of seizure freedom but unfortunately these are less often maintained long-term9 and occur most often around the second year of life (ranged from 2 months to 11 years). Cortical visual impairment is common in CDD. The phenotypic traits analyzed in this study did not correlate with gene grouping or gender. Furthermore, we were able to create a simple developmental outcome scale using 7 milestones that are relevant to the spectrum of outcomes observed in CDD. This scale correlated with CVI and age suggesting utility as an outcome measure. Repeat use of this scale in future cohorts will be necessary to assess repeatability and validity.
Seizures presenting as the previously described hypermotor-tonic spasms sequence24 were seen in a minority of patients, however this data suggest that the more common feature is multiple seizure types that cluster in phases with the most common being tonic seizures followed by a cluster of epileptic spasms. While seizures occurring in phases is not unique to CDD as it has been described in a variety of types of epilepsy, it is quite characteristic in this population26. Providers should consider CDD when a patient presents with clustering of epileptic spasms and tonic seizures (with or without hypsarrhythmia) starting in the first months of life.
In broader populations of patients with infantile spasms more than 80% of patients have hypsarrhythmia27 whereas only 47% of patients in our CDD cohort who had infantile spasms also had evidence of hypsarrhythmia. Prior reports have also supported that patients can have normal EEGs early in infancy despite having a severe epilepsy28; 29, though this can be quit variable. EEGs were not collected in a standardized fashion so there is some risk that patients may have developed hypsarrhythmia later, but it was not captured. A standardized assessment for hypsarrhythmia was not possible given the observational nature of the study. Future studies should try to address these limitations. Nevertheless, hypsarrhythmia was associated with final developmental score on univariate analysis although this association was not significant when accounting for age at last follow up. This should be evaluated further with better longitudinal outcome data. Importantly, development, as measured by CDS and as shown by others1, continues to increase with age, suggesting the ability to achieve new milestones even after 5 or 10 years of age. This is one of the goals of ongoing educational and therapeutic interventions in this and similar populations.
This is the first systematic assessment of cortical visual impairment (CVI) in CDD and was noted to be present in 75% of patients. While prior reports have noted poor visual attention with similar frequencies it was thought to be an autistic feature rather than cortical visual impairment5. In our cohort this assessment was based on both parents reporting that the patient does not see well and detailed eye exams. Established CVI assessments were utilized including: visual attention, ability to fix and follow, assessment of abnormal eye movements or response to optokinetic nystagmus maneuvers30. These signs were used to develop a global assessment of whether the patient had CVI. Strikingly, this was the variable most clearly associated with achievement of developmental milestones. Even after accounting for age of last visit and other relevant variables, patients without CVI achieved 1.4 more milestones out of 7 possible milestones. Indeed, it is unclear if this is a causative relationship or an association, but CVI may provide a useful marker of severity, prognosis or need for more intensive and specific therapies as it can be assessed in infancy. Because of their potential association, CVI and developmental impairment deserve consideration as outcomes in future clinical trials that specifically target epilepsy. Furthermore, this may have utility in preclinical models creating parity between preclinical models and meaningful disease outcomes in patients13. More detailed future exploration of this relationship is important including evaluation of this population with visual evoked potentials and a more detailed evaluation of the severity of CVI.
The current gene variant grouping did not show any correlation with the variables explored in this study. CDD does have clinical variability but this is relatively small spectrum compared to other developmental and epileptic encephalopathy genes such as SCN1A31 or KCNQ232; 33. This may suggest an overall intolerance to functional changes that result from these gene variants. A better understanding of the functional role of CKDL5 may improve genotype-phenotype correlation efforts. Contrary to general perception, the males in this study did not demonstrate a different or worse phenotype based on the variables analyzed. While generalized tonic clonic seizures were less common, this p-value (p=0.03) must be interpreted within the context of multiple comparisons. While the general perceptions is that males are more severe, the mean CDS for males was very similar compared to females. However, it is possible that there is simply not enough power to see a difference with only 18 male patients in our cohort. Finally, the genotype-phenotype and gender analysis did not exclude known mosaic patients. Even if patients are mosaic it is not known what the degree of mosaicism is in the brain and we therefore felt that exclusion from the analysis was not justified. As our dataset grows, it will be important to compare mosaic patients in a more focused fashion. We have included a list of our variants for more specific review as it is also possible that there may be an alternative gene grouping that makes more sense (supplementary table 1). This includes ACMG classification of each of these variants and the criteria that determined that classification. Furthermore, a gene map that includes these variants as well as previously reported variants was recently published34.
There were a few important limitations to our data set. While data was collected prospectively and using standardized forms, there was still some variability in the way the data was collected at different sites. For example, not all eye exam techniques were used at all sites. Furthermore, the way that families were questioned evolved overtime and may have improved the quality of some variables collected later. Data were collected in standard clinical settings; while this may have altered the quantity of data, we do not feel it altered the quality and validates this setting as useful. Many of the patients traveled long distances reducing the opportunity to see patients multiple times and get high quality longitudinal data. Specifically, this reduced our ability to assess seizure frequency overtime. Alternative approaches in future may rely on telehealth style settings to overcome this barrier. Prior survey-based studies suggest that Type C variants may have fewer seizures than Type A and reduced seizures may be associated with spoken word and walking. Given that seizure frequency is well known to fluctuate over time35 longitudinal methods would be best to assess the contribution of seizure frequency which our study was not able to capture adequately. Future work will need to assess both seizure types and seizures frequency overtime to determine the relative importance of these features. Furthermore, the developmental scale that we developed and utilized here has not been validated, however, the milestones discussed are well recognized as fundamental milestones of human development. A validated scale was not used in this case because there are currently no existing validate scales that appropriately assess severe developmental and epileptic encephalopathy patients without saturating the lower end of the assessment. This is a critical area of future research.
This multicenter study clarifies the clinical phenotype in CDD. We demonstrate that epileptic spasms are common but hypsarrhythmia is relatively less common. Patients with CDD often have seizures with multiple phases that may have the components of a hypermotor tonic spasms sequence as previously described but these components may not always occur in the same order or with the same components. Cortical Visual impairment is a common feature of CDD and correlates with the number of developmental milestones achieved.
Supplementary Material
Supplemental Table 1 is a complete list of all patient variants in the CDKL5 gene represented in this study. The variants for all three clinical sites have been combined and organized by mutation grouping. The variant effect, inheritance, ACMG classification and supporting evidence categories for determining the ACMG classification have been provided for reference. A protein map of these mutations and other previously reported mutations was recently published34.
Key Points:
In CDD seizures typically start in the first months of life.
80% of patients develop infantile spasms but hypsarrhythmia is only present in half of those with infantile spasms.
Many patients do experience a seizure free period, but this is rarely maintained long-term.
Cortical visual impairment (CVI) occurs in at least 75% of patients with CDD.
CVI correlates with a simple CDD developmental scales. Scales like this may have value in assessing severe developmental and epileptic encephalopathies.
Acknowledgements
This research was supported by Rocky Mountain Rett Association, International Foundation for CDKL5 Research, Ponzio Family Chair in Neurology Research. NIH funding includes - Rett and Rett-related disorders Natural History Study consortium (U54 HD061222) as well as 1K23 NS107646-01, 5K12NS079414-02 and 1K12NS089417-01.
Footnotes
Disclosures of Conflicts of Interest
Dr. Demarest has consulted for Upsher Smith and BioMarin on subject matter unrelated to this project and sits on the advisory board for the SLC6A1 Connect family foundation. Dr. Benke has consulted for IFCR, LouLou foundation, Marinus, Avexis, Takeda, Ovid, Adadia and Neuren. Dr. Olson has consulted for Takeda. All other authors have no conflicts of interests or disclosures.
Ethical Publication Statement: We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
References
- 1.Fehr S, Leonard H, Ho G, et al. There is variability in the attainment of developmental milestones in the CDKL5 disorder. Journal of Neurodevelopmental Disorders 2015;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bahi-Buisson N, Bienvenu T. CDKL5-Related Disorders: From Clinical Description to Molecular Genetics. Molecular Syndromology 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moseley BD, Dhamija R, Wirrell EC, et al. Historic, clinical, and prognostic features of epileptic encephalopathies caused by CDKL5 mutations. Pediatr Neurol 2012;46:101–105. [DOI] [PubMed] [Google Scholar]
- 4.Weaving LS, Christodoulou J, Williamson SL, et al. Mutations of CDKL5 Cause a Severe Neurodevelopmental Disorder with Infantile Spasms and Mental Retardation. The American Journal of Human Genetics 2004;75:1079–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bahi-Buisson N, Nectoux J, Rosas-Vargas H, et al. Key clinical features to identify girls with CDKL5 mutations. Brain 2008;131:2647–2661. [DOI] [PubMed] [Google Scholar]
- 6.Jahn J, Caliebe A, von Spiczak S, et al. CDKL5 mutations as a cause of severe epilepsy in infancy: clinical and electroencephalographic long-term course in 4 patients. J Child Neurol 2013;28:937–941. [DOI] [PubMed] [Google Scholar]
- 7.Kothur K, Holman K, Farnsworth E, et al. Diagnostic yield of targeted massively parallel sequencing in children with epileptic encephalopathy. Seizure 2018;59:132–140. [DOI] [PubMed] [Google Scholar]
- 8.Lindy AS, Stosser MB, Butler E, et al. Diagnostic outcomes for genetic testing of 70 genes in 8565 patients with epilepsy and neurodevelopmental disorders. Epilepsia 2018;59:1062–1071. [DOI] [PubMed] [Google Scholar]
- 9.Fehr S, Wong K, Chin R, et al. Seizure variables and their relationship to genotype and functional abilities in the CDKL5 disorder. Neurology 2016;87:2206–2213. [DOI] [PubMed] [Google Scholar]
- 10.Fehr S, Downs J, Ho G, et al. Functional abilities in children and adults with the CDKL5 disorder. Am J Med Genet A 2016;170:2860–2869. [DOI] [PubMed] [Google Scholar]
- 11.Hagebeuk EE, van den Bossche RA, de Weerd AW. Respiratory and sleep disorders in female children with atypical Rett syndrome caused by mutations in the CDKL5 gene. Dev Med Child Neurol 2013;55:480–484. [DOI] [PubMed] [Google Scholar]
- 12.Mangatt M, Wong K, Anderson B, et al. Prevalence and onset of comorbidities in the CDKL5 disorder differ from Rett syndrome. Orphanet J Rare Dis 2016;11:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mazziotti R, Lupori L, Sagona G, et al. Searching for biomarkers of CDKL5 disorder: early-onset visual impairment in CDKL5 mutant mice. Hum Mol Genet 2017;26:2290–2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guzzetta F, Frisone MF, Ricci D, et al. Development of visual attention in West syndrome. Epilepsia 2002;43:757–763. [DOI] [PubMed] [Google Scholar]
- 15.Rando T, Bancale A, Baranello G, et al. Visual function in infants with West syndrome: correlation with EEG patterns. Epilepsia 2004;45:781–786. [DOI] [PubMed] [Google Scholar]
- 16.Bahi-Buisson N, Villeneuve N, Caietta E, et al. Recurrent mutations in the CDKL5 gene: genotype-phenotype relationships. Am J Med Genet A 2012;158A:1612–1619. [DOI] [PubMed] [Google Scholar]
- 17.Fisher RS, Cross JH, D’Souza C, et al. Instruction manual for the ILAE 2017 operational classification of seizure types. Epilepsia 2017;58:531–542. [DOI] [PubMed] [Google Scholar]
- 18.Chorna OD, Guzzetta A, Maitre NL. Vision Assessments and Interventions for Infants 0–2 Years at High Risk for Cerebral Palsy: A Systematic Review. Pediatr Neurol 2017;76:3–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rossi A, Gnesi M, Montomoli C, et al. Neonatal Assessment Visual European Grid (NAVEG): Unveiling neurological risk. Infant Behav Dev 2017;49:21–30. [DOI] [PubMed] [Google Scholar]
- 20.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hector RD, Kalscheuer VM, Hennig F, et al. CDKL5 variants: Improving our understanding of a rare neurologic disorder. Neurol Genet 2017;3:e200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Friedman E, Pampiglione G. Prognostic implications of electroencephalographic findings of hypsarrhythmia in first year of life. Br Med J 1971;4:323–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Selya AS, Rose JS, Dierker LC, et al. A Practical Guide to Calculating Cohen’s f(2), a Measure of Local Effect Size, from PROC MIXED. Front Psychol 2012;3:111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Klein KM, Yendle SC, Harvey AS, et al. A distinctive seizure type in patients with CDKL5 mutations: Hypermotor-tonic-spasms sequence. Neurology 2011;76:1436–1438. [DOI] [PubMed] [Google Scholar]
- 25.Müller A, Helbig I, Jansen C, et al. Retrospective evaluation of low long-term efficacy of antiepileptic drugs and ketogenic diet in 39 patients with CDKL5-related epilepsy. European Journal of Paediatric Neurology 2016;20:147–151. [DOI] [PubMed] [Google Scholar]
- 26.Sanchez Fernandez I, Ramgopal S, Powell C, et al. Clinical evolution of seizures: distribution across time of day and sleep/wakefulness cycle. J Neurol 2013;260:549–557. [DOI] [PubMed] [Google Scholar]
- 27.Demarest ST, Shellhaas RA, Gaillard WD, et al. The impact of hypsarrhythmia on infantile spasms treatment response: Observational cohort study from the National Infantile Spasms Consortium. Epilepsia 2017;58:2098–2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bahi-Buisson N, Kaminska A, Boddaert N, et al. The three stages of epilepsy in patients with CDKL5 mutations. Epilepsia 2008;49:1027–1037. [DOI] [PubMed] [Google Scholar]
- 29.Melani F, Mei D, Pisano T, et al. CDKL5 gene-related epileptic encephalopathy: electroclinical findings in the first year of life. Dev Med Child Neurol 2011;53:354–360. [DOI] [PubMed] [Google Scholar]
- 30.Good WV, Jan JE, DeSa L, et al. Cortical visual impairment in children. Surv Ophthalmol 1994;38:351–364. [DOI] [PubMed] [Google Scholar]
- 31.Gataullina S, Dulac O. From genotype to phenotype in Dravet disease. Seizure 2017;44:58–64. [DOI] [PubMed] [Google Scholar]
- 32.Miceli F, Soldovieri MV, Ambrosino P, et al. Genotype-phenotype correlations in neonatal epilepsies caused by mutations in the voltage sensor of K(v)7.2 potassium channel subunits. Proc Natl Acad Sci U S A 2013;110:4386–4391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Millichap JJ, Park KL, Tsuchida T, et al. KCNQ2 encephalopathy: Features, mutational hot spots, and ezogabine treatment of 11 patients. Neurol Genet 2016;2:e96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Olson HE, Demarest ST, Pestana-Knight EM, et al. Cyclin-Dependent Kinase-Like 5 Deficiency Disorder: Clinical Review. Pediatr Neurol 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Berg AT, Rychlik K, Levy SR, et al. Complete remission of childhood-onset epilepsy: stability and prediction over two decades. Brain 2014;137:3213–3222. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table 1 is a complete list of all patient variants in the CDKL5 gene represented in this study. The variants for all three clinical sites have been combined and organized by mutation grouping. The variant effect, inheritance, ACMG classification and supporting evidence categories for determining the ACMG classification have been provided for reference. A protein map of these mutations and other previously reported mutations was recently published34.