

Supplemental Digital Content is available in the text.
Keywords: arrhythmia, calcium signaling, electrophysiology, hypokalemia, ion channel
Abstract
Rationale:
Hypokalemia occurs in up to 20% of hospitalized patients and is associated with increased incidence of ventricular and atrial fibrillation. It is unclear whether these differing types of arrhythmia result from direct and perhaps distinct effects of hypokalemia on cardiomyocytes.
Objective:
To investigate proarrhythmic mechanisms of hypokalemia in ventricular and atrial myocytes.
Methods and Results:
Experiments were performed in isolated rat myocytes exposed to simulated hypokalemia conditions (reduction of extracellular [K+] from 5.0 to 2.7 mmol/L) and supported by mathematical modeling studies. Ventricular cells subjected to hypokalemia exhibited Ca2+ overload and increased generation of both spontaneous Ca2+ waves and delayed afterdepolarizations. However, similar Ca2+-dependent spontaneous activity during hypokalemia was only observed in a minority of atrial cells that were observed to contain t-tubules. This effect was attributed to close functional pairing of the Na+-K+ ATPase and Na+-Ca2+ exchanger proteins within these structures, as reduction in Na+ pump activity locally inhibited Ca2+ extrusion. Ventricular myocytes and tubulated atrial myocytes additionally exhibited early afterdepolarizations during hypokalemia, associated with Ca2+ overload. However, early afterdepolarizations also occurred in untubulated atrial cells, despite Ca2+ quiescence. These phase-3 early afterdepolarizations were rather linked to reactivation of nonequilibrium Na+ current, as they were rapidly blocked by tetrodotoxin. Na+ current-driven early afterdepolarizations in untubulated atrial cells were enabled by membrane hyperpolarization during hypokalemia and short action potential configurations. Brief action potentials were in turn maintained by ultra-rapid K+ current (IKur); a current which was found to be absent in tubulated atrial myocytes and ventricular myocytes.
Conclusions:
Distinct mechanisms underlie hypokalemia-induced arrhythmia in the ventricle and atrium but also vary between atrial myocytes depending on subcellular structure and electrophysiology.
Meet the First Author, see p 808
Hypokalemia is a common electrolyte disturbance, which is present in over 20% of hospitalized patients.1 Defined as serum [K+] <3.6 mmol/L, hypokalemia is associated with in-hospital mortality rates that are 10-fold higher than that of the entire hospitalized population.2 The increased mortality is linked to a 5-fold elevated incidence of ventricular fibrillation in patients with hypokalemia compared with patients with normokalemia.3 Patients with hypokalemia also have a higher risk of atrial fibrillation; an association that is independent of age, sex, serum [Mg2+], and other potential confounders.4 Understanding the proarrhythmic role of hypokalemia in these 2 conditions thus has important implications for preventative treatment in a large number of patients.
The arrhythmogenic effects of hypokalemia have been suggested to result from an increased propensity for ectopic (triggered) activity. While the underlying mechanisms in atrial cardiomyocytes are unclear, considerable evidence from ventricular myocytes has linked triggered activity during hypokalemia to dysregulated cellular Ca2+ homeostasis. Initially, reduction in extracellular K+ levels ([K+]o) decreases the magnitude of Ca2+ transients, as rapid hyperpolarization of the cell membrane augments driving force for Na+-Ca2+ exchanger (NCX)-mediated Ca2+ extrusion and temporarily reduces sarcoplasmic reticulum (SR) Ca2+ content and release.5,6 However, lowered K+ levels also inhibit the Na+, K+-ATPase (NKA), reducing NCX-mediated Ca2+ extrusion and leading to progressive cellular Ca2+ overload.7–10 Thus, the overall pattern of change in Ca2+ transients is biphasic, as the eventual increase in SR Ca2+ content at steady state produces larger Ca2+ transients than are present in normokalemic conditions.6 Spontaneous Ca2+ release from the overloaded SR is removed from the cell by NCX, resulting in a phasic depolarization classified as an early afterdepolarization (EAD) or delayed afterdepolarization (DAD), depending on whether it occurs during the action potential (AP) or between beats. EADs are additionally promoted by prolongation of the AP during hypokalemia, which occurs as repolarizing K+ currents are inhibited by lowered [K+]o.7 The longer AP allows recovery of L-type Ca2+ channels from inactivation, and recent evidence has suggested that activation of CaMKII (Ca2+-calmodulin kinase II) by rising Ca2+ levels may additionally favor L-type channel reactivation.11
Although hypokalemia is also associated with atrial fibrillation, it is uncertain if and how hypokalemia promotes triggered activity in atrial cardiomyocytes. Myocyte structure and Ca2+ homeostasis are fundamentally different in ventricular and atrial myocytes, as atrial cells generally exhibit a low, but variable density of t-tubules and associated Na+ and Ca2+ handling proteins.12,13 Thus, it is not intuitive that exposure of atrial cells to hypokalemic conditions will result in Ca2+ overload in a manner resembling ventricular cells. Furthermore, the AP is much briefer in atrial cells, which might deter EAD generation due to delayed repolarization and reactivation of L-type Ca2+ channels. However, recent work has indicated that cells with abbreviated APs may be susceptible to phase-3 EADs driven by nonequilibrium reactivation of Na+ current (INa).14,15 It is unknown if hypokalemia would facilitate this mechanism.
In the present work, we identify distinct mechanisms which lead to triggered activity in ventricular and atrial cardiomyocytes during hypokalemia. We show that Ca2+ overload and accompanying Ca2+ waves/DADs are robustly induced in hypokalemic rat ventricular cells, and in a subpopulation of atrial cells which contain t-tubules. These effects are attributed to a close functional pairing of NKA and NCX proteins within these structures. While EADs are induced by both Ca2+ waves and L-type channel reactivation in ventricular cells, we show that brief APs in untubulated hypokalemic atrial cells promote phase-3 EADs during hypokalemia driven by nonequilibrium INa reactivation.
Methods
The data that support the findings of this study are available from the corresponding author upon reasonable request.
A detailed methods description and Major Resources Table are provided in the Data Supplement.
This study was approved by the Norwegian Animal Research Authority (FDU application number 7786) under the Norwegian Animal Welfare Act and conformed with Directive 2010/63/EU of the European Parliament.
Recordings of Ca2+ Transients and Waves
Myocytes were isolated from the left ventricle and both left and right atrium of male Wistar rats, as described previously.12 Cells were then loaded with 20 μM fluo-4 AM (Molecular Probes, Eugene, OR) for 10 minutes, plated on laminin-coated coverslips, and placed in a superfusion chamber mounted on an inverted microscope. Characterization of whole-cell Ca2+ transients was performed during field-stimulation at 1 Hz. In an initial 2-minute control period, myocytes were superfused at 37°C with a solution containing (in millimolar): NaCl 140, KCl 5.0, MgCl2 0.5, NaH2PO4 0.4, CaCl2 1.0, Hepes 5, D-glucose 5.5, pH adjusted to 7.4 with NaOH. Hypokalemia was then simulated by rapidly reducing [K+]o from 5.0 to 2.7 mmol/L via a rapid-solution changer for the ensuing 3 minutes. In follow-up experiments, the incidence of Ca2+ waves was investigated during a 30-second pause in the stimulation. SR Ca2+ content was estimated by rapid application of 10 mmol/L caffeine (Sigma-Aldrich) and measuring the magnitude of the resulting Ca2+ transient. Ca2+ transients elicited by 1 Hz field stimulation during continuous caffeine superfusion were employed to examine rates of Ca2+ extrusion.16
T-Tubule Imaging in Isolated Cardiomyocytes
Quantification of t-tubule structure in isolated ventricular and atrial myocytes was performed by staining cells with 10 µmol/L di-8-ANEPPS (Invitrogen, Paisley, United Kingdom) or with CellMask Orange (1:1000 dilution; Thermo Fisher Scientific, Waltham, MA; C10045). For each cell, t-tubule density was determined by thresholding the image intensity of the entire cell by the Otsu method, using an automated algorithm in ImageJ (National Institutes of Health). The t-index17 was then calculated for the myocyte interior, defined as the percentage of the cellular cross-sectional area, excluding the nucleus, occupied by above-threshold pixels. Cells exhibiting a t-index of ≥ 2% were defined as being tubulated.12 Detubulation was performed using a protocol similar to that described by Kawai et al.18
Monitoring of Intracellular Na+
Intracellular Na+ levels were assessed in myocytes loaded with SBFI AM (Thermo Fischer) and 0.15% Pluronic F-127 for 45 minutes.16,19 Cardiomyocytes were field stimulated and superfused with the same normokalemic and hypokalemic solutions as used in the fluo-4 experiments (1 Hz pacing, 37°C).
Immunocytochemistry and Imaging
Isolated cardiomyocytes were seeded on coated coverglasses, before fixation with 4% paraformaldehyde, and permeabilization with 0.5% Triton X-100 (Sigma-Aldrich). The following primary and secondary antibodies were used at the indicated dilutions: NCX (Swant, R3F1, 1:100), NKA-α1 (Merck Millipore, 05-369, 1:100), NKA-α2 (Merck Millipore, 07-674, 1:100), F(ab’)2-goat anti-mouse IgG (H+L) secondary antibody (Alexa Fluor 488, Thermo Fisher, A-11017, 1:200), and F(ab’)2-goat anti-mouse IgG (H+L) secondary antibody (Alexa Fluor 546, Thermo Fisher, A-11071, 1:200). Cells were imaged on an LSM-800 confocal microscope (Carl Zeiss AG, Oberkochen, Germany) in Airyscan mode.
Immunocytochemistry was additionally performed on rapidly excised ventricular and atrial tissue, frozen in Tissue Tek O.C.T. compound (Sakura Fintek, Torrance, CA). Ten micrometer tissue sections were transferred to microscope slides, fixed in 4% paraformaldehyde, and permeabilized with 0.5% Triton X-100 (Merck). Subsequent antibody labeling and Airyscan imaging were performed as described above for isolated cardiomyocytes. T-tubules were labeled in intact cardiac tissue using caveolin-3 antibody (Abcam, ab2912) at 1:100 dilution. For all employed immunolabels, imaging was performed with secondary antibodies alone to exclude the influence of nonspecific fluorescence.
Patch-Clamp Experiments
Both current- and voltage-clamp experiments were performed using 1 to 2 MΩ pipettes, an Axoclamp-2B amplifier (Axon Instruments, Foster City, CA), and pCLAMP software (Axon Instruments). APs were recorded in bridge-mode and elicited by 3 ms supra-threshold current steps at 1 Hz. Effects of inhibition of ultra-rapid K+ current (IKur) on AP configuration were assessed during rapid application of 50 µmol/L 4-aminopyridine (4-AP). This concentration has been previously shown to almost completely block IKur while having minimal influence on other currents such as transient outward K+ current (ITO).20,21 EADs were defined as positive voltage deflections during the downstroke of AP repolarization with a minimum amplitude of 2 mV. DADs were defined as minimum 2 mV depolarizing deflections from resting potential. AP duration was measured as time from the upstroke to 25%, 50%, and 75% repolarization (APD25, APD50, APD75), and full duration was defined as repolarization to 5 mV above resting membrane potential.
Membrane currents were recorded in discontinuous voltage-clamp, using a switching rate of 8 kHz. NKA currents were recorded based on a described protocol,6 using hyperpolarizing voltage ramps from +70 to −120 mV, and defined as the reduction in current when extracellular K+ was rapidly removed (illustrated in Online Figure IA and IB). Steady-state K+ currents were measured at the end of 500 ms depolarizing voltage steps from −70 mV to a range of potentials (illustrated in Online Figure IIA). In further experiments, the contribution of ITO to steady-state current was inhibited using a 1 second inactivating voltage step from −70 mV to +50 mV, before test steps to a range of potentials (illustrated in Online Figure IIIA, described in study by Wang et al22). NCX activity was examined using the tail current elicited by repolarization following a 100 ms voltage step from −45 to 0 mV (illustrated in Online Figure IV).6
Western Blotting and PCR Analyses
Expression of KCNA5/Kv1.5 was examined by qPCR and Western blotting, as described in study by Røe et al23 and Lipsett et al24, respectively.
Mathematical Modeling
A computational simulation of the time-dependent effects of hypokalemia on Na+ and Ca2+ homeostasis was performed by adapting the model of Terkildsen et al25 wherein a set of ordinary differential equations describes the electrophysiology of the rat ventricular cardiomyocyte. For the purpose of this study, crosstalk between NCX and NKA activity was simulated by including a new dimensionless parameter NCXrev to scale the intracellular Na+ concentration (Nai) sensed by the exchanger:
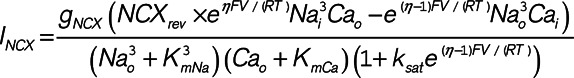 |
Increasing NCXrev thus effectively augmented the proportion of exchanger activity operating in reverse mode, as expected when NCX is localized closer to NKA and higher local Na+ levels. Hypokalemia experiments were simulated by instantaneously changing [K+]o from 5.0 to 2.7 mmol/L. After each modification of the model, steady state was reached by integrating the model equations over 10 000 beats.
Computational analysis of atrial myocyte electrophysiology was performed using our established human atrial cell model,26 recently updated with a Markov model of INa.14 Experimental voltage recordings were used as inputs in AP-clamp simulations to reveal underlying dynamics of Na+ and Ca2+ currents. Model code is available for download at: https://somapp.ucdmc.ucdavis.edu/Pharmacology/bers/ or http://elegrandi.wixsite.com/grandilab/downloads.
Statistics
All data were tested for normality of distribution using a Shapiro-Wilk test. Normally distributed data were compared with Student t-test or ANOVA with Bonferroni correction for multiple comparisons, as appropriate. Non-normal distributions were examined with a Wilcoxon signed-rank test, Mann-Whitney rank-sum test, or Kruskal-Wallis analysis on ranks with Dunn correction for multiple comparisons. Two-factor comparisons were performed by 2-way ANOVA with Bonferroni correction; results of post hoc comparisons are presented in the Online Tables. Differences in proportions were determined by z-test. P values <0.05 were considered statistically significant. All data were analyzed by Sigmaplot software (Systat Software, Chicago) and are presented as mean±SE.
Results
Effects of Hypokalemia on Ca2+ Transients and Waves in Ventricular and Atrial Cardiomyocytes
Effects of hypokalemia on isolated rat ventricular and atrial cardiomyocytes were simulated by lowering [K+]o from 5.0 to 2.7 mmol/L for 3 minutes, during continuous 1 Hz pacing. In agreement with previous work,6 we observed that ventricular myocytes exhibited a biphasic change in Ca2+ transient amplitude (Figure 1A). Lowering of [K+]o was associated with an initial depression of Ca2+ transients, followed by a rising phase which ultimately yielded larger transients than present in normokalemia. This second phase of the response was associated with an increased incidence of spontaneous Ca2+ waves when the stimulation was paused (Figure 1B).
Figure 1.
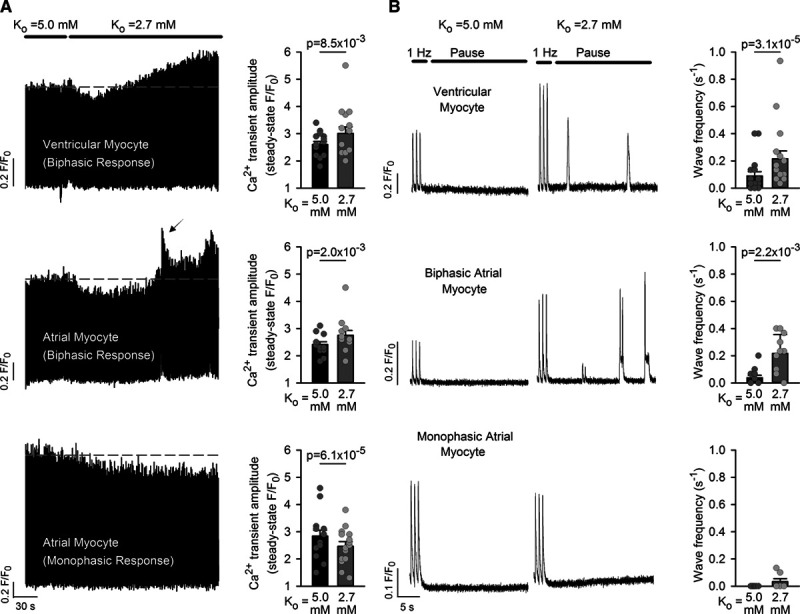
Hypokalemia promotes a steady-state increase in Ca2+ transients and Ca2+ waves in ventricular myocytes and a subpopulation of atrial myocytes. A, In field-stimulated ventricular cells, rapidly lowering [K+]o from 5.0 to 2.7 mmol/L produced an initial depression of Ca2+ transient magnitude. A secondary rising phase followed which ultimately yielded larger Ca2+ transients compared with control conditions (right, n=15 cells, 8 hearts). A similar biphasic response to hypokalemia was observed in some atrial cardiomyocytes (13 of 31 cells, 10 hearts), with associated over-activity (arrow). Other atrial cells exhibited only a monophasic reduction in Ca2+ transient amplitude. B, Ca2+ waves were assessed during pauses in the electrical stimulus. Ventricular cells and those atrial cells which exhibited a biphasic response demonstrated an increased frequency of Ca2+ waves during hypokalemia. For Ca2+ wave measurements, ncells=17, 7, 12; nhearts=10, 5, 11 in ventricular, biphasic atrial, and monophasic atrial populations. Statistics: Wilcoxon signed-rank test.
More variable effects of hypokalemia were observed in atrial cardiomyocytes; while some of these cells (13 of 31 cells) exhibited a biphasic response similar to that observed in ventricular cells, other cells showed only a monophasic decline, with a steady-state reduction in Ca2+ transient amplitude (Figure 1A). This variability in the response to lowered [K+]o was comparable in cells isolated from the left and right atria, as biphasic responses were observed in 59% and 38% of cells, respectively. In keeping with observations in ventricular cells, atrial cells which displayed a biphasic response leading to larger Ca2+ transients exhibited an increased incidence of spontaneous Ca2+ waves (Figure 1B). However, Ca2+ wave frequency was not increased in atrial cells which exhibited a monophasic reduction in Ca2+ transients.
The Biphasic Response to Hypokalemia Is Dependent on T-Tubules
As recent reports have shown that t-tubule organization is variable between individual atrial cells,12,27–29 we investigated whether such differences could account for the differing effects of hypokalemia on Ca2+ transients and waves. Imaging in intact tissue and isolated cells showed that while all ventricular myocytes exhibited a high density of well-organized t-tubules, atrial tissue contained distinct populations of tubulated and untubulated myocytes (Figure 2A and 2B). In those atrial cells that were tubulated, t-tubule density remained well below that of ventricular cells (Figure 2C), and tubules were generally disorganized in appearance. Paired imaging of t-tubules and Ca2+ revealed that only tubulated atrial myocytes showed a biphasic response to hypokalemia (ncells=12), while untubulated cells exhibited a monophasic, steady-state reduction in Ca2+ transient amplitude (ncells=7; Figure 2D). Furthermore, experimentally detubulating ventricular cells by osmotic shock reproduced the monophasic decline in Ca2+ transients observed in untubulated atrial cells (t-tubule density: 18.2±0.7% in control versus 3.4±0.3% in detubulated, ncells=40 control versus 40 detubulated, P=1.81×10−22 by paired t-test; Ca2+ transient amplitude: F/F0=2.35±0.19 in normokalemia versus 2.01±0.13 at 3-minute hypokalemia, ncells=10, P=5.12×10−3 by Wilcoxon signed-rank test). These data support that the presence of t-tubules promotes the proarrhythmic biphasic response of Ca2+ transients to hypokalemia.
Figure 2.
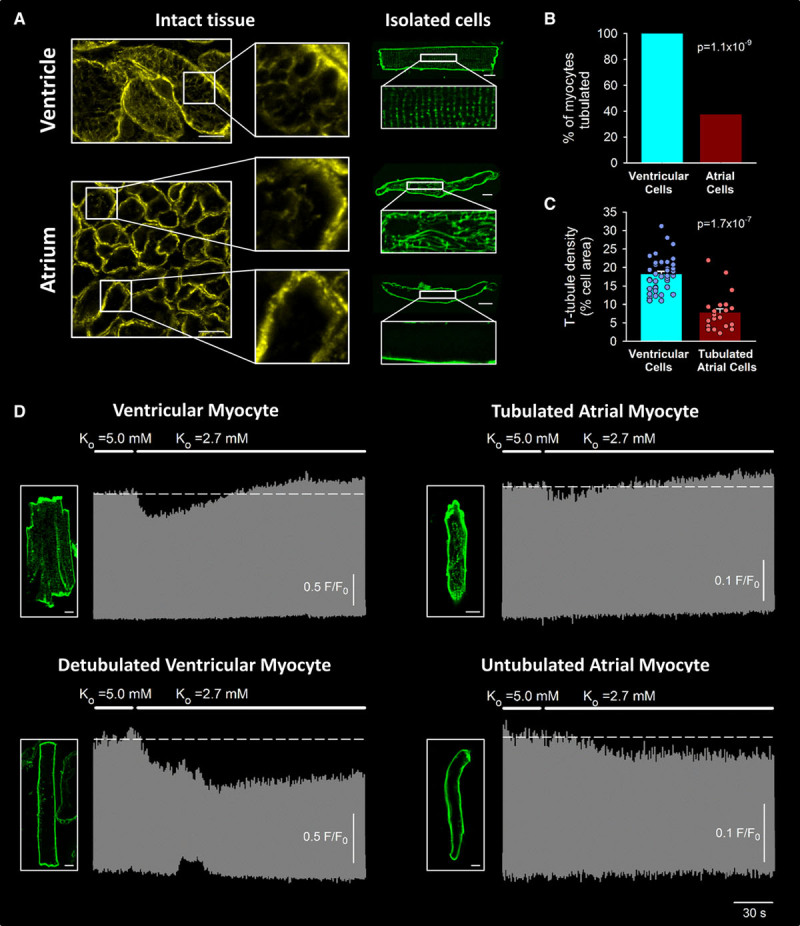
The biphasic Ca2+ transient response to hypokalemia requires t-tubules. A, T-tubule staining in intact tissue and isolated cells (caveolin-3 and di-8-ANEPPS, respectively) revealed a dense and well-organized t-tubule network in ventricular cells (scale bars=10 µm). T-tubules were only observed in approximately one-third of atrial cells (B, 21 of 56 cells, 4 hearts), and when present, these tubules were less well organized and at lower density (C) than in ventricular cells (40 cells, 3 hearts). D, Paired imaging of t-tubules and Ca2+ revealed that only tubulated atrial cells exhibited a biphasic response to hypokalemia (n=12 cells, 10 hearts), while untubulated cells exhibited a monophasic decline in Ca2+ transient amplitude (n=7 cells, 5 hearts). A similar monophasic decline in Ca2+ transients was reproduced in experimentally detubulated ventricular myocytes (n=10 cells, 3 hearts). Statistics: (B): z-test; (C): Mann-Whitney rank-sum test.
T-Tubular NKA-NCX Crosstalk Drives Ca2+-Dependent Arrhythmogenesis
Previous work in ventricular myocytes suggested that reduced activity of the NKA during hypokalemia promotes intracellular Na+ accumulation sensed by NCX, leading to reduced Ca2+ extrusion and gradual loading of the SR with Ca2+.6,7 While both NKA and NCX are highly localized in t-tubules in the ventricle, the distribution of these transporters in atrial cells is unknown. Immunolabeling and Airyscan imaging of intact tissue and isolated cells confirmed the presence of the α1 NKA isoform in both the surface membrane and t-tubules of ventricular cells and showed that this isoform was similarly localized in tubulated atrial cells (Figure 3A and 3B). Of note, untubulated atrial cells also expressed α1 NKA in the surface membrane. The α2 NKA isoform was robustly present in ventricular cells, with particular prevalence within t-tubules, as reported previously (Figure 3A).19,30 However, NKA α2 expression was very low in both tubulated and untubulated atrial myocytes; staining was limited to the intercalated disk regions adjoining neighbouring cells, and no expression was observed in t-tubules when they were present (Figure 3B). This finding suggests that the presence of the α2 NKA isoform in t-tubules is not a requirement for a biphasic response to hypokalemia. Indeed, preferential blockade of this isoform with low-dose ouabain (0.3 µmol/L, see study by Swift et al19) inhibited the biphasic response in ventricular cells but not in tubulated atrial cells (Figure 3C).
Figure 3.
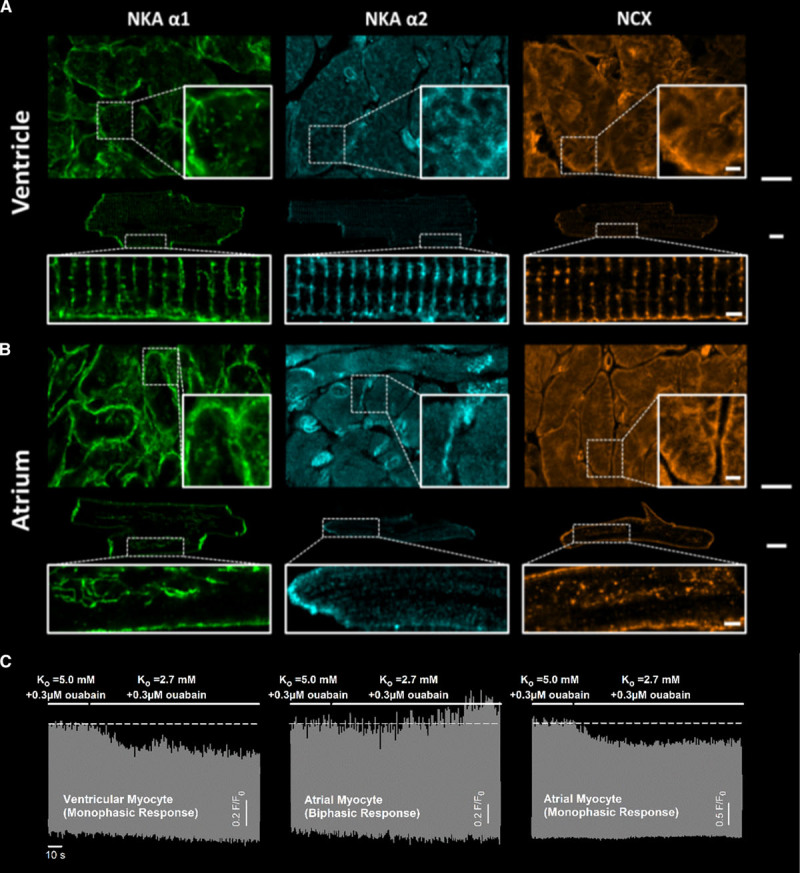
The NKA (Na+, K+-ATPase) and Na+-Ca2+ exchanger (NCX) are robustly expressed within the t-tubules of both ventricular and atrial cardiomyocytes. A, Immunolabeling in intact tissue and isolated ventricular cells showed a robust expression of the α1 NKA isoform in both the surface membrane and t-tubules, while the α2 isoform was preferentially expressed in t-tubules. B, In atrial cells, NKA α1 was expressed in both the surface membrane and, when present, the t-tubules. NKA α2 expression was low in atrial cells, never present in t-tubules, and appeared limited to the intercalated disks. NCX (right) was robustly expressed in both the surface membrane and t-tubules of ventricular and atrial cells. C, In ventricular cells, preferential blockade of NKA α2 with 0.3 µmol/L ouabain19 inhibited the biphasic response of Ca2+ transients to hypokalemia (5 of 5 cells). In atrial cells, both biphasic (7 cells) and monophasic responses (3 cells) continued to be observed in the presence of low-dose ouabain, in agreement with limited α2 expression in these cells. Scale bars in (A) and (B)=10 µm in zoomed-out images, 2 µm in enlargements.
Measurements of NKA activity also did not identify differences between tubulated and untubulated cells which could account for distinct responses to hypokalemia. NKA current, measured as the K+-sensitive current obtained during hyperpolarizing voltage ramps (protocol illustrated in Online Figure I), was similarly reduced during hypokalemia in ventricular, tubulated, and untubulated atrial cells (Figure 4A). This modest inhibition of NKA activity was insufficient to elevate global cytosolic Na+ levels during 3 minutes of hypokalemia, as assessed by experiments with the Na+-sensitive dye SBFI (Figure 4B, protocol illustrated in Online Figure VB). Maintenance of global [Na+] during hypokalemia paralleled a similar lack of increase in resting [Ca2+] in both ventricular and atrial myocytes (resting FKo=2.7/resting FKo=5.0=1.02±0.02 in ventricle, 1.03±0.01 in atria, ncells=15, 30; P=0.542, 0.054 by Wilcoxon signed-rank test, paired t-test). Experiments with low-dose ouabain, as described in Figure 3C, also failed to increase global [Na+], despite a demonstrated ability of SBFI to detect concentration changes as small as 1 mmol/L (Online Figure VA).
Figure 4.
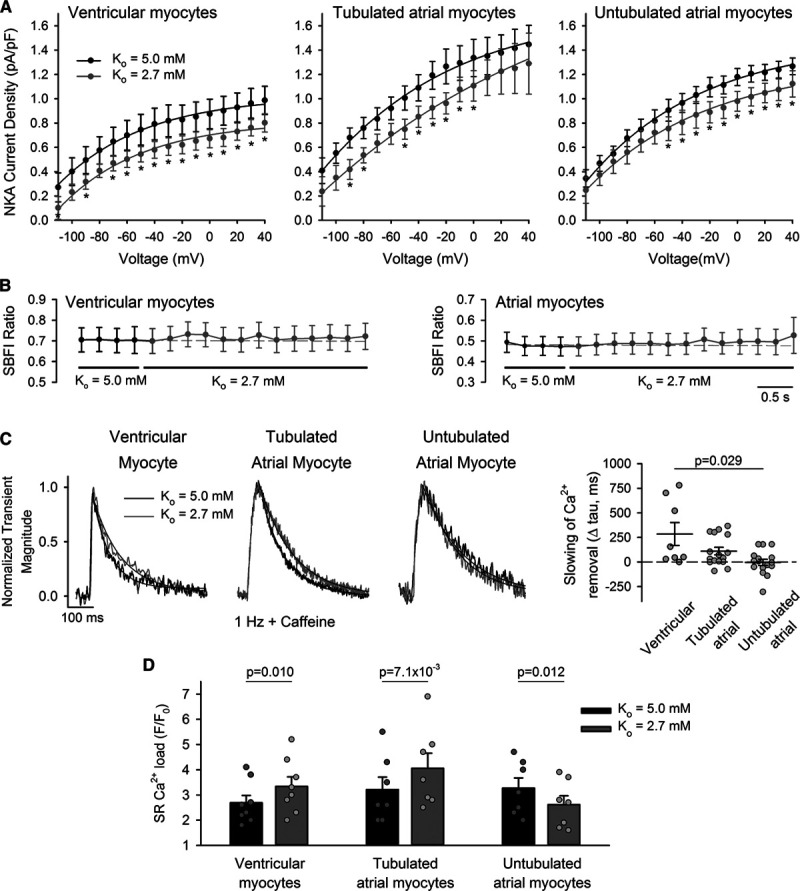
Decreased Na+, K+-ATPase (NKA) activity during hypokalemia inhibits Ca2+ extrusion by t-tubular Na+-Ca2+ exchanger (NCX), elevating sarcoplasmic reticulum (SR) Ca2+ content. A, NKA activity was measured during hyperpolarizing voltage ramps and calculated as the K+-sensitive current (see Online Figure I for protocol and representative traces). Hypokalemia induced a modest and similar reduction in NKA current in ventricular myocytes and atrial cells, regardless of the presence of t-tubules (ncells=10, 7, 9; nhearts=4, 4, 5 in ventricular, tubulated atrial, untubulated atrial cells). B, SBFI experiments revealed no change in global cytosolic [Na+] during the protocol (ncells=12, 8; nhearts=4, 4 for ventricular, atrial cells). C, However, tubulated cells showed slowed Ca2+ removal by NCX during hypokalemia, as indicated by the declining phase of Ca2+ transients stimulated in the continuous presence of 10 mmol/L caffeine (change in tau values shown at right, ncells=8, 16; nhearts=3, 5, in ventricular, tubulated atrial cells, P=0.016, 0.011 vs Ko=5.0 by Wilcoxon signed-rank test). No change in Ca2+ removal rate was observed in untubulated atrial cells (ncells=16; nhearts=5; P=0.912). Transient magnitude during Ko=2.7+caffeine: F/F0=1.46±0.07, 1.66±0.12, 1.57±0.08 in ventricular, tubulated, and atrial cells. D, Ventricular cells and tubulated atrial cells exhibited increased SR Ca2+ content during hypokalemia, as assessed by the magnitude of caffeine-induced Ca2+ release (ncells=8, 7, 7; nhearts=6, 6, 5 in ventricular, tubulated atrial, untubulated atrial cells; representative traces illustrated in Online Figure VI). Statistics: (A): 2-way repeated measures ANOVA with Bonferroni correction (see Online Table II for full results); (B): Kruskal-Wallis test (difference in medians: P=1.000, 1.000 in ventricular, atrial cells); (C): Kruskal-Wallis test with Dunn correction (difference in medians: P=0.023); (D): paired t-test. *P<0.05 vs Ko=5.0.
Since NKA function alone could not account for the key role of t-tubules in promoting Ca2+ overload during hypokalemia, we next examined NCX localization and function. As expected, immunolabeling in intact ventricular tissue and isolated cells revealed dense NCX localization in surface membrane and t-tubules (Figure 3A). Similar staining was observed in atrial cells, with robust staining in the t-tubules, when they were present (Figure 3B). NCX activity was assessed by fitting the decay phase of the Ca2+ transients stimulated in the continuous presence of caffeine. Ventricular cells exhibited slowed Ca2+ extrusion by NCX during hypokalemia (Figure 4C, left, P=0.016 versus Ko=5.0 mmol/L by Wilcoxon signed-rank test). A similarly slowed time course of NCX-mediated Ca2+ removal was observed in patch-clamped ventricular myocytes, measured during the tail current following triggering of L-type Ca2+ current (Online Figure IV). This finding suggests that although no change in global cytosolic Na+ levels was detected, NKA inhibition during hypokalemia leads to Na+ accumulation which is sensed by t-tubular NCX, perhaps due to close co-localization of the proteins. A resulting reduction in NCX-mediated Ca2+ removal during hypokalemia was associated with elevation of SR Ca2+ content (Figure 4D, left, protocol illustrated in Online Figure VIA), providing the basis for the observed steady-state increase in Ca2+ transients and Ca2+ waves in ventricular cells. Similar observations were made in tubulated atrial cells, as reduced Ca2+ removal by NCX during hypokalemia (Figure 4C) was also associated with elevation of SR Ca2+ content (Figure 4D). In untubulated atrial cells, however, NCX-mediated Ca2+ removal was not slowed at the conclusion of the hypokalemic period. Any local elevation of Na+ was therefore not sensed by NCX, while augmented Ca2+ removal by NCX, particularly during early hypokalemia, lead to reduced SR Ca2+ content (Figure 4D), small steady-state Ca2+ transients (monophasic response, Figure 1A), and low incidence of Ca2+ waves (Figure 1B).
We further investigated the role of t-tubular NKA-NCX crosstalk in driving Ca2+ overload during hypokalemia by employing a mathematical model of the rat ventricular cardiomyocyte (adapted from study by Terkildsen et al25). In agreement with observations in cellular experiments, reducing [K+]o from 5.0 to 2.7 mmol/L in the model produced a biphasic response of both SR Ca2+ content and Ca2+ transient magnitude (Figure 5A). However, under baseline conditions, the time course of the rising phase of the response was markedly slower than that observed experimentally, perhaps reflecting the lack of accommodation for protein colocalization in the model. To simulate a closer relationship between NKA and NCX with a shared local pool of Na+, the fraction of NCX operating in reverse-mode (NCXrev) was increased in a stepwise manner. This intervention resulted in a stepwise augmentation and acceleration of the biphasic response (Figure 5A and 5B) and slowing of the decay phase of the Ca2+ transient (Figure 5D), as the modest rise in cytosolic Na+ levels induced by NKA inhibition was sensed by NCX. The necessity for such sensing was further demonstrated by removing reverse-mode NCX function in the model, which resulted in a monophasic decline in Ca2+ transient magnitude and no change in Ca2+ decay kinetics (Figure 5A and 5D), resembling responses in untubulated myocytes. Sensitivity analyses indicated that the biphasic response could not be similarly inhibited by reducing other t-tubular ion fluxes (Na+ and L-type Ca2+ currents; Figure 5C), further supporting a cooperative and central role of NKA and NCX in driving Ca2+ overload. A schematic summarizing these findings is shown in Figure 8.
Figure 5.
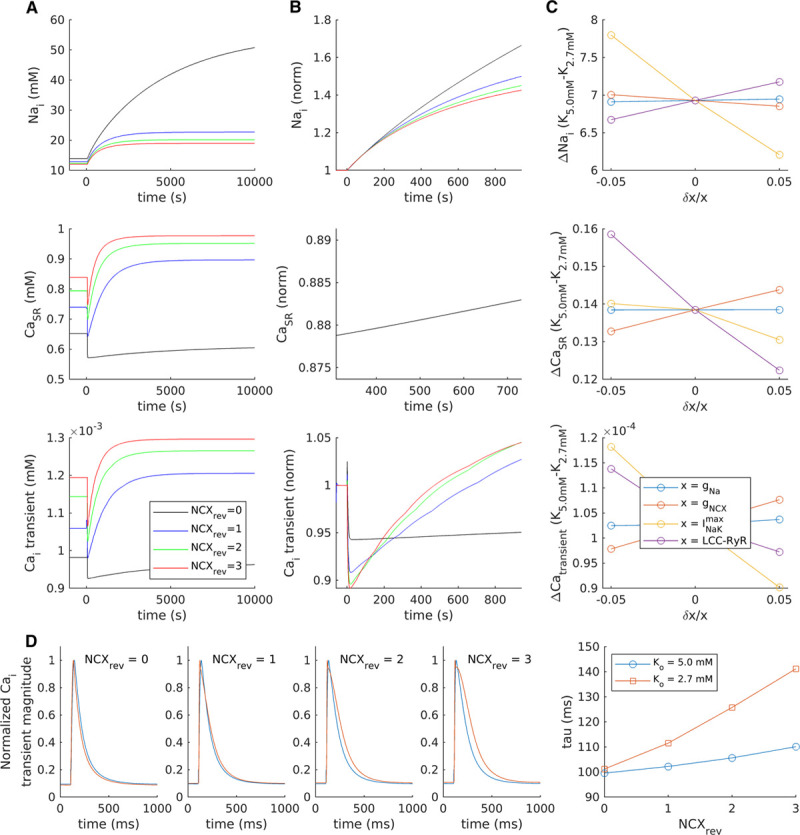
Close Na+, K+-ATPase (NKA)-Na+-Ca2+ exchanger (NCX) crosstalk drives the biphasic Ca2+ transient response to hypokalemia. A mathematical model of the rat ventricular cardiomyocyte was employed to investigate the contribution of various ion channels and transporters to changes in cellular [Na+], SR Ca2+ content, and Ca2+ transients. A, With baseline model settings (blue lines), reducing [K+]o from 5.0 to 2.7 mmol/L triggered a modest accumulation of intracellular [Na+], and a biphasic response of SR Ca2+ content and Ca2+ transients which occurred over a markedly longer time course than that observed experimentally. To simulate greater crosstalk between NCX and NKA, the fraction of NCX operating in reverse mode (NCXrev) was increased in a step-wise manner, which produced an augmented and accelerated biphasic response (see also enlargement of the first 1000 s in [B], presented normalized to [K+]o=5.0). Removing the NCXrev contribution prevented any secondary rise in Ca2+ transients, in resemblance to experiments in untubulated cells. C, Sensitivity analyses were performed to examine the effects of modulating individual ion fluxes±5%. Reducing t-tubule-associated fluxes such as INa (gNa) or the number of L-type-RyR units (LCC-RYR) did not inhibit the biphasic response, while changing NCX and NKA fluxes had proportionally opposite effects. D, In addition to augmenting the biphasic response to hypokalemia, increasing NCXrev slowed Ca2+ transient decay at steady state.
Figure 8.
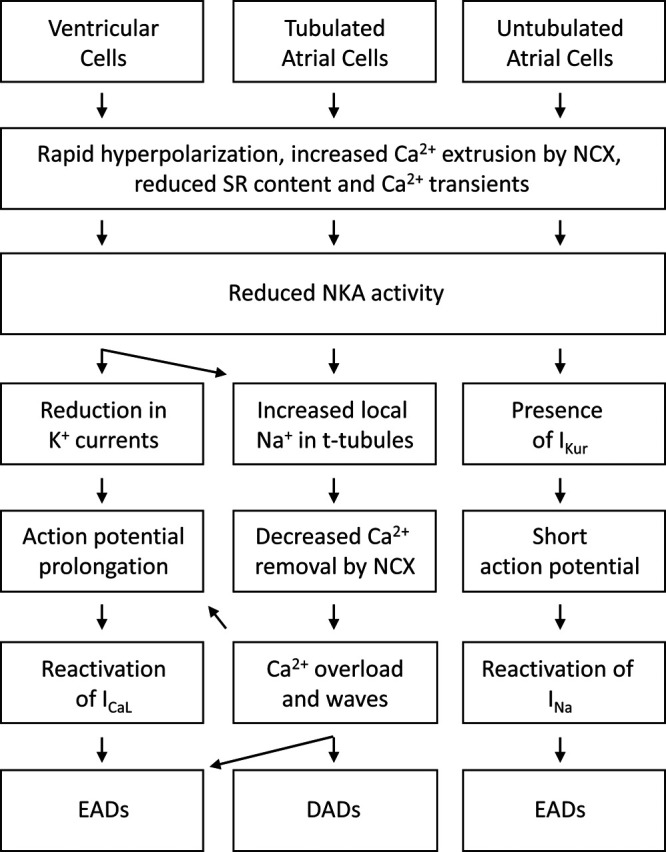
Schematic overview. Distinct mechanisms were observed to promote arrhythmogenesis in ventricular and atrial cardiomyocytes. DAD indicates delayed afterdepolarization; EAD, early afterdepolarization; NCX, Na+-Ca2+ exchanger; and NKA, Na+, K+-ATPase.
EADs Are Driven by Distinct Mechanisms in Subpopulations of Atrial Cardiomyocytes
The above data indicate that the presence of t-tubules predisposes ventricular cardiomyocytes and tubulated atrial myocytes to developing Ca2+ waves during hypokalemia. While spontaneous Ca2+ release can drive DADs from resting potential and spontaneous APs, these events may also drive EADs if they occur during the repolarizing phase of the AP. Indeed, in field-stimulated ventricular cells, we observed that hypokalemia induced Ca2+ oscillations during the late phase of the Ca2+ transient which appeared to be consistent with EADs (Figure 6A, arrows in top). These events were even more frequent in atrial cells (68% of cells versus 33% ventricular cells; P=0.026 by z-test), and were common in untubulated atrial myocytes (Figure 6A, bottom) despite an absence of signs of Ca2+ overload and waves in these cells (described in Figure 1B).
Figure 6.
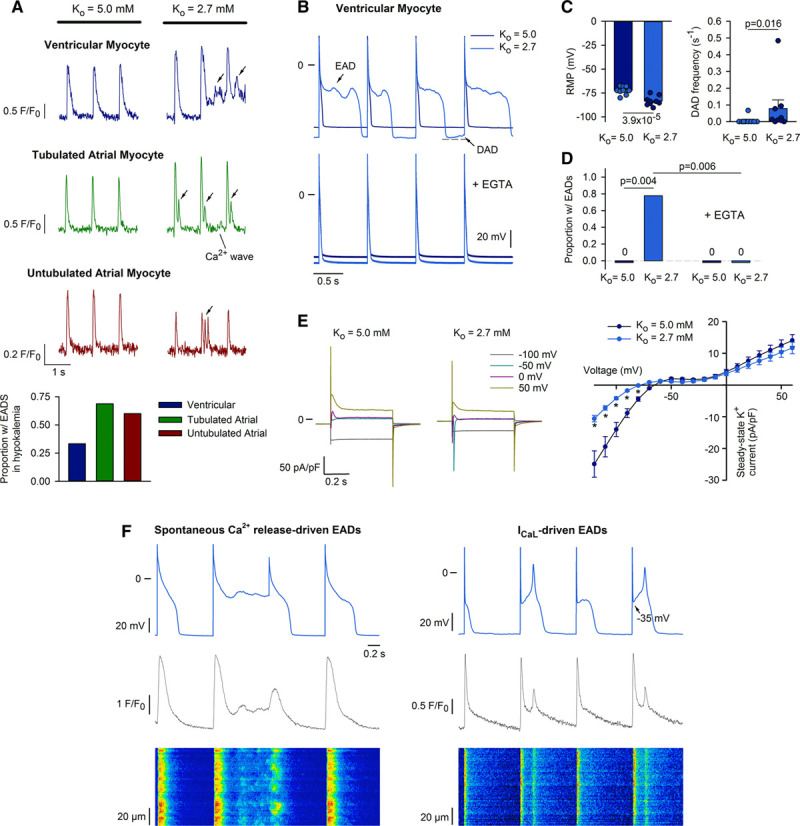
Early afterdepolarization (EAD) generation during hypokalemia: mechanisms in ventricular cells. A, In electrically stimulated ventricular and atrial myocytes, Ca2+ oscillations were commonly observed during the late phase of the Ca2+ transient, consistent with EAD generation (arrows, ncells=24, 16, 10 in ventricular, tubulated, untubulated atrial cells during 3-min hypokalemia). B, Patch-clamp recordings of action potentials (APs) confirmed the occurrence of EADs in ventricular cells during hypokalemia (7 of 9 cells from 5 hearts), which were prevented when Ca2+ was buffered by EGTA in the patch pipette (lower, 8 of 8 cells from 4 hearts). C, Mean changes in resting membrane potential (RMP) and delayed afterdepolarization (DAD) incidence (ncells=9 from 5 hearts). D, Proportion of cells exhibiting EADs. E, Steady-state K+ currents were reduced at negative potentials during hypokalemia (ncells=8, 7 in normo-, hypokalemia from 4, 4 hearts). F, Pairing AP recordings with Ca2+ imaging (fluo-4, confocal line-scans) revealed EADs associated with spontaneous Ca2+ release (left) or reactivation of L-type Ca2+ current (right). Statistics: (A): z-test; (C): paired t-test for RMP, Wilcoxon signed-rank test for DAD frequency; (D): z-test; (E): 2-way repeated measures ANOVA with Bonferroni correction (see Online Table III for full results). *P<0.05 vs Ko=5.0.
We employed the patch-clamp technique to investigate the underlying mechanisms. In ventricular cells, current-clamp recordings (Figure 6B) confirmed that hypokalemia promoted marked hyperpolarization of resting membrane potential and a robust increase in the occurrence of both DADs and EADs (Figure 6C and 6D). In agreement with previous work,7 EADs manifested as stereotypical oscillations during the plateau phase of the prolonged AP during hypokalemia (AP duration=108±18, 162±34 ms in Ko=5.0, 2.7, n=9, P=4.2×10−3 by Wilcoxon signed-rank test). AP prolongation was linked to decreased steady-state K+ currents at negative potentials, consistent with reduction of IK1 (Figure 6E; protocol presented in Online Figure IIA). Inclusion of 60 µmol/L EGTA in the patch pipette to buffer intracellular Ca2+ fully prevented EAD generation in ventricular myocytes (Figure 6B, lower panel, Figure 6D), indicating that these events are Ca2+ dependent. The Ca2+ dependence of EADs was further illustrated by pairing AP recordings with Ca2+ imaging by confocal linescans. Spontaneous Ca2+ release events observed during repolarization were temporally associated with the upstroke of EADs (Figure 6F, left). In other cases, EADs were associated with L-type Ca2+ channel re-opening, as evidenced by takeoff potentials clearly within the range for L-type activation, and a uniform increase in Ca2+ across the cell (Figure 6F, right).
Distinct mechanisms of EAD generation were observed in tubulated and untubulated atrial myocytes. Both types of atrial cells demonstrated significant membrane hyperpolarization during hypokalemia (tubulated cells: −11.5±1.2 mV, n=21; untubulated cells: −11.5±0.8 mV, n=48; P=3.38×10−9 and P=6.46×10−18, respectively, by paired t-test). As observed in ventricular myocytes, tubulated atrial cells exhibited an increased incidence of DADs during hypokalemia (0.047±0.007 events/s versus 0 in Ko=5.0 mmol/L, P=0.044 by paired t-test), and EADs associated with spontaneous Ca2+ release events (Figure 7A). Buffering of intracellular Ca2+ with EGTA significantly reduced the occurrence of these EADs (Figure 7B, top, Figure 7C). In contrast, untubulated atrial myocytes showed no significant increase in DAD frequency during hypokalemia (0.063±0.021 events/second versus 0 in Ko=5.0 mmol/L, P=0.069 by paired t-test), which paralleled measurements of Ca2+ wave frequency (Figure 1B). However, EADs were frequent in untubulated cells, even after treatment with EGTA to further ensure Ca2+ quiescence (Figure 7B, bottom, Figure 7C). Importantly, the EADs observed in these cells were triggered during phase-3 of the AP as the membrane potential rapidly repolarized (mean time to initiation=38±1 ms following AP peak), from membrane potentials below the activation range for L-type Ca2+ current (mean take-off potential=−50.9±0.4 mV, n=128 events from 10 cells). Rather, we observed that phase-3 EADs in untubulated atrial myocytes were rapidly inhibited by application of 1 µmol/L tetrodotoxin, consistent with triggering by INa (Figure 7E, upper). We observed that these EADs were also inhibited by rapid application of 10 mmol/L caffeine to inhibit Ca2+ release during the AP (Figure 7E, lower). This finding is in agreement with previous work indicating that inward NCX current generated by Ca2+ extrusion during the Ca2+ transient can collaborate with nonequilibrium INa to drive phase-3 EADs.14,15
Figure 7.
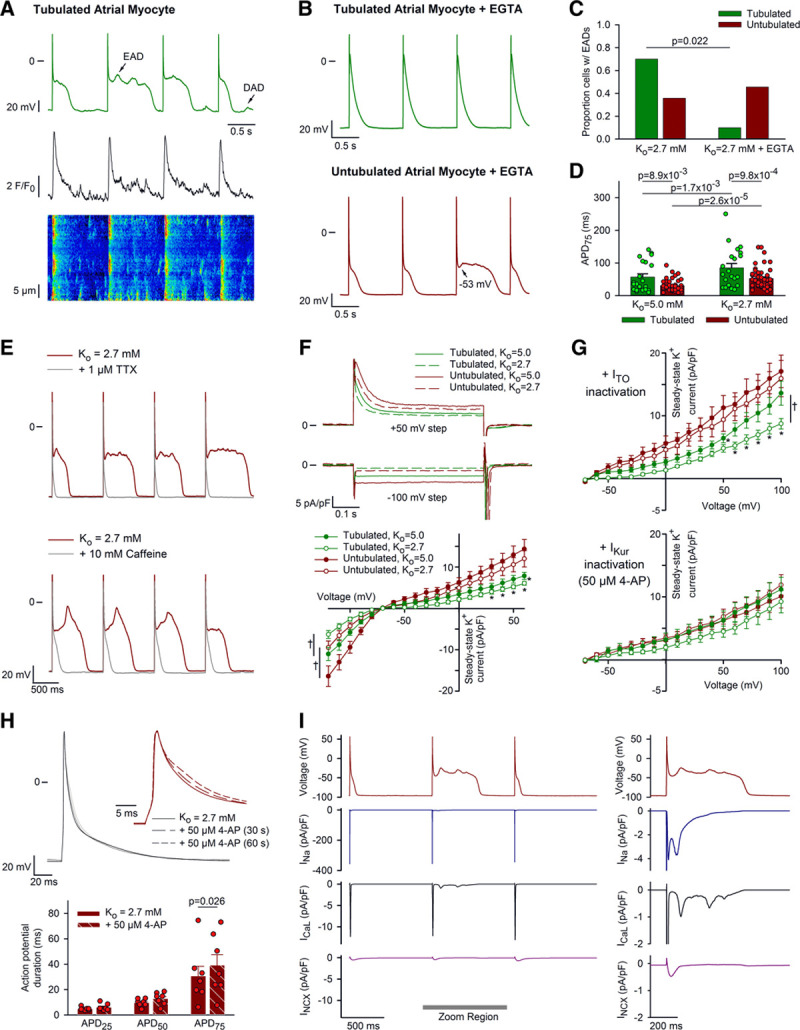
IKur maintains brief action potential (AP) configuration in untubulated atrial myocytes, promoting early afterdepolarization (EADs) driven by nonequilibrium INa during hypokalemia. A, In tubulated atrial myocytes, hypokalemia-induced EADs were frequently associated with Ca2+ waves. B, Buffering of intracellular Ca2+ with patch pipettes containing 60 µmol/L EGTA reduced EAD incidence in tubulated cells. However, in untubulated cells, EADs remained present and were observed to be triggered during rapid AP repolarization from negative potentials (arrow). C, Incidence of EADs in tubulated and untubulated atrial myocytes (−EGTA: ncells=10, 14 from 7, 8 hearts; +EGTA: ncells=10, 11 from 6, 9 hearts). D, AP configuration in tubulated and untubulated atrial cells (ncells=21, 48 from 12, 20 hearts). E, EADs observed in hypokalemic, untubulated atrial myocytes were inhibited by rapid application of 1 µmol/L TTX (top) or 10 mmol/L caffeine (bottom). F, Short AP configuration in untubulated atrial myocytes was associated with larger outward steady-state K+ current than in tubulated cells (protocol described in Online Figure IIA; ncells=11, 22 in tubulated, untubulated from 6, 11 hearts). G, Top: steady-state current remained larger in untubulated cells after inhibition of ITO (1 s prepulse to +50 mV, see Online Figure III; ncells=9, 9 in tubulated, untubulated from 5, 6 hearts), but not following addition of 50 µmol/L 4-AP to inhibit IKur (bottom, Ko=5.0: ncells=6, 8 from 5, 4 hearts; Ko=2.7: ncells=6, 6 from 4, 3 hearts). H, IKur inhibition prolonged APD75 in untubulated atrial cells during hypokalemia (ncells=7 from 3 hearts). I, Inclusion of AP waveforms in a mathematical model of the human atrial cardiomyocyte linked EADs triggered during Ca2+ quiescence to reactivation of nonequilibrium INa, parallel involvement of a small, forward-mode NCX current (INCX), and subsequent recruitment of L-type Ca2+ current. Statistics: (C): z-test; (D, F, G, H): 2-way ANOVA with Bonferroni correction. D, Difference in means: P=1.19×10−3 for tubulated vs untubulated, P=1.77×10−7 for Ko=5.0 vs Ko=2.7. H, Difference in means: P=0.114. Full results of (F) and (G) are presented in Online Tables IV and V. *P<0.05 vs untubulated and †P<0.05 vs Ko=5.0.
Triggering of phase-3 EADs by INa requires brief AP configuration, which allows rapid recovery of Na+ channels from inactivation.14,15 Indeed, untubulated atrial myocytes, where INa-driven EAD generation was most prominent, exhibited significantly shorter APs than their tubulated counterparts (Figure 7D; Online Figure VII). Rapid AP repolarization in untubulated cells was linked to larger outward K+ current, in comparison with tubulated atrial cells (Figure 7F; protocol described in Online Figure IIA). This difference remained present during hypokalemia and also when a voltage-clamp protocol was employed to inactivate ITO (Figure 7G, upper; protocol described in Online Figure III). However, application of 50 µmol/L 4-AP, which selectively inhibits ultra-rapid K+ current (IKur),20,21 significantly reduced current only in untubulated atrial cells (Online Figure IIB and IIC). Remaining 4-AP-insensitive current was similar in tubulated and untubulated cells (Figure 7G, bottom, Online Figure IIIC), supporting that the larger outward K+ current observed in untubulated atrial myocytes is due to the exclusive presence of IKur in these cells. In keeping with this finding, 4-AP application was also observed to rapidly prolong AP repolarization in untubulated atrial myocytes, both in normo- and hypokalemic conditions (Figure 7H; Online Figure IID), but not ventricular cells (APD50=34.7±11.6, 29.3±7.1 ms in Ko=2.7, Ko=2.7+4-AP, n=8, P=0.41 by paired t-test).
The above findings suggest that the presence of IKur and a brief AP increases Na+ channel availability in untubulated hypokalemic atrial cells, allowing for phase-3 EADs driven by nonequilibrium reactivation of the fast INa. To further examine this hypothesis, we introduced representative AP waveforms into a mathematical model of the human atrial cardiomyocyte.14 The model confirmed that the upstroke of the EAD was initiated by nonequilibrium INa reactivation and was paralleled by a small inward NCX current (Figure 7I). These INa-driven events are only possible when AP configuration is very brief, and INa availability is greatest (Online Figure VIII). Indeed, we observed that prolonging AP repolarization in the model inhibited the generation of INa-driven EADs (Online Figure IX). The model additionally demonstrated that hyperpolarization of the resting membrane potential during hypokalemia is also critical, as it further increases Na+ channel availability (Online Figure VIII). These findings support that the brief and hyperpolarized AP configuration is key to induction of phase-3 EADs in untubulated atrial myocytes during hypokalemia, while tubulated atrial and ventricular cells are susceptible to Ca2+-dependent EADs during phase 2. These mechanisms are summarized in Figure 8.
Discussion
We have presently identified distinct mechanisms which promote triggered activity in ventricular and atrial myocytes exposed to hypokalemia. We observed that hypokalemia induces progressive Ca2+ overload and associated Ca2+ waves and DADs, both in ventricular cells and in a subpopulation of atrial cells which contain t-tubules. This effect is attributed to a functional coupling of NKA and NCX proteins within these structures, where inhibited Na+ extrusion by NKA slows Ca2+ removal by NCX and elevates SR Ca2+ content. Although untubulated atrial cells were not susceptible to DADs during hypokalemia, EADs were common. These events were found to be driven by nonequilibrium INa, enabled by membrane hyperpolarization and a brief AP configuration which yielded rapid recovery of Na+ channels from inactivation. Prompt AP repolarization in untubulated atrial myocytes was linked to the presence of IKur; a current observed to be absent in tubulated atrial cells. This mechanism is distinct from that which occurs in hypokalemic ventricular cells, where AP prolongation predisposes for EADs driven by reactivation of L-type Ca2+ current. Despite these differing mechanisms, our results show that lowered [K+]o markedly increases susceptibility to afterdepolarizations in both cell types; actions which may contribute to an increased incidence of atrial and ventricular arrhythmias previously demonstrated in patients with hypokalemia.3,4
Both atrial and ventricular cells show an early reduction in Ca2+ transient magnitude when [K+]o is lowered, as the resting membrane potential becomes rapidly hyperpolarized.5,6,8,31 This hyperpolarization favors forward-mode NCX function, leading to an initial reduction in SR Ca2+ content and release.5,6 The subsequent rising phase of the biphasic response to hypokalemia is also dependent on NCX activity, as NKA inhibition slows removal of Ca2+ by NCX, leading to a progressive increase in SR Ca2+ content and release. We presently show that this secondary response requires t-tubules, as it was absent in untubulated atrial cells and detubulated ventricular cells (Figure 2D). It should be noted that we were not able to maintain viable atrial cells during the detubulation procedure, which is why experiments were instead performed with paired t-tubule imaging.
Previous work in ventricular cardiomyocytes has shown that NKA and NCX have higher activity in t-tubules than the surface sarcolemma, and that these currents balance each other within the two locations.19,32–34 Our data support this view, as NKA and NCX activity were paired in the different cell types; overall activity of both transporters was the largest in ventricular cells, smallest in untubulated atrial cells, and intermediate in tubulated atrial cells (Online Figures IC and VIB). However, mathematical modeling showed that the mere presence of NKA and NCX at higher densities is not sufficient to elicit a biphasic response to hypokalemia (Figure 5C). In fact, altering the activity of the 2 transporters in parallel has opposing effects which cancel out. We instead show that there must be a close functional pairing of NKA and NCX within t-tubules, which enables sensing of a shared local Na+ pool. Indeed, tubulated cells exhibited progressive Ca2+ gain during hypokalemia without a detectable increase in global [Na+] measured by SBFI; a dye which we observed can report changes in [Na+] as small as 1 mmol/L (Online Figure VA).35 This effect did not occur in untubulated atrial cells despite a similar reduction in NKA current (Figure 4A). Furthermore, we observed that preferential blockade of NKA activity in t-tubules using low-dose ouabain19 did not increase global [Na+] (Online Figure VC) but inhibited the biphasic response in ventricular cells (Figure 3C). This suggests that ouabain-induced Na+ accumulation was sufficient to favor nearby reverse-mode NCX activity at baseline, limiting further Ca2+ entry during hypokalemia. In the absence of a shared local Na+ pool, mathematical modeling in fact predicted opposite effects of NKA blockade (Figure 5C). However, NCX-NKA crosstalk could be simulated by increasing the fraction of NCX operating in reverse mode, and this effectively slowed Ca2+ extrusion and augmented Ca2+ gain during hypokalemia (Figure 5A, 5B, and 5D).
Others have previously reported a close functional pairing of NCX and NKA, reliant on a shared local Na+ domain.19,36,37 A common explanation for these observations is that there is restricted diffusion of Na+ around NKA, creating a subsarcolemmal fuzzy space with higher local Na+ levels.19,33,36,38,39 It has been suggested that the α2 NKA isoform is particularly important in regulating [Na+] within this microdomain and thereby fine-tunes cardiac contractility.19,40 However, recent super-resolution imaging data indicate that α2 is not positioned in closer proximity to ryanodine receptors than the α1 isoform.30 Instead, the authors proposed that the preferential localization of α2 within the t-tubules of ventricular cells is simply sufficient to enable local control of Na+ levels near NCX. Our present findings support this view, as imaging data in ventricular cells also showed preferential positioning of α2 within t-tubules (Figure 3A), and since α2 blockade inhibited NCX-mediated Ca2+ gain during hypokalemia (Figure 3C). However, we observed that even in the absence of the α2 isoform, the presence of α1 in atrial t-tubules was sufficient to mediate NKA-NCX crosstalk, and a biphasic response to hypokalemia was observed which persisted in the presence of low-dose ouabain (Figure 3C). Thus, our data support that NKA-NCX crosstalk is facilitated within t-tubules, but is not limited to the NKA α2 isoform.
In addition to driving Ca2+ waves and DADs, Ca2+ overload during hypokalemia has important implications for EAD generation. Ca2+ waves occurring during the repolarizing phase of the AP are a well-established driver of EADs, and we presently confirmed the robust presence of such events in hypokalemic ventricular and tubulated atrial cells. Previous work in ventricular myocytes has implicated a key involvement of CaMKII activation in triggering these events, as Ca2+-dependent activation of the kinase enhances late INa and Ca2+ current in a positive feedback loop.11 The increase in these currents prolongs the AP, enabling recovery of L-type Ca2+ channels from inactivation. In keeping with this mechanism, we observed L-type current-driven EADs in hypokalemic ventricular cells, characterized by uniform patterns of Ca2+ release across the cell and take-off potentials within the activation range of the current (Figure 6F). We further observed that EADs were absent in ventricular cells patch-clamped with EGTA-containing internal solution (Figure 6B and 6D). Reduction in several K+ currents is also reported to contribute to AP prolongation during hypokalemia.7 Indeed, we observed a marked reduction in K+ currents at negative potentials consistent with reduced IK1 and a tendency toward reduced current at positive potentials (Figure 6E). Thus, there are complex changes in Ca2+ handling and electrophysiology which promote EAD generation in hypokalemic ventricular cells, as summarized in Figure 8.
In contrast to EADs observed in hypokalemic ventricular cells and tubulated atrial cells, these events remained present in untubulated atrial cells when cytosolic Ca2+ was buffered by EGTA (Figure 7B and 7C). Both our experimental and modeling data indicated that these EADs were generated during phase-3 of the AP when membrane hyperpolarization and short AP duration increased Na+ channel availability and enabled recruitment of nonequilibrium current (Figure 7I; and Online Figures VIII and IX). Others have previously shown that short APs, such as those present in mouse ventricular cells15 or human atrial cells,14,41 are similarly prone to EADs triggered by nonequilibrium INa, under conditions where INa availability is comparable to that presently observed (Online Figure VIIID). In keeping with a key role of INa in triggering phase-3 EADs, we observed that these events were rapidly blocked by tetrodotoxin treatment (Figure 7E). Notably, previous work has shown that NCX-mediated Ca2+ removal and current during the peak of the Ca2+ transient can be synchronized with reactivation of INa, leading to synergistic depolarization and triggering of a phase-3 EAD.14,15 Our present work supports this view, since inhibition of triggered Ca2+ release by caffeine treatment blocked phase-3 EAD generation experimentally (Figure 7E). Our AP clamp simulations also showed that a small inward NCX current was temporally aligned with nonequilibrium INa (Figure 7I).
Further investigation indicated that the brief AP was maintained in untubulated atrial cells by the presence of IKur; a current which activates rapidly upon depolarization and is resistant to inactivation. Indeed, rapid application of 50 µmol/L 4-AP, which selectively blocks IKur,20,21 resulted in abrupt protraction of early AP repolarization (Figure 7H; Online Figure IID). Interestingly, IKur was observed to be absent in tubulated atrial and ventricular myocytes (Online Figure IIB and IIC), despite robust expression of Kv1.5 in both the atria and ventricles (Online Figure X). The reason for this apparent mismatch between channel expression and current in ventricle is unclear but seems to be in keeping with previous observations in rat myocardium (reviewed in study by Ravens et al42). Previous work has indicated that expression of Kv1.5 channels is largely restricted to the intercalated disk region of ventricular myocytes, while in atrial myocytes there is also significant localization longitudinally along the surface membrane.43,44 It is unclear if these channels have a different function at these sites, distinct from carrying IKur.43 In contrast, the presence of Kv1.5 and IKur in atrial myocytes is well established, and there has been considerable recent interest in employing IKur blockade for the treatment of atrial fibrillation, aimed at prolonging the AP and refractory period and reducing the risk of reentry (reviewed in study by Ravens45). Our results suggest that IKur blockade may be particularly beneficial in patients with hypokalemic atrial fibrillation, where brief APs are expected to favor both triggered and reentrant arrhythmia.
Taken together, the present results suggest that while ventricular myocytes are susceptible to Ca2+ overload-induced afterdepolarizations during hypokalemia, such overactivity in atrial cells is limited to those cells which contain t-tubules. We expect this mechanism of EAD and DAD generation to be particularly prominent in the atrial epicardium, where cells are the most frequenty tubulated.12 Untubulated myocytes, which are predominantly localized in the endocardium,12 are nevertheless prone to EADs during Ca2+ quiescence, due to the presence of IKur and short AP configurations. We speculate that elevated risk of atrial fibrillation in patients with hypokalemia4 may reflect these proarrhythmic mechanisms. Such a cause-and-effect relationship seems to be supported by recent clinical results showing that K+ infusion can revert patients with hypokalemic atrial fibrillation to sinus rhythm.46 These findings underscore an emerging role for K+ homeostasis in maintaining electrical stability of both the ventricles and atria.
Acknowledgments
We thank the Section for Comparative Medicine at Oslo University Hospital Ullevål for expert animal care.
Sources of Funding
This work was financially supported by the European Union’s Horizon 2020 research and innovation progamme (Consolidator grant, W.E. Louch) under grant agreement No. 647714, The South-Eastern Norway Regional Health Authority (K. Tazmini, E. Øie, W.E. Louch), Andre Jahre’s Fund for the Promotion of Science (W.E. Louch), The Norwegian Institute for Public Health (M. Frisk, W.E. Louch), Oslo University Hospital (W.E. Louch), and the University of Oslo (W.E. Louch). This work was further supported by the American Heart Association grant 15SDG24910015 (E. Grandi), the National Heart, Lung, and Blood Institute grants R01HL131517 (E. Grandi), and K99HL138160 (S. Morotti).
Disclosures
None.
Supplemental Materials
Expanded Methods
Online Figures I–X
Online Tables I–VII
Supplementary Material
{kind=link}
Footnotes
Nonstandard Abbreviations and Acronyms
- AP
- action potential
- CaMKII
- Ca2+-calmodulin kinase II
- DAD
- delayed afterdepolarization
- EAD
- early afterdepolarization
- IKur
- ultra-rapid K+ current
- NCX
- Na+-Ca2+ exchanger
- NKA
- Na+, K+-ATPase
- SR
- sarcoplasmic reticulum
K.T. and M.F. contributed equally to this article.
For Sources of Funding and Disclosures, see page 904.
The Data Supplement is available with this article at https://www.ahajournals.org/doi/suppl/10.1161/CIRCRESAHA.119.315641.
Novelty and Significance
What Is Known?
Hypokalemia is a common electrolyte disturbance particularly in hospitalized patients and is associated with higher mortality rates as well as elevated risk of both ventricular and atrial fibrillation.
In ventricular fibrillation, the association with hypokalemia has been attributed to the direct effects of reduced extracellular potassium levels on ventricular myocytes, as it promotes development of calcium overload and associated electrical instability (early and delayed afterdepolarizations).
It has been unclear whether a similar mechanistic relationship exists in atrial myocytes, linking hypokalemia to increased risk of atrial fibrillation.
What New Information Does This Article Contribute?
We observed that calcium overload and accompanying calcium waves/delayed afterdepolarizations are robustly induced in hypokalemic ventricular cells, and in a subpopulation of atrial cells that contain t-tubules; membrane invaginations where calcium extrusion is slowed during hypokalemia.
However, in untubulated atrial cardiomyocytes, hypokalemia induces early afterpolarizations due to sodium current reactivation, enabled by brief action potentials and membrane hyperpolarization.
This important mechanistic distinction seems to hinge on the exclusive presence of ultra-rapid K+ current (IKur) in untubulated atrial cells, which maintains brief action potential configuration.
Why do hypokalemic individuals have a higher risk of developing atrial fibrillation? Our results show that lowered extracellular potassium levels in the physiological range predispose atrial myocytes to electrical instability. We demonstrate two distinct mechanisms, depending on the presence or absence of t-tubules in these cells. Since t-tubule density varies across the atrium, our findings suggest that there may be complex regional differences in arrhythmia generation during hypokalemia. Furthermore, the realization that untubulated atrial myocytes develop over-activity by a mechanism which is distinct from the ventricle suggests that ventricular and atrial fibrillation may be differentially targeted by tailored treatments. Indeed, hypokalemic individuals with atrial fibrillation may constitute a unique patient pool that would benefit from personalized therapy. Given the atria-specific role of IKur, our results suggest that existing IKur inhibitors may be of unrealized benefit in this group of patients. Alternatively, potassium infusion may be considered as recent results indicate that this treatment can revert hypokalemic atrial fibrillation patients to sinus rhythm. These findings support a growing appreciation of the role of potassium homeostasis in maintaining electrical stability of both the ventricles and atria.
References
- 1.Gennari FJ. Hypokalemia. N Engl J Med. 1998;339:451–458. doi: 10.1056/NEJM199808133390707. doi: 10.1056/NEJM199808133390707. [DOI] [PubMed] [Google Scholar]
- 2.Paltiel O, Salakhov E, Ronen I, Berg D, Israeli A. Management of severe hypokalemia in hospitalized patients: a study of quality of care based on computerized databases. Arch Intern Med. 2001;161:1089–1095. doi: 10.1001/archinte.161.8.1089. doi: 10.1001/archinte.161.8.1089. [DOI] [PubMed] [Google Scholar]
- 3.Kjeldsen K. Hypokalemia and sudden cardiac death. Exp Clin Cardiol. 2010;15:e96–e99. [PMC free article] [PubMed] [Google Scholar]
- 4.Krijthe BP, Heeringa J, Kors JA, Hofman A, Franco OH, Witteman JC, Stricker BH. Serum potassium levels and the risk of atrial fibrillation: the Rotterdam Study. Int J Cardiol. 2013;168:5411–5415. doi: 10.1016/j.ijcard.2013.08.048. doi: 10.1016/j.ijcard.2013.08.048. [DOI] [PubMed] [Google Scholar]
- 5.Bouchard R, Clark RB, Juhasz AE, Giles WR. Changes in extracellular K+ concentration modulate contractility of rat and rabbit cardiac myocytes via the inward rectifier K+ current IK1. J Physiol. 2004;556:773–790. doi: 10.1113/jphysiol.2003.058248. doi: 10.1113/jphysiol.2003.058248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aronsen JM, Skogestad J, Lewalle A, Louch WE, Hougen K, Stokke MK, Swift F, Niederer S, Smith NP, Sejersted OM, et al. Hypokalaemia induces Ca2+ overload and Ca2+ waves in ventricular myocytes by reducing Na+,K+-ATPase α2 activity. J Physiol. 2015;593:1509–1521. doi: 10.1113/jphysiol.2014.279893. doi: 10.1113/jphysiol.2014.279893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Osadchii OE. Mechanisms of hypokalemia-induced ventricular arrhythmogenicity. Fundam Clin Pharmacol. 2010;24:547–559. doi: 10.1111/j.1472-8206.2010.00835.x. doi: 10.1111/j.1472-8206.2010.00835.x. [DOI] [PubMed] [Google Scholar]
- 8.Eisner DA, Lederer WJ, Ojeda C. Arrhythmogenic effects of hypokalaemia on mammalian ventricular muscle [proceedings]. J Physiol. 1978;280:74P–75P. [PubMed] [Google Scholar]
- 9.Skogestad J, Aronsen JM. Hypokalemia-induced arrhythmias and heart failure: new insights and implications for therapy. Front Physiol. 2018;9:1500. doi: 10.3389/fphys.2018.01500. doi: 10.3389/fphys.2018.01500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chan YH, Tsai WC, Ko JS, Yin D, Chang PC, Rubart M, Weiss JN, Everett TH, 4th, Lin SF, Chen PS. Small-conductance calcium-activated potassium current is activated during hypokalemia and masks short-term cardiac memory induced by ventricular pacing. Circulation. 2015;132:1377–1386. doi: 10.1161/CIRCULATIONAHA.114.015125. doi: 10.1161/CIRCULATIONAHA.114.015125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pezhouman A, Singh N, Song Z, Nivala M, Eskandari A, Cao H, Bapat A, Ko CY, Nguyen T, Qu Z, et al. Molecular basis of hypokalemia-induced ventricular fibrillation. Circulation. 2015;132:1528–1537. doi: 10.1161/CIRCULATIONAHA.115.016217. doi: 10.1161/CIRCULATIONAHA.115.016217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Frisk M, Koivumäki JT, Norseng PA, Maleckar MM, Sejersted OM, Louch WE. Variable t-tubule organization and Ca2+ homeostasis across the atria. Am J Physiol Heart Circ Physiol. 2014;307:H609–H620. doi: 10.1152/ajpheart.00295.2014. doi: 10.1152/ajpheart.00295.2014. [DOI] [PubMed] [Google Scholar]
- 13.Trafford AW, Clarke JD, Richards MA, Eisner DA, Dibb KM. Calcium signalling microdomains and the t-tubular system in atrial mycoytes: potential roles in cardiac disease and arrhythmias. Cardiovasc Res. 2013;98:192–203. doi: 10.1093/cvr/cvt018. doi: 10.1093/cvr/cvt018. [DOI] [PubMed] [Google Scholar]
- 14.Morotti S, McCulloch AD, Bers DM, Edwards AG, Grandi E. Atrial-selective targeting of arrhythmogenic phase-3 early afterdepolarizations in human myocytes. J Mol Cell Cardiol. 2016;96:63–71. doi: 10.1016/j.yjmcc.2015.07.030. doi: 10.1016/j.yjmcc.2015.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Edwards AG, Grandi E, Hake JE, Patel S, Li P, Miyamoto S, Omens JH, Heller Brown J, Bers DM, McCulloch AD. Nonequilibrium reactivation of Na+ current drives early afterdepolarizations in mouse ventricle. Circ Arrhythm Electrophysiol. 2014;7:1205–1213. doi: 10.1161/CIRCEP.113.001666. doi: 10.1161/CIRCEP.113.001666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Louch WE, Hougen K, Mørk HK, Swift F, Aronsen JM, Sjaastad I, Reims HM, Roald B, Andersson KB, Christensen G, et al. Sodium accumulation promotes diastolic dysfunction in end-stage heart failure following serca2 knockout. J Physiol. 2010;588:465–478. doi: 10.1113/jphysiol.2009.183517. doi: 10.1113/jphysiol.2009.183517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Heinzel FR, Bito V, Biesmans L, Wu M, Detre E, von Wegner F, Claus P, Dymarkowski S, Maes F, Bogaert J, et al. Remodeling of T-tubules and reduced synchrony of Ca2+ release in myocytes from chronically ischemic myocardium. Circ Res. 2008;102:338–346. doi: 10.1161/CIRCRESAHA.107.160085. doi: 10.1161/CIRCRESAHA.107.160085. [DOI] [PubMed] [Google Scholar]
- 18.Kawai M, Hussain M, Orchard CH. Excitation-contraction coupling in rat ventricular myocytes after formamide-induced detubulation. Am J Physiol. 1999;277:H603–H609. doi: 10.1152/ajpheart.1999.277.2.H603. doi: 10.1152/ajpheart.1999.277.2.H603. [DOI] [PubMed] [Google Scholar]
- 19.Swift F, Tovsrud N, Enger UH, Sjaastad I, Sejersted OM. The Na+/K+-ATPase α2-isoform regulates cardiac contractility in rat cardiomyocytes. Cardiovasc Res. 2007;75:109–117. doi: 10.1016/j.cardiores.2007.03.017. doi: 10.1016/j.cardiores.2007.03.017. [DOI] [PubMed] [Google Scholar]
- 20.Amos GJ, Wettwer E, Metzger F, Li Q, Himmel HM, Ravens U. Differences between outward currents of human atrial and subepicardial ventricular myocytes. J Physiol. 1996;491(pt 1):31–50. doi: 10.1113/jphysiol.1996.sp021194. doi: 10.1113/jphysiol.1996.sp021194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bertaso F, Sharpe CC, Hendry BM, James AF. Expression of voltage-gated K+ channels in human atrium. Basic Res Cardiol. 2002;97:424–433. doi: 10.1007/s00395-002-0377-4. doi: 10.1007/s00395-002-0377-4. [DOI] [PubMed] [Google Scholar]
- 22.Wang Z, Fermini B, Nattel S. Delayed rectifier outward current and repolarization in human atrial myocytes. Circ Res. 1993;73:276–285. doi: 10.1161/01.res.73.2.276. doi: 10.1161/01.res.73.2.276. [DOI] [PubMed] [Google Scholar]
- 23.Røe ÅT, Ruud M, Espe EK, Manfra O, Longobardi S, Aronsen JM, Nordén ES, Husebye T, Kolstad TRS, Cataliotti A, et al. Regional diastolic dysfunction in post-infarction heart failure: role of local mechanical load and SERCA expression. Cardiovasc Res. 2019;115:752–764. doi: 10.1093/cvr/cvy257. doi: 10.1093/cvr/cvy257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lipsett DB, Frisk M, Aronsen JM, Nordén ES, Buonarati OR, Cataliotti A, Hell JW, Sjaastad I, Christensen G, Louch WE. Cardiomyocyte substructure reverts to an immature phenotype during heart failure. J Physiol. 2019;597:1833–1853. doi: 10.1113/JP277273. doi: 10.1113/JP277273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Terkildsen JR, Niederer S, Crampin EJ, Hunter P, Smith NP. Using Physiome standards to couple cellular functions for rat cardiac excitation-contraction. Exp Physiol. 2008;93:919–929. doi: 10.1113/expphysiol.2007.041871. doi: 10.1113/expphysiol.2007.041871. [DOI] [PubMed] [Google Scholar]
- 26.Grandi E, Pandit SV, Voigt N, Workman AJ, Dobrev D, Jalife J, Bers DM. Human atrial action potential and Ca2+ model: sinus rhythm and chronic atrial fibrillation. Circ Res. 2011;109:1055–1066. doi: 10.1161/CIRCRESAHA.111.253955. doi: 10.1161/CIRCRESAHA.111.253955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Richards MA, Clarke JD, Saravanan P, Voigt N, Dobrev D, Eisner DA, Trafford AW, Dibb KM. Transverse tubules are a common feature in large mammalian atrial myocytes including human. Am J Physiol Heart Circ Physiol. 2011;301:H1996–H2005. doi: 10.1152/ajpheart.00284.2011. doi: 10.1152/ajpheart.00284.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gadeberg HC, Bond RC, Kong CH, Chanoit GP, Ascione R, Cannell MB, James AF. Heterogeneity of T-Tubules in pig hearts. PLoS One. 2016;11:e0156862. doi: 10.1371/journal.pone.0156862. doi: 10.1371/journal.pone.0156862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arora R, Aistrup GL, Supple S, Frank C, Singh J, Tai S, Zhao A, Chicos L, Marszalec W, Guo A, et al. Regional distribution of T-tubule density in left and right atria in dogs. Heart Rhythm. 2017;14:273–281. doi: 10.1016/j.hrthm.2016.09.022. doi: 10.1016/j.hrthm.2016.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yuen GK, Galice S, Bers DM. Subcellular localization of Na/K-ATPase isoforms in ventricular myocytes. J Mol Cell Cardiol. 2017;108:158–169. doi: 10.1016/j.yjmcc.2017.05.013. doi: 10.1016/j.yjmcc.2017.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Christé G. Effects of low K+o on the electrical activity of human cardiac ventricular and purkinje cells. Cardiovasc Res. 1983;17:243–250. doi: 10.1093/cvr/17.4.243. doi: 10.1093/cvr/17.4.243. [DOI] [PubMed] [Google Scholar]
- 32.Thomas MJ, Sjaastad I, Andersen K, Helm PJ, Wasserstrom JA, Sejersted OM, Ottersen OP. Localization and function of the Na+/Ca2+-exchanger in normal and detubulated rat cardiomyocytes. J Mol Cell Cardiol. 2003;35:1325–1337. doi: 10.1016/j.yjmcc.2003.08.005. doi: 10.1016/j.yjmcc.2003.08.005. [DOI] [PubMed] [Google Scholar]
- 33.Despa S, Brette F, Orchard CH, Bers DM. Na/Ca exchange and Na/K-ATPase function are equally concentrated in transverse tubules of rat ventricular myocytes. Biophys J. 2003;85:3388–3396. doi: 10.1016/S0006-3495(03)74758-4. doi: 10.1016/S0006-3495(03)74758-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Orchard CH, Pásek M, Brette F. The role of mammalian cardiac t-tubules in excitation-contraction coupling: experimental and computational approaches. Exp Physiol. 2009;94:509–519. doi: 10.1113/expphysiol.2008.043984. doi: 10.1113/expphysiol.2008.043984. [DOI] [PubMed] [Google Scholar]
- 35.Baartscheer A, Schumacher CA, Fiolet JW. Small changes of cytosolic sodium in rat ventricular myocytes measured with SBFI in emission ratio mode. J Mol Cell Cardiol. 1997;29:3375–3383. doi: 10.1006/jmcc.1997.0567. doi: 10.1006/jmcc.1997.0567. [DOI] [PubMed] [Google Scholar]
- 36.Su Z, Zou A, Nonaka A, Zubair I, Sanguinetti MC, Barry WH. Influence of prior Na+ pump activity on pump and Na+/Ca2+ exchange currents in mouse ventricular myocytes. Am J Physiol. 1998;275:H1808–H1817. doi: 10.1152/ajpheart.1998.275.5.H1808. doi: 10.1152/ajpheart.1998.275.5.H1808. [DOI] [PubMed] [Google Scholar]
- 37.Swift F, Tovsrud N, Sjaastad I, Sejersted OM, Niggli E, Egger M. Functional coupling of α2-isoform Na+/K+-ATPase and Ca2+ extrusion through the Na+/Ca2+-exchanger in cardiomyocytes. Cell Calcium. 2010;48:54–60. doi: 10.1016/j.ceca.2010.06.006. doi: 10.1016/j.ceca.2010.06.006. [DOI] [PubMed] [Google Scholar]
- 38.Skogestad J, Lines GT, Louch WE, Sejersted OM, Sjaastad I, Aronsen JM. Evidence for heterogeneous subsarcolemmal Na+ levels in rat ventricular myocytes. Am J Physiol Heart Circ Physiol. 2019;316:H941–H957. doi: 10.1152/ajpheart.00637.2018. doi: 10.1152/ajpheart.00637.2018. [DOI] [PubMed] [Google Scholar]
- 39.Verdonck F, Mubagwa K, Sipido KR. Na+ in the subsarcolemmal ‘fuzzy’ space and modulation of Ca2+i and contraction in cardiac myocytes. Cell Calcium. 2004;35:603–612. doi: 10.1016/j.ceca.2004.01.014. doi: 10.1016/j.ceca.2004.01.014. [DOI] [PubMed] [Google Scholar]
- 40.Despa S, Lingrel JB, Bers DM. Na+/K+-ATPase α2-isoform preferentially modulates Ca2+ transients and sarcoplasmic reticulum Ca2+ release in cardiac myocytes. Cardiovasc Res. 2012;95:480–486. doi: 10.1093/cvr/cvs213. doi: 10.1093/cvr/cvs213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Antzelevitch C, Belardinelli L, Zygmunt AC, Burashnikov A, Di Diego JM, Fish JM, Cordeiro JM, Thomas G. Electrophysiological effects of ranolazine, a novel antianginal agent with antiarrhythmic properties. Circulation. 2004;110:904–910. doi: 10.1161/01.CIR.0000139333.83620.5D. doi: 10.1161/01.CIR.0000139333.83620.5D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ravens U, Wettwer E. Ultra-rapid delayed rectifier channels: molecular basis and therapeutic implications. Cardiovasc Res. 2011;89:776–785. doi: 10.1093/cvr/cvq398. doi: 10.1093/cvr/cvq398. [DOI] [PubMed] [Google Scholar]
- 43.Mays DJ, Foose JM, Philipson LH, Tamkun MM. Localization of the Kv1.5 K+ channel protein in explanted cardiac tissue. J Clin Invest. 1995;96:282–292. doi: 10.1172/JCI118032. doi: 10.1172/JCI118032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Eldstrom J, Van Wagoner DR, Moore ED, Fedida D. Localization of Kv1.5 channels in rat and canine myocyte sarcolemma. FEBS Lett. 2006;580:6039–6046. doi: 10.1016/j.febslet.2006.09.069. doi: 10.1016/j.febslet.2006.09.069. [DOI] [PubMed] [Google Scholar]
- 45.Ravens U. Atrial-selective K+ channel blockers: potential antiarrhythmic drugs in atrial fibrillation? Can J Physiol Pharmacol. 2017;95:1313–1318. doi: 10.1139/cjpp-2017-0024. doi: 10.1139/cjpp-2017-0024. [DOI] [PubMed] [Google Scholar]
- 46.Tazmini K, Fraz MSA, Nymo SH, Stokke MK, Louch WE, Øie E. Potassium infusion increases the likelihood of conversion of recent-onset atrial fibrillation-A single-blinded, randomized clinical trial. Am Heart J. 2020;221:114–124. doi: 10.1016/j.ahj.2019.12.014. doi: 10.1016/j.ahj.2019.12.014. [DOI] [PubMed] [Google Scholar]
- 47.Louch WE, Mørk HK, Sexton J, Strømme TA, Laake P, Sjaastad I, Sejersted OM. T-tubule disorganization and reduced synchrony of Ca2+ release in murine cardiomyocytes following myocardial infarction. J Physiol. 2006;574:519–533. doi: 10.1113/jphysiol.2006.107227. doi: 10.1113/jphysiol.2006.107227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Eisner DA, Trafford AW, Díaz ME, Overend CL, O’Neill SC. The control of Ca release from the cardiac sarcoplasmic reticulum: regulation versus autoregulation. Cardiovasc Res. 1998;38:589–604. doi: 10.1016/s0008-6363(98)00062-5. doi: 10.1016/s0008-6363(98)00062-5. [DOI] [PubMed] [Google Scholar]
- 49.Mørk HK, Sjaastad I, Sejersted OM, Louch WE. Slowing of cardiomyocyte Ca2+ release and contraction during heart failure progression in postinfarction mice. Am J Physiol Heart Circ Physiol. 2009;296:H1069–H1079. doi: 10.1152/ajpheart.01009.2008. doi: 10.1152/ajpheart.01009.2008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.