

Abstract
An amphipathic peptide has been engineered and is capable of penetrating the blood–brain barrier as well as possessing a potent antiviral activity against Zika and other mosquito-borne viruses.
Subject terms: Peptide delivery, Drug development, Infection, Antiviral agents
One third of deaths from infectious diseases worldwide are of viral origins. Although hundreds of viruses are known to cause human diseases, the mainstay of antiviral research and development remains focused on direct antiviral agents (DAA) that inhibit specific viral proteins of individual viruses. This ‘one lock, one key’ approach has resulted in two major drawbacks of the current antiviral therapy: narrow spectrum of therapeutic coverage and emergence of drug resistance. These drawbacks are further inflamed by two daunting challenges. First, the development of a new drug is slow and expensive, taking at least ten years at an average cost of over US$2 billion dollars1. Second, the nature of virus emergence is very unpredictable. The one lock, one key approach is impossible to provide a rapid countermeasure when responding to a global crisis of virus emergence, as evidenced during the recent epidemics of severe acute respiratory syndrome (SARS), Middle East respiratory syndrome (MERS), chikungunya, Ebola and Zika viruses. Thus, new antiviral approaches are urgently needed. Writing in Nature Materials, Joshua Jackman and colleagues2 report an antiviral approach, termed lipid envelope antiviral disruption (LEAD), for development of broad-spectrum antiviral therapy. The authors demonstrated that an amphipathic α-helical peptide (AH-D), derived from the first 27 amino acids of hepatitis C virus (HCV) non-structural protein 5A (NS5A; Fig. 1a), has potent antiviral activities against Zika, dengue, yellow fever, Japanese encephalitis and chikungunya viruses, with EC50s (a concentration required to inhibit 50% of viral replication; 12–206 nM) and CC50 (a concentration required to cause 50% cytotoxicity; 63 μM). Remarkably, treatment of Zika virus-infected mice with this peptide, even when starting the therapy on day three post-infection, protected against disease and death without adverse effects.
Fig. 1. AH-D peptide and its membrane-disruptive activity against liposomes.
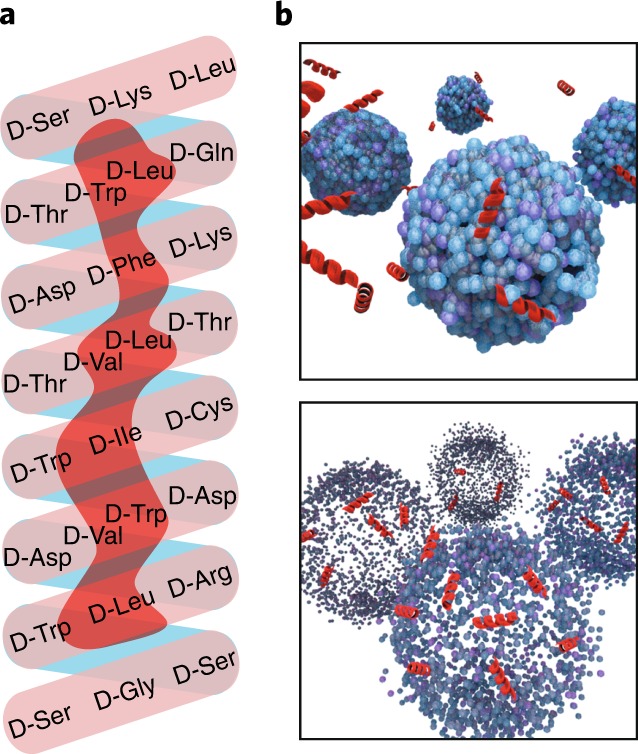
a, Helix net diagram of AH-D peptide. The N-terminus of the peptide starts at the bottom of the helix. b, Illustration of AH-D peptide (red) disrupting the membrane of liposomes (blue) with diameters of <160 nm.
The N-terminal region of HCV NS5A is known to associate with the endoplasmic reticulum membrane in infected cells. When studying the interaction of the N-terminal α-helix of this protein with lipid membranes, Cho et al. serendipitously discovered that the AH peptide could potently rupture liposomes3. Importantly, the AH peptide selectively ruptures liposomes with diameters of <160 nm. Such size-dependent liposome disruption is determined by the ability of the AH peptide to sense different degrees of membrane curvature4. Specifically, liposomes of <160 nm have high membrane curvatures that are susceptible to AH peptide-mediated pore formation (Fig. 1b, top); once a critical density of pores is formed, membrane lysis occurs (Fig. 1b, bottom). In contrast, liposomes of >160 nm have low membrane curvatures that are not susceptible to AH peptide-mediated pore formation, thus no lysis occurs5. In agreement with the liposome results, tested viruses prone to AH peptide inhibition fall within the disruptive range of liposome size: flaviviruses, 40–60 nm; chikungunya virus, 60–70 nm; bunyaviruses, 90–100 nm; and human immunodeficiency virus, 120 nm. In contrast, viruses of larger sizes, such as vaccinia virus, 360 × 270 × 250 nm3, as well as cell membranes are less sensitive to the peptide inhibition2,6. These results suggest that the AH peptide may also inhibit other untested enveloped viruses, as long as their virions are <160 nm.
Besides AH peptide, Chisari and colleagues also found that C5A peptide, representing amino acids 3–20 of HCV NS5A, has a broad spectrum of antiviral activity7,8. The potency of C5A peptide (EC50s 0.6–4.5 μM against different viruses) is much weaker than the current AH peptide, possibly due to the differences in peptide lengths and amino acid sequences. Indeed, mutagenesis analysis showed that the amphipathic α-helical structure as well as the amino acid composition are critical for the peptide’s antiviral activity7. Since chirality is not essential for antiviral activity, the current AH-D peptide was synthesized with D-amino acids rather than L-amino acids that are more susceptible to proteolytic degradation9. The D-chirality improves the peptide’s pharmacokinetic stability to a half-life time of 7 h in mice. In addition, the AH-D peptide can cross blood–brain barrier2. This brain-penetrating property is ideal for potential treatment of neurotrophic viruses. Indeed, the improved potency, pharmacokinetics and brain-penetrating property of the AH-D peptide have translated into excellent efficacy in vivo. Treatment of Zika virus-infected mice with AH-D peptide reduced viral loads in blood and organs, as well as reduction in inflammatory cytokines, weight loss and deaths (Fig. 2). Specifically, treatment with AH-D peptide decreased the death rate from 100% to 17%. The peptide’s ability to access the brain significantly enhanced its antiviral activity in the central nervous system and protected the brain function of infected mice, as indicated by a normal level of myeloperoxidase2.
Fig. 2. In vivo efficacy of AH-D peptide in mice.

a,b, Type-I interferon receptor knockout mice (IFN-α/βR–/–) were intravenously infected with 4 × 103 plaque-forming units (PFU) of Zika virus (ZIKV). The infected mice were treated with 25 mg kg–1 AH-D peptide or phosphate-buffered saline (PBS) mock on days three to six post-infection. The mice were monitored for survival (a). The levels of infectious virus within the brain (b) were measured on days three to seven post-infection. The AH-D treatment significantly decreased viral loads in serum and various organs, leading to improved survival. Adapted from ref. 2, Springer Nature Ltd.
Targeting host factors essential for virus life cycle is a valid antiviral approach. This approach has led to the development of maraviroc, a clinically approved HIV drug that inhibits host protein CCR5, a co-receptor for HIV entry10. Compared with direct antiviral agents, host-targeting antiviral approach offers the advantage of higher barriers for resistance emergence, since viral mutations are less able to compensate for loss of essential host cofactors. In addition, because different viruses share common host factors/pathways for viral replication, compounds targeting the shared host factors/pathways could have broad antiviral activities. For further development of the AH-D peptide, understanding the structure of how the peptide senses and disrupts the virions will help design future inhibitors. The ability to optimize and shorten the peptide, while keeping its potency, will reduce the drug cost. Most importantly, achieving no observed adverse effect level (NOAEL) in preclinical safety tests will advance the AH-D peptide to clinical trials. Overall, the current study represents a breakthrough in advancing the LEAD concept as a new class of broad-spectrum antiviral therapy.
References
- 1.DiMasi JA, Grabowski HG, Hansen RW. J. Health Econ. 2016;47:20–33. doi: 10.1016/j.jhealeco.2016.01.012. [DOI] [PubMed] [Google Scholar]
- 2.Jackman JA, et al. Nat. Mater. 2018 doi: 10.1038/s41563-018-0194-2. [DOI] [Google Scholar]
- 3.Cho NJ, Cho SJ, Cheong KH, Glenn JS, Frank CW. J. Am. Chem. Soc. 2007;129:10050–10051. doi: 10.1021/ja0701412. [DOI] [PubMed] [Google Scholar]
- 4.Tabaei SR, Rabe M, Zhdanov VP, Cho NJ, Höök F. Nano Lett. 2012;12:5719–5725. doi: 10.1021/nl3029637. [DOI] [PubMed] [Google Scholar]
- 5.Jackman JA, Goh HZ, Zhdanov VP, Knoll W, Cho NJ. J. Am. Chem. Soc. 2016;138:1406–1413. doi: 10.1021/jacs.5b12491. [DOI] [PubMed] [Google Scholar]
- 6.Cho NJ, et al. ACS Chem. Biol. 2009;4:1061–1067. doi: 10.1021/cb900149b. [DOI] [PubMed] [Google Scholar]
- 7.Cheng G, et al. Proc. Natl Acad. Sci. USA. 2008;105:3088–3093. doi: 10.1073/pnas.0712380105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bobardt MD, et al. Proc. Natl Acad. Sci. USA. 2008;105:5525–5530. doi: 10.1073/pnas.0801388105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Garton M, et al. Proc. Natl Acad. Sci. USA. 2018;115:1505–1510. doi: 10.1073/pnas.1711837115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abel S, Back DJ, Vourvahis M. Antivir. Ther. 2009;14:607–618. doi: 10.3851/IMP1297. [DOI] [PubMed] [Google Scholar]