

Abstract
Background
Randomised clinical trials have addressed the question whether propylthiouracil has any beneficial effects in patients with alcoholic liver disease.
Objectives
To assess the beneficial and harmful effects of propylthiouracil for patients with alcoholic liver disease.
Search methods
We searched The Cochrane Hepato‐Biliary Group Controlled Trials Register (April 2011), The Cochrane Central Register of Controlled Trials (CENTRAL) in The Cochrane Library (April 2011), MEDLINE (1948 to April 2011), EMBASE (1980 to April 2011), and Science Citation Index Expanded (1900 to April 2011). These electronic searches were combined with full text searches. Manufacturers and researchers in the field were also contacted.
Selection criteria
Randomised clinical trials studying patients with alcoholic steatosis, alcoholic fibrosis, alcoholic hepatitis, and/or alcoholic cirrhosis were included irrespective of blinding, publication status, or language. Interventions encompassed propylthiouracil at any dose versus placebo or no intervention.
Data collection and analysis
All analyses were performed according to the intention‐to‐treat method in RevMan Analyses. The risk of bias of the randomised clinical trials was evaluated by bias risk domains such as generation of allocation sequence, allocation concealment, blinding, incomplete outcome data, selective outcome reporting, academic bias, and source of funding.
Main results
Combining the results of six randomised clinical trials with high risk of bias which included 710 patients demonstrated no significant effects of propylthiouracil versus placebo on all‐cause mortality (risk ratio (RR) 0.93, 95% confidence interval (CI) 0.66 to 1.30), liver‐related mortality (RR 0.90, 95% CI 0.58 to 1.40), or complications of the liver disease. Although propylthiouracil was not associated with a significant increased risk of non‐serious adverse events, there were occasional instances of serious adverse events such as leukopenia and generalised bullous eruption.
Authors' conclusions
We could not demonstrate any significant beneficial effect of propylthiouracil on all‐cause mortality, liver‐related mortality, liver complications, or liver histology of patients with alcoholic liver disease. Propylthiouracil was associated with adverse events. Confidence intervals were wide. Thus, the risk of random errors and systematic errors was high. Accordingly, there is no evidence for using propylthiouracil for alcoholic liver disease outside randomised clinical trials.
Plain language summary
Propylthiouracil for alcoholic liver disease
The majority of liver diseases are caused by alcohol in the Western world. Propylthiouracil ‐ an antithyroid drug that is used for patients with raised metabolism ‐ has been suggested as a potential treatment for alcoholic liver disease. Six randomised clinical trials with a total of 710 patients were included in this systematic review. The trials were generally with high risk of bias. We could not demonstrate any significant effect of propylthiouracil on all‐cause mortality, liver‐related mortality, liver complications, or liver histology of patients with alcoholic liver disease. Although propylthiouracil was not associated with a significant increased risk of non‐serious adverse events, there were occasional instances of serious adverse events (leukopenia, generalized bullous eruption). The trials included a small number of patients, and so, the risk of random error (error due to play of chance) is high. There seems to be no evidence for using propylthiouracil for alcoholic liver disease outside randomised clinical trials.
Background
Liver fibrosis and liver cirrhosis are common reactions to a number of hepatotoxic substances, hepatotropic viruses, autoimmune liver diseases, metabolic liver diseases, etc. Alcohol and hepatotropic viruses are the cause of the majority of liver fibrosis and cirrhosis in the Western world. For example, the attributable risk for symptomatic liver cirrhosis in Italy explained by alcohol consumption, hepatitis B virus, and hepatitis C virus was 98.1% in men and 67.0% in women (Corrao 1998a).
Alcohol is a major hepatotoxin (Morgan 1999). Alcohol leads to fatty liver (Rubin 1968), alcoholic hepatitis, fibrosis, and cirrhosis (Morgan 1999). Alcoholic hepatitis is associated with peripheral leukocytosis and marked hepatic portal and parenchymal inflammatory infiltration predominantly by neutrophils (Hill 1993; Sheron 1993). Data from long‐term studies in which patients with alcoholic fatty change and alcoholic hepatitis were followed for up to 13 years demonstrates that alcoholic hepatitis is a predictor of a later development of liver fibrosis and cirrhosis (Sørensen 1984; Marbet 1987). Alcohol‐induced necrosis and inflammation may trigger the scarring and the development of fibrosis, and later on of the development of cirrhosis. In fact, about 70% of patients with clinical alcoholic hepatitis also have alcoholic cirrhosis at the time of diagnosis (Mendenhall 1984). Five‐year survival rates in patients with alcoholic cirrhosis who stop drinking are of order of 50% to 75%; whereas survival rates in patients continuing to drink rarely exceed 40% (Powell 1968). The progression of liver fibrosis and cirrhosis in patients with alcohol problems is enhanced by the presence of hepatitis B and hepatitis C virus markers (Chang 1994; Corrao 1998b).
Propylthiouracil (PTU) may reduce alcohol induced hepatocyte damage by acting as an antioxidant (Hicks 1992) and suppressing alcohol induced hepatic necrosis (Israel 1975a). Studies have found a 'hypermetabolic state', with an increase in hepatic oxygen consumption in rats chronically treated with alcohol (Israel 1975a) as well as in alcoholic patients (Iturriaga 1980). PTU, an antithyroid drug (Kampmann 1981; Klein 1994), reacts with some of the oxidizing species derived from the respiratory burst in neutrophils (Imamura 1986; Carmichael 1993; Ross 1998). PTU protects rat liver and isolated hepatocytes from ischaemic damage (Israel 1975b; Younes 1987; Gonzalez‐Reimers1988). Therefore, PTU could slow the progression of alcoholic liver disease.
Several randomised clinical trials have addressed the question whether PTU has any efficacy in patients with alcoholic liver disease. The results of these trials have been contradictory (Orrego 1979a; Hallé 1982a; Orrego 1987a). Some investigators found beneficial effects of PTU on all‐cause mortality, complications, and biochemistry (Orrego 1979a; Orrego 1987a). Others found no significant effect on all‐cause mortality (Hallé 1982a). Based on a questionnaire survey among European hospital‐based specialists in gastroenterology/hepatology, 15% of the specialists considered using PTU for alcoholic hepatitis (Gluud 1993). The present systematic review examines the beneficial and harmful effects of PTU for alcoholic liver disease.
Objectives
To assess the beneficial and harmful effects of PTU versus placebo or no intervention for patients with alcoholic liver disease based on the results of randomised clinical trials.
Methods
Criteria for considering studies for this review
Types of studies
Only randomised clinical trials were included, irrespective of blinding, publication status, or language. Trials using quasi‐randomisation were excluded.
Types of participants
Patients with alcoholic steatosis, alcoholic fibrosis, alcoholic hepatitis, and/or alcoholic cirrhosis were included.
Types of interventions
Peroral or parenteral administration of PTU at any dose versus placebo or no intervention. Additional interventions were allowed, as long as both intervention groups in the individual trial received the additional intervention.
Types of outcome measures
Primary outcomes
1. Number of patients dying (total and liver‐related deaths).
Secondary outcomes
1. Development of clinical symptoms and complications (ie, ascites, variceal bleeding, hepatic encephalopathy, hepato‐renal syndrome, hepato‐cellular carcinoma). 2. Liver biopsy findings. 3. Number and type of adverse events (non‐serious and serious). Adverse events were defined as any untoward medical occurrence that did not have a causal relationship with the treatment. Serious adverse events were defined according to the ICH guidelines (ICH‐GCP 1997) as any event that would increase all‐cause mortality; was life‐threatening; required in‐patient hospitalisation; resulted in a persistent or significant disability; or any important medical event, which may jeopardise the patient or required intervention to prevent it. 4. Quality‐of‐life. 5. Health economics.
Search methods for identification of studies
Electronic searches
Relevant randomised clinical trials were identified by searching The Cochrane Hepato‐Biliary Group Controlled Trials Register (Gluud 2011), The Cochrane Central Register of Controlled Trials (CENTRAL) in The Cochrane Library, MEDLINE, EMBASE, and Science Citation Index Expanded (Royle 2003). The search strategies applied to the individual electronic databases and the time span of the searches are given in Appendix 1.
Searching other resources
Further trials were identified by reading the reference lists of the identified studies.
The principal authors of the identified randomised clinical trials were approached and inquired about additional randomised clinical trials they might know. Pharmaceutical companies involved in the production of PTU were contacted in order to obtain unpublished randomised clinical trials.
Data collection and analysis
The meta‐analyses were conducted according to the published protocol (Rambaldi 2001a) as recommended by The Cochrane Collaboration (Higgins 2011) and The Cochrane Hepato‐Biliary Group Module (Gluud 2011).
Patient characteristics, diagnosis, and treatments The following data were recorded from the individual randomised clinical trials: mean (or median) age, sex ratio, alcohol consumption, form of liver disease, etiology of liver disease, duration of liver disease, severity of liver disease at entry, type and dose of PTU intervention, and type of intervention in the control group. The diagnostic work‐up before entry was registered, specifically if hepatitis markers were evaluated and the types of alcoholic liver disease excluded were specified. Development of clinical symptoms and complications, liver biochemistry, liver function, liver biopsy findings, alcohol consumption, quality‐of‐life, health economics (ie, length of hospital stay, cost of medication, and cost of additional follow‐up weighted against any gains in health), and adverse events during follow‐up were registered.
Selection and data‐extraction bias All randomised clinical trials considered for inclusion were analysed by the contributors, who planned to confer with an 'ombudsman' in case disagreements could not be solved. Such cases did not occur.
All randomised clinical trials had the pertinent data extracted by the contributors.
All identified trials were listed and trials excluded from the meta‐analysis of the review were identified with the reason for exclusion.
Assessment of risk of bias The risk of bias in the randomised clinical trials was assessed using generation of allocation sequence, allocation concealment, blinding, incomplete outcome data, selective outcome reporting, baseline imbalance, early stopping, academic bias, source of funding (Schulz 1995; Moher 1998; Kjaergard 2001; Wood 2008; Gurusamy 2009; Gluud 2011; Higgins 2011). Quality components were classified as follows:
Sequence generation
Low risk of bias (the methods used are either adequate (eg, computer generated random numbers, table of random numbers) or unlikely to introduce confounding).
Uncertain risk of bias (there is insufficient information to assess whether the method used is likely to introduce confounding).
High risk of bias (the method used (eg, quasi‐randomised studies) is improper and likely to introduce confounding).
Allocation concealment
Low risk of bias (the method used (eg, central allocation) is unlikely to induce bias on the final observed effect).
Uncertain risk of bias (there is insufficient information to assess whether the method used is likely to induce bias on the estimate of effect).
High risk of bias (the method used (eg, open random allocation schedule) is likely to induce bias on the final observed effect).
Blinding of participants, personnel, and outcome assessors
Low risk of bias (blinding was performed adequately, or the outcome measurement is not likely to be influenced by lack of blinding).
Uncertain risk of bias (there is insufficient information to assess whether the type of blinding used is likely to induce bias on the estimate of effect).
High risk of bias (no blinding or incomplete blinding, and the outcome or the outcome measurement is likely to be influenced by lack of blinding).
Incomplete outcome data
Low risk of bias (the underlying reasons for missingness are unlikely to make treatment effects departure from plausible values, or proper methods have been employed to handle missing data).
Uncertain risk of bias (there is insufficient information to assess whether the missing data mechanism in combination with the method used to handle missing data is likely to induce bias on the estimate of effect).
High risk of bias (the crude estimate of effects (eg, complete case estimate) will clearly be biased due to the underlying reasons for missingness, and the methods used to handle missing data are unsatisfactory).
Selective outcome reporting
Low risk of bias (the trial protocol is available and all of the trial's pre‐specified outcomes that are of interest in the review have been reported or similar; if the trial protocol is not available, all the primary outcomes in this review are reported).
Uncertain risk of bias (there is insufficient information to assess whether the magnitude and direction of the observed effect is related to selective outcome reporting).
High risk of bias (not all of the trial's pre‐specified primary outcomes have been reported or similar).
Other bias
Academic bias
Low risk of bias (the author of the trial has not conducted previous trials addressing the same interventions).
Uncertain risk of bias (It is not clear if the author has conducted previous trials addressing the same interventions).
High risk of bias (the author of the trial has conducted previous trials addressing the same interventions).
Source of funding bias
Low risk of bias (the trial's source(s) of funding did not come from any parties that might have conflicting interest (eg, drug manufacturer).
Uncertain risk of bias (the source of funding was not clear).
High risk of bias (the trial was funded by a drug manufacturer).
We classified trials as trials with low risk of bias if they were judged with low risk of bias in all the above domains. Otherwise, the trials were classified as trials with high risk of bias. Statistical methods All analyses were performed according to the intention‐to‐treat method including all randomised patients. Patients without the outcome variable were included in two analyses; one in which patients without the outcome were considered as failures, and one in which patients without the outcome were considered as successes.
The statistical package Review Manager provided by The Cochrane Collaboration was used (RevMan 2011). For all analyses we used both random‐effects (DerSimonian 1986) and fixed‐effect (DeMets 1987) models. In case of discrepancy between the two models (one showing a significant intervention effect and the other no significant intervention effect) we reported both results. Otherwise, we reported only the results from the fixed‐effect model. Discrepancy only occurred when there was heterogeneity (please see below). In case of discrepancy between the two models, we put most weight on the results of the fixed‐effect model if the meta‐analysis included one or more large trials with adequate methodology. Large trials were defined as trials that included more than half of all included events and participants in the meta‐analysis. Otherwise, we put most weight on the random‐effects model. The reason for this is that the random‐effects model puts more weight on small trials. Small trials are more often than large trials conducted with unclear or inadequate methods (Kjaergard 2001).
Dichotomous data were analysed by calculating the relative risks (RR) and continuous outcomes as mean difference (MD) both with 95% confidence intervals.
We conducted trial sequential analysis in order to estimate how far we had come in our development of the cumulative evidence (Wetterslev 2008; Thorlund 2009; Brok 2009). We based our trial sequential analysis on all‐cause mortality using an outcome proportion of 15% in the control group; an assumed relative risk reduction of 20%; an alpha of 5%; a beta of 20%; and a heterogeneity correction for the calculation of the required information size (Wetterslev 2008).
Heterogeneity and funnel plot asymmetry Heterogeneity in the results of the trials was initially assessed by the inspection of graphical presentations and by calculating a test of heterogeneity (Chi‐square) as well as level of inconsistency (I2). We anticipated between‐trial variation in estimation of morbidity and all‐cause mortality for those patients who presented with advanced liver disease (DerSimonian 1986; DeMets 1987). Subgroup analyses were performed in order to assess the impact of these possible sources of heterogeneity on the main results.
Potential causes for heterogeneity were explored by performing sensitivity analyses. We performed sensitivity analyses with regard to methodological quality of included randomised clinical trials (analysing separately randomised clinical trials with adequate quality components, ie, low risk of bias, and inadequate quality components, high risk of bias, and duration of treatment. We also planned analyses regarding way of administration of PTU as well as preparation and dose of PTU, but all trials used per oral PTU 300 mg per day.
Due to the risk of chance statistical findings, such findings were interpreted conservatively.
Potential publication bias (Vickers 1998) and other sources of bias were planned to be investigated by funnel plots (Egger 1997), but due to the few randomised clinical trials identified, we did not perform such analyses.
Results
Description of studies
Searches Electronic searches (through April 2011) of The Cochrane Hepato‐Biliary Group Controlled Trials Register (n = 17 publications), The Cochrane Central Register of Controlled Trials (CENTRAL) in The Cochrane Library (n = 19 publications), MEDLINE (n = 16 publications), EMBASE (n = 48 publications), and Science Citation Index Expanded (n = 13 publications) identified a total of 113 publications. Out of these publications, three randomised clinical trials described in ten publications were identified (Orrego 1979; Hallé 1982; Orrego 1987). By reading bibliographies we identified four further abstracts on three randomised clinical trials (Serrano‐Cancino 1981; Pierrugues 1989; Rodriguez 1993), which were not identified by the electronic searches. Included studies The individual randomised clinical trials are described in the table of 'Characteristics of included randomised clinical trials'. In total, six randomised clinical trials reported the random allocation of patients with alcoholic liver disease (n = 710) to PTU versus placebo in 14 publications. No randomised clinical trials comparing PTU versus no intervention were identified.
The entry criteria in the randomised clinical trials varied, but the inclusion criteria were generally of good quality making it highly likely that all patients did in fact have alcoholic liver disease.
The dosage of PTU was 300 mg orally per day in all the trials. The duration of the treatment was within 46 days in five of the trials (Orrego 1979; Serrano‐Cancino 1981; Hallé 1982; Pierrugues 1989; Rodriguez 1993), and the treatment duration was 24 months in the remaining trial (Orrego 1987) .
Excluded studies A total of two studies with reasons of exclusion are listed under 'Characteristics of excluded studies'.
Risk of bias in included studies
The method to generate the allocation sequence was considered adequate in three randomised clinical trials (Orrego 1979; Orrego 1987; Rodriguez 1993).
The method to conceal the allocation sequence was considered adequate in two randomised clinical trials (Orrego 1979; Rodriguez 1993).
All randomised clinical trials were described as 'double blind'. In four randomised clinical trials (Orrego 1979; Hallé 1982; Orrego 1987; Rodriguez 1993) placebo was described as having an identical presentation, making it likely that both investigators and patients were blinded.
Four randomised clinical trials were considered free of incomplete outcome data (Orrego 1979; Hallé 1982; Orrego 1987; Rodriguez 1993), and in the other two (Serrano‐Cancino 1981; Pierrugues 1989) there was not sufficient information regarding this item.
All randomised clinical trials were considered free of selective reporting, and academic bias.
None of the authors declared the source of funding of the included trials.
Effects of interventions
All‐cause mortality Combining the results of the six randomised clinical trials demonstrated no significant effect of PTU versus placebo on all‐cause mortality (risk ratio (RR) 0.93, 95% confidence interval (CI) 0.66 to 1.30). In the PTU group, 50/353 (14.2%) patients died versus 54/357 (15.1%) patients in the placebo group (Analysis 1.1).
1.1. Analysis.
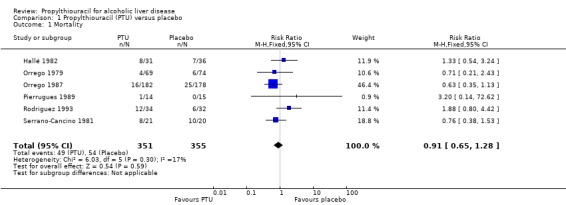
Comparison 1 Propylthiouracil (PTU) versus placebo, Outcome 1 Mortality.
Sensitivity analysis stratifying the randomised clinical trials according to the risk of bias in the trials (trials at low risk of bias versus high risk of bias for each domain, excluding the report 'free of source of funding' which was unclear in all trials) did not demonstrate differences regarding the intervention efficacy of PTU on all‐cause mortality between randomised clinical trials at low risk versus high risk of bias (Analysis 3.1).
3.1. Analysis.
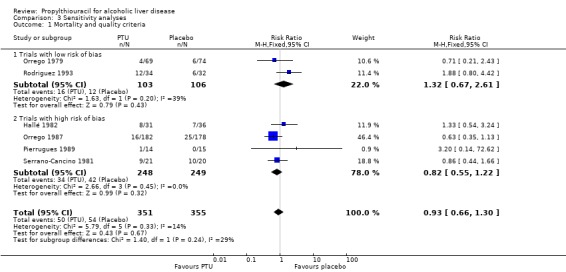
Comparison 3 Sensitivity analyses, Outcome 1 Mortality and quality criteria.
Sensitivity analysis stratifying the randomised clinical trials according to the duration of treatment did not change this estimate significantly. The RR of death of the randomised trials with a short‐term treatment (within 46 days) was 1.19 (95% CI 0.78 to 1.81) and the RR of the randomised trial with a treatment duration of 24 months was 0.63 (95% CI 0.35 to 1.13) (Analysis 3.2). Stratifying the randomised clinical trials according to a worst‐best case scenario analysis (all patients who dropped‐out or were withdrawn were considered dead), or a per‐protocol analysis did not change this estimate significantly (Analysis 3.3; Analysis 3.4).
3.2. Analysis.
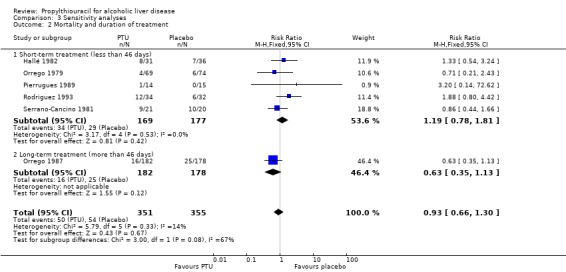
Comparison 3 Sensitivity analyses, Outcome 2 Mortality and duration of treatment.
3.3. Analysis.
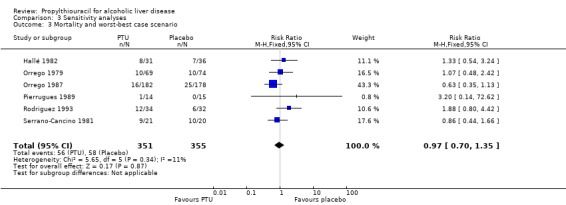
Comparison 3 Sensitivity analyses, Outcome 3 Mortality and worst‐best case scenario.
3.4. Analysis.
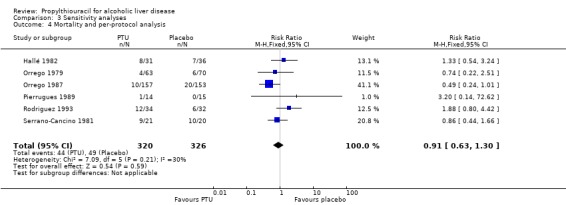
Comparison 3 Sensitivity analyses, Outcome 4 Mortality and per‐protocol analysis.
Trial sequential analysis based on a heterogeneity‐corrected information size of 4908 patients shows that with only 706 patients randomised, we are still very early in the development of evidence regarding this intervention (Figure 1).
1.
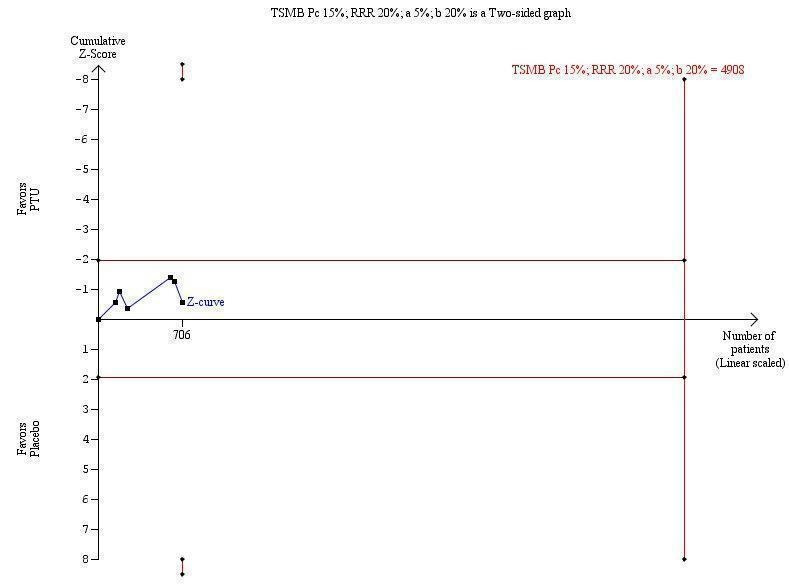
Trial sequential analysis of all‐course mortality of patients with alcoholic liver disease included in randomised clinical trials on propylthiouracil versus placebo. The heterogeneity‐corrected required information size of 4908 patients is based on an event proportion of 15% in the control group (Pc); a relative risk reduction (RRR) of 20% (to an event proportion of 13%) in the experimental group; an alpha of 5%; a beta of 20%; and a heterogeneity of 17%.
Liver‐related mortality Combining the results of four randomised clinical trials, which provided data on liver mortality, showed no significant effect of PTU versus placebo on this outcome (RR 0.90, 95% CI 0.58 to 1.40) (Orrego 1979; Hallé 1982; Orrego 1987; Rodriguez 1993). In the PTU group, 33/316 (10.4%) patients died a liver‐related death versus 37/320 (11.6%) patients in the placebo group (Analysis 1.2).
1.2. Analysis.
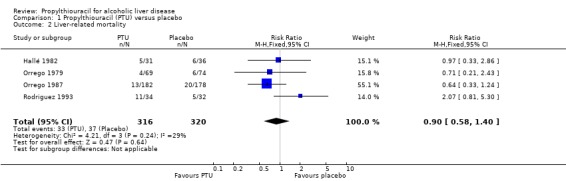
Comparison 1 Propylthiouracil (PTU) versus placebo, Outcome 2 Liver‐related mortality.
Liver complications No significant effect of PTU versus placebo could be demonstrated on hepatic encephalopathy (RR 0.84, 95% CI 0.39 to 1.83) (Hallé 1982) (Analysis 1.3).
1.3. Analysis.
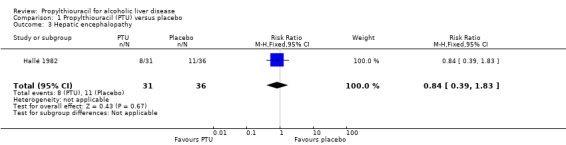
Comparison 1 Propylthiouracil (PTU) versus placebo, Outcome 3 Hepatic encephalopathy.
No significant effect of PTU versus placebo could be demonstrated on ascites (RR 2.32, 95% CI 0.22 to 24.40) (Hallé 1982) (Analysis 1.4).
1.4. Analysis.
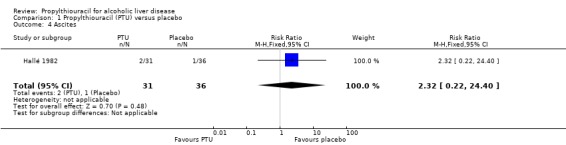
Comparison 1 Propylthiouracil (PTU) versus placebo, Outcome 4 Ascites.
No significant effect of PTU versus placebo could be demonstrated on variceal bleeding (RR 1.48, 95% CI 0.63 to 3.47) (Hallé 1982) (Analysis 1.5).
1.5. Analysis.
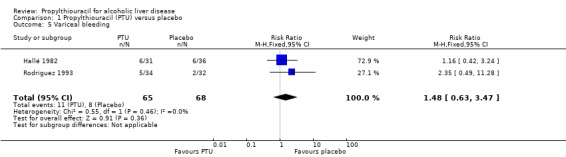
Comparison 1 Propylthiouracil (PTU) versus placebo, Outcome 5 Variceal bleeding.
No significant effect of PTU versus placebo could be demonstrated on hepato‐renal syndrome (RR 0.84, 95% CI 0.39 to 1.83) (Hallé 1982) (Analysis 1.6).
1.6. Analysis.
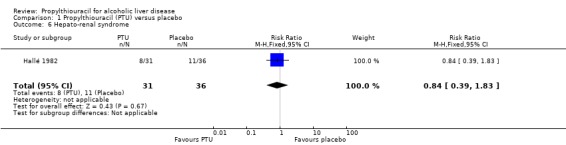
Comparison 1 Propylthiouracil (PTU) versus placebo, Outcome 6 Hepato‐renal syndrome.
Liver histology Due to the paucity of data on liver histology we did not assess this outcome. Only one trial reported as an abstract, provided data on histological changes (which were similar in PTU and placebo group) (Pierrugues 1989). In three trials, a liver biopsy was done at the beginning of the trial or at some time after in a subgroup of patients, but no data on liver histology at the end of the follow‐up were reported (Orrego 1979; Hallé 1982; Orrego 1987).
Adverse events Combining the results of the five randomised clinical trials demonstrated no significant effect of PTU on serious adverse events (RR 1.67, 95% CI 0.28 to 10.03). In the PTU group 2/100 (2%) patients had serious adverse events (marked leukopenia, generalized bullous eruption) versus 1/110 (0.9%) patients in the placebo group (leukopenia) (Analysis 2.1).
2.1. Analysis.
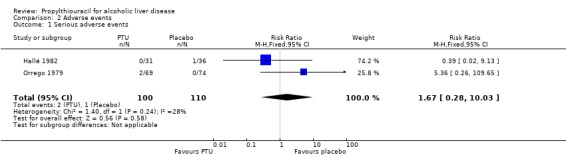
Comparison 2 Adverse events, Outcome 1 Serious adverse events.
Combining the results of the same five randomised clinical trials demonstrated no significant effects of PTU on non‐serious adverse events (RR 1.41, 95% CI 0.40 to 5.01). In the PTU group 5/100 (5%) patients had non‐serious adverse events versus 4/110 (3.6%) patients in the placebo group (Analysis 2.2). The adverse events included rash and hypothyroidism.
2.2. Analysis.
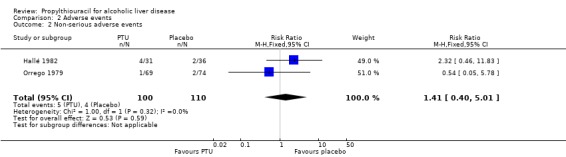
Comparison 2 Adverse events, Outcome 2 Non‐serious adverse events.
In one trial, 24 adverse events were reported (15 rashes, 8 leukopenia, thrombocytopenia); 15 adverse events occurred in the PTU group and 9 in the placebo group (the differences between groups were not statistically significant), but the number of serious and non‐serious adverse events in each group was not specified (Orrego 1987).
Quality‐of‐life and health economics None of the randomised clinical trials examined quality‐of‐life or health economics.
Funnel plot asymmetry Due to the paucity of randomised clinical trials and observed outcome measures reported in the included trials, we did not try to analyse for funnel plot asymmetry.
Discussion
We could not demonstrate any significant effects of PTU on all‐cause mortality, liver‐related mortality, and liver complications when tested against placebo in patients with alcoholic liver disease. However, absence of evidence is not evidence of absence of effect.
The lack of effect of PTU on all‐cause mortality was robust to sensitivity analyses taking the risk of bias in the randomised clinical trials into consideration. The sensitivity analyses contrasting trials of low risk of bias versus trials of high risk of bias as judged by the single domain did not reveal any significant influence on the RR of all‐cause mortality. This is in contrast to studies examining the association between intervention effects and risk of bias of randomised clinical trials (Schulz 1995; Moher 1998; Kjaergard 2001; Wood 2008). However, one should notice that two randomised clinical trials at high risk of bias were only reported as abstracts. First, the brevity of abstracts may make it difficult to report the risk of bias in a trial in sufficient detail. Second, the association between the risk of bias and intervention effects previously reported rests mainly or exclusively on trials reported as full articles (Schulz 1995; Moher 1998; Kjaergard 2001; Wood 2008).
Sensitivity analysis taking duration of treatment into consideration showed no significant difference on the RR of all‐cause mortality. The worst‐best case scenario analysis and the per‐protocol analysis did not show any significant effect of PTU on all‐cause mortality.
Our trial sequential analysis demonstrates that we are very early in our development of evidence on PTU for all‐cause mortality in alcoholic liver disease with only 706 patients randomised. We are still far from obtaining conclusive evidence on the effect of PTU for patients (Figure 1).
We were unable to detect any significant influence of PTU on liver histology due to the paucity of data. Only one trial (Pierrugues 1989), reported as an abstract, provided data on histological changes (which were similar in PTU and placebo group). In three studies (Orrego 1979; Hallé 1982; Orrego 1987), a liver biopsy was done at the beginning of the trial or at some time after in a subgroup of patients, but no data on liver histology at the end of the follow‐up were reported.
Although propylthiouracil was not associated with a significant increased risk of non‐serious adverse events, there were occasional instances of serious adverse events: one patient with leukopenia and one patient with generalised bullous eruption, and the latter patient died during the randomised clinical trial. Few patients with PTU induced fulminant hepatitis and a number of adverse events have been reported in the literature during PTU treatment for hyperthyroidism (Deidiker 1996; Ichiki 1998), eg, transient asymptomatic PTU hepatotoxicity occurs in one‐third of patients (Huang 1994) together with acute cases of interstitial nephritis, vasculitis, Stevens‐Johnson syndrome, and other adverse events (Dysseleer 2000; Morita 2000). Indeed, hypothyroidism was a non‐serious adverse event in some patients, and it could have been the reason of some dropouts in the trials (Hallé 1982).
The rationale behind PTU for alcoholic liver disease has been said to be via an effect on hepatic oxygen consumption (eg, Yuki 1982; Carmichael 1993). Contrary to observations in normal rats (Kawasaki 1989), however, PTU 300 mg or 600 mg intravenously were without significant effects on arterial and venous oxygen content in patients with alcoholic cirrhosis with or without alcoholic hepatitis (Sogni 1997). Furthermore, the latter study was unable to demonstrate any effects of PTU on systemic and splanchnic haemodynamics of these patients (Sogni 1997). These findings are contradictory to some extent to the Rojeter et al study (Rojter 1995), which examined haemodynamics measured by the Doppler technique. According to the latter study, PTU administration caused a significant increase in portal blood flow in patients with alcoholic cirrhosis.
We could not demonstrate any significant effect of PTU on any clinically important outcomes of patients with alcoholic liver disease. Accordingly, there seems to be no evidence for using PTU for alcoholic liver disease. The absence of evidence for an effect of PTU on clinically relevant outcome variables, however, does not exclude the possibility that PTU may possess effects. Ioannidis and Lau recently applied 'recursive cumulative meta‐analyses' of randomised clinical trials to evaluate the relative change in the pooled treatment effect over time for 60 medical interventions within pregnancy/perinatal medicine and cardiology (Ioannidis 2000). With 500 accumulated patients, the pooled relative risk may change by about 0.6 to 1.7 fold in the immediate future. When 2000 patients have been randomised, the pooled relative risk may change by 0.7 to 1.3 fold. With only 710 patients with alcoholic liver disease randomised to PTU versus placebo and the wide confidence intervals of the estimates we cannot rule out a potential efficacy of PTU for alcoholic liver disease. However, if clinicians wish to treat patients with alcoholic liver disease with PTU, they must first conduct new randomised clinical trials. Such randomised clinical trials ought to be large, conducted with adequate methodology, the treatment period ought to be several years, and efficacy and harmful effects ought to be closely monitored by an independent data monitoring and safety committee.
A number of medical interventions has been used for alcoholic liver disease (Gluud 1993), including colchicine (Rambaldi 2005a), glucocorticosteroids (Christensen 1995; Gluud 2001), anabolic‐androgenic steroids (Gluud 1988; Rambaldi 2006a), insulin/glucagon (Trinchet 1992), milk thistle (Flora 1998, Rambaldi 2007), parenteral amino acid supplementation (Mezey 1991), S‐adenosyl‐L‐methionine (Mato 1999), and polyenylphosphatidylcholine (Lieber 2000; Lieber 2003). None of these interventions have been demonstrated effective in systematic reviews of randomised clinical trials. S‐adenosyl‐L‐methionine may be a promising intervention for alcoholic liver disease (Mato 1999), but more randomised clinical trials are needed before this treatment can be recommended (Rambaldi 2006b). Pentoxiphylline has only been assessed in one small trial (Akriviadis 2000), and liver transplantation has never been assessed in randomised trials. Based on a matched and simulated control study, liver transplantation seems to work for patients with Child‐Pugh C class cirrhosis, but not significantly so for Child‐Pugh A and B class cirrhosis (Poynard 1994; Poynard 1999). The results of more randomised clinical trials must be awaited before we may have an efficient medical intervention for alcoholic liver disease.
Authors' conclusions
Implications for practice.
This systematic review could not demonstrate any significant beneficial effect of PTU on any clinically meaningful outcomes (all‐cause mortality, liver‐related mortality, and liver complications) of patients with alcoholic liver disease. PTU is associated with serious adverse events. Accordingly, there is no indication for using PTU for alcoholic liver disease outside randomised clinical trials.
Implications for research.
The absence of evidence for an effect of PTU on clinically relevant outcome variables, however, does not exclude the possibility of an effect. If researchers wish to conduct new randomised clinical trials they ought to be large, conducted with adequate methodology, the treatment period ought to be several years, and efficacy and harmful effects ought to be closely monitored by an independent data monitoring and safety committee. Such trials ought to follow the Consolidated Standards for Reporting Trials (CONSORT) Statement (www.consort‐statement.org).
What's new
| Date | Event | Description |
|---|---|---|
| 28 January 2011 | New citation required but conclusions have not changed | Conclusions did not change. No new trials were found for inclusion. |
| 22 January 2011 | New search has been performed | 1. We changed the reporting of Peto odds ratio (OR) to relative risk (RR) as the latter is more easily understood. 2. We removed the Jadad scoring system for evaluation of trial quality since methodological quality is better evaluated by bias risk domains. 3. We now use both random‐effects and fixed‐effect models to analyse our data following the most recent guidelines. |
| 21 January 2011 | New search has been performed | New team of authors. |
History
Protocol first published: Issue 4, 2000 Review first published: Issue 4, 2001
| Date | Event | Description |
|---|---|---|
| 23 August 2005 | New search has been performed | Conclusions changed. |
Notes
We have contacted Merck Frosst Canada Inc, Kirkland, Quebec (Canada) in order to obtain additional data, published or unpublished.
Acknowledgements
We acknowledge Andrea Rambaldi who wrote the protocol and the previous version of the review, Robert Sutton, Contact Editor, for helpful comments, and Dimitrinka Nikolova and Sarah Louise Klingenberg for expert assistance with the retrieval of publications.
Peer Reviewers: Alastair Burt, UK; Saboor A Khan, UK. Contact Editor: Robert Sutton, UK.
Appendices
Appendix 1. Search strategies
| Database | Time of search | Search strategy |
| The Cochrane Hepato‐Biliary Group Controlled Trials Register | April 2011 | propylthiouracil OR PTU |
| Cochrane Central Register of Controlled Trials (CENTRAL) in The Cochrane Library (Wiley) | Issue 2, April 2011 | #1 MeSH descriptor Propylthiouracil explode all trees in MeSH products #2 propylthiouracil OR PTU in All Fieldsin all products #3 (#1OR #2) #4 MeSH descriptor Liver Diseases, Alcoholic explode all trees in MeSH products #5 alcoholic and (liver disease* or steatosis or fibrosis or hepatitis or cirrhosis) in All Fields in all products #6 (#4 OR #5) #7 (#3 AND #6) |
| MEDLINE (Ovid SP) | 1948 to April 2011 |
#1 exp Propylthiouracil/ #2 (propylthiouracil or PTU).mp. [mp=protocol supplementary concept, rare disease supplementary concept, title, original title, abstract, name of substance word, subject heading word, unique identifier] #3 1 or 2 #4 exp Liver Diseases, Alcoholic/ #5 (alcoholic and (liver disease* or steatosis or fibrosis or hepatitis or cirrhosis)).mp. [mp=protocol supplementary concept, rare disease supplementary concept, title, original title, abstract, name of substance word, subject heading word, unique identifier] #6 4 or 5 #7 3 and 6 #8 (random* or placebo* or blind* or meta‐analysis).mp. [mp=protocol supplementary concept, rare disease supplementary concept, title, original title, abstract, name of substance word, subject heading word, unique identifier] #9 7 and 8 |
| EMBASE (OvidSP) | 1980 to April 2011 | #1 exp PROPYLTHIOURACIL/ #2 (propylthiouracil or PTU).mp. [mp=title, abstract, subject headings, heading word, drug trade name, original title, device manufacturer, drug manufacturer] #3 1 or 2 #4 exp alcohol liver disease/ #5 (alcoholic and (liver disease* or steatosis or fibrosis or hepatitis or cirrhosis)).mp. [mp=title, abstract, subject headings, heading word, drug trade name, original title, device manufacturer, drug manufacturer] #6 4 or 5 #7 3 and 6 #8 (random* or blind* or placebo* or meta‐analysis).mp. [mp=title, abstract, subject headings, heading word, drug trade name, original title, device manufacturer, drug manufacturer] #9 7 and 8 |
| Science Citation Index Expanded (ISI Web of Knowledge) | 1900 to April 2011 | #1 TS=(propylthiouracil or PTU) #2 TS=(alcoholic and (liver disease* or steatosis or fibrosis or hepatitis or cirrhosis)) #3 #2 AND #1 #4 TS=(random* or placebo* or blind* or meta‐analysis) #5 #4 AND #3 |
Data and analyses
Comparison 1. Propylthiouracil (PTU) versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Mortality | 6 | 706 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.91 [0.65, 1.28] |
| 2 Liver‐related mortality | 4 | 636 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.90 [0.58, 1.40] |
| 3 Hepatic encephalopathy | 1 | 67 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.84 [0.39, 1.83] |
| 4 Ascites | 1 | 67 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.32 [0.22, 24.40] |
| 5 Variceal bleeding | 2 | 133 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.48 [0.63, 3.47] |
| 6 Hepato‐renal syndrome | 1 | 67 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.84 [0.39, 1.83] |
Comparison 2. Adverse events.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Serious adverse events | 2 | 210 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.67 [0.28, 10.03] |
| 2 Non‐serious adverse events | 2 | 210 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.41 [0.40, 5.01] |
Comparison 3. Sensitivity analyses.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Mortality and quality criteria | 6 | 706 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.93 [0.66, 1.30] |
| 1.1 Trials with low risk of bias | 2 | 209 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.32 [0.67, 2.61] |
| 1.2 Trials with high risk of bias | 4 | 497 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.82 [0.55, 1.22] |
| 2 Mortality and duration of treatment | 6 | 706 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.93 [0.66, 1.30] |
| 2.1 Short‐term treatment (less than 46 days) | 5 | 346 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.19 [0.78, 1.81] |
| 2.2 Long‐term treatment (more than 46 days) | 1 | 360 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.63 [0.35, 1.13] |
| 3 Mortality and worst‐best case scenario | 6 | 706 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.97 [0.70, 1.35] |
| 4 Mortality and per‐protocol analysis | 6 | 646 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.91 [0.63, 1.30] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Hallé 1982.
| Methods | Randomised clinical trial. | |
| Participants | Sixty‐seven patients with severe alcoholic hepatitis. Thirty‐one patients (29 males and two females, mean age 40 ± 2 years (y)) received PTU, while 36 patients (32 males and four females, mean age 38.9 ± 1 y) received placebo. Inclusion criteria: heavy ethanol ingestion and clinical diagnosis of ALD. All had serum bilirubin > 5 mg/dl and at least one of the following: hepatic tenderness, fever above 100 degrees Fahrenheit, or leukocytosis above 12,000 per mm3. Exclusion criteria: serious bacterial infection, massive gastrointestinal bleeding, preexisting renal failure, and previous or current thyroid disease. |
|
| Interventions | Experimental group:
PTU 75 mg orally every six hours. Control group: placebo. Duration of the treatment: six weeks. Duration of follow‐up: eight weeks. |
|
| Outcomes | All‐cause mortality. Complications. Biochemistry. Liver histology. Adverse events. | |
| Notes | Sent letter in 2001.
Dr. Reynolds answered, but no additional data were obtained. Seventy‐one patients were randomised, but two patients refused participation and two patients were withdrawn as s‐bilirubin was < 5 mg/dl at randomisation. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Comment: double blind with placebo of identical presentation. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | The post‐randomisation drop‐outs unlikely to result in a change in the effect estimate. |
| Selective reporting (reporting bias) | Low risk | All the important outcomes were reported. |
| Free of academic bias | Low risk | Comment: No previous trial of the same comparison by the same authors was identified. |
Orrego 1979.
| Methods | Randomised clinical trial. | |
| Participants | A total of 143 patients (69 in the PTU and 74 in placebo group) were included in the trial. Inclusion criteria: excessive drinking, and well‐documented history of spree‐drinking. The criteria for diagnosing liver disease required one or more of the following clinical findings: hepatomegaly, tender liver, jaundice, ascites, collateral circulation, spider nevi, and splenomegaly. Further, at least two of the following abnormal laboratory tests were required: s‐aspartate aminotransferase, s‐alanine aminotransferase, s‐gamma glutamyltranspeptidase, s‐alkaline phosphatase, s‐total bilirubin. In 79 patients in whom the prothrombin time permitted, liver biopsies were performed at 7.6 ± 0.1 day after admission. All biopsy specimens were classified in: fatty liver, alcoholic hepatitis, and cirrhosis without hepatitis. The three histologically diagnosed groups were analysed separately. Exclusion criteria: hypothyroidism; diabetes; other therapies that contraindicated the use of PTU; congestive heart failure. The indications for being withdrawn from the study were massive gastrointestinal bleeding, incapacity to ingest drug, leukopenia, or adverse reactions. |
|
| Interventions | Experimental group:
PTU 300 mg orally every day. Control group: placebo. Maximum period of treatment and of follow‐up: 46 days. Patients could be discharged before this period if clinical improvement made further stay in the hospital unnecessary. |
|
| Outcomes | All‐cause mortality.
Biochemistry.
Liver histology.
Adverse events.
Thyroid function. A Composite Clinical and Laboratory Index was developed for assessing efficacy. The scoring system was based on the concept that the severity of the disease was proportional to the number of abnormal clinical and laboratory findings. The index included signs and symptoms (hepatomegaly, splenomegaly, ascites, encephalopathy, bleeding tendency, spider naevi, palmar erythema, collateral circulation, peripheral edema, anorexia, and weakness) and laboratory tests (s‐bilirubin, prothrombin time, s‐albumin, s‐gamma glutamyltranspeptidase, s‐glutamic oxalacetic transaminase) and depending on the finding, a score (0 to 27) was added for each patient. See also Orrego et al (Orrego 1987b). |
|
| Notes | Sent letter in 2001. No reply. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Comment: generation of allocation sequence by computer. |
| Allocation concealment (selection bias) | Low risk | Comment: allocation concealment involved an independent pharmacist |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Comment: double blind with placebo of identical presentation. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | The post‐randomisation drop‐outs unlikely to result in a change in the effect estimate. |
| Selective reporting (reporting bias) | Low risk | All the important outcomes were reported. |
| Free of academic bias | Low risk | Comment: No previous trial of the same comparison by the same authors was identified. |
Orrego 1987.
| Methods | Randomised clinical trial. | |
| Participants | A total of 360 patients were included in the study, 182 in PTU group (152 males and 30 females, mean age (standard error of the mean (SEM)) 49.2 ± 0.8 y and 178 in the placebo group (139 males and 39 females), mean age (SEM) 49.6 ± 0.8 y. Inclusion criteria: a) alcoholism, defined as excessive drinking or spree drinking consisting of repeated prolonged inebriations or a well documented history of > 80 g of ethanol per day; and b) a clinical and laboratory evidence of liver disease. The severity of disease in each patient were determined with use of the clinical and laboratory index. Exclusion criteria: hepatoma, presence of the hepatitis B surface antigen, contraindications to PTU therapy, and a history of hypothyroidism. |
|
| Interventions | Experimental group:
PTU 150 mg every 12 h orally every day. Because of the risk of severe hypothyroidism, patients in the PTU group were automatically switched every three months by the pharmacy to the placebo for one month, after which they were again given PTU. Control group: placebo. Additional treatment: 15 mg of riboflavin, a fluorescent compound that was used as a marker of compliance. Most of the urine samples contained the riboflavin marker (93.2 ± 0.8 percent in the placebo group, while 93.1 ± 0.7 percent in the PTU group). Maximum period of treatment: 24 months. |
|
| Outcomes | All‐cause mortality.
Biochemistry.
Liver histology.
Adverse events.
Alcohol consumption.
Thyroid function. This trial also evaluated efficacy with the Combined Clinical and Laboratory Index similar to that of Orrego 1979, but not identical to it (score range 0 to 25). |
|
| Notes | Sent letter in 2001. No reply. Only 310 compliant patients form the basis of the reports of the trial. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Comment: generation of allocation sequence by computer. |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Comment: double blind with placebo of identical presentation. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | The post‐randomisation drop‐outs unlikely to result in a change in the effect estimate. |
| Selective reporting (reporting bias) | Low risk | All the important outcomes were reported. |
| Free of academic bias | Low risk | Comment: No previous trial of the same comparison by the same authors was identified. |
Pierrugues 1989.
| Methods | Randomised clinical trial. | |
| Participants | Twenty‐nine patients were included in the study (17 males and 12 females, mean age 45.8 years ± 11) with alcoholic hepatitis, 14 in the PTU group and 15 in the placebo group. Diagnostic assessment: alcoholic hepatitis with liver biopsy. |
|
| Interventions | Experimental group:
PTU 300 mg/day orally. Control group: placebo. Duration of treatment: 28 days. |
|
| Outcomes | All‐cause mortality. Biochemistry. Liver histology. Thyroid function. | |
| Notes | Sent letter in 2001. No reply. Only published as abstract. A composite clinical and laboratory index was used to evaluate the effect of PTU, but details on which index that was used are not given. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Selective reporting (reporting bias) | Low risk | All the important outcomes were reported. |
| Free of academic bias | Low risk | Comment: No previous trial of the same comparison by the same authors was identified. |
Rodriguez 1993.
| Methods | Randomised clinical trial. | |
| Participants | The trial was stopped when 66 patients with acute alcoholic hepatitis were included, 34 in PTU and 32 in placebo group, because a trend was observed to higher all‐cause mortality rates in the PTU group (35.3% in the PTU versus 18.8% in the placebo group), despite nearly identical Maddrey's discriminant function (51.78 ± 34.9 in PTU group versus 53.72± 34.9 in the placebo group) and Child Pugh's score (11.06 ± 1.98 in the PTU group versus 10.72± 1.91 in the placebo group) at entry. Inclusion criteria: patients were all heavy drinkers presenting with s‐bilirubin > 4 mg/dl and fever or hepatic tenderness or more than 12000 leukocytes/mm3 in the absence of acute infection. |
|
| Interventions | Experimental group:
PTU 300 mg/day orally. Control group: placebo. Duration of treatment: 40 days. |
|
| Outcomes | All‐cause mortality. Complications Biochemistry. Liver histology. Adverse events. Thyroid function. | |
| Notes | Sent letter in 2001. Dr. Gonzalez‐Reimers answered, providing additional data. Only published as abstract. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Comment: generation of allocation sequence by computer. |
| Allocation concealment (selection bias) | Low risk | Comment: allocation concealment involved an independent observer. |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Comment: double blind with placebo of identical presentation. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | The post‐randomisation drop‐outs unlikely to result in a change in the effect estimate. |
| Selective reporting (reporting bias) | Low risk | All the important outcomes were reported. |
| Free of academic bias | Low risk | Comment: No previous trial of the same comparison by the same authors was identified. |
Serrano‐Cancino 1981.
| Methods | Randomised clinical trial. | |
| Participants | Forty‐one patients were studied with severe alcoholic hepatitis (21 in the PTU and 20 in the placebo group). At entry there were no significant difference in the clinical severity (ascites, encephalopathy, s‐bilirubin, s‐albumin, s‐creatinin, white blood cell count) between the placebo and the PTU group. | |
| Interventions | Experimental group:
PTU 100 mg every eight hours orally. Control group: placebo. Duration of treatment: 17.0 ± 13.3 days. Additional treatment in both groups: standard nutritional and supportive diet. |
|
| Outcomes | All‐cause mortality. Biochemistry. Adverse events. Duration of hospital stay. | |
| Notes | Sent letter in 2001. No reply. Only published as abstract. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Selective reporting (reporting bias) | Low risk | All the important outcomes were reported. |
| Free of academic bias | Low risk | Comment: No previous trial of the same comparison by the same authors was identified. |
PTU = propylthiouracil. y = year(s). h = hour(s). > = more than, greater than. < = less than.
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Rojter 1995 | The study is observational (case series). In eight patients with alcoholic liver cirrhosis mean arterial pressure and portal blood flow were measured before and after placebo and PTU administration. PTU administration caused a significant increase in portal blood flow in patients with alcoholic cirrhosis. |
| Sogni 1997 | The study is observational (case series). Systemic haemodynamics and splanchnic haemodynamics were not modified after the administration of PTU to 12 patients with alcoholic cirrhosis. |
Differences between protocol and review
Differences between current updated review and previous version
The methodological quality of the randomised clinical trials and type of outcomes were assessed using recent published recommendations (Gurusamy 2009; Higgins 2011).
Contributions of authors
GF identified trials, extracted data, analysed the data, drafted the review. GG performed second data extraction. CG and KG made critical comments. AKB checked and revised all these processes.
Sources of support
Internal sources
The Copenhagen Trial Unit, Denmark.
External sources
The Danish Medical Research Council Grant on Getting Research into Practice (GRIP), Denmark.
The 1991 Pharmacy Foundation, Denmark.
Copenhagen Hospital Corporation' Research Grant on Getting Research into Practice (GRIP), Denmark.
Declarations of interest
None known.
New search for studies and content updated (no change to conclusions)
References
References to studies included in this review
Hallé 1982 {published data only}
- Halle P, Pare P, Kaptein E, Kanel G, Redeker AG, Reynolds TB. Propylthiouracil therapy in severe acute alcoholic hepatitis. Gastroenterology 1980;79(5 II):1024. [MEDLINE: ] [PubMed] [Google Scholar]
- Hallé P, Paré P, Kaptein E, Kanel G, Redeker AG, Reynolds TB. Double‐blind, controlled trial of propylthiouracil in patients with severe acute alcoholic hepatitis. Gastroenterology 1982;82(5):925‐31. [PubMed] [Google Scholar]
Orrego 1979 {published data only}
- Israel Y, Walfish PG, Orrego H, Blake J, Kalant H. Thyroid hormones in alcoholic liver disease. Effect of treatment with 6‐n‐propylthiouracil. Gastroenterology 1979;76:116‐22. [PubMed] [Google Scholar]
- Orrego H, Israel Y. Propylthiouracil treatment for alcoholic hepatitis: the case of the missing thirty. Gastroenterology 1982;83(3):945‐6. [PubMed] [Google Scholar]
- Orrego H, Kalant H, Israel Y, Blake J, Medline A, Rankin JC, et al. Effect of short‐term therapy with propylthiouracil in patients with alcoholic liver disease. Gastroenterology 1979;76(1):105‐15. [PubMed] [Google Scholar]
Orrego 1987 {published data only}
- Orrego H, Blake JE, Blendis LM, Compton KV, Israel Y. Chronic propylthiouracil treatment increases survival in patients with alcoholic liver disease. Hepatology 1986;6(5):1125. [Google Scholar]
- Orrego H, Blake JE, Blendis LM, Compton KV, Israel Y. Long term treatment of alcoholic liver disease with propylthiouracil. The Lancet 1988;1(8590):892. [DOI] [PubMed] [Google Scholar]
- Orrego H, Blake JE, Blendis LM, Compton KV, Israel Y. Long‐term treatment of alcoholic liver disease with propylthiouracil. New England Journal of Medicine 1987;317:1421‐7. [DOI] [PubMed] [Google Scholar]
- Orrego H, Blake JE, Blendis LM, Compton KV, Israel Y. Propylthiouracil for alcoholic liver disease. New England Journal of Medicine 1988;318(2):1471‐2. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Orrego H, Blake JE, Blendis LM, Compton KV, Volpe R, Israel Y. Long‐term treatment of alcoholic liver disease with propylthiouracil. Part 2: influence of drop‐out rates and of continued alcohol consumption in a clinical trial. Journal of Hepatology 1994;20:343‐9. [DOI] [PubMed] [Google Scholar]
Pierrugues 1989 {published data only}
- Pierrugues R, Blanc P, Barneon G, Bories P, Michel H. Short‐term therapy with propylthiouracile (PTU) for alcoholic hepatitis (AH). A clinical, biochemical and histological randomized trial about 25 patients. Gastoenterology 1989;96(5):A644. [Google Scholar]
- Pierrugues R, Blanc P, Barneon G, Bories P, Michel H. Short‐term therapy with propylthiouracile (PTU) for alcoholic hepatitis (AH). A clinical, biochemical and histological randomized trial about 29 patients. Journal of Hepatology 1989;9:S72. [Google Scholar]
Rodriguez 1993 {published data only}
- Rodriguez‐Rodriguez E, Gonzalez‐Reimers E, Santolaria‐Fernandez F, Rodriguez‐Moreno F, Conde‐Martel A, Martinez‐Riera A, et al. Propylthiouracil in acute alcoholic hepatitis. II United European Gastroenterology Week. Barcelona, 1993:A339.
Serrano‐Cancino 1981 {published data only}
- Serrano‐Cancino H, Botero R, Jeffers L, Mariani A, Cowen G, Ravenhran N, et al. Treatment of severe alcoholic hepatitis with propylthiouracil (PTU). American Journal of Gastroenterology 1981;76(2):194A. [Google Scholar]
References to studies excluded from this review
Rojter 1995 {published data only}
- Rojter S, Tessler J, Alvarez D, Persico R, Lopez P, Bandi JC, et al. Vasodilatatory effects of propylthiouracil in patients with alcoholic cirrhosis. Journal of Hepatology 1995;22:184‐8. [DOI] [PubMed] [Google Scholar]
- Rojter S, Tessler J, Alvarez D, Persico R, Lopez P, Macias C, et al. Vasodilatatory effects of propylthiouracil (PTU) in patients with alcoholic cirrhosis (AC). Journal of Hepatology 1992;16:S64‐S65. [DOI] [PubMed] [Google Scholar]
Sogni 1997 {published data only}
- Sogni P, Hadengue A, Moreau R, Moine O, Soupison T, Oberti F, et al. Acute effects of propylthiouracil on hemodynamics and oxygen content in patients with alcoholic cirrhosis. Journal of Hepatology 1997;26:628‐33. [DOI] [PubMed] [Google Scholar]
Additional references
Akriviadis 2000
- Akriviadis E, Botla R, Briggs W, Han S, Reynolds T, Shakil O. Pentoxifylline improves short‐term survival in severe acute alcoholic hepatitis: a double‐blind, placebo‐controlled trial. Gastroenterology 2000;119:1637‐48. [DOI] [PubMed] [Google Scholar]
Brok 2009
- Brok J, Thorlund K, Wetterslev J, Gluud C. Apparently conclusive meta‐analyses may be inconclusive‐‐Trial sequential analysis adjustment of random error risk due to repetitive testing of accumulating data in apparently conclusive neonatal meta‐analyses. International Journal of Epidemiology 2009;38(1):287‐98. [DOI] [PubMed] [Google Scholar]
Carmichael 1993
- Carmichael FJ, Orrego H, Saldivia V, Israel Y. Effect of propylthiouracil on the ethanol‐induced increase in liver oxygen consumption in awake rats. Hepatology 1993;18:415‐21. [PubMed] [Google Scholar]
Chang 1994
- Chang TT, Lin CY, Chow NH, Hsu PI, Yang CC, Lin XZ, et al. Hepatitis B and hepatitis C virus infection among chronic alcoholic patients with liver disease in Taiwan. Journal of the Formosan Medical Association 1994;93(2):128‐33. [PubMed] [Google Scholar]
Christensen 1995
- Christensen E, Gluud C. Glucocorticosteroids are ineffective in alcoholic hepatitis: a meta‐analysis adjusting for confounding variables. Gut 1995;37(1):113‐8. [PMID: 7672658] [DOI] [PMC free article] [PubMed] [Google Scholar]
Corrao 1998a
- Corrao G, Zambon A, Torchio P, Arico S, Vecchia C, Iorio F. Attributable risk for symptomatic liver cirrhosis in Italy. Collaborative Groups for the Study of Liver Diseases in Italy. Journal of Hepatology 1998;28(4):608‐14. [DOI] [PubMed] [Google Scholar]
Corrao 1998b
- Corrao G, Arico S. Independent and combined action of hepatitis C virus infection and alcohol consumption on the risk of symptomatic liver cirrhosis. Hepatology 1998;27(4):914‐9. [DOI] [PubMed] [Google Scholar]
Deidiker 1996
- Deidiker R, Mello DE. Propylthiouracil‐induced fulminant hepatitis: case report and review of the literature. Pediatric Pathology 1996;16:845‐52. [PubMed] [Google Scholar]
DeMets 1987
- DeMets DL. Methods of combining randomized clinical trials: strengths and limitations. Statistics in Medicine 1987;6(3):341‐50. [DOI] [PubMed] [Google Scholar]
DerSimonian 1986
- DerSimonian R, Laird N. Meta‐analysis in clinical trials. Controlled Clinical Trials 1986;7(3):177‐88. [DOI] [PubMed] [Google Scholar]
Dysseleer 2000
- Dysseleer A, Buysschaert M, Fonck C, Ginder Deuren K, Jadoul M, Tennstedt D. Acute interstitial nephritis and fatal Stevens‐Johnson syndrome after propylthiouracil therapy. Thyroid 2000;10(8):713‐6. [PMID: 11014318] [DOI] [PubMed] [Google Scholar]
Egger 1997
- Egger M, Davey Smith G, Schneider M, Minder C. Bias in meta‐analysis detected by a simple graphical test. BMJ (Clinical Research Ed.) 1997;315:629‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Flora 1998
- Flora K, Hahn M, Rosen H, Benner K. Milk thistle (Silybum marianum) for the therapy of liver disease. The American Journal of Gastroenterology 1998;93(2):139‐43. [PMID: 9468229] [DOI] [PubMed] [Google Scholar]
Gluud 1988
- Gluud C. Testosterone and alcoholic cirrhosis. Epidemiologic, pathophysiologic and therapeutic studies in men. Danish Medical Bullettin 1988;35(6):564‐75. [PubMed] [Google Scholar]
Gluud 1993
- Gluud C, Afroudakis A, Caballeria J, Laskus T, Morgan MY, Rueff B, et al. Diagnosis and treatment of alcoholic liver disease in Europe. Gastroenterology International 1993;6(4):221‐30. [Google Scholar]
Gluud 2001
- Gluud C. Alcoholic hepatitis: no glucocorticosteroids?. Steatohepatitis (NASH and ASH). FALK Symposium 121. Leuschner U, James O, Dancygier H. Dordrecht: Kluwer Academic Publishers, 2001. [Google Scholar]
Gluud 2011
- Gluud C, Nikolova D, Klingenberg SL, Alexakis N, Als‐Nielsen B, Colli A, et al. Cochrane Hepato‐Biliary Group. About The Cochrane Collaboration (Cochrane Review Groups (CRGs)). 2011, Issue 5. Art. No.: LIVER.
Gonzalez‐Reimers1988
- Gonzalez‐Reimers CE, Santolaria‐Fernandez FJ, Gonzalez‐Hernandez T, Batista‐Lopez N, Gomez‐Sirvent JL, Perez‐Delgado MM, et al. Effect of propylthiouracil on liver cell development in the male albino mouse: protective effect against ethanol‐induced alterations. Drug and Alcohol Dependence 1988;21:11‐8. [DOI] [PubMed] [Google Scholar]
Gurusamy 2009
- Gurusamy KS. Assessment of risk of bias in randomized clinical trials in surgery. British Journal of Surgery 2009;96:342‐9. [DOI] [PubMed] [Google Scholar]
Hallé 1982a
- Hallé P, Paré P, Kaptein E, Kanel G, Redeker AG, Reynolds TB. Double‐blind, controlled trial of propylthiouracil in patients with severe acute alcoholic hepatitis. Gastroenterology 1982;82(5):925‐31. [PubMed] [Google Scholar]
Hicks 1992
- Hicks M, Wong LS, Day RO. Antioxidant activity of propylthiouracil. Biochemical Pharmacology 1992;43(3):439‐44. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Hill 1993
- Hill DB, Marsano LS, McClain CJ. Increased plasma interleukin‐8 concentrations in alcoholic hepatitis. Hepatology 1993;18(3):576‐80. [PubMed] [Google Scholar]
Huang 1994
- Huang MJ, Li KL, Wei JS, Wu SS, Fan KD, Liaw YF. Sequential liver and bone biochemical changes in hyperthyroidism: prospective controlled follow‐up study. The American Journal of Gastroenterology 1994;89(7):1071‐6. [PubMed] [Google Scholar]
ICH‐GCP 1997
- International Conference on Harmonisation Expert Working Group. International conference on harmonisation of technical requirements for registration of pharmaceuticals for human use. ICH harmonised tripartite guideline. Guideline for good clinical practice1997 CFR & ICH Guidelines. Vol. 1, PA 19063‐2043, USA: Barnett International/PAREXEL, 1997. [Google Scholar]
Ichiki 1998
- Ichiki Y, Akahoshi M, Yamashita N, Morita C, Maruyama T, Horiuchi T, et al. Propylthiouracil‐induced severe hepatitis: a case report and review of the literature. Journal of Gastroenterology 1998;33:747‐50. [DOI] [PubMed] [Google Scholar]
Imamura 1986
- Imamura M, Aoki N, Saito T, Ohno Y, Maruyama Y, Yamaguchi J, et al. Inhibitory effects of antithyroid drugs on oxygen radical formation in human neutrophils. Acta Endocrinologica 1986;112:210‐6. [DOI] [PubMed] [Google Scholar]
Ioannidis 2000
- Ioannidis JPA, Lau J. What certainty can there be on how much treatment work? Empirical insight from recursive cumulative meta‐analyses. 8th International Cochrane Colloquium, October 2000:21.
Israel 1975a
- Israel Y, Videla L, Berstein J. Liver hypermetabolic state after chronic ethanol consumption: hormonal interrelationships and pathogenic implications. Federation Proceedings 1975;34:2052‐9. [PubMed] [Google Scholar]
Israel 1975b
- Israel Y, Kalant H, Orrego H, Khanna JM, Videla L, Philips JM. Experimental alcohol‐induced hepatic necrosis: suppression by propylthiouracil. Proceedings of the National Academy of Sciences of the United States of America 1975;72:1137‐41. [DOI] [PMC free article] [PubMed] [Google Scholar]
Iturriaga 1980
- Iturriaga M, Ugarte G, Israel Y. Hepatic vein oxygenation liver blood flow and the rate of ethanol metabolism in recently abstinent patients. European Journal of Clinical Investigation 1980;10:211‐8. [DOI] [PubMed] [Google Scholar]
Kampmann 1981
- Kampmann JP, Hansen JM. Clinical pharmacokinetics of antithyroid drugs. Clinical Pharmacokinetics 1981;6:401‐28. [DOI] [PubMed] [Google Scholar]
Kawasaki 1989
- Kawasaki T, Carmichael FJ, Giles G, Saldivia V, Israel Y, Orrego H. Effects of propylthiouracil and methimazole on splanchnic hemodynamics in awake and unrestrained rats. Hepatology 1989;10:273‐4. [DOI] [PubMed] [Google Scholar]
Kjaergard 2001
- Kjaergard LL, Villumsen J, Gluud C. Reported methodological quality and discrepancies between large and small randomized trials in meta‐analyses. Annals of Internal Medicine 2001;135:982‐9. [DOI] [PubMed] [Google Scholar]
Klein 1994
- Klein I, Becker DV, Levey GS. Treatment of hyperthyroid disease. Annals of Internal Medicine 1994;121:281‐8. [DOI] [PubMed] [Google Scholar]
Lieber 2000
- Lieber CS. Alcoholic liver disease: new insights in pathogenesis lead to new treatments. Journal of Hepatology 2000;32(Suppl 1):113‐28. [DOI] [PubMed] [Google Scholar]
Lieber 2003
- Lieber CS, Weiss DG, Groszmann R, Paronetto F, Schenker S, For the Veterans Affairs Cooperative Study 391 Group. II. Veterans Affairs Cooperative Study of polyenylphosphatidylcholine in alcoholic liver disease. Alcohol Clinical and Experimental Research 2003;27:1765‐72. [DOI] [PubMed] [Google Scholar]
Marbet 1987
- Marbet UA, Bianchi L, Meury U, Stalder GA. Long‐term histological evaluation of the natural history and prognostic factors of alcoholic liver disease. Journal of Hepatology 1987;4(3):364‐72. [DOI] [PubMed] [Google Scholar]
Mato 1999
- Mato JM, Cámara J, Fernández de Paz J, Caballeria L, Coll S, Cabballero A, et al. S‐Andenosylmethionine in alcoholic cirrhosis: a randomised, placebo‐controlled, double‐blind, multicenter clinical trial. Journal of Hepatology 1999;30:1081‐9. [DOI] [PubMed] [Google Scholar]
Mendenhall 1984
- Mendenhall CL, Anderson S, Garcia‐Pont P, Goldenberg S, Kiernan T, Seeff LB, et al. Short‐term and long‐term survival in patients with alcoholic hepatitis treated with oxandrolone and prednisolone. New England Journal of Medicine 1984;311(23):1464‐70. [DOI] [PubMed] [Google Scholar]
Mezey 1991
- Mezey E, Caballeria J, Mitchell MC, Pares A, Herlong HF, Rodes J. Effect of parenteral amino acid supplementation on short‐term and long‐term outcomes in severe alcoholic hepatitis: a randomised controlled trial. Hepatology 1991;14(6):1090‐6. [PMID: 1959859] [PubMed] [Google Scholar]
Moher 1998
- Moher D, Pham B, Jones A, Cook DJ, Jadad AR, Moher M, et al. Does quality of reports of randomised trials affect estimates of intervention efficacy reported in meta‐analyses. The Lancet 1998;352:609‐13. [DOI] [PubMed] [Google Scholar]
Morgan 1999
- Morgan MY. Alcoholic liver disease: natural history, diagnosis, clinical features, evaluation, management, prognosis, and prevention. In: Bircher J, Benhamou J‐P, McIntyre N, Rizzetto M, Rodés J editor(s). Oxford Textbook of Clinical Hepatology. Second Edition. Vol. 2, Oxford: Oxford Medical Publications, 1999:1185‐238. [Google Scholar]
Morita 2000
- Morita S, Ueda Y, Eguchi K. Anti‐thyroid drug‐induced ANCA‐associated vasculitis: a case report and review of the literature. Endocrine Journal 2000;47(4):467‐70. [PMID: 11075728] [DOI] [PubMed] [Google Scholar]
Orrego 1979a
- Orrego H, Kalant H, Israel Y, Blake J, Medline A, Rankin JC. Effect of short‐term therapy with propylthiouracil in patients with alcoholic liver disease. Gastroenterology 1979;76(1):105‐15. [PubMed] [Google Scholar]
Orrego 1987a
- Orrego H, Blake JE, Blendis LM, Compton KV, Israel Y. Long‐term treatment of alcoholic liver disease with propylthiouracil. New England Journal of Medicine 1987;317:1421‐7. [DOI] [PubMed] [Google Scholar]
Orrego 1987b
- Orrego H, Blake JE, Blendis LM, Israel Y. Prospective validation of the relationship of the combined clinical and laboratory index, the CCLI, with mortality in alcoholic liver disease. Hepatology 1987;7(5):1083. [Google Scholar]
Powell 1968
- Powell WJ Jr, Klatskin G. Duration of survival in patients with Laennec´s cirrhosis. Influence of alcohol withdrawal, and possible effects of recent changes in general management of the disease. American Journal of Medicine 1968;44(3):406‐20. [DOI] [PubMed] [Google Scholar]
Poynard 1994
- Poynard T, Barthelemy P, Fratte S, Boudjema K, Doffoel M, Vanlemmens C, et al. Evaluation of efficacy of liver transplantation in alcoholic cirrhosis by a case‐control study and simulated controls. The Lancet 1994;344(8921):502‐7. [PMID: 7914613] [DOI] [PubMed] [Google Scholar]
Poynard 1999
- Poynard T, Naveau S, Doffoel M, Boudjema K, Vanlemmens C, Mantion G, et al. Evaluation of efficacy of liver transplantation in alcoholic cirrhosis using matched and simulated controls: 5‐year survival. Multi‐centre group. Journal of Hepatology 1999;30(6):1130‐7. [DOI] [PubMed] [Google Scholar]
Rambaldi 2005a
- Rambaldi A, Gluud C. Colchicine for alcoholic and non‐alcoholic liver fibrosis or cirrhosis. Cochrane Database of Systematic Reviews 2005, Issue 2. [DOI: 10.1002/14651858.CD002148.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Rambaldi 2006a
- Rambaldi A, Gluud C. Anabolic‐androgenic steroids for alcoholic liver disease. Cochrane Database of Systematic Reviews 2006, Issue 4. [DOI: 10.1002/14651858.CD003045.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Rambaldi 2006b
- Rambaldi A, Gluud C. S‐adenosyl‐L‐methionine for alcoholic liver diseases. Cochrane Database of Systematic Reviews 2006, Issue 2. [DOI: 10.1002/14651858.CD002235.pub2] [DOI] [PubMed] [Google Scholar]
Rambaldi 2007
- Rambaldi A, Jacobs BP, Gluud C. Milk thistle for alcoholic and/or hepatitis B or C virus liver diseases. Cochrane Database of Systematic Reviews 2007, Issue 4. [DOI: 10.1002/14651858.CD003620.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
RevMan 2011 [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.1. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2011.
Ross 1998
- Ross AD, Dey I, Janes N, Israel Y. Effect of antithyroid drugs on hydroxyl radical formation a alpha‐1‐proteinase inhibitor inactivation by neutrophils: therapeutic implications. The Journal of Pharmacology and Experimental Therapeutics 1998;285(3):1233‐8. [PubMed] [Google Scholar]
Royle 2003
- Royle P, Milne R. Literature searching for randomized controlled trials used in Cochrane reviews: rapid versus exhaustive searches. International Journal of Technology Assessment in Health Care 2003;19(4):591‐603. [DOI] [PubMed] [Google Scholar]
Rubin 1968
- Rubin E, Lieber CS. Alcohol‐induced hepatic injury in non alcoholic volunteers. New England Journal of Medicine 1968;278(16):869‐76. [DOI] [PubMed] [Google Scholar]
Schulz 1995
- Schulz KF, Chalmers I, Haeys RJ, Altman DG. Empirical evidence of bias. Dimensions of methodological quality associated with estimates of treatment effects in controlled trials. Journal of American Medical Association 1995;273(5):408‐12. [PMID: 7823387] [DOI] [PubMed] [Google Scholar]
Sheron 1993
- Sheron N, Bird G, Koskinas J, Portmann B, Ceska M, Lindley, et al. Circulating and tissue levels of the neutrophil chemotaxin interleukin‐8 are elevated in severe acute alcoholic hepatitis, and tissue levels correlate with neutrophil infiltration. Hepatology 1993;18(1):41‐6. [PubMed] [Google Scholar]
Sørensen 1984
- Sørensen TIA, Orholm M, Bentsen KD, Høybye G, Eghøje K, Christoffersen P. Prospective evaluation of alcohol abuse and alcoholic liver injury in men as predictors of development of cirrhosis. The Lancet 1984;2(8397):241‐4. [DOI] [PubMed] [Google Scholar]
Thorlund 2009
- Thorlund K, Devereaux PJ, Wetterslev J, Guyatt G, Ioannidis JP, Thabane L, et al. Can trial sequential monitoring boundaries reduce spurious inferences from meta‐analyses. International Journal of Epidemiology 2009;38(1):276‐86. [DOI] [PubMed] [Google Scholar]
Trinchet 1992
- Trinchet JC, Balkau B, Poupon RE, Heintzmann F, Callard P, Gotheil C, et al. Treatment of severe alcoholic hepatitis by infusion of insulin and glucagon: a multicenter sequential trial. Hepatology 1992;15(1):76‐81. [PMID: 1277803] [DOI] [PubMed] [Google Scholar]
Vickers 1998
- Vickers A, Goyal N, Harland R, Rees R. Do certain countries produce only positive results? A systematic review of controlled trials. Controlled Clinical Trials 1998;19(2):159‐66. [DOI] [PubMed] [Google Scholar]
Wetterslev 2008
- Wetterslev J, Thorlund K, Brok J, Gluud C. Trial sequential analysis may establish when firm evidence is reached in cumulative meta‐analysis. Journal of Clinical Epidemiology 2008;61(1):64‐75. [DOI] [PubMed] [Google Scholar]
Wood 2008
- Wood L, Egger M, Gluud LL, Schulz KF, Jüni P, Altman GD, et al. Empirical evidence of bias in treatment effect estimates in controlled trials with different interventions and outcomes: meta‐epidemiological study. BMJ (Clinical Research Ed.) 2008;336:601‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Younes 1987
- Younes M, Strubelt O. Enhancement of hypoxic liver damage by ethanol: involvement of xanthine oxidase and the role of glycolysis. Biochemical Pharmacology 1987;36:2973‐7. [DOI] [PubMed] [Google Scholar]
Yuki 1982
- Yuki T, Israel Y, Thurman RG. The swift increase in alcohol metabolism. Inhibition by propylthiouracil. Biochemical Pharmacology 1982;31:2403‐7. [DOI] [PubMed] [Google Scholar]
References to other published versions of this review
Rambaldi 2001a
- Rambaldi A, Gluud C. Propylthiouracil for alcoholic liver disease. Cochrane Database of Systematic Reviews 2001, Issue 4. [DOI: 10.1002/14651858.CD002800.pub2] [DOI] [PubMed] [Google Scholar]
Rambaldi 2001b
- Rambaldi A, Gluud C. Meta‐analysis of propylthiouracil for alcoholic liver disease ‐ a Cochrane Hepato‐Biliary Group Review. Liver 2001;21:398‐404. [DOI] [PubMed] [Google Scholar]
Rambaldi 2002
- Rambaldi A, Gluud C. Propylthiouracil for alcoholic liver disease. Cochrane Database of Systematic Reviews 2002, Issue 2. [DOI: 10.1002/14651858.CD002800.pub2] [DOI] [PubMed] [Google Scholar]
Rambaldi 2005b
- Rambaldi A, Gluud C. Propylthiouracil for alcoholic liver disease. Cochrane Database of Systematic Reviews 2005, Issue 4. [DOI: 10.1002/14651858.CD002800.pub2] [DOI] [PubMed] [Google Scholar]