

Abstract
Hepatotoxicity is a leading cause of attrition in the drug development process. Traditional preclinical and clinical studies to evaluate hepatotoxicity liabilities are expensive and time consuming. With the advent of critical advancements in high-throughput screening, there has been a rapid accumulation of in vitro toxicity data available to inform the risk assessment of new pharmaceuticals and chemicals. To this end, we curated and merged all available in vivo hepatotoxicity data obtained from the literature and public resources, which yielded a comprehensive database of 4089 compounds that includes hepatotoxicity classifications. After dividing the original database of chemicals into modeling and test sets, PubChem assay data were automatically extracted using an in-house data mining tool and clustered based on relationships between structural fragments and cellular responses in in vitro assays. The resultant PubChem assay clusters were further investigated. During the cross-validation procedure, the biological data obtained from several assay clusters exhibited high predictivity of hepatotoxicity and these assays were selected to evaluate the test set compounds. The read-across results indicated that if a new compound contained specific identified chemical fragments (ie, Molecular Initiating Event) and showed active responses in the relevant selected PubChem assays, there was potential for the chemical to be hepatotoxic in vivo. Furthermore, several mechanisms that might contribute to toxicity were derived from the modeling results including alterations in nuclear receptor signaling and inhibition of DNA repair. This modeling strategy can be further applied to the investigation of other complex chemical toxicity phenomena (eg, developmental and reproductive toxicities) as well as drug efficacy.
Keywords: big data, hepatotoxicity, read-across, risk assessment, mechanism-driven
Drug hepatotoxicity is a critical concern of the pharmaceutical industry and the public. Drug-Induced Liver Injury (DILI) is one of the leading causes of liver failure cases (Reuben et al., 2010). One of the reasons for the postmarketing withdrawal of a drug is due to unexpected hepatotoxicity in patients, which is not fully recognized in the preclinical and clinical trials (Kaplowitz, 2005). Furthermore, traditional preclinical and clinical studies to evaluate drug hepatotoxicity are expensive and time consuming (Hartung, 2009). With the advent of critical advancements in in vitro testing approaches as the alternatives to animal testing, in particular high-throughput screening (HTS), there has been a rapid accumulation of chemical toxicity data which can be used to better identify and prioritize chemical hazards (Ciallella and Zhu, 2019; Zhang et al., 2014). However, data obtained solely from available in vitro protocols have low correlation to hepatotoxicity risk and any single in vitro test cannot fully replace in vivo hepatotoxicity testing.
As an alternative technique to animal testing for toxicological assessment (Schultz et al., 2015), read-across is a promising low-cost method to evaluate the toxicity potential of new compounds (Ball et al., 2016). In a read-across study, the toxicity potential of a new compound will be evaluated by its most “similar” compound that has an experimental toxicity result (Ball et al., 2016). The similarity of compounds can be defined from chemical and/or biological properties. Based on the hypothesis that chemically similar compounds have similar bioactivities (Tropsha, 2012), quantitative structure-activity relationship models, which have been widely used for read-across studies, were developed by various machine learning approaches and chemical descriptors calculated from chemical structures (Solimeo et al., 2012; Zhang et al., 2013; Zhu and Kruhlak, 2014). Due to the inherent complexity of biological systems, covering all potential factors contributing to multifaceted in vivo outcomes, such as hepatotoxicity, is difficult using available quantitative structure-activity relationship models. (Muster et al., 2008). Using only chemical similarity in read-across studies for complex toxicity endpoints has proved to be error-prone due to “activity cliffs” (ie, structural similar compounds have different toxicity) (Medina-Franco et al., 2009; Stumpfe and Bajorath, 2012).
In addition to chemical structural properties, the inclusion of biosimilarity rankings based on biological data adds extra strength to the utility of read-across (Zhu et al., 2016). There have been previous studies that used biological data to support read-across, such as the toxicants profiled by ToxCast biological data, in which read-across was performed using chemical responses from a set of in vitro bioassays (Martin et al., 2011; Reif et al., 2010; Rotroff et al., 2013; Sipes et al., 2011, 2013). Because these bioassays were designed to reveal specific toxicity mechanisms, the predictions of new compounds can also be interpretable. Hewitt et al. (2013) presented this read-across scheme in a review of 2013 and several studies following this strategy were performed. For example, Liu et al. (2015a) used selected ToxCast assays and chemical structures to predict hepatotoxicity. Low et al. first used the combination of selected toxicogenomics data and chemical descriptors to create a hybrid model (Low et al., 2011) then extended this study by including gene expression data and cytotoxicity data (Low et al., 2013). However, the disadvantage of previous studies is that the read-across was limited by manually selected biological data, which only include limited well-known toxicity mechanisms. Thus, they are not able to cover all potential mechanisms relevant to in vivo animal toxicity. The key in the current toxicity big data scenario is to use an automatic data mining method to explore all relevant biological data, which is not limited to preselected in-house data, and perform read-across studies based on the biological data with high sparsity and variety.
We have reported several toxicity modeling studies that capitalize on the availability of big data (Kim et al., 2016; Russo et al., 2019; Zhang et al., 2014). In one of these studies, Kim et al. (2016) developed a virtual Adverse Outcome Pathway (vAOP) model for around 1300 drugs with classified liver injury results. The vAOP model reported in this study consists of 4 oxidative stress assays that were automatically identified from millions of PubChem assays for target compounds. However, the vAOP model developed in this study yielded relatively low accuracy (around 60%) due to limited hepatotoxicity data available at that time. All compounds used for modeling were obtained from a single resource, which was the U.S. FDA DILI data (Chen et al., 2011).
In the present study, a much larger database for hepatotoxicity was generated by summarizing and merging all current publicly available hepatotoxicity datasets, which consists of 4089 unique compounds with their hepatotoxicity categories defined in original sources. According to our best knowledge, so far this is the largest hepatotoxicity database curated for modeling purpose. An in-house automatic data mining portal was used to extract biological data from PubChem for all the compounds (Russo et al., 2017). The PubChem assays were analyzed and clustered using a novel approach developed in one of our recent studies (Russo et al., 2019). The read-across study was performed by calculating compound biosimilarity according to PubChem assay clusters, which were formed by calculating chemical fragment-in vitro relationships and selected by their predictivity for hepatotoxicity. Furthermore, several vAOP models were developed by identifying compounds with the same chemical fragments, which were defined as initial molecular events of toxicity pathways, within the PubChem assay clusters. The resultant vAOP models not only have good predictivity of hepatotoxicity but also indicate new hepatotoxicity mechanisms.
MATERIALS AND METHODS
Hepatotoxicity database
Hepatotoxicity data for chemicals were obtained from individual datasets in the literature as well as public database resources (Table 1). These datasets include various compounds with in vivo hepatotoxicity data defined using different standards. Compounds in datasets 1 (Ekins et al., 2010), 2 (Fourches et al., 2010), 6 (Kim et al., 2016), 7 (Mulliner et al., 2016), and 8 (Liew et al., 2011) were classified by 2 categories as hepatotoxic and nontoxic. Compounds in datasets 3 (Liu et al., 2015b) and 5 (Chen et al., 2011) were classified by 3 categories as hepatotoxic, possible hepatotoxic, and nontoxic. Compounds in dataset 4 (Greene et al., 2010) were classified by 4 categories as HH (evidence for human hepatotoxicity), NE (no evidence for hepatotoxicity in any species), WE (weak evidence for human hepatotoxicity), and AH (evidence for animal hepatotoxicity but not tested in humans). The category standards for hepatotoxicity can be found in detail in the references for each dataset (Table 1). We harmonized various hepatotoxicity classifications into binary categories of 1 (hepatotoxic) and 0 (nontoxic) according to the standards described in these datasets. The details of criterion used for harmonization are listed in Table 1.
Table 1.
General Information of Hepatotoxicity Datasets
| Dataset | Size | Original Categories | Species | Rules of Harmonization | Reference |
|---|---|---|---|---|---|
| 1 | 534 | 1 or 0 (hepatotoxic or not) | Humans | Same | Ekins et al. (2010) |
| 2 | 951 | 1 or 0 (hepatotoxic or not) | Humans, rodents, nonrodents | Only human data were used | Fourches et al. (2010) |
| 3 | 605 | 1, −1, or 0 (hepatotoxic or not, and inconclusive) | Humans | Excluding inconclusives | Liu et al. (2015b) |
| 4 | 627 | HH, NE, WE, AH | Humans, animals | HH, WE as 1; NE as 0 (AH were excluded) | Greene et al. (2010) |
| 5 | 287 | Most, less and no concern for DILI | Humans | Most and less concern as 1; no concern as 0 | Chen et al. (2011) |
| 6 | 1314 | 1 or 0 (hepatotoxic or not) | Humans | Same | Kim et al. (2016) |
| 7 | 3712 | 1 or 0 (hepatotoxic or not) | Humans, animals | Only human data were used | Mulliner et al. (2016) |
| 8 | 1274 | 1 or 0 (hepatotoxic or not) | Humans | Same | Liew et al. (2011) |
Abbreviations: HH, evidence for human hepatotoxicity; NE, no evidence for hepatotoxicity in any species; WE, weak evidence (<10 case reports) for human hepatotoxicity; AH, evidence for animal hepatotoxicity but not tested in humans.
The curation of chemical structures for individual datasets was performed using the chemical structure standardizer tool CASE Ultra DataKurator 1.6.0.3 to remove inorganic compounds and mixtures. Then, duplicates within each dataset were removed by using the Python RDKit Chem module and CASE Ultra DataKurator. Finally, overlapping compounds were identified among individual datasets. These overlapping compounds may yield different hepatotoxicity classifications in various sources. In this study, if there were different classifications from different sources for a compound, this chemical was then categorized according to the majority classification from these source datasets. If there was no majority classification for an overlapping compound (ie, the same count of records for both hepatotoxic and nontoxic), the compound was excluded from modeling.
Overall read-across workflow
The overall read-across workflow was shown in Figure 1. After data curation, the hepatotoxicity database was randomly split into a modeling set (66.7%) and a test set (33.3%). The bioprofile for compounds in this database was generated using the in-house profiling tool CIIPro (Russo et al., 2017). Then, mechanistically similar PubChem assays were identified using chemical fragment-in vitro relationships (Russo et al., 2019) to form multiple assay clusters. The assay clusters were selected for read-across based on their cross-validation predictivity of hepatotoxicity within the modeling set. The predictions of test set compounds by read-across were performed based on biosimilarity calculations within the prioritized PubChem assay clusters. Furthermore, several chemical fragments were identified and integrated into read-across as Molecular Initiating Events (MIEs) (see the following sections for details). The resultant vAOP models were also used to predict hepatotoxicity of the test set compounds.
Figure 1.

Virtual Adverse Outcome Pathway (vAOP) read-across workflow.
PubChem assay clusters
To perform a mechanism-driven read-across, it is critical to identify mechanistic-related assays. To this end, we first generated chemical fragments for compounds in the whole database using ToxPrint Chemotypes from ChemoTyper, which yielded toxicity-related chemical fingerprints for compounds. Then, all compounds were profiled using an in-house automatic data mining tool CIIPro (Russo et al., 2017) to search against the PubChem database for all available biological data, and a bioprofile was generated for each compound. The chemical fragment-in vitro relationships were generated using a novel method described in a recent study (Russo et al., 2019). Briefly, the relationship between each chemical fragment and PubChem assay was determined using Fisher’s exact test. The output of this test is a p value denoting the statistical significance of the relationship between the fragment and assay activity. Any relationships between a fragment and assay with a p < .05 were considered to be statistically significant. PubChem assays sharing many significant fragments could be mechanistic related and/or unveil potential novel mechanisms of hepatotoxicity for specific chemical toxicants. To group similar assays, the Jaccard similarity between each assay was calculated based on the profile of the fragment assay relationships calculated above. Clusters of PubChem assays were determined by using an overlapping network detecting algorithm OSLOM (Lancichinetti et al., 2011). The implementing package used for our analysis is available online (http://www.oslom.org/), and all parameters were set by default. Then, the PubChem assay clustering results were imported into a software package Gephi (v. 0.9.1, www.gephi.org/) to visualize all assay clusters by applying the force-based layout algorithm ForceAtlas 2 with default parameters (Figure 2).
Figure 2.

PubChem assay clusters based on chemical fragment-in vitro response relationships. Each dot indicates a unique PubChem assay. The assays (indicated by dots) with the same color belong to the same cluster except 78 assays (indicated by black dots) belong to more than 1 cluster.
Read-across study
In this study, a bioprofile-based read-across (Figure 1) was performed within each PubChem assay cluster. Briefly, for an assay cluster, the similarity between any 2 compounds was calculated based on the bioprofiles consisting of the PubChem assays that formed this cluster. This biosimilarity calculation utilized the equation published in our recent study (Russo et al., 2017) as following:
| (1) |
Because the bioprofile has missing data for most compounds in our datasets, an extra parameter confidence support was used to evaluate the biosimilarity confidence to avoid compounds only have few responses:
| (2) |
All PubChem assay clusters were used for read-across within the modeling set and the results were evaluated by the 5-fold cross-validation procedure. During this procedure, the modeling set was randomly divided into 5 equivalent subsets. Each time, 4 subsets (80% of the modeling set compounds) were combined as the training set and the remaining 1 subset (20% of the modeling set compounds) was used as a test set to validate the selected PubChem assays in this cluster. The compounds in the test set were predicted by their bio-nearest neighbors in the training set using the selected PubChem assays in the cluster. This procedure was repeated 5 times so that each modeling set compound was used for prediction once. Various statistical parameters were calculated to describe the read-across results, such as sensitivity, specificity, Correct Classification Rate (CCR), and Positive Predictive Value (ppv). All the formulas of these universal statistical parameters are shown in the following:
| (3) |
| (4) |
| (5) |
| (6) |
where TP represents the number of true positives (toxic compounds correctly predicted as toxic), FP represents the number of false positives (nontoxic compounds incorrectly predicted as toxic), TN represents the number of true negatives (nontoxic compounds correctly predicted as nontoxic), and FN represents the number of false negatives (toxic compounds incorrectly predicted as nontoxic).
Furthermore, ChemoTyper chemical fragments, which were identified from toxic compounds within each assay cluster, were evaluated for their ability to improve hepatotoxicity predictions. In this effort, the read-across analysis was performed for a subset of compounds containing a specific fragment within each cluster. If the result showed significant improvement, the relevant chemical fragment was considered as a MIE of a vAOP model.
Predicting new compounds
The hepatotoxicity of a new compound (eg, a test set compound) was evaluated by its nearest neighbor compound in the modeling set defined by biosimilarity within a selected assay cluster. Furthermore, if a new compound contained an identified MIE, its biosimilarity was calculated with the modeling set compounds containing the same MIE within the relevant assay cluster for vAOP model predictions.
RESULTS
Hepatotoxicity Database Overview
In this study, a large and diverse hepatotoxicity database was curated from various data sources. Because the original datasets contain in vivo hepatotoxicity data classified with different standards, it is necessary to harmonize the data into a binary classification (ie, hepatotoxic and nontoxic) for model development (Table 1). However, among 1639 compounds that were found in more than one original data source, 277 of them showed conflicting hepatotoxicity results. Then, to merge compounds with conflicting results from different sources, a majority rule was applied to define hepatotoxicity classifications for these compounds. Among the 4089 unique compounds in the original database, 3790 compounds remained in the curated database and were categorized as hepatotoxic (1549 compounds) or nontoxic (2241 compounds). The whole database (details of all compounds listed in Supplementary Table 1) was randomly split into modeling and test sets, which consist of 2522 and 1268 compounds, respectively. To show the chemical space of all the compounds, we performed a Principal Component Analysis study using 206 Molecular Operating Environment (MOE) 2D descriptors. The top 3 principal components, which account for 57.4% variance, were used to construct the chemical space. Except several structural outliers, the modeling and test compounds cover a large and diverse space (Supplementary Figure 1).
The relevant biological data for these 3790 compounds were extracted from PubChem. The resulted bioprofile consisted of 43 224 PubChem assays, which contained 880 449 data points. Furthermore, based on the chemical structures of these compounds, 729 ChemoTyper chemical fragments were identified. The chemical fragments and biological data were both large and diverse, thereby yielding useful information for the read-across studies described later.
PubChem Assay Clusters Result
Among the initial 43 224 assays within the bioprofile, 883 assays exhibited significant correlations (p < .05) with at least 1 ChemoTyper chemical fragment, resulting in a total of 19 039 significant relationships between chemical structural fragments and in vitro responses. The Jaccard similarity score between any 2 assays was calculated based on “chemical fragment-in vitro response” relationships. Two assays were defined as “mechanistic-related” to each other if they have a Jaccard similarity score higher than 0.75. In Figure 2, 2 mechanistic-related assays, which are shown as dots, were connected by an edge. There were 804 assays with a Jaccard similarity score to their nearest neighbor assays of over 0.75 and their relationships were further analyzed using the overlapping network detecting algorithm OSLOM (Lancichinetti et al., 2011). OSLOM can estimate and distinguish statistical significant clusters from pseudo-clusters, and it also allows overlapping among various clusters. An assay cluster, which was generated by OSLOM analysis, contains a group of assays that are mechanistic related. An overlapped assay in 2 clusters represented a potential receptor existing in 2 different biological mechanisms. There were 32 unique clusters with 3–87 assays per cluster that were identified using the OSLOM algorithm, as shown by different colors in Figure 2. The overlapping assays were colored as black. Information regarding the clustered assays is summarized in Supplementary Table 2.
Hepatotoxicity Predictions of New Compounds by Read-Across
All 32 PubChem assay clusters obtained from the above step were used for the read-across study of hepatotoxicity. The predictivity of hepatotoxicity using assays in each cluster was first evaluated by the 5-fold cross-validation within the modeling set. Sensitivity, specificity, CCR, and ppv were calculated for all clusters and these parameters were used to analyze the predictivity of hepatotoxicity. The predictivity, shown by the ppv (Supplementary Figure 2), indicates the potential applicability of using the assays within each cluster to evaluate chemical hepatotoxicity. The reason to use ppv as the major evaluation parameter is that the underlying mechanisms responsible for hepatotoxicity are vast and complicated, and thus, it is unlikely to expect a few PubChem assays to explain all potential hepatotoxic phenomena. When using PubChem assays for toxicity prediction, it is reasonable to expect a relatively high false negative rate (ie, compounds inactive in a particular assay, yet active in other toxicity tests). Furthermore, the active data of a bioassay mean a specific chemical biological phenomenon (eg, binding to a receptor and inhibition of an enzyme), which is more meaningful than inactive data when correlating to toxicity phenomena. Not surprisingly, most clusters have relatively low predictivity of hepatotoxicity (ppv <0.6), but several clusters showed higher predictivity (ppv > 0.7). However, the read-across within cluster 5 showed ppv = 1.0 from cross-validation assessments. This is due to the model overfitting, which could be indicated by their sensitivity, specificity, and CCR (Supplementary Table 3). The sensitivity, specificity, and CCR are 1.00, 0.00, and 0.50, respectively. These results indicated that only using ppv for model selection is flawed when the data size is small (ie, the number of compounds tested by the associated cluster is small).
ChemoTyper chemical fragments were further evaluated for their ability to improve hepatotoxicity predictions (details can be found in the Materials and Methods section). A chemical fragment was considered to be a MIE of a hepatotoxicity pathway if modeling set compounds containing this fragment showed improved cross-validation predictivity within an assay cluster. To minimize the effects of missing data on the model selection, only the clusters in which the read-across models have confidence support values larger than 5 were investigated. 4 criteria were applied to select chemical fragments as potential MIEs: (1) there are at least 5 hepatotoxic compounds containing the selected fragments, (2) ppv of cross-validation was above 60%, (3) ppv of cross-validation was improved by read-across within the compounds containing the fragment, and (4) the bioprofile of the compounds containing the fragment have < 65% missing data. Thus, several chemical fragments were selected and considered as MIEs. A compound containing a selected MIE will be predicted as hepatotoxic when it also shows active responses in the relevant pathway assays. The inclusion of MIEs into a pathway can not only improve the predictivity of read-across but also derive useful toxicity mechanisms based on the resultant vAOP models. However, due to the nature of ChemoTyper fingerprints, the resultant MIEs are general chemical fragments, which exist in many organic compounds. This issue is partially resolved in the current vAOP models with extra validation by biological testing against the assays selected for the pathways. This issue can be permanently resolved when more hepatotoxicity data are available in the future and more diverse chemical fragments were selected as MIEs, such as the structural alerts described in other studies with large amounts of data (Alves et al., 2016; Liu et al., 2015b; Stepan et al., 2011).
The top selected fragment, which was identified from cluster 1 as a MIE, is a 6-membered aromatic ring containing up to 1 nitrogen. The compounds containing this fragment and their corresponding bioprofiles, which were used for the read-across predictions, are shown in Figure 3. The assays (represented using PubChem AID in the following) in this cluster (Table 2) could be classified into 3 groups including (1) drug screening assays (AID 1876, 1877, 1883, 1886), (2) receptor binding assays (AID 485345, 488953, 720572, 720692, 720725, 743239), and (3) biomarkers (AID 463097, 485298, 493107). As shown, if a compound containing this identified chemical fragment (all compounds structures shown in Supplementary Figure 3) and showed active results in these assays, it can be predicted as hepatotoxic (Figure 3). A portion of these hepatotoxic compounds included calcium channel blockers, such as felodipine (compound 633), nimodipine (compound 751), and nifedipine (compound 2996). As shown in Table 2, a number of the bioassays that informed the cluster 1 vAOP model align with plausible mechanisms of hepatotoxicity. These include altered signaling through the farnesoid X receptor (FXR) (AID 743239) and glucocorticoid receptor (AID 720692 and 720725) as well as inhibition of DNA repair pathways (AID 493107). Interestingly, there were some other compounds within this cluster, which neither containing this structural fragment nor have active responses in these assays, but are in fact hepatotoxic. For example, loxoprofen (compound 2831), which only has inactive results in the assays in cluster 1, can induce hepatotoxicity in humans (Greig and Garnock-Jones, 2016). According to the information on LiverTox®, the mechanism of loxoprofen produce hepatic injury is considered an idiosyncratic reaction likely involving an immunologic reaction (Shrestha et al., 2018) that cannot be detected by the in vitro assays in cluster 1.
Figure 3.

The vAOP model developed from cluster 1. A compound (highlighted by yellow) was identified as toxic when it containing the chemical fragment (Molecular Initiating Event) and shows active responses (orange) in the selected PubChem assays.
Table 2.
General Information Regarding Bioassays Used in the vAOP Model From Cluster 1
| Bioassay AID | Bioassay Title | Bioassay Type | Overlap a |
|---|---|---|---|
| 1876 | qHTS for Differential Inhibitors of Proliferation of Plasmodium Falciparum Line 3D7 | Drug screen assays | Yes |
| 1877 | qHTS for Differential Inhibitors of Proliferation of Plasmodium Falciparum Line D10 | Yes | |
| 1883 | qHTS for Differential Inhibitors of Proliferation of Plasmodium Falciparum Line W2 | Yes | |
| 1886 | qHTS for Differential Inhibitors of Proliferation of Plasmodium Falciparum Line HB3 | Yes | |
| 485345 | qHTS Validation Assay to Find Inhibitors of Chronic Active B-Cell Receptor Signaling | Receptor binding assays | No |
| 488953 | qHTS Validation Assay for Inhibitors of HP1-beta Chromodomain Interactions with Methylated Histone Tails | Yes | |
| 720572 | qHTS for Activators of Parkin Expression: LOPAC Validation Assay (NLuc Reporter) | No | |
| 720692 | qHTS Assay to Identify Small Molecule Antagonists of the Glucocorticoid Receptor (GR) Signaling Pathway | No | |
| 720725 | qHTS Assay to Identify Small Molecule Antagonists of the Glucocorticoid Receptor (GR) Signaling Pathway: Summary | No | |
| 743239 | qHTS Assay to Identify Small Molecule Agonists of the Farnesoid-X-Receptor (FXR) Signaling Pathway: Summary | No | |
| 463097 | Validation Screen for Small Molecules That Induce DNA Rereplication in MCF 10A Normal Breast Cells | Biomarkers | No |
| 485298 | qHTS Assay for Small Molecule Inhibitors of Mitochondrial Division or Activators of Mitochondrial Fusion | No | |
| 493107 | Validation Screen for Small Molecules That Inhibit ELG1-Dependent DNA Repair in Human Embryonic Kidney (HEK293T) Cells Expressing Luciferase-Tagged ELG1 | Yes |
Overlapped assays exist in other clusters identified by OSLOM analysis.
There are 2 other structural fragments that were identified from clusters 3 and 17, respectively. The fragment from cluster 3 includes a phenoxyl group (Supplementary Figure 4). Hepatotoxicants containing this fragment include dichlorophene (compound 588), oxyquinoline sulfate (compound 766), pentachlorophenol (compound 1689), curcumin (compound 2579), and benzbromarone (compound 3427). According to the data on LIVERTOX, the mechanism of hepatotoxicity induced by benzbromarone arises primarily from 2 processes; first, benzbromarone undergoes hepatic metabolism by CYP2C9 and second, the parent compound or its metabolites alter mitochondrial function (Kaufmann et al., 2005). The fragment from cluster 17 represents a pyrimidine scaffold (Supplementary Figure 5). Azathioprine (compound 2374) which is an imidazolyl derivative and prodrug of mercaptopurine that inhibits cellular function by antagonism of purine metabolism contains this fragment. Azathioprine is associated with several forms of hepatotoxicity, including rises in serum aminotransferase levels, an acute cholestatic injury, and a chronic hepatic injury according to LIVERTOX (Corley et al., 1966; Mackay et al., 1964; Sparberg et al., 1969). The mechanism is not clear yet but is likely due to an immunological response to a metabolic byproduct (Aithal, 2011; Romagnuolo et al., 1998). Another drug containing this fragment, 6-mercaptopurine (compound 3088), is effective both as an anticancer and an immunosuppressive drug and is used to treat leukemia and autoimmune diseases (Björnsson et al., 2017). 6-Mercaptopurine causes direct, reproducible, dose-related hepatotoxicity in animal models (Clark et al., 1960; Einhorn and Davidsohn, 1964). The toxic effects of mercaptopurine, and particularly the myelotoxicity, have been linked to higher levels of methyl-mercaptopurine, a mercaptopurine metabolite (Nygaard et al., 2004).
The derived vAOP model can be applied to evaluate hepatotoxicants in the test set. There were a total of 369 test set compounds that contained this MIE and 61 of them showed at least 1 active response in the cluster 1 assays. Among these compounds, 12 of them had active responses in at least 4 assays (Figure 4) and were predicted to be hepatotoxic based on the resultant vAOP model with a predictive rate of 83.3%. As a comparison, these compounds were predicted using 2 DILI deep learning models recently developed (Xu et al., 2015) and the predictive rates are 50% for DL-combined model and 70% for DL-Liew model. Although only predicting 12 compounds is not statistically sufficient, this benchmark study showed the potential advantage of the vAOP model developed in this study compared to other hepatotoxicity models. Structures for all 12 compounds are shown in Supplementary Figure 3B. Among them, only 2 compounds, pimozide (compound 2561) and apigenin (compound 2841), were false positives. However, apigenin was reported to induce hepatotoxicity in Swiss mice (Singh et al., 2012), indicating a potential issue (ie, experimental error) in the database constructed in this study.
Figure 4.

Predicting new test set compounds using the vAOP model from cluster 1. The active responses were counted by the positive results of the selected PubChem assays within the cluster (as listed in Table 2).
There is an alternate way to also validate the vAOP model. The PubChem compounds, which were not in our hepatotoxicity database but were tested against those cluster 1 assays, were used for this validation purpose. There were 126 total compounds containing the MIE structure and 70 of them have at least 1 active response among these assays. Among these compounds, 21 compounds were predicted as hepatotoxic because they showed active responses in most of these assays. Literature searches were performed to investigate the toxicity potential of these prioritized compounds. There were 6 compounds (Table 3, represented using PubChem CID) with reported toxicity in previous studies. Among them, 2 are hepatotoxicants: clotrimazole (CID 2812 [Zhang et al., 2002]) and niclosamide (CID 4477 [Vliet et al., 2018]). The other 4 compounds, eliprodil (CID 60703), 2-chloro-5-nitro-N-phenylbenzamide (CID 644213), 1,10-phenanthroline (CID 1318), and ritanserin (CID 5074), exhibit toxicity effects other than hepatotoxicity as shown as in Table 3.
Table 3.
Prioritized Potential Hepatotoxicants Using the vAOP Model From Cluster 1
| PubChem CID | Name | Structure | Active Counts | Literature Supporting Hepatotoxicity |
|---|---|---|---|---|
| 2812 | Clotrimazole |
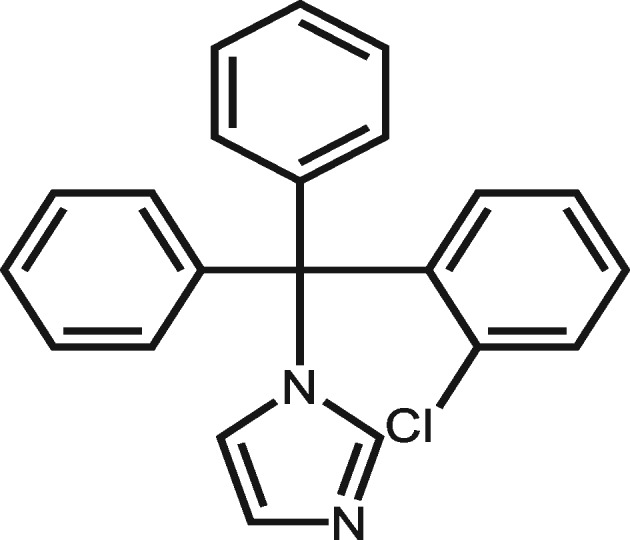
|
9 | The hepatotoxicity mechanism of clotrimazole is unknown. According to the report on LIVERTOX, the liver injury of clotrimazole might be caused by a toxic or immunogenic intermediate. |
| 1318 | 1,10-Phenanthroline |
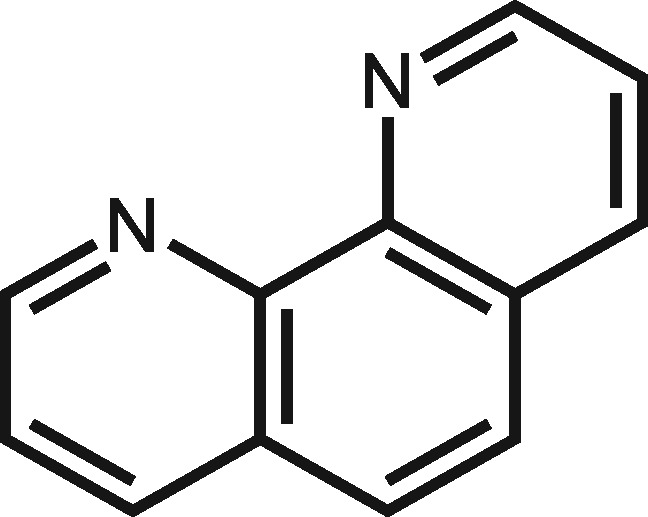
|
7 | 1,10-Phenanthroline could induce acute toxicity (Wijayanti et al., 2006). |
| 60703 | Eliprodil |

|
5 | Very toxic to aquatic life with long lasting effects from European Chemicals Agency (ECHA) data (https://echa.europa.eu/information-on-chemicals/cl-inventory-database/-/discli/details/167070). |
| 4477 | Niclosamide |
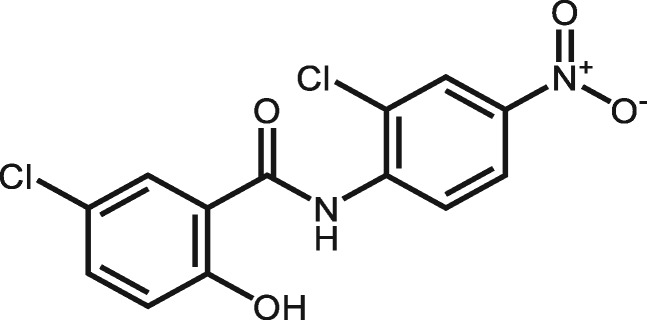
|
5 | Niclosamide may cause toxicity effect through interaction with DNA (Abreu et al., 2002) and induces epiboly delay during early Zebrafish embryogenesis (Vliet et al., 2018). |
| 644213 | 2-Chloro-5-nitro-N-phenylbenzamide |
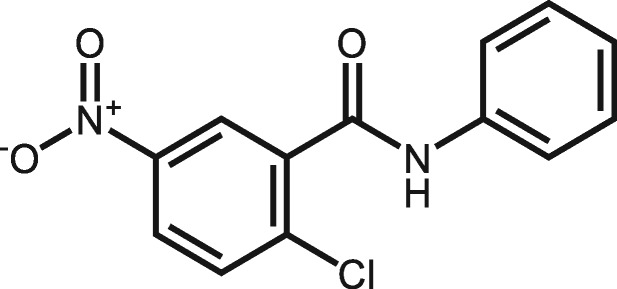
|
5 | No severe toxicity effects reported. But it causes an allergic skin reaction and serious eye irritation from European Chemicals Agency (ECHA) data (https://echa.europa.eu/information-on-chemicals/cl-inventory-database/-/discli/details/169195). |
| 5074 | Ritanserin |
|
5 | No severe toxicity effects. But it causes skin irritation, eye irritation, and respiratory irritation from European Chemicals Agency (ECHA) data (https://echa.europa.eu/information-on-chemicals/cl-inventory-database/-/discli/details/168593). |
DISCUSSION
We constructed a comprehensive hepatotoxicity database, automatically extracted relevant biological data from PubChem, and performed read-across studies for chemical hepatotoxicity. The key component of this study was to identify chemical fragment-in vitro-in vivo relationships, which were used to group PubChem assays that are mechanism similar and capable of evaluating hepatotoxicity. Furthermore, vAOP models were developed by integrating several chemical fragments as MIEs and PubChem assays as potential receptors biomarkers and cellular responses. New compounds containing the MIEs can be tested using the relevant assays to assess potential hepatotoxicity. Active responses from these assays indicate potential hepatotoxicity induced by pathway perturbations. Although the vAOP models developed in this study will not be sufficient to cover all the hepatotoxicity toxicity mechanisms, this work clearly indicates the benefits of using both chemical (ie, chemical structure) and biological (in vitro bioassays) data into the read-across process. Hepatotoxicity mechanisms could be indicated from these models, including alterations in nuclear receptor signaling and inhibition of DNA repair. With more data accumulated in the future, this workflow could be applied to other read-across studies for toxicity assessment.
DECLARATION OF CONFLICTING INTERESTS
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
FUNDING
National Institute of Environmental Health Sciences (R15ES023148), the Colgate-Palmolive Grant for Alternative Research, and the Johns Hopkins Center for Alternatives to Animal Testing (CAAT) grant.
Supplementary Material
REFERENCES
- Abreu F., Goulart M., Brett A. O. (2002). Detection of the damage caused to DNA by niclosamide using an electrochemical DNA-biosensor. Biosens. Bioelectron. 17, 913–919. [DOI] [PubMed] [Google Scholar]
- Aithal G. P. (2011). Hepatotoxicity related to antirheumatic drugs. Nat. Rev. Rheumatol. 7, 139–150. [DOI] [PubMed] [Google Scholar]
- Alves V. M., Muratov E. N., Capuzzi S. J., Politi R., Low Y., Braga R. C., Zakharov A. V., Sedykh A., Mokshyna E., Farag S., et al. (2016). Alarms about structural alerts. Green Chem. 18, 4348–4360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball N., Cronin M. T. D., Shen J., Blackburn K., Booth E. D., Bouhifd M., Donley E., Egnash L., Hastings C., Juberg D. R., et al. (2016). T4 report: Toward good read-across practice (GRAP) guidance. ALTEX 33, 149–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Björnsson E. S., Gu J., Kleiner D. E., Chalasani N., Hayashi P. H., Hoofnagle J. H. (2017). Azathioprine and 6-mercaptopurine induced liver injury: Clinical features and outcomes. J. Clin. Gastroenterol. 51, 63–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M., Vijay V., Shi Q., Liu Z., Fang H., Tong W. (2011). FDA-approved drug labeling for the study of drug-induced liver injury. Drug Discov. Today 16, 697–703. [DOI] [PubMed] [Google Scholar]
- Ciallella H. L., Zhu H. (2019). Advancing computational toxicology in the big data era by artificial intelligence: Data-driven and mechanism-driven modeling for chemical toxicity. Chem. Res. Toxicol. 32, 536–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark P., Hsia Y., Huntsman R. (1960). Toxic complications of treatment with 6-mercaptopurine. Br. Med. J. 1, 393–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corley C. C. Jr, Lessner H. E., Larsen W. E. (1966). Azathioprine therapy of “autoimmune” diseases. Am. J. Med. 41, 404–412. [DOI] [PubMed] [Google Scholar]
- Einhorn M., Davidsohn I. (1964). Hepatotoxicity of mercaptopurine. JAMA 188, 802–806. [DOI] [PubMed] [Google Scholar]
- Ekins S., Williams A. J., Xu J. J. (2010). A predictive ligand-based Bayesian model for human drug-induced liver injury. Drug Metab. Dispos. 38, 2302–2308. [DOI] [PubMed] [Google Scholar]
- Fourches D., Barnes J. C., Day N. C., Bradley P., Reed J. Z., Tropsha A. (2010). Cheminformatics analysis of assertions mined from literature that describe drug-induced liver injury in different species. Chem. Res. Toxicol. 23, 171–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene N., Fisk L., Naven R. T., Note R. R., Patel M. L., Pelletier D. J. (2010). Developing structure–activity relationships for the prediction of hepatotoxicity. Chem. Res. Toxicol. 23, 1215–1222. [DOI] [PubMed] [Google Scholar]
- Greig S. L., Garnock-Jones K. P. (2016). Loxoprofen: A review in pain and inflammation. Clin. Drug Investig. 36, 771–781. [DOI] [PubMed] [Google Scholar]
- Hartung T. (2009). Toxicology for the twenty-first century. Nature 460, 208–212. [DOI] [PubMed] [Google Scholar]
- Hewitt M., Enoch S., Madden J., Przybylak K., Cronin M. (2013). Hepatotoxicity: A scheme for generating chemical categories for read-across, structural alerts and insights into mechanism(s) of action. Crit. Rev. Toxicol. 43, 537–558. [DOI] [PubMed] [Google Scholar]
- Kaplowitz N. (2005). Idiosyncratic drug hepatotoxicity. Nat. Rev. Drug Discov. 4, 489–499. [DOI] [PubMed] [Google Scholar]
- Kaufmann P., Török M., Hänni A., Roberts P., Gasser R., Krähenbühl S. (2005). Mechanisms of benzarone and benzbromarone-induced hepatic toxicity. Hepatology 41, 925–935. [DOI] [PubMed] [Google Scholar]
- Kim M. T., Huang R., Sedykh A., Wang W., Xia M., Zhu H. (2016). Mechanism profiling of hepatotoxicity caused by oxidative stress using antioxidant response element reporter gene assay models and big data. Environ. Health Perspect. 124, 634–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancichinetti A., Radicchi F., Ramasco J. J., Fortunato S. (2011). Finding statistically significant communities in networks. PLoS One 6, e18961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liew C. Y., Lim Y. C., Yap C. W. (2011). Mixed learning algorithms and features ensemble in hepatotoxicity prediction. J. Comput. Aided Mol. Des. 25, 855–871. [DOI] [PubMed] [Google Scholar]
- Liu J., Mansouri K., Judson R. S., Martin M. T., Hong H., Chen M., Xu X., Thomas R. S., Shah I. (2015a). Predicting hepatotoxicity using ToxCast in vitro bioactivity and chemical structure. Chem. Res. Toxicol. 28, 738–751. [DOI] [PubMed] [Google Scholar]
- Liu R., Yu X., Wallqvist A. (2015b). Data-driven identification of structural alerts for mitigating the risk of drug-induced human liver injuries. J. Cheminform. 7, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low Y., Sedykh A., Fourches D., Golbraikh A., Whelan M., Rusyn I., Tropsha A. (2013). Integrative chemical–biological read-across approach for chemical hazard classification. Chem. Res. Toxicol. 26, 1199–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low Y., Uehara T., Minowa Y., Yamada H., Ohno Y., Urushidani T., Sedykh A., Muratov E., Kuz’min V., Fourches D., et al. (2011). Predicting drug-induced hepatotoxicity using QSAR and toxicogenomics approaches. Chem. Res. Toxicol. 24, 1251–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay I., Weiden S., Ungar B. (1964). Treatment of active chronic hepatitis and lupoid hepatitis with 6-mercaptopurine and azothioprine. Lancet 1, 899–902. [DOI] [PubMed] [Google Scholar]
- Martin M. T., Knudsen T. B., Reif D. M., Houck K. A., Judson R. S., Kavlock R. J., Dix D. J. (2011). Predictive model of rat reproductive toxicity from ToxCast high throughput screening. Biol. Reprod. 85, 327–339. [DOI] [PubMed] [Google Scholar]
- Medina-Franco J. L., Martínez-Mayorga K., Bender A., Marín R. M., Giulianotti M. A., Pinilla C., Houghten R. A. (2009). Characterization of activity landscapes using 2D and 3D similarity methods: Consensus activity cliffs. J. Chem. Inf. Model. 49, 477–491. [DOI] [PubMed] [Google Scholar]
- Mulliner D., Schmidt F., Stolte M., Spirkl H.-P., Czich A., Amberg A. (2016). Computational models for human and animal hepatotoxicity with a global application scope. Chem. Res. Toxicol. 29, 757–767. [DOI] [PubMed] [Google Scholar]
- Muster W., Breidenbach A., Fischer H., Kirchner S., Müller L., Pähler A. (2008). Computational toxicology in drug development. Drug Discov. Today 13, 303–310. [DOI] [PubMed] [Google Scholar]
- Nygaard U., Toft N., Schmiegelow K. (2004). Methylated metabolites of 6-mercaptopurine are associated with hepatotoxicity. Clin. Pharmacol. Ther. 75, 274–281. [DOI] [PubMed] [Google Scholar]
- Reif D. M., Martin M. T., Tan S. W., Houck K. A., Judson R. S., Richard A. M., Knudsen T. B., Dix D. J., Kavlock R. J. (2010). Endocrine profiling and prioritization of environmental chemicals using ToxCast data. Environ. Health Perspect. 118, 1714–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuben A., Koch D. G., Lee W. M. (2010). Drug-induced acute liver failure: Results of a U.S. multicenter, prospective study. Hepatology 52, 2065–2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romagnuolo J., Sadowski D., Lalor E., Jewell L., Thomson A. (1998). Cholestatic hepatocellular injury with azathioprine: A case report and review of the mechanisms of hepatotoxicity. Can. J. Gastroenterol. Hepatol. 12, 479–483. [DOI] [PubMed] [Google Scholar]
- Rotroff D. M., Dix D. J., Houck K. A., Knudsen T. B., Martin M. T., McLaurin K. W., Reif D. M., Crofton K. M., Singh A. V., Xia M., et al. (2013). Using in vitro high throughput screening assays to identify potential endocrine-disrupting chemicals. Environ. Health Perspect. 121, 7–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo D. P., Kim M. T., Wang W., Pinolini D., Shende S., Strickland J., Hartung T., Zhu H. (2017). CIIPro: A new read-across portal to fill data gaps using public large-scale chemical and biological data. Bioinformatics 33, 464–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo D. P., Strickland J., Karmaus A. L., Wang W., Shende S., Hartung T., Aleksunes L. M., Zhu H. (2019). Nonanimal models for acute toxicity evaluations: Applying data-driven profiling and read-across. Environ. Health Perspect. 127, 047001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz T. W., Amcoff P., Berggren E., Gautier F., Klaric M., Knight D. J., Mahony C., Schwarz M., White A., Cronin M. T. (2015). A strategy for structuring and reporting a read-across prediction of toxicity. Regul. Toxicol. Pharmacol. 72, 586–601. [DOI] [PubMed] [Google Scholar]
- Shrestha R., Cho P. J., Paudel S., Shrestha A., Kang M. J., Jeong T. C., Lee E. S., Lee S. (2018). Exploring the metabolism of loxoprofen in liver microsomes: The role of cytochrome P450 and UDP-glucuronosyltransferase in its biotransformation. Pharmaceutics 10, 112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh P., Mishra S. K., Noel S., Sharma S., Rath S. K. (2012). Acute exposure of apigenin induces hepatotoxicity in Swiss mice. PLoS One 7, e31964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sipes N. S., Martin M. T., Kothiya P., Reif D. M., Judson R. S., Richard A. M., Houck K. A., Dix D. J., Kavlock R. J., Knudsen T. B. (2013). Profiling 976 ToxCast chemicals across 331 enzymatic and receptor signaling assays. Chem. Res. Toxicol. 26, 878–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sipes N. S., Martin M. T., Reif D. M., Kleinstreuer N. C., Judson R. S., Singh A. V., Chandler K. J., Dix D. J., Kavlock R. J., Knudsen T. B. (2011). Predictive models of prenatal developmental toxicity from ToxCast high-throughput screening data. Toxicol. Sci. 124, 109–127. [DOI] [PubMed] [Google Scholar]
- Solimeo R., Zhang J., Kim M., Sedykh A., Zhu H. (2012). Predicting chemical ocular toxicity using a combinatorial QSAR approach. Chem. Res. Toxicol. 25, 2763–2769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparberg M., Simon N., del Greco F. (1969). Intrahepatic cholestasis due to azathioprine. Gastroenterology 57, 439–441. [PubMed] [Google Scholar]
- Stepan A. F., Walker D. P., Bauman J., Price D. A., Baillie T. A., Kalgutkar A. S., Aleo M. D. (2011). Structural alert/reactive metabolite concept as applied in medicinal chemistry to mitigate the risk of idiosyncratic drug toxicity: A perspective based on the critical examination of trends in the top 200 drugs marketed in the United States. Chem. Res. Toxicol. 24, 1345–1410. [DOI] [PubMed] [Google Scholar]
- Stumpfe D., Bajorath J. (2012). Exploring activity cliffs in medicinal chemistry miniperspective. J. Med. Chem. 55, 2932–2942. [DOI] [PubMed] [Google Scholar]
- Tropsha A. (2012). Recent trends in statistical QSAR modeling of environmental chemical toxicity. Exp. Suppl. 101, 381–411. [DOI] [PubMed] [Google Scholar]
- Vliet S. M., Dasgupta S., Volz D. C. (2018). Niclosamide induces epiboly delay during early zebrafish embryogenesis. Toxicol. Sci. 166, 306–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wijayanti M. A., Sholikhah E. N., Tahir I., Hadanu R., Jumina, Supargiyono, Mustofa (2006). Antiplasmodial activity and acute toxicity of N-alkyl and N-benzyl-1,10-phenanthroline derivatives in the mouse malaria model. J. Health Sci. 52, 794–799. [Google Scholar]
- Xu Y. J., Dai Z. W., Chen F. J., Gao S. S., Pei J. F., Lai L. H. (2015). Deep learning for drug-induced liver injury. J. Chem. Inf. Model. 55, 2085–2093. [DOI] [PubMed] [Google Scholar]
- Zhang J., Hsieh J. H., Zhu H. (2014). Profiling animal toxicants by automatically mining public bioassay data: A big data approach for computational toxicology. PLoS One 9, [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L. Y., Sedykh A., Tripathi A., Zhu H., Afantitis A., Mouchlis V. D., Melagraki G., Rusyn I., Tropsha A. (2013). Identification of putative estrogen receptor-mediated endocrine disrupting chemicals using QSAR- and structure-based virtual screening approaches. Toxicol. Appl. Pharm. 272, 67–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W. J., Ramamoorthy Y., Kilicarslan T., Nolte H., Tyndale R. F., Sellers E. M. (2002). Inhibition of cytochromes P450 by antifungal imidazole derivatives. Drug Metab. Dispos. 30, 314–318. [DOI] [PubMed] [Google Scholar]
- Zhu H., Bouhifd M., Donley E., Egnash L., Kleinstreuer N., Kroese E. D., Liu Z., Luechtefeld T., Palmer J., Pamies D., et al. (2016). Supporting read-across using biological data. ALTEX 33, 167–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X., Kruhlak N. L. (2014). Construction and analysis of a human hepatotoxicity database suitable for QSAR modeling using post-market safety data. Toxicology 321, 62–72. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.