

Abstract
Fosmidomycin and related molecules comprise a family of phosphonate natural products with potent antibacterial, antimalarial and herbicidal activities. To understand the biosynthesis of these compounds, we characterized the fosmidomycin producer, Streptomyces lavendulae, using biochemical and genetic approaches. Surprisingly, we were unable to elicit production of fosmidomycin, instead observing the unsaturated derivative dehydrofosmidomycin, which we showed potently inhibits 1-deoxy-D-xylulose 5-phosphate reductoisomerase and has bioactivity against a number of bacteria. The genes required for dehydrofosmidomycin biosynthesis were established by heterologous expression experiments. Bioinformatics analyses, characterization of intermediates, and in vitro biochemistry show that the biosynthetic pathway involves conversion of a two-carbon phosphonate precursor into the unsaturated three-carbon product via a highly unusual rearrangement reaction, catalyzed by the 2-oxoglutarate dependent dioxygenase DfmD. The required genes and biosynthetic pathway for dehydrofosmidomycin differ substantially from that of the related natural product FR-900098, suggesting that the ability to produce these bioactive molecules arose via convergent evolution.
Graphical Abstract
Resistance to bioactive small molecules lies at the heart of many pressing societal concerns. Antibiotic-resistant bacterial infections have risen to crisis levels, with approximately 2 million cases and 23,000 deaths a year in the United States alone (1). Similarly, resistance of Plasmodium falciparum to widely used combination therapies has been observed in at least 5 countries in southeast Asia, raising the frightening prospect of untreatable malaria (currently 200 million cases and 400,000 deaths a year) (2); whereas resistance to commonly used herbicides and pesticides threatens the food supply, costing US farmers an estimated $1 billion per year in lost crops (3). To deal with these threats, society must implement more effective policies and strategies governing the use of these essential molecules so as to minimize the acquisition of resistance. It is equally clear that improved discovery pipelines will be required to deliver novel bioactive agents that are effective against resistant organisms that already exist, as well as those that will inevitably arise in the future. Historically, natural products have been among the best sources for these bioactive molecules. Although private sector research in this area has diminished, recent advances in genomics, chemistry and bioinformatics suggest that natural products will remain a critical source of bioactive compounds in the future (4–6).
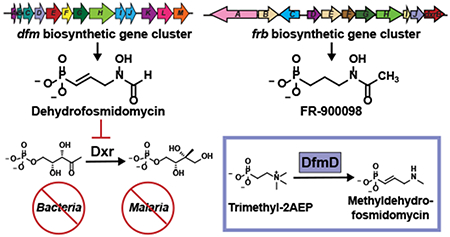
A particularly promising family of natural products, with great potential in both agriculture and human health, was discovered by researchers at Fujisawa Pharmaceutical in 1979 (Fig. 1a) (7–10). These molecules share a common hydroxyaminopropylphosphonic acid scaffold and include fosmidomycin (1, originally designated FR-31564) and an unsaturated derivative designated FR-32863 (2, hereafter dehydrofosmidomycin) produced by Streptomyces lavendulae (7–9), as well as FR-900098 and its hydroxylated congener FR-33289 produced by Streptomyces rubellomurinus (7–10). Three of the four molecules (FR-33289 is the exception) have potent antibacterial activity against both Gram-positive and Gram-negative organisms, including clinical isolates of multidrug-resistant Mycobacterium tuberculosis, Pseudomonas aeruginosa, Klebsiella pneumoniae, Escherichia coli, and Enterobacter cloacae (8, 11). Both FR-900098 and fosmidomycin also display substantial antimalarial activity (12), with promising results in early clinical trials (13). The bioactivity of FR-900098 and fosmidomycin is based on their inhibition of 1-deoxy-D-xylulose 5-phosphate reductoisomerase (DXR) (14), the rate-limiting step of the methylerythritol phosphate (MEP) pathway for biosynthesis of isoprenoid precursors. The MEP pathway is an attractive target for development of antibiotic and antimalarial medicines due to the absence of the pathway in humans and other mammals, which instead utilize the mevalonate pathway for isoprenoid biosynthesis (15). Importantly, isoprenoid biosynthesis in plant chloroplasts also occurs via the MEP pathway, and inhibition of DXR is lethal to plants (16, 17). Thus, the fosmidomycin family of natural products also possess herbicidal activity (18). In this regard, S. rubellomurinus encodes an FR-900098-resistant allele of DXR within the biosynthetic gene cluster, which could be used to genetically engineer crops with specific resistance to these herbicides (19).
Figure 1. The fosmidomycin family of phosphonate natural products.
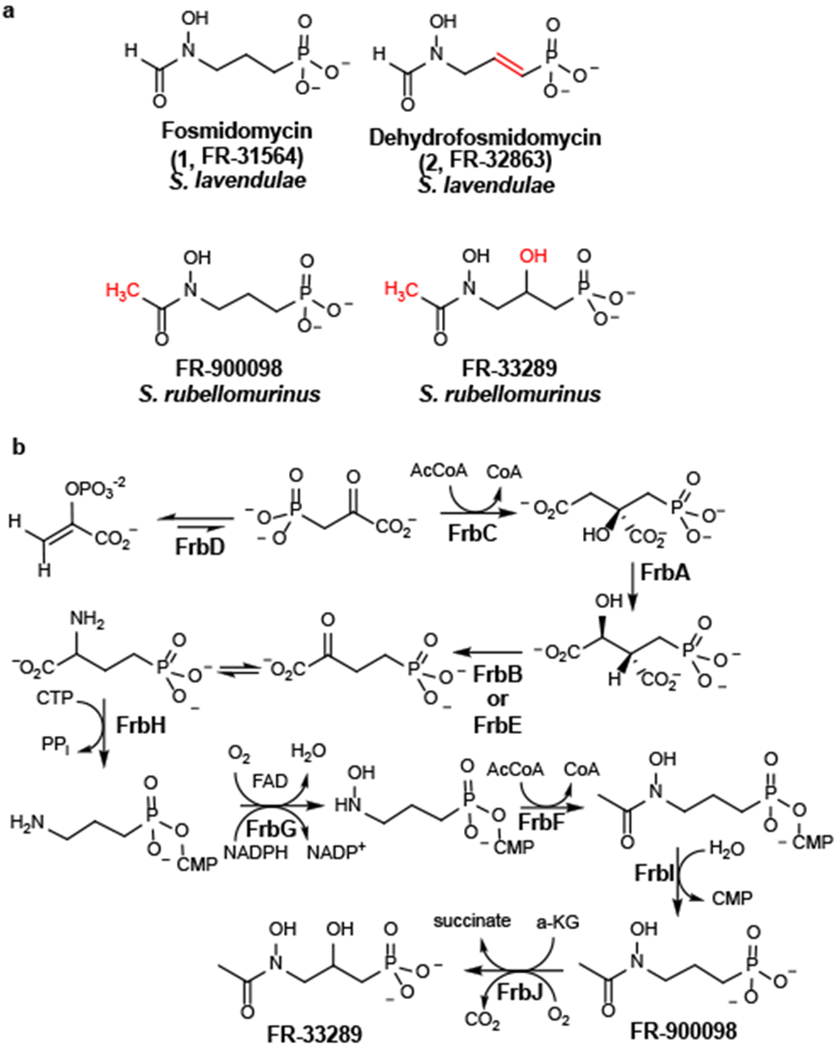
(A) Structures of fosmidomycin and related antibiotics. Key chemical differences are noted in red. (B) The biosynthetic pathway for FR-900098.
Although the biosynthetic machinery needed for production of fosmidomycin has yet to be established, the genes and metabolic pathway required for synthesis of FR-900098 have been characterized in great detail (19, 20) (Fig. 1b & Supplementary Fig. 1A). The FR-900098 biosynthetic gene cluster is comprised of eleven genes designated frbABCDEFGHI-dxrB. The function of each gene, as well as the enzymatic reactions catalyzed by the encoded proteins, has been definitively established in a series of genetic and biochemical studies (19, 20). Moreover, knowledge of this pathway has enabled the construction of engineered strains that produce FR-900098 at substantially higher levels than the native producer S. rubellomurinus (20). Biosynthesis of FR-900098 begins with conversion of phosphoenolpyruvate (3) to phosphonopyruvate (4) by the enzyme PEP mutase, a step common to nearly all phosphonate biosynthetic pathways (6). Because synthesis of phosphonopyruvate is highly unfavorable, net synthesis of phosphonic acids via PEP mutase requires a thermodynamically favorable driving reaction following the PEP mutase reaction. In the case of FR-900098, this driving reaction involves condensation of phosphonopyruvate with acetyl-CoA, a reaction similar to that catalyzed by citrate synthase. Subsequent steps in the pathway are also similar to those of the TCA cycle, with the four-carbon phosphonic acid analogue of 2-oxoglutarate ultimately being converted to the three-carbon final product via decarboxylation of a nucleotide-activated intermediate (20, 21). Notably, the FR-900098 pathway differs from most other known phosphonate biosynthetic pathways, which decarboxylate phosphonopyruvate to phosphonoacetaldehyde (5) to provide the thermodynamic driving force needed for phosphonate synthesis (e.g. biosynthesis of the common phosphonate natural product 2-aminoethylphopshonate (2AEP; 6), Supplementary Fig. 1B) (22–24). Thus, canonical phosphonate biosynthetic pathways proceed via an early two-carbon intermediate rather than the four-carbon intermediate found in the FR-900098 pathway. The frb gene cluster also encodes an FR-900098-resistant allele of Dxr (dxrB), which is responsible for self-resistance in S. rubellomurinus and a 2-oxoglutarate-dependent dioxygense (encoded by frbJ) that converts FR-900098 to FR-33289 (20).
Given the similarity of their chemical structures, biological targets and producing microorganisms, one might expect that the biosynthetic pathways of fosmidomycin and FR-900098 would be nearly identical. Thus, it was surprising to find that the S. lavendulae genome lacks homologs of most of the frb genes. Instead, the organism encodes a putative biosynthetic gene cluster that shares only four of thirteen genes with the frb cluster, but which is otherwise more similar to canonical phosphonate biosynthetic clusters. Here, we show that this divergent gene cluster is indeed responsible for phosphonate biosynthesis in S. lavendulae, producing an unsaturated member of the fosmidomycin family via an unusual biochemical pathway that is quite different from the FR-900098 pathway found in S. rubellomurinus.
RESULTS
Streptomyces lavendulae produces dehydrofosmidomycin.
To facilitate our studies, we attempted to optimize production of fosmidomycin by S. lavendulae by growing the organism for varying amounts of time in five different media, including the original fosmidomycin production medium (8). Spent media from these cultures was tested for inhibition of DXR activity using an in vitro enzymatic assay and for inhibition of growth using a phosphonate-specific bioassay strain of E. coli (19). Positive results were obtained in both assays for numerous samples (Supplementary Table 1). Subsequent analysis of these samples by 31P NMR revealed the presence of numerous peaks in the range that is typical for phosphonic acids (5 to 30 ppm, (6, 25)), with a notable peak at ~11 ppm found in all active samples (Supplementary Table 1, Fig. 2). This peak differs substantially from that of fosmidomycin (~25 ppm), suggesting that this compound was not produced at levels detectable by 31P NMR under any of the growth conditions examined, a conclusion supported by the appearance of a new peak at 25 ppm upon spiking of these samples with authentic fosmidomycin standards (Fig. 2). Because fosmidomycin is a potent inhibitor of E. coli DXR (IC50 ca. 100 nM) (14), we considered the possibility that it was produced at levels high enough to inhibit DXR, but too low to be detected by NMR. To address this, we established the IC50 of the inhibitory compound via dilution of extracts analyzed by 31P NMR (Supplementary Fig. 2A,B). Based on these experiments, we conclude that had the activity been caused by fosmidomycin, it would have been present at ~3 mM, a concentration easily observable by 31P NMR (Supplementary Fig. 2C,D). Lastly, we were unable to detect fosmidomycin in any sample using sensitive high-resolution mass spectrometric (HRMS) detection, which easily detects fosmidomycin at 25 μM (Supplementary Fig. 2E). Thus, S. lavendulae does not appear to produce detectable levels of fosmidomycin under the conditions examined here. The reason for this discrepancy from previously published work is unclear at this time.
Figure 2. S. lavendulae produces dehydrofosmidomycin.
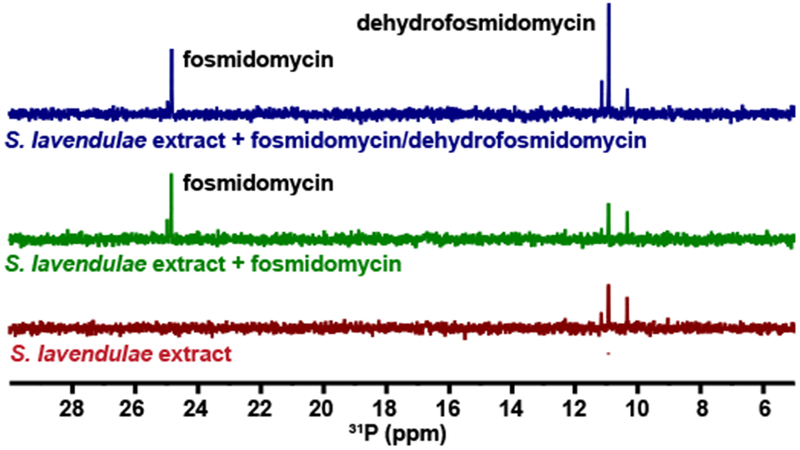
The bottom (red) spectrum shows the 31P NMR spectrum of spent media from S. lavendulae. The middle (green) spectrum shows the same spent media from S. lavendulae after addition of 1 mM fosmidomycin. The top (blue) spectrum shows the spent media from S. lavendulae after addition both 1 mM fosmidomycin and 1 mM dehydrofosmidomycin. Representative data from three independent replicates.
S. lavendulae has also been reported to produce dehydrofosmidomycin, an unsaturated phosphonate with antibiotic activity similar to fosmidomycin (8). To determine whether the 31P NMR peak at 11 ppm is dehydrofosmidomycin, we chemically synthesized the compound using a streamlined route based on a previously published synthesis (26) (Supplementary Fig. 3). Addition of this standard to active extracts caused an increase in the 11 ppm signal, suggesting that this NMR peak represents dehydrofosmidomycin (Fig. 2). This conclusion is supported by HRMS detection of a molecule with the exact mass of dehydrofosmidomycin in the original samples (Supplementary Fig. 2F). We were also able to purify dehydrofosmidomycin from S. lavendulae extracts and confirm its structure via 1H NMR (Supplementary Fig. 4). Thus, we conclude that the major bioactive phosphonate produced by S. lavendulae under each of the conditions examined here is dehydrofosmidomycin rather than fosmidomycin.
Bioactivity of dehydrofosmidomycin.
Few studies have examined the bioactivity of dehydrofosmidomycin (8, 27), possibly due to the challenge of isolating or synthesizing adequate amounts of the compound. Using our streamlined synthesis, we were able to access sufficient quantities of pure dehydrofosmidomycin to allow in vitro inhibition studies of bacterial DXR, revealing it to be a potent inhibitor of E. coli DXR, with IC50 values slightly lower than fosmidomycin (Supplementary Fig. 5). Synthetic dehydrofosmidomycin also showed antibiotic activity against Gram-negative bacteria, including K. pneumoniae, P. aeruginosa, and E. coli, while having no activity against Gram-positive bacteria that lack the MEP pathway, such as S. aureus and E. faecalis (Supplementary Table 2).
The dehydrofosmidomycin biosynthetic gene cluster.
The genome of S. lavendulae was recently sequenced as part of a large scale genome-mining project for discovery of phosphonate natural products (6). Because virtually all known phosphonate biosynthetic pathways begin with the enzyme PEP mutase (encoded by the pepM gene), and because genes encoding natural product biosynthetic pathways are typically clustered, it is usually possible to identify phosphonate biosynthetic gene clusters by searching genome sequences for pepM (5). Only one pepM-containing gene cluster was identified in the S. lavendulae genome sequence (Fig. 3A and Supplementary Table 3). This 15-kb locus contains 13 genes, which we designated dfmA through dfmM.
Figure 3. Characterization of the dfm gene cluster.
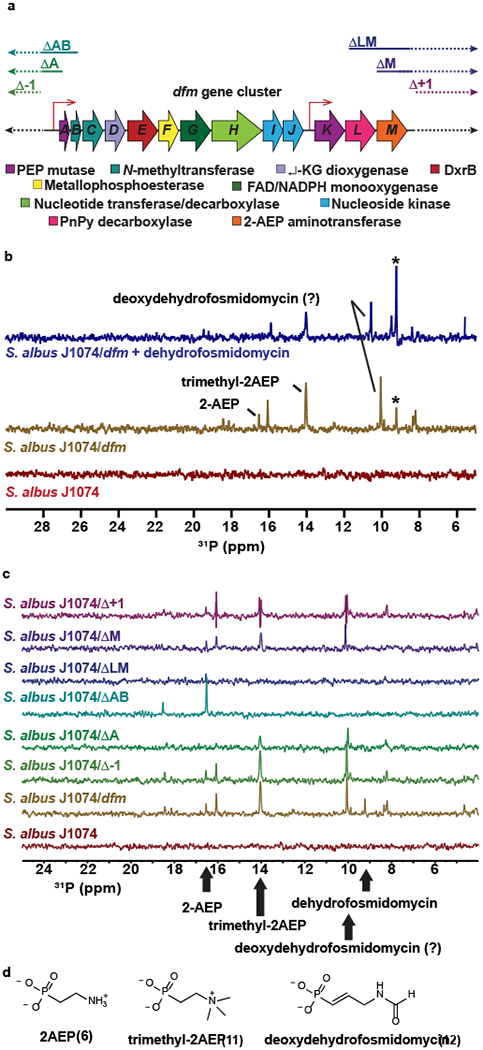
(A) The dehydrofosmidomycin (dfm) gene cluster from S. lavendulae. The putative function of each gene is shown. A Genbank-formatted file of an integrative plasmid with the full dfm cluster, as well ca. 15 kb of flanking sequences upstream and downstream of the cluster is provided in a Supplementary Note. The extent of the various deletion derivatives used for heterologous expression studies is indicated above the diagram. Putative promoter regions are indicated by the thin red arrows. (B) 31P NMR spectra for extracts from the parent strain (S. albus J1074, red spectrum), the heterologous expression strain (S. albus J1704/dfm, mustard spectrum), and the heterologous expression strain spiked with 1 mM dehydrofosmidomycin (blue spectrum). The asterisk indicates dehydrofosmidomycin. Other compounds that were verified by 31P NMR and HRMS are indicated (see Supplementary Fig. 7). The predicted deoxdehydrofosmidomycin NMR peak was not confirmed due to lack of a synthetic standard. Representative data from three independent experiments. (C) 31P NMR spectra for extracts from heterologous expression strains carrying the indicated plasmids. The position of peaks corresponding to specific metabolites is indicated. Representative data from three independent experiments. HRMS analysis of these extracts confirming the presence of the presence of these metabolites is presented in Supplementary Fig. 7. (D) Structures of additional phosphonates produced by the heterologous expression strain.
Surprisingly, the S. lavendulae dfm locus is very different from the S. rubellomurinus frb locus, which directs synthesis of the nearly identical natural product FR-900098. Comparison of the two biosynthetic gene clusters reveals only four homologous gene products: PEP mutase (frbD/dfmK), a putative self-resistance gene encoding a phosphonate-resistant DXR (dxrB/dfmE), a flavin-dependent amine monooxygenase (frbG/dfmG) and a nucleotide transferase/decarboxylase (frbH/dfmH). Notably, these genes have relatively low similarities (<45% identity, Supplementary Table 3), suggesting that they diverged from a common ancestor long ago. Homologs of the FR-900098 biosynthetic genes frbA, frbB, frbC, frbE, frbF, frbI and frbJ are not found within the dfm cluster. The dfm gene cluster also encodes a putative phosphonopyruvate decarboxylase (dfmL) and 2-aminoethylphosphonate aminotransferase (dfmM). Based on the well-characterized biosynthetic pathway (Supplementary Fig. 1B), the enzymes encoded by dfmKLM are likely to produce 2-AEP, suggesting that this compound is an intermediate in dehydrofosmidomycin biosynthesis. If so, the biosynthesis of dehydrofosmidomycin via an early two-carbon intermediate differs substantially from the four-carbon intermediate that is produced in the third step of FR-900098 biosynthesis (Fig. 1B). In addition to these genes, the dfm gene cluster encodes two putative S-adenosyl-methionine (SAM)-dependent N-methyltransferases (dfmB and dfmC) with substantial homology to ones that convert phosphoethanolamine to phosphocholine (i.e. N-trimethyl-phosphoethanolamine), a putative 2-oxoglutarate dioxygenase (dfmD) similar to γ-butyrobetaine dioxygenase, and two nucleoside-diphosphate kinases (dfmI and dfmJ). Finally, the gene cluster encodes a second, truncated homolog of PEP mutase (dfmA); however, the protein encoded by this gene lacks key PepM catalytic residues and is therefore likely to be a degenerate pseudogene.
Heterologous expression of the dfm gene cluster.
To establish whether the dfm gene cluster is responsible for dehydrofosmidomycin biosynthesis, we performed a series of heterologous expression studies using S. albus J1074. A recombinant strain (S. albus J1074/dfm) carrying the full dfm gene cluster and several flanking genes on a chromosomal integration vector produced a compound with a 31P-NMR peak not seen in the untransformed S. albus host (Fig. 3A,B). This molecule was confirmed to be dehydrofosmidomycin by spiking with a synthetic standard (Fig. 3B), as well as by phosphonate-specific bioactivity assays, DXR inhibition assays, and HRMS experiments (Supplementary Fig. 6). Based on NMR and/or HRMS, the heterologous expression strain produced several additional phosphonates that could be biosynthetic intermediates; these include 2AEP, trimethyl-2AEP (11) and deoxydehydrofosmidomycin (12, Fig. 3B–D and Supplementary Fig. 7).
To determine the boundaries of the cluster, we used a series of plasmids with progressively larger deletions for heterologous production experiments (Fig. 3A,C and Supplementary Fig. 8). Deletion of genes upstream of dfmA (Δ−1) has no effect on dehydrofosmidomycin production, thus defining the 5’-end of the cluster. A larger deletion that removes the PepM pseudogene dfmA (ΔA) does not abrogate phosphonate production, although strains carrying this plasmid produced lower levels of phosphonates, suggesting that the deletion alters expression levels of required downstream genes, perhaps by altering the native dfm promoter. Upon removal of dfmB (ΔAB), which encodes the first of the two N-methyltransferases, neither trimethyl-2AEP nor dehydrofosmidomycin are produced. Instead, a buildup of 2AEP is observed, suggesting that dfmB (and/or a downstream gene whose expression is reduced by removal of a shared promoter) is necessary to convert 2-AEP to trimethyl-2AEP, which is then further modified to generate dehydrofosmidomycin. In this regard, the biosynthesis of similar trimethylated amines (e.g. phosphocholine and betaine) often involves two SAM-dependent methyltransferases with one enzyme catalyzing the first (and sometimes second) methylation event and the second enzyme catalyzing the formation of the trimethylated product (28–30). Therefore, it is likely that the second SAM-dependent methyltransferase gene (dfmC) is also involved in the synthesis of trimethyl-2AEP. As discussed above, the dfmKLM genes are probably responsible for synthesis of 2AEP. The fact that 2AEP is produced by recombinant strains carrying the ΔAB plasmid suggests that the dfmKLM genes have their own promoter.
Deletions of genes downstream of dfmM (Δ+1) did not affect heterologous production of dehydrofosmidomycin in S. albus, defining the 3’-end of the cluster. When the 3’-deletion (ΔLM) included both dfmL, which encodes phosphonopyruvate decarboxylase, and dfmM, which encodes 2-AEP aminotransferase, no phosphonates were observed. This strongly suggests that phosphonoacetaldehyde (5; Supplementary Fig. 1B) is an early intermediate in the dehydrofosmidomycin biosynthetic pathway. Interestingly, the aminotransferase dfmM can be deleted (ΔM) without affecting production of any phosphonate. This is likely because other aminotransferases in S. albus J1074 are capable of converting phosphonoacetaldehyde into 2AEP. Similar results have been observed in the biosynthesis of phosphinothricin by Streptomyces hygroscopicus, which also accumulates 2AEP despite the absence of an AEP aminotransferase in the biosynthetic gene cluster (31). The genome of S. albus J1074 encodes two aminotransferases with moderate identity to DfmM (WP_003947938 at 38% and WP_003951764 at 31%) that could be responsible for this activity.
Identification of biosynthetic intermediates.
The heterologous expression data suggested that 2AEP is an intermediate in the dehydrofosmidomycin biosynthetic pathway. To provide further evidence, we grew S. albus J1074/ΔLM, with and without addition of 2AEP to the medium. Several phosphonates, including trimethyl-2AEP and dehydrofosmidomycin, were observed only when 2AEP was added to the medium. Because the deletion mutant is unable to produce any phosphonates, this provides strong evidence that 2AEP is taken up and converted to dehydrofosmidomycin in this strain (Figure 4A,B and Supplementary Fig. 9A,B).
Figure 4. Feeding studies with 2AEP and L-methionine-(methyl-13C).
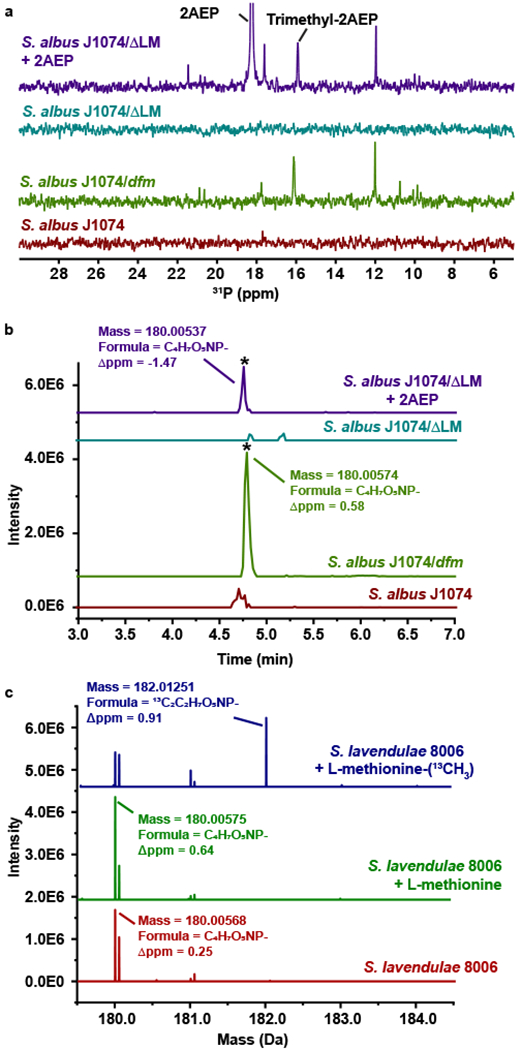
(A) 31P NMR spectra for extracts from the parent strain (S. albus J1074, red spectrum), the heterologous expression strain carrying the full dfm gene cluster (S. albus J1074/dfm, green spectrum), and an expression strain incapable of 2AEP biosynthesis due to deletion of phosphonopyruvate decarboxylase (S. albus J1074/ΔLM) grown without (teal spectrum) and with 1 mM 2AEP (purple spectrum). Representative data from three independent experiments. (B) HRMS analysis of the same three strains showing the EIC for dehydrofosmidomycin (180.00–180.01 Da). Peaks corresponding to dehydrofosmidomycin are indicated with an asterisk. Representative data from three independent experiments. (C) HRMS analysis for S. lavendulae grown with no additives (red) or with added unlabeled L-methionine (green) or 13C-labeled methionine (blue). Peaks corresponding to unlabeled and doubly labeled dehydrofosmidomycin are indicated. Representative data from three independent experiments.
Similarly, deletion of the SAM-dependent methyltransferase genes abrogates trimethyl-2AEP production with concomitant accumulation of 2AEP, suggesting that the latter is a precursor for the former. To test this, we grew the native producer S. lavendulae in media with 13CH3-labeled methionine. Because methionine is the direct precursor of methyl moiety in SAM, this results in 13C-methyl-labeling of molecules produced by SAM-dependent methyltransferases (e.g. DfmB and DfmC). Comparison of spent media with labeled and unlabeled methionine revealed the incorporation of three 13C into trimethyl-2AEP (Supplementary Figure 9C). Dehydrofosmidomycin containing two labeled carbons is also produced in this experiment, suggesting that two of the three methyl groups on trimethyl-2AEP are retained in the final compound (Figure 4C). To localize the position of these labeled carbons, we analyzed the ions produced by fragmentation of dehydrofosmidomycin using HRMS/MS (Supplementary Figure 9D,E). These data are consistent with one label being incorporated into the N-formyl carbon and the other between the nitrogen and the double bond within the propenyl carbon backbone of the molecule (Supplementary Figure 9D,E). Because it seems likely that multiple steps are required to convert trimethyl-2AEP to dehydrofosmidomycin, we also compared the metabolites in the extracts of S. lavendulae grown in media with labeled and unlabeled methionine, which revealed doubly labeled 13C-methyldehydrofosmidomycin (13) in extracts of the native producer S. lavendulae. This potential biosynthetic intermediate is also observed in the recombinant S. albus J1074/dfm strain, suggesting that enzyme responsible for production of methyldehydrofosmidomycin is encoded with the cluster (Supplementary Fig. 10).
In vitro reconstitution of DfmD.
The SAM-dependent 13C-labeling pattern of dehydrofosmidomycin suggests that unusual biochemistry occurs after synthesis of trimethyl-2AEP. While the incorporation of 13C into the formyl group of dehydrofosmidomycin can be easily explained by direct oxidation of one of the N-methyl moieties during biosynthesis, the incorporation of a methyl-derived 13C into the propenyl carbon skeleton is unexpected. This finding suggests that a highly unusual rearrangement reaction, in which an N-methyl moiety is inserted into the carbon skeleton of final product. Given that this labeling pattern is retained in methyldehydrofosmidomycin, we propose that the substrate for this rearrangement reaction is trimethyl-2AEP. The dehydration reaction required to produce methyldehydrofosmidomycin could occur during, or subsequent to, the rearrangement reaction. This reaction series also requires the loss of one of the N-methyl groups of trimethyl-2AEP, which we suggest would occur via oxidation to formaldehyde, based on well-established precedent for the metabolism of N-methyl compounds (32).
A candidate for all three of these reactions is DfmD, a putative α-ketoglutarate dependent dioxygenase. Members of this large enzyme family are known to catalyze desaturation of molecules such as carbapenem and glucoraphasatin (33, 34), demethylation of N-methyl moieties by their oxidation to formaldehyde (35), and methylene insertion reactions such as that catalyzed by deacetoxycephalosporin C synthase (36). In order for DfmD to catalyze the three reactions that are proposed here, it would need to perform sequential oxidation reactions requiring two equivalents of molecular oxygen. Such a reaction would involve a four-electron oxidation of the phosphonate substrate and two, two-electron oxidations of the co-substrate α-ketoglutarate. Precedent for sequential oxidations by an α-ketoglutarate dependent dioxygenase can be found in the reaction catalyzed by clavaminate synthase, which oxidatively cyclizes and oxidatively dehydrogenates proclavaminic acid to give clavaminic acid (37, 38). The closest biochemically characterized homologs of DfmD are γ-butyrobetaine dioxygenases, which catalyze the oxidation of a trimethylamine substrate during synthesis of carnitine (39). γ-Butyrobetaine dioxygenases have been shown to catalyze an unusual rearrangement reaction resulting in a methylene insertion when incubated with the enzyme inhibitor trimethylhydrazine-propionate (THP). This non-physiological reaction results in the 1,2-rearrangement of the quaternary ammonium compound to give the carbon chain extension product,3-amino-4-(methylamino)butanoic acid (AMBA) (Fig. 5A).(40, 41)
Figure 5. Conversion of trimethyl-2AEP to methyldehydrofosmidomycin by DfmD.
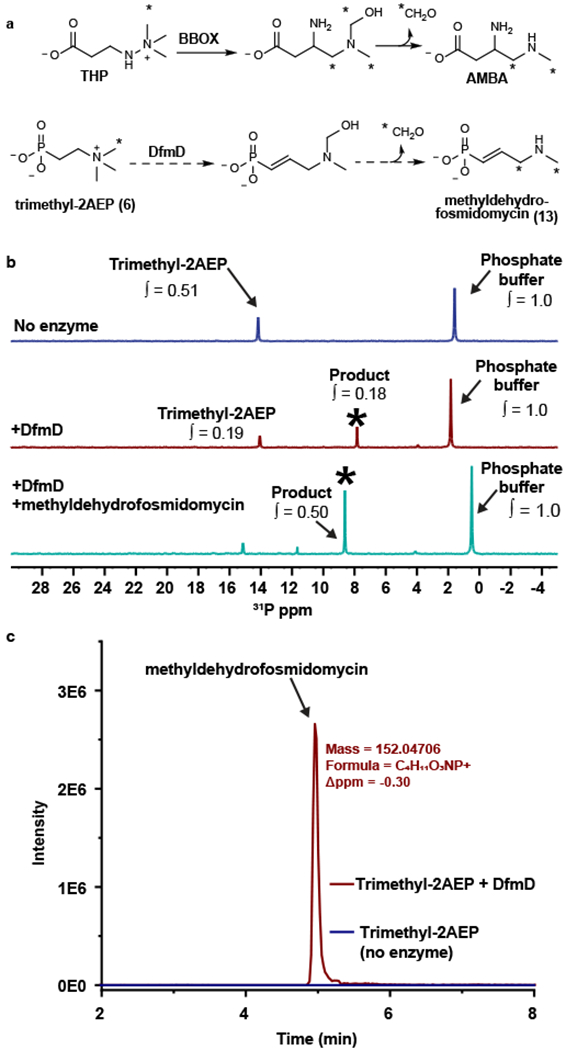
(A) The rearrangement of trimethylhydrazine-propionate (THP) to 3-amino-4-(methylamino)butanoic acid (AMBA) catalyzed by γ-butyrobetaine dioxygenase (BBOX) is shown at the top. The analogous reaction catalyzed by DfmD is shown below. (B) 31P NMR analysis of the DfmD reaction. The blue spectrum is from a no-enzyme control. The red spectrum is from a reaction containing purified DfmD. The teal spectrum is from the DfmD-containing reaction spiked with synthetic methyldehydrofosmidomycin. The asterisk marks the putative methyldehydrofosmidomycin peak. The integration values (∫) show the abundance of each compound relative to the phosphate buffer. Representative data from three independent experiments. (C) The EIC for methyldehydrofosmidomycin from the DfmD-containing reaction (red) and no-enzyme control (blue). Representative data from three independent experiments.
To test the idea that DfmD is responsible for conversion of trimethyl-2AEP to methyldehydrofosmidomycin, we purified His6-tagged DfmD after heterologous expression in E. coli (Supplementary Fig. 11). The purified enzyme was then incubated with trimethyl-2AEP, α-ketoglutarate, and iron(II) prior to analysis by 31P and 1H NMR. Reactions containing DfmD produced a novel phosphorus-containing peak at ca. 8 ppm that was not observed in reactions lacking the enzyme (Fig. 5B). Addition of a chemically synthesized methyldehydrofosmidomycin standard (Supplementary Fig. 12) to this reaction caused an increase in the 8-ppm signal, supporting the idea that the new peak represents methyldehydrofosmidomycin (Fig. 5B). This conclusion is fully supported by the 1H NMR data (Supplementary Fig. 13) and HRMS detection of a molecule with the exact mass of methyldehydrofosmidomycin (Fig. 5C and Supplementary Fig. 14A,B). Based on the rearrangement reaction catalyzed by γ-butyrobetaine dioxygenase, we expect that the third methyl moiety from trimethyl-2AEP is oxidized to formaldehyde. To test this, we employed a colorimetric assay, which showed enzyme dependent production of formaldehyde during the reaction (Supplementary Fig. 14C). These data fully support the unusual DfmD-catalyzed transformation of trimethyl-2AEP to methyldehydrofosmidomycin and formaldehyde shown in Figure 5A.
DISCUSSION
Novel bioactive natural products, such as dehydrofosmidomycin and FR-900098, are desperately needed to combat the rise of resistant organisms, while knowledge of their biosynthetic pathways enables the design of rational approaches for cost-effective production. The data presented here allow us to suggest a reasonable, although highly unusual biosynthetic pathway for dehydrofosmidomycin that differs substantially from that of the related molecule FR-900098 (Fig. 6). The first three steps of the pathway involve conversion of phosphoenolpyruvate to 2AEP via DfmK (PepM), DfmL (phosphonopyruvate decarboxylase), and DfmM (aminotransferase). 2AEP is then converted to trimethyl-2AEP by the SAM-dependent methyltransferases DfmB and/or DfmC, with the most likely scenario involving the action of both enzymes in a sequential fashion. DfmD then catalyzes a series of unusual reactions to convert trimethyl-2AEP to methyldehydrofosmidomycin, releasing formaldehyde in the process. Due to their homology with characterized enzymes in the FR-900098 biosynthetic pathway, we propose that DfmH then catalyzes the transfer of a CMP group to methyldehydrofosmidomycin, followed by hydroxylation at the nitrogen by DfmG. All that then remains is removal of the CMP group, which is probably performed by the metallophosphoesterase DfmF, and oxidation of the methyl group to a formyl group. Regarding the latter, only two genes in the dfm cluster (dfmG and dfmD) encode proteins with homology to known oxidoreductases. Alternatively, this oxidation reaction could be performed by one of the remaining enzymes encoded in the cluster, DfmI or DfmJ, but we view this as unlikely, given their homology to nucleoside diphosphate kinases. Instead, we suggest that these genes are involved in self-resistance to phosphonate metabolites based on the role of a similar pair of nucleoside diphosphate kinases required for biosynthesis of the phosphonate antibiotic fosfomycin (42). In this regard, we also expect that DfmE will catalyze a dehydrofosmidomycin-resistant DXR reaction, providing an additional self-resistance mechanism. Finally, it should be noted that none of the genes in the dfm cluster appear to encode enzymes that would be able to reduce dehydrofosmidomycin to fosmidomycin. While this is consistent with our inability to detect fosmidomycin in either the native or the heterologous producers, it deepens the mystery of how this molecule was originally discovered.
Figure 6. Proposed biosynthetic pathway for dehydrofosmidomycin.
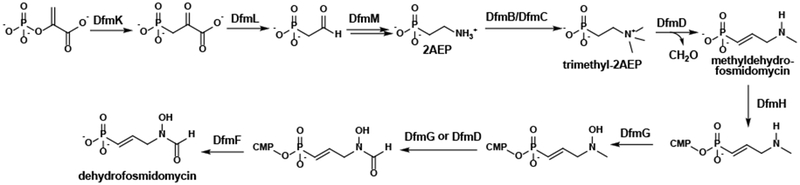
Putative intermediates and enzymes are shown. See text for detailed discussion.
The strikingly different biosynthetic pathways for FR-900098 and dehydrofosmidomycin show that the ability to produce these related molecules arose by convergent evolution, a rare occurrence in biology. Moreover, the observation that nature has independently arrived at the hydroxyaminophosphonate scaffold speaks to the potency of these bioactive molecules and the efficacy of DXR as a natural product target. Interestingly, this is the second documented occurrence of convergent evolution towards a bioactive phosphonate natural product, with fosfomycin being the first (43). Like FR-900098 and dehydrofosmidomycin, fosfomycin is also a three-carbon phosphonate. This peptidoglycan biosynthesis inhibitor is synthesized via shortening of a four-carbon intermediate in Pseudomonas and by extension of a two-carbon intermediate in Streptomyces, which echoes the pattern seen during biosynthesis of FR-900098 and dehydrofosmidomycin. The initial step in all four pathways is synthesis of the three-carbon phosphonopyruvate. Due to the poor thermodynamics of this reaction, a subsequent, thermodynamically favorable reaction, which shortens or lengthens the carbon skeleton of the initial phosphonate product, is required to achieve net phosphonate synthesis. This, in turn, required the evolution of pathways to regenerate the three-carbon skeleton of the final product via interesting and unusual transformations.
The observation that dehydrofosmidomycin, rather than fosmidomycin, is the main product of the phosphonate biosynthetic gene cluster encoded by S. lavendulae, suggests that unsaturated congeners of this molecular family merit further study. Indeed, our data show that dehydrofosmidomycin is a potent antibiotic, with activity against important drug-resistant pathogens. During the course of this work, others developed a similar synthesis for dehydrofosmidomycin and showed that it is also a potent in vitro inhibitor of P. falciparum DXR (IC50 = 0.09 μM) with nearly identical activity to fosmidomycin (IC50 = 0.06 μM)(27). Excitingly, they found that dehydrofosmidomycin is >50-fold more active in P. falciparum growth inhibition assays compared to fosmidomycin, likely due to its improved membrane permeability (27). These data suggest that dehydrofosmidomycin is a very promising antibacterial and antimalarial candidate, perhaps with greater potential than the previously clinically investigated fosmidomycin.
ONLINE METHODS
Chemicals.
General chemical reagents were purchased from Sigma Aldrich. Diethyl allylphosphonate (7, CAS: 5954–65-4) was purchased from Alfa Chemistry. L-methionine methyl-13C was purchased from Cambridge Isotope Laboratories, Inc. (CAS: 49705-26-2). Fosmidomycin and 1-deoxy-D-xylulose 5-phosphate were purchased from Echelon Biosciences. Solvents were dried by passage through columns packed with activated alumina.
Media.
All ingredients are given per liter
AGS medium:
12.5 mL glycerol, 1.0 g arginine-HCl, 1.0 g NaCl, 1.0 g K2HPO4, 0.5 g MgSO4-7H2O, 0.01 g Fe2(SO4)3-6H2O, 1 mg CuSO4-5H2O, 1 mg MnCl2-4H2O, 1 mg ZnSO4-7H2O and 16.0 g agar; adjust pH to 7.0.
ATCC 172 medium:
10 g dextrose, 20 g soluble starch, 5 g yeast extract, 5 g N-Z Amine Type A, 1 g CaCO3, adjust to pH 7.3 prior to autoclaving.
ISP2 medium:
10 g malt extract, 4 g yeast extract, 4 g dextrose.
ISP4-N medium:
ISP medium 4 with 10 mM N-acetylglucosamine; 10.0 g soluble starch, 1.0 g K2HPO4, 1.0 g MgSO4-7H2O, 1.0 g NaCl, 2.0 g (NH4)2SO4, 2.0 g CaCO3, 1 mg FeSO4-7H2O, 1 mg MnCl2-4H2O, 1 mg ZnSO4-7H2O and 15.0 g agar; adjust pH to 7.2; after autoclaving add 2.21 g N-acetylglucosamine as a sterile solution in 10 mL of dH2O.
GUBC medium:
5 ml glycerol, 10.0 g sucrose, 5.0 g beef extract, 5.0 g casamino acids, 5 mL 1 M Na2HPO4-KH2PO4 buffer (pH 7.3), 2 mL Hunter’s Concentrated Base (20 g Nitrilotriacetic acid, 59.3 g MgSO4-7H20, 6.67 g CaCl2-2H2O, 18.5 mg (NH4)6Mo7O24-4H20, 0.198 g FeSO4-7H2O, 0.25 g EDTA sodium salt, 1.095 g ZnSO4-7H20, 0.50 g FeSO4-7H2O, 0.154 g MnSO4-H2O, 39.2 mg CuSO4-5H2O, 25.0 mg Co(NO3)2–6H2O, 17.7 mg Na2B4O7-10H2O, pH 6.8), 15.0 g agar, 10 mL of sterile Balch’s Vitamins (5 mg p-aminobenzoic acid, 2 mg folic acid, 2 mg biotin, 5 mg nicotinic acid, 5 mg calcium pantothenate, 5 mg riboflavin, 5 mg thiamin HCl, 10 mg pyridoxine HCl (B6), 100 μg cyanocobalamin (B12), 5 mg thioctic acid (lipoic acid), pH to 7.0 with NaOH) added after autoclaving.
MS medium:
20.0 g mannitol, 20.0 g soya flour and 20.0 g agar
R2AS medium:
0.5 g yeast extract, 0.5 g peptone, 0.5 g casamino acids, 0.5 g dextrose, 0.5 g soluble starch, 0.3 g sodium pyruvate, 0.3 g K2HPO4, and 0.05 g of MgSO4-7H2O; after autoclaving, add sodium succinate to 20 mM final.
R2AS-N medium:
same as R2AS; after autoclaving add N-acetylglucosamine to 10 mM final.
Nuclear Magnetic Resonance (NMR).
1H-NMR, 13C-NMR, and 31P-NMR were recorded on either an Agilent DD2 600 MHz spectrometer (600 MHz for 1H, 150 MHz for 13C and 243 MHz for 31P) or a Bruker 500 MHz spectrometer equipped with a CryoProbe (500 MHz for 1H, 126 MHz for 13C and 203 MHz for 31P). Spectra generated from a solution of CDCl3 were referenced to residual chloroform (1H: δ 7.26 ppm, 13C: δ 77.16 ppm). Spectra generated from a solution of D2O were referenced to residual water (1H: δ 4.79 ppm).
Liquid Chromatography High Resolution Mass Spectrometry (LC-HRMS).
HRMS for synthetic molecules was performed by the UIUC Mass Spectrometry Laboratory. Unless otherwise noted, all LC-HRMS experiments on spent media were performed at the UIUC Metabolomics Center using the following protocol. Briefly, samples were re-suspended in 75% ACN. Samples were vortexed and sonicated before filtering through a 0.2 μm syringe filter. For high resolution LC/MS, the samples were analyzed by using the Q-Exactive MS system (Thermo. Bremen, Germany) in the Metabolomics Laboratory of Roy J. Carver Biotechnology Center, University of Illinois at Urbana-Champaign. Software Xcalibur 4.1.31.9 was used for data acquisition and analysis. The Dionex Ultimate 3000 series HPLC system (Thermo, Germering, Germany) used includes a degasser, an autosampler, and a binary pump. The LC separation was performed on a 250 x 4.6 mm inner diameter, 4-μm particle size Hydro-RP column (Phenomenex) with mobile phase A (water) and mobile phase B (acetonitrile). The flow rate was 0.5 mL/min. The linear gradient was as follows: 0 to 1 min, 0% B, 1 to 5 min, gradient to 10% B, 5 to 35 min, gradient to 100% B, 35 to 45 min, 100% B, 46 min, to 0% B, 55 min, 0% B. The autosampler was set to 10°C. The injection volume was 10 μL. Mass spectra were acquired under both positive (sheath gas flow rate, 52; aux gas flow rate: 13.5; sweep gas flow rate, 3.5; spray voltage, 3.5 kV; capillary temp, 268 °C; Aux gas heater temp, 430 °C) and negative electrospray ionization (sheath gas flow rate, 52; aux gas flow rate: 13.5; sweep gas flow rate, 3.5; spray voltage, −2.5 kV; capillary temp, 268 °C; Aux gas heater temp, 430 °C). The full scan mass spectrum resolution was set to 70,000 with the scan range of m/z 230 ~ m/z 1,600, and the AGC target was 1E6 with a maximum injection time of 200 ms. For MS/MS scan, the mass spectrum resolution was set to 17,500. AGC target was 5E4 with a maximum injection time of 50 ms. Loop count was 4. Isolation window was 1.0 m/z with NCE of 30 and 40 eV.
Synthetic Chemistry.
Synthesis of compounds used in the study is reported in the Supplementary Note: Synthetic Procedures.
Isolation and purification of dehydrofosmidomycin.
S. lavendulae 8006 was revived from a frozen DMSO stock by streaking onto solid ATCC 172 media and grown for 3 days at 30°C. A single colony was then used to inoculate 5 mL of ATCC 175 liquid in a 20 x 150 mm test tube and grown on an angled roller drum (75 rpm) at 30°C for 3 days. 5 mL was then used to inoculate 500 mL Fernbach flasks containing 100 mL of ATCC 172. These were grown for three days on a platform shaker (200 rpm). 40 mL of this culture was then used to inoculate 4 L Fernbach flasks containing 1 L of ISP4-N medium. These cultures were grown for seven days on a platform shaker (200 rpm). This process was performed for a total of 25 L of growth media.
After growth, the cells were pelleted, and the supernatant was frozen and dried via lyophilization. The cell pellet was washed with methanol (~5 L total). The dried supernatant was re-suspended in water and the methanol from the cell pellet was added so that the final solution was 90% methanol. The sample was stored at −20°C overnight and then centrifuged to remove the precipitant. The supernatant was dried via rotary evaporator until nearly dry and then re-suspended in 600 mL of dH2O. The molecule of interest was present in the supernatant as evidenced by both 31P NMR and DXR inhibition assay. At each subsequent step of purification, the presence of the molecule of interest was ensured via 31P NMR.
Amberlite XAD-16 resin (500 g) was washed with dH2O (~4 L), 25% methanol (2 L), and then dH2O (2 L). The supernatant was then added to the resin and incubated on at room temperature with gentle mixing for four hours. The resin was recovered by filtration and washed with 800 mL of dH2O. The filtrate and water washes were combined, dried via rotary evaporation, and re-suspended in 200 mL dH2O.
To the sample was added 1.8 L of methanol (90% methanol), and the sample was incubated at −20°C overnight. The sample was centrifuged to remove the precipitant, and the supernatant was dried via rotary evaporator until nearly dry. The sample was re-suspended in 80 mL of dH2O and 720 mL of ethanol was added (90% ethanol). Product was found in the pellet. Pellet was re-dissolved in ~200 mL dH2O.
Oasis HLB resin (50 g) was washed with methanol (500 mL) and then dH2O (500 mL) and loaded into a column (2.5 x 8.5 in). The sample was then loaded onto the column and washed with dH2O (500 mL), 5% methanol (500 mL), and 100% methanol (500 mL). The flow through and water washes were combined and dried via rotary evaporation. Sample was re-suspended in dH2O (50 mL).
Approximately 10% of the material was taken forward for further purification using a 5.7 g RediSep SAX column and a Combiflash Rf+ (Teledyne ISCO). The column was pre-equilibrated with 20 column volumes (CV) of 5% acetic acid in dH2O followed by 20 CV of dH2O and 20 CV of 90% methanol. The sample was then loaded onto the column. The following gradient was then run (Flow rate = 18 mL/min): 3.3 min at 100% solvent A (90% methanol) then a linear gradient to 100% solvent B (5% acetic acid in dH2O) over 15 min then 100% B for 6 min followed by 100% A for 3.5 min. Upon completion of the column, fractions were neutralized using concentrated NH4OH. The samples were then directly injected onto an Agilent 1200 LC/MSD Quad SL System and analyzed via selected-ion monitoring (SIM) for dehydrofosmidomycin ([M]− = 180 Da). Fractions containing the mass of interest were combined and dried via rotary evaporation.
The material was dissolved in 700 μL of 66% methanol and further purified using an Atlantis Silica HILIC column (10 x 250 mm, 5 μm particle size) using a gradient elution. Chromatography was performed at a flow rate of 4 mL/min using dH2O (solvent A) and acetonitrile (solvent B) with the following gradient: 7 min at 90% solvent B then a linear gradient to 55% B over 29 min then 55% B for 15 min then a linear gradient to 90% B over 14 min and 43 min at 90% B. The samples were then directly injected onto an Agilent 1200 LC/MSD Quad SL System and analyzed via SIM for dehydrofosmidomycin ([M]− = 180 Da). Fractions containing the mass of interest were combined and dried via rotary evaporation yielding 13 mg.
The material (13 mg) was then dissolved in 500 μL of 5 mM H2SO4 and further purified using a Bio-Rad Aminex HPX-87H column (7.8 x 300 mm, 9 μm particle size) using an isocratic elution. Chromatography was performed at a rate of 0.6 mL/min using 5 mM sulfuric acid as the eluent. The run was monitored at 210 nm, and 1 min fractions were collected. Fractions were neutralized using 1 M NaOH, frozen, and dried via lyophilization. Fractions were screened for product via 1H NMR. Fractions containing product were combined and dried via rotary evaporation to yield 4 mg of nearly clean product.
The material (4 mg) was dissolved in 500 μL of dH2O and run on a Sephadex LH-20 column (2 cm x 165 cm) with water as the eluent (flow rate = 0.18 mL/min) to yield 175 1 mL fractions. Fractions were dried via lyophilization and screened by 1H NMR. Fractions containing product were combined and dried via rotary evaporation.
The material was dissolved in 600 μL of 50% methanol and further purified using an Atlantis Silica HILIC column (10 x 250 mm, 5 μm particle size) using a gradient elution. Chromatography was performed at a flow rate of 4 mL/min using dH2O (solvent A) and acetonitrile (solvent B) with the following gradient: Linear gradient from 90% solvent B to 75% B over 36 min then a linear gradient to 55% B over 15 min then 55% B for 7 min then a linear gradient to 90% B over 7 min and 43 min at 90% B. The samples were then directly injected onto an Agilent 1200 LC/MSD Quad SL System and analyzed via SIM for dehydrofosmidomycin ([M]− = 180 Da). Fractions containing the mass of interest were combined and dried via rotary evaporation yielding 1.2 mg. This material was pure dehydrofosmidomycin, other than residual inorganic phosphate.
1H NMR (600 MHz, D2O) δ 8.36 (minor rotamer, s, 0.3H), 8.03 (major rotamer, s, 0.9H), 6.23 (major rotamer, m, 1H), 6.15 (minor rotamer, m, 0.14H), 6.07 (major rotamer, t, 0.8H, J = 17 Hz), 6.03 (minor rotamer, t, 0.3H, J = 17 Hz), 4.30 (minor rotamer, d, 0.5 H, J = 5 Hz), 4.26 (major rotamer, d, 1.1 H, J = 4 Hz). Due to the very low concentration of compound, it was necessary to run solvent suppression on this NMR.
31P NMR (243 MHz, D2O) δ 9.47
HRMS (ESI) calculated for C4H7NO5P (M)−: 180.0062 , found: 180.0066, Δppm: 2.2.
Functional annotation of genes in the biosynthetic gene cluster of dehydrofosmidomycin.
The genome sequence of Streptomyces lavendulae 8006 (Genbank accession ASM71562v1) was downloaded and analyzed in house. The dfm gene cluster can be found on contig 2.1 (Genbank accession JNXL01000002). The protein sequences encoded by each gene in the dfm gene cluster from Streptomyces lavendulae were compared to proteins in the non-redundant Genbank database using BlastP (44). Proteins with the highest percent identity with high confidence functions were used to assign the putative functions shown in Table S3. This analysis was performed on May 23, 2018.
Screen for fosmidomycin production.
Streptomyces lavendulae 8006 was revived from a frozen stock by streaking onto solid ATCC 172 media and grown for 3 days at 30°C. A single colony was then used to inoculate 5 mL of ATCC 175 liquid in a 20 x 150 mm test tube and grown on an angled roller drum (75 rpm) at 30°C for 3 days. 5 mL was then used to inoculate 500 mL Fernbach flasks containing 100 mL of ATCC 172. These were grown for three days on a platform shaker (200 rpm). This culture was then used to inoculate four liquid and four solid medium types (GUBC, ISP4-N, R2AS, and R2AS-N). These media were chosen based on previous screens of the same strain. For the solid media, each 100 x 15 mm petri dish received 500 μL of culture and was grown at 30°C for varying times (see Table S2). For the liquid media, 500 mL Fernbach flasks containing 100 mL of media were inoculated with 5 mL of culture and grown on a platform shaker (200 rpm) for varying times (see Table S2). To harvest the solid media, plates were frozen at −20°C for 24 h and then thawed to room temperature and compressed through non-gauze milk filters to yield ~15 mL per plate. To harvest the liquid media, 25 mL was taken from the flask and centrifuged at 4500xg for 10 min. The supernatant was transferred to a new 50 mL conical tube. A 500 μL aliquot was removed for DXR inhibition and phosphonate bioactivity testing. The rest of the sample was frozen, dried via lyophilization, and dissolved in 1 mL D2O for NMR analysis.
Purification of E. coli 1-deoxy-D-xylulose 5-phosphate reductoisomerase (DXR).
E. coli DXR was purified using a previously published method.(21) Briefly, E. coli Rosetta (DE3) cells were transformed with a pET15b vector with the E. coli dxr gene inserted into the NdeI and BglII sites. The cells were grown in LB broth with 12 μg/mL chloramphenicol and 50 μg/mL ampicillin at 37°C until OD600 = 0.7. The cells were cooled to 30°C, and expression of the dxr gene was induced by addition of 0.5 mM IPTG. The cells were grown at 30°C with shaking (250 rpm) for 4h and then harvested via centrifugation (4500 x g for 10 min at 4°C). The pellets were frozen at −80°C overnight. The cells were thawed on ice and re-suspended in lysis buffer (25 mL of 50 mM NaH2PO4, 300 mM NaCl, 10 mM imidazole, and 10% glycerol, pH 8.0) with 1 mg/mL lysozyme, 100 μM PMSF, and 300 units of DNase-1. The cells were incubated with intermittent mixing at 4°C for 30 minutes. The suspension was passed twice through a chilled French pressure cell and clarified via centrifugation (20,000 x g for 30 min at 4°C). Supernatant was then incubated with Ni-NTA agarose (Qiagen) for 1h at 4°C with gentle agitation and then loaded into an empty column. The resin was washed with 20 mL of wash buffer 1 (25 mL of 50 mM NaH2PO4, 300 mM NaCl, 34 mM imidazole, and 10% glycerol, pH 8.0) and 20 mL of wash buffer 2 (25 mL of 50 mM NaH2PO4, 300 mM NaCl, 70 mM imidazole, and 10% glycerol, pH 8.0). The protein was eluted with 25 mL of elution buffer (25 mL of 50 mM NaH2PO4, 300 mM NaCl, 250 mM imidazole, and 10% glycerol, pH 8.0). SDS-PAGE revealed that the protein was pure (a single band at the expected molecular weight, 45.4 kDa). The protein was then concentrated using an Amicon Ultra-15 filter device (MWCO = 30 kDa) and buffer exchanged to 20 mM Tris HCl, 0.2 mM DTT, 10% glycerol (pH 8.0) using a PD10 desalting column (GE Healthcare). Protein concentration was determined to be 6.5 mg/mL via Bradford assay, and yield was 23 mg/L.
DXR Inhibition Assay.
The DXR inhibition assay was performed using a previously published method.(21) Each reaction contained 100 mM Tris-HCl (pH 8.0), 2.5 mM MgCl2, 1 mg/mL BSA, 100 μM 1-deoxy-D-xylulose 5-phosphate (DXP; Echelon Biosciences Inc.), 150 μM NADPH, and 60 nM DXR in a total volume of 200 μL. Enzyme, inhibitor or spent media, and NADPH were pre-incubated in the assay buffer at 25°C for 2 min prior to initiation of the reaction with DXP. Activity was measured spectrophotometrically by observing the decrease of absorbance at 340 nm due to the oxidation of NADPH every 2 s using a Cary 4000 UV-vis. Data were fitted using OriginPro 2016 or 2018 (OriginLab).
Phosphonate Bioactivity Assay.
E. coli WM6242 is an E. coli strain that expresses the phosphonate uptake system phnCDE under the control of a Ptac promoter (21). Inducing the expression of phnCDE by addition of IPTG increases sensitivity to most phosphonate antibiotics. The phosphonate specific bioassay, which uses E. coli WM6242, was previously described (21). Briefly, WM6252 was grown to an OD600 of 0.8 in LB at 37°C. 6 mL of LB top agar, with and without 1 mM IPTG, was inoculated with 50 μL of the culture and then added to the corresponding LB plates with and without 1 mM IPTG. Test solutions (9 μL) were added to filter discs (6 mm, Becton Dickinson Company), and the discs were placed on the surface of the solidified plates. Plates were incubated for 12 h, and zones of inhibition were measured. When activity is greater on plates containing IPTG, it indicates the presence of a phosphonate antibiotic. Synthetic fosmidomycin (100 μM) was used as a positive control.
Generation of heterologous expression strains.
A clone containing the full dfm cluster was obtained from a genomic library of S. lavendulae that was constructed and screened as previously described (21). This plasmid designated cosmid 14A8 (hereafter simply referred to as dfm) contains all of contig 2.1 from Genbank accession JNXL01000002, as well as sequences from several smaller contigs, cloned in the fosmid pJK050. The full, annotated sequence of the genomic DNA inserted into cosmid 14A8 is presented in the supplementary materials. To determine the boundaries of the dehydrofosmidomycin biosynthetic gene cluster, multiple deletions were made using λ Red recombinase (45). Primer sequences used to generate the deletion mutants and to check for successful deletion can be found in Table S4 and S5, respectively. Transfer of these to S. albus J1074 after retrofitting with plasmid pAE4 was as described (21).
Antibacterial Testing.
Susceptibility testing for all bacteria was performed using the microdilution broth method as outlined by the Clinical and Laboratory Standards Institute (CLSI) (46). Mueller Hinton Broth 2 (MH, Sigma-Aldrich, 90922) was used. Briefly, 2 μL of 50X compound stocks were added to the wells of a 96-well round well plate. Live and dead controls received 2 μL of vehicle. 88 μL of MH broth was added to all wells except the dead control wells, which received 98 μL of MH broth. 100 μL of an overnight culture of bacteria was then added to 10 mL of MH broth and grown until the culture reached a turbidity equal to 1 X 107 – 2 x 108 cfu/mL (based on a previously determined calibration curve). The culture was then diluted to 5 X 106 cfu/mL and 10 μL of this solution was added to each well except the dead controls for a final of 5 X 105 cfu/mL. Plates were incubated at 37 °C for 16-20 h. Absorbance was then read on a Tecan infinite M200 Pro plate reader at λ = 600 nm. Minimum Inhibitory Concentration (MIC) values were defined as the lowest concentration of compound that resulted in ≥ 90% growth inhibition.
Heterologous expression of dehydrofosmidomycin by S. albus J1074/dfm.
S. albus J1074/dfm was revived from a frozen DMSO stock by streaking onto solid ATCC 172 media with 50 μg/mL apramycin and grown for 7 days at 30°C. A single colony was then used to inoculate 5 mL of ATCC 175 liquid in a 20 x 150 mm test tube and grown on an angled roller drum (75 rpm) at 30°C for 5 days. The culture was then used to inoculate seven solid media (AGS, ATCC 172, GUBC, ISP2, ISP4-N, R2AS, MS). Each standard petri dish received 500 μL of culture and was grown at 30°C for 10 days. Plates were then frozen overnight at −20 °C, thawed to room temperature, and compressed through non-gauze milk filters to yield ~15 mL per plate. The production media was then frozen and dried via lyophilization. Samples were re-suspended in 1 mL dH2O, and 9 mL methanol was added. After incubation at −20°C overnight, the precipitants were removed via centrifugation and the supernatant was dried via rotary evaporation. The samples along with samples from the parent strain S. albus J1074 were then analyzed by NMR, LC-HRMS, DXR inhibition assay, and phosphonate bioactivity assay for production of dehydrofosmidomycin. ISP2 was the best medium and was used for all subsequent studies with the heterologous expression strain.
Deletion analysis of the biosynthetic gene cluster.
To determine the boundaries of the dehydrofosmidomycin biosynthetic gene cluster, multiple deletions were made using λ Red recombinase.(45) Electrocompetent BW26678 (lacIq rrnBT14 ΔlacZWJ16 hsdR514 ΔaraBADAH33 ΔrhaBADLD78/pKD46) (45, 47) were transformed with the dehydrofosmidomycin fosmid and allowed to recover in SOC for 1 h at 30°C on a roller drum. The bacteria were then plated onto selective media (LB + 100 μg/mL ampicillin + 50 μg/mL apramycin) and allowed to grow overnight at 30°C. An overnight of BW26678 with the dehydrofosmidomycin fosmid was grown and used to inoculate 1 L of SOB with 100 μg/mL ampicillin and 25 μg/mL apramycin. The culture was grown until OD600 = 0.2 at which time 10 mM arabinose was added. The culture was then grown until OD600 = 0.6. Cells were put on ice, washed three times with ice-cold 10% glycerol and then flash frozen.
Primers with homology to the gene to be inactivated and the kanamycin resistance gene aminoglycoside 3′-phosphotransferase (APH) from pKD4(45) and were designed for each deletion (see Table S4). PCR amplification was performed with Q5 polymerase. Each reaction contained the following: 10 μL of 5X Q5 buffer, 200 μM dNTPs, 500 nM of forward and reverse primer, 41 ng of pKD4, 10 μL of Q5 High GC Enhancer, 0.5 μL Q5 polymerase, and nuclease free water to a final volume of 50 μL. Thermocyclers were operated under the following program: (i) initial denaturation at 98 °C for 30 sec; (ii) 98 °C for 10 sec; (iii) 52 °C for 30 sec; (iv) 72 °C for 50 sec; (v) Repeat ii-iv for a total of 35 cycles; (vi) 72 °C for 2 min; (vii) hold at 4 °C. PCR products were cleaned up using the NEB Monarch PCR cleanup kit using the standard protocol. DNA was eluted with 45 μL of dH2O. Cutsmart buffer (5 μL) and DpnI (5U) were added and incubated at 37 °C for 1h. The products were separated by electrophoresis through a 1.5% agarose gel with the NEB 2-log ladder used as a control. The band at 1.6 kbp was extracted using the EZNA gel extraction kit using the standard protocol.
The electrocompetent BW26678 carrying the dehydrofosmidomycin fosmid were then transformed with the PCR products described above. SOC (1 mL) was added to the culture, and it was allowed to recover for 4 h at 37 °C on an angled roller drum before plating onto LB with 50 μg/mL kanamycin and 25 μg/mL apramycin. Plates were incubated overnight at 37 °C. C, and colonies were then screened by PCR for the presence of the KanR gene using the k2 and kt primers (see Table S5). Colonies were re-suspended in 10 μL of dH2O. Each reaction contained 12.5 μL of 2X GoTaq Green Master Mix (Promega), 800 nM primers, 1 μL of colony resuspension, and nuclease free water to a final volume of 25 μL. Thermocyclers were operated under the following program: (i) initial denaturation at 95 °C for 10 min; (ii) 95 °C for 30 sec; (iii) 52 °C for 30 sec; (iv) 72 °C for 60 sec; (v) Repeat ii-iv for a total of 35 cycles; (vi) 72 °C for 5 min; (vii) hold at 4 °C. The products were separated by electrophoresis through a 1.5% agarose gel with the NEB 2-log ladder used as a control. Those that were successful (band at 471 bp) were then screened via PCR for the proper insertion site using the Ftest/k1 and Rtest/k2 primers using a similar procedure. If those were successful (bands at 604 and 997 bp, respectively), the colony was used to inoculate 5 mL of LB + 50 μg/mL kanamycin + 25 μg/mL apramycin that was grown overnight at 37 °C. The fosmid was isolated using a miniprep kit (Qiagen). Usually this gave quite low yields of the fosmid due to its low copy number. WM4489(21, 43) was transformed with the fosmid, and plated on LB + 50 μg/mL kanamycin. An overnight culture from a colony was then grown with 10 mM rhamnose to access greater quantities of the fosmid. The conjugal donor WM6029(48) was then transformed with the fosmid and following recovery with SOC + 25 μg/mL diaminopimelic acid was plated on LB + 25 μg/mL diaminopimelic acid + 50 μg/mL apramycin. Successful transformants were identified using the same PCR described above.
Upon successful transformation of the conjugal donor WM6029 with the mutant fosmid, conjugation with S. albus J1074 was performed. A single colony of WM6029 carrying each mutant fosmid was grown in LB + 25 μg/mL diaminopimelic acid + 50 μg/mL apramycin was grown overnight at 37 °C. A spore stock of S. albus J1074 (100 μL, ~108 spores) was heat shocked at 56 °C for 10 min. After the heat shock, 100 μL of 2XYT (per liter: 16 g tryptone, 10 g yeast extract, 5 g NaCl) was added to the spore stock. The overnight culture of WM6029 with mutant fosmid was then diluted 1:10 in 2XYT. 4 μL of the diluted WM6029 culture was then added to the spore stock. The mixed culture was centrifuged briefly to concentrate the cells in the bottom of the Eppendorf tube and allowed to incubate at room temperature for 10 min. The cells were then re-suspended and plated on R2 plates without sucrose (0.25 g K2SO4, 10.12 g MgCl2-6H2O, 10 g dextrose, and 0.1 g Difco Casamino acids dissolved in 800 mL of dH2O and 80 mL aliquoted into 250 mL Erlenmeyer flasks with 2.2 g agar. At time of use, agar was melted and the following autoclaved solutions were added in the order listed: 1 mL of 0.5% KH2PO4, 8 mL of 3.68% CaCl2-2H2O, 1.5 mL of 20% proline, 10 mL of 5.73% TES buffer (pH 7.2), 0.2 mL of trace element solution (per liter, 40 mg ZnCl2, 200 mg FeCl3-6H2O, 10 mg CuCl2-2H2O, 10 mg MnCl2-4H2O, 10 mg Na2B4O7-10H2O, 10 mg (NH4)6Mo7O24-4H2O), and 0.5 mL 1 N NaOH (unsterilized)). Plates were incubated at 30 °C for 16 h. Then, 2 mL of 1 mg/mL apramycin was added to each plate and allowed to dry. The plates were then incubated at 30 °C until colonies appeared (usually ~1 week). Colonies that appeared were then plated onto ATCC 172 plates with 50 μg/mL apramycin.
Colonies that regrew were then tested for successful conjugation via colony PCR. Half a colony was picked and re-suspended in 10 μL of DMSO. Each reaction contained the following: 10 μL of 5X Q5 buffer, 200 μM dNTPs, 500 nM of forward and reverse primer, 1 μL of the colony suspension, 10 μL of Q5 High GC Enhancer, 0.5 μL Q5 polymerase, and nuclease free water to a final volume of 50 μL. Thermocyclers were operated under the following program: (i) initial denaturation at 98 °C for 10 min; (ii) 98 °C for 10 sec; (iii) 52 °C for 30 sec; (iv) 72 °C for 50 sec; (v) Repeat ii-iv for a total of 35 cycles; (vi) 72 °C for 2 min; (vii) hold at 4 °C. The products were separated by electrophoresis through a 1.5% agarose gel with the NEB 2-log ladder used as a control. The appropriate bands were extracted using the EZNA gel extraction kit using the standard protocol. The PCR products were then submitted to UIUC Core Sequencing Facility where they were sequenced via Sanger sequencing. The sequences were then compared to the expected sequences.
After generation of the strains, they were frozen as DMSO mycelial stocks. For production tests, each strain was revived by streaking onto solid ATCC 172 media with 50 μg/mL apramycin and grown for 7 days at 30°C. A single colony was then used to inoculate 5 mL of ATCC 172 liquid in a 20 x 150 mm test tube and grown on an angled roller drum (75 rpm) at 30°C for 5 days. The culture was then used to inoculate solid ISP2 media and grown for 17 days along with S. albus J1074 WT and S. albus J1074 with the full dehydrofosmidomycin fosmid. After growth, plates were frozen overnight, thawed, and spent media was obtained via squeezing through non-gauze milk filters. Spent media was frozen and dried via lyophilization prior to analysis via 31P NMR and LC-HRMS.
Feeding studies with 2-AEP.
Each strain was revived by streaking onto solid ATCC 172 media and grown for 7 days at 30°C. For S. albus J1074, no additional additive was used. For S. albus J1074/dfm and S. albus J1074/ΔLM, the media also contained 50 μg/mL apramycin. A single colony was then used to inoculate 5 mL of ATCC 172 liquid in a 20 X 150 mm test tube and grown on an angled roller drum (75 rpm) at 30°C for 5 days. The culture (0.5 mL) was then used to inoculate solid ISP2 media with or without 1 mM 2AEP and grown for 17 days. After growth, plates were frozen overnight, thawed, and spent media was obtained via squeezing through non-gauze milk filters. Spent media was frozen and dried via lyophilization prior to analysis via 31P NMR and LC-HRMS.
Feeding studies with L-methionine.
S. lavendulae 8006 was revived by streaking onto solid ATCC 172 media and grown for 7 days at 30°C. A single colony was then used to inoculate 5 mL of ATCC 172 liquid in a 20 x 150 mm test tube and grown on an angled roller drum (75 rpm) at 30°C for 5 days. The culture (0.5 mL) was then used to inoculate solid ISP4-N media with or without 1 mM L-methionine or 1 mM L-methionine-(methyl-13C) and grown for 13 days. After growth, plates were frozen overnight, thawed, and spent media was obtained via squeezing through non-gauze milk filters. Spent media was frozen and dried via lyophilization prior to analysis via 31P NMR and LC-HRMS.
DfmD cloning.
The gene for DfmD was amplified via PCR using primer pair 8006_DioxF (5’- AGCGGCCTGGTGCCGCGCGGCAGCCATatgactacaacagccgaagcatatctgtcgat-3’) and 8006_DioxR (5’- ATCTCAGTGGTGGTGGTGGTGGTGCTCGAGtcactacaccgtttcgtcggcgaaacgcga -3’) from S. lavendulae 8006 genomic DNA. The uppercase letters represent sequence homology to the plasmid pET28B, and the lowercase letters represent sequence homology to the gene to be amplified. An extra stop codon was inserted at the end of 8006_DioxR (italicized letters) to ensure termination of the protein. PCR amplification was performed with Q5 polymerase. Each reaction contained the following: 10 μL of 5X Q5 buffer, 200 μM dNTPs, 500 nM of forward and reverse primer, 200 ng genomic DNA, 10 μL of Q5 High GC Enhancer, 0.5 μL Q5 polymerase, and nuclease free water to a final volume of 50 μL. Thermocyclers were operated under the following program: (i) initial denaturation at 98 °C for 30 sec; (ii) 98 °C for 10 sec; (iii) 72 °C for 30 sec; (iv) 72 °C for 40 sec; (v) Repeat ii-iv for a total of 35 cycles; (vi) 72 °C for 2 min; (vii) hold at 4 °C. The products were separated by electrophoresis through a 1.5% agarose gel with the NEB 2-log ladder used as a control. The band at 1.2 kbp was extracted using the EZNA gel extraction kit using the standard protocol. The plasmid pET28b was PCR amplified using primers pET28-NdeI-R (5′-CATATGGCTGCCGCGCGGCACCAGGCCGCT-3′) and pET28B-XhoI-F (5′-CTCGAGCACCACCACCACCACCACTGAGAT-3′). PCR amplification was performed with Q5 polymerase. Each reaction contained the following: 10 μL of 5X Q5 buffer, 200 μM dNTPs, 500 nM of forward and reverse primer, 50 ng pET28b, 10 μL of Q5 High GC Enhancer, 0.5 μL Q5 polymerase, and nuclease free water to a final volume of 50 μL. Thermocyclers were operated under the following program: (i) initial denaturation at 98 °C for 30 sec; (ii) 98 °C for 10 sec; (iii) 72 °C for 30 sec; (iv) 72 °C for 2 min 45 sec; (v) Repeat ii-iv for a total of 35 cycles; (vi) 72 °C for 2 min; (vii) hold at 4 °C. The products were separated by electrophoresis through a 1.5% agarose gel with the NEB 2-log ladder used as a control. The band at 5.3 kbp was extracted using the EZNA gel extraction kit using the standard protocol. The gene fragment was then cloned into pET28b using NEBuilder HiFi DNA Assembly Master Mix to yield pEIP001. Specifically, 0.058 pmol (5.3 μL) of vector was combined with 0.14 pmol (2.3 μL) of insert. This was combined with an equal volume of Master mix and incubated at 50 °C for 30 min. NEB High Efficiency cells were then transformed with 2 μL of the mix before plating on Luria-Bertani (LB) plates supplemented with kanamycin (50 μg/mL). Colonies were then grown overnight in 5 mL of LB supplemented with kanamycin at 37 °C on a roller drum. Plasmids were isolated using the QIAprep Spin Miniprep Kit and verified by sequencing at the University of Illinois Urbana-Champaign Core Sequencing Facility. Primers for sequencing included T7-pro (5’- TAA TAC GAC TCA CTA TAG GG-3’), T7-term (5’- GCT AGT TAT TGC TCA GCG G-3’), 8006_DioxSeq1 (5’-CCTGGAGATCGTCTGGGAGG-3’), and 8006_DioxSeq2 (5’-GTTGTCAAAGAACATCTTGG-3’).
DfmD overexpression and purification for activity assays.
pEIP001 was introduced into Rosetta(DE3)pLysS Competent Cells (EMD Millipore) by chemical transformation. Transformed cells were selected for on an LB plate supplemented with chloramphenicol and kanamycin (12.5 μg/mL and 50 μg/mL, respectively). Single colonies from a fresh plate were used to inoculate 10 mL LB with 12.5 μg/mL chloramphenicol and 50 μg/mL kanamycin and grown for 16 h at 37 °C on a roller drum. The overnight culture was then used to inoculate 1 L of LB with 12.5 μg/mL chloramphenicol and 50 μg/mL kanamycin. The culture was grown at 37 °C with shaking (250 rpm) until OD600 = 0.6. The culture was then cooled on ice before addition of IPTG (0.1 mM). The culture was then grown at 18 °C with shaking (250 rpm) for 18 h. Cells were collected via centrifugation (4500Xg for 10 min at 4 °C) and frozen overnight at −80 °C. Cell pellets were thawed on ice for 30 min prior to addition of 25 mL of Lysis buffer (50 mM NaH2PO4, 300 mM NaCl, 10 mM imidazole, 10% glycerol (v/v) pH 8.0). The cell suspension was mixed with 1 mg/mL lysozyme, 85 μg RNase A, 300 units of DNaseI, and 400 μM phenylmethylsulfonyl fluoride for 30 min at 4 °C. The cell suspension was passed twice through a French press cell disruptor (Thermo Electron Corp., Waltham, MA) at 1000 psi. The lysate was cleared by centrifugation at 17,000 × g for 30 min. The supernatant was batch loaded onto 2.5 mL of Ni-NTA resin (Qiagen) at 4 °C for 30 min with inversion. The resin was washed with 20 mL wash buffer (50 mM NaH2PO4, 300 mM NaCl, 33 mM imidazole, 10% glycerol (v/v) pH 8.0). Protein was eluted with 25 mL of elution buffer (50 mM NaH2PO4, 300 mM NaCl, 250 mM imidazole, 10% glycerol (v/v) pH 8.0), and 1.3 mL fractions were collected. Eluted fractions were assessed by SDS-PAGE using a 4-20% TGX Mini-PROTEAN gel (Bio-Rad) and stained using Coomassie stain (see Figure S2). Fractions containing pure protein were pooled and concentrated using an Amicon Ultra-15 centrifugal filter with 30 kDa cutoff (EMD Millipore). Samples were then exchanged into storage buffer (50 mM NaH2PO4, 300 mM NaCl, 10% glycerol (v/v) pH 8.0) using a PD-10 desalting column (GE Healthcare Life Sciences). Protein concentration was determined using the Pierce Coomassie Plus Protein Assay Reagent (Thermo Scientific) and BSA as the control.
DfmD activity assays by NMR and LC-HRMS/MS.
NMR experiments were performed on an Agilent 600 MHz spectrometer equipped with a OneNMR probe. Each sample (500 μL) contained 5 μM enzyme, 200 μM Fe(II)(NH4)2(SO4)2, 10 mM α-ketoglutarate, 1 mM L-ascorbate, and 5 mM substrate in phosphate-buffered saline (PBS, 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, and 1.8 mM KH2PO4, pH 7.4). Assays were performed in 15 mL conical tubes left open on their side for maximum oxygen exchange. Assays were performed at room temperature for 24 h unless otherwise noted. The reaction was then treated with Chelex (~100 μL) for 5 min, and the protein was removed using a 30 kDa cutoff Amicon Ultra-0.5 mL centrifugal filter (EMD Millipore). D2O (100 μL) was added prior to analysis by 31P and 1H NMR. Any spiking with known standards was performed after the reaction. Spectra were analyzed using MestReNova version 10.0.2. An aliquot of each samples (5 μL) was diluted into dH2O (45 μL) and ACN (150 μL), mixed, and filtered through 0.2 um syringe filters prior to analysis via LC-HRMS/MS.
Formaldehyde quantification assay.
The samples used in the NMR experiments were also used in the formaldehyde quantification assay. The assay is based on a previously published assay.(49) An acetylacetone solution was prepared by combining 15 g ammonium acetate, 0.2 mL acetylacetone, 0.3 mL glacial acetic acid, and dH2O to 100 mL. It was stored in the dark at 4 °C for up to 1 week. For each assay, a calibration curve of formaldehyde in dH2O was freshly prepared and used for quantification of the samples (0, 0.016, 0.032, 0.079, 0.16, 0.32, 0.47, 0.63 mM). For each sample, 100 μL of sample was mixed with 100 μL of acetylacetone solution in a glass vial and heated at 65 °C for 10 min. 100 μL was then transferred to a 96 well plate, and absorbance at 435 nm was read using a Tecan infinite M200PRO. The samples that had absorbances higher than the calibration curve were diluted 1/10 with dH2O and read again.
DATA AVAILABILITY
The authors declare that all relevant data supporting the findings of this study are available within the paper and its Supplementary Information.
Supplementary Material
ACKNOWLEDGMENTS
We thank M. Goettge and K. Wang for helpful discussions, X. Guan for his assistance with NMR experiments, Z. Li for his assistance with HRMS experiments, the UIUC Core Sequencing Facility for sequencing, and P. Hergenrother (Chemistry Department, UIUC) for providing strains. This work was funded by the National Institutes of Health (P01 GM077596 and R01GM127659 to WWM and F32GM120999 to EIP). NMR spectra were recorded on an instrument purchased with support from NIH grant S10 RR028833.
Footnotes
COMPETING INTERESTS
WWM has financial interest in Microbial Pharmaceuticals.
REFERENCES
- 1.Antibiotic Resistance Threats in the United States, 2013 | Antibiotic/Antimicrobial Resistance | CDC (2013) Available at: http://www.cdc.gov/drugresistance/threat-report-2013/ [Accessed November 19, 2015].
- 2.Health Organization W (2016) World Malaria Report 2016 Available at: http://apps.who.int/iris/bitstream/handle/10665/252038/9789241511711-eng.pdf;jsessionid=5B7F07446008DD504C3BFBA2ADD0AAC8?sequence=1 [Accessed June 13, 2018].
- 3.Bain C, Selfa T, Dandachi T, Velardi S (2017) ‘Superweeds’ or ‘survivors’? Framing the problem of glyphosate resistant weeds and genetically engineered crops. J Rural Stud 51:211–221. [Google Scholar]
- 4.Newman DJ, Cragg GM (2016) Natural Products as Sources of New Drugs from 1981 to 2014. J Nat Prod 79(3):629–661. [DOI] [PubMed] [Google Scholar]
- 5.Doroghazi JR, et al. (2014) A roadmap for natural product discovery based on large-scale genomics and metabolomics. Nat Chem Biol 10(11):963–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ju K-S, et al. (2015) Discovery of phosphonic acid natural products by mining the genomes of 10,000 actinomycetes. Proc Natl Acad Sci U S A 112(39):12175–12180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Iguchi E, Okuhara M, Kohsaka M, Aoki H, Imanaka H (1980) Studies on new phosphonic acid antibiotics. II. Taxonomic studies on producing organisms of the phosphonic acid and related compounds. J Antibiot (Tokyo) 33(1):19–23. [PubMed] [Google Scholar]
- 8.Okuhara M, et al. (1980) Studies on new phosphonic acid antibiotics. III. Isolation and characterization of FR-31564, FR-32863 and FR-33289. J Antibiot (Tokyo) 33(1):24–8. [DOI] [PubMed] [Google Scholar]
- 9.Kuroda Y, et al. (1980) Studies on new phosphonic acid antibiotics. IV. Structure determination of FR-33289, FR-31564 and FR-32863. J Antibiot (Tokyo) 33(1):29–35. [DOI] [PubMed] [Google Scholar]
- 10.Okuhara M, et al. (1980) Studies on new phosphonic acid antibiotics. I. FR-900098, isolation and characterization. J Antibiot (Tokyo) 33(1):13–7. [DOI] [PubMed] [Google Scholar]
- 11.Davey MS, et al. (2011) A promising target for treatment of multidrug-resistant bacterial infections. Antimicrob Agents Chemother 55(7):3635–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jomaa H, et al. (1999) Inhibitors of the nonmevalonate pathway of isoprenoid biosynthesis as antimalarial drugs. Science 285(5433):1573–6. [DOI] [PubMed] [Google Scholar]
- 13.Fernandes JF, et al. (2015) Fosmidomycin as an antimalarial drug: a meta-analysis of clinical trials. Future Microbiol 10(8):1375–90. [DOI] [PubMed] [Google Scholar]
- 14.Kuzuyama T, Shimizu T, Takahashi S, Seto H (1998) Fosmidomycin, a specific inhibitor of 1-deoxy-d-xylulose 5-phosphate reductoisomerase in the nonmevalonate pathway for terpenoid biosynthesis. Tetrahedron Lett 39(43):7913–7916. [Google Scholar]
- 15.Coppens I (2013) Targeting lipid biosynthesis and salvage in apicomplexan parasites for improved chemotherapies. Nat Rev Microbiol 11(12):823–835. [DOI] [PubMed] [Google Scholar]
- 16.Hartmann M, et al. (2013) The effect of MEP pathway and other inhibitors on the intracellular localization of a plasma membrane-targeted, isoprenylable GFP reporter protein in tobacco BY-2 cells. F1000Research 2:170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hunter WN (2007) The Non-mevalonate Pathway of Isoprenoid Precursor Biosynthesis. J Biol Chem 282(30):21573–21577. [DOI] [PubMed] [Google Scholar]
- 18.Lichtenthaler HK, Zeidler J, Schwender J, Müller C The non-mevalonate isoprenoid biosynthesis of plants as a test system for new herbicides and drugs against pathogenic bacteria and the malaria parasite. Z Naturforsch C 55(5–6):305–13. [DOI] [PubMed] [Google Scholar]
- 19.Eliot AC, et al. (2008) Cloning, Expression, and Biochemical Characterization of Streptomyces rubellomurinus Genes Required for Biosynthesis of Antimalarial Compound FR900098. Chem Biol 15(8):765–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Johannes TW, et al. (2010) Deciphering the Late Biosynthetic Steps of Antimalarial Compound FR-900098. Chem Biol 17(1):57–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eliot AC, et al. (2008) Cloning, expression, and biochemical characterization of Streptomyces rubellomurinus genes required for biosynthesis of antimalarial compound FR900098. Chem Biol 15(8):765–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Peck SC, van der Donk WA (2013) Phosphonate biosynthesis and catabolism: a treasure trove of unusual enzymology. Curr Opin Chem Biol 17(4):580–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Metcalf WW, van der Donk WA (2009) Biosynthesis of phosphonic and phosphinic acid natural products. Annu Rev Biochem 78:65–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Horsman GP, Zechel DL (2017) Phosphonate Biochemistry. Chem Rev 117(8):5704–5783. [DOI] [PubMed] [Google Scholar]
- 25.Sindt M, Stephan B, Schneider M, Mieloszynski JL (2001) CHEMICAL SHIFT PREDICTION OF 31 P-NMR SHIFTS FOR DIALKYL OR DIARYL PHOSPHONATES. Phosphorus Sulfur Silicon Relat Elem 174(1):163–175. [Google Scholar]
- 26.Hemmi K, Takeno H, Hashimoto M, Kamiya T (1982) Studies on phosphonic acid antibiotics. IV. Synthesis and antibacterial activity of analogs of 3-(N-acetyl-N-hydroxyamino)-propylphosphonic acid (FR-900098). Chem Pharm Bull (Tokyo) 30(1):111–8. [DOI] [PubMed] [Google Scholar]
- 27.Wang X, et al. (2018) MEPicides: α,β-Unsaturated Fosmidomycin Analogues as DXR Inhibitors against Malaria. J Med Chem 61(19):8847–8858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Waditee R, et al. (2003) Isolation and Functional Characterization of N-Methyltransferases That Catalyze Betaine Synthesis from Glycine in a Halotolerant Photosynthetic Organism Aphanothece halophytica. J Biol Chem 278(7):4932–4942. [DOI] [PubMed] [Google Scholar]
- 29.Martinez-Morales F, Schobert M, López-Lara IM, Geiger O (2003) Pathways for phosphatidylcholine biosynthesis in bacteria. Microbiology 149(12):3461–3471. [DOI] [PubMed] [Google Scholar]
- 30.Geiger O, López-Lara IM, Sohlenkamp C (2013) Phosphatidylcholine biosynthesis and function in bacteria. Biochim Biophys Acta - Mol Cell Biol Lipids 1831(3):503–513. [DOI] [PubMed] [Google Scholar]
- 31.Blodgett JAV, et al. (2007) Unusual transformations in the biosynthesis of the antibiotic phosphinothricin tripeptide. Nat Chem Biol 3(8):480–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Loenarz C, Schofield CJ (2011) Physiological and biochemical aspects of hydroxylations and demethylations catalyzed by human 2-oxoglutarate oxygenases. Trends Biochem Sci 36(1):7–18. [DOI] [PubMed] [Google Scholar]
- 33.Herr CQ, Hausinger RP (2018) Amazing Diversity in Biochemical Roles of Fe(II)/2-Oxoglutarate Oxygenases. Trends Biochem Sci 43(7):517–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Islam MS, Leissing TM, Chowdhury R, Hopkinson RJ, Schofield CJ (2018) 2-Oxoglutarate-Dependent Oxygenases. Annu Rev Biochem 87(1):585–620. [DOI] [PubMed] [Google Scholar]
- 35.Dong C, Zhang H, Xu C, Arrowsmith CH, Min J (2014) Structure and function of dioxygenases in histone demethylation and DNA/RNA demethylation. IUCrJ 1(6):540–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Valegård K, et al. (1998) Structure of a cephalosporin synthase. Nature 394(6695):805–809. [DOI] [PubMed] [Google Scholar]
- 37.Salowe SP, Marsh EN, Townsend CA (1990) Purification and characterization of clavaminate synthase from Streptomyces clavuligerus: an unusual oxidative enzyme in natural product biosynthesis. Biochemistry 29(27):6499–6508. [DOI] [PubMed] [Google Scholar]
- 38.Zhang Z, et al. (2002) Crystal structure of a clavaminate synthase-Fe(II)-2-oxoglutarate-substrate-NO complex: evidence for metal centered rearrangements. FEBS Lett 517(1–3):7–12. [DOI] [PubMed] [Google Scholar]
- 39.Rydzik AM, et al. (2014) Comparison of the substrate selectivity and biochemical properties of human and bacterial γ-butyrobetaine hydroxylase. Org Biomol Chem 12(33):6354–8. [DOI] [PubMed] [Google Scholar]
- 40.Henry L, Leung IKH, Claridge TDW, Schofield CJ (2012) γ-Butyrobetaine hydroxylase catalyses a Stevens type rearrangement. Bioorg Med Chem Lett 22(15):4975–4978. [DOI] [PubMed] [Google Scholar]
- 41.Leung IKH, et al. (2010) Structural and Mechanistic Studies on γ-Butyrobetaine Hydroxylase. Chem Biol 17(12):1316–1324. [DOI] [PubMed] [Google Scholar]
- 42.Kobayashi S, Kuzuyama T, Seto H (2000) Characterization of the fomA and fomB gene products from Streptomyces wedmorensis, which confer fosfomycin resistance on Escherichia coli. Antimicrob Agents Chemother 44(3):647–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim SY, et al. (2012) Different Biosynthetic Pathways to Fosfomycin in Pseudomonas syringae and Streptomyces Species. Antimicrob Agents Chemother 56(8):4175–4183. [DOI] [PMC free article] [PubMed] [Google Scholar]
METHODS REFERENCES
- 44.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215(3):403–410. [DOI] [PubMed] [Google Scholar]
- 45.Datsenko KA, Wanner BL (2000) One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci 97(12):6640–6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Patel JB, et al. (2015) Methods for Dilution Antimicrobial Susceptibilities Tests for Bacteria That Grow Aerobically; Approved Standard--Tenth Edition (Clinical and Laboratory Standards Institute, Wayne, PA: ). CLSI docum Available at: http://shop.clsi.org/site/Sample_pdf/M07A10_sample.pdf. [Google Scholar]
- 47.Blodgett JAV, Zhang JK, Metcalf WW (2005) Molecular Cloning, Sequence Analysis, and Heterologous Expression of the Phosphinothricin Tripeptide Biosynthetic Gene Cluster from Streptomyces viridochromogenes DSM 40736. Antimicrob Agents Chemother 49(1):230–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yu X, Price NPJ, Evans BS, Metcalf WW (2014) Purification and characterization of phosphonoglycans from Glycomyces sp. strain NRRL B-16210 and Stackebrandtia nassauensis NRRL B-16338. J Bacteriol 196(9):1768–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang S, Cui X, Fang G (2007) Rapid determination of formaldehyde and sulfur dioxide in food products and Chinese herbals. Food Chem 103(4):1487–1493. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that all relevant data supporting the findings of this study are available within the paper and its Supplementary Information.