

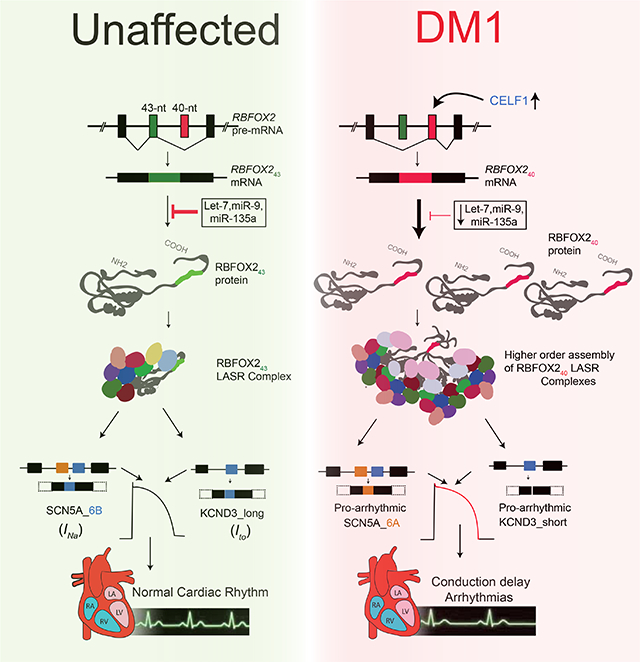
SUMMARY
Myotonic dystrophy type 1 (DM1) is a multisystemic genetic disorder caused by the CTG repeat expansion in the 3′-untranslated region of DMPK gene. Heart dysfunctions occur in ~80% of DM1 patients and are the second leading cause of DM1-related deaths. Herein, we report that upregulation of a non-muscle splice isoform of RNA-binding-protein RBFOX2 in DM1 heart tissue—due to altered splicing factor and microRNA activities—induces cardiac conduction defects in DM1 individuals. Mice engineered to express the non-muscle RBFOX240 isoform in heart via tetracycline-inducible transgenesis, or CRISPR/Cas9-mediated genome editing, reproduced DM1-related cardiac-conduction delay and spontaneous episodes of arrhythmia. Further, by integrating RNA binding with cardiac transcriptome datasets from DM1 patients and mice expressing the non-muscle RBFOX2 isoform, we identified RBFOX240-driven splicing defects in voltage-gated sodium and potassium channels, which alter their electrophysiological properties. Thus, our results uncover a trans-dominant role for an aberrantly expressed RBFOX240 isoform in DM1 cardiac pathogenesis.
Keywords: Myotonic dystrophy, alternative splicing, cardiac arrhythmias, genome editing, Protein-RNA interactions, microRNA, ion channels, genomics, molecular dynamics
In brief
Cardiac dysfunction is a mortality cause in myotonic dystrophy, yet the underlying mechanisms are poorly understood. Misra et al. identify a switch from muscle to non-muscle splice isoforms of the RNA-binding-protein RBFOX2 in DM1 tissue. The non-muscle isoform induces missplicing of channel transcripts, causing cardiac conduction delay and spontaneous arrhythmogenesis.
Graphical Abstract
INTRODUCTION
DM1 is an autosomal-dominant disorder and the most commonly inherited form of adult onset muscular dystrophy (Day and Ranum, 2005; Yum et al., 2017). The disease arises due to an expansion of trinucleotide CTG repeat in the 3’-UTR of DMPK gene (Brook et al., 1992; Fu et al., 1992; Mahadevan et al., 1992), which produces mutant RNAs with long tracts of CUG repeats r(CUG)exp forming stable hairpin loops that aggregate in the nucleus (Davis et al., 1997; Mooers et al., 2005). Although DMPK is broadly expressed, and the disease affects multiple tissues, the predominant causes of mortality are muscle wasting and sudden cardiac death ( Heatwole et al., 2012; Phillips and Harper, 1997). More than two-thirds of DM1 individuals experience severe heart dysfunctions including cardiac-conduction delay, fatal sinoatrial and atrioventricular blocks, atrial fibrillation, and ventricular arrhythmias (Groh et al., 2008; Lazarus et al., 2002; Tokgozoglu et al., 1995). Affected individuals often display atrophy of the conduction system as well as fibrofatty infiltration of the myocardium and the Bundle of His. Further, some patients develop dilated cardiomyopathy along with systolic/diastolic dysfunctions (Bhakta et al., 2010; Nguyen et al., 1988; Phillips and Harper, 1997).
The two major mechanisms by which r(CUG)exp RNAs cause toxicity and thereby disease pathogenesis are: (i) the accumulated r(CUG)exp RNAs sequester muscleblind-like proteins (MBNL1, MBNL2 and MBNL3) with high affinity, resulting in their depletion and loss-of-function (Mankodi et al., 2001; Miller et al., 2000); (ii) r(CUG)exp RNAs activate the protein kinase C pathway and suppress the expression of a specific set of MEF2-regulated micro (mi)RNAs causing upregulation of CELF1 protein (Kalsotra et al., 2014; Kalsotra et al., 2010; Kuyumcu-Martinez et al., 2007). MBNL and CELF family of RNA binding proteins direct a large number of developmentally-regulated alternative splicing and polyadenylation decisions ( Ho et al., 2004; Kalsotra et al., 2008; Lin et al., 2006; Wang et al., 2015). Therefore, disruption of their normal activities in DM1 shifts RNA processing of target pre-mRNAs towards fetal patterns (Batra et al., 2014; Kanadia et al., 2003; Koshelev et al., 2010; Thomas et al., 2017), many of which induce key features of the disease (Charlet et al., 2002; Fugier et al., 2011; Mankodi et al., 2002; Savkur et al., 2001). The exact molecular basis for the electrophysiological and cardiac contractility abnormalities in DM1, however, is still undetermined.
Here, we demonstrate that the non-muscle splice isoform of RNA-binding protein FOX2 (RBFOX240) is overexpressed in DM1 human heart tissues, and that this overexpression results from a combination of elevated CELF1 and reduced miRNA activities. Modeling the increase in non-muscle RBFOX240 isoform in mouse hearts was sufficient to trigger DM1-related cardiac conduction defects. By integrating RBFOX2-RNA interactions with cardiac transcriptome datasets—from DM1 patients, CELF1 overexpressing mice, MBNL1 knockouts, and mice expressing the non-muscle RBFOX240 isoform—we identified a unique set of RBFOX240-driven mRNA splicing defects that are dysregulated in the DM1 hearts. Specifically, the RBFOX240 isoform caused missplicing of voltage-gated sodium and potassium channel transcripts directing generation of pro-arrhythmic variants that elicit altered rates of ion transport and electrophysiological channel properties. Thus, upregulation of non-muscle RBFOX240 isoform augments the production of pathogenic ion channel splice variants that may directly contribute to DM1-related cardiac conduction delay and arrhythmogenesis.
RESULTS
Selective upregulation of the non-muscle RBFOX240 isoform in DM1 heart tissue
Considered as master regulators of tissue-specific alternative splicing (Conboy, 2017; Jangi and Sharp, 2014), RBFOX proteins (RBFOX1, RBFOX2, and RBFOX3) utilize a highly conserved RRM domain to bind (U)GCAUG motifs in pre-mRNAs and control splicing in a position-dependent manner (Jin et al., 2003; Singh et al., 2014; Yeo et al., 2009). We have determined that RBFOX2 protein levels are significantly increased in the autopsied heart samples of DM1 patients but not of individuals with a history of arrhythmias caused due to left ventricular tachycardia (Fig. 1A, B). Parallel examination of RBFOX2 mRNA unexpectedly showed only a modest increase in abundance in DM1 heart tissues (Supplementary Fig. 1A).
Figure 1. Selective upregulation of the non-muscle RBFOX240 isoform in DM1 heart tissue.

(A) Immunoblot analysis of RBFOX2 protein in unaffected (n=8), DM1 (n=5), and arrhythmic non-DM (n=3) human heart samples. (B) Quantification of relative band intensities for RBFOX2 from A, normalized to GAPDH levels. (C) Schematic of amino acid residues encoded by RBFOX2 non-muscle (40-nt) and muscle-specific (43-nt) exons. Three tyrosine (Y) residues specific to the 40-nt exon are highlighted in red. (D) RT-PCR analysis monitoring the inclusion of RBFOX2 40-nt exon in unaffected (n=8), DM1 (n=9), and arrhythmic non-DM [SVA: Sustained Ventricular Arrhythmia (n=3); and VT: Ventricular Tachycardia (n=3)] human heart samples. (E) Percent Spliced In (PSI) values of Rbfox2 40-nt exon from D. (F) Immunoblot analysis of RBFOX2 protein (top) and RT-PCR analysis of Rbfox2 43-nt and 40-nt exons (bottom) in HL-1 cells transfected with DT0 or DT960 plasmids for 48h. (G) Relative quantification of immunoblot and RT-PCR data from F. n=4 independent transfections. (H) RT-PCR analysis of Rbfox2 43-nt and 40-nt exons (top), and immunoblot analysis of RBFOX2 protein (bottom) from HL-1 cells treated with control ASO or with an ASO targeting the 5’ss of Rbfox2 43-nt exon for 48h. (I) Relative quantification of RT-PCR and immunoblot data from H. n=4 independent transfections. All data are mean ± s.d., and p-values were derived from parametric t-test (two-sided, unpaired), with Welch’s correction.
RBFOX2 gene has a complex architecture comprising multiple promoters and alternative exons (Supplementary Fig. 1B), which direct tissue-specific expression of various RBFOX2 isoforms that are functionally distinct in their RNA binding activities, subcellular localization, and interactions with regulatory co-factors (Conboy, 2017). Amongst them, a pair of 43-nucleotide (nt) and 40-nt exons in the c-terminal domain encode the muscle and non-muscle isoforms that are expressed in a mutually exclusive, developmentally regulated, and evolutionarily conserved manner. Interestingly, the 40-nt exon contains three tyrosine residues (Fig. 1C) that promote a higher-order RBFOX2 complex of proteins called the Large assembly of Splicing Regulators (LASR) complex, which enhances RBFOX2’s splicing activity (Damianov et al., 2016; Ying et al., 2017). The 43-nt exon lacks these tyrosine residues (Fig. 1C), and therefore, fails to form the higher-order LASR complex (Ying et al., 2017). Both humans and mice predominantly express the 40-nt exon containing RBFOX2 isoform in fetal hearts, which is replaced by the 43-nt exon containing isoform in adult hearts, specifically within cardiomyocytes (Supplementary Fig. 1C–E). Strikingly, we noticed a significant splicing shift—from 43-nt to 40-nt exon—within RBFOX2 transcripts in DM1 patient heart samples (ΔPSI ranging from 12% to 95%), which was not detected in the hearts of individuals diagnosed with non-DM related arrhythmias or heart failure (Fig. 1D, E). We inspected three other RBFOX2 alternative exons in unaffected fetal, adult, and DM1 heart tissues, however, their splicing pattern was unaltered during development, or in DM1 (Supplementary Fig. 1F).
To determine if the increase in RBFOX2 protein abundance and change in its splicing were direct effects of the CTG expansion, we transfected HL-1 cardiac cell cultures with plasmids expressing zero (DT0) or 960 CTG repeats (DT960). Similar to DM1 heart tissues, transient transfection of HL-1 cells with the DT960 plasmid led to a significant elevation in RBFOX2 steady-state protein levels and a simultaneous shift in splicing from 43-nt to the 40-nt exon (Fig. 1F–G and Supplementary Fig. 1G). To explore potential coupling between the increase in RBFOX2 protein and its isoform switch, we redirected Rbfox2 splicing in HL-1 cells using splice-site (ss) blocking anti-sense oligonucleotides (ASO) (Bangru et al., 2018). Relative to control, HL-1 cells treated with ASO targeting the 5’ss of 43-nt exon caused a near complete switch to the 40-nt containing isoform but had little effect on RBFOX2 protein abundance (Fig. 1H–I). These data indicate that increase in RBFOX2 and its splice isoform switch in DM1 are two separate events, and that upregulation of RBFOX2 protein is not a consequence of higher stability of the non-muscle isoform.
Reduced expression of miRNAs de-represses RBFOX2 in DM1 cardiac cultures
Given that RBFOX2 protein levels decrease during postnatal heart development (Kalsotra et al., 2008) and the decrease is quickly reversed upon Dicer deletion in the adult myocardium (Kalsotra et al., 2010), we reasoned that RBFOX2 upregulation in DM1 might result from miRNA deregulation (Fernandez-Costa et al., 2013; Kalsotra et al., 2014; Rau et al., 2011). Consistent with this notion, we found conserved binding sites for a set of miRNAs—Let-7g/7b, miR-135a, miR-9, and miR-186—within the 3’-UTR of Rbfox2 (Fig. 2A), and expression of these miRNAs is reduced in DM1 heart tissue (Kalsotra et al., 2014). Transient expression of r(CUG)960 RNA was sufficient to downregulate all five miRNAs in HL-1 cells (Fig. 2B); and likewise, transfections with Let-7g, miR-186, Let-7b, miR-9, and miR-135a mimics reduced RBFOX2 protein abundance compared to scrambled control or the non-targeting miR-30 mimic (Fig. 2C), without affecting Rbfox2 mRNA levels (Supplementary Fig. 2A).
Figure 2. Reduced expression of miRNAs de-represses RBFOX2 in DM1 cardiac cultures.

(A) Schematic of putative miRNAs targeting Rbfox2 3’-UTR that are downregulated in the hearts of DM1 patients (Kalsotra et al., 2014). (B) Relative expression of indicated miRNAs in HL-1 cells following transfection with DT0 or DT960 plasmids for 48h. n=6 independent transfections. (C) Immunoblot analysis of RBFOX2 protein in HL-1 cells following treatment with scrambled control or indicated miRNA mimics. n=4 independent transfections. (D) Relative luciferase activity derived from Rbfox2 3’-UTR reporter transfected in HEK 293T cells following treatment with scrambled control or indicated miRNA mimics. Direct binding of miRNAs to the seed sequences in Rbfox2 3’-UTR was examined by co-transfecting indicated miRNA mimics and reporters with mutations that would disrupt the predicted miRNA interactions. n=4 independent transfections. (E) Relative luciferase activity derived from Rbfox2 3’-UTR reporter in HEK 293T cells, and (F) Immunoblot analysis of RBFOX2 protein in HL-1 cells after co-transfection with DT0 or DT960 plasmids, and scrambled control or a cocktail of indicated miRNA mimics. n=4 independent transfections. Data are mean ± s.d., and p-values were derived from a parametric t-test (two-sided, unpaired), with Welch’s correction.
To identify which of these miRNAs regulate RBFOX2 through direct interactions, we constructed dual luciferase reporters (Chorghade et al., 2017) wherein the 3’-UTR of Rbfox2 was cloned downstream from Renilla luciferase ORF, with or without mutations in seed sequences that would disrupt the predicted miRNA interactions (Supplementary Fig. 2B). Co-transfection of HEK293T cells with the reporter plasmids and miRNA mimic(s) individually or in combination revealed that relative to control, Let-7g, miR-9, and miR-135a could each repress the luciferase (Rluc/Fluc) activity of wildtype, but not the mutant constructs (Fig. 2D). Despite lowering RBFOX2 protein levels, miR-186, did not alter the reporter activity (Fig. 2C, D). We next asked whether restoring the activity of select miRNAs can normalize RBFOX2 levels in a DM1 cardiac cell culture model. Owing to the reduction of Rbfox2 targeting miRNAs, co-transfection of HL-1 cells with DT960, relative to DT0 plasmid, increased the Rbfox2 3’-UTR luciferase reporter activity (Fig. 2B, E). Notably, supplementing the r(CUG)960 RNA expressing HL-1 cells with a cocktail of Let-7g, miR-9 and miR-135a mimics normalized not only the reporter activity (Fig. 2E) but also endogenous RBFOX2 protein levels (Fig. 2F). Rbfox2 mRNA levels were unchanged in these experiments (Supplementary Fig. 2C). Altogether, these findings illustrate that multiple miRNAs directly or indirectly repress RBFOX2 protein expression in cardiac cells, their downregulation de-represses RBFOX2 in DM1 heart, and when supplemented, a mixture of miRNA mimics can reverse this de-repression in a DM1 cardiac cell culture model.
CELF1 upregulation promotes the production of RBFOX240 isoform in the heart
We next sought to address the mechanism(s) promoting generation of RBFOX240 splice isoform in DM1. Because DMPK RNAs containing r(CUG)exp repeats disrupt alternative splicing through MBNL sequestration and/or CELF1 upregulation, we assessed Rbfox2 splicing pattern in the hearts of Mbnl1ΔE3/ΔE3, tetracycline (tet)-inducible TRE-CELF1; MHCrtTA, and littermate control mice (Kalsotra et al., 2008; Kanadia et al., 2003). Although CELF1 overexpression in the adult heart did not perturb RBFOX2 protein abundance (Supplementary Fig. 3A), it evoked a significant shift from 43-nt to 40-nt exon within Rbfox2 transcripts, mimicking the splicing pattern observed in DM1 heart tissue (Fig. 3A, B). Deletion of MBNL1 had no effect on Rbfox2 splicing. We analyzed published time-series RNA-seq data following CELF1 overexpression in mouse hearts (Wang et al., 2015) and found a progressive shift in Rbfox2 splicing, which was detected as early as 24h after CELF1 induction (Fig. 3C). Furthermore, forced expression of CELF1 in HL-1 cells was sufficient to induce a switch from 43-nt to 40-nt exon without affecting RBFOX2 protein levels (Fig. 3D, E and Supplementary Fig. 3B). As expected, r(CUG)960 RNA expression resulted in CELF1 upregulation, and a consequent shift in Rbfox2 splicing in these cells (Fig. 3F, G). To determine whether increased CELF1 levels were required to generate the RBFOX240 isoform in DM1, we used siRNA(s) to offset CELF1 upregulation in r(CUG)960 RNA expressing HL-1 cells. Relative to scrambled control, Celf1-targeting siRNAs normalized the CELF1 protein levels and partially abrogated the r(CUG)960 RNA-induced switch in Rbfox2 splicing (Fig. 3F, G). These data imply that CELF1 promotes the production of non-muscle RBFOX240 isoform in the heart, and that high CELF1 activity is in part required for producing this isoform in a DM1 cardiac cell culture model.
Figure 3. CELF1 upregulation promotes the production of non-muscle RBFOX240 isoform in the heart.

(A) RT-PCR analysis of Rbfox2 43-nt and 40-nt exons in hearts of tet-inducible, heart-specific CELF1 bitransgenics (TRE-CELF1; MHCrtTA) and littermate control mice induced with 6g/kg Dox for ten days, as well as in hearts of wildtype (WT) and Mbnl1ΔE3/ΔE3 mice. (B) PSI values of Rbfox2 40-nt exon from the indicated genotypes in A. n=4 mice for each genotype. (C) RNA-seq read density across Rbfox2 43-nt and 40-nt exons from mouse hearts following CELF1 overexpression at indicated time points (Wang et al., 2015). (D) RT-PCR analysis of Rbfox2 43-nt and 40-nt exons (top), and immunoblot analysis of CELF1 protein normalized to GAPDH (bottom) in HL-1 cells following infection with GFP or CELF1 expressing adenoviruses. (E) PSI values of Rbfox2 40-nt exon from D. n=4 independent infections. (F) RT-PCR analysis of Rbfox2 43-nt and 40-nt exons (top) and immunoblot analysis of CELF1 protein normalized to GAPDH (bottom) in HL-1 cells after co-transfection with scrambled control or Celf1 targeting siRNA(s), and DT0 or DT960 plasmids. (G) PSI values of Rbfox2 40-nt exon from F. n=4 independent transfections. All data are mean ± s.d., and p-values were derived from a parametric t-test (two-sided, unpaired), with Welch’s correction.
RBFOX240 isoform induces DM1-like cardiac pathology in mice
Next, we characterized the functional consequences of RBFOX240 isoform in DM1 cardiac pathogenesis. To distinguish the effects of overexpression from splice isoform switch, we generated two separate mice models: a tet-inducible transgenic mouse model that overexpresses FLAG-tagged RBFOX240 isoform in the adult heart; and a CRISPR/Cas9 derived Rbfox2 43-nt exon deletion mouse model (Supplementary Fig. 4A–D). When induced with doxycycline (Dox), the hemizygous TRE-RBFOX240; MHCrtTA bitransgenic mice produced approximately 8-fold higher RBFOX2 protein compared to the uninduced littermate controls (Fig. 4A, B and Supplementary Fig. 4E). On the other hand, homozygous deletion of 43-nt exon by CRISPR/Cas9 in Rbfox2Δ43/Δ43 mice redirected splicing to 40-nt exon generating the non-muscle isoform in cardiac and skeletal muscles, but without any change in RBFOX2 mRNA or protein abundance (Fig. 4C, D and Supplementary Fig. 4E, F).
Figure 4. RBFOX240 isoform expression induces DM1-like cardiac pathology in mice.

(A) Immunoblot analysis of RBFOX2 and GAPDH proteins, and (B) RT-PCR analysis of Rbfox2 43-nt and 40-nt exons in the hearts of hemizygous MHCrtTA and TRE-RBFOX240; MHCrtTA bitransgenic mice fed regular or 0.5g/kg Doxycycline (Dox) containing Chow for 3 days. Quantifications of relative band intensities of RBFOX2 normalized to GAPDH, and the PSI values of Rbfox2 40-nt exon are shown below the gel images. n=5 mice for each genotype. (C) Immunoblot analysis of RBFOX2 and GAPDH proteins, and (D) RT-PCR analysis of Rbfox2 43-nt and 40-nt exons in the hearts from wildtype (WT) and Rbfox2Δ43/Δ43 mice. Quantifications of relative band intensities of RBFOX2 normalized to GAPDH, and the PSI values of Rbfox2 40-nt exon are shown below the gel images. n=4–6 mice for each genotype. (E) Kaplan-Meier survival curves of mice from indicated genotypes fed regular or 0.5g/kg Dox containing Chow for 45 days. (F) ECG analysis of ambulatory TRE-RBFOX240; MHCrtTA bitransgenic mice 48h before and 9 days after RBFOX240 overexpression with 0.5g/kg Dox containing Chow. (G) Representative ECG traces from F recorded before and after RBFOX240 overexpression. PVC: premature ventricular contraction, AV block: atrioventricular block. (H) Surface ECG analysis of WT and Rbfox2Δ43/Δ43 mice. (I) Representative ECG traces from H. PR, and QT intervals in F and H are indicated below the traces. All data are mean ± s.d., and p-values were derived from a parametric t-test (two-sided, paired for F and unpaired for H), with Welch’s correction.
Strikingly, ~20% of TRE-RBFOX240; MHCrtTA bitransgenic mice died within two to three weeks of transgene expression whereas no mortality was seen in Rbfox2Δ43/Δ43 or littermate control mice (Fig. 4E). Because no apparent differences in heart-to-body weight ratios, atrial and ventricular size or histological abnormalities such as myofiber disarray, inflammation, and fibrosis were evident (Supplementary Fig. 4G–I), we suspected that sudden death following RBFOX240 overexpression in the heart might be related to electrical phenotypes. Hence, we performed ECG telemetry on un-anaesthetized TRE-RBFOX240; MHCrtTA bitransgenic mice—continuously recording their ECG waveforms—before (for 48h) and after (for nine days) Dox administration. In addition, we also did surface ECGs on a separate cohort of wildtype, Rbfox2Δ43/Δ43, TRE-RBFOX240; MHCrtTA bitransgenics and littermate control mice following three days of Dox administration.
Compared to the baseline recordings, all mice following RBFOX240 overexpression exhibited a significant increase in PR intervals, indicating slowed conduction from the atria to the ventricles as well as an elongation in QT intervals, representing prolonged ventricular repolarization (Fig. 4F and Supplementary Fig. 5A, B). Importantly, while the uninduced mice displayed a normal sinus rhythm, progressive increases in the number of pauses and arrhythmic events were detected following RBFOX240 overexpression. Heart rate variability and sinus pauses indicated sinus node dysfunction, sometimes resulting in ventricular escape beats. Additionally, we observed premature ventricular contractions and frequent atrioventricular conduction blocks—evidenced by non-conducting P-waves (Fig. 4G). Furthermore, nine days after transgene induction, RBFOX240 overexpressing mice showed a high variability in RR interval duration (Supplementary Fig. 5C). Echocardiography studies revealed that abnormal conductivity in these mice is accompanied by reduced ejection fraction, fractional shortening and systolic/diastolic alterations (Supplementary Fig. 5D). Thus, overexpression of RBFOX240 isoform in mouse heart recapitulates DM1 cardiac phenotypes including conduction delay, spontaneous episodes of arrhythmias and contractile dysfunctions.
Although the electrophysiological defects in Rbfox2Δ43/Δ43 mice were milder relative to RBFOX240 overexpressing mice, ECG analysis of Rbfox2Δ43/Δ43 mice reproduced certain pathologic features of DM1. For instance, we noted a clear trend of prolonged PR interval with narrower QRS morphology in Rbfox2Δ43/Δ43 mice (Fig. 4H), which reflects supraventricular origin of QRS complex. The slow conductivity was also accompanied by an increase in sinus pauses, atrioventricular blocks and ventricular electric events (Fig. 4I). Histological and echocardiography studies, however, showed no differences in the systolic/diastolic ventricular dimensions or contractility including fractional shortening between Rbfox2Δ43/Δ43 and wildtype mice (Supplementary Fig. 4I, 5D). Collectively, these results demonstrate that inappropriate expression of the non-muscle RBFOX240 isoform in mouse heart disrupts the cardiac conduction, rhythm and function; and analogous to DM1 patients, triggers progressive heart blocks and sudden death in a subset of mice.
RBFOX240 isoform driven transcriptome alterations in DM1 heart tissue
To further investigate the mechanism(s) by which RBFOX240 isoform provokes DM1-like cardiac pathology, we deep-sequenced poly(A) selected RNAs prepared freshly from the ventricular cardiomyocytes isolated from Dox-induced TRE-RBFOX240; MHCrtTA and littermate control mice (Supplementary Table 2 and Supplementary Fig. 6A, B). In order to minimize any secondary effects of arrhythmias on the transcriptome, that are not directly dependent on RBFOX240, we induced the mice for only 72h. Remarkably, even a short-term induction of RBFOX240 transgene produced extensive changes in mRNA splicing and abundance (Fig. 5A and Supplementary Fig. 6C). The differentially expressed mRNAs following RBFOX240 overexpression were enriched in gene ontologies that grouped into major functional clusters especially cardiac conduction, immune response, cell proliferation, apoptosis, and metabolic processes, whereas the gene set with altered splicing formed clusters related to transcriptional regulation, mRNA processing, cytoskeletal organization and transport functions (Supplementary Fig. 6D).
Figure 5. RBFOX240 isoform driven transcriptome alterations in DM1 heart tissue.

(A) Overlap of differentially expressed (p<0.05, Wald test as described DESeq2; Log2[Fold Change]>1, and TPM>4) with differentially spliced mRNAs (p<0.05, FDR<0.10 adjusted for multiple testing, Difference in Percent Spliced Index [ΔPSI]≥20%, and Junction Counts≥10) in cardiomyocytes isolated from hemizygous MHCrtTA, and TRE-RBFOX240; MHCrtTA bitransgenic mice after administration of 0.5g/kg Dox containing Chow for 3 days. (B) Position and relative enrichment of RBFOX-binding motif near RBFOX240 regulated cassette exons. (C) Breakup of 3144 RBFOX240 regulated splicing events in cardiomyocytes with or without RBFOX240 binding peaks and a GCAUG motif within those peaks from iCLIP data in mouse brain (Damianov et al., 2016). (D-E) Top 3 pentamers enriched within the binding peaks near RBFOX240 regulated exons with (D) or without (E) GCAUG motifs. Fractional enrichments of (D) UGUGU and (E) UUUUU sequences aligning at each nucleotide relative to the RBFOX240 crosslink site are also plotted. Overlap of (G) mRNA abundance, and (F) alternative splicing changes among cardiac transcriptomes of DM1 patients (Freyermuth et al., 2016), RBFOX240 overexpressing, CELF1 overexpressing, and Mbnl1ΔE3/ΔE3 mice (Wang et al., 2015). (H) Directionality of mRNA abundance and alternative splicing changes in DM1 patient hearts and RBFOX240 overexpressing cardiomyocytes. (I) Distribution of changes in mRNA abundance and splicing events co-regulated in DM1 patient hearts and RBFOX240 overexpressing cardiomyocytes. (J) p-values of top Gene Ontology terms for alternatively spliced mRNAs in DM1 patient hearts (hypergeometric test; Benjamini method for multiple testing), and corresponding numbers of genes for each category that are similarly misspliced in the cardiomyocytes of RBFOX240 overexpressing, and hearts of CELF1 overexpressing, or Mbnl1ΔE3/ΔE3 mice.
Consistent with earlier studies (Singh et al., 2014; Yeo et al., 2009), the RBFOX2 binding motif GCAUG was significantly overrepresented in the exonic and surrounding intronic regions of RBFOX240-regulated exons (Fig. 5B). We next integrated the RBFOX240 overexpression RNA-seq data with previously published RBFOX240 iCLIP data from mouse brain (Damianov et al., 2016). Overlay of these two datasets revealed that ~50% of misspliced pre-mRNAs had a robust RBFOX2-binding cluster in the regulated exon or surrounding introns, but only half of those clusters contained a GCAUG motif (Fig. 5C). Because the RBFOX240 isoform in LASR complex can be recruited to RNA directly or indirectly through binding of hnRNP proteins (Ying et al., 2017), we searched for pentamers near RBFOX240-iCLIP crosslink sites bearing a GCAUG motif, or not (Supplementary Fig. 6E, F). Besides the expected overrepresentation of GCAUG sequences, a clear enrichment of potential hnRNP M-binding motif UGUGU was noted directly next to the GCAUG containing crosslink sites (Fig. 5D and Supplementary Fig. 6E). On the contrary, the polyU-rich hnRNP C-binding motifs were the most enriched pentamers detected adjacent to non-GCAUG containing crosslink sites (Fig. 5E and Supplementary Fig. 6F). Thus, our cardiomyocyte data are in agreement with previous studies (Damianov et al., 2016; Ying et al., 2017), demonstrating that in addition to direct interactions, the non-muscle RBFOX240 isoform can target pre-mRNAs through RNA contacts of hnRNP M and C components of the LASR complex.
To determine the respective contribution of RBFOX240 overexpression towards DM1—in relation to misregulation of MBNL and CELF proteins—we analyzed cardiac transcriptomes of DM1 patients (Freyermuth et al., 2016), CELF1 overexpressing and Mbnl1ΔE3/ΔE3 mice (Wang et al., 2015), and compared them with the cardiac transcriptome of RBFOX240 overexpressing mice. Amongst the three mice models, RBFOX240 overexpression reproduced the most number of mRNA abundance and splicing defects observed in heart tissue of DM1 patients (Fig. 5F, G). A large proportion of these mRNA abundance (74%) and splicing (67%) defects were shared in directionality (Fig. 5H); however, relative to DM1 hearts, the overall magnitude of change was higher in RBFOX240 overexpressing mouse cardiomyocytes (Fig. 5I). For the coregulated sets of splicing events, we found a positive correlation of CELF1 with RBFOX240 and a negative correlation of MBNL1 with CELF1 and RBFOX240 proteins (Supplementary Fig. 6G). Thus, our analysis confirmed the antagonistic splicing regulation by CELF1 and MBNL1 proteins (Kalsotra et al., 2008; Wang et al., 2015) while revealing a synergistic mode of regulation between RBFOX240 and CELF1 proteins. The involvement of RBFOX240 in DM1 cardiac pathology was further strengthened by the large overlap of gene ontologies between DM1 patients and RBFOX240 overexpressing mice (Fig. 5J). Altogether, our comparative transcriptomic findings illustrate that upregulation of non-muscle RBFOX240 isoform in DM1 alters the mRNA abundance and splicing of a large group of genes via direct and/or indirect pre-mRNA interactions through the LASR complex.
Structure-function analysis of DM1-associated alternatively spliced isoforms of voltage-gated ion channels
Further analysis of the RBFOX240-driven splicing alterations in DM1 identified twenty-two genes encoding components of conduction system and/or contractile apparatus—many of which are related to the NCBI MeSH terms: “Ventricular Tachycardia,” “Brugada and Long QT Syndromes,” “Atrial Fibrillation” and “Sudden Cardiac Death” (Fig. 6A). Of these, we elected to focus on SCN5A, and KCND3 because they encode the major voltage-gated sodium, and potassium channels in the heart respectively; and they switch to producing their pro-arrhythmic splice isoforms (Freyermuth et al., 2016; Pang et al., 2018; Wang and Wehrens, 2012) in DM1 patients. Additionally, multiple genome-wide association studies have identified variants of SCN5A and KCND3 among genetic determinants of prolonged PR interval and QRS duration (Pfeufer et al., 2010; Verweij et al., 2014), which are distinctive features of cardiac-conduction delay in DM1 patients.
Figure 6. Structure-function analysis of normal and DM1-related splice isoforms of voltage-gated sodium, calcium, and potassium channels.

(A) The subset of cardiac arrhythmia-associated genes misspliced following RBFOX240 overexpression in cardiomyocytes with their respective Gene Ontology terms (Left) and MeSH disease headings (Right). Genes misspliced similarly in the hearts of DM1 patients are highlighted in bold letters. (B) Schematic representation of action potentials from atria and ventricles, and their correlation with the surface electrocardiogram (ECG). The sodium channel SCN5A (NaV1.5) mediated Ina current, calcium channel CACNA1C (CaV1.2) mediated Ica current, and potassium channel KCND3 (KV4.3) mediated Ito current with their respective functions in propagation and duration of a typical action potential are depicted. Cartoons representing topology, relative locations and amino acid sequences for the two alternatively spliced exons in (C) SCN5A, and (D) KCND3 proteins. (E) Molecular dynamics (MD) simulation showing modelled structure of NaV1.5:SCN5A (6B) and (6A) exon containing isoforms embedded in a 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) lipid bilayer. Domains I-IV of the channel are represented by yellow, red, black and blue colors respectively. Differences in protein sequence are highlighted on the structure by magenta/cyan spheres and sticks. (F) Secondary structure content of domain I segment S3-S4 observed in MD simulations for the two isoforms. (G) Representative snapshots of MD simulations highlighting differential interactions of D211 in 6B (Left) and K211 in 6A (Right) with the lipid head group. The non-conserved residues between the two isoforms are shown in Van der Waals (VdW) representation. The protein is colored based on the secondary structure, yellow: α-helix, blue: 310-helix, gray: turn, white: coil.
For each cardiac cycle, when the pacemaker cells in the sinoatrial node fire, the action potential spreads through the atrial and ventricular myocardium via gap junctions that allow it to propagate along the myocytes. Once the action potential arrives, the membrane potential of myocytes increases, which activates the voltage-gated SCN5A sodium channels (NaV1.5) to mediate a rapid influx of Na+ ions (INa) across the cell membrane, resulting in a fast depolarization phase (Veerman et al., 2015) (Fig. 6B). As the membrane potential continues to increase, the voltage-gated L-type calcium channels (CaV1.2) open, and Ca2+ ions flow into the cells as an inward current (ICa), causing additional release of Ca2+ from the sarcoplasmic reticulum, which is crucial for coupling the electrical excitation to the physical contraction of the myocyte. After the membrane potential peaks, the voltage-gated KCND3 potassium channels (KV4.3) open and potassium ions flow out of the cells generating a rapidly inactivating transient outward current (Ito), which is important for the eventual repolarization of cardiac myocytes (Dixon et al., 1996) (Fig. 6B). Thus, proper impulse propagation and duration of the action potential in heart is highly contingent on the precise function of these ion channels.
Structurally, the voltage-gated ion channels are made up of either four separate subunits (K+ channels) or one polypeptide with four homologous domains (Na+ channels) (Fig. 6C, D). Each subunit or domain has six transmembrane segments (S1-S6) and a pore loop. The voltage sensing transmembrane element in SCN5A lies within segments S3 and S4, and that of domain I is encoded by a pair of conserved, developmentally regulated, mutually exclusive exons that are each 93-nts long. The fetal exon 6A differs from the adult exon 6B by 31-nts creating seven amino acid substitutions in Nav1.5 protein (Gellens et al., 1992), which includes replacement of a positively charged lysine (K211) in exon 6A with a negatively charged aspartate (D211) in exon 6B (Fig. 6C). Because a shift from exon 6B to 6A is linked to DM1-associated cardiac-conduction delay and heart arrhythmias (Freyermuth et al., 2016; Pang et al., 2018), we investigated functionally relevant structural and dynamical differences between the two alternatively spliced Nav1.5 isoforms using molecular dynamics (MD) simulations (Fig. 6E).
Our simulations revealed that in both channels, the S4 helix of domain I maintains its α-helical secondary structure (Fig. 6F), but with different tendencies to form a more tightly wound, longer, and thinner 310-helix at different segments of S4. Analysis of structural dynamics during the simulations showed that, while similar propensities for 310-helix formation were evident in the middle of S4 helix for both 6A and 6B isoforms, the former exhibited a higher 310-helix content near the top portion of S4, which can facilitate the downward S4 transition from the activated (up) state (Schwaiger et al., 2011) (Fig. 6F, G). Frequent transitions observed between α- and 310-helix further indicate that the activated state of S4 in the 6A isoform is destabilized and it may deactivate more readily due to a reduction in energy barrier, which is consistent with a depolarized shift in the voltage-dependence of channel activation for the pro-arrhythmic 6A isoform (Freyermuth et al., 2016; Pang et al., 2018). Additionally, we note that although D211 in 6B and K211 in 6A are both positioned close to the lipid headgroups, unlike D211, which preferably interacts with the choline moiety at a slightly higher position with respect to the lipid bilayer, K211—due to its longer side-chain—can reach down into the headgroup region and coordinate closely with the nearby phosphate group of POPC lipids (Fig. 6G, Supplementary Fig. 7). The K211D variant shifts the voltage-dependence of activation of the 6A containing Nav1.5 isoform back towards the hyperpolarized direction (Onkal et al., 2008). We propose that the observed persistent lipid-interaction of K211 in 6A isoform is crucial for stabilizing a 310-helical conformation near the top portion of S4 to allow rapid deactivation that would slow the activation kinetics (Supplementary video 1, 2). Indeed, forced expression of the exon 6A containing NaV1.5 isoform in adult mouse heart reduces excitability, slows conduction velocity, prolongs PR and QRS intervals, and increases susceptibility to arrhythmias (Freyermuth et al., 2016; Pang et al., 2018).
The cardiac isoform of the α subunit of potassium channel KCND3 (KV4.3) includes a 57-nt alternative exon, which encodes 19 amino acids at the C-terminal end (Fig. 6D). Amongst these amino acids is a conserved threonine residue, which is phosphorylated by protein kinase C to regulate the timing of the transient outward current Ito (Kong et al., 1998). The KV4.3 isoform lacking the 57-nt exon has been shown to promote faster inactivation of the channel, which slows down the ion flux and lengthens the duration of action potential—particularly affecting the early recovery and plateau phases of repolarization (Xie et al., 2009).
RBFOX240 expression in the heart reproduces DM1-related missplicing of voltage-gated sodium and potassium channels
Owing to such drastic effects of ion channel variants on cardiac rhythm and conductivity, we assessed their splicing patterns in the heart tissues of unaffected and DM1 individuals as well as in wildtype, Rbfox2Δ43/Δ43, TRE-RBFOX240; MHCrtTA bitransgenics and littermate control mice. Notably, the indicated exons in SCN5A and KCND3 ion channel transcripts were similarly misspliced in DM1 patients and RBFOX240 overexpressing mice (Fig. 7A–C) but not in individuals diagnosed with other forms of arrhythmias (Supplementary Fig. 8A). Further, in DM1 patient samples, we detected a strong positive correlation between SCN5A and KCND3 splicing defects and the relative expression of the non-muscle RBFOX240 isoform (Fig. 7B). We noticed that Scn5a and Kcnd3 were also misspliced in the hearts of Rbfox2Δ43/Δ43 mice, albeit to a lesser extent (Fig. 7C), which might explain their milder arrhythmia phenotypes compared to RBFOX240 overexpressing mice. We compared the effects of muscle-specific RBFOX243 and non-muscle RBFOX240 isoforms on SCN5A splicing in HL-1 cardiac cultures and found that only RBFOX240 overexpression stimulated missplicing in Scn5a transcripts (Supplementary Fig. 8b). Kcnd3 mRNA was undetectable in HL-1 cells. Redirection of Rbfox2 splicing with ASOs further confirmed that a switch from RBFOX243 to RBFOX240 isoform is sufficient to induce Scn5a missplicing in HL-1 cells (Supplementary Fig. 8C). Additionally, we detected large clusters of RBFOX240 footprints upstream of the Kcnd3 alternative exon in the iCLIP data (Damianov et al., 2016), indicating that the non-muscle RBFOX240 isoform directly binds these intronic regions and, when expressed at high levels, causes skipping of the downstream exon (Fig. 7D). As expected, no RBFOX240 binding on the Scn5a transcript was detected in the brain iCLIP dataset because Scn5a is a cardiac-specific sodium channel and is not expressed in the brain (Fig. 7D and Supplementary Fig. 8D, E).
Figure 7. RBFOX240 isoform expression in the heart reproduces DM1-related missplicing of voltage-gated sodium and potassium channels.

(A) RT-PCR analysis comparing the inclusion of 57-nt exon in KCND3 and 93-nt mutually exclusive exons (6A and 6B) in SCN5A transcripts in the hearts of unaffected individuals (n=4) and DM1 patients (n=9). (B) Scatter plot showing correlations between RBFOX240 isoform expression and misregulation of SCN5A or KCND3 splicing in DM1 patients. (C) RT-PCR analysis of Scn5a and Kcnd3 splicing patterns in hemizygous MHCrtTA, and TRE-RBFOX240; MHCrtTA bitransgenics induced with 0.5g/kg Dox for 3 days, as well as wildtype (WT) and Rbfox2Δ43/Δ43 mice. n=3 for all mouse samples. (D) UCSC genome browser snapshots demonstrating RBFOX240 footprints and splicing patterns of indicated alternative exons in Kcnd3 and Scn5a transcripts from 0.5g/kg Dox-induced TRE-RBFOX240; MHCrtTA bitransgenics (Orange panel) and littermate control mice (blue panel). X-axis: the position of RBFOX240 iCLIP data from mouse brain (Damianov et al., 2016) (top track), or RNA-seq reads across indicated transcripts (bottom two tracks). Y-axis: normalized read density scaled for each track. (E) Immunoblot analysis of RBFOX2 protein levels and RT-PCR analysis of Rbfox2, Kcnd3, and Scn5a alternative exons in the hearts of TRE-RBFOX240; MHCrtTA and littermate control (MHCrtTA) mice induced with 0.5g/kg Dox for 24h followed by a 72h recovery period. PSI values are shown below the gel images. n=3 mice for each genotype/condition. (F) Surface ECG analysis of arrhythmogenic events and PR intervals in TRE-RBFOX240; MHCrtTA and littermate control (MHCrtTA) mice induced with 0.5g/kg Dox for 72h followed by a 72h and 21-days recovery period. All data are mean ± s.d., and p-values were derived from a parametric t-test (two-sided, unpaired), with Welch’s correction, and from one-way ANOVA plus Dunnett’s post-hoc test. *p<0.05 (between MHCrtTA and TRE-RBFOX240; MHCrtTA), and #p<0.05 (between TRE-RBFOX240; MHCrtTA (induced) and TRE-RBFOX240; MHCrtTA (recovered).
To further investigate whether the missplicing of ion channels following RBFOX240 overexpression can be reversed, we first induced TRE-RBFOX240; MHCrtTA mice with Dox for 24h and then silenced the transgene in a subset of mice by withdrawing Dox for a period of 72h. In the induced mice, expression of RBFOX240 non-muscle isoform was rapidly increased (Fig. 7E), leading to a switch from exon 6B to 6A in Scn5a and skipping of the 57-nt exon in Kcnd3 transcripts (Fig. 7E). Turning off the transgene led to a marked reduction in RBFOX240 levels (Fig. 7E), and near-complete restoration of both Scn5a and Kcnd3 splicing to their normal patterns (Fig. 7E). Additionally, silencing the RBFOX240 transgene following a 72h induction period reduced the number of arrhythmic episodes and completely normalized the duration of the PR interval after 21 days of Dox withdrawal (Fig 7F). Overall, these data demonstrate that the non-muscle RBFOX240 isoform directs the production of pro-arrhythmic splice variants of sodium and potassium channels that elicit altered activation-deactivation kinetics, rates of ion transport, and electrophysiological properties. Furthermore, quick reversal of the ion channel missplicing and rescue of the conduction defects upon silencing of RBFOX240 transgene suggests that eliminating the aberrant RBFOX240 isoform from the heart might mitigate DM1 cardiac phenotypes.
DISCUSSION
Cardiac arrhythmias are a prominent cause of mortality in DM1; but, our understanding of the molecular events triggering electrophysiological abnormalities in DM1 heart is limited. While the degeneration of the conduction system, especially the bundle of His-Purkinje system is common, pathology within the parenchyma of the heart, such as diffuse interstitial fibrosis and myocardial dysfunctions also contribute to conduction abnormalities (Bhakta et al., 2010; Phillips and Harper, 1997). Cardiac imaging has indicated that nearly 20% of DM1 patients exhibit structural abnormalities including left ventricular hypertrophy, mitral valve prolapse, and left atrial dilatation (Bhakta et al., 2004). Overall it is likely that several cell types within the heart are affected and contribute to disease manifestations.
In this study, we provide multiple lines of evidence that inappropriate expression of non-muscle RBFOX240 isoform in the myocardium leads to DM1-like cardiac pathology, including conduction delay, atrioventricular heart blocks, and spontaneous arrhythmogenesis. RBFOX family represents a multifunctional group of sequence-specific RNA binding proteins that are critical regulators of splicing in multiple tissues including skeletal muscle, heart, and brain (Conboy, 2017; Gehman et al., 2012; Gehman et al., 2011; Jacko et al., 2018; Jangi and Sharp, 2014; Kalsotra et al., 2008; Singh et al., 2018; Singh et al., 2014). Loss-of-function mutations in RBFOX2 gene have been associated with congenital heart disease in humans (Homsy et al., 2015; McKean et al., 2016; Verma et al., 2016); whereas RBFOX2 activity in diabetic hearts is inhibited due to upregulation of its dominant negative splice isoform (Damianov and Black, 2010; Nutter et al., 2016). Recently, RBFOX1 and RBFOX2 proteins were shown to compete with MBNL1 for binding to expanded r(CCUG)exp repeats in DM2 but not to r(CUG)exp repeats in DM1 muscle cells (Sellier et al., 2018). However, unlike MBNL family of proteins, RBFOX binding to the r(CCUG)exp repeats does not result in sequestration or loss of splicing activity (Sellier et al., 2018).
We demonstrate that in DM1 patient hearts, levels of a highly active non-muscle RBFOX240 splice isoform are elevated. Our data support a model where combined effects of increased CELF1 activity and reduced expression of specific miRNAs allow for selective upregulation of RBFOX240 isoform in DM1 heart tissue. We further propose that compared to the muscle isoform, RBFOX240 stimulates the higher-order assembly of LASR complex, which boosts its splicing activity, resulting in altered isoform expression of proteins that can disrupt the normal cardiac rhythm and function. Indeed, we demonstrated that RBFOX240 promotes the generation of pathogenic splice variants of voltage-gated sodium and potassium channels that are known to exhibit slower conduction velocity and increased susceptibility to arrhythmias (Freyermuth et al., 2016; Kong et al., 1998; Pang et al., 2018; Xie et al., 2009). It is likely that RBFOX240-mediated missplicing of other components of the conduction system also contribute towards DM1-related cardiac dysfunctions. Nonetheless, our MD simulations show that a RBFOX240-mediated splicing switch in Nav1.5 transcript changes the amino acid composition of the voltage sensing element within domain I that promotes persistent lipid-interactions and stabilizes the 310-helical conformation of S4 helix, which reduces the energy barrier and allows for rapid deactivation of the channel. Accordingly, we found that tet-inducible RBFOX240 overexpression or targeted deletion of the 43-nt muscle-specific exon in Rbfox2 gene results in prolonged PR and QT intervals, inadvertent sinus pauses, atrioventricular heart blocks, and sudden death in mice. Notably, in a recent large-scale transcriptome study of human DM1 patients, RBFOX2 mRNA levels were inversely correlated with the strength of tibialis muscle (Wang et al., 2018). In the future, it would be interesting to determine whether increased RBFOX2 mRNA abundance in DM1 skeletal muscle also accompanies a corresponding splicing switch and whether the non-muscle RBFOX240 isoform acts as a disease modifier in skeletal muscle as well.
The RNA toxicity hypothesis for DM1 predicts that longer CUG repeats will lead to greater sequestration of MBNL proteins, resulting in increased severity of the disease. Although the severity tends to rise with repeat length, extreme variability in penetrance of specific symptoms is observed across DM1 patient population (Yum et al., 2017). For instance, cardiac involvement is reported in more than 80% of individuals, and conduction abnormalities can cause up to 30% of DM1 fatalities (Groh et al., 2008; Heatwole et al., 2012; Tokgozoglu et al., 1995). However, which patients have the highest risk for sudden cardiac death and what molecular events dictate these outcomes are currently unknown. Previous studies have indicated that high NKX2–5 and PKC activities can modify DM1-associated RNA toxicity in the heart (Wang et al., 2009; Yadava et al., 2008). In our study, we noticed that RBFOX240 isoform levels varied considerably among the hearts of DM1 patients. Whether the degree of RBFOX240 upregulation depends on the size of CTG repeat expansion and/or if a threshold for RBFOX240 levels exists that increases the susceptibility of DM1 patients to cardiac arrhythmias remain topics for future investigations. Nonetheless, the fact that normal cardiac rhythm and splicing patterns for both sodium and potassium channels could be restored within days of silencing the RBFOX240 expression highlights the potential of RBFOX240 isoform as a therapeutic target and a biomarker for DM1 cardiac pathology.
STAR*METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Auinash Kalsotra (kalsotra@illinois.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Experimental Animals
For generating Rbfox240 non-muscle isoform overexpression mouse model, the mouse RBFOX240 cDNA, harboring a N-terminus sequence coding for FLAG-tag, was expressed from a transgene with a TRE/minimal CMV promoter, RBFOX2 ORF containing non-muscle 40-nt (B40) exon and bovine growth hormone polyadenylation site and 3’ flanking genomic segment for proper mRNA 3’-end formation. The linearized transgene construct was subjected to pronuclear injection in the transgenic mouse facility at UIUC, using standard methods to generate TRE-RBFOX240 transgenic mice that were maintained on a C57BL/6J background. MHCrtTA transgenic mice (FVB/N-Tg (Myh6rtTA)1Jam) expressing a codon-optimized rtTA variant specifically in cardiomyocytes were commercially obtained (RRID: MMRRC_010478) (Valencik and McDonald, 2001). Henceforth, mice reported were the F1 progeny of TRE-RBFOX240 and MHCrtTA mating and were, therefore, hemizygous for one or both transgenes. Primers used to genotype both transgenes are listed in Supplementary Table 1. RBFOX240 isoform expression in 8- to 12-week-old bitransgenic animals was induced through doxycycline (Dox) in the food (0.5g Dox/kg food, Harlan, KY).
To delete the muscle-specific 43-nt exon in Rbfox2, two single guide RNAs (sgRNAs) were purchased (Sigma-Aldrich, St. Louis, Missouri) that flank the genomic region across the 43-nt exon of Rbfox2 (5’sgRNA: 5’GAGTGAGAAGAAGTCGCTCGGG3’ and 3’sgRNA: 5’GAGGCCAAACATTAGACCCTGG3’). After complex formation with clustered regularly interspaced short palindromic repeat (CRISPR)-associated protein 9 (Cas9) (CP01–20, Newbury Park, California), the Cas9-sgRNA mixture was delivered into the cytoplasm of ~100 pronuclear stage zygotes (C57BL/6J). Injected zygotes were transferred into pseudo-pregnant ICR females (25–30 zygotes/female). The sgRNAs produced double-stranded cuts, which removed the targeted Rbfox2 genomic sequence (Supplementary Figure 4). To identify the positive founder animals wherein, double‐stranded breaks induced by Cas9 resulted in non-homologous end joining (NHEJ) repair and created a null allele, mice were genotyped using standard PCR on tail-clip derived genomic DNA. Primers flanking the 2 sgRNA sites were designed (Supplementary Table 1) to amplify a smaller deletion amplicon compared with the WT amplicon. An additional primer pair was designed within the Rbfox2 43-nt exon to confirm deletion by the absence of the amplicon in PCR reactions (Supplementary Table 1). The germline deletion of 43-nt exon was further confirmed by sanger sequencing of the F1 progeny mice homozygous for the 43-nt deletion (Rbfox2Δ43/Δ43).
Use and care of laboratory animals were followed according to the National Institutes of Health (NIH) and guidelines set by the Institutional Animal Care and Use Committee (IACUC) at University of Illinois, Urbana-Champaign (UIUC). Animals used in the study were identified using ear tags and genotyped before weaning age. Experiments within the study were not gender-specific and specimens included both male and female animals. Whole heart tissues and cardiomyocytes were isolated from mice following the IACUC guidelines for euthanasia and anesthesia.
Human Samples
Human fetal (22-week old) heart RNAs were purchased from Clonetech Laboratories, Inc., (Mountain View, CA). Three unaffected human adult heart RNAs were from 51-year-old Caucasian male were purchased from Clonetech Laboratories, Inc., (Mountain View, CA), pooled from 3 male Caucasians, ages: 30–39 purchased from Takara Bio USA, Inc., (Mountain View, CA) and 24-year-old male RNA from Bio chain Institute Inc., (Newark, CA). Human DM1 RNA samples and other unaffected samples were from a 50-year-old male (respiratory failure [RF]), 48-year-old female (1,500 repeats, RF), 55-year-old male (pneumonia [PN]), 52-year-old female (>1,000 repeats, RF), 46-year-old male (PN), 50-year-old female (RF), 53-year-old male (unknown cause), 26-year-old male (glioma), 55-year-old male (pulmonary embolism). Unaffected heart samples were pooled autopsy samples ranging from 21- to 55-year-old individuals. Human non-DM cardiac arrhythmia samples (severe) were from 26-year-old male (Ventricular septal defects since infancy, sudden death), 47-year-old male (non-ischemic cardiomyopathy, >20 Ejection fraction [EF]), 56-year-old male (Sinus tachycardia, left posterior fascicular block, abnormal ECG). All three severe arrhythmic patients suffered from sudden cardiac arrest. Three less severe non-DM arrhythmia patient samples had a history of Ventricular tachycardia but ECG within normal limits at the time of death. All individuals in the less severe group also suffered from ischemic heart failure. They were from 50-year-old male (>15 EF), 56-year-old female (>15 EF), 64-year-old male (>24 EF).
METHOD DETAILS
Conscious telemetry recordings and Surface ECGs
Telemetry studies were performed in Mouse Cardiovascular Phenotyping Core, Washington University School of Medicine in St. Louis, with 3-month old TRE-RBFOX240; MHCrtTA (n=2 male, n=2 female) mice. ETA-F10 implantable radio frequency transmitters for ECG (Data Sciences International Inc.) were implanted subcutaneously in the posterior neck of adult mice. Leads were tunneled to the anterior chest in lead II position. After a post-implant recovery period of one week, ambulatory heart rhythm and heart rate (HR) were monitored in unrestrained, caged mice. After baseline recording for 24h, 0.1g/kg Dox containing diet was administered to induce expression of the RBFOX240 transgene. Recordings were obtained continuously at 1 kHz for nine days. The ECG Analysis module for Lab Chart (Data Sciences International Inc.) was used to automatically quantify heart rate, PR, RR, QRS, and QT intervals and the voltages of ECG recordings. Lab Chart was used to manually identify ECG events, including, premature ventricular contractions (PVCs), sinus pause (lasting at least 1.5-times as long as the preceding RR interval) and atrioventricular conduction block. Surface ECGs were captured from leads I, II and III with a MP150 data acquisition system (BIOPAC Systems Inc., Goleta, California). Two to three month‐old wild type C57BL/6J (n=6 males, n=4 females ), Rbfox2Δ43/Δ43 (n=8 female, n=6 female), MHCrtTA with Dox (n=3 male, n=2 female), TRE-RBFOX240; MHCrtTA with and without Dox (n=6 male, n=4 female) ), or after withdrawal of Dox (n=4 male, n=3 female) were anesthetized in an induction chamber using 2.5% isoflurane in 100% oxygen and maintained during data collection with 1.5% isoflurane. Body temperature was monitored by a rectal probe. Data were collected for 2 minutes per mouse and annotated for analysis using ACQ Knowledge software (BIOPAC Systems Inc., Goleta, California).
Echocardiography
In vivo cardiac function and morphology were evaluated using a Vevo 2100 ultrasound machine equipped with a 40 MHz transducer‐MS550S (VisualSonics, Toronto, Ontario, Canada) at the Beckman Institute for Advanced Science and Technology (UIUC). Mice were first anesthetized in an induction chamber using 2.0% isoflurane in 100% oxygen and then the anesthesia (1% Isoflurane) maintained by nose cone delivery. During the imaging procedure, mice were transferred to a heated ECG platform for heart rate monitoring. Body temperature was maintained at 37°C, monitored through a rectal probe. Two-dimensional M-mode echocardiography images were taken in the short‐axis position for each animal. Data analysis was performed using the VisualSonics Vevo Lab analysis package. Three M‐mode tracings were analyzed and averaged for every animal.
Histology
Heart tissues from Dox-induced MHCrtTA and TRE-RBFOX240; MHCrtTA, as well as wildtype and Rbfox2Δ43/Δ43 mice were harvested and fixed overnight in 10% neutral-buffered formalin, embedded in paraffin, and sectioned (5 μm thickness). Unstained slides were deparaffinized in xylene (two treatments, 5 min each), rehydrated sequentially in ethanol (2 min each in 100%, 95%, and 80%), and washed for 3 min in water. For Hematoxylin and eosin (H&E) staining, sections were washed in Hematoxylin (2 min) and Eosin (1 min) successively, cover slipped with Permount medium, and imaged on Hamamatsu Nanozoomer. For Sirius Red staining, sectioned tissues were stained in picro-sirius red for an hour, washed in acidified water, successively dehydrated in 100% ethanol, finally cleared in xylene and mounted.
Isolation of neonatal and adult cardiomyocytes
Neonatal cardiomyocytes were isolated from wildtype C57BL/6J mice with a neonatal mouse cardiomyocyte isolation kit (Cellutron Life Tech Highland Park, NJ, USA; nc-6031) as previously described (Chorghade et al., 2017). Cells from 12 to 18 hearts were pooled, pre-plated for 2h on an uncoated dish to separate fibroblasts from cardiomyocytes. Adult cardiomyocytes from MHCrtTA and TRE-RBFOX240; MHCrtTA mice—induced with 0.5g/kg Dox containing diet for 3 days—were isolated by cannulating the hearts through the aorta and perfusing on a Langendorff apparatus for 4 min at 3 ml/min with perfusion buffer (NaCl 58.4mM, KCl 74.55mM, MgSO4 120.4mM, Na2HPO4 142mM, KH2PO4 136.1mM, NaHCO3 84 12mM, KHCO3 101.12mM, Taurine 125.1mM, Phenol red 376.4mM, Hepes 10mM) and then with digestion buffer (perfusion buffer containing 2.7 mg Liberase TM (Roche Life Sciences, Germany) for ~8 to 10mins at 37 °C. Mice were treated with anticoagulant (500U heparin i.p.) 30 min prior to heart extractions. After perfusion, the atria were removed, and the heart was minced in the digestion buffer. Minced tissue was gently pipetted up and down on ice to release the cells, which were filtered through a 100μm pluristrainer mesh (pluriSelect Life Science, Leipzig, Germany) and then centrifuged for 5 min at 3,000 g. Cardiomyocytes and fibroblasts were separated using the pre-plating method and their purity was determined by qRT-PCR analysis. Cardiomyocytes isolated from two hearts were pooled and RNA was immediately extracted with Qiagen RNeasy kit (QIAGEN Inc., Germantown, Maryland).
RNA sequencing and computational analysis
Total RNA was purified from freshly isolated cardiomyocytes using RNeasy tissue mini-kit (Qiagen). RNA quality was analyzed with Agilent Bioanalyzer and quantified using a Qubit fluorimeter at the Functional Genomics Core at the Roy J. Carver Biotechnology Center, UIUC. All RNA samples had at least A260nm/A280nm≥1.8, A260nm/A230nm≥1.4, r28S:16S≥1.5, and RNA integrated number (RIN)≥6.5. Hi-Seq4000 libraries were prepared, and 100-bp paired-end Illumina sequencing was performed on a HiSeq platform at the High Throughput Sequencing and Genotyping Unit, UIUC. Computational analysis of RNA-seq experiments for this study, and of previously published datasets (Supplementary Table 2) was performed as previously described (Bangru et al., 2018). Briefly, the sequencing reads were processed for quality and read length filters with Trimmomatic (version 0.38)(Bolger et al., 2014). RNA-seq reads were further aligned to the mouse (mm10) or human (hg38) genomes using STAR (version 2.4.2a) (Dobin et al., 2013). Mapping percentages and sample details are provided in Supplementary Table 2. Gene expression levels were determined as FPKM/TPM using count and differential expression values obtained from Cuffdiff(Trapnell et al., 2012). Genes were considered as having significantly different expression following imposed cutoff clearance (FDR (q-value) <0.05, log2(fold change) >1). Differential splicing analysis was performed using rMATS (version 3.2.5), and significant events were identified with imposed cutoffs (FDR <0.10, junction read counts ≥20, PSI ≥20%)(Shen et al., 2014). Motif analysis for differentially spliced exons was performed using rMAPS with default parameters, and by adding putative motifs as described previously(Bhate et al., 2015; Park et al., 2016). Gene ontology analysis was performed with DAVID version 6.8 (Dennis et al., 2003), and mapped using the ‘Enrichment Maps’ plugin in Cytoscape (Merico et al., 2010). All expressed genes with TPM>1 served as background, and the biological function category was analyzed with three pathways (Biocarta, Kegg, and Panther). Functional clustering was performed and top clusters (P value <0.05) represented.
To identify co-regulated exons between RNA-seq samples from mouse and human experiments, the analysis was limited to mm10 annotated mouse cassette exons, which were converted to corresponding human exons in the hg38 annotation using UCSC liftover with minimum ratio of bases matching as 0.8. To identify RBFOX240-regulated exons in mouse cardiomyocytes that also contained RBFOX240 binding peaks, previously published iCLIP data for heavy molecular weight fraction (HMW) from mouse brains was used (Damianov et al., 2016). RBFOX240 binding clusters near these regulated exons were identified with the HOMER pipeline using its standard parameters (Heinz et al., 2010). To associate alternative exons with RBFOX240 binding, custom scripts were used to search for binding peaks present either in the exon or the surrounding upstream or downstream introns. To identify the crosslink sites for peaks associated with alternative splicing events, the CTK pipeline from CITS algorithm was used with default parameters (Shah et al., 2017; Weyn-Vanhentenryck et al., 2014). After crosslink sites were identified, strand-specific sequences within a 70-nt window were obtained using getfasta script in the bedtools suite, which was followed by motif enrichment analysis using the MEME suite with standard parameters (Bailey et al., 2009).
Protein isolation and western blot analysis
Protein lysates were prepared from indicated frozen heart tissues or freshly isolated cardiomyocytes by homogenization in bullet blender followed by sonication. Homogenization buffer (10 mM HEPES-KOH, pH 7.5, 0.32 M sucrose, 5 μM MG132, 5 mM EDTA, and Pierce proteinase inhibitor tablet (1 tablet/ 20 mL buffer volume) was used to isolate protein from heart tissues. Prior to sonication 20% Sodium Dodecyl sulfate (SDS) to a final concentration of 1% (v/v) was added. Protein concentrations were measured using Thermo scientific BCA assay kit. Approximately 50 μg of total protein sample was loaded onto a 10% SDS-PAGE gel, and then transferred onto a PVDF membrane overnight at 4°C. Membranes were blocked using 5% milk powder (w/v) in TBST (Tris-buffered saline, 0.1% Tween 20) for 2 hours at room temperature (RT). Blots were incubated in primary antibodies at pre-determined concentrations for 2 hours. Blots were washed in TBST, and then incubated in HRP conjugated secondary antibodies for 1 hour at RT. Blots were developed using Clarity Western ECL kit (Bio-Rad). All antibodies used, and their respective dilutions are listed in Key Resource Table.
Gene expression and splice isoform analysis
Total RNA from mouse hearts and cardiomyocytes, were isolated using either RNeasy kit (QIAGEN Inc., Germantown, Maryland) or TRIzol reagent (Thermo Fisher Scientific, Waltham, Massachusetts). Upon DNase I treatment, 2 μg of RNA was reverse transcribed to cDNA using random hexamers and Maxima Reverse transcriptase kit (Thermo Fisher Scientific, Waltham, Massachusetts). The cDNA was diluted to a final concentration of 25 ng/μL and Real-time q-RTPCR and RT-PCR based alternative splicing assays were performed as described previously (Kalsotra et al., 2010). For splicing assays, the PCR products were analyzed by electrophoresis on a 6.5% polyacrylamide gel, stained with ethidium bromide and quantified using a Chemidoc XRS+ imaging system (Bio-Rad, Hercules, CA). PSI values for the variably spliced region were calculated with ImageLab software (BioRad) as [(exon inclusion band intensity) / (exon inclusion band intensity+exon exclusion band intensity) × 100]. All primers used for alternative splicing are listed in Supplementary Table 1. SCN5A exons 6A and 6B are of similar size (93-nts), therefore, PCR products were digested by BstbI enzyme before loading on 6.5% polyacrylamide gel, as previously described (Freyermuth et al., 2016).
qRT-PCR was performed using the PerfeCTa SYBR® Green SuperMix (Quanta Biosciences, Beverly, Massachusetts) in an ABI QuantStudio3 (Thermo Fisher Scientific, Waltham, Massachusetts) with 10 min at 95 °C followed by 40 cycles of 15 s at 95 °C, 1:00min at 60°C using primers described in the Supplementary Table 1. Gapdh mRNA was used as a loading control and data were analyzed using the 2–ΔΔCt method as previously described (Kalsotra et al., 2008).
miRNA profiling
To quantify mature miRNA expression, we used TaqMan stem-loop RT-PCR MicroRNA Assays (Applied Biosystems). Briefly, 100 ng of total RNA from each sample was reverse-transcribed using specific miRNA primers from the TaqMan MicroRNA Assays and the TaqMan miRNA reverse transcription kit. Quantitative real-time PCR reactions were performed in triplicate on an ABI QuantStudio3 (Thermo Fisher Scientific, Waltham, Massachusetts) Real-Time PCR System using miRNA-specific TaqMan probe and ABI TaqMan Universal PCR Master Mix. An initial denaturation step of 10 min at 95°C was followed by 40 cycles of 95°C for 15 s and 60°C for 1 min. U6 snRNA was used to n ormalize the miRNA expression, and Fold change of miRNA expression was calculated as previously described (Kalsotra et al., 2014).
Cell culture and transfection
HL-1 cardiomyocytes were cultured on gelatin (0.02%, w/v)/fibronectin (10μg/ml) coated plates with Claycomb Medium supplemented with 10% FBS (Sigma-Aldrich Corporation, St. Louis, Missouri) 2 mM L-glutamine, 0.1 mM Norepinephrine, 100 units/mL penicillin and 100 μg/mL streptomycin and were maintained at 37°C in 5% CO2 as previously descried (Kalsotra et al., 2014). After the cells reached 80–90% confluency and started beating, the cultures were split into new T75 flasks for maintenance or 12 well plates for further experiments. HL-1 cells were transiently transfected with DT0 and DT960 plasmids with Lipofectamine 3000 (Thermo Fisher Scientific, Waltham, Massachusetts) using manufacturer’s protocol. To increase the transfection efficiency, we applied reverse transfection technique, where the plasmid DNA and transfection reagent complex is assembled in the tissue culture plate and then the cells are seeded into the wells. Forty-eight hours later cells were harvested to isolate total RNA and protein. Celf1 siRNA delivery was performed 12 h after DT0 or DT960 plasmid transfections, using Lipofectamine RNAi-Max (Thermo Fisher Scientific, Waltham, Massachusetts) according to manufacturer’s recommendations. Final concentration of siRNAs was 10nM. The sequences/reference numbers of siRNAs used are listed in Supplementary Table 1. To induce Rbfox2 43-nt skipping, an antisense morpholino oligomer (Rbfox2-43 ASO) sequence was used: 5’-CACTGGCACTCCTAC CTGAGGTATT-3’, which binds to the 5’ss of Rbfox2 exon 43 pre-mRNA sequence. As a control, a non-target standard control sequence was used:5’-CCTCTTACCTCAGTTACAATTTATA-3’ (Gene Tools, LLC, Philomath, Oregon, USA). Morpholinos were delivered at 10uM final concentration using Endo-porter reagent in HL-1 cells for 48h and cells were harvested to prepare total RNA and protein lysates. For isoform-specific overexpression studies, the FLAG-RBFOX240 and FLAG-RBFOX243 plasmids were transfected in HL-1 cells for 48h and cells were harvested to prepare total RNA and protein lysates.
Luciferase activity assays
The 3’-UTR of mouse Rbfox2 was cloned at the Not1 and Xho1 site of the psiCHECK™−2 plasmid (Promega), downstream of Renilla Luciferase (hRLuc). The primers used to amplify the Rbfox2 3’-UTR are listed in Supplementary Table 1. To generate the indicated luciferase constructs, In-Fusion HD cloning system (Clontech) was used. Next, we introduced mutations in Let-7, miR-9 and miR-135a binding sites within the Rbfox2 3’-UTR construct by using the QuikChangeII Mutagenesis Kit (Stratagene, La Jolla, CA, USA). A combined Let-7+miR-9+miR-135a mutant construct was also generated in which all three miRNAs binding sites were mutated simultaneously. The primers used for incorporating mutations were designed using Quick-change primer design software and are listed in Supplementary Table 1. HEK293T cells were seeded on twelve-well plates at approximately 70–80% confluence and after 24h, 1μg of luciferase expression construct and 20nM of miRIDIAN microRNA Mimics (Dharmacon, USA) were co-transfected with TransIT-X2 (Mirus Bio LLC, USA). A Non-targeting mimic was used as control (miRIDIAN microRNA Mimic Negative Controls, CN-001000–01-05, Dharmacon, USA). After 48h from transfection, the cells were lysed and assayed with Dual Luciferase Assay (Promega) according to the manufacturer’s instructions. Wildtype and mutant Rbfox2 3’-UTR constructs were tested in three independent experiments. Renilla luciferase activity was normalized with the Firefly luciferase activity levels and expressed as relative luciferase units (RLU). For the rescue experiments, HL-1 cells were reverse-transfected with DT0 or DT960 plasmids. Twelve hours later, Rbfox2 3’-UTR reporter plasmid was co-transfected with a cocktail of 10nM Let-7g+miR-9+miR-135a miRNA mimics with TransIT-X2 (Mirus Bio LLC, USA). Cells were incubated for 48h, collected for RNA and protein extractions, and luciferase activity assays.
Adenovirus production
cDNAs encoding FLAG-tagged human CELF1 were sub-cloned into the p-Adeno-X-ZsGreen1 vector (Clonetech, 632267) using the In-Fusion kit (Clonetech, 639646) as per the manufacturer’s instructions. High-titer adenoviruses were generated and purified as previously described (Bhate et al., 2015). To determine the effect of CELF1 overexpression, six-wells containing ~70% confluent HL-1 cells were infected with 2.0 × 109 o.p.u. (optical particle units) of the CELF1 or GFP adenovirus for 48h and cells were harvested to extract RNA and protein for further analysis.
Homology modeling
The cryo-EM structures of eukaryotic sodium channels Nav1.4 (pdb:6agf) and NavPas (pdb:6a95) were used as templates for modeling a human sodium channel Nav1.5 (SCN5A, UniProtKB: Q14524). The sequence segments corresponding to the domain I-II linker and domain II-III linker were truncated from the full human SCN5A protein sequence to allow better alignment with the templates. Multi-sequence alignment between the templates and target sequence was performed using Clustal Omega (Sievers and Higgins, 2014) and the homology modeling was performed using MODELLER v9.21 (Eswar et al., 2006). 50 models were generated for both the adult and fetal isoforms of SCN5A and the model with the best objective function score was selected for each isoform to perform molecular dynamics (MD) simulations.
MD simulations
Given that the three-dimensional structure of Nav1.5 channel is unavailable, we built homology models of the exon 6A or exon 6B containing human Nav1.5 isoforms using existing cryo-EM structures of the activated sodium channels Nav1.4 and NavPas (Pan et al., 2018; Shen et al., 2018), which share 80% and 47% of sequence identity with the human Nav1.5 respectively. The transmembrane sequence segments of human Nav1.5 (SCN5A, UniProtKB: Q14524) was aligned with that of the template structures and the homology modeling was performed using MODELLER (Eswar, N. et al., 2006). The resulting human Nav1.5 model has a virtually identical sequence (94.5% identity) as the mouse Nav1.5. These models were then used in subsequent MD simulations, where three independent 100ns-long equilibrium simulations were carried out on 6A or 6B containing Nav1.5 isoforms.
The selected homology models for both isoforms were embedded in a POPC lipid bilayer and solvated with 0.15 M of NaCl and TIP3P water (William L. Jorgensen, 1983) using CHARMM-GUI Membrane Builder (Wu et al., 2014). The dimension for both systems is 140 Å×140 Å×145 Å. Both systems underwent 1 ns NPT initial equilibration with the standard protocol described in the CHARMM-GUI Membrane Builder, which involves gradually releasing positional and dihedral restraints on the protein and lipid molecules. Thereafter, 10 ns of NPT equilibration with dihedral restraints (k=100 kcal/mol/rad2) on the protein secondary structure were performed. The last frame of each system was then used for three independent (different initial velocities) NPT simulations. To remove any initial structural bias from the initial model, each independent trajectory was further equilibrated without restraint for 20 ns, followed by 100 ns production run. All simulations were carried out with NAMD 2.13 (Phillips et al., 2005), using CHARMM36m protein (Huang et al., 2017) and CHARMM36 lipid (Klauda et al., 2010) parameters. SHAKE algorithm (Jean-PaulRyckaert, 1977) was employed to constrain hydrogen bond lengths to allow 2 fs time steps for the integrator. Constant temperature of 310 K was maintained by Langevin thermostat (Martyna, 1994) with a damping coefficient of 1 ps−1. Nosé-Hoover Langevin piston (Scott E. Feller, 1995) with a period of 200 ps and decay time of 50 ps was employed to maintain constant pressure at 1 atm. Periodic boundary condition and a nonbonded cutoff of 12 Å (with 10 Å switching distance and vdW force switching) were used. Long-range electrostatics were calculated using the particle mesh Ewald method (Tom Darden, 1993) with 1 Å grid spacing.
QUANTIFICATION AND STATISTICAL ANALYSIS
All experiments have at least three independent biological repeats. Differences between groups were examined for statistical significance using Student’s t-test with Welch’s correction (for two groups), or one-way ANOVA plus Dunnett’s post-hoc test (for more than two groups) using the GraphPad Prism 7 Software. Results were expressed as mean ± s.d., unless otherwise specified. *p<0.05, **p<0.001, were considered statistically significant.
DATA AND CODE AVAILABILITY
All raw RNA-seq data files are available for download from NCBI Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE126771.
Supplementary Material
Supplementary Video 1. Interactions of D211 in SCN5A_6B with the lipid head group. This supplemental video is related to Figure 6.
Supplementary Video 2. Interactions of K211 in SCN5A_6A with the lipid head group. This supplemental video is related to Figure 6.
Highlights.
Aberrant expression of the non-muscle RBFOX240 isoform in hearts of DM1 patients.
Forced expression of RBFOX240 isoform reproduces DM1-like cardiac pathology in mice
RBFOX240 isoform induces DM1-related splicing defects in voltage-gated ion channels
Silencing RBFOX240 restores the normal cardiac rhythm and splicing of ion channels
ACKNOWLEDGEMENTS
This research was supported through NIH (R01HL126845) and Muscular Dystrophy Association (MDA514335) grants to A.K., NIH (R01HL134824; R35HL135754) to P.J.M., NIH (P41-GM104601, R01-GM122420, and R01-GM123455) to E.T. and NIH (R01AR045653 and R01HL045565) to T.A.C. C.M. was supported by American Heart Association post-doctoral fellowship (16POST29950018). S.B. was supported by the NIH Tissue microenvironment training program (T32-EB019944). J.H. was supported by Beckman-Brown postdoctoral fellowship. All simulations were performed using the Blue Waters PRAC allocation (grant ACI1713784 to E.T.) of the National Center for Supercomputing Applications at UIUC. Three cores at UIUC supported this project: Transgenic mouse facility core, High-throughput sequencing and genotyping core and Histology-microscopy core. Cardiovascular phenotyping was carried out in part in the Biomedical Imaging Center at the Beckman Institute for Advanced Science and Technology at the University of Illinois at Urbana-Champaign and in the Mouse Cardiovascular Phenotyping Core at the Washington University School of Medicine, St. Louis, MO.
Footnotes
COMPETING INTERESTS
The authors declare no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, and Noble WS (2009). MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res 37, W202–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bangru S, Arif W, Seimetz J, Bhate A, Chen J, Rashan EH, Carstens RP, Anakk S, and Kalsotra A (2018). Alternative splicing rewires Hippo signaling pathway in hepatocytes to promote liver regeneration. Nat Struct Mol Biol 25, 928–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batra R, Charizanis K, Manchanda M, Mohan A, Li M, Finn DJ, Goodwin M, Zhang C, Sobczak K, Thornton CA, et al. (2014). Loss of MBNL Leads to Disruption of Developmentally Regulated Alternative Polyadenylation in RNA-Mediated Disease. Molecular cell 56, 311–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhakta D, Groh MR, Shen C, Pascuzzi RM, and Groh WJ (2010). Increased mortality with left ventricular systolic dysfunction and heart failure in adults with myotonic dystrophy type 1. Am Heart J 160, 1137–1141, 1141 e1131. [DOI] [PubMed] [Google Scholar]
- Bhakta D, Lowe MR, and Groh WJ (2004). Prevalence of structural cardiac abnormalities in patients with myotonic dystrophy type I. Am Heart J 147, 224–227. [DOI] [PubMed] [Google Scholar]
- Bhate A, Parker DJ, Bebee TW, Ahn J, Arif W, Rashan EH, Chorghade S, Chau A, Lee JH, Anakk S, et al. (2015). ESRP2 controls an adult splicing programme in hepatocytes to support postnatal liver maturation. Nat Commun 6, 8768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, and Usadel B (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brook JD, McCurrach ME, Harley HG, Buckler AJ, Church D, Aburatani H, Hunter K, Stanton VP, Thirion JP, Hudson T, et al. (1992). Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3’ end of a transcript encoding a protein kinase family member. Cell 68, 799–808. [DOI] [PubMed] [Google Scholar]
- Charizanis K, Lee KY, Batra R, Goodwin M, Zhang C, Yuan Y, Shiue L, Cline M, Scotti MM, Xia G, et al. (2012). Muscleblind-like 2-mediated alternative splicing in the developing brain and dysregulation in myotonic dystrophy. Neuron 75, 437–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlet BN, Savkur RS, Singh G, Philips AV, Grice EA, and Cooper TA (2002). Loss of the muscle-specific chloride channel in type 1 myotonic dystrophy due to misregulated alternative splicing. Molecular cell 10, 45–53. [DOI] [PubMed] [Google Scholar]
- Chorghade S, Seimetz J, Emmons R, Yang J, Bresson SM, Lisio M, Parise G, Conrad NK, and Kalsotra A (2017). Poly(A) tail length regulates PABPC1 expression to tune translation in the heart. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conboy JG (2017). Developmental regulation of RNA processing by Rbfox proteins. Wiley interdisciplinary reviews. RNA 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damianov A, and Black DL (2010). Autoregulation of Fox protein expression to produce dominant negative splicing factors. Rna 16, 405–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damianov A, Ying Y, Lin CH, Lee JA, Tran D, Vashisht AA, Bahrami-Samani E, Xing Y, Martin KC, Wohlschlegel JA, et al. (2016). Rbfox Proteins Regulate Splicing as Part of a Large Multiprotein Complex LASR. Cell 165, 606–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis BM, McCurrach ME, Taneja KL, Singer RH, and Housman DE (1997). Expansion of a CUG trinucleotide repeat in the 3’ untranslated region of myotonic dystrophy protein kinase transcripts results in nuclear retention of transcripts. Proc Natl Acad Sci U S A 94, 7388–7393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day JW, and Ranum LP (2005). Genetics and molecular pathogenesis of the myotonic dystrophies. Current neurology and neuroscience reports 5, 55–59. [DOI] [PubMed] [Google Scholar]
- Dennis G Jr., Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, and Lempicki RA (2003). DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol 4, P3. [PubMed] [Google Scholar]
- Dixon JE, Shi W, Wang HS, McDonald C, Yu H, Wymore RS, Cohen IS, and McKinnon D (1996). Role of the Kv4.3 K+ channel in ventricular muscle. A molecular correlate for the transient outward current. Circulation research 79, 659–668. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eswar N, Webb B, Marti-Renom MA, Madhusudhan MS, Eramian D, Shen MY, Pieper U, and Sali A (2006). Comparative protein structure modeling using Modeller. Curr Protoc Bioinformatics Chapter 5, Unit-5 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Costa JM, Garcia-Lopez A, Zuniga S, Fernandez-Pedrosa V, Felipo-Benavent A, Mata M, Jaka O, Aiastui A, Hernandez-Torres F, Aguado B, et al. (2013). Expanded CTG repeats trigger miRNA alterations in Drosophila that are conserved in myotonic dystrophy type 1 patients. Human molecular genetics 22, 704–716. [DOI] [PubMed] [Google Scholar]
- Freyermuth F, Rau F, Kokunai Y, Linke T, Sellier C, Nakamori M, Kino Y, Arandel L, Jollet A, Thibault C, et al. (2016). Splicing misregulation of SCN5A contributes to cardiac-conduction delay and heart arrhythmia in myotonic dystrophy. Nat Commun 7, 11067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu YH, Pizzuti A, Fenwick RG Jr., King J, Rajnarayan S, Dunne PW, Dubel J, Nasser GA, Ashizawa T, de Jong P, et al. (1992). An unstable triplet repeat in a gene related to myotonic muscular dystrophy. Science 255, 1256–1258. [DOI] [PubMed] [Google Scholar]
- Fugier C, Klein AF, Hammer C, Vassilopoulos S, Ivarsson Y, Toussaint A, Tosch V, Vignaud A, Ferry A, Messaddeq N, et al. (2011). Misregulated alternative splicing of BIN1 is associated with T tubule alterations and muscle weakness in myotonic dystrophy. Nature medicine 17, 720–725. [DOI] [PubMed] [Google Scholar]
- Gehman LT, Meera P, Stoilov P, Shiue L, O’Brien JE, Meisler MH, Ares M Jr., Otis TS, and Black DL (2012). The splicing regulator Rbfox2 is required for both cerebellar development and mature motor function. Genes Dev 26, 445–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehman LT, Stoilov P, Maguire J, Damianov A, Lin CH, Shiue L, Ares M Jr., Mody I, and Black DL (2011). The splicing regulator Rbfox1 (A2BP1) controls neuronal excitation in the mammalian brain. Nature genetics 43, 706–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gellens ME, George AL Jr., Chen LQ, Chahine M, Horn R, Barchi RL, and Kallen RG (1992). Primary structure and functional expression of the human cardiac tetrodotoxin-insensitive voltage-dependent sodium channel. Proc Natl Acad Sci U S A 89, 554–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giudice J, Xia Z, Wang ET, Scavuzzo MA, Ward AJ, Kalsotra A, Wang W, Wehrens XH, Burge CB, Li W, et al. (2014). Alternative splicing regulates vesicular trafficking genes in cardiomyocytes during postnatal heart development. Nat Commun 5, 3603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groh WJ, Groh MR, Saha C, Kincaid JC, Simmons Z, Ciafaloni E, Pourmand R, Otten RF, Bhakta D, Nair GV, et al. (2008). Electrocardiographic abnormalities and sudden death in myotonic dystrophy type 1. N Engl J Med 358, 2688–2697. [DOI] [PubMed] [Google Scholar]
- Heatwole C, Bode R, Johnson N, Quinn C, Martens W, McDermott MP, Rothrock N, Thornton C, Vickrey B, Victorson D, et al. (2012). Patient-reported impact of symptoms in myotonic dystrophy type 1 (PRISM-1). Neurology 79, 348–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Molecular cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho TH, Charlet BN, Poulos MG, Singh G, Swanson MS, and Cooper TA (2004). Muscleblind proteins regulate alternative splicing. The EMBO journal 23, 3103–3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homsy J, Zaidi S, Shen Y, Ware JS, Samocha KE, Karczewski KJ, DePalma SR, McKean D, Wakimoto H, Gorham J, et al. (2015). De novo mutations in congenital heart disease with neurodevelopmental and other congenital anomalies. Science 350, 1262–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Rauscher S, Nawrocki G, Ran T, Feig M, de Groot BL, Grubmuller H, and MacKerell AD Jr. (2017). CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat Methods 14, 71–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacko M, Weyn-Vanhentenryck SM, Smerdon JW, Yan R, Feng H, Williams DJ, Pai J, Xu K, Wichterle H, and Zhang C (2018). Rbfox Splicing Factors Promote Neuronal Maturation and Axon Initial Segment Assembly. Neuron 97, 853–868 e856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jangi M, and Sharp PA (2014). Building robust transcriptomes with master splicing factors. Cell 159, 487–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jean-PaulRyckaert G, Herman J Berendsen C (1977). Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. Journal of Computational Physics 23, 327–341. [Google Scholar]
- Jin Y, Suzuki H, Maegawa S, Endo H, Sugano S, Hashimoto K, Yasuda K, and Inoue K (2003). A vertebrate RNA-binding protein Fox-1 regulates tissue-specific splicing via the pentanucleotide GCAUG. The EMBO journal 22, 905–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalsotra A, Singh RK, Gurha P, Ward AJ, Creighton CJ, and Cooper TA (2014). The Mef2 transcription network is disrupted in myotonic dystrophy heart tissue, dramatically altering miRNA and mRNA expression. Cell Rep 6, 336–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalsotra A, Wang K, Li PF, and Cooper TA (2010). MicroRNAs coordinate an alternative splicing network during mouse postnatal heart development. Genes Dev 24, 653–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalsotra A, Xiao X, Ward AJ, Castle JC, Johnson JM, Burge CB, and Cooper TA (2008). A postnatal switch of CELF and MBNL proteins reprograms alternative splicing in the developing heart. Proc Natl Acad Sci U S A 105, 20333–20338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanadia RN, Johnstone KA, Mankodi A, Lungu C, Thornton CA, Esson D, Timmers AM, Hauswirth WW, and Swanson MS (2003). A muscleblind knockout model for myotonic dystrophy. Science 302, 1978–1980. [DOI] [PubMed] [Google Scholar]
- Klauda JB, Venable RM, Freites JA, O’Connor JW, Tobias DJ, Mondragon-Ramirez C, Vorobyov I, MacKerell AD Jr., and Pastor RW (2010). Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J Phys Chem B 114, 7830–7843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong W, Po S, Yamagishi T, Ashen MD, Stetten G, and Tomaselli GF (1998). Isolation and characterization of the human gene encoding Ito: further diversity by alternative mRNA splicing. The American journal of physiology 275, H1963–1970. [DOI] [PubMed] [Google Scholar]
- Koshelev M, Sarma S, Price RE, Wehrens XH, and Cooper TA (2010). Heart-specific overexpression of CUGBP1 reproduces functional and molecular abnormalities of myotonic dystrophy type 1. Human molecular genetics 19, 1066–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuyumcu-Martinez NM, Wang GS, and Cooper TA (2007). Increased steady-state levels of CUGBP1 in myotonic dystrophy 1 are due to PKC-mediated hyperphosphorylation. Molecular cell 28, 68–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarus A, Varin J, Babuty D, Anselme F, Coste J, and Duboc D (2002). Long-term follow-up of arrhythmias in patients with myotonic dystrophy treated by pacing: a multicenter diagnostic pacemaker study. J Am Coll Cardiol 40, 1645–1652. [DOI] [PubMed] [Google Scholar]
- Li G, Wang J, Liao P, Bartels P, Zhang H, Yu D, Liang MC, Poh KK, Yu CY, Jiang F, et al. (2017). Exclusion of alternative exon 33 of CaV1.2 calcium channels in heart is proarrhythmogenic. Proc Natl Acad Sci U S A 114, E4288–E4295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin X, Miller JW, Mankodi A, Kanadia RN, Yuan Y, Moxley RT, Swanson MS, and Thornton CA (2006). Failure of MBNL1-dependent post-natal splicing transitions in myotonic dystrophy. Human molecular genetics 15, 2087–2097. [DOI] [PubMed] [Google Scholar]
- Mahadevan M, Tsilfidis C, Sabourin L, Shutler G, Amemiya C, Jansen G, Neville C, Narang M, Barcelo J, O’Hoy K, et al. (1992). Myotonic dystrophy mutation: an unstable CTG repeat in the 3’ untranslated region of the gene. Science 255, 1253–1255. [DOI] [PubMed] [Google Scholar]
- Mankodi A, Lin X, Blaxall BC, Swanson MS, and Thornton CA (2005). Nuclear RNA foci in the heart in myotonic dystrophy. Circ Res 97, 1152–1155. [DOI] [PubMed] [Google Scholar]
- Mankodi A, Takahashi MP, Jiang H, Beck CL, Bowers WJ, Moxley RT, Cannon SC, and Thornton CA (2002). Expanded CUG repeats trigger aberrant splicing of ClC-1 chloride channel pre-mRNA and hyperexcitability of skeletal muscle in myotonic dystrophy. Molecular cell 10, 35–44. [DOI] [PubMed] [Google Scholar]
- Mankodi A, Urbinati CR, Yuan QP, Moxley RT, Sansone V, Krym M, Henderson D, Schalling M, Swanson MS, and Thornton CA (2001). Muscleblind localizes to nuclear foci of aberrant RNA in myotonic dystrophy types 1 and 2. Human molecular genetics 10, 2165–2170. [DOI] [PubMed] [Google Scholar]
- Martyna GJ (1994). Constant pressure molecular dynamics algorithms. The Journal of Chemical Physics. [Google Scholar]
- McKean DM, Homsy J, Wakimoto H, Patel N, Gorham J, DePalma SR, Ware JS, Zaidi S, Ma W, Patel N, et al. (2016). Loss of RNA expression and allele-specific expression associated with congenital heart disease. Nat Commun 7, 12824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merico D, Isserlin R, Stueker O, Emili A, and Bader GD (2010). Enrichment map: a network-based method for gene-set enrichment visualization and interpretation. PLoS One 5, e13984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JW, Urbinati CR, Teng-Umnuay P, Stenberg MG, Byrne BJ, Thornton CA, and Swanson MS (2000). Recruitment of human muscleblind proteins to (CUG)(n) expansions associated with myotonic dystrophy. The EMBO journal 19, 4439–4448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mooers BH, Logue JS, and Berglund JA (2005). The structural basis of myotonic dystrophy from the crystal structure of CUG repeats. Proc Natl Acad Sci U S A 102, 16626–16631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen HH, Wolfe JT 3rd, Holmes DR Jr., and Edwards WD (1988). Pathology of the cardiac conduction system in myotonic dystrophy: a study of 12 cases. J Am Coll Cardiol 11, 662–671. [DOI] [PubMed] [Google Scholar]
- Nutter CA, Jaworski EA, Verma SK, Deshmukh V, Wang Q, Botvinnik OB, Lozano MJ, Abass IJ, Ijaz T, Brasier AR, et al. (2016). Dysregulation of RBFOX2 Is an Early Event in Cardiac Pathogenesis of Diabetes. Cell Rep 15, 2200–2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onkal R, Mattis JH, Fraser SP, Diss JK, Shao D, Okuse K, and Djamgoz MB (2008). Alternative splicing of Nav1.5: an electrophysiological comparison of ‘neonatal’ and ‘adult’ isoforms and critical involvement of a lysine residue. J Cell Physiol 216, 716–726. [DOI] [PubMed] [Google Scholar]
- Pang PD, Alsina KM, Cao S, Koushik AB, Wehrens XHT, and Cooper TA (2018). CRISPR -Mediated Expression of the Fetal Scn5a Isoform in Adult Mice Causes Conduction Defects and Arrhythmias. J Am Heart Assoc 7, e010393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JW, Jung S, Rouchka EC, Tseng YT, and Xing Y (2016). rMAPS: RNA map analysis and plotting server for alternative exon regulation. Nucleic Acids Res 44, W333–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeufer A, van Noord C, Marciante KD, Arking DE, Larson MG, Smith AV, Tarasov KV, Muller M, Sotoodehnia N, Sinner MF, et al. (2010). Genome-wide association study of PR interval. Nature genetics 42, 153–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, and Schulten K (2005). Scalable molecular dynamics with NAMD. J Comput Chem 26, 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips MF, and Harper PS (1997). Cardiac disease in myotonic dystrophy. Cardiovasc Res 33, 13–22. [DOI] [PubMed] [Google Scholar]
- Rau F, Freyermuth F, Fugier C, Villemin JP, Fischer MC, Jost B, Dembele D, Gourdon G, Nicole A, Duboc D, et al. (2011). Misregulation of miR-1 processing is associated with heart defects in myotonic dystrophy. Nat Struct Mol Biol 18, 840–845. [DOI] [PubMed] [Google Scholar]
- Savkur RS, Philips AV, and Cooper TA (2001). Aberrant regulation of insulin receptor alternative splicing is associated with insulin resistance in myotonic dystrophy. Nature genetics 29, 40–47. [DOI] [PubMed] [Google Scholar]
- Schwaiger CS, Bjelkmar P, Hess B, and Lindahl E (2011). 3(1)(0)-helix conformation facilitates the transition of a voltage sensor S4 segment toward the down state. Biophysical journal 100, 1446–1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feller Scott E., Y.Z., and Pastor Richard W. (1995). Constant pressure molecular dynamics simulation: The Langevin piston method. The Journal of Chemical Physics. [Google Scholar]
- Sellier C, Cerro-Herreros E, Blatter M, Freyermuth F, Gaucherot A, Ruffenach F, Sarkar P, Puymirat J, Udd B, Day JW, et al. (2018). rbFOX1/MBNL1 competition for CCUG RNA repeats binding contributes to myotonic dystrophy type 1/type 2 differences. Nat Commun 9, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah A, Qian Y, Weyn-Vanhentenryck SM, and Zhang C (2017). CLIP Tool Kit (CTK): a flexible and robust pipeline to analyze CLIP sequencing data. Bioinformatics 33, 566–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen S, Park JW, Lu ZX, Lin L, Henry MD, Wu YN, Zhou Q, and Xing Y (2014). rMATS: robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc Natl Acad Sci U S A 111, E5593–5601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sievers F, and Higgins DG (2014). Clustal Omega, accurate alignment of very large numbers of sequences. Methods Mol Biol 1079, 105–116. [DOI] [PubMed] [Google Scholar]
- Singh RK, Kolonin AM, Fiorotto ML, and Cooper TA (2018). Rbfox-Splicing Factors Maintain Skeletal Muscle Mass by Regulating Calpain3 and Proteostasis. Cell Rep 24, 197–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh RK, Xia Z, Bland CS, Kalsotra A, Scavuzzo MA, Curk T, Ule J, Li W, and Cooper TA (2014). Rbfox2-coordinated alternative splicing of Mef2d and Rock2 controls myoblast fusion during myogenesis. Molecular cell 55, 592–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sznajder LJ, Michalak M, Taylor K, Cywoniuk P, Kabza M, Wojtkowiak-Szlachcic A, Matloka M, Konieczny P, and Sobczak K (2016). Mechanistic determinants of MBNL activity. Nucleic Acids Res 44, 10326–10342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas JD, Sznajder LJ, Bardhi O, Aslam FN, Anastasiadis ZP, Scotti MM, Nishino I, Nakamori M, Wang ET, and Swanson MS (2017). Disrupted prenatal RNA processing and myogenesis in congenital myotonic dystrophy. Genes Dev 31, 1122–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokgozoglu LS, Ashizawa T, Pacifico A, Armstrong RM, Epstein HF, and Zoghbi WA (1995). Cardiac involvement in a large kindred with myotonic dystrophy. Quantitative assessment and relation to size of CTG repeat expansion. JAMA 274, 813–819. [PubMed] [Google Scholar]
- Tom Darden DY, and Pedersen Lee (1993). Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. The Journal of Chemical Physics. [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, and Pachter L (2012). Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 7, 562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valencik ML, and McDonald JA (2001). Codon optimization markedly improves doxycycline regulated gene expression in the mouse heart. Transgenic Res 10, 269–275. [DOI] [PubMed] [Google Scholar]
- Veerman CC, Wilde AA, and Lodder EM (2015). The cardiac sodium channel gene SCN5A and its gene product NaV1.5: Role in physiology and pathophysiology. Gene 573, 177–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma SK, Deshmukh V, Nutter CA, Jaworski E, Jin W, Wadhwa L, Abata J, Ricci M, Lincoln J, Martin JF, et al. (2016). Rbfox2 function in RNA metabolism is impaired in hypoplastic left heart syndrome patient hearts. Sci Rep 6, 30896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verweij N, Mateo Leach I, van den Boogaard M, van Veldhuisen DJ, Christoffels VM, LifeLines Cohort S, Hillege HL, van Gilst WH, Barnett P, de Boer RA, et al. (2014). Genetic determinants of P wave duration and PR segment. Circ Cardiovasc Genet 7, 475–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ET, Treacy D, Eichinger K, Struck A, Estabrook J, Olafson H, Wang TT, Bhatt K, Westbrook T, Sedehizadeh S, et al. (2018). Transcriptome alterations in myotonic dystrophy skeletal muscle and heart. Human molecular genetics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ET, Ward AJ, Cherone JM, Giudice J, Wang TT, Treacy DJ, Lambert NJ, Freese P, Saxena T, Cooper TA, et al. (2015). Antagonistic regulation of mRNA expression and splicing by CELF and MBNL proteins. Genome Res 25, 858–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang GS, Kuyumcu-Martinez MN, Sarma S, Mathur N, Wehrens XH, and Cooper TA (2009). PKC inhibition ameliorates the cardiac phenotype in a mouse model of myotonic dystrophy type 1. J Clin Invest 119, 3797–3806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, and Wehrens XH (2012). Enhanced impact of SCN5A mutation associated with long QT syndrome in fetal splice isoform. Heart Rhythm 9, 598–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei C, Qiu J, Zhou Y, Xue Y, Hu J, Ouyang K, Banerjee I, Zhang C, Chen B, Li H, et al. (2015). Repression of the Central Splicing Regulator RBFox2 Is Functionally Linked to Pressure Overload-Induced Heart Failure. Cell Rep. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weyn-Vanhentenryck SM, Mele A, Yan Q, Sun S, Farny N, Zhang Z, Xue C, Herre M, Silver PA, Zhang MQ, et al. (2014). HITS-CLIP and integrative modeling define the Rbfox splicing-regulatory network linked to brain development and autism. Cell Rep 6, 1139–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen William L., J.C., and Madura Jeffry D. (1983). Comparison of simple potential functions for simulating liquid water. The Journal of Chemical Physics, 926–935. [Google Scholar]
- Wu EL, Cheng X, Jo S, Rui H, Song KC, Davila-Contreras EM, Qi Y, Lee J, Monje-Galvan V, Venable RM, et al. (2014). CHARMM-GUI Membrane Builder toward realistic biological membrane simulations. J Comput Chem 35, 1997–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie C, Bondarenko VE, Morales MJ, and Strauss HC (2009). Closed-state inactivation in Kv4.3 isoforms is differentially modulated by protein kinase C. Am J Physiol Cell Physiol 297, C1236–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadava RS, Frenzel-McCardell CD, Yu Q, Srinivasan V, Tucker AL, Puymirat J, Thornton CA, Prall OW, Harvey RP, and Mahadevan MS (2008). RNA toxicity in myotonic muscular dystrophy induces NKX2–5 expression. Nature genetics 40, 61–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo GW, Coufal NG, Liang TY, Peng GE, Fu XD, and Gage FH (2009). An RNA code for the FOX2 splicing regulator revealed by mapping RNA-protein interactions in stem cells. Nat Struct Mol Biol 16, 130–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying Y, Wang XJ, Vuong CK, Lin CH, Damianov A, and Black DL (2017). Splicing Activation by Rbfox Requires Self-Aggregation through Its Tyrosine-Rich Domain. Cell 170, 312–323 e310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yum K, Wang ET, and Kalsotra A (2017). Myotonic dystrophy: disease repeat range, penetrance, age of onset, and relationship between repeat size and phenotypes. Curr Opin Genet Dev 44, 30–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Video 1. Interactions of D211 in SCN5A_6B with the lipid head group. This supplemental video is related to Figure 6.
Supplementary Video 2. Interactions of K211 in SCN5A_6A with the lipid head group. This supplemental video is related to Figure 6.
Data Availability Statement
All raw RNA-seq data files are available for download from NCBI Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE126771.