

Abstract
All-carbon tetrasubstituted olefins have been found in numerous biologically important compounds and organic materials. However, regio- and stereocontrolled construction of this structural motif still constitutes a significant synthetic challenge. Here, we show that a modular and regioselective synthesis of all-carbon tetrasubstituted olefins can be realized via alkenyl halide- or triflate-mediated palladium/norbornene (Pd/NBE) catalysis, which is enabled by a modified NBE containing a C2 amide moiety. This new NBE co-catalyst effectively suppressed undesired cyclopropanation pathways, which have previously been a main obstacle for developing such reactions. Diverse cyclic and acyclic alkenyl bromides or triflates with a wide range of functional groups can be employed as substrates. Various substituents can be introduced at the alkene C1 and C2 positions regioselectively simply by changing the coupling partners. Initial mechanistic studies provide insights on the rate-limiting step as well as the structure of the actual active ligand in this system.
Graphical Abstract
All-carbon tetrasubstituted olefins are commonly found in natural products (Fig. 1), pharmaceuticals and organic materials, and often serve as the precursors for preparing highly substituted epoxides, aziridines and cyclopropanes1. However, synthesis of all-carbon tetrasubstituted olefins has constituted significant challenges, particularly in a regio- and stereocontrolled fashion1. Among various approaches, the one involving carbometallation of alkynes appears to be most efficient and modular, resulting from a rapid assembly of three common components2-11. However, controlling regioselectivity with electronically or sterically unbiased alkynes is nontrivial. In addition, the product scope is somewhat limited due to the accessibility of cyclic alkynes12. Hence, alternative, regioselective and general methods for preparing unsymmetrical all-carbon tetrasubstituted olefins remain highly sought after.
Figure 1∣. Representative Natural Products Containing All-carbon Tetrasubstituted Olefins.

All-carbon tetrasubstituted olefins are often found in natural products and other bioactive compounds. Regio- and stereoselective synthesis of this structural motif still poses a significant challenge.
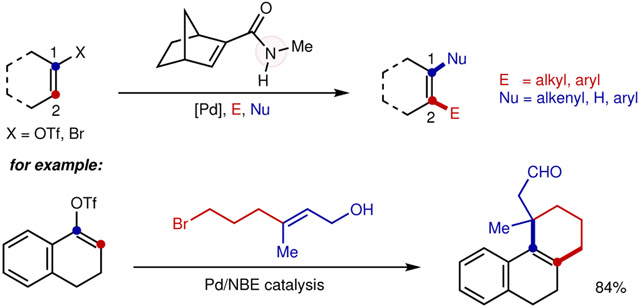
The palladium/norbornene (Pd/NBE) cooperative catalysis, originally discovered by Catellani13, offers streamlined synthesis of poly-substituted arenes14-19. Taking advantage of a unique aryl-norbornyl-palladacycle (ANP) intermediate, an electrophile could be introduced at the ortho position, while a nucleophile could be coupled at the ipso position, thus allowing vicinal difunctionalization of aryl halides (Fig. 2a)13,20-22. One would imagine that, the corresponding reaction with alkenyl halides as substrates would afford distinct access to all-carbon tetrasubstituted olefins in a regioselective manner. However, such an alkenyl Catellani reaction remains underexplored, and the corresponding alkenyl ANP intermediate has been elusive primarily owing to the high reactivity of the olefin π bond compared to a more stable aryl structure.
Figure 2∣. Alkenyl Catellani-type Reactions.

a, Classical Pd/NBE catalysis employs aryl halides as substrates through the aryl-norbornyl-palladacycle (ANP) intermediate. b, Prior works showed that reaction of alkenyl halides with NBE majorly led to cyclopropanation side-products without forming the analogous alkenyl ANP intermediate. c, Related examples with partially aromatic alkenyl halides could gave Catellani-type products; however, non-aromatic alkenyl halides are more challenging substrates. d, This work realizes the alkenyl Catellani reaction enabled by an amide-substituted NBE, which in turn leads to regioselective synthesis of all-carbon tetrasubstituted alkenes.
In the 1980s, Catellani and co-workers found that reactions of alkenyl bromides with NBE gave rise to two types of cyclopropanation products, through a 3-exo-trig reaction pathway (Fig. 2b)23. Recently, Van Vranken and co-workers further extended the reaction scope for such cyclopropanation reactions24. The 3-exo-trig reaction is likely to be kinetically facile, partially due to a favored geometry of the olefin complex towards migratory insertion. On the other hand, Lautens25 and later Yamamoto26 demonstrated that uracil and 2-quinolone-derived iodides successfully delivered the Catellani products, likely benefited by the partial aromaticity of these substrates (Fig. 2c). To the best of our knowledge, the Pd/NBE catalysis with non-aromatic, regular alkenyl halides has not been reported to date. Here, we describe our initial development of an alkenyl Catellani reaction that employs regular alkenyl bromides and triflates as substrates as a unique strategy for preparing unsymmetrical all-carbon tetrasubstituted olefins (Fig. 2d).
Compared to the aryl Catellani reactions, several new challenges can be envisaged for using alkenyl substrates (Fig. 2c): (1) the complete loss of aromaticity would greatly enhance the reactivity of the olefin π bond towards the undesired cyclopropanation reaction; (2) the presence of allylic β-Hs would make the cyclopropanation reaction irreversible through following β-H elimination; (3) the absence of ortho substituent may hinder the NBE de-insertion step27; (4) while alkenyl bromides or triflates28 are more available than the corresponding iodides, they are clearly more challenging substrates29.
The anticipated catalytic cycle indicates that two steps can potentially lead to cyclopropanation side-products (Fig. 2d). First, after NBE migratory insertion into the vinyl Pd species, if the C−H metalation to give the alkenyl-ANP (II) is slower than the alkyl migratory insertion into the olefin, a 3-exo-trig cyclopropanation would dominate (vide infra, Table 1). Second, after the C2 (ortho) functionalization with the electrophile (E−X), if the NBE extrusion via β-carbon elimination is slow, 3-exo-trig insertion could again take place to give the undesired side-products. Therefore, the key for the success of the reaction would be suppress the 3-exo-trig pathway and/or promote the C−H metalation and β-carbon elimination processes.
Table 1.
NBE Effect for the Alkenyl Catellani Reactiona
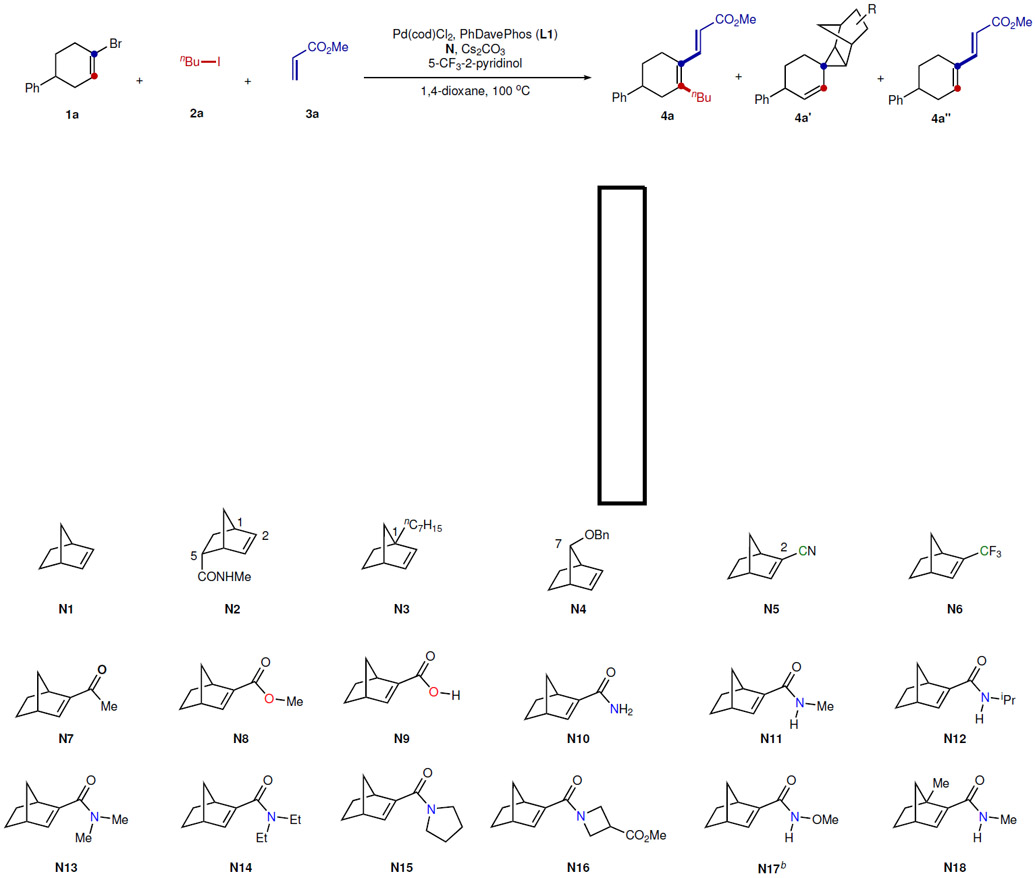 |
Reaction conditions: 1a (0.10 mmol), 2a (0.30 mmol), 3a (0.15 mmol), Pd(cod)Cl2 (0.01 mmol), L1 (0.01 mmol), N (0.15 mmol), 5-trifluomethyl-2-pyridinol (0.02 mmol), Cs2CO3 (0.30 mmol), 100 °C, 16 h. Yield determined by 1H NMR using 1,1,2,2-tetrachloroethane as the internal standard.
The conversion was 19%.
Results and discussion
We hypothesized that use of substituted NBEs may hamper the cyclopropane formation due to the increased bulkiness of the migrating alkyl group, and could simultaneously promote the β-carbon elimination27. To test this hypothesis, ortho alkylation/ipso Heck reaction13,20,30,31 of alkenyl bromide 1a was chosen as the model reaction and a range of substituted NBEs were employed as the cofactor (Table 1). Not surprisingly, simple NBE (N1) or remotely substituted NBE (N2)32 only gave cyclopropanation product 4a’. C1 and C7-substituted NBEs (N3 and N4) inhibited cyclopropanation to some extent, but still provided no desired product. It is likely that these substituents also hampered forming the alkenyl-ANP intermediate. The C2-substiutted NBEs (N5-N18) were found more effective on suppressing cyclopropane side products. While NBEs having cyano (N5), trifluoromethyl (N6), and methyl ketone (N7) groups at 2-positions were not reactive, majorly affording direct Heck product 4a’’, methyl ester-substituted NBE (N8), pioneered by the Yu group33, gave the desired product 4a in 23% yield along with 16% cyclopropane product 4a’. In addition, the free carboxylic acid-derived NBE (N9) shows similar reactivity as N8. It was surprising that primary amide-substituted NBE (N10) exhibited remarkable selectivity and reactivity to provide the desired tetrasubstituted olefin in 54% yield with almost no cyclopropane formation. Further modification on N10 showed that N-methyl amide-substituted NBE (N11) was most efficient with 74% yield. Further increasing the sterics on the amide moiety (N12) reduced the reactivity, therefore giving more direct Heck product 4a’’. The trend was clearly observed for the N,N-dialkylamide-substituted NBEs (N13−N16): the less sterically hindered azetidine-derived NBE showed better reactivity. N-Methoxy amide-substituted NBE (N17) gave a low conversion, though the reason is unclear. Unsurprisingly, an additional methyl substituent at the bridgehead position (N18) reduced the reactivity.
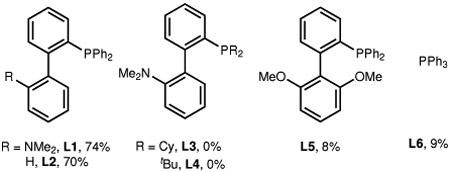
The reaction was further investigated with alkenyl triflate 5a as the substrate, given the ease of preparing vinyl triflates from simple ketones. Under the “standard” conditions (entry 1, Table 2), 74% yield of the desired product (4a) was obtained using 50 mol% of N11, 10 mol% of Pd(cod)Cl2 and 10 mol% of Buchwald’s Ph-DavePhos (L1)34. A relatively high loading of N11 was used to suppress direct ipso functionalization19. A series of control experiments were subsequently conducted to understand the role of each reactant. In the absence of the palladium, phosphine ligand or N11, no desired product was formed (entries 2-4). Pd(cod)Cl2 was found to be slightly more efficient than Pd(OAc)2 (entry 5). A survey of ligands showed that Ph-DavePhos (L1) and Ph-JohnPhos (L2) were the best choices (entry 6), while the use of more electron-rich DavePhos (L3) or tBu-DavePhos (L4) shut down the reaction (for mechanistic studies, vide infra). The 2,6-dimethoxy analogue of L1 and PPh3 (L5 and L6) were much less effective. The use of potassium carbonate as the base dramatically decreased the yield compared to cesium carbonate (entry 7). In addition, 5-trifluoromethyl-2-pyridinol (20 mol%) was previously discovered by Yu35,36 to be an excellent co-catalyst to promote concerted metalation deprotonation (CMD); use of pivalic acid instead gave a low yield (entry 8). Besides 1,4-dioxane, toluene is also a suitable solvent (entry 9). Finally, a lower reaction temperature slightly decreased the yield (entry 10).
Table 2.
Control Experiments
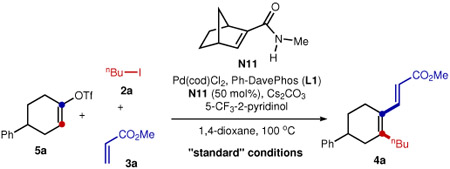 | ||
|---|---|---|
| Entry | Variations from the "standard" conditions | Yield of 4a (%)a |
| 1 | None | 74 |
| 2 | No Pd(cod)Cl2 | 0 |
| 3 | No Ph-DavePhos | 0 |
| 4 | No N11 | 0 |
| 5 | Pd(OAc)2 instead of Pd(cod)Cl2 | 70 |
| 6 | L2-L6 instead of L1 | Listed below |
| 7 | K2CO3 instead of Cs2CO3 | 12 |
| 8 | PivOH instead of 5-CF3-2-pyridinol | 12 |
| 9 | Toluene instead of 1,4-dioxane | 74 |
| 10 | 85 °C instead of 100 °C | 62 |
 | ||
Reaction conditions: 5a (0.10 mmol), 2a (0.30 mmol), 3a (0.15 mmol), Pd(cod)Cl2 (0.01 mmol), L1 (0.01 mmol), N11 (0.05 mmol), 5-trifluomethyl-2-pyridinol (0.02 mmol), Cs2CO3 (0.30 mmol), 100 °C, 16 h.
Yield determined by 1H NMR using 1,1,2,2-tetrachloroethane as the internal standard.
With the optimized conditions in hand, the substrate scope was studied (Table 3). Besides methyl acrylate, other Michael acceptors, such as tert-butyl acrylate, N,N-dimethylacrylamide, ethylvinyl ketone, and N-tert-butylacrylamide, can all be smoothly coupled at the ipso-position (4b−4e). Less electron-deficient olefins, such as 2-vinylpyridine (4f) and regular styrene (4g), can also be employed. Regarding the scope of the alkyl electrophiles, alkyl iodides with various functional groups, such as silyl protected alcohol (4i), alkyl nitrile (4j), alkyl chloride (4k), and methyl ester (4m), were suitable. The bulkier isobutyl iodide gave a lowered yield (4n), while secondary alkyl iodides were unreactive, likely due to the steric congestion of the alkenyl-ANP species. Gratifyingly, the use of phenyltrimethylammonium salt (2o’) was found to serve as a mild electrophile for delivering the methylation product (4o)37. To the best of our knowledge, ammonium salts have not been used as electrophiles in Catellani-type reactions before.
Table 3.
Reaction scope
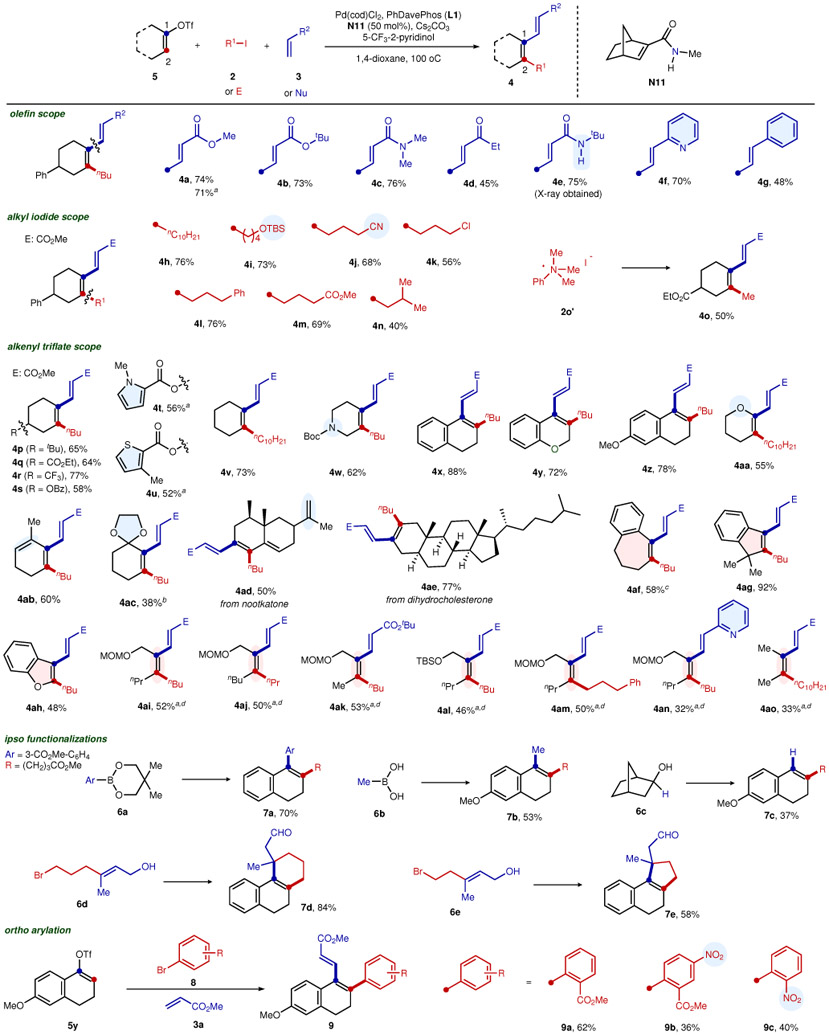 |
*Reaction conditions: 5 (0.30 mmol), 2 (0.90 mmol), 3 (0.45 mmol), Pd(cod)Cl2 (0.03 mmol), L1 (0.03 mmol), N11 (0.15 mmol), 5-trifluomethyl-2-pyridinol (0.06 mmol), Cs2CO3 (0.90 mmol), 1,4-dioxane (6 mL), 100 °C, 16 h.
The corresponding alkenyl bromides were used instead of 5.
The corresponding alkenyl iodide was used instead of 5.
L2 (0.03 mmol) was used instead of L1.
N11 (0.30 mmol) was used, K3PO4 (0.90 mmol) was used instead of Cs2CO3, and a mixed solvent of 1,4-dioxane (3 mL) and toluene (3 mL) was used instead of 1,4-dioxane alone.
The scope with respect to different alkenyl triflates or bromides was examined next. Good functional group compatibility was observed with tolerance of ester (4q), trifluoromethyl (4r), benzoyl-protected alcohol (4s), N-methyl pyrrole (4t), thiophene (4u), Boc-protected amine (4w), ketal (4ac) and regular olefin (4ad). The benzofused vinyl triflates gave higher efficiencies (4x−4z), probably due to an easier NBE extrusion in the presence of ortho substituents. Vinyl triflates directly derived from a lactone (4aa) or an enone (4ab) could also be employed. In addition, vinyl triflates derived from ketone-containing natural products, such as nootkatone and dihydrocholestrone, delivered the desired products (4ad and 4ae) in good to moderate yields. Besides six-membered ring substrates, preliminary successes have been obtained with other types of alkenyl substrates. Seven-membered (4af) and five-membered alkenyl triflates (4ag and 4ah) proved to be competent for this transformation. With slightly modified reaction conditions, acyclic alkenyl bromides also successfully delivered the desired products (4ai−4ao).
To demonstrate the generality of this reaction, different couplings at the C1 (ipso) position were then investigated. In addition to the Heck termination, Suzuki coupling38 (7a and 7b) and hydrogenation39-42 (7c) can also be used to install an aryl group, a methyl group, or hydrogen at the C1 position. Besides intermolecular couplings, an intramolecular ortho alkylation/redox-Heck annulation was achieved43,44, which provides a rapid synthesis of tricycles containing an all-carbon tetrasubstituted olefin and an adjacent quaternary stereocenter (7d and 7e). Furthermore, beyond alkylation at the vicinal C2 position, this catalytic system was also effective for C2-arylation using aryl bromides21,22 as the external electrophile (9a-9c).
The preliminary mechanistic study started with measuring the kinetic profiles of the reaction with substrate 5a. The initial-rate method was employed to determine the reaction order of each component (Fig. 3a). Not surprisingly, the dependence of the initial rate on the concentration of [Pd/L1] was found to be first-order. Moreover, the rate of reaction shows zero-order dependences on [5a], [2a], [3a] and [N11], indicating that oxidative addition of aryl triflate 5a, migratory insertion into N11, the reaction between alkenyl-ANP and electrophile 2a, and migratory insertion into acrylate 3a are not the turnover-limiting step. Interestingly, the turnover rate for the formation of side-product 4a’’ increases with lower loadings of N11, indicating that migratory insertion into N11 is a pre-equilibrium before the turnover-limiting step (see Supplementary Fig. 13 for a detailed discussion). In addition, the kinetic isotopic effect (KIE) was found to be 1.5 using two parallel reactions (Fig. 3b); while a competition KIE study employing a mixture of 5a and 5a-d3 revealed a KIE of 1.6. Taken together, these values suggest that the C–H cleavage step is partially turnover-limiting and that another elementary step either before or after the C–H cleavage (e.g., subsequent ligand exchange step) also contributes to the catalytic turnover rate.
Figure 3∣. Mechanistic studies and synthetic utility.

a, Determination of the reaction order: zero-order kinetics for 5a, 2a, 3a and N11, and first-order kinetics for [Pd/L1] were observed, indicating that oxidative addition of 5a, migratory insertion into N11, the reaction with 2a, and migratory insertion into 3a are not the turnover-limiting step. b, The parallel and competition kinetic isotopic effects (KIE) were measured, indicating that the C–H cleavage step is only partially turnover-limiting. c, The observation of cyclized phosphafluorene oxide 10 and by-product 11, together with the parallel kinetic study between L2 and L7, indicate that the actual ligand in this system is likely the corresponding phosphafluorene. d. Synthesis of tricyclic compound 14 is illustrated using this method, which uses fewer steps than the prior route.
As an interesting observation during our investigation, cyclized phosphafluorene oxides (10) were isolated from the reaction mixture when using Ph-DavePhos (L1) and Ph-JohnPhos (L2) (Fig. 3c). Presumably, a sequential C−H and C−P bond activation would transform, i.e., L2 into phosphafluorene L7, along with the formation of by-product 11 (see Supplementary Fig. 16 for the proposed mechanism)45. To examine whether phosphafluorene L7 was the “real” ligand (instead of L2) in the alkenyl Catellani reaction, a parallel kinetic study was conducted. First, the reaction with L2 exhibited a notable induction period, while the one with cyclic phosphine L7 did not. Second, the reaction with L7 had an almost identical initial reaction rate to that with L2. Altogether, these results indicated that L7 was likely the actual ligand in this system and, during the induction period, L1 or L2 was transformed into cyclized phosphafluorenes. These phosphafluorene ligands are less bulky, less σ-donating and more π-accepting than PPh346, which could be beneficial to generate a more π-acidic Pd species thereby promoting the alkenyl-ANP formation (see Supplementary Table 10 for a survey of ligand effect). This finding may also explain why the structurally similar ligand L5 was significantly less effective due to the inability of forming cyclic phosphines. Finally, to show utility of this method, a tricyclic compound 14, previously synthesized in 7 steps47, can now be accessed in a concise manner from the known vinyl triflate 6y (prepared in one step from commercially available chemicals) (Fig. 3d).
In summary, a new approach for regioselective preparation of all-carbon tetrasubstituted olefins from readily available alkenyl bromides/triflates is realized, which is enabled by Pd-catalyzed alkenyl Catellani reactions. A class of amide-substituted NBEs has been identified and plays a pivotal role in preventing undesired cyclopropanation pathways. The first use of tetraalkylammonium salts as electrophiles and phosphafluorenes as ligands in Pd/NBE catalysis may have further implications. The broad functional group tolerance could make the reaction attractive for complex molecule synthesis. Future efforts will focus on understanding the unique function of the amine moiety in N11 and expanding the reaction scope to more acyclic alkenyl halides and other types of C1 and C2 functionalizations.
Supplementary Material
Acknowledgements
Financial supports from the University of Chicago and NIGMS (1R01GM124414-01A1) are acknowledged. Mr. Ki-Young Yoon is thanked for X-ray crystallography.
Footnotes
Data availability. The data supporting the findings of this study are available within the paper and its Supplementary Information. Crystallographic data for compound 4e have been deposited at the Cambridge Crystallographic Data Centre (CCDC) under deposition no. CCDC 1908383. These data can be obtained free of charge from the CCDC (http://www.ccdc.cam.ac.uk/data_request/cif).
Competing interests
The authors declare no competing interests.
Supplementary information and chemical compound information are available in the online version of the paper.
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- (1).Flynn AB & Ogilvie WW Stereocontrolled Synthesis of Tetrasubstituted Olefins. Chem. Rev 107, 4698–4745 (2007). [DOI] [PubMed] [Google Scholar]
- (2).Normant JF & Alexakis A Carbometallation (C-Metallation) of Alkynes - Stereospecific Synthesis of Alkenyl Derivatives. Synthesis 841–870 (1981). [Google Scholar]
- (3).Doyle MP In Comprehensive Organometallic Chemistry II; Abel EW, Stone FGA, Wilkinson G, Hegedus L, Eds.; Pergamon Press: Oxford, 1995; Vol. 12, pp 387–420. [Google Scholar]
- (4).Muller DS & Marek I Copper Mediated Carbometalation Reactions. Chem. Soc. Rev 45, 4552–4566 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Negishi E, Zhang Y, Cederbaum FE & Webb MB A Selective Method for the Synthesis of Stereodefined Exocyclic Alkenes Via Allylmetalation of Propargyl Alcohols. J. Org. Chem 51, 4080–4082 (1986). [Google Scholar]
- (6).Itami K, Kamei T & Yoshida J Diversity-Oriented Synthesis of Tamoxifen-type Tetrasubstituted Olefins. J. Am. Chem. Soc 125, 14670–14671 (2003). [DOI] [PubMed] [Google Scholar]
- (7).Zhou CX & Larock RC Regio- and Stereoselective Route to Tetrasubstituted Olefins by the Palladium-Catalyzed Three-Component Coupling of Aryl Iodides, Internal Alkynes, and Arylboronic Acids. J. Org. Chem 70, 3765–3777 (2005). [DOI] [PubMed] [Google Scholar]
- (8).Zhang D-H & Ready JM Iron-catalyzed Carbometalation of Propargylic and Homopropargylic Alcohols. J. Am. Chem. Soc 128, 15050–15051 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Gericke KM, Chai DI, Bieler N & Lautens M The Norbornene Shuttle: Multicomponent Domino Synthesis of Tetrasubstituted Helical Alkenes through Multiple C−H Functionalization. Angew. Chem. Int. Ed 48, 1447–1451 (2009). [DOI] [PubMed] [Google Scholar]
- (10).Zhou Y-Q, You W, Smith KB & Brown MK Copper-Catalyzed Cross- Coupling of Boronic Esters with Aryl Iodides and Application to the Carboboration of Alkynes and Allenes. Angew. Chem. Int. Ed 53, 3475–3479 (2014). [DOI] [PubMed] [Google Scholar]
- (11).Xue F, Zhao J, Hor TSA & Hayashi T Nickel-Catalyzed Three-Component Domino Reactions of Aryl Grignard Reagents, Alkynes, and Aryl Halides Producing Tetrasubstituted Alkenes. J. Am. Chem. Soc 137, 3189–3192 (2015). [DOI] [PubMed] [Google Scholar]
- (12).Gampe CM & Carreira EM Arynes and Cyclohexyne in Natural Product Synthesis. Angew. Chem. Int. Ed 51, 3766–3778 (2012). [DOI] [PubMed] [Google Scholar]
- (13).Catellani M, Frignani F & Rangoni A A Complex Catalytic Cycle Leading to a Regioselective Synthesis of o,o'-Disubstituted Vinylarenes. Angew. Chem. Int. Ed. Engl 36, 119–122 (1997). [Google Scholar]
- (14).Catellani M, Motti E & Della Ca' N Catalytic Sequential Reactions Involving Palladacycle-Directed Aryl Coupling Steps. Acc. Chem. Res 41, 1512–1522 (2008). [DOI] [PubMed] [Google Scholar]
- (15).Martins A, Mariampillai B & Lautens M Synthesis in the Key of Catellani: Norbornene-Mediated ortho C−H Functionalization. Top. Curr. Chem 292, 1–33 (2010). [DOI] [PubMed] [Google Scholar]
- (16).Ye J & Lautens M Palladium-Catalysed Norbornene-Mediated C−H Functionalization of Arenes. Nat. Chem 7, 863–870 (2015). [DOI] [PubMed] [Google Scholar]
- (17).Della Ca' N, Fontana M, Motti E & Catellani M Pd/Norbornene: A Winning Combination for Selective Aromatic Functionalization via C−H Bond Activation. Acc. Chem. Res 49, 1389–1400 (2016). [DOI] [PubMed] [Google Scholar]
- (18).Liu Z-S, Gao Q, Cheng H-G & Zhou Q The Alkylating Reagents Employed in Catellani-Type Reactions. Chem. Eur. J 24, 15461–15476 (2018). [DOI] [PubMed] [Google Scholar]
- (19).Wang J & Dong G Palladium/Norbornene Cooperative Catalysis. Chem. Rev 119, 7478–7528 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Lautens M & Piguel S A New Route to Fused Aromatic Compounds by Using a Palladium-Catalyzed Alkylation-Alkenylation Sequence. Angew. Chem. Int. Ed 39, 1045–1046 (2000). [DOI] [PubMed] [Google Scholar]
- (21).Catellani M, Motti E & Baratta S A Novel Palladium-Catalyzed Synthesis of Phenanthrenes from ortho-Substituted Aryl Iodides and Diphenyl- or Alkylphenylacetylenes. Org. Lett 3, 3611–3614 (2001). [DOI] [PubMed] [Google Scholar]
- (22).Faccini F, Motti E & Catellani M A New Reaction Sequence Involving Palladium-Catalyzed Unsymmetrical Aryl Coupling. J. Am. Chem. Soc 126, 78–79 (2004). [DOI] [PubMed] [Google Scholar]
- (23).Catellani M & Chiusoli GP Competitive Processes in Palladium-Catalyzed C−C Bond Formation. J. Organomet. Chem 233, C21–C24 (1982). [Google Scholar]
- (24).Khanna A, Premachandra IDUA, Sung PD & Van Vranken DL Palladium-Catalyzed Catellani Aminocyclopropanation Reactions with Vinyl Halides. Org. Lett 15, 3158–3161 (2013). [DOI] [PubMed] [Google Scholar]
- (25).Blaszykowski C, Aktoudianakis E, Bressy C, Alberico D & Lautens M Preparation of Annulated Nitrogen-Containing Heterocycles via a One-Pot Palladium-Catalyzed Alkylation/Direct Arylation Sequence. Org. Lett 8, 2043–2045 (2006). [DOI] [PubMed] [Google Scholar]
- (26).Yamamoto Y, Murayama T, Jiang J, Yasui T & Shibuya M The Vinylogous Catellani Reaction: A Combined Computational and Experimental Study. Chem. Sci 9, 1191–1199 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Wang J, Li R, Dong Z, Liu P & Dong G Complementary Site-Selectivity in Arene Functionalization Enabled by Overcoming the ortho Constraint in Palladium/Norbornene Catalysis. Nat. Chem 10, 866–872 (2018). [DOI] [PubMed] [Google Scholar]
- (28).Blanchot M, Candito DA, Larnaud F & Lautens M Formal Synthesis of Nitidine and NK109 via Palladium-Catalyzed Domino Direct Arylation/N-Arylation of Aryl Triflates. Org. Lett 13, 1486–1489 (2011). [DOI] [PubMed] [Google Scholar]
- (29).Dong Z, Lu G, Wang J, Liu P & Dong G Modular ipso/ortho Difunctionalization of Aryl Bromides via Palladium/Norbornene Cooperative Catalysis. J. Am. Chem. Soc 140, 8551–8562 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Zhang H, Chen P & Liu G Palladium-Catalyzed Cascade C−H Trifluoroethylation of Aryl Iodides and Heck Reaction: Efficient Synthesis of ortho-Trifluoroethylstyrenes. Angew. Chem. Int. Ed 53, 10174–10178 (2014). [DOI] [PubMed] [Google Scholar]
- (31).Qureshi Z, Schlundt W & Lautens M Introduction of Hindered Electrophiles via C−H Functionalization in a Palladium-Catalyzed Multicomponent Domino Reaction. Synthesis 47, 2446–2456 (2015). [Google Scholar]
- (32).Dong Z, Wang J, Ren Z & Dong G Ortho C−H Acylation of Aryl Iodides by Palladium/Norbornene Catalysis. Angew. Chem. Int. Ed 54, 12664–12668 (2015). [DOI] [PubMed] [Google Scholar]
- (33).Shen P-X, Wang X-C, Wang P, Zhu R-Y & Yu J-Q Ligand-Enabled meta-C−H Alkylation and Arylation using a Modified Norbornene. J. Am. Chem. Soc 137, 11574–11577 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Surry DS & Buchwald SL Biaryl Phosphane Ligands in Palladium-Catalyzed Amination. Angew. Chem. Int. Ed 47, 6338–6361 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Wang P et al. Ligand-Promoted meta-C−H Arylation of Anilines, Phenols, and Heterocycles. J. Am. Chem. Soc 138, 9269–9276 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Wang P et al. Ligand-Accelerated Non-directed C–H Functionalization of Arenes. Nature 551, 489 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Uemura T, Yamaguchi M & Chatani N Phenyltrimethylammonium Salts as Methylation Reagents in the Nickel-Catalyzed Methylation of C−H Bonds. Angew. Chem. Int. Ed 55, 3162–3165 (2016). [DOI] [PubMed] [Google Scholar]
- (38).Catellani M, Motti E & Minari M Symmetrical and Unsymmetrical 2,6-Dialkyl-1,1'-biaryls by Combined Catalysis of Aromatic Alkylation via Palladacycles and Suzuki-Type Coupling. Chem. Commun 0, 157–158 (2000). [Google Scholar]
- (39).Catellani M & Fagnola MC Palladacycles as Intermediates for Selective Dialkylation of Arenes and Subsequent Fragmentation. Angew. Chem. Int. Ed 33, 2421–2422 (1994). [Google Scholar]
- (40).Wilhelm T & Lautens M Palladium-Catalyzed Alkylation-Hydride Reduction Sequence: Synthesis of meta-Substituted Arenes. Org. Lett 7, 4053–4056 (2005). [DOI] [PubMed] [Google Scholar]
- (41).Martins A & Lautens M Aromatic ortho-Benzylation Reveals an Unexpected Reductant. Org. Lett 10, 5095–5097 (2008). [DOI] [PubMed] [Google Scholar]
- (42).Deledda S, Motti E & Catellani M Palladium-Catalysed Synthesis of Nonsymmetrically Disubstituted-1,1'-Biphenyls from o-Substituted Aryl Iodides through Aryl Coupling and Delayed Hydrogenolysis. Can. J. Chem 83, 741–747 (2005). [Google Scholar]
- (43).Catellani M et al. A New Catalytic Method for the Synthesis of Selectively Substituted Biphenyls Containing an Oxoalkyl Chain. J. Organomet. Chem 687, 473–482 (2003). [Google Scholar]
- (44).Liu Z-S et al. Palladium/Norbornene Cooperative Catalysis To Access Tetrahydronaphthalenes and Indanes with a Quaternary Center. ACS Catal. 8, 4783–4788 (2018). [Google Scholar]
- (45).Baba K, Tobisu M & Chatani N Palladium-Catalyzed Direct Synthesis of Phosphole Derivatives from Triarylphosphines through Cleavage of Carbon−Hydrogen and Carbon−Phosphorus Bonds. Angew. Chem. Int. Ed 52, 11892–11895 (2013). [DOI] [PubMed] [Google Scholar]
- (46).Fourmy K, Nguyen DH, Dechy-Cabaret O & Gouygou M Phosphole-Based Ligands in Catalysis. Catal. Sci. Technol 5, 4289–4323 (2015). [Google Scholar]
- (47).Trost BM & Murayama E An Approach to the Phenanthrene Nucleus Via Thionium Ions and Epoxyketone Cyclizations. Tetrahedron Lett. 23, 1047–1050 (1982). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.