

Abstract
Background
Osteoporosis is a complex disease with a strong genetic contribution. A recently published genome-wide association study (GWAS) for estimated bone mineral density (eBMD) identified 1103 independent genome-wide significant association signals. Most of these variants are non-coding, suggesting that regulatory effects may drive many of the associations. To identify genes with a role in osteoporosis, we integrate the eBMD GWAS association results with those from our previous osteoclast expression quantitative trait locus (eQTL) dataset.
Results
We identify sixty-nine significant cis-eQTL effects for eBMD GWAS variants after correction for multiple testing. We detect co-localisation of eBMD GWAS and osteoclast eQTL association signals for 21 of the 69 loci, implicating a number of genes including CCR5, ZBTB38, CPE, GNA12, RIPK3, IQGAP1 and FLCN. Summary-data-based Mendelian Randomisation analysis of the eBMD GWAS and osteoclast eQTL datasets identifies significant associations for 53 genes, with TULP4 presenting as a strong candidate for pleiotropic effects on eBMD and gene expression in osteoclasts. By performing analysis using the GARFIELD software, we demonstrate significant enrichment of osteoporosis risk variants among high-confidence osteoclast eQTL across multiple GWAS P value thresholds. Mice lacking one of the genes of interest, the apoptosis/necroptosis gene RIPK3, show disturbed bone micro-architecture and increased osteoclast number, highlighting a new biological pathway relevant to osteoporosis.
Conclusion
We utilise a unique osteoclast eQTL dataset to identify a number of potential effector genes for osteoporosis risk variants, which will help focus functional studies in this area.
Keywords: Osteoclast, Osteoporosis, GWAS, eQTL, BMD, Fracture, RIPK3, RIP3, FBN2, SNP
Background
Genome-wide association studies (GWAS) have successfully identified thousands of genetic variants associated with complex traits and diseases in humans. However, the vast majority of these variants lie in non-coding regions of the genome such as intronic or intergenic DNA, and it is thought that many of them influence the phenotype through regulatory effects on nearby genes. For many of these variants, the identity of the effector gene responsible for the association remains to be determined. This is further complicated by the fact that the closest gene to the GWAS association signal is not always the effector gene, and in some cases, neither is the most plausible biological candidate gene from the region. Expression quantitative trait locus (eQTL) studies in disease-relevant cell/tissue types provide valuable information on this issue by characterising associations between genetic variants and expression of nearby genes. The Genotype-Tissue Expression (GTEx) project provides extensive eQTL data for many tissues [1]; however, not all cell types are represented in that resource and many eQTL studies have used complex tissues containing a mixture of cell types. Apart from an eQTL study performed on human osteoblasts cultured from surgical explants of bone [2], there has historically been a lack of eQTL studies performed in cell types relevant to osteoporosis. We recently generated a unique osteoclast-specific eQTL resource using cells obtained from 158 subjects to fill the void of eQTL data for that cell type and for the study of human bone disease [3, 4].
Bone is a dynamic tissue that is constantly being remodelled through the coupled actions of osteoblasts and osteoclasts. Osteoporosis is the most common metabolic bone disease in humans and is characterised by reduced bone mineral density (BMD), micro-architectural deterioration of the bone tissue and an increased risk of fracture. Excess mortality caused by osteoporotic fracture has been estimated at 9% in women and 24% in men at 1 year postfracture, and 24% in women and 26% in men at 5 years postfracture [5]. Postmenopausal women are at particularly high risk of developing osteoporosis due to reduced oestrogen levels. Certain environmental factors such as dietary calcium intake and exercise have also been shown to influence an individual’s risk of developing the disease [6, 7]. In addition to these factors, osteoporosis has a strong heritable component, with twin and family studies generating heritability estimates of 0.46–0.84 for BMD [8–10]. Individuals with an affected first-degree relative have an elevated estimated familial relative risk for fragility fracture of 1.31–4.24 [11, 12].
GWAS have proven to be very successful at identifying genetic variants associated with bone parameters [13–18]. A recently published GWAS for estimated BMD (eBMD), a trait calculated using quantitative ultrasound measurements at the heel, included 426,824 individuals from the UK Biobank and is the largest GWAS ever performed for a bone density phenotype [19]. The study identified 1103 conditionally independent genome-wide significant association signals located in 518 loci, of which 301 are novel loci [19]. Like most complex disease GWAS, the majority of these signals are led by non-coding variants. In order to identify putative effector genes and genetic regulatory mechanisms with a role in osteoporosis, particularly those that are relevant to bone resorbing cells, we integrated the results from that study with our osteoclast eQTL dataset and bone phenotypes from knockout mice.
Results
Population characteristics of the 158 women recruited into the osteoclast eQTL study cohort are presented in Additional file 1: Table S1. All participants had self-reported European ancestry, and principal components analysis failed to identify any ethnic outliers. After genotype imputation and QC, genotype data was available for 5,373,348 variants in the study sample with a minor allele frequency (MAF) ≥ 5% and an IMPUTE2 info score ≥ 0.4. After applying QC criteria to the gene expression dataset, a total of 15,688 expressed gene transcripts were identified in the osteoclast-like cells.
Identification of eQTL associations for eBMD GWAS variants
Of the 1103 lead genetic variants recently identified as independently associated with eBMD at the genome-wide significance level, 929 were present in the osteoclast eQTL dataset (MAF ≥ 5% in the cohort and had an expressed gene located within the ± 1 Mb analysis window). The vast majority of those missing were low frequency variants with a MAF < 5% in the cohort. For the 929 variants present in the dataset, a mean of 15.0 genes was present within the ± 1 Mb analysis window.
After correction for multiple testing, we observed a total of 69 significant eQTL associations for the eBMD GWAS variants (Table 1, Fig. 1 and Additional file 1: Table S2), with 61 of the variants associated with the expression of 68 genes. Since variants with regulatory effects would be expected to be located in close proximity to their respective eQTL-gene (eGene) transcription start site (TSS), we examined this in our data. Of the 69 eQTL associations, 51 (74%) were for variants located within 150 kb of the eGene TSS, with the average distance being 154.6 kb. The eQTL associations were also stronger for variants located in close proximity to the associated gene, with the 10 strongest eQTL associations identified all representing variants located within 155 kb of the associated eGene TSS. eQTL associations were observed for 8 of the genes identified in a previous smaller study [3], including PIGV, ST7L, TRMT61B, DYSF, FADS2, GLDN, CYP19A1 and IQGAP1. Many of the eQTL variants demonstrated significant associations with expression of more than one gene, including rs11677953 (RP11-378A13.1 and TMBIM1), rs2432236 (CTD-2376I4.1, CTD-2376I4.2 and FCHO2), rs42916 (LINC01184 and SLC12A2), rs11814082 (DDX10P1 and ZNF438), rs7147775 (EIF2B2 and ACYP1), rs2899472 (GLDN and CYP19A1) and rs12325187 (ZNF200 and ZNF174).
Table 1.
Genetic loci demonstrating co-localised eBMD GWAS and osteoclast cis-eQTL associations
| Variant | Location | EA | OA | EAF | βGWAS | Gene | Expressiona | Distance to TSS | PeQTL | βeQTL |
|---|---|---|---|---|---|---|---|---|---|---|
| rs4683184 | chr3:46146215 | G | A | 0.37 | − 0.01 | CCR5 | 22.96 ± 7.33 | − 224,638 | 2.06E−04 | − 0.47 |
| rs13072536 | chr3:52827195 | T | A | 0.22 | 0.02 | SFMBT1 | 2.87 ± 0.59 | − 219,556 | 1.43E−04 | 0.50 |
| rs1991431 | chr3:141414608 | A | G | 0.44 | − 0.02 | ZBTB38 | 7.13 ± 1.57 | 90,395 | 7.68E−05 | 0.44 |
| rs1550270 | chr4:165340648 | C | T | 0.33 | 0.02 | CPE | 4.64 ± 3.7 | − 20,546 | 7.29E−06 | 0.51 |
| rs798545 | chr7:2722752 | T | C | 0.23 | 0.01 | GNA12 | 21.9 ± 4.75 | − 121,573 | 7.94E−05 | 0.53 |
| rs2551769 | chr7:135453583 | A | G | 0.28 | 0.01 | CALD1 | 1.23 ± 1.05 | 709,328 | 1.38E−05 | − 0.58 |
| rs11245388 | chr10:124850559 | T | G | 0.47 | − 0.01 | METTL10 | 1.06 ± 0.19 | 58,688 | 1.47E−11 | 0.72 |
| rs3212240 | chr14:24341692 | C | T | 0.43 | − 0.01 | RIPK3 | 4.51 ± 1.07 | 1646 | 1.47E−06 | − 0.55 |
| rs7147775 | chr14:75203508 | G | T | 0.44 | − 0.02 | EIF2B2 | 4.62 ± 0.58 | 200,597 | 2.16E−09 | − 0.62 |
| rs7147775 | chr14:75203508 | G | T | 0.44 | − 0.02 | ACYP1 | 1.8 ± 0.56 | 134,024 | 4.75E−06 | 0.55 |
| rs11073930 | chr15:90503480 | G | C | 0.46 | − 0.02 | IQGAP1 | 66.34 ± 9.26 | 115,262 | 1.46E−10 | 0.66 |
| rs1736213 | chr17:17231214 | T | G | 0.43 | 0.01 | FLCN | 4.06 ± 0.89 | − 5975 | 1.82E−09 | − 0.54 |
| rs1661725 | chr17:75564053 | C | T | 0.44 | 0.01 | LLGL2 | 0.7 ± 0.24 | 38,973 | 9.98E−07 | − 0.55 |
| rs77420750 | chr19:15868934 | A | C | 0.31 | 0.01 | TMEM38A | 0.49 ± 0.29 | − 792,194 | 1.43E−04 | 0.45 |
| rs314675 | chr19:46692822 | C | T | 0.1 | − 0.02 | FKRP | 1.45 ± 0.27 | − 53,224 | 5.88E−06 | 0.92 |
| rs73066226 | chr19:58496846 | C | T | 0.17 | 0.01 | UBE2M | 22.85 ± 3.39 | − 62,115 | 1.71E−06 | 0.66 |
| rs5754387 | chr22:21620414 | C | G | 0.17 | − 0.02 | UBE2L3 | 20.1 ± 2.88 | 70,967 | 1.82E−05 | 0.46 |
| rs2294358 | chr22:36375211 | C | G | 0.07 | 0.04 | FOXRED2 | 2.44 ± 1.37 | − 131,893 | 1.55E−06 | 1.11 |
| rs932536 | chr22:49866824 | A | G | 0.13 | − 0.02 | ZBED4 | 5.67 ± 0.67 | 12,982 | 8.39E−06 | 0.71 |
| rs5770908 | chr22:50439289 | A | G | 0.29 | − 0.02 | PPP6R2 | 10.17 ± 1.8 | 95,985 | 1.36E−04 | 0.41 |
| rs5914035 | chrX:56981783 | C | T | 0.23 | 0.02 | LINC01420 | 13.14 ± 2.09 | 252,542 | 4.91E−06 | 0.62 |
EA effect allele, OA other allele, EAF effect allele frequency (derived from the osteoclast eQTL cohort), eBMD estimated BMD, TSS transcription start site; variant locations derived from dbSNP build 150 (GRCh38/hg38), βGWAS values are relevant to the effect allele and were obtained from Morris et al. [19], βeQTL values are given as the normalised effect size on gene expression for the effect allele. eQTL associations are significant using a multiple-testing corrected FDR of 5%
aExpression levels are stated as mean reads per kilobase million (RPKM) ± standard deviation
Fig. 1.
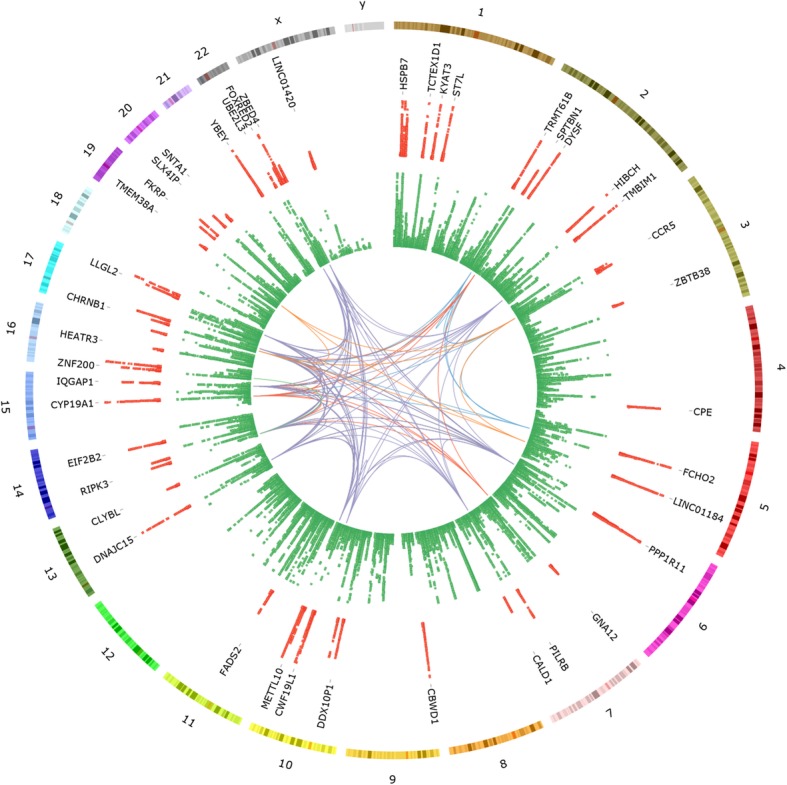
CIRCOS plot [20] displaying (from outside to inside) chromosome numbers, chromosome ideograms, scatterplot complete with gene labels representing the osteoclast eQTL associations presented in Table 1 and Additional file 1: Table S2 (red), scatterplot representing the eBMD GWAS results (green) and gene ontology (GO) biological process groupings relevant to osteoclast biology. Osteoclast eQTL and eBMD GWAS associations are displayed as −log10P values ranging from 0.01 > P > 1.0 × 10−15 (osteoclast eQTL) and 0.001 > P > 1.0 × 10−50 (eBMD GWAS). GO biological process groupings displayed include membrane organisation (blue), regulation of cell migration (red), regulation of catalytic activity (purple), cation transport (orange) and I-kappaB kinase/NF-kappaB signalling (green)
Co-localisation analysis
Since it is possible for independent GWAS and eQTL associations to exist within a genetic locus, we decided to assess co-localisation of eBMD GWAS and osteoclast eQTL association signals for each of the 69 significant eQTL loci using the coloc package in R [21]. If the association signals within the two datasets are shown to co-localise, it can be interpreted as strong evidence that the GWAS association is mediated through regulatory effects on the eQTL gene. Evidence of co-localisation of GWAS and eQTL association signals was identified (posterior probability > 50%) in 19 of the 69 loci, including CCR5 (82.1%), ZBTB38 (83.3%), CPE (54.9%), GNA12 (50.1%), CALD1 (91.3%), METTL10 (96.6%), RIPK3 (89.8%) (Additional file 1: Fig. S1), EIF2B2 (65.9%), ACYP1 (83.4%), IQGAP1 (75.2%), FLCN (95.6%), LLGL2 (78.1%), TMEM38A (71.1%), FKRP (84.0%), UBE2M (82.6%), UBE2L3 (79.5%), FOXRED2 (55.0%), PPP6R2 (73.1%) and LINC01420 (87.2%). The eBMD GWAS association signal at 14q24.3 led by rs7147775 demonstrated strong evidence for co-localisation with eQTL effects for both the EIF2B2 and ACYP1 genes (Additional file 1: Fig. S2). Co-localisation analysis was also performed using a newer, but as yet less widely evaluated version of the coloc R functions, named coloc2, which is thought to more fully address allelic heterogeneity when present in the data [22]. Apart from CPE (3.0%) and LLGL2 (1.0%), all of the genetic loci demonstrating co-localisation in the coloc analysis also presented with co-localisation using coloc2 (posterior probabilities 98.3–100%). Two additional co-localised genetic loci were identified by coloc2, SFMBT1 (98.2%) and ZBED4 (99.1%). eBMD GWAS and osteoclast eQTL results for all co-localised loci are presented in Table 1, with those demonstrating independent (non-co-localised) association signals presented in Additional file 1: Table S2. An example of a locus with independent eBMD GWAS and osteoclast eQTL association signals, LINC01184 (posterior probability of co-localisation < 0.01% using coloc [21] and coloc2 [22]), is displayed in Additional file 1: Fig. S3.
Tissue-shared and osteoclast-specific eQTL associations
To establish the proportion of the identified eQTL associations that have supporting evidence from other tissue types and to identify those that are potentially osteoclast-specific, we analysed each of the 69 eQTL associations for evidence of replication in the 53 tissues of the GTEx V7 dataset [1]. Only GTEx tissues with > 70 samples were included in the analysis, with a multiple-testing corrected false discovery rate (FDR) threshold of 0.05 used to identify significant associations. Of the 69 eQTL associations, 45 (65.2%) demonstrated evidence of association in at least one GTEx tissue, leaving 24 (34.8%) as potentially specific to osteoclasts. These included the eQTL associations for the genes HSPB7, NBPF3, ZNF593, PIGV, KYAT3, HIBCH, CCR5, CPE, SLC12A2, PPP1R11, CALD1, CBWD1, ZNF438, RP11-517P14.2, GLDN, ZNF174, NLRC3, HEATR3, RP11-104 N10.2, TMEM38A, UBE2M, SLX4IP, SNTA1 and LINC01420.
Summary-data-based Mendelian Randomisation analysis
In order to identify genes whose expression levels in osteoclasts are associated with the eBMD trait, we performed an integrative analysis of the eBMD GWAS and osteoclast eQTL datasets using the Summary-data-based Mendelian Randomisation (SMR) software [23]. A total of 1070 genes with an eQTL association significant at P < 5 × 10−8 were identified and included in the analysis. Using a multiple-testing corrected significance threshold of PSMR < 4.7 × 10−5, the SMR test identified significant associations for 53 genes (Additional file 1: Table S3). Heterogeneity (PHEIDI < 0.05) was detected for 52 of these, leaving a single locus TULP4 as a strong candidate for harbouring shared genetic effects (i.e. pleiotropy) on gene expression in osteoclasts and eBMD (Additional file 1: Fig. S4). Expression of TULP4 was found to be positively associated with eBMD (βSMR = 0.02, Additional file 1: Table S3).
Enrichment of osteoporosis risk variants within osteoclast eQTL
We used the GWAS Analysis of Regulatory or Functional Information Enrichment with LD correction (GARFIELD) software [24] to determine whether osteoporosis risk variants in the eBMD GWAS summary result dataset are enriched among high-confidence osteoclast eQTL variants identified using a multiple-testing corrected FDR threshold of 0.05. Using eBMD GWAS P value thresholds of P < 1 × 10−5, 1 × 10−6, 1 × 10−7 and 1 × 10−8, we observed significant enrichment of osteoporosis risk variants within osteoclast eQTL (P = 3.25 × 10−40, 8.83 × 10−28, 6.93 × 10−24 and 7.79 × 10−18, respectively, Fig. 2). As a comparison, we also performed enrichment analysis using GWAS results from a similarly powered study for a trait not considered to be related to bone biology, neuroticism [25]. We observed nominally significant enrichment of neuroticism GWAS variants within the osteoclast eQTL dataset at the P < 1 × 10−7 and 1 × 10−8 thresholds (P = 0.01 and 0.007, respectively, Fig. 2); however, these were not significant after correction for multiple testing.
Fig. 2.

Analysis of the eBMD GWAS and osteoclast eQTL datasets using the GARFIELD software [24] demonstrated significant enrichment of osteoporosis risk variants among osteoclast eQTL across four GWAS P value thresholds (P < 1 × 10−5, 1 × 10−6, 1 × 10−7 and 1 × 10−8), with the enrichment results for the trait neuroticism [25] included for comparison. The upper panels present the −log10 enrichment P values for each threshold, while the lower panels present the natural logarithm of the odds ratios for each threshold with 95% confidence intervals
Analysis of Ripk3 knockout mice
Among the genes identified in this study as potentially relevant to osteoporosis is RIPK3. According to data available in GTEx V7, the eBMD GWAS variant rs3212240 is an eQTL for RIPK3 only in sun-exposed skin (P = 2.0 × 10−5); however, data from this study suggests that it is also relevant to bone cells. The C allele at rs3212240, associated with a lower eBMD measurement, was found to be associated with reduced expression of the RIPK3 gene in our osteoclast eQTL dataset (Table 1). Because of the strong evidence for co-localisation of eBMD GWAS and osteoclast eQTL association signals within this locus, combined with the known role of RIPK3 in cellular necroptosis in response to the tumour necrosis factor (TNF)-alpha family of death-inducing cytokines and potential involvement in bone resorption in osteoporosis [26], we performed in-depth skeletal phenotyping of Ripk3-deficient mice. Analysis of the distal femur of 15-week-old male Ripk3−/− mice using micro-CT revealed increased subperiosteal perimeter, increased endosteal perimeter and increased bone marrow volume compared to wildtype (WT) animals (Fig. 3). There was no difference in cortical bone volume or trabecular indices between Ripk3−/− and WT mice (Additional file 1: Fig. S5). However, the extent of trabecularisation along the diaphysis was significantly reduced in the Ripk3−/− mice (Fig. 3a).
Fig. 3.

Micro-CT assessment of the distal femur from 15-week-old male Ripk3−/− and WT mice. Representative 3D images of the distal femur demonstrate the following: a reduced extension of the trabecular network into the diaphysis, b expansion of the medullary cavity and c increased size of the trabecular bone compartment with no change in trabecular bone density in the Ripk3-/- mice. Quantitative analysis of micro-CT parameters (mean + standard deviation) displays the following: d cortical volume, e bone marrow volume, f endosteal perimeter, g periosteal perimeter, h trabecular bone volume fraction and i trabecular extension in the Ripk3−/− mice relative to WT mice. N = 5 for each group; WT, wildtype. **P < 0.01
Decalcified bone sections were stained for TRAP activity and with haematoxylin and eosin (H&E) to further characterise the cellular features of the Ripk3−/− mice by histomorphometry (Fig. 4a–f). Analysis of the TRAP-stained bone sections revealed a significant increase in the total number of osteoclasts, number of osteoclasts relative to bone surface and osteoclast surface relative to bone surface in the Ripk3−/− bones relative to those from the WT mice (Fig. 4a–c). No significant differences in osteoblast parameters were observed between the Ripk3−/− and WT bones (Fig. 4d, e). These results suggest that Ripk3−/− mice present with increased osteoclast number in vivo.
Fig. 4.

Quantitative histomorphometric analysis of femora from 15-week-old male WT and Ripk3−/− mice (mean + standard deviation). a Osteoclast number (N.Oc). b Osteoclast surface relative to bone surface (Oc.S/BS). c Number of osteoclasts relative to bone surface (N.Oc/BS(mm−1)). d Osteoblast number (N.Ob). e Number of osteoblasts relative to bone surface (N.Ob/BS(mm−1)). f Representative low-power images (× 40 magnification) of TRAP-stained sections of the femur just below the growth plate. N = 4 and 5 for the WT and Ripk3−/− groups, respectively; WT, wildtype
Discussion
In this study of human osteoclast-like cells, we have identified a large number of genetic regulatory effects for osteoporosis risk variants. The majority of the eQTLs identified in this study are located in close proximity to their effector gene TSS, consistent with findings from the GTEx project [27]. A significant proportion (34.8%) of the eQTL associations identified in this study are potentially osteoclast-specific when compared with data from the 53 tissues of the GTEx V7 dataset [1], demonstrating the value of performing such investigations in disease-relevant cell/tissue types. We also found that high-confidence osteoclast eQTLs are strongly enriched for osteoporosis risk variants, with an average 3.5-fold enrichment seen across the four GWAS significance thresholds tested.
An osteoporosis-relevant eQTL association was identified in this study for the eBMD GWAS variant rs3212240, which was found to be significantly associated with expression of the RIPK3 gene, a member of the receptor-interacting protein (RIP) family of serine/threonine protein kinases. Co-localisation analysis of the locus suggested that the eBMD GWAS and osteoclast eQTL association signals may be driven by the same causal variant. Analysis of Ripk3−/− mice revealed a variety of bone effects, primarily characterised by expansion of the femoral shaft with reduced extension of the trabecular network and an increase in marrow volume. The effects observed on trabecular extension could potentially account for the association with eBMD in humans, a phenotype derived using quantitative ultrasound measurements at the heel and hence primarily influenced by bone structural parameters including the orientation of the trabecular bone [28], as opposed to BMD. The lead eBMD GWAS variant from the human RIPK3 locus, rs3212240, does not demonstrate even nominal associations with total body [14], femoral neck or lumbar spine [18] BMD derived using dual-energy X-ray absorptiometry. The product of the RIPK3 gene is a component of the TNF receptor 1 signalling complex and has an established role in necroptosis [29, 30], a type of programmed cell death thought to cause inflammation through the release of intracellular factors [31]. Our histomorphometric analysis of the Ripk3−/− bones revealed increased numbers of osteoclasts relative to WT bones, suggesting that this gene may have a role in osteoclast necroptosis and that loss of this mechanism may lead to an excess of osteoclasts. If this is the case, it could potentially explain why the C allele at rs3212240 is associated with reduced expression of the RIPK3 gene in our osteoclast eQTL dataset and a reduced eBMD [19]. The variant rs3212240 is located ~ 1.5 kb upstream of the RIPK3 TSS and is in moderate linkage disequilibrium (LD) (r2 = 0.50) with a nearby body height GWAS variant (rs1950500) [32]. It is also in moderate LD (r2 = 0.79) with a missense variant in an adjacent gene, NFATC4 (rs2229309), and we cannot rule out the possibility that the association at this locus with eBMD is partially mediated through the effects of this variant. However, rs2229309 is classified as benign by various pathogenicity prediction algorithms [33–35]. Considering the strong regulatory effect on RIPK3 demonstrated for rs3212240, combined with the altered bone phenotypes observed in Ripk3−/− mice, we consider that the association at this locus with eBMD is likely to be mediated through regulatory effects on the RIPK3 gene.
A novel eQTL association relevant to osteoporosis was identified in this study for the CPE gene. This locus also presented with evidence for co-localisation of eBMD GWAS and osteoclast eQTL association signals using the coloc package [21] and was identified as a significant locus in the SMR analysis. Of note, the association seen in our data between the eBMD GWAS variant rs1550270 and expression of CPE was not observed in any of the 53 tissues of the GTEx V7 dataset [1]. The product of the CPE gene, carboxypeptidase E, is a prohormone-processing enzyme that cleaves carboxy-terminal basic residues from neuropeptide intermediates and peptide hormones to produce mature bioactive products, including insulin [36]. The CPE gene has been strongly implicated in diabetes, with mutation in the human CPE gene identified as the likely cause of a genetic disease characterised by obesity, type 2 diabetes, intellectual disability and hypogonadotrophic hypogonadism [37]. A deactivating mutation in the murine Cpe gene also results in a pronounced obesity-diabetes syndrome [38]. Cpe-knockout mice present with obesity and reduced BMD with increased plasma levels of osteocalcin and carboxy-terminal collagen crosslinks, indicating the presence of increased bone turnover [39]. Expression of receptor activator of nuclear factor kappa-B ligand (RANKL) was found to be elevated approximately 2-fold relative to osteoprotegerin in the femora of Cpe-deficient mice, potentially leading to increased osteoclast activity [39]. It has also been established that expression of Cpe is highly upregulated in mouse osteoclasts compared to their precursor cells and that this upregulation is associated with increased formation of TRAP-positive multinucleated osteoclasts and expression of key genes involved with osteoclastogenesis [40]. Our osteoclast eQTL data indicates that the human CPE gene has a role in bone density regulation and highlights this gene as a potential pleiotropic locus relevant to diabetes and osteoporosis.
We observed some interesting eQTL effects in this study for several genes implicated in monogenic disorders. An example of this is the FBN2 gene, encoding fibrillin 2, which is an extracellular protein that is a major component of connective tissue microfibrils. Mutation in this gene has been identified as the cause of congenital contractural arachnodactyly (CCA) [41], a disorder phenotypically similar to Marfan syndrome (caused by mutation in the related gene FBN1). CCA is characterised by a variety of skeletal abnormalities including congenital contractures, arachnodactyly, deformities of the chest wall, scoliosis and elongated limbs. Another example is the gene FLCN, encoding folliculin, which was identified as a significant locus in the SMR analysis and demonstrated strong evidence for co-localisation of eBMD GWAS and osteoclast eQTL association signals. Folliculin has recently been shown to regulate osteoclastogenesis through metabolic effects on oxidative phosphorylation and purine metabolism [42]. Induced deletion of the murine Flcn gene in osteoclast precursors was found to result in severe osteoporosis caused by excess osteoclastogenesis [42]. Mutation in the human FLCN gene has been identified as the cause of Birt-Hogg-Dubé syndrome [43], an autosomal dominant condition characterised by early onset renal tumours, hair follicle hamartomas, pulmonary cysts and spontaneous pneumothoraces. However, we are not aware of any significant bone or skeletal phenotypes that have been reported in instances of this condition to date.
The SMR analysis identified a significant association for the TULP4 gene, with the HEIDI test suggesting that the locus may exhibit pleiotropic effects on TULP4 expression and eBMD. The product of the TULP4 gene has not been extensively characterised with regard to a role in bone biology; however, it is thought to be a cytoplasmic protein characterised by a large amino terminus containing a WD40 repeat region and a suppressor of cytokines signalling domain [44]. TULP4 has been implicated in the formation of orofacial clefts [45] and is a body height GWAS locus [32, 46]. Mutation in the related gene WDR35 has been implicated in cranioectodermal dysplasia, also known as Sensenbrenner syndrome, an autosomal recessive condition characterised by craniofacial and skeletal abnormalities [47]. The top eQTL variant identified for TULP4 is rs341106, which is only in weak LD with the lead eBMD GWAS variant from the locus rs12206717 (r2 = 0.02 in Europeans [48]). rs12206717 is a missense variant in exon 9 of TULP4, which causes a change of amino acid 522 from serine to asparagine. It is possible that there are 2 independent association signals for eBMD arising from the locus, one potentially causing a structural change in the TULP4 protein and the other influencing expression of the gene.
It is worth noting that the differentiated osteoclast-like cells used in this study likely express a smaller panel of genes when compared to complex tissues containing a mixture of cell types such as those used in the GTEx project. When this is considered in the context of a relatively modest sample size, it potentially explains why a greater number of co-localised genetic loci were not identified in this study. It is also possible that the eBMD GWAS data are primarily reflective of bone accrual and osteoblast function and that resorption and osteoclast activity are not the primary drivers of those results. It should also be noted that the threshold used to identify co-localised loci (posterior probability > 50%) is merely a guideline and that probabilities below this value do not necessarily indicate independent association signals, particularly when considering the sample size of the osteoclast eQTL cohort. The SMR software may also be susceptible to small-sample biases, which could potentially explain the large proportion of significant HEIDI test scores seen in the study.
Conclusions
We have used a unique osteoclast-specific eQTL dataset to identify a number of genetic regulatory effects relevant to osteoporosis. Approximately one third of these regulatory effects were not seen in the 53 tissues of the GTEx V7 dataset [1] and are potentially osteoclast-specific. A number of the genes highlighted in this study present strongly as having a potential role in bone biology, including CPE, FBN2, FLCN and RIPK3, and our results support the growing body of evidence suggesting that the CPE gene may exert pleiotropic effects on type 2 diabetes and osteoporosis. We performed thorough skeletal phenotyping of Ripk3-deficient mice, which presented with altered bone micro-architectural characteristics potentially driven through effects on osteoclast necroptosis. The results from this study have identified a number of potential effector genes for GWAS loci relevant to osteoporosis, which will help target future translational studies.
Materials and methods
Subject recruitment and generation of osteoclast-like cells
Details of the recruitment process and cell culture procedures used in this study have been described previously [3]. Briefly, the osteoclast eQTL study cohort comprises 158 women aged 30–70 years with self-reported European ancestry who attended the Bone Density Unit at Sir Charles Gairdner Hospital in Western Australia for a dual-energy X-ray absorptiometry BMD scan in 2016 (Hologic, Bedford, MA, USA). Exclusion criteria used during recruitment included presence of medical conditions or use of medications that are likely to influence osteoclastic bone resorption or the process of osteoclastogenesis. Osteoclast-like cells were generated using the conventional procedure; peripheral blood mononuclear cells were first isolated from blood samples obtained from each individual by density gradient centrifugation using protocols well established in our laboratory [3, 49]. These cells were cultured in triplicate for 2 days in α-MEM supplemented with 25 ng/ml macrophage colony stimulating factor (M-CSF), then for a further 12 days in α-MEM supplemented with 25 ng/ml M-CSF and 100 ng/ml RANKL while osteoclastogenesis occurred. The osteoclastic character of the cultures was verified by staining for tartrate-resistant acid phosphatase (TRAP) as described previously [3] and by gene expression profiling (Additional file 1: Fig. S6).
Nucleic acid extraction
Genomic DNA for each participant was extracted from whole blood using the QIAamp DNA Blood Mini Kit (QIAGEN) according to the manufacturer’s instructions. At day 14 of culture, RNA and DNA were harvested from each set of triplicate osteoclast-like cell cultures using the AllPrep DNA/RNA Mini Kit (QIAGEN) according to the manufacturer’s instructions, with on-column DNase digestion for the RNA fraction. High-quality RNA was obtained, with all samples recording RNA integrity numbers (RINs) ≥ 9.7.
Genotyping and imputation
Genome-wide array genotyping was performed on the genomic DNA samples using the Illumina Infinium OmniExpress-24 BeadChip array. QC criteria applied to the genotype data included removal of individuals with a call rate < 90%, variants that were monomorphic, unmapped, had a MAF < 5%, Hardy-Weinberg equilibrium P < 5 × 10−8 or call rate < 90%, leaving 572,898 variants for imputation. Genotype imputation was then performed by the Sanger Imputation Service using the Haplotype Reference Consortium (HRC) release 1.1 reference panel [50]. Any variants with an IMPUTE2 info score < 0.4 were removed from the imputed dataset. Relatedness testing and principal components analysis was performed on the genotype dataset using Plink v1.9 [51], with 10 principal components generated for use as covariates in the eQTL analysis to correct for population stratification.
Generation and processing of gene expression data
Transcriptome-wide quantitation of gene expression was performed on the osteoclast RNA samples using 50 bp single-end RNA-Seq on an Illumina HiSeq 2500. Raw read counts were generated for each gene (GENCODE v25), while those displaying a read count < 1 per million or expressed in < 10 individuals were removed. Trimmed mean of M value (TMM) normalisation and correction for total read count by conversion to counts per million (CPM) was performed on the gene expression data using the edgeR package [52]. Reads per kilobase per million (RPKM) values were also calculated for each gene using edgeR [52] to enable comparison of expression levels between different genes.
eQTL association analysis
The top genetic variant at each of the 1103 recently identified independent association signals for eBMD [19] was investigated for cis- (local) eQTL effects in the osteoclast samples. The eQTL analysis was performed on the TMM-normalised CPM gene expression values using the FastQTL software [53], which performs linear regressions between genotypes and gene expression values. Quantile normalisation was implemented using FastQTL (based on the rntransform function of the GenABEL package [54]) to ensure the gene expression values were normally distributed with a mean of 0 and standard deviation of 1. Only variants with a MAF ≥ 5% were included in the analysis, which was adjusted for the covariates patient age, RNA-Seq batch and 10 genomic principal components. Each variant was tested for association with the expression of any gene with a TSS located within a 1 Mb window on either side of the variant, consistent with the methods used by the GTEx project. Correction for multiple testing was performed using the Benjamini-Hochberg procedure [55], utilising a FDR of 5%.
Co-localisation analysis
To assess the probability that eBMD GWAS and osteoclast eQTL association signals residing in the same locus share the same causal variant, we performed a co-localisation analysis of the two datasets using the coloc package in R (default settings) [21]. This software uses a Bayesian framework to calculate posterior probabilities for 5 different scenarios (hypotheses) regarding the presence and sharing of causal variants between two genetic association datasets using summary statistics. A posterior probability of > 50% for hypothesis H4 for a specific locus (association with trait 1 and trait 2, one shared variant) indicates that co-localisation of the association signals in the two datasets due to a shared causal variant is the most likely of the 5 scenarios. We also performed co-localisation analysis using an updated version of the coloc software, named coloc2 [22]. Coloc2 performs alignment of the GWAS and eQTL summary results for each cis-eQTL region as a pre-processing step and includes optional changes implemented in the gwas-pw algorithm [56].
Summary-data-based Mendelian Randomisation analysis
To further characterise functionally relevant genes for the eBMD GWAS associations and to identify potential pleiotropic effects on gene expression and eBMD, we performed integration of the complete set of eBMD GWAS summary results with the osteoclast eQTL dataset (± 1 Mb) using the SMR software [23]. This package applies the principles of Mendelian randomisation [57] to test for association between gene expression and a trait due to a shared variant at a genetic locus. The software performs an SMR test, which detects dual association signals in the GWAS and eQTL datasets by testing for association between gene expression and the trait of interest at the top associated eQTL for each gene. The software also performs a heterogeneity in dependent instruments (HEIDI) test, which compares the association signals for nearby co-inherited markers in the GWAS and eQTL datasets. A significant HEIDI test indicates heterogeneity in the association profiles of the two datasets, thereby suggesting that the association signals seen in each dataset are less likely to be driven by the same causal variant. The osteoclast eQTL cohort genotype dataset was used as the reference panel in these analyses for estimation of LD, and only genes with at least 1 cis-eQTL association at P < 5 × 10−8 were included (N = 1070). A Bonferroni multiple-testing corrected significance threshold of P < 4.7 × 10−5 was used for the SMR test (PSMR), while a conservative significance threshold of P < 0.05 was set for the HEIDI test (PHEIDI) as an indicator of heterogeneity.
Osteoclast eQTL enrichment analysis
The GARFIELD software [24] was used to test for enrichment of osteoporosis risk variants among high-confidence osteoclast eQTL. GARFIELD performs LD pruning (r2 > 0.1) of GWAS association results to generate an independent set of variants and then integrates this with annotations containing regulatory or functional information. Each variant is annotated to a functional category or regulatory feature if the variant or an LD proxy (r2 > 0.8) is part of that annotation group. Fold enrichment is calculated using odds ratios at specified GWAS P value thresholds with significance assessed using a generalised linear model while accounting for MAF, distance to nearest TSS and number of LD proxies. A custom annotation category was generated containing all significant osteoclast eQTL associations identified at FDR 5%. Enrichment of eBMD GWAS variants in this annotation was assessed at four GWAS significance thresholds: P < 1 × 10−5, 1 × 10−6, 1 × 10−7 and 1 × 10−8. The UK10K variant set was used as the reference population for these analyses, with correction for multiple testing performed using the Bonferroni method.
X-ray microcomputed tomography analysis of mouse bones
Ripk3-deficient mice were generated and phenotyped as described previously [58]. Femora were cleaned of soft tissue and fixed in 10% neutral buffered formalin for 48 h, followed by 24 h immersion in phosphate-buffered saline (PBS). Five pairs of 15-week-old male Ripk3−/− mice and WT controls were used for the micro-CT analysis. Fixed femora were immersed in PBS and immobilised in a 2-ml tube prior to scanning of the distal femur using a Skyscan 1176 micro-CT instrument (Bruker, Kontich, Belgium). Scan parameters were as follows: 50 kV, 500 μA, 0.5 mm Al filter, exposure time 1 s, 0.4° rotation step, frame averaging of 2 and pixel resolution 8.89 μm. Scans were reconstructed using NRecon software (Bruker, Kontich, Belgium) using a constant threshold value and then analysed using the CTAn software (Bruker, Kontich Belgium). A trabecular region of interest was defined 0.5 mm below the base of the growth plate and 1 mm in height. A cortical region of interest was defined 3 mm below the growth plate and 1 mm in height. Trabecular extent was defined as the distance from the base of the growth plate to the furthest definable continuous trabecular element. Reported trabecular values were bone volume/tissue volume (BV/TV), trabecular thickness (Tb.Th), trabecular separation (Tb.Sp), trabecular number (Tb.N), trabecular extension (Tb.Ex) and BMD; reported cortical values were cortical thickness (Ct.Th), cortical bone volume (Ct.BV), marrow volume (Ma.V), endocortical perimeter (Ec.Pm), periosteal perimeter (Ps.Pm) and tissue mineral density (TMD). Differences between WT and Ripk3−/− samples were assessed using an unpaired t test.
Histomorphometric analysis of mouse bones
Mouse femur samples were formalin-fixed, decalcified and embedded in paraffin before being subjected to staining with H&E and a chromogenic TRAP substrate to characterise in vivo osteoblast and osteoclast parameters. Stained bone sections were scanned using a Scanscope XT machine (Aperio) at × 20 objective, with histomorphometric analysis performed using the BioQuant Osteo software (BioQuant). To characterise the trabecular bone, a region of interest located approximately 500 μm below the growth plate at the distal femur and 1 mm in height was defined. For the cortical bone analysis, we used a region located approximately 4 mm below the growth plate with a height of 1 mm. Two sections were analysed for each femur, with the mean of the two sets of measurements used for statistical analysis. Differences between the WT and Ripk3−/− groups were assessed using an unpaired t test.
Supplementary information
Additional file 1: Tables S1-S3 and Fig. S1-S6.
Acknowledgements
We are very grateful to the women who kindly participated in the study and the staff involved including nursing staff, bone density technologists and clerical staff. The authors would also like to acknowledge the facilities and scientific and technical assistance of the National Imaging Facility, a National Collaborative Research Infrastructure Strategy (NCRIS) capability, at the Centre for Microscopy, Characterisation and Analysis, The University of Western Australia.
Review history
The review history is available as Additional file 2.
Peer review information
Anahita Bishop was the primary editor on this article and managed its editorial process and peer review in collaboration with the rest of the editorial team.
Authors’ contributions
BM, KZ, JW and SW conceived and designed the study. BM, KZ and SM performed the patient recruitment and cell culture. NP, EM, JT, JX and JK performed the analysis of the animal samples. BM, SB, JT and FD performed the data analysis. BM, KZ, JX, SB, JT, NP, FD, JW and SW contributed to the interpretation of the data. BM and SW drafted the manuscript. BM, KZ, JX, SB, SM, JT, NP, FD, JW, JK and SW contributed to revising the content of the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by the Australian National Health and Medical Research Council (project grants 1010494, 1048216, 1087407, 1107828, 1127156, 1163933), the Sir Charles Gairdner Osborne Park Health Care Group (SCGOPHCG) Research Advisory Committee (grant 2016-17/017) and the iVEC/Pawsey Supercomputing Centre (with funding from the Australian Government and the Government of Western Australia; project grants: Pawsey0260 (S.G.W.), Director2025 (S.G.W.)). The salary of B.H.M. was supported by a Raine Medical Research Foundation Priming Grant.
Availability of data and materials
The GTEx V7 eQTL dataset can be accessed at https://gtexportal.org/home/datasets. The osteoclast eQTL data described in this study relevant to the genes listed in Table 1 are publicly available from the figshare repository [59].
Ethics approval and consent to participate
Written informed consent was obtained from all participants in the osteoclast eQTL cohort, and the study was approved by the Sir Charles Gairdner and Osborne Park Health Care Group Human Research Ethics Committee. All mouse work was performed using methods approved by the University of Western Australia Animal Ethics Committee (RA/3/100/779). The experimental methods used in this study comply with the Helsinki Declaration.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
John P. Walsh, Jiake Xu and Scott G. Wilson contributed equally to this work.
Supplementary information
Supplementary information accompanies this paper at 10.1186/s13059-020-01997-2.
References
- 1.Battle A, Brown CD, Engelhardt BE, Montgomery SB. Genetic effects on gene expression across human tissues. Nature. 2017;550(7675):204–213. doi: 10.1038/nature24277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Grundberg E, Kwan T, Ge B, Lam KC, Koka V, Kindmark A, et al. Population genomics in a disease targeted primary cell model. Genome Res. 2009;19(11):1942–1952. doi: 10.1101/gr.095224.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mullin BH, Zhu K, Xu J, Brown SJ, Mullin S, Tickner J, et al. Expression quantitative trait locus study of bone mineral density GWAS variants in human osteoclasts. J Bone Miner Res. 2018;33(6):1044-51. [DOI] [PubMed]
- 4.Mullin BH, Zhu K, Brown SJ, Mullin S, Tickner J, Pavlos NJ, et al. Genetic regulatory mechanisms in human osteoclasts suggest a role for the STMP1 and DCSTAMP genes in Paget’s disease of bone. Sci Rep. 2019;9(1):1052. doi: 10.1038/s41598-018-37609-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Robbins JA, Biggs ML, Cauley J. Adjusted mortality after hip fracture: from the cardiovascular health study. J Am Geriatr Soc. 2006;54(12):1885–1891. doi: 10.1111/j.1532-5415.2006.00985.x. [DOI] [PubMed] [Google Scholar]
- 6.Specker BL. Evidence for an interaction between calcium intake and physical activity on changes in bone mineral density. J Bone Miner Res. 1996;11(10):1539–1544. doi: 10.1002/jbmr.5650111022. [DOI] [PubMed] [Google Scholar]
- 7.Uusi-Rasi K, Sievanen H, Vuori I, Pasanen M, Heinonen A, Oja P. Associations of physical activity and calcium intake with bone mass and size in healthy women at different ages. J Bone Miner Res. 1998;13(1):133–142. doi: 10.1359/jbmr.1998.13.1.133. [DOI] [PubMed] [Google Scholar]
- 8.Arden NK, Baker J, Hogg C, Baan K, Spector TD. The heritability of bone mineral density, ultrasound of the calcaneus and hip axis length: a study of postmenopausal twins. J Bone Miner Res. 1996;11(4):530–534. doi: 10.1002/jbmr.5650110414. [DOI] [PubMed] [Google Scholar]
- 9.Krall EA, Dawson-Hughes B. Heritable and life-style determinants of bone mineral density. J Bone Miner Res. 1993;8(1):1–9. doi: 10.1002/jbmr.5650080102. [DOI] [PubMed] [Google Scholar]
- 10.Gueguen R, Jouanny P, Guillemin F, Kuntz C, Pourel J, Siest G. Segregation analysis and variance components analysis of bone mineral density in healthy families. J Bone Miner Res. 1995;10(12):2017–2022. doi: 10.1002/jbmr.5650101223. [DOI] [PubMed] [Google Scholar]
- 11.Deng HW, Chen WM, Recker S, Stegman MR, Li JL, Davies KM, et al. Genetic determination of Colles’ fracture and differential bone mass in women with and without Colles’ fracture. J Bone Miner Res. 2000;15(7):1243–1252. doi: 10.1359/jbmr.2000.15.7.1243. [DOI] [PubMed] [Google Scholar]
- 12.Keen RW, Hart DJ, Arden NK, Doyle DV, Spector TD. Family history of appendicular fracture and risk of osteoporosis: a population-based study. Osteoporos Int. 1999;10(2):161–166. doi: 10.1007/s001980050211. [DOI] [PubMed] [Google Scholar]
- 13.Estrada K, Styrkarsdottir U, Evangelou E, Hsu YH, Duncan EL, Ntzani EE, et al. Genome-wide meta-analysis identifies 56 bone mineral density loci and reveals 14 loci associated with risk of fracture. Nat Genet. 2012;44(5):491–501. doi: 10.1038/ng.2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Medina-Gomez C, Kemp JP, Trajanoska K, Luan J, Chesi A, Ahluwalia TS, et al. Life-course genome-wide association study meta-analysis of total body BMD and assessment of age-specific effects. Am J Hum Genet. 2018;102(1):88–102. doi: 10.1016/j.ajhg.2017.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mullin BH, Walsh JP, Zheng HF, Brown SJ, Surdulescu GL, Curtis C, et al. Genome-wide association study using family-based cohorts identifies the WLS and CCDC170/ESR1 loci as associated with bone mineral density. BMC Genomics. 2016;17(1):136. doi: 10.1186/s12864-016-2481-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mullin BH, Zhao JH, Brown SJ, Perry JRB, Luan J, Zheng HF, et al. Genome-wide association study meta-analysis for quantitative ultrasound parameters of bone identifies five novel loci for broadband ultrasound attenuation. Hum Mol Genet. 2017;26(14):2791–2802. doi: 10.1093/hmg/ddx174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Richards JB, Rivadeneira F, Inouye M, Pastinen TM, Soranzo N, Wilson SG, et al. Bone mineral density, osteoporosis, and osteoporotic fractures: a genome-wide association study. Lancet. 2008;371(9623):1505–1512. doi: 10.1016/S0140-6736(08)60599-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zheng HF, Forgetta V, Hsu YH, Estrada K, Rosello-Diez A, Leo PJ, et al. Whole-genome sequencing identifies EN1 as a determinant of bone density and fracture. Nature. 2015;526(7571):112–117. doi: 10.1038/nature14878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morris JA, Kemp JP, Youlten SE, Laurent L, Logan JG, Chai RC, et al. An atlas of genetic influences on osteoporosis in humans and mice. Nat Genet. 2019;51(2):258–266. doi: 10.1038/s41588-018-0302-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, et al. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19(9):1639–1645. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Giambartolomei C, Vukcevic D, Schadt EE, Franke L, Hingorani AD, Wallace C, et al. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet. 2014;10(5):e1004383. doi: 10.1371/journal.pgen.1004383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dobbyn A, Huckins LM, Boocock J, Sloofman LG, Glicksberg BS, Giambartolomei C, et al. Landscape of conditional eQTL in dorsolateral prefrontal cortex and co-localization with schizophrenia GWAS. Am J Hum Genet. 2018;102(6):1169–1184. doi: 10.1016/j.ajhg.2018.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhu Z, Zhang F, Hu H, Bakshi A, Robinson MR, Powell JE, et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat Genet. 2016;48(5):481–487. doi: 10.1038/ng.3538. [DOI] [PubMed] [Google Scholar]
- 24.Iotchkova V, Ritchie GRS, Geihs M, Morganella S, Min JL, Walter K, et al. GARFIELD classifies disease-relevant genomic features through integration of functional annotations with association signals. Nat Genet. 2019;51(2):343–353. doi: 10.1038/s41588-018-0322-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nagel M, Jansen PR, Stringer S, Watanabe K, de Leeuw CA, Bryois J, et al. Meta-analysis of genome-wide association studies for neuroticism in 449,484 individuals identifies novel genetic loci and pathways. Nat Genet. 2018;50(7):920–927. doi: 10.1038/s41588-018-0151-7. [DOI] [PubMed] [Google Scholar]
- 26.Cui H, Zhu Y, Jiang D. The RIP1-RIP3 complex mediates osteocyte necroptosis after ovariectomy in rats. PLoS One. 2016;11(3):e0150805. doi: 10.1371/journal.pone.0150805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gamazon ER, Segre AV, van de Bunt M, Wen X, Xi HS, Hormozdiari F, et al. Using an atlas of gene regulation across 44 human tissues to inform complex disease- and trait-associated variation. Nat Genet. 2018;50(7):956–967. doi: 10.1038/s41588-018-0154-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lin L, Lin W, Qin YX. Enhanced correlation between quantitative ultrasound and structural and mechanical properties of bone using combined transmission-reflection measurement. J Acoust Soc Am. 2015;137(3):1144–1152. doi: 10.1121/1.4906830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Linkermann A, Green DR. Necroptosis. N Engl J Med. 2014;370(5):455–465. doi: 10.1056/NEJMra1310050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kaiser WJ, Upton JW, Long AB, Livingston-Rosanoff D, Daley-Bauer LP, Hakem R, et al. RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature. 2011;471(7338):368–372. doi: 10.1038/nature09857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kaczmarek A, Vandenabeele P, Krysko DV. Necroptosis: the release of damage-associated molecular patterns and its physiological relevance. Immunity. 2013;38(2):209–223. doi: 10.1016/j.immuni.2013.02.003. [DOI] [PubMed] [Google Scholar]
- 32.Lango Allen H, Estrada K, Lettre G, Berndt SI, Weedon MN, Rivadeneira F, et al. Hundreds of variants clustered in genomic loci and biological pathways affect human height. Nature. 2010;467(7317):832–838. doi: 10.1038/nature09410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4(7):1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 34.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46(3):310–315. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cawley NX, Wetsel WC, Murthy SR, Park JJ, Pacak K, Loh YP. New roles of carboxypeptidase E in endocrine and neural function and cancer. Endocr Rev. 2012;33(2):216–253. doi: 10.1210/er.2011-1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Alsters SI, Goldstone AP, Buxton JL, Zekavati A, Sosinsky A, Yiorkas AM, et al. Truncating homozygous mutation of carboxypeptidase E (CPE) in a morbidly obese female with type 2 diabetes mellitus, intellectual disability and hypogonadotrophic hypogonadism. PLoS One. 2015;10(6):e0131417. doi: 10.1371/journal.pone.0131417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Naggert JK, Fricker LD, Varlamov O, Nishina PM, Rouille Y, Steiner DF, et al. Hyperproinsulinaemia in obese fat/fat mice associated with a carboxypeptidase E mutation which reduces enzyme activity. Nat Genet. 1995;10(2):135–142. doi: 10.1038/ng0695-135. [DOI] [PubMed] [Google Scholar]
- 39.Cawley NX, Yanik T, Woronowicz A, Chang W, Marini JC, Loh YP. Obese carboxypeptidase E knockout mice exhibit multiple defects in peptide hormone processing contributing to low bone mineral density. Am J Physiol Endocrinol Metab. 2010;299(2):E189–E197. doi: 10.1152/ajpendo.00516.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim HJ, Hong J, Yoon HJ, Yoon YR, Kim SY. Carboxypeptidase E is a novel modulator of RANKL-induced osteoclast differentiation. Mol Cells. 2014;37(9):685–690. doi: 10.14348/molcells.2014.0179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Putnam EA, Zhang H, Ramirez F, Milewicz DM. Fibrillin-2 (FBN2) mutations result in the Marfan-like disorder, congenital contractural arachnodactyly. Nat Genet. 1995;11(4):456–458. doi: 10.1038/ng1295-456. [DOI] [PubMed] [Google Scholar]
- 42.Baba M, Endoh M, Ma W, Toyama H, Hirayama A, Nishikawa K, et al. Folliculin regulates osteoclastogenesis through metabolic regulation. J Bone Miner Res. 2018;33(10):1785–1798. doi: 10.1002/jbmr.3477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nickerson ML, Warren MB, Toro JR, Matrosova V, Glenn G, Turner ML, et al. Mutations in a novel gene lead to kidney tumors, lung wall defects, and benign tumors of the hair follicle in patients with the Birt-Hogg-Dube syndrome. Cancer Cell. 2002;2(2):157–164. doi: 10.1016/s1535-6108(02)00104-6. [DOI] [PubMed] [Google Scholar]
- 44.Li QZ, Wang CY, Shi JD, Ruan QG, Eckenrode S, Davoodi-Semiromi A, et al. Molecular cloning and characterization of the mouse and human TUSP gene, a novel member of the tubby superfamily. Gene. 2001;273(2):275–284. doi: 10.1016/s0378-1119(01)00582-0. [DOI] [PubMed] [Google Scholar]
- 45.Vieira AR, de Carvalho FM, Johnson L, DeVos L, Swailes AL, Weber ML, et al. Fine mapping of 6q23.1 identifies TULP4 as contributing to clefts. Cleft Palate-Craniofacial J. 2015;52(2):128–134. doi: 10.1597/13-023. [DOI] [PubMed] [Google Scholar]
- 46.Wood AR, Esko T, Yang J, Vedantam S, Pers TH, Gustafsson S, et al. Defining the role of common variation in the genomic and biological architecture of adult human height. Nat Genet. 2014;46(11):1173–1186. doi: 10.1038/ng.3097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gilissen C, Arts HH, Hoischen A, Spruijt L, Mans DA, Arts P, et al. Exome sequencing identifies WDR35 variants involved in Sensenbrenner syndrome. Am J Hum Genet. 2010;87(3):418–423. doi: 10.1016/j.ajhg.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Machiela MJ, Chanock SJ. LDlink: a web-based application for exploring population-specific haplotype structure and linking correlated alleles of possible functional variants. Bioinformatics (Oxford) 2015;31(21):3555–3557. doi: 10.1093/bioinformatics/btv402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mullin BH, Mamotte C, Prince RL, Wilson SG. Influence of ARHGEF3 and RHOA knockdown on ACTA2 and other genes in osteoblasts and osteoclasts. PLoS One. 2014;9(5):e98116. doi: 10.1371/journal.pone.0098116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McCarthy S, Das S, Kretzschmar W, Delaneau O, Wood AR, Teumer A, et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat Genet. 2016;48(10):1279–1283. doi: 10.1038/ng.3643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ. Second-generation PLINK: rising to the challenge of larger and richer datasets. GigaSci. 2015;4:7. doi: 10.1186/s13742-015-0047-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics (Oxford) 2010;26(1):139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ongen H, Buil A, Brown AA, Dermitzakis ET, Delaneau O. Fast and efficient QTL mapper for thousands of molecular phenotypes. Bioinformatics (Oxford) 2016;32(10):1479–1485. doi: 10.1093/bioinformatics/btv722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Aulchenko YS, Ripke S, Isaacs A, van Duijn CM. GenABEL: an R library for genome-wide association analysis. Bioinformatics (Oxford) 2007;23(10):1294–1296. doi: 10.1093/bioinformatics/btm108. [DOI] [PubMed] [Google Scholar]
- 55.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B Methodol. 1995;57(1):289–300. [Google Scholar]
- 56.Pickrell JK, Berisa T, Liu JZ, Segurel L, Tung JY, Hinds DA. Detection and interpretation of shared genetic influences on 42 human traits. Nat Genet. 2016;48(7):709–717. doi: 10.1038/ng.3570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Smith GD, Ebrahim S. ‘Mendelian randomization’: can genetic epidemiology contribute to understanding environmental determinants of disease? Int J Epidemiol. 2003;32(1):1–22. doi: 10.1093/ije/dyg070. [DOI] [PubMed] [Google Scholar]
- 58.Newton K, Sun X, Dixit VM. Kinase RIP3 is dispensable for normal NF-kappa Bs, signaling by the B-cell and T-cell receptors, tumor necrosis factor receptor 1, and Toll-like receptors 2 and 4. Mol Cell Biol. 2004;24(4):1464–1469. doi: 10.1128/MCB.24.4.1464-1469.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mullin BH. Osteoclast-specific eQTL results for osteoporosis risk variants (co-localised loci). Dataset Figshare Repository. 2020. 10.6084/m9.figshare.11953674.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Tables S1-S3 and Fig. S1-S6.
Data Availability Statement
The GTEx V7 eQTL dataset can be accessed at https://gtexportal.org/home/datasets. The osteoclast eQTL data described in this study relevant to the genes listed in Table 1 are publicly available from the figshare repository [59].