

Abstract
Membrane contact sites (MCSs) are specialized subcellular compartments formed by closely apposed membranes from two organelles. The intermembrane gap is separated by a distance ranging from 10 to 35 nm. MCSs are typically maintained through dynamic protein–protein and protein–lipid interactions. These intermembrane contact sites constitute important intracellular signalling hotspots to mediate a plethora of cellular processes, including calcium homeostasis, lipid metabolism, membrane biogenesis and organelle remodelling. In recent years, a series of genetically encoded probes and chemogenetic or optogenetic actuators have been invented to aid the visualization and interrogation of MCSs in both fixed and living cells. These molecular tools have greatly accelerated the pace of mechanistic dissection of membrane contact sites at the molecular level. In this review, we present an overview on the latest progress in this endeavour, and provide a general guide to the selection of methods and molecular tools for probing interorganellar membrane contact sites.
Keywords: Membrane contact sites, MCSs visualization, MCSs manipulation, Calcium homeostasis, lipid metabolism, organelle remodeling, fluorescence, optogenetics
Schematic diagram showing the interorganellar membrane contact sites (MCSs), which are intimately involved in the regulation of lipid and ion exchange, organelle biogenesis and subcellular positioning. Innovative methods and genetically encoded molecular tools have been developed to aid the visualization of MCSs and non‐invasive manipulation of interorganellar communication with high spatiotemporal precision.
Introduction
Membrane contact sites (MCSs) are defined as specialized subcellular zones where membranes of two different organelles come into close apposition. Typically, the gap between the two membranes ranges from 10 to 35 nm and there is no direct membrane fusion. The first hint of MCSs can be dated back to electron microscopic studies performed on muscle cells in the 1950s (Porter, 1953; Porter & Palade, 1957), in which close contacts between the endoplasmic/sarcoplasmic reticulum (ER/SR) and plasma membrane (PM) invaginations were noted and later characterized as skeletal muscle triads in the 1980s (Kawamoto et al. 1986). The ER in eukaryotic cells forms an extensive and highly dynamic elastic intracellular membrane network. The ER facilitates the frequent contact and communication with various subcellular membranous structures (Fig. 1 A), such as the PM and the membranes of the vacuole, endosome, lysosome, peroxisome, mitochondrion, lipid droplet and Golgi apparatus (Wu et al. 2018). Well‐characterized MCSs are also found at the outer membrane interfaces between peroxisome and mitochondrion (Shai et al. 2018), peroxisome and lysosome (Chu et al. 2015), as well as mitochondrion and endosome (Das et al. 2016). MCSs form crucial signalling platforms to coordinate a plethora of physiological processes, including calcium (Ca2+) signalling, lipid metabolism, organelle biogenesis and subcellular trafficking (Carrasco & Meyer, 2011; Rowland & Voeltz, 2012; Phillips & Voeltz, 2016; Wu et al. 2018).
Figure 1. Intracellular membrane contact sites (MCSs).
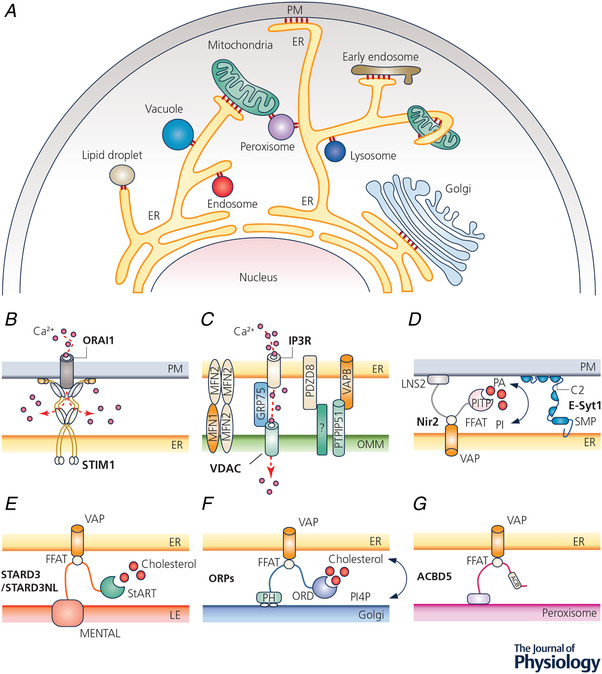
A, diagram depicting MCSs formed between intracellular organelles (red circles). B–G, examples of MCSs and their tentative molecular compositions. B, STIM1–ORAI1 coupling that mediates store‐operated calcium entry at ER–PM junctions; C, the makeup of the ER–mitochondria junctions. IP3R, VDAC, VAPB, PTPIP51, MFN1/2 and PDZD8 are essential for mitochondrial Ca2+ uptake and/or the formation of ER–OMM contacts. D–G, lipid transfer by LTPs at MCSs between ER and other organelles. LTPs can establish intermembrane tethering via PH domains, C2 domains, or FFAT‐motif‐dependent interactions with VAP. Abbreviations: ACB, acyl‐coenzyme A binding domain; ACBD5, acyl‐coenzyme A binding domain protein 5; ER, endoplasmic reticulum; E‐syt1, extended synaptotagmin‐1; FFAT, phenylalanine–phenylalanine–acid tract; GRP75, 75 kDa glucose‐regulated proteins; IP3R, inositol 1,4,5‐trisphosphate receptor; LTP, lipid transfer protein; MFN, mitofusin; MEMTAL, MLN64 N‐terminal; Nir2, also called PITPnm1, membrane‐associated phosphatidylinositol transfer protein 1; OMM, outer mitochondrial membrane; ORD, oxysterol binding protein related domain; PA, phosphatidic acid; PI, phosphatidylinositol; PDZD8, PDZ domain‐containing protein 8; PITP, phosphatidylinositol transfer protein; PH, pleckstrin homology; PM, plasma membrane; PTPIP51, protein tyrosine phosphatase interacting protein 51; STARD3, StAR related lipid transfer domain‐3; STARD3NL, STARD3 N‐terminal like; SMP, synaptotagmin‐like mitochondrial lipid binding protein; STIM1, stromal interaction molecule 1; VAP, vesicle‐associated membrane protein (VAMP)‐associated protein; VDAC, voltage‐dependent anion selective channel.
MCSs as signalling hotspots for Ca2+ homeostasis and lipid metabolism
Ca2+ is a versatile intracellular second messenger that regulates a vast array of biological activities, including short‐term processes such as muscle contraction and neurotransmitter release, as well as long‐term cell metabolism and programmed cell death (Zhou et al. 2013; Trebak & Kinet, 2019). In non‐excitable cells, store‐operated Ca2+ entry (SOCE) constitutes a primary Ca2+ entry route that uniquely connects the internal Ca2+ store with Ca2+ channels in the plasma membrane. As a prototypical example of SOCE, the calcium release‐activated channel (CRAC) in cells of the immune system is formed by the dynamic coupling between stromal interaction molecules (STIM1 and STIM2) and ORAI Ca2+ channels (ORAI1, ORAI2 and ORAI3) (Hogan et al. 2010; Soboloff et al. 2012; Prakriya & Lewis, 2015; Nguyen, Han, et al. 2018). Upon store depletion, STIM1 and ORAI1 accumulate at ER–PM MCSs to form the elementary unit of SOCE (Fig. 1 B). STIM1 is an ER‐localized protein, containing a Ca2+‐binding EF‐hand motif in its ER luminal domain and an ORAI‐activating domain in its cytoplasmic region (Liou et al. 2005; Roos et al. 2005; Zhang et al. 2005; Luik et al. 2006; Kawasaki et al. 2009; Park et al. 2009; Yuan et al. 2009), whereas ORAI1 serves as a highly Ca2+ selective ion channel in the PM (Feske et al. 2006; Vig et al. 2006). When the ER Ca2+ store is full at rest, STIM1 and ORAI1 are evenly distributed across the ER and PM, respectively. Following Ca2+ depletion within the ER lumen, STIM1 is activated by the release of Ca2+ from the luminal EF‐hand, which triggers a series of conformational changes to enable its oligomerization and translocation toward the PM. Activated STIM1 directly engages and gates ORAI1 Ca2+ channel (Park et al. 2009; Yuan et al. 2009; Zhou et al. 2010). STIM1 and ORAI1 can thus redistribute within their respective membranes and co‐accumulate in clusters at ER–PM contact sites to mediate Ca2+ entry. In addition to STIM–ORAI coupling at ER–PM MCSs, contacts between the ER/SR and the mitochondria provide a site for inositol 1,4,5‐trisphosphate receptor (IP3R)/ryanodine receptor (RyR)‐mediated Ca2+ oscillations and an avenue for localized Ca2+ microdomain (Szalai et al. 2000; De Stefani et al. 2012; Rizzuto et al. 2012). MCSs between the ER and mitochondria are essential for rapid and sustained Ca2+ movement from the ER lumen to mitochondria (Rizzuto et al. 1998; Csordas et al. 2006). Many proteins participate in the formation and maintenance of ER–mitochondria MCSs (Fig. 1 C). Notable examples include (i) the 75 kDa glucose‐regulated protein (GRP75), which mediates the interaction between IP3R and mitochondrion‐anchored voltage‐dependent anion selective channel 1 (VDAC1) (Szabadkai et al. 2006); (ii) mitofusin 2 (MFN2), which mediates homotypic interactions and heterotypic interactions with MFN1 (de Brito & Scorrano, 2008); and (iii) PDZ domain‐containing protein 8 (PDZD8), which is a recently discovered scaffold to facilitate the assembly of ER–mitochondria MCSs (de Brito & Scorrano, 2008; Hirabayashi et al. 2017).
Lipids are major components of cellular membranes. Maintaining the proper lipid distribution of the membranes is critical for numerous cellular processes. Hydrophobic lipids form permeability barriers of cells and subcellular organelles in the form of lipid bilayers, which are essential for the embedding of proteins in the membrane as receptors, transporters and enzymes (Spector & Yorek, 1985). Meanwhile, biochemical reactions for lipid synthesis and degradation are often responsible for initiating or terminating signalling cascades of intracellular organelles. Many types of lipids can only be synthesized in the ER and are subsequently delivered to other membranes via vesicular or non‐vesicular transport mechanisms at MCSs (Levine, 2004). Phosphatidylinositol (PI), for instance, is synthesized in the ER, where phosphatidic acid (PA) acts as a precursor for PI. The transport of PI from the ER to the PM could be mediated by vesicular transport or via phosphatase‐interacting proteins (PTPIs). In the PM, PI is further phosphorylated to yield PI(4)P and PI(4,5)P2, the latter of which can be hydrolysed to generate two important second messengers, IP3 and diacylglycerol (DAG). DAG is subsequently converted to PA, which serves as a precursor for the synthesis of many phospholipids in the ER, including PI. The transfer of PA from the PM to the ER is believed to be primarily mediated by Nir2, a key protein that is essential for the maintenance of the ‘PI‐cycle’ via non‐vesicular PA/PI exchange at ER–PM contact sites (Chang & Liou, 2015; Kim et al. 2015, 2016; Muallem et al. 2017). Therefore, interorganellar contact sites are regarded as crucial sites for non‐vesicular trafficking of lipids from the synthesis sites to their functional destinations (van Meer & Sprong, 2004; Helle et al. 2013). Lipid transport proteins (LTPs) play important roles during this process by providing hydrophobic pockets to shield the bound lipids from the aqueous phase and thus guarantee the proper trafficking (Lev, 2010). In addition to the core lipid binding domains, most LTPs also harbour pleckstrin homology (PH) domains, or C2 domains, or phenylalanine–phenylalanine–acid tract (FFAT) motifs to mediate the interaction with the second organellar membrane (Lahiri et al. 2015) (Fig. 1 D–G). Moreover, other physiological processes can occur at MCSs. For instance, lipid rafts localized at ER–mitochondria MCSs can facilitate the formation of autophagosome (Garofalo et al. 2016). MCSs are also involved in organelle division, such as mitochondrial and endosomal fission. The contact sites where ER tubules wrap around the mitochondria or endosomes often define the fission positions, which is crucial for maintaining proper turnover of mitochondria and endosome (Phillips & Voeltz, 2016; Wu et al. 2018).
Dysregulation of MCS assembly and maintenance has been linked to several human diseases. Recent studies have suggested that defective assembly of ER–mitochondria contact sites might be associated with neurodegenerative diseases, including Alzheimer's disease, Parkinson's disease and amyotrophic lateral sclerosis (Paillusson et al. 2016). Dysfunction of the ER–peroxisomes contact sites is also correlated with the development of peroxisome‐related disorders (Castro et al. 2018). Despite the aforementioned exciting progress, the molecular composition of MCSs (i.e. intermembrane connectome or imConnectome) and how MCSs contribute to (patho)physiological processes remain incompletely defined. This is partially attributable to the lack of appropriate methods and convenient tools to probe and perturb MCSs in situ. Although electron microscopy (EM) serves as the gold standard for the visualization of MCSs in fixed cells, a number of innovative approaches and tools now enable real‐time monitoring and manipulation of MCS dynamics in living cells. In this review, we provide a brief overview of the latest progress in these exciting new directions, with an emphasis on genetically encoded molecular probes and optogenetic MCS actuators that can be conveniently used in any laboratory equipped with a fluorescence microscope.
MCS visualization by electron microscopy in fixed samples
Electron microscopy (EM) is regarded as the ‘gold standard’ for MCS visualization by providing the static snapshot of the MCS architecture at the nanoscale often by utilizing horseradish peroxidase (HRP) to localize proteins (Fig. 2 A) (Wu et al. 2006). When coupled with the immunogold labelling technique (Orci et al. 2009; Eisenberg‐Bord et al. 2016), it is commonly used to determine whether a protein of interest is localized within or outside a defined interorganellar contact site. Furthermore, focused ion beam‐scanning EM (FIB‐SEM) uses an electron beam to capture the SEM image of the sample, followed by an ion beam to remove a thin layer of the material (15–50 nm) and thus expose a new section for imaging. The series of SEM images are then merged to form a three‐dimensional (3D) visualization of the sample. With this technique, a resolution of ∼20–40 nm is achieved for thick 3D samples without the need to manually generate serial sections (Fig. 2 B) (Bushby et al. 2011; Ohta et al. 2014; Hirabayashi et al. 2017; Cohen et al. 2018). However, maintaining the samples in their native states after fixation poses a challenge to these EM‐based techniques. Because the preparation of samples often involves chemical fixation, dehydration and plastic embedment, intracellular organelles tend to be damaged or undergo structural rearrangements to disrupt the intact architecture of MCSs (Tocheva et al. 2010).
Figure 2. MCS visualization by electron microscopy and fluorescence microscopy.
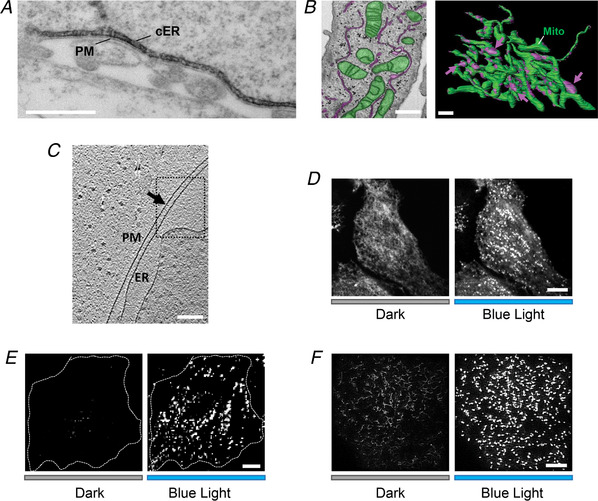
A, a representative electron micrograph (EM) of HRP–STIM1 in HEK293 cells following Ca2+ store depletion. Scale bar, 500 nm. B, MCSs formed between ER (magenta) and mitochondria (green) in HeLa cells visualized by FIB‐SEM (left). The 3D model was built based on images acquired by reconstructing FIB‐SEM images (right). MCSs are indicated by magenta arrows. Scale bar, 1 µm. This image from Hirabayashi et al. (2017) is republished with permission from the American Association for the Advancement of Science. C, a representative Cryo‐ET image of ER–PM contact sites in COS‐7 cell. Scale bars, 200 nm. This image from Collado & Fernandez‐Busnadiego (2017) is republished with permission from Elsevier Science and Technology Journals. D–F, light‐induced alterations in the distribution of mRuby2‐OptoPBer at the footprint of HeLa cells imaged by confocal microscopy (D), TIRFM (E) and SIM (F), respectively. Transfected cells were either shielded (black bar) or exposed to blue light illumination for 60 s (blue bar). Scale bar, 10 µm.
Electron cryo‐tomography (Cryo‐ET) solves this problem by immobilizing samples in non‐crystalline ice so that one can carry out imaging experiments under cryogenic conditions to avoid dehydration and chemical fixation procedures (Tocheva et al. 2010). Cryo‐ET has been gaining increasing interest in the cell biology field by providing fine MCS structures at a high resolution of approximately 4–10 nm. Most impressively, a series of two‐dimensional images can be combined to generate a 3D structure of MCSs. Friedman et al. utilized this powerful technique to reveal that ER tubules tightly wrap around mitochondria or endosomes in eukaryotic cells (Friedman et al. 2011, 2013). Taking advantage of Cryo‐ET, Fernandez‐Busnadiego et al. analysed the 3D architecture of ER–PM junctions (Fig. 2 C) and found that the junctions could be mediated by extended synaptotagmins (E‐Syts) and STIM1, with each bridging a different gap distance and contributing to the maintenance of ER–PM MCSs in response to intracellular Ca2+ mobilization (Fernandez‐Busnadiego et al. 2015; Collado & Fernandez‐Busnadiego, 2017). Clearly, the advent of Cryo‐ET enables the visualization of the 3D structures of MCSs without chemical fixation at a near‐native state with high resolution.
MCS visualization with fluorescence microscopy in living cells
To enable MCS visualization in living cells, genetically encoded fluorescent proteins are often attached to MCS‐resident proteins so that the MCS structure becomes visible under multispectral fluorescence microscopy (Fig. 2 D). The lateral resolution of conventional epifluorescence or confocal microscopy is approximately 200 nm, while the axial resolution is even worse, approaching approximately 500 nm (Schulz et al. 2013; MacDonald et al. 2015). This diffraction limit makes it difficult to image the biological processes occurring at or near the plasma membrane. Higher axial resolution can be achieved (less than 200 nm) by taking advantage of the total internal reflection fluorescence microscopy (TIRFM; Fig. 2 E). TIRFM is also called ‘evanescent wave microscopy’, because it utilizes the complete reflection of a ray of light at the glass–water interface to generate an evanescent wave to selectively illuminate the fluorophores only at the surface regions. The wave penetrates to a depth of approximately 100 nm into the sample without exciting fluorescent proteins far away from the surface, thereby leading to a significant reduction of the background noise (Axelrod, 2001; Mattheyses et al. 2010). TIRFM has been extremely useful in monitoring the kinetics and dynamics of proteins residing at the ER–PM contact sites since these MCSs are formed within 100 nm above the bottom of a culture dish. For instance, STIM1 puncta formation at ER–PM junctions can be readily visualized by TIRFM within 1 min after Ca2+ store depletion (Liou et al. 2005; Wu et al. 2006; Jing et al. 2015).
Although confocal microscopy or TIRFM can provide low‐resolution dynamic pictures of MCSs, these methods cannot resolve the fine architecture of MCSs (10–35 nm). Super‐resolution microscopes become a more suitable choice to pinpoint the exact localization of a target protein at MCSs. When coupled with fluorophore labelling of MCS‐resident proteins, super‐resolution light microscopy methods such as structured illumination microscopy (SIM; Hirabayashi et al. 2017; Fig. 2 F), photoactivation localization microscopy (PALM; Sengupta et al. 2014), and stochastic optical reconstruction microscopy (STORM; Shim et al. 2012) provide alternative non‐EM methods to observe the behaviour of MCSs in situ with a higher lateral and axial resolution (Table 1; Cohen et al. 2018). A brief comparison of the resolution among all these imaging techniques is summarized in Table 2. However, acquisition speed is an inherent limitation of PALM and STORM techniques. These techniques require the sequential sampling of thousands of raw frames to generate a stochastic image, and therefore tend to compromise the temporal resolution (Shcherbakova et al. 2014). The recently reported multi‐colour grazing incidence (GI)‐SIM provides a potential solution to this problem. GI‐SIM permits the acquisition of one to two images per second and is most suitable for monitoring the highly dynamic interactions of ER with its neighbouring organelles (MacDonald et al. 2015; Guo et al. 2018). Although these techniques are capable of improving resolution in both lateral and axial dimensions, they are not readily available in a standard laboratory because of the high cost.
Table 1.
Exemplary approaches and molecular tools for MCS visualization and perturbation
| MCS location | Design/binding partners | Approach or tool | Reference |
|---|---|---|---|
| MCS visualization and labelling | |||
| ER–mitochondria |
|
BiFC | Yang et al. (2018) |
| ER–mitochondria |
|
PLA | Gomez‐Suaga et al. (2017) |
| ER–LE |
|
PLA | Alpy et al. (2013) |
| ER–PM | Engineered ER‐resident Rit‐PB with spacers and linkers; GFP‐tagged | MAPPER | Chang et al. (2013) |
| Chemical or optogenetic actuators for tethering and MCS assembly | |||
| ER–mitochondria tethering |
|
Rapamycin‐inducible chemical dimerizer | Csordas et al. (2010) |
| ER–mitochondria tethering |
|
iLID‐based optical dimerizer | Shi et al. (2018) |
| ER–PM MCS | ER‐anchored photosensitive GFP‐tagged Rit‐PB | LOV2‐based LiMETER | Jing et al. (2015) |
| ER–PM MCS | ER‐anchored photosensitive GFP‐ or mRuby2‐tagged STIM1‐PB and its variants | LOV2‐based OptoPBer | He et al. (2017) |
Abbreviations: ActA, actin assembly inducing protein; BiFC, bimolecular fluorescence complementation; CB5, cytochrome b 5; ER, endoplasmic reticulum; FKBP, FK506 binding protein; FRB domain, FKBP12‐rapamycin binding domain; iLID, improved light‐induced dimer; LE, late endosome; LiMETER, light‐inducible membrane tethered peripheral ER; LOV2, light‐oxygen‐voltage sensing domain 2; MAPPER, membrane‐attached peripheral ER; MCS, membrane contact site; PB, polybasic domain; PLA, proximity ligation assay; PM, plasma membrane; PTPIP51, protein tyrosine phosphatase interacting protein; STARD3, StAR related lipid transfer domain‐3; STARD3NL, STARD3 N‐terminal like; VAP, vesicle‐associated membrane protein‐associated protein.
Table 2.
Summary of the resolution of different imaging techniques
| Technique | Lateral resolution | Axial resolution | Reference |
|---|---|---|---|
| Confocal | 200 nm | 500–700 nm | Schulz et al. (2013), MacDonald et al. (2015) |
| TIRFM | 200 nm | Up to 100 nm | Axelrod (2001), Mattheyses et al. (2010) |
| SIM | 100 nm | 250 nm | Gustafsson (2000) |
| PALM | Up to 20 nm | 50 nm | Betzig et al. (2006) |
| STORM | Up to 20 nm | 50 nm | Rust et al. (2006) |
Abbreviations: PALM, photoactivation localization microscopy; SIM, structured illumination microscopy; STORM, stochastic optical reconstruction microscopy; TIRFM: total internal reflection fluorescence microscopy.
MCS labelling by proximity ligation assay
Classic EM and Cryo‐ET are useful techniques for MCS visualization after cell fixation. However, these methods require special equipment and laborious sample preparation steps, making them non‐ideal for daily use. To label endogenous protein complexes in fixed cells and visualize them with fluorescence microscopy, a highly sensitive approach termed in situ proximity ligation assay (PLA) has been developed, which is frequently used for detecting endogenous protein–protein interactions in fixed cells or tissues (Soderberg et al. 2006). PLA relies on probe‐based targeting and labelling of target proteins. Proteins of interest can be probed with primary antibodies, followed by secondary antibodies coupled to specific oligonucleotides (Soderberg et al. 2008). If two target proteins harbouring the oligonucleotides are situated in close proximity, a third connector oligonucleotide can be hybridized with both oligonucleotides to generate circular DNA strands, which serve as templates for rolling circular amplification (Fig. 3 A). Using fluorophore‐labelled complementary nucleotides, PLA allows for the microscopic visualization of two targets in close proximity at approximately 30 nm (Soderberg et al. 2006).
Figure 3. MCS visualization and manipulation with chemogenetic or optogenetic tools.
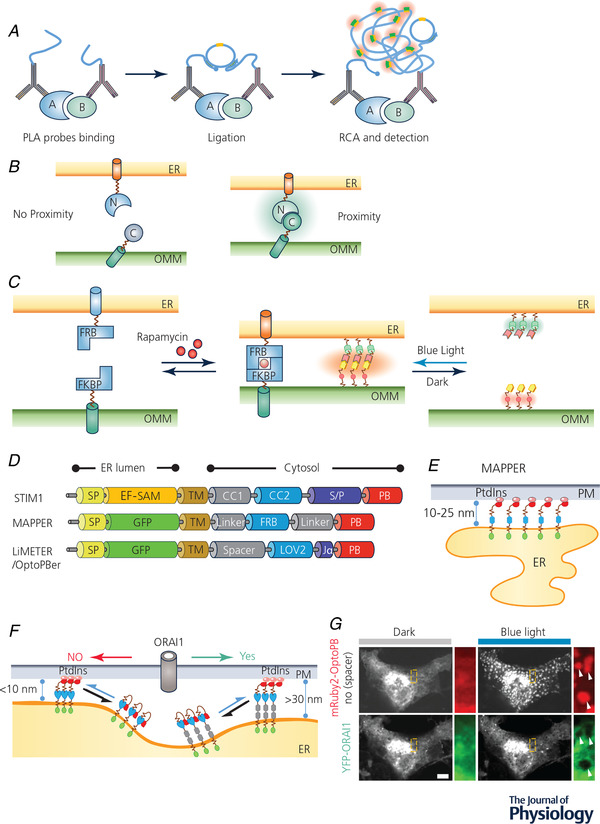
A, cartoon illustration of the proximity ligation assay (PLA). RCA, rolling circle amplification. B, detection of MCS‐resident proteins by bimolecular fluorescence complementation (BiFc). Two non‐fluorescent fragments of a fluorescent protein (FP) were individually anchored to the opposing membranes of different organelles. When the two organellar membranes are positioned in close proximity, an intact FP will be reassembled to emit fluorescence. C, inducible intermembrane tethering between ER and mitochondria. The tethering can be achieved by either using a rapamycin/FRB/FKBP‐based chemical dimerization module (left) or an iLID‐based optical dimerizer (right). ER, endoplasmic reticulum; FKBP, FK506 binding protein; FRB domain, FKBP12‐rapamycin binding domain; OMM, outer mitochondrial membrane. D, scheme illustrating the domain architecture of MAPPER, LiMETER and OptoPBer, which can be used to visualize or inducibly control MCS formation at ER–PM junctions. CC, coiled coild region; EF‐SAM: EF‐hand and sterile alpha domain; FRB, FKBP‐rapamycin binding domain; LOV2, light‐oxygen‐voltage domain 2; PB, polybasic domain; SP, ER‐targeting signal peptide; S/P, serine/proline‐rich region; TM, transmembrane domain. E, schematic illustration of MAPPER at ER–PM junctions. F, cartoon illustrating light‐inducible assembly of ER–PM MCS mediated by LiMETER or OptoPBer. By tuning the spacer within these optogenetic constructs, the ER–PM gap distance can be tuned to manipulate the diffusion of PM‐resident proteins (e.g. ORAI1) into ER–PM contact sites. G, a confocal image showing the exclusion of ORAI1–YFP (green) from ER–PM contact sites marked by mRuby2–OptoPBer (red) when the gap distance was shortened to less than 10 nm in response to blue light stimulation. White arrows indicate ORAI1 exclusion from ER–PM MCSs. Scale bar, 5 µm.
Taking advantage of PLA, Alpy et al. discovered that both late‐endosomal membrane‐anchored proteins, StAR‐related lipid transfer domain‐3 (STARD3) and STARD3 N‐terminal like protein (STARD3NL), are involved in the MCS assembly between late endosomes (LE) and ER (Alpy et al. 2013). In a second study, PLA was used to examine whether vesicle‐associated membrane protein‐associated protein B (VAPB) and protein tyrosine phosphatase interacting protein 51 (PTPIP51) would impact the assembly of ER–mitochondria MCSs marked by the IP3R–VDAC1 pair. Overexpression of VAPB or PTPIP51 was found to enhance PLA signals, thus implying an indispensable role of these two new players in shaping ER–mitochondria MCSs to facilitate Ca2+ exchange and autophagy (Gomez‐Suaga et al. 2017). Because PLA relies on antibodies, it is suitable for detecting endogenous protein interactions in both cell lines and tissue sections ex vivo. However, for MCS‐resident proteins lacking appropriate antibodies, PLA may not be the first choice for MCS visualization.
MCS labelling by bimolecular fluorescence complementation
Another method relying on the spatial proximity to label MCSs is based on biomolecular fluorescence complementation (BiFC) systems, including split Venus (Toulmay & Prinz, 2012) or spGFP1–10/spGFP11 (Feinberg et al. 2008). In a typical BiFC configuration, a fluorescent protein (FP) is split into two non‐fluorescent parts, with either the N‐ or C‐terminal fragment fused to each of the two proteins residing within the opposing membranes (Fig. 3 B). If the two proteins of interest interact with each other, they may bring the two FP fragments into close proximity (∼10–30 nm) to restore the fluorescence of the chromophore (Shai et al. 2018). Given the straightforward readout and its compatibility with high‐throughput screening platforms, BiFC can be further applied to identify unknown binding partners within MCSs. In order to discover new interorganellar contact sites in yeast, Shai et al. generated a series of constructs, with various membrane proteins from two different organelles tagged with either half of a split Venus (Shai et al. 2018; Yang et al. 2018). They discovered at least four potential new contacts: PM–vacuole, PM–lipid droplet, PM–peroxisome and peroxisome–vacuole MCSs. Moreover, they found two novel tether proteins, Fzo1 and Pex34, at the mitochondria–peroxisome contact sites. They found that the contact site expansion triggered by Pex34 overexpression, but not by Fzo1 expression, contributed to fatty acids β‐oxidation in yeast (Shai et al. 2018). Hence, the contact site machinery and functions can be systematically investigated by using BiFC. The extension of a similar method to probe MCSs in various types of mammalian cells is anticipated to expand the connectome by unravelling previously unrecognized MCS‐resident proteins.
The split FP system can also be utilized to monitor MCS dynamics in real time. A split green fluorescent protein (GFP), composed of spGFP1–10 and spGFP11, can be anchored at ER membrane and mitochondrial outer membrane, respectively. This reporter makes it is possible to analyse ER–mitochondria contact site distribution and dynamics during the cell cycle and under different stressful cellular conditions. With this method, Yang et al. demonstrated that the contacts sites between ER and mitochondria are dynamic structures that undergo active remodelling under conditions of different cellular needs (Yang et al. 2018). Thus, BiFC can be broadly applied to analyse native MCS distribution and MCS dynamics under various (patho)physiological conditions.
Three caveats, nonetheless, have to be taken into consideration for BiFC‐related applications. First, because the orientation and positioning of the fused FP fragments might affect the efficiency of fluorescence restoration, repeated rounds of trial and error studies are often needed to find the best combination. The absence of fluorescence signal, sometimes, may not reflect the absence of protein–protein interactions within a particular MCS. Second, because fluorescence complementation seems to be irreversible in vitro, BiFC could potentially stabilize intermembrane coupling to perturb the size and dynamics of MCSs. Third, given that the FP partners tend to self‐assemble at high local concentrations, a negative control group is essential to draw a clear conclusion in each experiment. The most appropriate negative control is the fusion in which the interaction sites have been mutated, thereby abolishing interaction with the partner (Kudla & Bock, 2016). An inducible expression system is most desirable under the scenario that an unspecific interaction is foreseen.
Inducible intermembrane tethering
Apart from reporters or markers to aid MCS visualization, molecular tools that allow remote manipulation of intermembrane tethering and MCS assembly are equally needed to study interorganellar communication (Table 1). These perturbative tools parallel the gain‐of‐function manipulation seen in a typical genetic study. To this end, pairs of chemical‐ or light‐inducible dimerization modules can be tethered to proteins residing on either side of the opposing membranes of MCSs (Fig. 3 C). Forced intermembrane tethering can be achieved by simple addition of chemicals or via light illumination. The FK506 binding protein (FKBP) and the FKBP‐rapamycin binding (FRB) domain based chemical dimerization modules are most frequently used to trigger protein–protein interactions upon the addition of rapamycin or its analogues (Crabtree & Schreiber, 1996; Rivera et al. 1996; Castellano & Chavrier, 2000; Castellano et al. 2000). Rapamycin binds to FKBP and the FRB fragment of the mechanistic target of rapamycin (mTOR) protein to mediate the heterodimer formation (Inobe & Nukina, 2016). Fluorophore‐conjugated FRB and FKBP modules have been engineered into the cytoplasmic side of an ER‐resident protein and the inner half of PM, respectively, to bring ER membrane and PM into close proximity following rapamycin treatment. The distance between ER and PM can thus be adjusted by introducing flexible linker with varying lengths. By taking advantage of this rapamycin‐induced dimerization system, Varnai et al. demonstrated that an optimized gap distance is required to enable efficient ORAI–STIM coupling at ER–PM contact sites. They estimated that a distance of at least 6 nm is required for STIM1 diffusion into ER–PM junctions, whereas ORAI1 needs at least 10–14 nm to be accommodated within this specialized contact site (Varnai et al. 2007).
In a separate study, Csordas et al. (2010) applied a similar chemical‐inducible system to regulate calcium dynamics at the ER–mitochondria contact sites. In brief, the outer mitochondrial membrane (OMM) targeting sequence from mouse a‐kinase anchor protein 1 (mAKAP1) (residues 34–63) was added to the monomer red fluorescent protein (mRFP)–FKBP fusion. For ER targeting, human Sac1 (residues 521–587) was fused to cyan fluorescent protein (CFP)–FRB. Following rapamycin treatment, a stable linkage was formed between the FKBP and FRB domains to drive the close apposition between ER and OMM. In order to visualize the ER–mitochondria contact sites, Csordas et al. (2010) designed a Förster resonance energy transfer (FRET)‐based reporter that targets the ER–mitochondria interface. FRET is a non‐radiative energy transfer process between two chromophores. FRET is initiated by the electronically excited donor chromophore, which transfers its energy to the nearby acceptor chromophore. The excitation spectrum of the acceptor chromophore is required to overlap with the emission spectrum of the donor chromophore (Sekar & Periasamy, 2003). An advantage of this approach is that FRET offers a higher spatial resolution (about 1–10 nm) than fluorescence colocalization (Loura & Prieto, 2011). With this design, FRET signals were detected on almost all the mitochondrial surface upon rapamycin‐induced dimerization between the ER‐targeted FRET donor and the mitochondria‐anchored acceptor. This chemical‐inducible FRET reporter system has been utilized to demonstrate that the existence of an optimal gap width is required for efficient Ca2+ transfer between ER and mitochondria (Csordas et al. 2010). Collectively, chemical‐inducible intermembrane tethering provides a non‐conventional approach to modulate cellular events occurring at MCSs. Because rapamycin‐induced artificial tethering lacks strict spatial control, this chemogenetic approach tends to cause a massive apposition between two opposing membranes, rather than the formation of spatially restricted bona fide contact sites.
To enable both spatial and temporal control over tethering, Shi et al. modified the chemical‐inducible ER–mitochondria system by replacing the FRB/FKBP components with an optical dimerizer termed iLID (improved light‐induced dimer) (Shi et al. 2018). The iLID system consists of a bacterial peptide, SsrA, which is originally caged by the photoswitch LOV2 (light‐oxygen‐voltage sensing domain 2), and its binding partner, SspB (Guntas et al. 2015). In the dark, SsrA is docked to the LOV2 core body via a Jα helix. Following blue light illumination, SsrA is released from LOV2 and selectively associates with its natural partner, SspB, resulting in a light‐inducible heterodimer formation. mKate2‐tagged SspB is anchored toward the ER membrane via the ER‐resident peptide sequence of cytochrome b 5 (CB5), whereas the light‐responsive LOV2‐SsrA component is anchored to OMM by an OMM linker peptide sequence derived from actin assembly inducing protein (ActA). When both components were co‐expressed in mammalian cells, SspB–mKate2–CB5 retained a tubular ER distribution and Venus–iLID–ActA was anchored on mitochondria, respectively. Upon blue light stimulation, SspB–mKate2–CB5 translocated toward mitochondria, and subsequently colocalized with Venus–iLID–ActA to form puncta and tubular structures within approximately 90 s. The movement of SspB–mKate2–CB5 toward mitochondria could be manipulated by repeatedly switching on and off the light source without significant loss of the photo‐induced responses. In addition to temporal control, the iLID system also displays a superior spatial resolution. For instance, focal blue light can be applied to illuminate only a selected region of interest to induce local ER–OMM tethering without eliciting global responses outside the region. This synthetic device proves to be highly reversible with high spatial and temporal precision. Nonetheless, whether or not such optogenetic manipulations recapitulate the behaviour and functional consequences of bona fide membrane contacts sites between ER and OMM is still questionable. To bridge this technical gap, chemical biological tools engineered from naturally existing MCS‐resident proteins are most desirable, but have yet to be developed in the near future.
Light‐inducible labelling and manipulation of MCSs
To overcome the disadvantages of the above‐mentioned inducible systems that tend to form massive intermembrane ‘patches’ rather than discrete contact ‘sites’, optogenetic tools have been developed for selective manipulation of MCS assembly, particularly at the ER–plasma membrane contact sites. Compared with other methods, the optogenetic approach excels in its reversibility, genetic encodability, high spatiotemporal precision and non‐invasiveness (Liu & Tucker, 2017).
ER–PM MCSs are among the most abundant intermembrane appositions in a mammalian cell, and serve as a specialized subcellular compartment for regulating Ca2+ homeostasis and lipid exchange (Carrasco & Meyer, 2011; Saheki & De Camilli, 2017). To selectively label ER–PM MCSs in living cells, Chang et al. developed a genetically encoded ER–PM tether, termed ‘MAPPER’ (for membrane‐attached peripheral ER) by taking advantage of a phospholipid‐binding motif from a small GTPase (Chang et al. 2013) (Fig. 3 D and E). An ER‐targeting signal peptide and the single transmembrane domain derived from STIM1 are used in MAPPER to ensure its ER localization, with GFP inserted in between to make MAPPER visible under fluorescence microscope. The cytoplasmic side of MAPPER contains flexible linkers and tunable spacers with varying lengths to span a 10–25 nm gap distance, as well as a polybasic domain (PB) of the small G protein Rit. Rit‐PB is rich in positively charged residues and interacts with the negatively charged phosphoinositides (e.g. phosphatidylinositol 4,5‐ bisphosphate or phosphatidylinositol 3,4,5‐trisphosphate) in the inner half of the lipid bilayer of PM (Heo et al. 2006; Fig. 3 E). One limitation of the MAPPER system is that this artificial construct may constitutively induce the formation of MCSs to perturb the phosphoinositide homeostasis in the host cells.
To avoid the possibility of the constitutive localization of MAPPER at ER–PM MCSs and make this system inducible, Jing et al. inserted a light switch derived from the LOV2 domain of oat phototropin 1 upstream of Rit‐PB (Jing et al. 2015). Through multiple rounds of optimization, the best performing construct, designated LiMETER (for light‐inducible membrane tethered peripheral ER tool), was created to enable light‐inducible assembly of membrane contacts sites between ER and the plasma membrane (Figs 2 D and E and 3 F). In the dark, LiMETER is distributed evenly across the ER network with the C‐terminal PB tightly docked toward LOV2, thereby preventing its association with PM‐resident phosphoinositides. Upon blue light stimulation, the photo‐sensing cofactor flavin mononucleotide (FMN) forms a photoadduct with the neighbouring cysteine residue to trigger conformational changes that lead to the unwinding and unfolding of the Jα helix. As a consequence, the C‐terminally appended Rit‐PB was exposed and its function restored to engage phosphoinositides and bridge the distance between ER and PM within tens of seconds. Given that the photoreaction is totally reversible, the assembly and disassembly of ER–PM MCSs can be mimicked by simply switching on and off the light source. Compared with MAPPER, which constitutively makes stable connections between ER and PM, LiMETER can be used to transiently mark ER–PM MCSs with minimal perturbation of the host physiology. Both MAPPER and LiMETER can be employed to perform functional rescue experiments under circumstances in which a hypothetical MCS tether at ER–PM junctions is genetically depleted.
Very recently, a further evolved version of LiMETER with an enhanced signal‐to‐noise ratio and tunable helical spacers has been developed (He et al. 2017; Nguyen et al. 2018). By replacing the Rit‐PB with a poly basic C‐tail from STIM1, He et al. developed the OptoPBer system to inducibly generate ER–PM MCSs with varying gap distance (Fig. 3 F). By introducing mutations into the STIM1‐PB domain, OptoPBer has been successfully used to identify key residues responsible for protein–phosphoinositide interactions in living cells (He et al. 2017). Likewise, this modular system can serve as a scaffold to validate if any predicted phospholipid binding domain can indeed mediate protein–lipid interactions in a cellular context. To tune the gap distances between ER and PM, zero to eight α‐helical spacers composed of (EAAAR)4, with each spanning an estimated distance of 3 nm, were insert between the transmembrane domain and the LOV2 domain to span 10–35 nm at ER–PM junctions. This tunable system was used to gauge the distance requirement for the free diffusion of ORAI1 channels into ER–PM puncta marked by OptoPBer. With a gap distance of approximately 10 nm, yellow fluorescent protein (YFP)–ORAI1 diffusion into the puncta seemed to be disrupted (Fig. 3 G). When the gap distance was gradually increased up to 30 nm, He et al. observed a step‐wise increase of YFP–ORAI1 diffusion into ER–PM MCSs, a finding that is consistent with results obtained using the chemical‐inducible strategy (Varnai et al. 2007). Ideally, the LiMETER or OptoPBer systems can be repurposed to deliver proteins of interests into ER–PM MCSs to assess their contributions to MCS assembly, maintenance and dynamics. Clearly, these exciting examples have illustrated the prowess of optogenetics in the molecular dissection of MCSs in situ with high spatiotemporal resolution.
Conclusions
Tight contacts between two opposing membranes from different organelles regulate a number of fundamental physiological processes. Over the past two decades, we have witnessed technological advances in both electron microscopy and fluorescence microscopy, both of which are widely adopted to aid MCS visualization. Fluorophore‐based MCS labelling methods, when coupled with advanced high‐content imaging platforms, now open new avenues to discovery and systematically dissect the molecular composite of individual MCSs within different cell types in a high‐throughput manner. Complementary to existing MCS labelling methods or tools, a series of genetically encoded MCS actuators, exemplified by LiMETER and OptoPBer as ER–PM MCS marker, have enabled remote and reversible control of MCS assembly with unprecedented spatial and temporal precision. Such optogenetic manipulations make it possible to design and perform loss‐of‐function or gain‐of‐function experiments typically seen in classical genetic studies.
These new techniques and tools will likely find uses to solve some controversies in the field of membrane contact sites. A prominent example is the debated functional role of mitofusin 2 (MFN2) proposed by the Scorrano (de Brito & Scorrano, 2008) and Pozzan/Pizzo (Filadi et al. 2015) groups. Mitofusins, including MFN1 and MFN2, are GTPases located in the outer mitochondria membrane (OMM), and are thought to be essential for the fusion of OMM (Santel & Fuller, 2001). In addition to its undisputed role in mitochondrial fusion, MFN2 is found to be critical for the formation of ER–mitochondria juxtaposition. In one study by the Scorrano group, the ER–mitochondria interactions were disrupted upon genetic ablation or silencing of MFN2 in mouse embryonic fibroblasts (MEFs) and HeLa cells (de Brito & Scorrano, 2008). In a second study, the Pozzan/Pizzo group, nevertheless, reported an increase in the ER–mitochondria coupling in MFN2 −/− MEFs or MFN2 knockdown MEFs (Filadi et al. 2015). Both groups utilized traditional EM and confocal imaging approaches to monitor the morphological changes at ER–mitochondria interfaces. To reconcile this controversy, it is imperative to compare the ER–mitochondria contact sites at near‐native states in both normal and MFN2 −/− cells. This can be achieved by using either super‐resolution fluorescence microscopy or the Cryo‐ET technique that obviates the need for dehydration or chemical fixation. In parallel, chemogenetic or optogenetic tools can be applied to precisely perturb ER–mitochondria tethering in MFN2 −/− MEFs, mimicking functional rescue to elucidate the role of mitofusions at ER–mitochondria junctions.
Conceivably, the aforementioned chemogenetic and optogenetic engineering approaches can be broadly extended to photo‐manipulate other types of interorganellar contact sites, such as the ER–endosome, ER–lysosome, ER–peroxisome, ER–lipid droplet and ER–mitochondria MCSs, thus exerting remote control over interorganellar communication to achieve tailored function non‐invasively. With the rapid expansion of molecular tools designed for visualization and interrogation of MCSs, we look forward to exciting times for the burgeoning field of MCS biology.
Additional information
Competing interests
The authors declare no competing interests.
Author contributions
J.J. and Y.Z. wrote the manuscript. All authors have read and approved the final version of this manuscript and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
Y.Z.’s and Y.H.’s labs are supported by grants from the National Institutes of Health (R01GM112003, R01HL146852, R21GM126532, R01CA232017 and R01HL134780); the Cancer Prevention and Research Institute of Texas (RP170660 and RR140053); the John S. Dunn Foundation Collaborative Research Award, the Welch Foundation (BE‐1913); and the American Cancer Society (ACS) (RSG‐16‐215‐01‐TBE and RSG‐18‐043‐01‐LIB).
Biographies
Ji Jing obtained his PhD in biomedical science from Texas A&M University. He studied calcium signalling and optogenetics in Dr Yubin Zhou's lab at Texas A&M, as well as the mechanisms of pexophagy in Dr. Cheryl Walker's lab at Baylor College of Medicine. His ongoing research focuses on designing optogenetic tools to study intracellular organelles and their communications.

Yubin Zhou is an associate professor at the Institute of Biosciences and Technology, Texas A&M University. He received his medical training from Zhejiang University School of Medicine and obtained his master and PhD degrees in Chemistry from Georgia State University. He thereafter pursued his postdoctoral training at Harvard Medical School and La Jolla Institute for Immunology. His research interests focus on calcium and lipid signalling at membrane contact sites, optogenetics, chemical biology and immunoengineering.

Edited by: Ole Petersen & Calum Wilson
This review was presented at the symposium ‘Calcium and Cell Function: From Mechanisms to Disease’, which took place at Granlibakken Resort, Tahoe City, CA, USA, 10–15 June 2018.
This is an Editor's Choice article from the 1 May 2020 issue.
References
- Alpy F, Rousseau A, Schwab Y, Legueux F, Stoll I, Wendling C, Spiegelhalter C, Kessler P, Mathelin C, Rio MC, Levine TP & Tomasetto C (2013). STARD3 or STARD3NL and VAP form a novel molecular tether between late endosomes and the ER. J Cell Sci 126, 5500–5512. [DOI] [PubMed] [Google Scholar]
- Axelrod D (2001). Total internal reflection fluorescence microscopy in cell biology. Traffic 2, 764–774. [DOI] [PubMed] [Google Scholar]
- Betzig E, Patterson GH, Sougrat R, Lindwasser OW, Olenych S, Bonifacino JS, Davidson MW, Lippincott‐Schwartz J & Hess HF (2006). Imaging intracellular fluorescent proteins at nanometer resolution. Science 313, 1642–1645. [DOI] [PubMed] [Google Scholar]
- Bushby AJ, P'Ng KM, Young RD, Pinali C, Knupp C & Quantock AJ (2011). Imaging three‐dimensional tissue architectures by focused ion beam scanning electron microscopy. Nat Protoc 6, 845–858. [DOI] [PubMed] [Google Scholar]
- Carrasco S & Meyer T (2011). STIM proteins and the endoplasmic reticulum‐plasma membrane junctions. Annu Rev Biochem 80, 973–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellano F & Chavrier P (2000). Inducible membrane recruitment of small GTP‐binding proteins by rapamycin‐based system in living cells. Methods Enzymol 325, 285–295. [DOI] [PubMed] [Google Scholar]
- Castellano F, Montcourrier P & Chavrier P (2000). Membrane recruitment of Rac1 triggers phagocytosis. J Cell Sci 113, 2955–2961. [DOI] [PubMed] [Google Scholar]
- Castro IG, Schuldiner M & Zalckvar E (2018). Mind the organelle gap – peroxisome contact sites in disease. Trends Biochem Sci 43, 199–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CL, Hsieh TS, Yang TT, Rothberg KG, Azizoglu DB, Volk E, Liao JC & Liou J (2013). Feedback regulation of receptor‐induced Ca2+ signaling mediated by E‐Syt1 and Nir2 at endoplasmic reticulum‐plasma membrane junctions. Cell Rep 5, 813–825. [DOI] [PubMed] [Google Scholar]
- Chang CL & Liou J (2015). Phosphatidylinositol 4,5‐bisphosphate homeostasis regulated by Nir2 and Nir3 proteins at endoplasmic reticulum‐plasma membrane junctions. J Biol Chem 290, 14289–14301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu BB, Liao YC, Qi W, Xie C, Du X, Wang J, Yang H, Miao HH, Li BL & Song BL (2015). Cholesterol transport through lysosome‐peroxisome membrane contacts. Cell 161, 291–306. [DOI] [PubMed] [Google Scholar]
- Cohen S, Valm AM & Lippincott‐Schwartz J (2018). Interacting organelles. Curr Opin Cell Biol 53, 84–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collado J & Fernandez‐Busnadiego R (2017). Deciphering the molecular architecture of membrane contact sites by cryo‐electron tomography. Biochim Biophys Acta Mol Cell Res 1864, 1507–1512. [DOI] [PubMed] [Google Scholar]
- Crabtree GR & Schreiber SL (1996). Three‐part inventions: intracellular signaling and induced proximity. Trends Biochem Sci 21, 418–422. [DOI] [PubMed] [Google Scholar]
- Csordas G, Renken C, Varnai P, Walter L, Weaver D, Buttle KF, Balla T, Mannella CA & Hajnoczky G (2006). Structural and functional features and significance of the physical linkage between ER and mitochondria. J Cell Biol 174, 915–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csordas G, Varnai P, Golenar T, Roy S, Purkins G, Schneider TG, Balla T & Hajnoczky G (2010). Imaging interorganelle contacts and local calcium dynamics at the ER‐mitochondrial interface. Mol Cell 39, 121–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das A, Nag S, Mason AB & Barroso MM (2016). Endosome‐mitochondria interactions are modulated by iron release from transferrin. J Cell Biol 214, 831–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Brito OM & Scorrano L (2008). Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 456, 605–610. [DOI] [PubMed] [Google Scholar]
- De Stefani D, Bononi A, Romagnoli A, Messina A, De Pinto V, Pinton P & Rizzuto R (2012). VDAC1 selectively transfers apoptotic Ca2+ signals to mitochondria. Cell Death Differ 19, 267–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg‐Bord M, Shai N, Schuldiner M & Bohnert M (2016). A tether is a tether is a tether: tethering at membrane contact sites. Dev Cell 39, 395–409. [DOI] [PubMed] [Google Scholar]
- Feinberg EH, Vanhoven MK, Bendesky A, Wang G, Fetter RD, Shen K & Bargmann CI (2008). GFP Reconstitution Across Synaptic Partners (GRASP) defines cell contacts and synapses in living nervous systems. Neuron 57, 353–363. [DOI] [PubMed] [Google Scholar]
- Fernandez‐Busnadiego R, Saheki Y & De Camilli P (2015). Three‐dimensional architecture of extended synaptotagmin‐mediated endoplasmic reticulum‐plasma membrane contact sites. Proc Natl Acad Sci U S A 112, E2004–E2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feske S, Gwack Y, Prakriya M, Srikanth S, Puppel SH, Tanasa B, Hogan PG, Lewis RS, Daly M & Rao A (2006). A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature 441, 179–185. [DOI] [PubMed] [Google Scholar]
- Filadi R, Greotti E, Turacchio G, Luini A, Pozzan T & Pizzo P (2015). Mitofusin 2 ablation increases endoplasmic reticulum‐mitochondria coupling. Proc Natl Acad Sci U S A 112, E2174–E2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman JR, Dibenedetto JR, West M, Rowland AA & Voeltz GK (2013). Endoplasmic reticulum‐endosome contact increases as endosomes traffic and mature. Mol Biol Cell 24, 1030–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman JR, Lackner LL, West M, DiBenedetto JR, Nunnari J & Voeltz GK (2011). ER tubules mark sites of mitochondrial division. Science 334, 358–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garofalo T, Matarrese P, Manganelli V, Marconi M, Tinari A, Gambardella L, Faggioni A, Misasi R, Sorice M & Malorni W (2016). Evidence for the involvement of lipid rafts localized at the ER‐mitochondria associated membranes in autophagosome formation. Autophagy 12, 917–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez‐Suaga P, Paillusson S, Stoica R, Noble W, Hanger DP & Miller CCJ (2017). The ER‐mitochondria tethering complex VAPB‐PTPIP51 regulates autophagy. Curr Biol 27, 371–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guntas G, Hallett RA, Zimmerman SP, Williams T, Yumerefendi H, Bear JE & Kuhlman B (2015). Engineering an improved light‐induced dimer (iLID) for controlling the localization and activity of signaling proteins. Proc Natl Acad Sci U S A 112, 112–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Li D, Zhang S, Yang Y, Liu JJ, Wang X, Liu C, Milkie DE, Moore RP, Tulu US, Kiehart DP, Hu J, Lippincott‐Schwartz J, Betzig E & Li D (2018). Visualizing intracellular organelle and cytoskeletal interactions at nanoscale resolution on millisecond timescales. Cell 175, 1430–1442.e17. [DOI] [PubMed] [Google Scholar]
- Gustafsson MG (2000). Surpassing the lateral resolution limit by a factor of two using structured illumination microscopy. J Microsc 198, 82–87. [DOI] [PubMed] [Google Scholar]
- He L, Jing J, Zhu L, Tan P, Ma G, Zhang Q, Nguyen NT, Wang J, Zhou Y & Huang Y (2017). Optical control of membrane tethering and interorganellar communication at nanoscales. Chem Sci 8, 5275–5281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helle SC, Kanfer G, Kolar K, Lang A, Michel AH & Kornmann B (2013). Organization and function of membrane contact sites. Biochim Biophys Acta 1833, 2526–2541. [DOI] [PubMed] [Google Scholar]
- Heo WD, Inoue T, Park WS, Kim ML, Park BO, Wandless TJ & Meyer T (2006). PI(3,4,5)P3 and PI(4,5)P2 lipids target proteins with polybasic clusters to the plasma membrane. Science 314, 1458–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirabayashi Y, Kwon SK, Paek H, Pernice WM, Paul MA, Lee J, Erfani P, Raczkowski A, Petrey DS, Pon LA & Polleux F (2017). ER‐mitochondria tethering by PDZD8 regulates Ca2+ dynamics in mammalian neurons. Science 358, 623–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogan PG, Lewis RS & Rao A (2010). Molecular basis of calcium signaling in lymphocytes: STIM and ORAI. Annu Rev Immunol 28, 491–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inobe T & Nukina N (2016). Rapamycin‐induced oligomer formation system of FRB‐FKBP fusion proteins. J Biosci Bioeng 122, 40–46. [DOI] [PubMed] [Google Scholar]
- Jing J, He L, Sun A, Quintana A, Ding Y, Ma G, Tan P, Liang X, Zheng X, Chen L, Shi X, Zhang SL, Zhong L, Huang Y, Dong MQ, Walker CL, Hogan PG, Wang Y & Zhou Y (2015). Proteomic mapping of ER–PM junctions identifies STIMATE as a regulator of Ca2+ influx. Nat Cell Biol 17, 1339–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamoto RM, Brunschwig JP, Kim KC & Caswell AH (1986). Isolation, characterization, and localization of the spanning protein from skeletal muscle triads. J Cell Biol 103, 1405–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki T, Lange I & Feske S (2009). A minimal regulatory domain in the C terminus of STIM1 binds to and activates ORAI1 CRAC channels. Biochem Biophys Res Commun 385, 49–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YJ, Guzman‐Hernandez ML, Wisniewski E & Balla T (2015). Phosphatidylinositol‐phosphatidic acid exchange by Nir2 at ER‐PM contact sites maintains phosphoinositide signaling competence. Dev Cell 33, 549–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YJ, Guzman‐Hernandez ML, Wisniewski E, Echeverria N & Balla T (2016). Phosphatidylinositol and phosphatidic acid transport between the ER and plasma membrane during PLC activation requires the Nir2 protein. Biochem Soc Trans 44, 197–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudla J & Bock R (2016). Lighting the way to protein‐protein interactions: recommendations on best practices for bimolecular fluorescence complementation analyses. Plant Cell 28, 1002–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahiri S, Toulmay A & Prinz WA (2015). Membrane contact sites, gateways for lipid homeostasis. Curr Opin Cell Biol 33, 82–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lev S (2010). Non‐vesicular lipid transport by lipid‐transfer proteins and beyond. Nat Rev Mol Cell Biol 11, 739–750. [DOI] [PubMed] [Google Scholar]
- Levine T (2004). Short‐range intracellular trafficking of small molecules across endoplasmic reticulum junctions. Trends Cell Biol 14, 483–490. [DOI] [PubMed] [Google Scholar]
- Liou J, Kim ML, Heo WD, Jones JT, Myers JW, Ferrell JE Jr & Meyer T (2005). STIM is a Ca2+ sensor essential for Ca2+‐store‐depletion‐triggered Ca2+ influx. Curr Biol 15, 1235–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q & Tucker CL (2017). Engineering genetically‐encoded tools for optogenetic control of protein activity. Curr Opin Chem Biol 40, 17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loura LM & Prieto M (2011). FRET in membrane biophysics: an overview. Front Physiol 2, 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luik RM, Wu MM, Buchanan J & Lewis RS (2006). The elementary unit of store‐operated Ca2+ entry: local activation of CRAC channels by STIM1 at ER‐plasma membrane junctions. J Cell Biol 174, 815–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald L, Baldini G & Storrie B (2015). Does super‐resolution fluorescence microscopy obsolete previous microscopic approaches to protein co‐localization? Methods Mol Biol 1270, 255–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattheyses AL, Simon SM & Rappoport JZ (2010). Imaging with total internal reflection fluorescence microscopy for the cell biologist. J Cell Sci 123, 3621–3628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muallem S, Chung WY, Jha A & Ahuja M (2017). Lipids at membrane contact sites: cell signaling and ion transport. EMBO Rep 18, 1893–1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen NT, Han W, Cao WM, Wang Y, Wen S, Huang Y, Li M, Du L & Zhou Y (2018). Store‐operated calcium entry mediated by ORAI and STIM. Compr Physiol 8, 981–1002. [DOI] [PubMed] [Google Scholar]
- Nguyen NT, Ma G, Lin E, D'Souza B, Jing J, He L, Huang Y & Zhou Y (2018). CRAC channel‐based optogenetics. Cell Calcium 75, 79–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta K, Okayama S, Togo A & Nakamura K (2014). Three‐dimensional organization of the endoplasmic reticulum membrane around the mitochondrial constriction site in mammalian cells revealed by using focused‐ion beam tomography. Microscopy (Oxf) 63(Suppl 1), i34. [DOI] [PubMed] [Google Scholar]
- Orci L, Ravazzola M, Le Coadic M, Shen WW, Demaurex N & Cosson P (2009). STIM1‐induced precortical and cortical subdomains of the endoplasmic reticulum. Proc Natl Acad Sci U S A 106, 19358–19362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paillusson S, Stoica R, Gomez‐Suaga P, Lau DHW, Mueller S, Miller T & Miller CCJ (2016). There's something wrong with my MAM; the ER‐mitochondria axis and neurodegenerative diseases. Trends Neurosci 39, 146–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park CY, Hoover PJ, Mullins FM, Bachhawat P, Covington ED, Raunser S, Walz T, Garcia KC, Dolmetsch RE & Lewis RS (2009). STIM1 clusters and activates CRAC channels via direct binding of a cytosolic domain to Orai1. Cell 136, 876–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips MJ & Voeltz GK (2016). Structure and function of ER membrane contact sites with other organelles. Nat Rev Mol Cell Biol 17, 69–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter KR (1953). Observations on a submicroscopic basophilic component of cytoplasm. J Exp Med 97, 727–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter KR & Palade GE (1957). Studies on the endoplasmic reticulum. III. Its form and distribution in striated muscle cells. J Biophys Biochem Cytol 3, 269–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakriya M & Lewis RS (2015). Store‐operated calcium channels. Physiol Rev 95, 1383–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera VM, Clackson T, Natesan S, Pollock R, Amara JF, Keenan T, Magari SR, Phillips T, Courage NL, Cerasoli F Jr, Holt DA & Gilman M (1996). A humanized system for pharmacologic control of gene expression. Nat Med 2, 1028–1032. [DOI] [PubMed] [Google Scholar]
- Rizzuto R, De Stefani D, Raffaello A & Mammucari C (2012). Mitochondria as sensors and regulators of calcium signalling. Nat Rev Mol Cell Biol 13, 566–578. [DOI] [PubMed] [Google Scholar]
- Rizzuto R, Pinton P, Carrington W, Fay FS, Fogarty KE, Lifshitz LM, Tuft RA & Pozzan T (1998). Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 280, 1763–1766. [DOI] [PubMed] [Google Scholar]
- Roos J, DiGregorio PJ, Yeromin AV, Ohlsen K, Lioudyno M, Zhang S, Safrina O, Kozak JA, Wagner SL, Cahalan MD, Veliçelebi G & Stauderman KA (2005). STIM1, an essential and conserved component of store‐operated Ca2+ channel function. J Cell Biol 169, 435–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowland AA & Voeltz GK (2012). Endoplasmic reticulum‐mitochondria contacts: function of the junction. Nat Rev Mol Cell Biol 13, 607–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rust MJ, Bates M & Zhuang X (2006). Sub‐diffraction‐limit imaging by stochastic optical reconstruction microscopy (STORM). Nat Methods 3, 793–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saheki Y & De Camilli P (2017). Endoplasmic reticulum‐plasma membrane contact sites. Annu Rev Biochem 86, 659–684. [DOI] [PubMed] [Google Scholar]
- Santel A & Fuller MT (2001). Control of mitochondrial morphology by a human mitofusin. J Cell Sci 114, 867–874. [DOI] [PubMed] [Google Scholar]
- Schulz O, Pieper C, Clever M, Pfaff J, Ruhlandt A, Kehlenbach RH, Wouters FS, Großhans J, Bunt G & Enderlein J (2013). Resolution doubling in fluorescence microscopy with confocal spinning‐disk image scanning microscopy. Proc Natl Acad Sci U S A 110, 21000–21005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekar RB & Periasamy A (2003). Fluorescence resonance energy transfer (FRET) microscopy imaging of live cell protein localizations. J Cell Biol 160, 629–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta P, van Engelenburg SB & Lippincott‐Schwartz J (2014). Superresolution imaging of biological systems using photoactivated localization microscopy. Chem Rev 114, 3189–3202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shai N, Yifrach E, van Roermund CWT, Cohen N, Bibi C, IJlst L, Cavellini L, Meurisse J, Schuster R, Zada L, Mari MC, Reggiori FM, Hughes AL, Escobar‐Henriques M, Cohen MM, Waterham HR, Wanders RJA, Schuldiner M & Zalckvar E (2018). Systematic mapping of contact sites reveals tethers and a function for the peroxisome‐mitochondria contact. Nat Commun 9, 1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shcherbakova DM, Sengupta P, Lippincott‐Schwartz J & Verkhusha VV (2014). Photocontrollable fluorescent proteins for superresolution imaging. Annu Rev Biophys 43, 303–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi F, Kawano F, Park SE, Komazaki S, Hirabayashi Y, Polleux F & Yazawa M (2018). Optogenetic control of endoplasmic reticulum‐mitochondria tethering. ACS Synth Biol 7, 2–9. [DOI] [PubMed] [Google Scholar]
- Shim SH, Xia C, Zhong G, Babcock HP, Vaughan JC, Huang B, Wang X, Xu C, Bi G‐Q & Zhuang X (2012). Super‐resolution fluorescence imaging of organelles in live cells with photoswitchable membrane probes. Proc Natl Acad Sci U S A 109, 13978–13983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soboloff J, Rothberg BS, Madesh M & Gill DL (2012). STIM proteins: dynamic calcium signal transducers. Nat Rev Mol Cell Biol 13, 549–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soderberg O, Gullberg M, Jarvius M, Ridderstrale K, Leuchowius KJ, Jarvius J, Wester K, Hydbring P, Bahram F, Larsson LG & Landegren U (2006). Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat Methods 3, 995–1000. [DOI] [PubMed] [Google Scholar]
- Soderberg O, Leuchowius KJ, Gullberg M, Jarvius M, Weibrecht I, Larsson LG & Landegren U (2008). Characterizing proteins and their interactions in cells and tissues using the in situ proximity ligation assay. Methods 45, 227–232. [DOI] [PubMed] [Google Scholar]
- Spector AA & Yorek MA (1985). Membrane lipid composition and cellular function. J Lipid Res 26, 1015–1035. [PubMed] [Google Scholar]
- Szabadkai G, Bianchi K, Varnai P, De Stefani D, Wieckowski MR, Cavagna D, Nagy AI, Balla T & Rizzuto R (2006). Chaperone‐mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J Cell Biol 175, 901–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szalai G, Csordas G, Hantash BM, Thomas AP & Hajnoczky G (2000). Calcium signal transmission between ryanodine receptors and mitochondria. J Biol Chem 275, 15305–15313. [DOI] [PubMed] [Google Scholar]
- Tocheva EI, Li Z & Jensen GJ (2010). Electron cryotomography. Cold Spring Harb Perspect Biol 2, a003442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toulmay A & Prinz WA (2012). A conserved membrane‐binding domain targets proteins to organelle contact sites. J Cell Sci 125, 49–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trebak M & Kinet JP (2019). Calcium signalling in T cells. Nat Rev Immunol 19, 154–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Meer G & Sprong H (2004). Membrane lipids and vesicular traffic. Curr Opin Cell Biol 16, 373–378. [DOI] [PubMed] [Google Scholar]
- Varnai P, Toth B, Toth DJ, Hunyady L & Balla T (2007). Visualization and manipulation of plasma membrane‐endoplasmic reticulum contact sites indicates the presence of additional molecular components within the STIM1‐Orai1 complex. J Biol Chem 282, 29678–29690. [DOI] [PubMed] [Google Scholar]
- Vig M, Peinelt C, Beck A, Koomoa DL, Rabah D, Koblan‐Huberson M, Kraft S, Turner H, Fleig A, Penner R & Kinet JP (2006). CRACM1 is a plasma membrane protein essential for store‐operated Ca2+ entry. Science 312, 1220–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Carvalho P & Voeltz GK (2018). Here, there, and everywhere: The importance of ER membrane contact sites. Science 361, eaan5835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu MM, Buchanan J, Luik RM & Lewis RS (2006). Ca2+ store depletion causes STIM1 to accumulate in ER regions closely associated with the plasma membrane. J Cell Biol 174, 803–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Zhao X, Xu J, Shang W & Tong C (2018). A novel fluorescent reporter detects plastic remodeling of mitochondria‐ER contact sites. J Cell Sci 131, jcs208686. [DOI] [PubMed] [Google Scholar]
- Yuan JP, Zeng W, Dorwart MR, Choi YJ, Worley PF & Muallem S (2009). SOAR and the polybasic STIM1 domains gate and regulate Orai channels. Nat Cell Biol 11, 337–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SL, Yu Y, Roos J, Kozak JA, Deerinck TJ, Ellisman MH, Stauderman KA & Cahalan MD (2005). STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature 437, 902–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Meraner P, Kwon HT, Machnes D, Oh‐hora M, Zimmer J, Huang Y, Stura A, Rao A & Hogan PG (2010). STIM1 gates the store‐operated calcium channel ORAI1 in vitro. Nat Struct Mol Biol 17, 112–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Xue S & Yang JJ (2013). Calciomics: integrative studies of Ca2+‐binding proteins and their interactomes in biological systems. Metallomics 5, 29–42. [DOI] [PMC free article] [PubMed] [Google Scholar]