

Key Points
Complement is a key component of immunity with crucial inflammatory and opsonic properties; inappropriate activation of complement triggers or exacerbates inflammatory disease.
Complement dysregulation is a core feature of some diseases and contributes to pathology in many others.
Approved agents have been developed for and are highly effective in some orphan applications, but their progress to use in more common diseases has been slow.
Numerous challenges, such as target concentration or high turnover, limit the efficacy of these agents in humans.
Numerous novel agents targeting different parts of the complement system in different ways are now emerging from pre-clinical studies and are entering Phase I/II trials; these agents bring the potential for more-effective and more-specific anti-complement therapies in disease.
Other agents, both biologic and small molecule, are in Phase II or III trials for both rare and common diseases — administration routes include localized (for example, intravitreal) and systemic routes.
There is an urgent need to develop biomarkers and imaging methods that enable monitoring of the effects and efficacy of anti-complement agents.
Supplementary information
The online version of this article (doi:10.1038/nrd4657) contains supplementary material, which is available to authorized users.
Subject terms: Complement cascade, Inflammatory diseases, Drug discovery
The complement cascade, a key regulator of innate immunity, is a rich source of potential therapeutic targets for diseases including autoimmune, inflammatory and degenerative disorders. Morgan and Harris discuss the progress made in modulating the complement system and the existing challenges, including dosing, localization of the drug to the target and how to interfere with protein–protein interactions.
Supplementary information
The online version of this article (doi:10.1038/nrd4657) contains supplementary material, which is available to authorized users.
Abstract
The complement system is a key innate immune defence against infection and an important driver of inflammation; however, these very properties can also cause harm. Inappropriate or uncontrolled activation of complement can cause local and/or systemic inflammation, tissue damage and disease. Complement provides numerous options for drug development as it is a proteolytic cascade that involves nine specific proteases, unique multimolecular activation and lytic complexes, an arsenal of natural inhibitors, and numerous receptors that bind to activation fragments. Drug design is facilitated by the increasingly detailed structural understanding of the molecules involved in the complement system. Only two anti-complement drugs are currently on the market, but many more are being developed for diseases that include infectious, inflammatory, degenerative, traumatic and neoplastic disorders. In this Review, we describe the history, current landscape and future directions for anti-complement therapies.
Supplementary information
The online version of this article (doi:10.1038/nrd4657) contains supplementary material, which is available to authorized users.
Main
The complement system includes more than 30 component proteins, regulators and receptors, present in plasma and on cells, which collaborate to provide defence against infection and to clear toxic materials. Complement is a key part of innate immunity and a modulator of the adaptive immune response; inherited deficiencies in complement proteins predispose individuals to bacterial infection and/or immune complex disease. The powerful cell-targeting and cell-killing properties of complement can be turned against self, a scenario that is prevented by the abundant expression of regulator proteins. In health, these regulator proteins maintain a fragile truce in which complement is continuously activated in what is known as tickover. Bacteria or other foreign surfaces lacking regulator proteins provoke massive activation and amplification of complement, generating activation products that label the invader, attract and activate phagocytic cells and directly cause lytic pathogen killing. Complement comprises a cluster of activation pathways that are triggered in various ways and progress through several amplifying enzymes to converge on a final common cell-killing pathway1,2. Surface-bound antibodies trigger the classical pathway, bacterial sugars initiate the lectin pathway and amplification is provided by the alternative pathway, regardless of initiating route (Fig. 1). Fluid-phase regulators prevent uncontrolled activation in plasma, whereas membrane-bound regulators restrict activation on cells. The protective (and pathological) effects of complement are mediated by its activation products — active fragments or complexes of complement proteins. C3, the most abundant complement protein, has numerous activities; some C3 fragments (for example, C3b, iC3b and C3dg) can covalently bind to targets and act as flags to instruct phagocytes to bind to and engulf the targets, or they can enhance humoral immunity by facilitating B cell activation. The small C3a fragment, among its many roles, can attract and activate phagocytes. Downstream of C3, the terminal pathway releases the proinflammatory fragment C5a and assembles a pore in target membranes — the membrane attack complex (MAC) — that effects the lysis of bacteria and other activating cells.
Figure 1. Targets for inhibition in the complement pathway.

The figure shows a highly simplified view of the complement system and highlights the targets for pathway inhibition. Activation is triggered through classical (antibody) or lectin (sugar) pathways that rapidly converge to form a complement C3-cleaving enzyme (C3 convertase), C4b2a. The alternative pathway can be independently activated to generate its own C3 convertase (C3bBb) but, more importantly, amplifies activation regardless of trigger. C3 fragments, both soluble and surface-attached, engage specific receptors on expressing cells to mediate key activities. The C5 convertase, formed by the recruitment of an additional C3b into the C3 convertase, cleaves C5 to release a small fragment, C5a, which binds to receptors on expressing cells to mediate activation events. Formation of C5b initiates the membrane attack pathway; sequential recruitment of components C6, C7, C8 and C9 creates a pore in the target membrane – the membrane attack complex (MAC) – that can activate or kill the targeted cell. The complement system presents many targets for inhibition with drugs. In the activation pathways these include the initiating complexes and enzymes, the initiators of the alternative pathway loop and the C3 convertases. In the C3–C5 axis, potential targets include the individual components (for example, C3 and C5), the activation fragments (for example, C3a and C5a) and the C5 convertases. In the terminal pathway, agents might target individual components (such as C5, C6 or C7) or intermediates (such as C5b6 and C5b67), block the functional MAC pore or inhibit the downstream signalling events that mediate cell activation or destruction.
The list of diseases in which complement has a role, either primary or secondary to other triggers, is long and growing (Table 1). Genetic deficiencies in complement components are relatively rare and associated with infections and immune complex diseases (Box 1); however, excessive or dysregulated complement activation contributes to many inflammatory, autoimmune and degenerative diseases, and is a much larger problem3,4. In a minority of these diseases, genetic changes, mutations or polymorphisms in complement proteins, regulators or receptors are the direct cause of dysregulation and disease; in most of these diseases, complement is not the primary cause but is recruited in response to tissue damage and serves to amplify and exacerbate inflammation and injury. Crucially, a small but growing group of conditions are now widely recognized as diseases caused by complement dysregulation, presenting obvious targets for complement-inhibiting drugs5,6. Lessons from disease-associated polymorphisms and highly penetrant disease-causing mutations in complement proteins have pinpointed the pathways and components driving pathology and guided the development of therapies for the numerous disease indications in which complement has an important role (Table 1).
Table 1. Complement and disease*.
| Process or disease | Evidence implicating complement‡ | Refs |
|---|---|---|
| Primary dysregulation | ||
| Hereditary angioedema | Genetic, clinical and therapeutic | 59,60,61,62,63,158,159,160 |
| PNH | Genetic, clinical and therapeutic | 6,7,24 |
| aHUS and TTP | Genetic, clinical and therapeutic | 28,147 |
| Thrombotic microangiopathy | Clinical and therapeutic | 161 |
| C3 glomerulopathy and MPGN | Genetic, clinical and therapeutic | 5 |
| Transplant rejection | Models, clinical and therapeutic | 38,89,125 |
| Autoimmune | ||
| Neuromyelitis optica | Clinical and therapeutic | 162 |
| Multiple sclerosis | Models, clinical and therapeutic | 163,164 |
| Guillain–Barré syndrome | Models, clinical and therapeutic | 165,166 |
| Myasthenia gravis | Models, clinical and therapeutic | 167,168 |
| Lupus nephritis | Models, clinical and therapeutic | 99,169 |
| IgA nephropathy | Genetic, clinical and therapeutic | 170,171 |
| Rheumatoid arthritis | Models and therapeutic | 172 |
| Crohn disease and ulcerative colitis | Models, clinical and genetic | 173,174 |
| Autoimmune haemolytic anemia | Clinical and therapeutic | 175 |
| Pemphigus and pemphigoid | Clinical and therapeutic | 143,176 |
| Anti-phospholipid syndrome | Models, clinical and therapeutic | 177,178 |
| Inflammatory | ||
| Macular degeneration | Models, genetic, clinical and therapeutic | 33,131,132,133,134,135 |
| Uveitis | Models, clinical and therapeutic | 179 |
| ANCA-associated vasculitis | Models, clinical and therapeutic | 180 |
| Atherosclerosis | Models and clinical | 181,182 |
| Mood disorders | Clinical | 183,184 |
| Asthma | Models, clinical and therapeutic | 185,186 |
| COPD | Models and clinical | 187 |
| Anaphylaxis | Models, clinical and therapeutic | 188,189 |
| Sepsis and ARDS | Models, clinical and therapeutic | 190,191 |
| Cerebral malaria | Models | 192 |
| Psoriatic arthropathy | Clinical and therapeutic | 193,194 |
| Dermatomyositis | Clinical | 195,196 |
| Degenerative | ||
| Osteoarthritis | Models, clinical and therapeutic | 197,198 |
| Dementia | Models | 199,200 |
| Glaucoma | Models and clinical | 201 |
| Diabetic angiopathy | Models and clinical | 202,203 |
| Ischaemia–reperfusion | ||
| Myocardial infarction | Models and clinical | 68,88,204 |
| Stroke | Models and clinical | 69,205,206 |
| Post-bypass | Models, clinical and therapeutic | 37 |
| Acute injuries | ||
| Polytrauma | Models and clinical | 207 |
| Neurotrauma | Models and clinical | 105,208 |
| Haemodialysis | Models, clinical and therapeutic | 209,210 |
| Post-infection HUS | Clinical and therapeutic | 211 |
*The table comprises an incomplete list of the many diseases in which complement has a role; the diseases are grouped by process, although these groupings are porous and many diseases fit more than one category.
‡Clinical indicates evidence from human studies including biomarker and pathological findings; genetic indicates evidence from gene linkage studies; models indicates preclinical evidence of complement involvement from animal models; and therapeutic indicates evidence from use of a therapeutic (in humans or in animal models).
aHUS, atypical haemolytic uremic syndrome; ANCA, anti-neutrophil cytoplasmic antibody; ARDS, adult respiratory distress syndrome; COPD, chronic obstructive pulmonary disease; IgA, immunoglobulin A; MPGN, membranoproliferative glomerulonephropathy; PNH, paroxysmal nocturnal haemoglobinuria; TTP, thrombotic thrombocytopenic purpura.
The merits of anti-complement therapy in preclinical models of disease have been explored in hundreds of papers; the earliest examples date back more than 50 years and describe the use of cobra venom factor (CVF) to inhibit disease in animal models7,8. Despite this long history, few drugs have entered clinical trials, and fewer still have progressed beyond Phase I. The recent interest in complement and disease has been fuelled by a number of factors — the demonstration of genetic associations that firmly link complement to common diseases was a key finding, but the biggest factor has been the success of a single drug, eculizumab (Soliris; Alexion Pharmaceuticals), in an orphan application: treatment of paroxysmal nocturnal haemoglobinuria (PNH) (Boxes 1,2).
PNH was among the first diseases to be treated with anti-complement therapeutics (Box 2); however, PNH is rare, and the identification of a much more common disease with complement dysregulation at its root was required to catapult complement into the spotlight. Age-related macular degeneration (AMD) is the commonest cause of blindness in the Western world; a perfect storm of genetic, pathological and clinical evidence has demonstrated beyond doubt that complement dysregulation is a crucial driver of inflammation and tissue damage in AMD and has catalysed an explosion of interest in controlling complement9,10. As a consequence, what began as a small industry targeting PNH and a few other orphan diseases has, over the past decade, grown into a major endeavour involving numerous companies ranging from small biotechnology companies to large pharmaceutical organizations.
As the field has grown, it has become increasingly clear that there will be no 'one size fits all' solution to complement therapies; agents that are effective in one disease might do nothing in, or even exacerbate, another. Rational design and selection of therapies will require in-depth understanding of the way complement works in each disease. In this Review, we consider the diverse disease targets, the many intervention points, the challenges associated with drugging these targets, and the multiple approaches to inhibition that are emerging as complement moves into the clinical mainstream. Below, before exploring pathology, we briefly revisit physiology.
Box 1: Genetics dictate the risk of complement-mediated injury.
Complement comprises a large number of interacting components and regulators, and common genetic variation in these proteins dictates whether a given individual possesses a complement system that is more or less active. We have reviewed this complotype concept previously129, and here we focus on its relevance to complement-mediated diseases and therapy. Put crudely, individuals who have inherited a more active complotype are better able to resist infection but are at greater risk of complement-driven inflammation. Some variant proteins directly affect the activity of the amplification loop (for example, complement C3 R102G (C3-R102G) and Factor B R32Q (FB-R32Q))130, and this is the primary reason for disease association, whereas others influence control local to the disease site, and risk is thus disease- or tissue-specific. This concept is well-illustrated by considering age-related macular degeneration (AMD): the most important genetic risk factor in AMD is a common polymorphism in the alternative pathway regulator FH that changes a single amino acid in the middle of the molecule (Y402H). The risk of developing AMD is at least threefold greater in His-variant homozygotes than in Tyr homozygotes131,132. The Y402H variant influences the localization of FH to retinal ligands, whereas other complement polymorphisms (including those in FB and C3) influence AMD risk by increasing complement alternative pathway activity133–135; common variants in just these three proteins together alter AMD risk up to 10-fold. In deciding whether and how to inhibit complement for therapy in an individual, the complotype should be an important consideration.
Mutations in complement components and regulators can also affect complement dysregulation and disease risk. Again, FH provides the best example. FH is a large, elongated molecule comprising a chain of 20 short consensus repeats. The 20th short consensus repeat (SCR20) is a hotspot for mutations that may be single amino acid substitutions, premature stops or more complicated changes; many of these mutations are strong risk factors for another disease of complement dysregulation, atypical haemolytic uremic syndrome (aHUS), which is typified by thrombosis, haemolysis and renal injury136,137. Onset of aHUS typically occurs in childhood or young adulthood and the frequency and severity of episodes is highly variable, with some patients having an indolent course requiring no therapy and others rapidly progressing to end-stage renal failure. All of the mutations in the SCR20 domain of FH that are associated with aHUS disrupt the capacity of FH to bind to and regulate complement on cells and other surfaces; regulation in the fluid phase is unaffected. Numerous other complement protein mutations have been described and shown to influence the risk of aHUS and/or other diseases in which complement plays a part. Many of these are coding mutations that change the amino acid sequence and affect protein activity, others are non-coding mutations and probably affect the biosynthesis and expression of the protein; some mutations trigger FH-specific autoantibody production138. An understanding of the influence of polymorphisms and mutations in complement proteins on complement activity in a particular disease is essential for the intelligent design and selection of therapeutic approaches. The figure summarises the effect of gain-of-function and loss-of-function polymorphisms and mutations in complement proteins and regulators that alter the risk of disease.
Box 2: The development of eculizumab for the treatment of paroxysmal nocturnal haemoglobinuria.
The first description of functional blocking of complement C5 with a monoclonal antibody (mAb) dates back almost 30 years; a C5-specific mAb, BB5.1, generated in C5-deficient mice, blocked haemolysis in mouse serum139. BB5.1 provided a powerful proof of principle for inhibition of the membrane attack complex (MAC) that was effective in numerous mouse models140; BB5.1 also provided a rationale for the development of blocking mAbs specific to human C5. One such mAb, 5G1.1, was selected by Alexion, humanized and further manipulated to generate Fab (antigen-binding) and scFv (single-chain variable) fragments141,142. By 2002, humanized 5G1.1, termed eculizumab, was in early clinical trials in a range of joint, kidney and skin conditions143; however, its remarkable efficacy in paroxysmal nocturnal haemoglobinuria (PNH) focused efforts in that area144. PNH is a clonal haematological disorder that involves chronic intravascular haemolysis, with complement lysis of glycosylphosphatidylinositol (GPI)-anchor-deficient erythrocytes as its principal feature. Patients with PNH are anaemic with resultant lethargy and at risk of thrombosis. Prior to the use of eculizumab, treatment involved frequent blood transfusions and symptomatic support. Eculizumab transformed the outlook for patients with PNH, preventing haemolysis, reversing anaemia and removing transfusion dependence in most patients, thereby improving quality of life145. Eculizumab therapy is effective over the long term (>5 years), and substantially reduces mortality146. The only other disease for which eculizumab is currently approved by the US Food and Drug Administration (FDA) is atypical haemolytic uremic syndrome (aHUS). As with PNH, there were no specific therapies for this potentially catastrophic disease and the evidence implicating complement was overwhelming. In patients with aHUS, eculizumab treatment increased platelet number and improved renal function over the course of 1 year and some patients were no longer dependent on dialysis147. Numerous other clinical trials in various conditions are in progress, and these are summarized in Table 2. A major concern of governing bodies in the United Kingdom and elsewhere is cost; at the time of writing, standard PNH maintenance therapy (900 mg eculizumab delivered intravenously every 2 weeks) costs approximately UK£340,000 per patient per year; therefore, even for a rare disease with only ~200 cases in the United Kingdom, the estimated cost to the National Health Service (NHS) is £58 million per year. A concise and up-to-date summary of the clinical data can be found on the electronic Medicines Compendium website (see Further information). It should be noted that an eculizumab biosimilar is already being developed by Epirus Biopharmaceuticals.
Table 2. Eculizumab in the clinic and in development*.
| Disease | Stage | Outcome | Clinical trials | Refs |
|---|---|---|---|---|
| PNH | Marketed | Major effect on patient survival and QoL in this rare disease | N/A | 24,143,144,145 |
| aHUS | Marketed | Major effect on patient survival and QoL in this rare disease | N/A | 25,147 |
| AMD (geographic atrophy) | Phase II completed | Systemic therapy well tolerated, no effect on disease progression | NCT00935883 (COMPLETE study) | 212 |
| Neuromyelitis optica | Phase II completed | Significantly reduced attack frequency; stabilized neurological disability | NCT00904826 | 169 |
| Phase III underway | NCT01892345 (PREVENT study) | |||
| Complement injury in kidney transplant | Phase I ongoing | N/A | NCT01327573 | – |
| Delayed kidney graft rejection | Phase II recruiting | N/A (due to complete in 2016) | NCT01919346 | – |
| Phase II recruiting | N/A (deceased donors) | NCT01403389 | ||
| Antibody-mediated kidney graft rejection | Phase II ongoing | N/A | NCT01895127 | 213 |
| Phase I/II terminated | N/A (ABO blood group incompatible) | NCT01095887 | ||
| Kidney graft reperfusion injury | Phase II recruiting | N/A | NCT01756508 | – |
| Primary MPGN | Phase II ongoing | N/A (due to complete in 2016) | NCT02093533 (EAGLE trial) | – |
| Kidney transplant in CAPS patients | Phase II recruiting | N/A | NCT01029587 | – |
| Dense deposit disease and C3 nephropathy | Phase I ongoing | N/A | NCT01221181 | – |
| Refractory myasthenia gravis | Phase II completed | Improved myasthenia score | NCT00727194 | 168 |
| Phase III recruiting | N/A (due to complete in 2016) | NCT01997229 (REGAIN study) | ||
| Guillain–Barré syndrome | Phase II recruiting | N/A | NCT02029378 (ICA–GBS study) | – |
| Cold agglutinin disease | Phase II completed | No results posted | NCT01303952 (DECADE study) | – |
| Severe thrombocytopenia | Phase 0 recruiting | Will test the effect on survival of transfused platelets | NCT02298933 | – |
| Shiga toxin positive HUS | Phase II/III completed | No results posted | NCT01410916 | – |
| Phase III recruiting | N/A (due to complete in 2017) | NCT02205541 (ECULISHU study) | ||
| Cardiac transplant rejection | Phase IV recruiting | N/A | NCT02013037 | – |
| Mild allergic asthma | Phase II completed | No results posted | NCT00485576 | – |
| Dermatomyositis | Phase II completed | No results posted | NCT00005571 | – |
The Table lists diseases and conditions in which eculizumab is already in reported clinical trials. Where trials have completed, the outcomes (when available) are listed. Trials for applications already in clinic, withdrawn trials, observational studies and case reports are not included. The unique identifier (NCT) number and, where appropriate, references to published trials are included. aHUS, atypical haemolytic uremic syndrome; AMD, age-related macular degeneration; CAPS, catastrophic antiphospholipid antibody syndrome; MPGN, membranoproliferative glomerulonephropathy; N/A, not available; PNH, paroxysmal nocturnal haemoglobinuria; QoL, quality of life. Data compiled from ClinicalTrials.gov and other sources.
Physiological roles of complement
Complement is a key part of innate immunity because it interacts with many other processes; for any therapy that seeks to block or modulate complement, the potential effects on these physiological roles must be considered to avoid iatrogenic injury.
Defence against infection. Patients deficient in complement proteins usually present with recurrent bacterial infections. C3 is a crucial source of opsonins, which label bacteria for removal by phagocytes; deficiency of C3 is rare and always marked by severe recurrent infections3. Patients deficient in classical pathway or lectin pathway proteins (C1, C4, mannose-binding lectin (MBL), MBL-associated serine protease 1 (MASP1) and MASP2) may also present with recurrent bacterial infections, and deficiencies of alternative pathway components or the positive regulator properdin also predispose to infection, although these deficiencies are associated with a strong bias towards Gram-negative bacterial infections. Deficiencies in terminal pathway proteins specifically predispose to infections with Neisseria species, typically those causing meningococcal meningitis or sepsis, as a consequence of the peculiar sensitivity of these pathogens to lysis by the MAC.
Protection against immune complexes. Immune complexes comprise antibodies bound to target antigens (cell debris) in a multi-molecular complex. Unhindered, immune complexes grow by acquisition and aggregation, eventually becoming large, insoluble aggregates that lodge in capillary beds and trigger inflammation and tissue damage. In healthy individuals, immune complexes activate the classical pathway and become coated with C1 and fragments of C4 and C3; these proteins mask antigens in the immune complex and disrupt the lattice, thereby limiting growth of the aggregates and simultaneously providing ligands for the receptors (complement receptor type 1 (CR1)) on erythrocytes, which sequester immune complexes, and for the receptors (CR1, CR3 and CR4) on phagocytic cells, which engulf and destroy immune complexes11. Patients with deficiencies in classical pathway components (in particular C1 and C4) usually present with an immune complex disease that closely resembles systemic lupus erythematosus (SLE), itself a disease of complement dysregulation12.
Priming adaptive immunity. The role of complement in stimulating the adaptive immune response was first demonstrated in complement-depleted mice 40 years ago13. Antigens coated with complement activation fragments provoke markedly greater antibody responses than do those that are uncoated; the C3dg fragment is crucial to this effect, ligating CR2 on B cells to deliver a powerful co-stimulatory signal that reduces the threshold for B cell receptor triggering and increases the amplitude of the response14. The smaller fragments of complement activation, C3a and C5a, enhance the ability of antigen presenting cells (APCs) to present antigens and stimulate T cell proliferation; together, complement products in the inflammatory microenvironment markedly influence T cell activation and the balance between effector and regulatory T cells15.
Other physiological roles. Many other roles of complement continue to emerge, including directing haematopoietic stem cells to marrow niches, regulating triglyceride uptake and storage in adipose tissue and the regeneration of damaged tissue after injury and during wound healing16. Each role in itself could warrant a review; therefore, here we merely highlight that these many roles may be affected by complement inhibitors.
Anti-complement drugs. Anti-complement drugs have the potential to affect each and all of the physiological roles discussed above (Fig. 1). It is inevitable that a drug that blocks any of the complement pathways will increase the risk of infections, either non-selectively or for certain groups of organisms. Any drug that stops activation of the classical pathway will affect the clearance of immune complexes and apoptotic cells. Inhibition of the activation pathways may disrupt an individual's capacity to mount an adaptive immune response, although therapeutic inhibition later in life is less detrimental as adaptive immunity is established and developed in older individuals. Anti-complement drugs may also disrupt normal lipid metabolism or interfere with the healing and resolution of injuries. All of these potentially damaging effects need to be considered when deciding whether and when to use an anti-complement drug and which part of the system to target. With appropriate prophylactic measures such as immunization and antibiotic therapy, and with careful consideration of the target, most of these effects can be managed.
Avoiding toxicity
A limiting factor in the use of anti-complement drugs has been an understandable concern regarding the harmful consequences of blocking the important physiological effects of complement described above, which are graphically illustrated in individuals with complement deficiencies. Deficiencies in the classical pathway prevent efficient clearance of immune complexes and apoptotic cells, leading to a high penetrance of lupus (93% penetrance in individuals with a C1q deficiency and 75% penetrance in individuals with a C4 deficiency)17, whereas individuals who are deficient in C3 or components of the alternative pathway or the terminal pathway have recurrent infections that can be life-threatening18. Although these risks can be managed with good clinical care, a guiding principle for any drug should be to cause as little disruption of physiological roles as possible, particularly if treatment is to be continued in the long term.
Researchers have devised several strategies to inhibit complement without risking iatrogenic injury: complement can be inhibited late in the pathway, wherein the only infection risk is from Neisseria species; complement can be inhibited transiently, which is sufficient for acute conditions, with minimal risk of immune complex disease; or therapy can be delivered directly to disease sites such that systemic inhibition is avoided. These strategies have spawned drugs that have entered the clinic, in some instances with dramatic effect. The C5-specific monoclonal antibody (mAb) eculizumab prevents MAC formation; opsonization is thus unaffected by eculizumab treatment and the risk of meningococcal infection can be mitigated through vaccination and antibiotics. The C3 blocker compstatin and an antibody antigen-binding fragment (Fab) directed against Factor D (FD), administered intravitreally, have undergone clinical trials for AMD19,20. The compstatin derivative APL-2 (Apellis Pharmaceuticals; a long half-life form) has just entered Phase I trials for add-on to standard-of-care in PNH; it will be interesting to see how systemic administration affects the risk of infection. Chimeric agents, comprising a complement ligand-targeting modality linked to a complement inhibitor, localize activity at the disease site. This concept has shown promise in murine models and has been developed as a therapy for PNH21,22. For any anti-complement drug, short-term inhibition for acute indications in a hospital setting is unlikely to incur serious risk of immune complex disease, and infection risk can be managed with tailored antibiotic cover.
Specific issues for complement-targeted drugs
Dosing. Experience gained from the first few drugs entering the clinic has added knowledge and identified issues that are specific to complement-targeted therapies. One major issue is dosing; most complement proteins are abundant in plasma and turn over rapidly, so adequate dosing of an inhibitor can be challenging. For example, C3 is present in plasma at ~1 g per l, and ~10–20% of total plasma C3 (~300–1000 mg) is consumed and replaced daily simply through the normal tickover process23. It is therefore obvious that large doses of complement-targeting drugs and frequent administration will be needed to block complement at the level of C3. Dosing is an issue for current drugs; the Cinryze (a plasma-derived C1 inhibitor; Shire Pharmaceuticals) dose for prophylaxis in hereditary angioedema (HAE) is 1,000 units (100–250 mg) delivered intravenously every 3 days; the eculizumab maintenance dose for adults with PNH is 900 mg every 2 weeks24,25, whereas in atypical haemolytic uremic syndrome (aHUS) the maintenance dose is 1,200 mg every 2 weeks. These huge and frequent doses contrast starkly with agents targeting cytokines, which are released de novo in disease and at much lower levels; for example, the tumour necrosis factor (TNF)-specific mAb adalimumab (Humira; AbbVie) is effective in rheumatoid arthritis at a dose of 40 mg every 2 weeks26. The plasma C5 concentration is ~80 mg per l and the turnover rate is ~60 hours27; as a consequence, even with such high doses, breakthrough activity can occur in some patients treated with eculizumab and monitoring of complement activation in plasma is required28. Alexion have recently launched two new agents that are in Phase I trials (ALXN1210 and ALXN5500; 'next-generation' eculizumab molecules) that were probably developed to overcome some of the issues with eculizumab. C5 is not limiting in the complement cascade; therefore, inhibition at the C5 stage requires near-complete blockade or depletion of C5; these threshold kinetics necessitate large doses to achieve an effect. Approaches to circumvent the problem of high levels of protein turnover have been described, using drugs that only bind to activated forms of complement proteins, which are present at much lower concentrations in plasma29,30; these approaches show potential but have not been tested in humans.
Apart from the problems arising from the sheer abundance of complement proteins, dosing is complicated because plasma complement levels vary widely in the population and because many are acute phase reactants, with synthesis increasing markedly in inflammation, which sometimes causes plasma levels to rise even in the face of increased consumption31. The plasma concentration of FD is relatively low (~5 mg per l) but it is turned over extremely rapidly (~1.33 mg per kg per day)32; it can therefore be predicted that large amounts of a drug will be needed to effectively inhibit FD in vivo. Studies in non-human primates support this — a 5 mg per kg intravenous dose of a FD-specific Fab (lampalizumab, formerly called FCFD4514S, Roche/Genentech) inhibited the alternative complement pathway for only 3 hours; in that time the plasma levels of FD increased 10-fold, which is probably reflective of the retention of FD–lampalizumab complexes in the plasma33. Lampalizumab, administered intravitreally, is currently in Phase III trials in AMD.
Owing to their chemical nature, small-molecule anti-complement agents tend to have short half-lives in vivo. This is not necessarily a limitation, as full coverage can often be achieved through repeated dosing in situations of long-term therapy. Daily subcutaneous dosing is manageable and is likely to be acceptable for patients with life-threatening diseases; oral bioavailability can often be achieved with these agents, which is a key advantage. In some conditions — such as traumatic brain injury, myocardial infarction, transplantation, stroke and other ischaemia–reperfusion injuries — transient complement inhibition for a few hours may be sufficient to prevent tissue damage. In these cases, small molecules have an added benefit in that once dosing is ceased, rapid clearance leads to speedy recovery of complement activity, restoring its infection-fighting and tissue-repairing properties. When developing small-molecule drugs for specific targets, their mode of action is crucial and target tractability becomes a key consideration; in some instances, the small size of the drugs may become a limitation, for example in blocking protein–protein interactions (see below). When a drug with a longer half-life is needed, agents such as antibodies come to the fore. Alternatively, small molecules may be modified to increase their mass by pegylation or by coupling to domains that extend their half-life in vivo: for example, albumin-binding or antibody crystallisable fragment (Fc) domains34,35.
In some diseases, complete inhibition of complement may not be needed for efficacy; agents such as soluble CR1 (sCR1; also known as TP10, Celldex Therapeutics) that downmodulate the complement enzymes through dynamic activities (such as accelerating their decay or increasing cofactor availability) can mediate dose-dependent partial inhibition of complement rather than the sharp threshold effect typical of blocking antibodies36,37. Such agents may enable therapy at scalable doses and in a safe and effective manner; however, the fact that even large biological agents such as sCR1 have relatively short half-lives will affect the choice of indication.
Getting to where the action is. Many pharmaceutical companies are now focused on the development and manufacture of biologics because of their historical success, good economic return and low rate of attrition. As a consequence, most anti-complement agents developed to date are biologics — either antibodies or recombinant proteins — that deliver inhibitory activity. Essential to all drugs is their ability to get to the sites of disease; some complement-mediated diseases manifest in the vasculature (for example, PNH and vasculitis), whereas in other diseases (such as AMD, neuromyelitis optica and arthritis) complement attacks tissues at sites remote from systemic circulation. The ability of systemically-administered drugs to diffuse to the site of tissue damage and inflammation will thus be essential for treatment success. The liver produces most of the complement components found in the circulation, and if diffusion of circulating components to disease sites drives inflammation and disease then systemic depletion or blocking of specific complement components may ameliorate the pathology. However, locally produced complement is known to have key roles in some indications. For example, in renal allograft rejection, locally produced and activated C3 drives the T cell response and immune rejection38; in this case, systemic anti-C3 therapy will be ineffective unless the drug gets into the tissues. Similar challenges exist in organs protected by tight barriers, such as the nervous system and the eye. In some diseases these barriers become damaged and leaky with disease progression, but it is likely that complement activation contributes to the early, pre-inflammatory stages of eye and brain diseases, so to be of value complement-inhibiting drugs are required in the relevant organ early in the disease when the barriers are still intact. Assuming that they have good bioavailability, small-molecule drugs may have advantages over biologics in these contexts because of their higher tissue penetrance; however, of the numerous small-molecule anti-complement agents that have been developed and tested in humans, none has yet progressed to the market39–41.
Getting in the way. Complement is a good target for biologics because the cascade involves multiple protein–protein interactions and conformational changes, exposing large surfaces that lend themselves to large blocking drugs (Box 3). By contrast, for a small molecule to block a protein–protein interaction it must target crucial sites and specific surface features — for example, charged or hydrophobic pockets42. The wealth of structural information now available in the field, including snapshots of convertase enzymes and MAC precursor complexes captured in active conformations, unmasks the precise nature of these protein–protein interactions and identifies sites that are key to the interaction that can be targeted with small molecules or biologicals using structure-based drug design43,44. Alternatively, the expansive small-molecule libraries that are now ubiquitous in pharmaceutical companies have been successfully screened to reveal agents that bind to and inhibit interactions within the cascade through competitive or allosteric mechanisms45,46. Small peptides selected from such libraries for binding components or inhibiting complexes are now numerous in the literature; for example, the cyclic peptide compstatin, which blocks C3 from binding to the convertase, has already entered clinical trials45,47.
Target validation in models. Validation in rodent models of disease can be challenging or even misleading, and care needs to be taken to ensure that the disease mechanism in the chosen model is the same as that in the human disease. To illustrate the differences in complement systems between mice and humans, consider the receptors for C3 fragments. In humans, CR1 and CR2 are encoded by separate genes and have different roles: CR2 binds to C3dg and enhances humoral immunity whereas CR1 acts as a cofactor for the FI-mediated cleavage of C3b and has a key role in transporting immune complexes48. In mice, CR1 and CR2 are generated from the splicing of one gene and have limited expression; the main cofactor for C3b cleavage and generation of C3dg in the mouse is CR1-related gene/protein Y (CRRY), a broadly expressed protein absent in humans49. Therefore, mice and humans respond to C3 fragments through different receptors and with different outcomes that will probably alter the effects of C3-targeted drugs. Nevertheless, in many models, disease mechanism and response to complement inhibition translate well and proof-of-principle is readily obtained. With current efforts to reduce animal use in scientific research, initiatives are emerging to better validate drug targets at an early stage of development without the use of animals, for example, using technologies such as microfluidic organs-on-chips50.
Box 3: Inhibiting protein–protein interactions.
Blocking protein–protein interactions offers numerous targets for drugs in the complement system; approaches may involve blocking the formation of an essential complex or receptor–ligand interaction, preventing a key conformational change or inhibiting a crucial enzymatic cleavage. Such activities underpin many of the current and evolving therapeutics, particularly the monoclonal antibody (mAb)-based agents. Several of the mAbs that target C3 convertase work in this way; either they prevent the complex from forming at all or they lock it in an inactive conformation. Similarly, the Staphylococcus aureus-derived inhibitor SCIN (staphylococcal complement inhibitor) holds the convertase in an inactive state148. The C5-specific mAb eculizumab binds to C5 remote from the cleavage site, at a site in the α-chain that prevents C5 from binding to C5 convertase24,25,149. Human experimental proof was recently provided: a C5 polymorphism, present in ~3.5% of the Japanese population, caused a single residue change (R885H) in the putative eculizumab binding site and rendered the protein refractory to inhibition150. In R885H heterozygotes treated with eculizumab, half of the circulating C5 remained active and paroxysmal nocturnal haemoglobinuria (PNH) therapy was ineffective.
Blocking protein–protein interactions using small molecules has attracted less interest because of the perceived difficulties of successfully interfering with large protein–protein interfaces151; for example, the footprint of the Factor H regulatory domains on the C3 convertase extends over an enormous 4,500 Å2 (Ref. 152). Nevertheless, some success has been achieved with peptides or other small-molecule inhibitors; the best example is that of compstatin — a 13 amino acid cyclic peptide that inhibits C3 — which, over the course of almost two decades, has been modified into a highly potent high-affinity inhibitor of C3 cleavage19,47,153,154,155. Compstatin binds to native C3 and prevents its interaction with the C3 convertase, thus blocking further complement activation. In vivo testing of compstatin has been limited by its specificity for human and primate C3; in primates, compstatin reduced drusen load and inhibited disease in an age-related macular degeneration (AMD) model19. Alcon (a Novartis company) has taken a compstatin analogue, POT-4 (al-78898a), through Phase I trials; Phase II trials in AMD, which started in 2010, are yet to report any results (see the Alcon website in Further information)156. For their compstatin analogue, AMY-101, Amyndas has gained European Medicines Agency (EMA) and US Food and Drug Administration (FDA) Orphan Drug status for use in PNH. Apellis Pharmaceuticals are developing a compstatin derivative in an inhaled short-acting form (APL-1) for the treatment of asthma and chronic obstructive pulmonary disease (COPD) (currently in Phase I as a disease modifying therapy for COPD). This derivative is also being developed in a long-acting form (APL-2) for the treatment of haemolytic disorders and AMD; APL-2 is currently in Phase I for PNH as an add-on to eculizumab therapy (ClinicalTrials.gov identifier: NCT02264639), and a Phase I trial has recently started for APL-2 as an intraocular therapy for AMD (NCT02461771).
What does the current toolbox contain?
Before describing specific tools, we first provide some guiding principles and suggestions for the classification of anti-complement agents. There are several discrete ways in which an agent might act to influence complement (Fig. 1). An agent might inhibit progression in the activation or terminal pathways, act as an antagonist of an activation product, or cause activation and consumption of complement. Another broad distinction can be drawn between those drugs that inhibit complement systemically and those that are directed to specific sites of pathology to localize their inhibitory activity. Anti-complement drugs can also be divided into categories based upon their molecular characteristics: purified or recombinant forms of naturally occurring human regulators; modified protein copies or mimics of natural regulators; copies of pathogen-derived regulators; mAbs or fragments of mAbs; naturally occurring or synthetic small-molecule inhibitors; or antisense or similar nucleic acid constructs. Finally, anti-complement agents can be grouped according to the part of the system they target, a convention that we follow in this Review.
Targeting the binding and assembly of the initiating complex. Initiation of classical or lectin pathway activation involves the recognition of an activating surface; in the classical pathway this activation occurs through a surface-bound antibody that ligates C1q, whereas in the lectin pathway this occurs via specific sugars on the surface that bind to MBL or collectins (Fig. 1). Agents that block these events would switch off the respective activation pathways at the very first step, an attractive prospect given the amplifying nature of the complement system51. A potential classical pathway inhibitor might block C1q-binding sites on the antibody or the antibody-binding sites on C1q. A lectin pathway inhibitor might mask surface MBL-binding sugars or the sugar-binding sites on MBL. In either case, the agent would need to bind the target with higher affinity than the physiological ligand to compete effectively. Alternatively, an agent could disrupt or prevent the assembly of the multi-molecular complex, which comprises the recognition unit (C1q and MBL in the classical and lectin pathways, respectively) and the associated enzymes (C1r and C1s in the classical pathway and MASP1 and MASP2 in the lectin pathway) that are essential for pathway activation52. Both complexes circulate preassembled in plasma, so any agent targeting the complex would need to dislodge the bound enzyme to be effective; surprisingly, the C1s- and MASP2-targeting mAbs from True North and Omeros, respectively (described below), both achieve this effect. Another approach to inhibiting activation of the complexes would be to develop a molecular lock that restricts the conformational changes in C1q or MBL that occur upon ligand binding and are essential for activation of the associated enzymes. Such approaches have been used successfully to develop small-molecule inhibitors of chemokine receptor activation53,54, but have not yet been reported for C1 or the MBL–MASP complex.
Several C1q function-blocking mAbs have been reported and were recently shown to be effective in a murine model of the antibody-mediated demyelinating disease, neuromyelitis optica55. Numerous peptide blockers of antibody–C1q interactions have been described, derived from the sequences of C1q binding sites in the antibody, C1q receptors or C1q itself, or selected from libraries. One such peptide blocker, termed peptide 2J, is a powerful inhibitor of the classical activation pathway in multiple species56; however, there have been no further reports of this agent. Other peptides have been described that interfere with the binding of C1r and C1s in the C1 complex, but none has progressed beyond in vitro studies. The binding of MBL to specific sugar residues on surfaces presents a potential drug target, but no agents acting in this manner are reported. A recent publication describes a virus-derived peptide, peptide inhibitor of complement C1 (PIC1), that binds to the collagenous regions in both C1q and MBL and inhibits both the lectin and classical pathways in vitro and in vivo in rodents57.
Targeting the enzymes of the initiating complexes. The enzymatic activity of the classical or lectin pathway-initiating complexes is provided by the serine proteases C1s and MASP2, respectively. In the C1 complex, immunoglobulin binding induces conformational changes in C1q, triggering auto-activation of C1r that in turn cleaves and activates C1s in the same complex. The precise steps involved in activation of the MBL–MASP complex are less clear but probably involve a similar multi-step process58. The serine protease enzymes in these complexes are potential targets for pharmacological inhibition. Both the pharmaceutical industry and nature itself have invested heavily in the development of serine protease inhibitors (SPIs), and numerous SPI drugs have been made, of differing specificities and compositions, to target the myriad serine proteases essential for digestion, haemostasis, immunity, reproduction and many other physiological and pathological processes. The biggest problem in designing an SPI drug to inhibit a particular protease is specificity — almost all SPI drugs have some 'off-target' effects that can limit their use. Even nature struggles — the sole natural inhibitor of C1r and C1s is an SPI, C1 inhibitor (C1INH; also known as SERPING1), but C1INH also controls MASPs and proteases in the coagulation and kinin systems. Indeed, deficiency of C1INH leads to dysregulation in all these proteolytic cascades that, in aggregate, cause the disease HAE59,60. C1INH removes activated C1r and C1s from C1q to form a stable complex in which C1INH itself is cleaved and inactivated.
C1INH has a long history of use as a drug; indeed, it can claim to be the 'first-in-man' complement drug and the first of the natural inhibitors to be recognized as a potential therapeutic. More than 40 years ago, patients with HAE were shown to be deficient in C1INH and to respond to plasma replacement, provoking efforts to purify C1INH for treatment of acute attacks59,61. Remarkable successes in these early studies rapidly led to the adoption of plasma-derived C1INH as the standard of care for acute episodes in HAE. The obvious advantage of this approach is that a plasma-derived molecule should be low risk (once infectious agents are eliminated) and non-immunogenic. The widespread availability of industrial-scale protein-fractionation facilities — established for the production of immunoglobulins, albumin and other plasma products — made this a viable approach for purifying C1INH. Unfortunately, concerns about viral transmission in plasma-derived proteins have led to its removal from the clinic in several countries, including the United States. Recent methodology improvements and new approaches to production of C1INH have changed this unsatisfactory situation; two ultra-pure plasma-derived C1INH products, Cinryze and Berinert (CSL Behring) are now approved by the US Food and Drug Administration (FDA) and other regulatory agencies. Some companies invested in better methods of purifying plasma C1INH, whereas others set about making it. A full-length recombinant C1INH was first reported a decade ago. This agent, termed Ruconest (Salix Pharmaceuticals), has been approved for therapy of acute attacks in Europe and, very recently (July 2014), also in the United States.
Early administration of C1INH reduces the duration and severity of acute attacks, findings confirmed in the large IMPACT 2 study of over 1,000 attacks62. Its use in prophylaxis is more controversial; a subset of HAE patients have frequent and severe attacks despite therapy with danazol (an anabolic steroid that increases C1INH synthesis) and/or ε-aminocaproic acid (a protease inhibitor) or cannot tolerate these agents. Regular intravenous administration of C1INH (every 3–7 days) to restore plasma levels is an effective therapy in this group63,64; however, in the United States it is now becoming accepted practice to treat all HAE patients in this manner, a practice limited elsewhere by cost and lack of evidence base65. Berinert has been approved for on-demand, patient-administered therapy in the United States and Europe.
Given its long history and known safety and efficacy in HAE, it is surprising that C1INH has not been more widely used in other conditions characterized by complement dysregulation. There are a number of published studies, mostly small, demonstrating that administration of C1INH reduces mortality in sepsis, and in some studies the effect is startling66,67. Other studies have reported positive effects of C1INH in myocardial infarction and in ischaemia–reperfusion injuries in both animal models and humans, but no large-scale clinical trials in these other applications have been published68,69.
Several broad-spectrum protease inhibitors already in clinical use have C1r, C1s and MASPs among their targets; perhaps the best example of these is nafamostat mesilate (also known as Futhan or FUT-175), a small-molecule protease inhibitor used to treat disseminated intravascular coagulation and acute pancreatitis that affects multiple plasma protease systems39. However, the many off-target effects and resultant toxicity of nafamostat mesilate make it a poor candidate for regulating complement in vivo. Other small-molecule inhibitors of C1s of varying degrees of specificity and strength have been described, including C1s-INH-248 (BASF Pharma) and BCX-1470 (BioCryst Pharmaceuticals), both no longer in development, and recently a family of biphenylsulphonyl thiophene derivatives that, when pegylated, displayed strong C1s inhibition and promising pharmacokinetics70. Modern methods of structure-based design offer the prospect of much more selective small-molecule inhibitors of the initiating complex enzymes, as demonstrated for other proteases71. Indeed, the 'directed evolution' of a sunflower-derived peptide, trypsin inhibitor 1, created specific inhibitors of MASP1 and MASP2 that await testing in vivo72.
An inhibitory mAb against C1r and C1s or MASPs might address the specificity issue that limits the clinical use of small-molecule agents. A mAb against C1s, TNT003 (True North Therapeutics), prevented erythrocyte haemolysis in an ex vivo study of autoimmune haemolytic anaemia41; a humanized analogue of this mAb, TNT009 (True North Therapeutics), is being fast-tracked to clinical trials in this disease. A recombinant mAb Fab was recently described that bound C1s with nanomolar affinity and potently inhibited classical pathway activation, but so far this has only been tested in vitro73. A MASP2-blocking human mAb (OMS721), which is an effective and long-lived (up to a week) lectin-pathway inhibitor that is delivered intravenously or subcutaneously, was recently awarded US FDA Orphan Drug status for use in aHUS and other thrombotic angiopathies and is currently in Phase II/III trials (NCT02222545). A recent (August 2015) Omeros press release described positive outcomes in aHUS from this trial.
Targeting the C3 convertases. The C3-cleaving enzymes of the classical, lectin and alternative pathways, C4b2a and C3bBb, are assembled in quite different ways despite their functional and structural similarities (Fig. 2). In the classical and lectin pathways, C4 is cleaved by activated C1s (or MASP2) in the surface-bound C1 (or MBL–MASP) complex; the large C4b fragment binds to adjacent surfaces via a nucleophilic attack on a thioester group, captures C2 and presents it for cleavage by C1s or MASP2 as above. The C4b2a complex is the C3 convertase of the classical and lectin pathways. C2a is a serine protease that cleaves C3 to form C3a and the opsonin C3b; C2a can also cleave C5 (when properly presented) to form C5a and C5b. These two complement proteins are the sole substrates of the C2a serine protease. Assembly of the alternative pathway C3 convertase requires C3b, generated through classical and lectin pathway activation or from tickover activation on surfaces. C3b binds to FB, triggering major conformational changes that expose FB to cleavage by FD, a unique plasma serine protease that has FB as its sole substrate. The C3bBb complex is the C3 convertase used in the alternative pathway; the Bb fragment, generated from FB cleavage, is a serine protease that cleaves C3 and C5 in precisely the same manner as C2a in the classical pathway. The key amplifying role of the alternative pathway makes it a very attractive source of drug targets that can be targeted in a variety of ways (Fig. 2).
Figure 2. Controlling the alternative-pathway amplification loop.
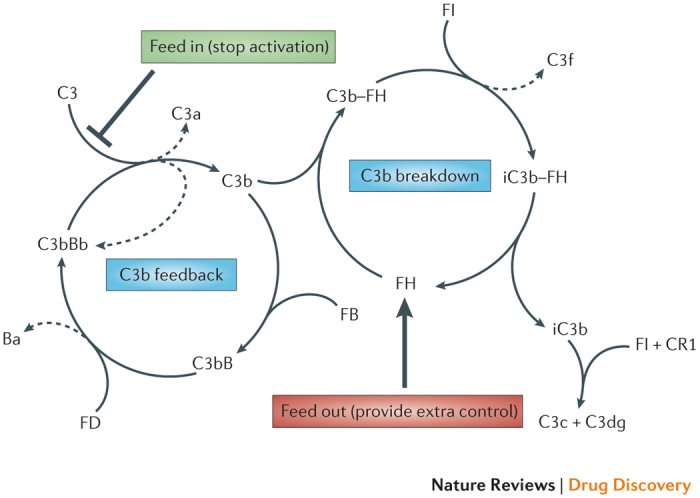
The amplification (C3b feedback) loop is a positive-feedback cycle that consumes complement C3 to generate more enzyme and activation products; if unregulated, it cycles until all available C3 is consumed. Tight regulation is provided in the plasma by enzymes and cofactors that remove the C3 fragment C3b from the feedback cycle for breakdown into smaller fragments. As a consequence, the C3b feedback cycle normally operates at a very low rate (tickover). The balance between activation and regulation is disturbed in disease; a healthy balance can be restored by providing extra control (for example, increasing regulation, such as that provided by the complement regulatory protein Factor H (FH), thereby increasing 'feed out' from the amplification loop) or by preventing the formation of C3b in the feedback cycle (for example, by blocking convertase enzyme, thereby decreasing 'feed in' to the amplification loop). Agents that target amplification of complement can have major therapeutic effects. Ba, non-catalytic fragment of FB; C3b–FH, complex between C3b and FH; CR1, complement receptor type 1; iC3b–FH, complex between inactive C3b and FH. Adapted with permission from Ref. 157, Elsevier.
It is obvious from the above description that the C3 convertases present a dizzying array of opportunities for regulation. Agents might target the assembly of one or more of the complexes, preventing complex formation and/or causing complexes to break up. They might target one or more of the serine proteases responsible for formation of the convertase (C1r, C1s, MASP2 or FD), convertase activity (C2a or Bb) or convertase regulation (FI). Alternatively, they might target the substrates of these enzymes, C3 or C5, by blocking binding to or cleavage by the convertase or acting as molecular locks to prevent conformational changes essential for activation. Agents might also target active fragments (C3a, C5a, C3b or C5b) to block their downstream effects. Indeed, nature has adopted some of these strategies to keep complement in check. The convertases are naturally labile and break up in a matter of minutes; regulatory proteins that accelerate this decay exist both in plasma and on membranes, and they function by binding to the complex and displacing the enzyme. Some of these regulatory proteins remain bound after decay and act as cofactors for the further cleavage and irreversible inactivation of C3b or C4b by the plasma serine protease FI.
Using the endogenous C3 convertase regulators as drugs or leads. The plasma regulators of the C3 convertases were obvious targets for drug development given the success of C1INH. To date, none of the other plasma regulators has entered the clinic despite a large amount of research and preclinical activity. In vitro studies more than 35 years ago showed that modest (around 10% above baseline) increases in the concentrations of the complement regulators FH and FI markedly reduced plasma alternative-pathway activity, provoking the suggestion that augmentation of these proteins might be of therapeutic benefit74; however, large (and probably frequent) doses will be required to substantially alter the levels of these abundant proteins. Despite this, there has been considerable recent interest in using plasma-derived FH as a therapy, particularly in those diseases strongly linked to FH polymorphisms and mutations. Deficiency of FH is rare and always associated with severe C3 dysregulation in plasma or on cell surfaces that leads to a range of renal diseases, including C3 glomerulopathy and aHUS; FH deficiency has been successfully treated with plasma exchange, strongly suggesting that FH would be an effective therapy in these diseases75. Indeed, in FH-deficient mice, therapy with human FH reversed renal injury76. AMD is strongly linked with the FH Y402H polymorphism, and this has provoked intense interest in giving at-risk individuals the protective Y402 form of FH to prevent or treat AMD. Several companies established programmes to produce plasma-derived or recombinant FH with this as the primary aim, but none of these programmes survive. Of note, some evidence from mouse models and human studies suggest that FH would need to be administered intra-ocularly in AMD to control complement dysregulation locally and alter the course of disease77,78.
In one study, instead of expressing full-length FH for use in therapy, this large molecule was stripped down to its key functional units and re-assembled to create a minimized FH, mini FH, which comprised four amino-terminal, complement-regulatory short consensus repeats and two carboxy-terminal, surface-binding short consensus repeats joined by a short linker. Mini FH proved to be an effective complement inhibitor in the fluid phase and on surfaces in multiple in vitro assays79,80, and it is now in preclinical development for therapy of PNH (AMY-201; see the Amyndas website). Other approaches aimed at targeting FH activity to surfaces are described in later sections.
C4BP, the fluid-phase regulator of the classical pathway C3 convertase, is a complex, oligomeric molecule that to date has received little attention in terms of therapeutic applications. C4BP deficiencies are extremely rare, restricted to a single case report81. In mouse models of arthritis, administration of plasma-derived C4BP reduced severity of disease82, but no human studies have been described.
FI is the key enzyme for regulation of the C3 convertases and deficiency causes complement consumption and consequent susceptibility to bacterial infections. Individuals with an FI deficiency present with repeated infections that may be life-threatening, and even heterozygotes may have an increased infection risk83. Plasma therapy to replace FI would, by analogy with FH deficiency, be expected to help, although a single small study found no clinical benefit84. A recent report showed that FI supplementation reduced complement activity in sera from individuals with more active complotypes, provoking the suggestion that FI therapy would reduce the risk of developing diseases linked to complement dysregulation85.
Properdin is a plasma protein that binds to and stabilizes the alternative pathway convertase; in other words, it acts as a positive regulator86. Properdin may also act as a pattern-recognition molecule, binding targets and initiating activation of the alternative pathway. Deficiency of properdin (X-linked) predisposes individuals to infections by bacteria, particularly Neisseria species. Supplementation of properdin in individuals who have a deficiency would be expected to reduce infection risk. A recent study suggested a broader role for properdin therapy — low-dose recombinant properdin markedly reduced susceptibility to bacterial infection in a mouse model, raising the possibility that supplementation might be effective in sepsis and other severe infections in humans87. The stabilizing role of properdin on the alternative pathway C3 convertase means that blocking this positive regulator may be therapeutic in inflammatory disease. A mAb targeting properdin (NM9401) and a small-molecule properdin antagonist have been reported by Novelmed Therapeutics for use in PNH and rheumatoid arthritis, respectively (see the Novelmed Therapeutics website).
Using the membrane C3 convertase regulators as drugs or leads. The membrane regulators of the C3 convertases present a different challenge compared to the plasma regulators in that they cannot readily be purified for testing as potential drugs. The only way to test whether a membrane regulator might work as a therapeutic is to make a recombinant soluble form. The first of these, described in 1990, was a soluble recombinant form of CR1 (known as sCR1 or TP10) comprising the entire extracellular sequence of thirty short consensus repeats (>1,800 amino acids). The protein was a powerful inhibitor of both classical and alternative pathway convertases in vitro in human and rodent sera; pre-treatment with sCR1 protected against reperfusion injury in a rat model of myocardial infarction88. Over the ensuing two decades, this agent provided an excellent proof-of-principle for complement inhibition in diverse models of human disease, effective in a wide range of disease models and in rodents, pigs and primates. After initial development work by SmithKline Beecham, Avant Immunotherapeutics took sCR1 into clinical trials in adult respiratory distress syndrome, acute lung injury and cardiopulmonary bypass; although some positive results were described37, there were also many failures, notably in large sepsis trials, and sCR1 has not progressed for these indications. More recently, guided by success in animal models, sCR1 entered clinical trials in dense deposit disease (CDX-1135; Celldex Therapeutics); regrettably, this study terminated in 2014 because of recruitment issues.
Numerous modified forms of sCR1 have been created to improve its therapeutic potential and/or simplify its production. TP20 incorporated multiple oligosaccharide sialyl Lewis X epitopes to bind to selectins, which are upregulated on endothelia at inflammatory sites. This simple change improved efficacy compared to unmodified sCR1 in rat ischaemia–reperfusion models89. The minimum complement regulatory unit from CR1 was known to be the three N-terminal short consensus repeats. A drug that comprised these three short consensus repeats attached to a nonspecific membrane-binding motif (APT-070; also known as Mirococept; AdProTech) proved remarkably effective as a complement inhibitor when delivered locally or systemically in rodent and pig models90,91. The agent was inexpensive to produce and proved safe in human studies. The MRC-funded EMPIRIKAL trial (UK Clinical Research Network (UKCRN) identifier: 16181), which is due to commence in 2015, tests the effect of Mirococept pretreatment of donor kidneys on their survival and function post-transplant.
Although recombinant soluble forms of the other membrane C3 convertase regulators, CD46 and CD55, have been produced and tested in animal models, most have stalled at the early preclinical stage. The exception was a chimeric CD55–CD46 molecule (CAB-2; also known as MLN-2222; Millenium Pharmaceuticals) that entered Phase I trials in cardiopulmonary bypass but was not progressed.
Inhibitors of the proteases involved in convertase formation and activity. There are eight protease targets that could be used for convertase regulation. The classical and lectin pathway initiating-complex enzymes (C1r, C1s, MASP1 and MASP2) are discussed above. Generation of the alternative pathway convertase requires FD, and deficiency of FD results in the absence of alternative pathway activity and marked susceptibility to bacterial infections92. The alternative pathway has a crucial role as an amplifier regardless of the initiating trigger, making it an attractive target for therapeutic modulation and bringing FD centre stage. The first FD-targeted therapeutic to reach the clinic, lampalizumab, is a humanized immunoglobulin G1 (IgG1) Fab fragment that inhibits FD activity by binding to the exosite to prevent substrate binding93. A published Phase I study of intraocular administration in patients with a severe form of AMD termed geographic atrophy (GA) demonstrated safety20. Phase II results of monthly treatments, announced but unpublished (MAHALO study), describe a 20% reduction in GA area compared to controls at 18 months; these startling results were even better in patients selected based on an undisclosed FI genetic biomarker, who showed a 44% reduction at 18 months. Phase III trials of therapy (10 mg intravitreal every 4–6 weeks) in patients with GA are ongoing.
Several broad-spectrum small-molecule protease inhibitors, including dichloroisocoumarin, bind to and inhibit FD; structures obtained from these complexes have identified unique properties of FD that offer the prospect of designing specific inhibitors94,95. Novartis recently reported the evolution of small-molecule inhibitors of FD that are active in the nanomolar range96. Very recently, Achillion have announced the development of orally active, potent and specific small-molecule inhibitors of FD that they propose for therapy in PNH. It remains to be seen whether the high natural turnover of FD will limit the use of these small molecules in humans.
A number of mAbs or mAb fragments have been described that inhibit FB cleavage in a variety of ways. One of these, termed mAb 1379, has been developed as a therapeutic by Taligen97; TA106 is a Fab fragment of mAb 1379 that blocks formation of the alternative pathway convertase. Taligen was acquired by Alexion in 2011 for US$111M, and the agent is currently undergoing humanization for further use in humans, with AMD and asthma as primary targets. Another FB-specific antibody, bikaciomab (NM9308; Novelmed), is also in development for use in AMD, and small molecule inhibitors of FB have been reported (in Novartis patents) although there is no evidence yet of their use in preclinical models of disease. Isis Pharmaceuticals have described the use of FB antisense oligonucleotides for therapy of murine lupus nephritis, but progression of this technique to humans has not been reported98. The functional homologue of FB in the classical pathway is C2; despite a more limited role in pathology, C2a-specific mAbs that block the classical pathway C3 convertase have been developed and patented by Tanox (now part of Genentech; Patent ref. WO 2001070818 A8); no trials are reported on these interesting drugs. An intriguing agent, a linear peptide (rH17d) derived from a Schistosoma species protein, bound C2 and inhibited C3 convertase formation, but has not been further developed to date99.
Targeting individual convertase components or regulators. Individual convertase components and regulators can be targeted in various ways to influence complement activity: components may be depleted in the plasma by increasing their consumption or decreasing their synthesis; regulator concentrations may be increased by supplementation or an increase in synthesis; components and/or regulators may be prevented from undergoing essential conformational changes or enzymatic cleavage; or components may be rendered more susceptible to spontaneous or accelerated decay.
One example of altering consumption is illustrated by CVF, a C3b analogue present in cobra venom that, when injected into mammals, binds to FB to form a stable C3 convertase that is resistant to normal regulatory processes and thus activates the system to exhaustion. This activity of cobra venom was recognized over a century ago, and purified CVF was first used as a tool to render the complement system of rodents deficient almost 50 years ago, making it the first anti-complement 'therapeutic' (Refs 100,101). Although the empiric use of cobra venom in therapy has a long history in everything from cancer to cosmetics, CVF as a drug has severe limitations: first because it is immunogenic, which renders repeat injections ineffective, and second because rapid and complete activation of complement by CVF generates high concentrations of C5a, causing circulating neutrophils to activate and aggregate in the lungs and other organs102. It is also unclear whether CVF can deplete complement locally in tissues. Attempts to address immunogenicity through humanization of CVF have created a hybrid that is 94% identical to human C3; small stretches of CVF-derived sequences in the C3 α-chain are sufficient to retain CVF-like complement consumption103. Although immunogenicity may be low, concerns over the toxic effects of continuous consumption of complement by CVF ensure that use will be restricted to acute conditions.
Supplementation of the plasma convertase regulators offers therapeutic possibilities above and beyond the restoration of plasma levels in rare cases of deficiency. Systemic modulation of the cascade using endogenous soluble regulators (or soluble versions of membrane-associated control proteins) is likely to cause downmodulation rather than complete blockade and effect therapy in a manner that is more measured. Efforts to modulate complement by administering extra FH or FI are described above. However, the amounts of protein needed to raise plasma levels sufficiently to affect convertase activity may be prohibitive; the feasibility of this approach will require careful dose prediction, and dosing will probably be disease-specific and perhaps individual-specific. Gene-therapy approaches to increase local synthesis are also now attracting attention — for example, the introduction of a construct that increases the production of FH or another complement regulator in the retina might prove effective in AMD, a disease in which other gene-therapy approaches have already been effective in models and have reached Phase I/II clinical trials104.
Decreased synthesis is the aim of a variety of antisense and related agents targeting complement components either systemically or locally, and these agents are the subject of a number of published patents. However, to date these strategies have only been tested in vitro and (for a minority) in animal models; for example, C6-targeted antisense oligonucleotides markedly reduced hepatic synthesis and plasma C6 levels, and ameliorated disease in a mouse model of traumatic brain injury105. C3 antisense is described in a number of patents over the past 10 years but has not progressed in vivo. Alnylam described an RNA interference (RNAi) molecule targeting C5, ALN-CC5, that reduced plasma C5 levels by >90% when administered monthly by subcutaneous injection to non-human primates; this agent, developed for therapy of PNH, is now in Phase I/II trials (as announced in a recent Alnylam press release).
Targeting the anaphylactic peptides. The polypeptides C3a and C5a, released owing to the action of the C3 and C5 convertase enzymes, are biologically active, and they bind specific receptors on phagocytes and many other cell types to exert effects. The range of cell targets and activities is large and reviewed well elsewhere106, but their pro-inflammatory activities are particularly relevant to disease. C5a is a powerful inflammatory trigger implicated in many diseases, whereas C3a exhibits a complex mix of pro- and anti-inflammatory activities107. Both C3a and C5a (and their metabolites) bind to seven-transmembrane-domain G-protein-coupled receptors (GPCRs), which are members of the large cytokine and chemokine receptor family. Similar to other GPCRs, those that bind C5a and C3a are attractive drug targets; many specific small-molecule blockers have been developed for cytokine receptors, although few have been developed for chemokine receptors, which more closely resemble the C3a anaphylatoxin chemotactic receptor (C3aR) and C5aR1 (Ref. 108). Dompé Pharmaceutical have described small-molecule C5aR1 blockers and claim potential for therapy of a wide panel of diseases; however, no trials are yet reported. Perhaps the most studied C5aR1 blocker is the cyclic hexapeptide PMX53, invented by Taylor and Woodruff and developed first by Peptech and then Arana Therapeutics109. This agent, modelled on the C terminus of C5a, showed high specificity and nanomolar affinity for C5aR1 and worked across many species; this latter property enabled the testing of PMX53 in a broad sweep of rodent models of inflammatory disease, and in many of these models this peptide had good efficacy. A number of Phase I and IIa clinical trials with PMX53 were initiated in rheumatoid arthritis, osteoarthritis, psoriasis and AMD; the molecule proved safe, but all trials were terminated because of poor efficacy. One possible reason for these failures was the poor oral bioavailability and tissue penetrance of PMX53 (Ref. 110). A modified version of PMX53, PMX205, shows improvements in these parameters and may re-surface in future trials, perhaps in combination with other agents. An orally active small molecule that blocks C5aR1 has also been developed by ChemoCentryx; this agent, CCX-168, has completed Phase II studies in anti-neutrophil cytoplasmic antibody (ANCA) vasculitis, in which it was successful in enabling the reduction and elimination of high-dose corticosteroids and demonstrated improvement in renal health parameters. In 2014, CCX-168 was granted Orphan Drug status in both ANCA-associated vasculitides and aHUS. Emerging evidence for the role of C3a as a 'modulator' of the C5a-driven inflammatory response highlights the potential for C3aR agonism in acute neutrophil-driven pathologies such as ischaemia–reperfusion injury106,111.
An alternative approach, again by analogy to the chemokine and cytokine field, is the use of blockers — either antibodies or others — that bind to C3a or C5a and prevent receptor engagement. Cytokine-specific antibodies and decoy receptors have evolved into blockbuster drugs. InflaRx have developed a blocking mAb against C5a (IFX-1) that is currently in a Phase II trial for early sepsis and septic shock. Noxxon described a series of L-RNA aptamers (Spiegelmer; NOX-D19 to NOX-D21) that block C5a; these agents were effective in models of sepsis and transplant rejection112,113, but have not yet progressed to humans. All of the above suggests that pharmacological blockade of C3a and/or C5a activities might be relatively straightforward; however, such an approach will only work in those situations in which these molecules are the major drivers of pathology, such as in neutrophil-mediated disorders including ANCA-vasculitis, chronic obstructive pulmonary disease (COPD) and lupus.
Targeting C5. As described above, C5 is activated to create two moieties with distinct activities. The small fragment, C5a, is a powerful chemoattractant, and therapeutic blockade of its activities is described above. The larger fragment, C5b, forms the foundation of the MAC. Assembly of the MAC involves the non-enzymatic aggregation of the five terminal pathway components (C5b and C6–C9) into a pore that inserts into and through the cell membrane. C5b, while still attached to the convertase, binds sequentially to C6 and C7; the C5b67 complex is released from the convertase and binds to the membrane via a labile hydrophobic site. C8 and multiple copies of C9 are recruited in turn to build the pore. MAC activity is naturally regulated by inefficiencies built into the assembly system and by regulators policing the membrane to block complexes that reach the surface. The hydrophobic binding site in C5b67 is intrinsically unstable, decaying by interaction with water in a fraction of a second. Additionally, there are several proteins in the plasma that bind to the MAC to further restrict its capacity to bind to the membrane: clusterin and vitronectin (also known as S protein) are multifunctional proteins that, among their many roles, inhibit MAC assembly in this way, and C8 is an efficient inhibitor if it binds to C5b67 in the fluid phase. Once bound to the membrane, C5b67 is protected from these plasma inhibitors and recruits C8 and C9 to form the MAC. The last line of defence is the membrane regulator CD59, a small glycosylphosphatidylinositol (GPI)-anchored protein that binds to the forming complex and prevents the completion of the MAC pore.
Opportunities for therapeutic inhibition are legion. The first obvious target is the first MAC component, C5b. Cutting off the supply of C5b by targeting the C3 and C5 convertases will inhibit MAC production; alternatively, C5 itself can be targeted to render it resistant to cleavage, which is the mode of action of the C5 monoclonal eculizumab, the most successful complement-specific drug to date. The success of eculizumab (Box 2) has triggered interest in other ways of preventing C5 cleavage. Novartis have taken a C5-specific mAb, LFG316, into Phase II trials of intraocular therapy in advanced AMD (completed but unpublished) and, more recently, in choroiditis and panuveitis114. In August 2015, Novartis announced Phase I trials to assess the safety and efficacy of LFG316 in patients with PNH (NCT02534909). A recombinant human C5-specific mAb fragment, Mubodina (Adienne Pharma & Biotech), that prevents cleavage of C5 has undergone early-stage trials in aHUS and other renal diseases, although no development has been reported recently. Ornithodoros moubata complement inhibitor (OmCI), a small (16 kDa) lipocalin present in the saliva of soft ticks, is a potent C5 inhibitor that probably protects the tick from complement in its blood meal115. Recombinant OmCI (Coversin; Volution Immuno Pharmaceuticals) blocked C5 in numerous species and inhibited disease in rodent models116,117. The drug bound C5 at a site remote from the convertase cleavage site, suggesting that it functioned by either blocking the interaction of C5 with the convertase or preventing conformational change essential for cleavage. Coversin completed Phase I trials in 2014 and is in development for PNH and other MAC-driven diseases. SOBI002 is a C5-blocker that is based on Affibody technology and developed by Swedish Orphan Biovitrum. Inclusion of an albumin-binding motif extends the plasma half-life of SOBI002. This small (~12 kDa) protein, derived using phage display, avidly binds to C5 and prevents cleavage; the simple structure of SOBI002 makes synthesis easy and inexpensive. SOBI002 is currently in a Phase I trial, which was recently listed as terminated (NCT02083666). Various non-biologic approaches have also been developed and are either soon to be tested or already in clinical trials. Ophthotech have developed a C5-specific aptamer (ARC1905, also known as Zimura) that, when given intraocularly in a Phase IIa study in combination with an antibody targeting vascular endothelial growth factor (VEGF), improved outcomes in patients with wet AMD. The RNAi approach taken by Alnylam is described above; at the 2014 American Society of Haematology (ASH) meeting, Alnylam reported effective knockdown in non-human primates using ALN-CC5 (see Further information), and Phase I trials are now underway with PNH as the primary indication. Interim Phase I data demonstrate substantial knock-down of C5 in single-ascending-dose studies in humans (as announced in a recent Alnylam press release). RaPharma have developed a cyclic peptide that binds to C5 and prevents cleavage and activation; promising preclinical data were reported at ASH 2014 (see Further information), and Phase I studies are proposed for the near future. As with many of the C5-targeting agents reported here, the primary indication is likely to be PNH. The crowded 'landscape' in the C5 space (Fig. 3) clearly illustrates the activity generated by the success of eculizumab and the desire to block the activities of this key component in the cascade.
Figure 3. Progress of complement therapeutics towards clinical use.

The figure graphically illustrates a current 'snapshot' of the different areas of complement that are being targeted and the progress of the drugs — both marketed and en route to approval. It is a rapidly moving field and, inevitably, some compounds will progress, others will fail and new drugs will emerge to take their positions in the landscape in the coming months and years. One certainty is that this already crowded field will become much more so in the near future. Among the monoclonal antibodies, TNT003 and TNT009 (True North Therapeutics) target complement fragment C1s; OMS721 (Omeros) targets MBL-associated serine protease 2 (MASP2); Lampalizumab (Roche/Genentech) targets Factor D (FD), Bikaciomab (Novelmed) targets FB; IFX-1 (InflaRx) targets C5a; eculizumab (Soliris; Alexion Pharmaceuticals), LFG316 (Novartis), ALXN1210 and ALXN550 (Alexion) all target C5. Regenesance are developing an antibody that targets C6, and Novelmed are developing an antibody (NM9401) against properdin. Among the protein biologics, Berinert (CSL Behring), Ruconest (Salix Pharmaceuticals) and Cinryze (Shire Pharmaceuticals) are all C1INH preparations that target C1; AMY-201 (Amyndas) is a mini FH; TT30 (ALXN1102; Alexion) is a CR2–FH hybrid; TP10 (CDX-1135; Celldex Therapeutics) is a soluble form of complement receptor type 1 (CR1); Coversin (Volution Immuno Pharmaceuticals) is a C5-binding protein; Mirococept is a targeted CR1 fragment developed by AdProTech and currently in clinical trials led by King's College London; and the affibody SOBI002 (Swedish Orphan Biovitrum) targets C5 (programme recently terminated). Small-molecule inhibitors include CCX-168 (ChemoCentryx) and DF2593A (Dompé Pharmaceutical), which target C5aR; FD inhibitors from Achillion and Novartis; and a properdin inhibitor from Novelmed. Among the peptide-based therapeutics, Cp40 (Amyndas), APL-1 and APL-2 (Apellis Pharmaceuticals) are compstatin derivatives targeting C3; and RA101348 (RaPharma) is a C5 blocking peptide. Nucleic acid-based drugs include ARC1905 (Zimura; Ophthotech), a C5 aptamer; ALN-CC5 (Alnylam Pharmaceuticals), a C5 RNA interference (RNAi) molecule; and the Spiegelmers NOX-D19 to NOX-D21 (Noxxon Pharma), targeting C5a. Regenesance are developing a C6 antisense molecule.
Targeting the terminal pathway: beyond C5. Blocking C5 cleavage by any of the above strategies will stop the production of C5a as well as the MAC and this may have important consequences if continued long term. To specifically target the MAC, agents with activities downstream of C5 cleavage are needed. Agents that mimic the ability of the natural inhibitors clusterin and vitronectin (and C8) to compete for the membrane binding site on C5b67 might further reduce the efficiency of membrane attack, whereas mimics of CD59 that block the final steps of MAC assembly might be effective blockers of lytic pore formation. Soluble recombinant CD59 (sCD59) has been generated and shown to inhibit lysis in vitro; however, in vivo, sCD59 binds to plasma proteins, markedly reducing its inhibitory effects118. Recombinant CD59, attached to the same membrane-binding motif as in Mirococept, has been generated and shown to protect PNH erythrocytes from lysis ex vivo and ameliorate arthritis development in a rat model when administered intra-articularly at the time of disease induction119,120. Alternatively, any one of the terminal pathway components, each of which is essential for MAC formation, could be targeted. Regenesance BV has targeted C6 with both blocking C6-specific mAb and antisense approaches, and the latter was effective in animal models105. Regenesance also described a small-molecule inhibitor of C6 in development for Guillain–Barré syndrome, but further development was not reported. A recent publication describes a small molecule (aurin tricarboxylic acid) that inhibits MAC assembly by blocking C9 binding; the agent prevented the lysis of PNH erythrocytes by serum ex vivo121.
Localizing therapeutics to sites of pathology
Inhibition of systemic complement activity is a major concern with most anti-complement drugs as it renders the recipient susceptible to infection. Delivery of the drug specifically to the site of disease will not only reduce risk but also enhance efficacy. At its simplest, local delivery could mean direct administration to the site of disease — into the inflamed joint, eye or brain, for example. More interesting are the many modifications that have been made to ensure the homing of an agent where needed. Modification of sCR1 to favour its accumulation on inflamed endothelia (TP20) increased efficacy in models89, and addition of a membrane-binding tag to a truncated form of sCR1 (Mirococept) converted an inactive fragment into a viable therapeutic90,91. Similarly, a recombinant truncated FH comprising the minimal active site (short consensus repeats 1–4) is essentially inactive in vivo, but adding in the FH membrane-binding short consensus repeats creates a powerful inhibitor (AMY-201)79,80.
Localising anti-complement agents to sites of complement activation has attracted abundant attention. Homing of the drug not only avoids systemic inhibition but also enhances efficacy by concentrating the drug in one site and by placing active sites in the correct orientation to inactivate target enzymes. These approaches tend to focus on the homing of agents that have regulatory functions, such as decay acceleration or cofactor activity, rather than the blocking activities typical of antibodies. It is important that the drug that is delivered to the site is able to 'recycle' and release its target once inactivated, otherwise the target surface will rapidly become saturated with useless agent. More than 20 years ago, we described a prodrug approach whereby recombinant forms of natural regulators were generated as Fc fusion proteins in which regulatory activity was absent because of steric hindrance. Engineered enzyme-cleavage sites enabled the release of active agent by matrix metalloproteinases and related enzymes that are abundant at inflammatory sites122. Similar strategies have proved successful in anticancer therapy123.
Complement fragments bind covalently to tissues at sites of activation and provide a potential ligand for localizing an anti-complement drug where needed. A hybrid molecule comprising the rodent C3 convertase regulator CRRY coupled to the C3dg-binding short consensus repeats of CR2 provided proof-of-principle of this approach; the hybrid molecule localized to sites of pathology and was 10-fold more active compared to CRRY alone in mouse models of ischaemia–reperfusion124. The same group also described a CR2–FH hybrid protein that was effective in several murine models125; this provided the impetus to create a human CR2–FH hybrid, TT30, which is currently under development by Alexion for therapy of PNH (ALXN1102 and ALXN1103)22,126. Potential advantages of this agent over eculizumab include a reduced risk of infection, a lower dose requirement and protection of PNH erythrocytes from C3 deposition and the resultant extravascular haemolysis. Progress towards the clinic has been slow, perhaps because of commercial rather than scientific considerations. A Phase I clinical trial was initiated (NCT01335165), although no data have been reported.
Looking to the future
Although the many roles of complement in driving pathology in diverse diseases have long been recognized, the capacity to treat these diseases by controlling complement activation has lagged behind. Today, we boast just two anti-complement drugs that are being marketed, approved only in rare orphan diseases (HAE, PNH and aHUS), which is a rather disappointing outcome given the investments made by researchers and companies. However, the future looks much brighter; new therapeutics are entering the clinic, targeting different parts of the complement system, with different half-lives, different routes of administration and each with its own range of activities and side effects (Fig. 3). Importantly, although the emphasis on orphan applications was pivotal in getting the first agents to the market, many of the new agents target common acute and chronic inflammatory diseases, for example AMD, arthritis and cardiovascular disease.
Hurdles to the commercialization of complement therapeutics still exist, including cost, routes of administration and infection risk. Short-duration complement inhibition, particularly in a controlled clinical environment, carries the least risk, and anti-complement drugs will probably first become mainstream for acute indications, such as reperfusion injuries. For chronic conditions, cost, ease of administration and safety may be substantial issues. Cost will become less of an issue as increased competition (the emergence of small-molecule inhibitors) and increased use should, theoretically, bring prices down rapidly. The experience of chronic therapy with agents that block complement is still relatively short-term, and it is possible that the elimination of molecules, such as C5a, may affect other activities of this multi-functional molecule, such as modulation of adaptive immunity, in the long term106. However, eculizumab was approved for use in PNH in 2007 and for aHUS in 2011 and, although the long-term consequences of C5 blockade remain uncertain, it is encouraging that after almost a decade of use no substantial adverse effects have been noted in carefully monitored and compliant recipients.
Frequent intravenous administration is not clinically acceptable for chronic use, and so agents will need either very long half-lives or to be delivered by routes not requiring medical intervention (for example, subcutaneous or oral). It may be that a therapy could be titrated to give partial inhibition of complement, sufficient to inhibit disease but sparing key protective functions. Alternatively, a drug may be engineered to deliver complement inhibition where it is needed at the site of disease. Inevitably, different diseases will require different approaches to complement inhibition — some may be entirely MAC-driven, others C5a-dependent, and most involve a complicated interplay between the various complement products and other inflammatory mediators.
Selecting the right agent or agents will be crucial to success in trials, and here a comprehensive understanding of the mechanisms by which complement drives pathology in the target disease is a sine qua non. Biomarkers that reflect complement activation in fluids and tissues can inform the selection of the right drug for the right patient, demonstrating engagement of the target and efficacy of the drug, and monitoring response to therapy. A lack of good biomarkers often handicaps early stage trials and simple readouts of effectiveness are crucial. In some cases this is obvious; for example, blockade of MAC formation can be demonstrated in serum through simple haemolytic assays. When activation fragments such as C3a, C5a, C4d, Bb and terminal complement complex (TCC) are present in the disease their levels can be monitored to demonstrate response to therapy and confirm target engagement. In some diseases, such as PNH, there is an obvious clinical readout of target engagement — eculizumab treatment prevents intravascular haemolysis in PNH. However, for diseases restricted to specific sites, for example, the retina, central nervous system or kidney glomerulus, plasma complement biomarkers may not reflect the response to therapy and are poor tools for assessing target engagement. There is thus a need for effective, non-invasive methods to detect complement activation and deposition within affected tissues without resorting to biopsy. Methods for imaging complement activation in vivo may fill this need; agents comprising the C3-binding domain of CR2 have been used to detect C3 fragments deposited in the kidney by magnetic resonance imaging in mouse models127. Antibodies against C3dg have also been developed as imaging agents and shown to identify retinal lesions in a mouse model of AMD128. Further development of the capacity to image complement activation in vivo will inform the conduct of future clinical trials by providing measurable outcomes and early proof of target engagement.
Acknowledgements
The authors thank K. Miller of GlaxoSmithKline, for her thoughtful review and discussion of the manuscript.
Glossary
- Tickover
The spontaneous low-level complement activation that occurs continuously in the plasma of normal individuals and assists in the rapid response to pathogens.
- Opsonins
Proteins and protein fragments that coat pathogens and dead or dying host cells to label them for recognition and elimination by phagocytes. The principle complement opsonins are C3b and iC3b.
- Short consensus repeats
Structural units comprising approximately 60 amino acids held in a globular domain by 2 invariant disulfide bonds; they are the key building blocks for the assembly of proteins in the regulators of complement activation (RCA) family — including Factor H (FH) and the FH-related (FHR) proteins, C4b-binding protein (C4BP), complement receptor type 1 (CR1) and CR2 — but are also found in other complement proteins and proteins unrelated to complement.
- Oligosaccharide sialyl Lewis X epitopes
Carbohydrate structures that are expressed on cell surfaces and act as ligands for cell adhesion molecules of the selectin family.
Biographies
B. Paul Morgan is Professor of Immunology and Director of the Systems Immunity University Research Institute at Cardiff University, UK. He is a chemical pathologist but has worked on the complement system for over 30 years. His Ph.D. explored the biology of the terminal pathway and the mechanisms of cell escape from complement killing, work that he consolidated during 2 years of post-doctoral training in US complement labs. He returned to Cardiff to complete clinical training and build a research group, generously supported by the Wellcome Trust. He has published over 400 papers and reviews exploring different aspects of the complement system, particularly the biology of complement regulation and the roles of complement in disease. In recent years he has focused primarily on the roles of complement in neurological diseases and novel approaches to therapy. 10.1038/nrd4657.
Claire L. Harris holds a Personal Chair at Cardiff University, UK, and is also Head of Complement in the Cytokine, Chemokine and Complement Discovery Performance Unit (C3 DPU) at GlaxoSmithKline. She graduated from the University of Cambridge with a PhD in the biochemistry of the complement system and moved to Cardiff in 1993 to join the Complement Biology Group at the University of Wales College of Medicine, Cardiff, UK. After a postdoctoral period working in the biochemistry of complement in humans and other species, she obtained a University Award from the Wellcome Trust and established a research group focused on structure–function relationships in complement activators and regulators. She has a longstanding interest in the mechanisms underlying complement dysregulation and disease and in designing improved and targeted therapies for diseases driven by complement. Her recent work includes functional characterization of complement protein variants linked to diseases such as age-related macular degeneration, this has led to the concept of the 'complotype' — the influence of complement genetic makeup on inflammation & infection. She joined the C3 DPU at GSK in June 2013 to drive development of a differentiated portfolio in complement therapies. Claire L. Harris's homepage.
Related links
FURTHER INFORMATION
Eculizumab in the electronic Medicines Compendium
RaPharma ASH presentation 2014
PowerPoint slides
Competing interests
B.P.M. has undertaken paid consultancy work and sponsored research for GlaxoSmithKline, Swedish Orphan Biovitrum and Achillion Pharma. C.L.H. is employed by GlaxoSmithKline.
Contributor Information
B. Paul Morgan, Email: morganbp@cardiff.ac.uk.
Claire L. Harris, Email: harriscl@cardiff.ac.uk
References
- 1.Morgan BP. Fundamental Immunology. 2012. pp. 850–862. [Google Scholar]
- 2.Kim DD, Song WC. Membrane complement regulatory proteins. Clin. Immunol. 2006;118:127–136. doi: 10.1016/j.clim.2005.10.014. [DOI] [PubMed] [Google Scholar]
- 3.Grumach AS, Kirschfink M. Are complement deficiencies really rare? Overview on prevalence, clinical importance and modern diagnostic approach. Mol. Immunol. 2014;61:110–117. doi: 10.1016/j.molimm.2014.06.030. [DOI] [PubMed] [Google Scholar]
- 4.de Cordoba SR, Tortajada A, Harris CL, Morgan BP. Complement dysregulation and disease: from genes and proteins to diagnostics and drugs. Immunobiology. 2012;217:1034–1046. doi: 10.1016/j.imbio.2012.07.021. [DOI] [PubMed] [Google Scholar]
- 5.Barbour TD, Pickering MC, Cook TH. Dense deposit disease and C3 glomerulopathy. Semin. Nephrol. 2013;33:493–507. doi: 10.1016/j.semnephrol.2013.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Risitano AM. Paroxysmal nocturnal hemoglobinuria and other complement-mediated hematological disorders. Immunobiology. 2012;217:1080–1087. doi: 10.1016/j.imbio.2012.07.014. [DOI] [PubMed] [Google Scholar]
- 7.Brodsky RA. Paroxysmal nocturnal hemoglobinuria. Blood. 2014;124:2804–2811. doi: 10.1182/blood-2014-02-522128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Linton SM, Morgan BP. Complement activation and inhibition in experimental models of arthritis. Mol. Immunol. 1999;36:905–914. doi: 10.1016/S0161-5890(99)00113-3. [DOI] [PubMed] [Google Scholar]
- 9.Gehrs KM, Anderson DH, Johnson LV, Hageman GS. Age-related macular degeneration — emerging pathogenetic and therapeutic concepts. Ann. Med. 2006;38:450–471. doi: 10.1080/07853890600946724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Troutbeck R, Al Qureshi S, Guymer RH. Therapeutic targeting of the complement system in age-related macular degeneration: a review. Clin. Experiment Ophthalmol. 2012;40:18–26. doi: 10.1111/j.1442-9071.2011.02581.x. [DOI] [PubMed] [Google Scholar]
- 11.Schifferli JA. Complement and immune complexes. Res. Immunol. 1996;147:109–110. doi: 10.1016/0923-2494(96)87183-5. [DOI] [PubMed] [Google Scholar]
- 12.Truedsson L, Bengtsson AA, Sturfelt G. Complement deficiencies and systemic lupus erythematosus. Autoimmunity. 2007;40:560–566. doi: 10.1080/08916930701510673. [DOI] [PubMed] [Google Scholar]
- 13.Pepys MB. Role of complement in induction of antibody production in vivo. Effect of cobra factor and other C3 reactive agents on thymus-dependent and thymus-independent antibody responses. J. Exp. Med. 1974;140:126–145. doi: 10.1084/jem.140.1.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carroll MC, Isenman DE. Regulation of humoral immunity by complement. Immunity. 2012;37:199–207. doi: 10.1016/j.immuni.2012.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Clarke EV, Tenner AJ. Complement modulation of T cell immune responses during homeostasis and disease. J. Leukoc. Biol. 2014;96:745–756. doi: 10.1189/jlb.3MR0214-109R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement — a key system for immune surveillance and homeostasis. Nat. Immunol. 2010;11:785–797. doi: 10.1038/ni.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lewis MJ, Botto M. Complement deficiencies in humans and animals: links to autoimmunity. Autoimmunity. 2006;39:367–378. doi: 10.1080/08916930600739233. [DOI] [PubMed] [Google Scholar]
- 18.Skattum L, van Deuren M, van der Poll T, Truedsson L. Complement deficiency states and associated infections. Mol. Immunol. 2011;48:1643–1655. doi: 10.1016/j.molimm.2011.05.001. [DOI] [PubMed] [Google Scholar]
- 19.Chi ZL, Yoshida T, Lambris JD, Iwata T. Suppression of drusen formation by compstatin, a peptide inhibitor of complement C3 activation, on cynomolgus monkey with early-onset macular degeneration. Adv. Exp. Med. Biol. 2010;703:127–135. doi: 10.1007/978-1-4419-5635-4_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Do DV, et al. A phase Ia dose-escalation study of the anti-factor D monoclonal antibody fragment FCFD4514S in patients with geographic atrophy. Retina. 2014;34:313–320. doi: 10.1097/IAE.0b013e3182979ddd. [DOI] [PubMed] [Google Scholar]
- 21.He C, Imai M, Song H, Quigg RJ, Tomlinson S. Complement inhibitors targeted to the proximal tubule prevent injury in experimental nephrotic syndrome and demonstrate a key role for C5b-9. J. Immunol. 2005;174:5750–5757. doi: 10.4049/jimmunol.174.9.5750. [DOI] [PubMed] [Google Scholar]
- 22.Risitano AM, et al. The complement receptor 2/factor H fusion protein TT30 protects paroxysmal nocturnal hemoglobinuria erythrocytes from complement-mediated hemolysis and C3 fragment. Blood. 2012;119:6307–6316. doi: 10.1182/blood-2011-12-398792. [DOI] [PubMed] [Google Scholar]
- 23.Swaak AJ, et al. Determination of the half-life of C3 in patients and its relation to the presence of C3 breakdown products and/or circulating immune complexes. Rheumatol. Int. 1982;2:161–166. doi: 10.1007/BF00286138. [DOI] [PubMed] [Google Scholar]
- 24.Rother RP, Rollins SA, Mojcik CF, Brodsky RA, Bell L. Discovery and development of the complement inhibitor Eculizumab for the treatment of paroxysmal nocturnal hemoglobinuria. Nat. Biotech. 2007;25:1256–1264. doi: 10.1038/nbt1344. [DOI] [PubMed] [Google Scholar]
- 25.Zuber J, et al. Use of Eculizumab for atypical haemolytic uraemic syndrome and C3 glomerulopathies. Nat. Rev. Nephrol. 2012;8:643–657. doi: 10.1038/nrneph.2012.214. [DOI] [PubMed] [Google Scholar]
- 26.Ternant D, et al. Pharmacokinetics and concentration-effect relationship of adalimumab in rheumatoid arthritis. Br. J. Clin. Pharmacol. 2015;79:286–297. doi: 10.1111/bcp.12509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sissons JG, Liebowitch J, Amos N, Peters DK. Metabolism of the fifth component of complement, and its relation to metabolism of the third component, in patients with complement activation. J. Clin. Invest. 1977;59:704–715. doi: 10.1172/JCI108689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Noris M, et al. Dynamics of complement activation in aHUS and how to monitor eculizumab therapy. Blood. 2014;124:1715–1726. doi: 10.1182/blood-2014-02-558296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Katschke KJ, Jr, et al. Structural and functional analysis of a C3b specific antibody that selectively inhibits the alternative pathway of complement. J. Biol. Chem. 2009;284:10473–10479. doi: 10.1074/jbc.M809106200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Paixão-Cavalcante D, et al. A humanized antibody that regulates the alternative pathway convertase: potential for therapy of renal disease associated with nephritic factors. J. Immunol. 2014;192:4844–4851. doi: 10.4049/jimmunol.1303131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Müller-Eberhard HJ. Complement. Annu. Rev. Biochem. 1975;44:697–724. doi: 10.1146/annurev.bi.44.070175.003405. [DOI] [PubMed] [Google Scholar]
- 32.Pascual M, et al. Metabolism of complement factor D in renal failure. Kidney Int. 1988;34:529–536. doi: 10.1038/ki.1988.214. [DOI] [PubMed] [Google Scholar]
- 33.Loyet KM, et al. Complement inhibition in cynomolgus monkeys by anti-factor D antigen-binding fragment for the treatment of an advanced form of dry age-related macular degeneration. J. Pharmacol. Exp. Ther. 2014;351:527–537. doi: 10.1124/jpet.114.215921. [DOI] [PubMed] [Google Scholar]
- 34.Kang JS, Deluca PP, Lee KC. Emerging PEGylated drugs. Expert Opin. Emerg. Drugs. 2009;14:363–380. doi: 10.1517/14728210902907847. [DOI] [PubMed] [Google Scholar]
- 35.Jazayeri JA, Carroll GJ. Fc based cytokines: prospects for engineering superior therapeutics. BioDrugs. 2008;22:11–26. doi: 10.2165/00063030-200822010-00002. [DOI] [PubMed] [Google Scholar]
- 36.Zimmerman JL, Dellinger RP, Straube RC, Levin JL. Phase I trial of the recombinant soluble complement receptor 1 in acute lung injury and acute respiratory distress syndrome. Crit. Care Med. 2000;28:3149–3154. doi: 10.1097/00003246-200009000-00004. [DOI] [PubMed] [Google Scholar]
- 37.Lazar HL, et al. Soluble human complement receptor 1 limits ischemic damage in cardiac surgery patients at high risk requiring cardiopulmonary bypass. Circulation. 2004;110(11 Suppl. 1):II274–II279. doi: 10.1161/01.CIR.0000138315.99788.eb. [DOI] [PubMed] [Google Scholar]
- 38.Sacks SH, Zhou W. The role of complement in the early immune response to transplantation. Nat. Rev. Immunol. 2012;12:431–442. doi: 10.1038/nri3225. [DOI] [PubMed] [Google Scholar]
- 39.Schwertz H, et al. Serine protease inhibitor nafamostat given before reperfusion reduces inflammatory myocardial injury by complement and neutrophil inhibition. Cardiovasc. Pharmacol. 2008;52:151–160. doi: 10.1097/FJC.0b013e318180188b. [DOI] [PubMed] [Google Scholar]
- 40.Gál P, Dobó J, Beinrohr L, Pál G, Závodszky P. Inhibition of the serine proteases of the complement system. Adv. Exp. Med. Biol. 2013;735:23–40. doi: 10.1007/978-1-4614-4118-2_2. [DOI] [PubMed] [Google Scholar]
- 41.Shi J, et al. TNT003, an inhibitor of the serine protease C1s, prevents complement activation induced by cold agglutinins. Blood. 2014;123:4015–4022. doi: 10.1182/blood-2014-02-556027. [DOI] [PubMed] [Google Scholar]
- 42.Watkins AM, Arora PS. Structure-based inhibition of protein–protein interactions. Eur. J. Med. Chem. 2014;94:480–488. doi: 10.1016/j.ejmech.2014.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ruiz-Gómez G, et al. Structure–activity relationships for substrate-based inhibitors of human complement factor B. J. Med. Chem. 2009;52:6042–6052. doi: 10.1021/jm900781m. [DOI] [PubMed] [Google Scholar]
- 44.Forneris F, et al. Structures of C3b in complex with factors B and D give insight into complement convertase formation. Science. 2010;330:1816–1820. doi: 10.1126/science.1195821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kocsis A, et al. Selective inhibition of the lectin pathway of complement with phage display selected peptides against mannose-binding lectin-associated serine protease (MASP)-1 and -2: significant contribution of MASP 1 to lectin pathway activation. J. Immunol. 2010;185:4169–4178. doi: 10.4049/jimmunol.1001819. [DOI] [PubMed] [Google Scholar]
- 46.Woodruff TM, et al. Species dependence for binding of small molecule agonist and antagonists to the C5a receptor on polymorphonuclear leukocytes. Inflammation. 2001;25:171–177. doi: 10.1023/A:1011036414353. [DOI] [PubMed] [Google Scholar]
- 47.Ricklin D, Lambris JD. Compstatin: a complement inhibitor on its way to clinical application. Adv. Exp. Med. Biol. 2008;632:273–292. doi: 10.1007/978-0-387-78952-1_20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jacobson AC, Weis JH. Comparative functional evolution of human and mouse CR1 and CR2. J. Immunol. 2008;181:2953–2959. doi: 10.4049/jimmunol.181.5.2953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Holers VM, Kinoshita T, Molina H. The evolution of mouse and human complement C3 binding proteins: divergence of form but conservation of function. Immunol. Today. 1992;13:231–236. doi: 10.1016/0167-5699(92)90160-9. [DOI] [PubMed] [Google Scholar]
- 50.Bhatia SN, Ingber DE. Microfluidic organs-on-chips. Nat. Biotech. 2014;32:760–772. doi: 10.1038/nbt.2989. [DOI] [PubMed] [Google Scholar]
- 51.Roos A, Ramwadhdoebé TH, Nauta AJ, Hack CE, Daha MR. Therapeutic inhibition of the early phase of complement activation. Immunobiology. 2002;205:595–609. doi: 10.1078/0171-2985-00157. [DOI] [PubMed] [Google Scholar]
- 52.Kjaer TR, Thiel S, Andersen GR. Toward a structure-based comprehension of the lectin pathway of complement. Mol. Imm. 2013;56:413–422. doi: 10.1016/j.molimm.2013.05.007. [DOI] [PubMed] [Google Scholar]
- 53.Bertini R, et al. Noncompetitive allosteric inhibitors of the inflammatory chemokine receptors CXCR1 and CXCR2: prevention of reperfusion injury. Proc. Natl Acad. Sci. USA. 2004;101:11791–11796. doi: 10.1073/pnas.0402090101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cavalieri B, et al. Neutrophil recruitment in the reperfused-injured rat liver was effectively attenuated by repertaxin, a novel allosteric noncompetitive inhibitor of CXCL8 receptors: a therapeutic approach for the treatment of post-ischemic hepatic syndromes. Int. J. Immunopathol. Pharmacol. 2005;18:475–486. doi: 10.1177/039463200501800307. [DOI] [PubMed] [Google Scholar]
- 55.Phuan PW, et al. C1q targeted monoclonal antibody prevents complement-dependent cytotoxicity and neuropathology in in vitro and mouse models of neuromyelitis optica. Acta Neuropathol. 2013;125:829–840. doi: 10.1007/s00401-013-1128-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Roos A, et al. Specific inhibition of the classical complement pathway by C1q binding peptides. J. Immunol. 2001;167:7052–7059. doi: 10.4049/jimmunol.167.12.7052. [DOI] [PubMed] [Google Scholar]
- 57.Mauriello CT, et al. A novel peptide inhibitor of classical and lectin complement activation including ABO incompatibility. Mol. Immunol. 2013;53:132–139. doi: 10.1016/j.molimm.2012.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sharp JA, Whitley PH, Cunnion KM, Krishna NK. Peptide inhibitor of complement C1, a novel suppressor of classical pathway activation: mechanistic studies and clinical potential. Front. Immunol. 2014;5:406. doi: 10.3389/fimmu.2014.00406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gadek JE, et al. Replacement therapy in hereditary angioedema: successful treatment of acute episodes of angioedema with partly purified C1 inhibitor. N. Engl. J. Med. 1980;302:542–546. doi: 10.1056/NEJM198003063021002. [DOI] [PubMed] [Google Scholar]
- 60.Zanichelli A, Mansi M, Periti G, Cicardi M. Therapeutic management of hereditary angioedema due to C1 inhibitor deficiency. Expert Rev. Clin. Immunol. 2013;9:477–488. doi: 10.1586/eci.13.22. [DOI] [PubMed] [Google Scholar]
- 61.Rosen FS, Alper CA, Pensky J, Klemperer MR, Donaldson VH. Genetically determined heterogeneity of the C1 esterase inhibitor in patients with hereditary angioneurotic edema. J. Clin. Invest. 1971;50:2143–2149. doi: 10.1172/JCI106708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Craig TJ, et al. C1 esterase inhibitor concentrate in 1085 hereditary angioedema attacks — final results of the I.M.P.A.C.T.2 study. Allergy. 2011;66:1604–1611. doi: 10.1111/j.1398-9995.2011.02702.x. [DOI] [PubMed] [Google Scholar]
- 63.Bork K, Witzke G. Long-term prophylaxis with C1 inhibitor (C1 INH) concentrate in patients with recurrent angioedema caused by hereditary and acquired C1 inhibitor deficiency. J. Allergy Clin. Immunol. 1989;83:677–682. doi: 10.1016/0091-6749(89)90082-1. [DOI] [PubMed] [Google Scholar]
- 64.Bernstein JA, et al. Escalating doses of C1 esterase inhibitor (CINRYZE) for prophylaxis in patients with hereditary angioedema. J. Allergy Clin. Immunol. Pract. 2014;2:77–84. doi: 10.1016/j.jaip.2013.09.008. [DOI] [PubMed] [Google Scholar]
- 65.Bork K. An evidence based therapeutic approach to hereditary and acquired angioedema. Curr. Opin. Allergy Clin. Immunol. 2014;14:354–362. doi: 10.1097/ACI.0000000000000082. [DOI] [PubMed] [Google Scholar]
- 66.Caliezi C, et al. C1 inhibitor in patients with severe sepsis and septic shock: beneficial effect on renal dysfunction. Crit. Care Med. 2002;30:1722–1728. doi: 10.1097/00003246-200208000-00008. [DOI] [PubMed] [Google Scholar]
- 67.Igonin AA, et al. C1 esterase inhibitor infusion increases survival rates for patients with sepsis. Crit. Care Med. 2012;40:770–777. doi: 10.1097/CCM.0b013e318236edb8. [DOI] [PubMed] [Google Scholar]
- 68.Lu F, Fernandes SM, Davis AE. 3rd The effect of C1 inhibitor on myocardial ischemia and reperfusion injury. Cardiovasc. Pathol. 2013;22:75–80. doi: 10.1016/j.carpath.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lu F, et al. The effect of C1 inhibitor on intestinal ischemia and reperfusion injury. Am. J. Physiol. Gastrointest. Liver Physiol. 2008;295:G1042–G1049. doi: 10.1152/ajpgi.90460.2008. [DOI] [PubMed] [Google Scholar]
- 70.Subasinghe NL, et al. Design and synthesis of biphenylsulphonyl thiophene carbxamide inhibitors of the complement component C1s. Bioorg. Med. Chem. Lett. 2012;22:5303–5307. doi: 10.1016/j.bmcl.2012.06.030. [DOI] [PubMed] [Google Scholar]
- 71.Coppens M, Eikelboom JW, Gustafsson D, Weitz JI, Hirsh J. Translational success stories: development of direct thrombin inhibitors. Circ. Res. 2012;111:920–929. doi: 10.1161/CIRCRESAHA.112.264903. [DOI] [PubMed] [Google Scholar]
- 72.Héja D, et al. Monospecific inhibitors show that both mannan-binding lectin-associated serine protease 1 (MASP 1) and -2 are essential for lectin pathway activation and reveal structural plasticity of MASP 2. J. Biol. Chem. 2012;287:20290–20300. doi: 10.1074/jbc.M112.354332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Carroll S, Georgiou G. Antibody-mediates inhibition of human C1s and the classical complement pathway. Immunobiology. 2013;218:1041–1048. doi: 10.1016/j.imbio.2013.01.001. [DOI] [PubMed] [Google Scholar]
- 74.Nydegger UE, Fearon DT, Austen KF. The modulation of the alternative pathway of complement in C2 deficient human serum by changes in concentration of the component and control proteins. J. Immunol. 1978;120:1404–1408. [PubMed] [Google Scholar]
- 75.Heinen S, et al. Hemolytic uremic syndrome: a factor H mutation (E1172Stop) causes defective complement control at the surface of endothelial cells. J. Am. Soc. Nephrol. 2007;18:506–514. doi: 10.1681/ASN.2006091069. [DOI] [PubMed] [Google Scholar]
- 76.Fakhouri F, et al. Treatment with human complement factor H rapidly reverses renal complement deposition in factor H deficient mice. Kidney Int. 2010;78:279–286. doi: 10.1038/ki.2010.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kim SJ, et al. Intravitreal human complement factor H in a rat model of laser-induced choroidal neovascularisation. Br. J. Ophthalmol. 2013;97:367–370. doi: 10.1136/bjophthalmol-2012-302307. [DOI] [PubMed] [Google Scholar]
- 78.Khandhadia S, et al. Age-related macular degeneration and modification of systemic complement factor H production through liver transplantation. Ophthalmology. 2013;120:1612–1618. doi: 10.1016/j.ophtha.2013.01.004. [DOI] [PubMed] [Google Scholar]
- 79.Schmidt CQ, et al. Rational engineering of a minimized immune inhibitor with unique triple-targeting properties. J. Immunol. 2013;190:5712–5721. doi: 10.4049/jimmunol.1203548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hebecker M, et al. An engineered construct combining complement regulatory and surface-recognition domains represents a minimal-size functional factor H. J. Immunol. 2013;191:912–921. doi: 10.4049/jimmunol.1300269. [DOI] [PubMed] [Google Scholar]
- 81.Trapp RG, Fletcher M, Forristal J, West CD. C4 binding protein deficiency in a patient with atypical Behçet's disease. J. Rheumatol. 1987;14:135–138. [PubMed] [Google Scholar]
- 82.Blom AM, Nandakumar KS, Holmdahl R. C4b binding protein (C4BP) inhibits development of experimental arthritis in mice. Ann. Rheum. Dis. 2009;68:136–142. doi: 10.1136/ard.2007.085753. [DOI] [PubMed] [Google Scholar]
- 83.Grumach AS, Leitão MF, Arruk VG, Kirschfink M, Condino-Neto A. Recurrent infections in partial complement factor I deficiency: evaluation of three generations of a Brazilian family. Clin. Exp. Immunol. 2006;143:297–304. doi: 10.1111/j.1365-2249.2005.02988.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Møller Rasmussen J, et al. Three cases of factor I deficiency: the effect of treatment with plasma. Clin. Exp. Immunol. 1988;74:131–136. [PMC free article] [PubMed] [Google Scholar]
- 85.Lay E, et al. Complotype affects the extent of down-regulation by Factor I of the C3b feedback cycle in vitro. Clin. Exp. Immunol. 2014;181:314–322. doi: 10.1111/cei.12437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lesher AM, Nilsson B, Song WC. Properdin in complement activation and tissue injury. Mol. Immunol. 2013;56:191–198. doi: 10.1016/j.molimm.2013.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ali YM, et al. Low-dose recombinant properdin provides substantial protection against Streptococcus pneumoniae and Neisseria meningitidis infection. Proc. Natl Acad. Sci. USA. 2014;111:5301–5306. doi: 10.1073/pnas.1401011111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Weisman HF, et al. Soluble human complement receptor type 1: in vivo inhibitor of complement suppressing post-ischemic myocardial inflammation and necrosis. Science. 1990;249:146–151. doi: 10.1126/science.2371562. [DOI] [PubMed] [Google Scholar]
- 89.Schmid RA, Hillinger S, Hamacher J, Stammberger U. TP20 is superior to TP10 in reducing ischemia/reperfusion injury in rat lung grafts. Transplant. Proc. 2001;33:948–949. doi: 10.1016/S0041-1345(00)02279-X. [DOI] [PubMed] [Google Scholar]
- 90.Linton SM, et al. Therapeutic efficacy of a novel membrane-targeted complement regulator in antigen-induced arthritis in the rat. Arthritis Rheum. 2000;43:2590–2597. doi: 10.1002/1529-0131(200011)43:11<2590::AID-ANR29>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 91.Banz Y, et al. Attenuation of myocardial reperfusion injury in pigs by Mirococept, a membrane-targeted complement inhibitor derived from human CR1. Cardiovasc. Res. 2007;76:482–493. doi: 10.1016/j.cardiores.2007.07.016. [DOI] [PubMed] [Google Scholar]
- 92.Sprong T, et al. Deficient alternative complement pathway activation due to factor D deficiency by 2 novel mutations in the complement factor D gene in a family with meningococcal infections. Blood. 2006;107:4865–4870. doi: 10.1182/blood-2005-07-2820. [DOI] [PubMed] [Google Scholar]
- 93.Katschke KJ, Jr, et al. Inhibiting alternative pathway complement activation by targeting the factor D exosite. J. Biol. Chem. 2012;287:12886–12892. doi: 10.1074/jbc.M112.345082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Cole LB, Kilpatrick JM, Chu N, Babu YS. Structure of 3,4 dichloro isocoumarin-inhibited factor D. Acta Crystallogr. D Biol. Crystallogr. 1998;54:711–717. doi: 10.1107/S0907444997010457. [DOI] [PubMed] [Google Scholar]
- 95.Sukuru SC, et al. A lead discovery strategy driven by a comprehensive analysis of proteases in the peptide substrate space. Protein Sci. 2010;19:2096–2109. doi: 10.1002/pro.490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Abdel-Magid AF. Inhibitors of factor D may provide a treatment for age-related macular degeneration. ACS Med. Chem. Lett. 2014;5:106–107. doi: 10.1021/ml500037a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hu X, et al. Therapeutic inhibition of the alternative complement pathway attenuates chronic EAE. Mol. Immunol. 2013;54:302–308. doi: 10.1016/j.molimm.2012.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Grossman, T. R. et al. Inhibition of the alternative complement pathway by antisense oligonucleotides targeting complement factor B improves lupus nephritis in mice. Immunobiology10.1016/j.imbio.2015.08.001 (2015). [DOI] [PubMed]
- 99.Oh S, Kweon MH, Rhee KH, Ho Lee K, Sung HC. Inhibition of complement by recombinant Sh-CRIT-ed1 analogues. Immunology. 2003;110:73–79. doi: 10.1046/j.1365-2567.2003.01706.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Maillard JL, Zarco RM. Decomplementation par un facteur extrait du venin de cobra. Ann. Inst. Pasteur. 1968;114:756–780. [PubMed] [Google Scholar]
- 101.Wright MC, Nelson RA, Finch SC. The effects of a cobra venom factor and ethyl palmitate on the prolongation of survival of heterologous erythrocytes. Yale J. Biol. Med. 1970;43:173–176. [PMC free article] [PubMed] [Google Scholar]
- 102.Till GO, Johnson KJ, Kunkel R, Ward PA. Intravascular activation of complement and acute lung injury. Dependency on neutrophils and toxic oxygen metabolites. J. Clin. Invest. 1982;69:1126–1135. doi: 10.1172/JCI110548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Vogel CW, Finnegan PW, Fritzinger DC. Humanized cobra venom factor: activity and therapeutic efficacy in preclinical disease models. Mol. Immunol. 2014;61:191–203. doi: 10.1016/j.molimm.2014.06.035. [DOI] [PubMed] [Google Scholar]
- 104.Kinnunen K, Yla-Herttuala S. Gene therapy in age-related macular degeneration and hereditary macular disorders. Front. Biosci. 2012;4:2546–2557. doi: 10.2741/e565. [DOI] [PubMed] [Google Scholar]
- 105.Fluiter K, Opperhuizen AL, Morgan BP, Baas F, Ramaglia V. Inhibition of the membrane attack complex of the complement system reduces secondary neuroaxonal loss and promotes neurologic recovery after traumatic brain injury in mice. J. Immunol. 2014;192:2339–2348. doi: 10.4049/jimmunol.1302793. [DOI] [PubMed] [Google Scholar]
- 106.Klos A, et al. The role of the anaphylatoxins in health and disease. Mol. Immunol. 2009;46:2753–2766. doi: 10.1016/j.molimm.2009.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Engelke C, et al. Distinct roles of the anaphylatoxins C3a and C5a in dendritic cell-mediated allergic asthma. J. Immunol. 2014;193:5387–5401. doi: 10.4049/jimmunol.1400080. [DOI] [PubMed] [Google Scholar]
- 108.Solari R, Pease JE, Begg M. Chemokine receptors as therapeutic targets: why aren't there more drugs? Eur. J. Pharmacol. 2014;746:363–367. doi: 10.1016/j.ejphar.2014.06.060. [DOI] [PubMed] [Google Scholar]
- 109.Woodruff TM, et al. Therapeutic activity of C5a receptor antagonists in a rat model of neurodegeneration. FASEB J. 2006;20:1407–1417. doi: 10.1096/fj.05-5814com. [DOI] [PubMed] [Google Scholar]
- 110.Vergunst CE, et al. Blocking the receptor for C5a in patients with rheumatoid arthritis does not reduce synovial inflammation. Rheumatology (Oxford) 2007;46:1773–1778. doi: 10.1093/rheumatology/kem222. [DOI] [PubMed] [Google Scholar]
- 111.Wu MC, et al. The receptor for complement component C3a mediates protection from intestinal ischemia-reperfusion injuries by inhibiting neutrophil mobilization. Proc. Natl Acad. Sci. USA. 2013;110:9439–9444. doi: 10.1073/pnas.1218815110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Hoehlig K, et al. A novel C5a neutralizing mirror-image (l-)aptamer prevents organ failure and improves survival in experimental sepsis. Mol. Ther. 2013;21:2236–2246. doi: 10.1038/mt.2013.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Khan MA, et al. Targeting complement component 5a promotes vascular integrity and limits airway remodeling. Proc. Natl Acad. Sci. USA. 2013;110:6061–6066. doi: 10.1073/pnas.1217991110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Singer M. Advances in the management of macular degeneration. F1000Prime Rep. 2014;6:29. doi: 10.12703/P6-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Nunn MA, et al. Complement inhibitor of C5 activation from the soft tick Ornithodoros moubata. J. Immunol. 2005;174:2084–2091. doi: 10.4049/jimmunol.174.4.2084. [DOI] [PubMed] [Google Scholar]
- 116.Fredslund F, et al. Structure of and influence of a tick complement inhibitor on human complement component 5. Nat. Immunol. 2008;9:753–760. doi: 10.1038/ni.1625. [DOI] [PubMed] [Google Scholar]
- 117.Hepburn NJ, et al. In vivo characterization and therapeutic efficacy of a C5 specific inhibitor from the soft tick Ornithodoros moubata. J. Biol. Chem. 2007;282:8292–8299. doi: 10.1074/jbc.M609858200. [DOI] [PubMed] [Google Scholar]
- 118.Vakeva A, et al. High-density lipoproteins can act as carriers of glycophosphoinositol lipid anchored CD59 in human plasma. Immunology. 1994;82:28–33. [PMC free article] [PubMed] [Google Scholar]
- 119.Fraser DA, et al. Generation of a recombinant, membrane-targeted form of the complement regulator CD59; activity in vitro and in vivo. J. Biol. Chem. 2003;278:48921–48927. doi: 10.1074/jbc.M302598200. [DOI] [PubMed] [Google Scholar]
- 120.Hill A, et al. Protection of erythrocytes from human complement-mediated lysis by membrane-targeted recombinant soluble CD59: a new approach to PNH therapy. Blood. 2006;107:2131–2137. doi: 10.1182/blood-2005-02-0782. [DOI] [PubMed] [Google Scholar]
- 121.Lee M, Narayanan S, McGeer EG, McGeer PL. Aurin tricarboxylic acid protects against red blood cell hemolysis in patients with paroxysmal nocturnal hemoglobinemia. PLoS ONE. 2014;29:e87316. doi: 10.1371/journal.pone.0087316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Harris CL, et al. Generation of anti-complement 'prodrugs': cleavable reagents for specific delivery of complement regulators to disease sites. J. Biol. Chem. 2003;278:36068–36076. doi: 10.1074/jbc.M306351200. [DOI] [PubMed] [Google Scholar]
- 123.Bagshawe KD, Sharma SK, Begent RH. Antibody-directed enzyme prodrug therapy (ADEPT) for cancer. Expert Opin. Biol. Ther. 2004;4:1777–1789. doi: 10.1517/14712598.4.11.1777. [DOI] [PubMed] [Google Scholar]
- 124.Atkinson C, et al. Targeted complement inhibition by C3d recognition ameliorates tissue injury without apparent increase in susceptibility to infection. J. Clin. Invest. 2005;115:2444–2453. doi: 10.1172/JCI25208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Atkinson C, et al. Targeted complement inhibitors protect against posttransplant cardiac ischemia and reperfusion injury and reveal an important role for the alternative pathway of complement activation. J. Immunol. 2010;185:7007–7013. doi: 10.4049/jimmunol.1001504. [DOI] [PubMed] [Google Scholar]
- 126.Fridkis-Hareli M, et al. Design and development of TT30, a novel C3d targeted C3/C5 convertase inhibitor for treatment of human complement alternative pathway-mediated diseases. Blood. 2011;118:4705–4713. doi: 10.1182/blood-2011-06-359646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Thurman JM, et al. Detection of complement activation using monoclonal antibodies against C3d. J. Clin. Invest. 2013;123:2218–2230. doi: 10.1172/JCI65861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Thurman JM, Rohrer B. Noninvasive detection of complement activation through radiologic imaging. Adv. Exp. Med. Biol. 2013;735:271–282. doi: 10.1007/978-1-4614-4118-2_19. [DOI] [PubMed] [Google Scholar]
- 129.Harris CL, Heurich M, Cordoba SR, Morgan BP. The complotype: dictating risk for inflammation and infection. Trends Immunol. 2012;33:513–521. doi: 10.1016/j.it.2012.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Heurich M, et al. Common polymorphisms in C3, factor B, and factor H collaborate to determine systemic complement activity and disease risk. Proc. Natl Acad. Sci. USA. 2011;108:8761–8766. doi: 10.1073/pnas.1019338108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Hageman GS, et al. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc. Natl Acad. Sci. USA. 2005;102:7227–7232. doi: 10.1073/pnas.0501536102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Sofat R, et al. Complement factor H genetic variant and age-related macular degeneration: effect size, modifiers and relationship to disease subtype. Int. J. Epidemiol. 2012;41:250–262. doi: 10.1093/ije/dyr204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Gold B, et al. Variation in factor B (BF) and complement component 2 (C2) genes is associated with age-related macular degeneration. Nat. Genet. 2006;38:458–462. doi: 10.1038/ng1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Yates YR, et al. Complement C3 variant and the risk of age-related macular degeneration. N. Engl. J. Med. 2007;357:19–27. doi: 10.1056/NEJMoa072618. [DOI] [PubMed] [Google Scholar]
- 135.Jager RD, Mieler WF, Miller JW. Age-related macular degeneration. N. Engl. J. Med. 2008;358:2606–2617. doi: 10.1056/NEJMra0801537. [DOI] [PubMed] [Google Scholar]
- 136.Kavanagh D, Goodship TH, Richards A. Atypical hemolytic uremic syndrome. Semin. Nephrol. 2013;33:508–530. doi: 10.1016/j.semnephrol.2013.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Manuelian T, et al. Mutations in factor H reduce binding affinity to C3b and heparin and surface attachment to endothelial cells in hemolytic uremic syndrome. J. Clin. Invest. 2003;111:1181–1190. doi: 10.1172/JCI16651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Moore I, et al. Association of factor H autoantibodies with deletions of CFHR1, CFHR3, CFHR4, and with mutations in CFH, CFI, CD46, and C3 in patients with atypical hemolytic uremic syndrome. Blood. 2010;115:379–387. doi: 10.1182/blood-2009-05-221549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Frei Y, Lambris JD, Stockinger B. Generation of a monoclonal antibody to mouse C5; application in an ELISA assay for detection of anti C5 antibodies. Mol. Cell. Probes. 1987;1:141–149. doi: 10.1016/0890-8508(87)90022-3. [DOI] [PubMed] [Google Scholar]
- 140.Wang Y, Rollins SA, Madri JA, Matis LA. Anti C5 monoclonal antibody therapy prevents collagen-induced arthritis and ameliorates established disease. Proc. Natl Acad. Sci. USA. 1995;92:8955–8959. doi: 10.1073/pnas.92.19.8955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Thomas TC, et al. Inhibition of complement activity by humanized anti C5 antibody and single-chain Fv. Mol. Immunol. 1996;33:1389–1140. doi: 10.1016/S0161-5890(96)00078-8. [DOI] [PubMed] [Google Scholar]
- 142.Adis International Limited Eculizumab: 5G1.1, h5G1.1, long-acting anti C5 monoclonal antibody 5G1 1, long-acting anti C5 monoclonal antibody 5G1.1. Drugs R. D.8, 61–68 (2007). [DOI] [PubMed]
- 143.Kaplan M. Eculizumab (Alexion) Curr. Opin. Investig. Drugs. 2002;3:1017–1023. [PubMed] [Google Scholar]
- 144.Hillmen P, et al. Effect of eculizumab on hemolysis and transfusion requirements in patients with paroxysmal nocturnal hemoglobinuria. N. Engl. J. Med. 2004;350:552–559. doi: 10.1056/NEJMoa031688. [DOI] [PubMed] [Google Scholar]
- 145.Brodsky RA, et al. Multicenter phase 3 study of the complement inhibitor eculizumab for the treatment of patients with paroxysmal nocturnal hemoglobinuria. Blood. 2008;111:1840–1847. doi: 10.1182/blood-2007-06-094136. [DOI] [PubMed] [Google Scholar]
- 146.Hillmen P, et al. Long-term safety and efficacy of sustained eculizumab treatment in patients with paroxysmal nocturnal haemoglobinuria. Br. J. Haematol. 2013;162:62–73. doi: 10.1111/bjh.12347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Legendre CM, et al. Terminal complement inhibitor eculizumab in atypical hemolytic-uremic syndrome. N. Engl. J. Med. 2013;368:2169–2181. doi: 10.1056/NEJMoa1208981. [DOI] [PubMed] [Google Scholar]
- 148.Ricklin D, et al. A molecular insight into complement inhibition by the Staphylococcal complement inhibitor family. J. Immunol. 2009;183:2565–2574. doi: 10.4049/jimmunol.0901443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Parker CJ, Kar S, Kirkpatrick P. Eculizumab. Nat. Rev. Drug Disc. 2007;6:515–516. doi: 10.1038/nrd2369. [DOI] [PubMed] [Google Scholar]
- 150.Nishimura J, et al. Genetic variants in C5 and poor response to Eculizumab. N. Engl. J. Med. 2014;370:632–639. doi: 10.1056/NEJMoa1311084. [DOI] [PubMed] [Google Scholar]
- 151.Mullard A. Protein–protein interaction inhibitors get into the groove. Nat. Rev. Drug Disc. 2012;11:173–175. doi: 10.1038/nrd3680. [DOI] [PubMed] [Google Scholar]
- 152.Wu J, et al. Structure of complement fragment C3b–factor H and implications for host protection by complement regulators. Nat. Immunol. 2009;10:728–733. doi: 10.1038/ni.1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Sahu A, Kay BK, Lambris JD. Inhibition of complement by a C3 binding peptide isolated from a phage-displayed random peptide library. J. Immunol. 1996;157:884–891. [PubMed] [Google Scholar]
- 154.Qu H, et al. New analogs of the clinical complement inhibitor compstatin with subnanomolar affinity and enhanced pharmacokinetic properties. Immunobiology. 2013;218:496–505. doi: 10.1016/j.imbio.2012.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Mastellos DC, et al. Compstatin: a C3 targeted complement inhibitor reaching its prime for bedside intervention. Eur. J. Clin. Invest. 2015;45:423–440. doi: 10.1111/eci.12419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Leung E, Landa G. Update on current and future novel therapies for dry age-related macular degeneration. Expert Rev. Clin. Pharmacol. 2013;6:565–579. doi: 10.1586/17512433.2013.829645. [DOI] [PubMed] [Google Scholar]
- 157.Lachmann PJ. The amplification loop of the complement pathways. Adv. Immunol. 2009;104:115–149. doi: 10.1016/S0065-2776(08)04004-2. [DOI] [PubMed] [Google Scholar]
- 158.Martinez-Saguer I, et al. Pharmacokinetics of plasma-derived C1 esterase inhibitor after subcutaneous versus intravenous administration in subjects with mild or moderate hereditary angioedema: the PASSION study. Transfusion. 2014;54:1552–1561. doi: 10.1111/trf.12501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.van Doorn MB, et al. A phase I study of recombinant human C1 inhibitor in asymptomatic patients with hereditary angioedema. J. Allergy Clin. Immunol. 2005;116:876–883. doi: 10.1016/j.jaci.2005.05.019. [DOI] [PubMed] [Google Scholar]
- 160.Riedl MA, et al. Recombinant human C1 esterase inhibitor relieves symptoms of hereditary angioedema attacks: phase 3, randomized, placebo-controlled trial. Ann. Allergy Asthma Immunol. 2014;112:163–169. doi: 10.1016/j.anai.2013.12.004. [DOI] [PubMed] [Google Scholar]
- 161.Riedl M, et al. Spectrum of complement-mediated thrombotic microangiopathies: pathogenetic insights identifying novel treatment approaches. Semin. Thromb. Hemost. 2014;40:444–464. doi: 10.1055/s-0034-1376153. [DOI] [PubMed] [Google Scholar]
- 162.Pittock SJ, et al. Eculizumab in AQP4 IgG-positive relapsing neuromyelitis optica spectrum disorders: an open-label pilot study. Lancet Neurol. 2013;12:554–562. doi: 10.1016/S1474-4422(13)70076-0. [DOI] [PubMed] [Google Scholar]
- 163.Ingram G, Hakobyan S, Robertson NP, Morgan BP. Complement in multiple sclerosis: its role in disease and potential as a biomarker. Clin. Exp. Immunol. 2009;155:128–139. doi: 10.1111/j.1365-2249.2008.03830.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Ingram G, et al. Systemic complement profiling in multiple sclerosis as a biomarker of disease state. Mult. Scler. 2012;18:1401–1411. doi: 10.1177/1352458512438238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Willison HJ, et al. The role of complement and complement regulators in mediating motor nerve terminal injury in murine models of Guillain–Barré syndrome. J. Neuroimmunol. 2008;201–202:172–182. doi: 10.1016/j.jneuroim.2008.05.028. [DOI] [PubMed] [Google Scholar]
- 166.Walgaard C, Jacobs BC, van Doorn PA. Emerging drugs for Guillain–Barré syndrome. Expert Opin. Emerg. Drugs. 2011;16:105–120. doi: 10.1517/14728214.2011.531699. [DOI] [PubMed] [Google Scholar]
- 167.Morgan BP, et al. The membrane attack pathway of complement drives pathology in passively induced experimental autoimmune myasthenia gravis in mice. Clin. Exp. Immunol. 2006;146:294–302. doi: 10.1111/j.1365-2249.2006.03205.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Howard JF, Jr, et al. A randomized, double-blind, placebo-controlled phase II study of eculizumab in patients with refractory generalized myasthenia gravis. Muscle Nerve. 2013;48:76–84. doi: 10.1002/mus.23839. [DOI] [PubMed] [Google Scholar]
- 169.Coppo R, et al. Dramatic effects of eculizumab in a child with diffuse proliferative lupus nephritis resistant to conventional therapy. Pediatr. Nephrol. 2015;30:167–172. doi: 10.1007/s00467-014-2944-y. [DOI] [PubMed] [Google Scholar]
- 170.Kiryluk K, Novak J. The genetics and immunobiology of IgA nephropathy. J. Clin. Invest. 2014;124:2325–2332. doi: 10.1172/JCI74475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Rosenblad T, et al. Eculizumab treatment for rescue of renal function in IgA nephropathy. Pediatr. Nephrol. 2014;29:2225–2228. doi: 10.1007/s00467-014-2863-y. [DOI] [PubMed] [Google Scholar]
- 172.Okroj M, Heinegård D, Holmdahl R, Blom AM. Rheumatoid arthritis and the complement system. Ann. Med. 2007;39:517–530. doi: 10.1080/07853890701477546. [DOI] [PubMed] [Google Scholar]
- 173.Sivaram G, et al. Association of genetic variants of mannan-binding (MBL) lectin 2 gene, MBL levels and function in ulcerative colitis and Crohn's disease. Innate Immun. 2011;17:526–531. doi: 10.1177/1753425910384531. [DOI] [PubMed] [Google Scholar]
- 174.Elvington M, Schepp-Berglind J, Tomlinson S. Regulation of the alternative pathway of complement modulates injury and immunity in a chronic model of DSS-induced colitis. Clin Exp Immunol. 2014;179:500–508. doi: 10.1111/cei.12464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Gupta N, Wang ES. Long-term response of refractory primary cold agglutinin disease to eculizumab therapy. Ann. Hematol. 2014;93:343–344. doi: 10.1007/s00277-013-1800-7. [DOI] [PubMed] [Google Scholar]
- 176.Lessey E, Li N, Diaz L, Liu Z. Complement and cutaneous autoimmune blistering diseases. Immunol. Res. 2008;41:223–232. doi: 10.1007/s12026-008-8028-y. [DOI] [PubMed] [Google Scholar]
- 177.Lim W. Complement and the antiphospholipid syndrome. Curr. Opin. Hematol. 2011;18:361–365. doi: 10.1097/MOH.0b013e3283497f3e. [DOI] [PubMed] [Google Scholar]
- 178.Lonze BE, et al. Eculizumab prevents recurrent antiphospholipid antibody syndrome and enables successful renal transplantation. Am. J. Transplant. 2014;14:459–465. doi: 10.1111/ajt.12540. [DOI] [PubMed] [Google Scholar]
- 179.Yang MM, et al. Association of C2 and CFB polymorphisms with anterior uveitis. Invest. Ophthalmol. Vis. Sci. 2012;53:4969–4974. doi: 10.1167/iovs.12-9478. [DOI] [PubMed] [Google Scholar]
- 180.Kallenberg CG, Stegeman CA, Abdulahad WH, Heeringa P. Pathogenesis of ANCA-associated vasculitis: new possibilities for intervention. Am. J. Kidney Dis. 2013;62:1176–1187. doi: 10.1053/j.ajkd.2013.05.009. [DOI] [PubMed] [Google Scholar]
- 181.Lewis RD, Jackson CL, Morgan BP, Hughes TR. The membrane attack complex of complement drives the progression of atherosclerosis in apolipoprotein E knockout mice. Mol. Immunol. 2010;47:1098–1105. doi: 10.1016/j.molimm.2009.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Hertle E, Stehouwer CD, van Greevenbroek MM. The complement system in human cardiometabolic disease. Mol. Immunol. 2014;61:135–148. doi: 10.1016/j.molimm.2014.06.031. [DOI] [PubMed] [Google Scholar]
- 183.Li Y, et al. Label-free quantitative proteomic analysis reveals dysfunction of complement pathway in peripheral blood of schizophrenia patients: evidence for the immune hypothesis of schizophrenia. Mol. Biosyst. 2012;8:2664–2671. doi: 10.1039/c2mb25158b. [DOI] [PubMed] [Google Scholar]
- 184.Håvik B, et al. The complement control-related genes CSMD1 and CSMD2 associate to schizophrenia. Biol. Psychiatry. 2011;70:35–42. doi: 10.1016/j.biopsych.2011.01.030. [DOI] [PubMed] [Google Scholar]
- 185.Zhang X, Köhl J. A complex role for complement in allergic asthma. Expert Rev. Clin. Immunol. 2010;6:269–277. doi: 10.1586/eci.09.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Khan MA, Nicolls MR, Surguladze B, Saadoun I. Complement components as potential therapeutic targets for asthma treatment. Respir. Med. 2014;108:543–549. doi: 10.1016/j.rmed.2014.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Marc MM, et al. Complement factors C3a, C4a, and C5a in chronic obstructive pulmonary disease and asthma. Am. J. Respir. Cell. Mol. Biol. 2004;31:216–219. doi: 10.1165/rcmb.2003-0394OC. [DOI] [PubMed] [Google Scholar]
- 188.Kodama T, et al. Role of complement in a murine model of peanut-induced anaphylaxis. Immunobiology. 2013;218:844–850. doi: 10.1016/j.imbio.2012.10.003. [DOI] [PubMed] [Google Scholar]
- 189.Khodoun M, et al. Peanuts can contribute to anaphylactic shock by activating complement. J. Allergy Clin. Immunol. 2009;123:342–351. doi: 10.1016/j.jaci.2008.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Rittirsch D, Redl H, Huber-Lang M. Role of complement in multiorgan failure. Clin. Dev. Immunol. 2012;2012:962927. doi: 10.1155/2012/962927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Huber-Lang M, et al. Double blockade of CD14 and complement C5 abolishes the cytokine storm and improves morbidity and survival in polymicrobial sepsis in mice. J. Immunol. 2014;192:5324–5331. doi: 10.4049/jimmunol.1400341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Ramos-Summerford TN, Barnum SR. Experimental cerebral malaria: the murine model provides crucial insight into the role of complement. Trends Parasitol. 2014;30:215–216. doi: 10.1016/j.pt.2014.03.002. [DOI] [PubMed] [Google Scholar]
- 193.Chimenti MS, et al. Imunomodulation in psoriatic arthritis: focus on cellular and molecular pathways. Autoimmun. Rev. 2013;12:599–606. doi: 10.1016/j.autrev.2012.10.002. [DOI] [PubMed] [Google Scholar]
- 194.Chimenti MS, et al. Complement system in psoriatic arthritis: a useful marker in response prediction and monitoring of anti-TNF treatment. Clin. Exp. Rheumatol. 2012;30:23–30. [PubMed] [Google Scholar]
- 195.De Bleecker JL, Creus KK, De Paepe B. Potential therapeutic targets for idiopathic inflammatory myopathies. Drug News Perspect. 2006;19:549–557. doi: 10.1358/dnp.2006.19.9.1050424. [DOI] [PubMed] [Google Scholar]
- 196.Gordon PA, Winer JB, Hoogendijk JE, Choy EH. Immunosuppressant and immunomodulatory treatment for dermatomyositis and polymyositis. Cochrane Database Syst. Rev. 2012;8:CD003643. doi: 10.1002/14651858.CD003643.pub4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Wang Q, et al. Identification of a central role for complement in osteoarthritis. Nat. Med. 2011;17:1674–1679. doi: 10.1038/nm.2543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Lepus CM, et al. Brief report: carboxypeptidase B serves as a protective mediator in osteoarthritis. Arthritis Rheumatol. 2014;66:101–106. doi: 10.1002/art.38213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Eikelenboom P, et al. Innate immunity and the etiology of late-onset Alzheimer's disease. Neurodegener. Dis. 2012;10:271–273. doi: 10.1159/000334287. [DOI] [PubMed] [Google Scholar]
- 200.Wyss-Coray T, Rogers J. Inflammation in Alzheimer disease a brief review of the basic science and clinical literature. Cold Spring Harb Perspect. Med. 2012;2:a006346. doi: 10.1101/cshperspect.a006346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Rosen AM, Stevens B. The role of the classical complement cascade in synapse loss during development and glaucoma. Adv. Exp. Med. Biol. 2010;703:75–93. doi: 10.1007/978-1-4419-5635-4_6. [DOI] [PubMed] [Google Scholar]
- 202.Flyvbjerg A. Diabetic angiopathy, the complement system and the tumor necrosis factor superfamily. Nat. Rev. Endocrinol. 2010;6:94–101. doi: 10.1038/nrendo.2009.266. [DOI] [PubMed] [Google Scholar]
- 203.Yanai R, Thanos A, Connor KM. Complement involvement in neovascular ocular diseases. Adv. Exp. Med. Biol. 2012;946:161–183. doi: 10.1007/978-1-4614-0106-3_10. [DOI] [PubMed] [Google Scholar]
- 204.Lindberg S, et al. Soluble form of membrane attack complex independently predicts mortality and cardiovascular events in patients with ST elevation myocardial infarction treated with primary percutaneous coronary intervention. Am. Heart J. 2012;164:786–792. doi: 10.1016/j.ahj.2012.08.018. [DOI] [PubMed] [Google Scholar]
- 205.Orsini F, et al. Targeting mannose-binding lectin confers long-lasting protection with a surprisingly wide therapeutic window in cerebral ischemia. Circulation. 2012;126:1484–1494. doi: 10.1161/CIRCULATIONAHA.112.103051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Heydenreich N, et al. C1 inhibitor protects from brain ischemia-reperfusion injury by combined antiinflammatory and antithrombotic mechanisms. Stroke. 2012;43:2457–2467. doi: 10.1161/STROKEAHA.112.660340. [DOI] [PubMed] [Google Scholar]
- 207.Huber-Lang M, Kovtun A, Ignatius A. The role of complement in trauma and fracture healing. Semin. Immunol. 2013;25:73–78. doi: 10.1016/j.smim.2013.05.006. [DOI] [PubMed] [Google Scholar]
- 208.Gorsuch WB, Chrysanthou E, Schwaeble WJ, Stahl GL. The complement system in ischemia-reperfusion injuries. Immunobiology. 2012;217:1026–1033. doi: 10.1016/j.imbio.2012.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.DeAngelis RA, Reis ES, Ricklin D, Lambris JD. Targeted complement inhibition as a promising strategy for preventing inflammatory complications in hemodialysis. Immunobiology. 2012;217:1097–1105. doi: 10.1016/j.imbio.2012.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Reis ES, et al. Therapeutic C3 inhibitor Cp40 abrogates complement activation induced by modern hemodialysis filters. Immunobiology. 2014;220:476–482. doi: 10.1016/j.imbio.2014.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Noris M, Mescia F, Remuzzi G. STEC-HUS, atypical HUS and TTP are all diseases of complement activation. Nat. Rev. Nephrol. 2012;8:622–633. doi: 10.1038/nrneph.2012.195. [DOI] [PubMed] [Google Scholar]
- 212.Yehoshua Z, et al. Systemic complement inhibition with eculizumab for geographic atrophy in age-related macular degeneration: the COMPLETE study. Ophthalmology. 2014;121:693–701. doi: 10.1016/j.ophtha.2013.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Stegall MD, et al. Terminal complement inhibition decreases antibody-mediated rejection in sensitized renal transplant recipients. Am. J. Transplant. 2011;11:2405–2413. doi: 10.1111/j.1600-6143.2011.03757.x. [DOI] [PubMed] [Google Scholar]