

Abstract
Aims
In vitro data show that talazoparib is a substrate for P‐glycoprotein (P‐gp) and breast cancer resistance protein transporters. This open‐label, 2‐arm, drug–drug interaction Phase 1 study in patients with advanced solid tumours assessed the effect of a P‐gp inhibitor (itraconazole) and a P‐gp inducer (rifampicin) on the pharmacokinetics of a single dose of talazoparib. The safety and tolerability of a single dose of talazoparib with and without itraconazole or rifampicin were also assessed.
Methods
Thirty‐six patients were enrolled (Arm A [itraconazole], n = 19; Arm B [rifampicin], n = 17). Patients in both arms received 2 single oral doses of talazoparib (0.5 mg, Arm A; 1 mg, Arm B) alone and with multiple daily oral doses of itraconazole (Arm A) or rifampicin (Arm B).
Results
Coadministration of itraconazole and talazoparib increased talazoparib area under the plasma concentration–time profile from time 0 extrapolated to infinity by ~56% and maximum observed plasma concentration by ~40% relative to talazoparib alone. Coadministration of rifampicin and talazoparib increased talazoparib maximum observed plasma concentration by approximately 37% (geometric mean ratio 136.6% [90% confidence interval 103.2–180.9]); area under the curve was not affected relative to talazoparib alone (geometric mean ratio 102.0% [90% confidence interval 94.0–110.7]). Talazoparib had an overall safety profile consistent with that observed in prior studies in which talazoparib was administered as a single dose.
Conclusion
Coadministration of itraconazole increased talazoparib plasma exposure compared to talazoparib alone. A reduced talazoparib dose is recommended if coadministration of potent P‐gp inhibitors cannot be avoided. Similar exposure was observed when talazoparib was administered alone and with rifampicin suggesting that the effect of rifampicin on talazoparib exposure is limited.
Keywords: breast cancer, cancer, drug interaction, P‐glycoprotein, pharmacokinetics
What is already known about this subject
Talazoparib is approved by the US Food and Drug Administration for treatment of germline BRCA‐mutated, HER2‐negative advanced breast cancer and is being investigated in multiple solid tumours.
In vitro studies confirmed that talazoparib is a substrate for P‐gp and breast cancer resistance protein.
Potent P‐gp inhibitors increased talazoparib's relative bioavailability based on population pharmacokinetics analysis.
What this study adds
Itraconazole increased talazoparib plasma exposure; rifampicin had limited effect on talazoparib exposure.
These findings support the current talazoparib dose recommendations to avoid coadministration of potent P‐gp inhibitors. If coadministration of a potent P‐gp inhibitor is necessary, talazoparib dose should be reduced from 1 to 0.75 mg once daily.
1. INTRODUCTION
The DNA damage repair (DDR) pathway is regulated by poly (ADP‐ribose) polymerase (PARP), and inhibition of PARP in DDR‐deficient cells leads to accumulation of irreparable DNA damage and cell death.1 PARP inhibitors have been approved for a variety of cancers with mutations in DNA repair genes.2 Talazoparib is a PARP inhibitor that inhibits PARP1/PARP2 and traps PARP on DNA, which can prevent DNA damage repair and result in cell death in cells with DDR gene mutations.3 Talazoparib was approved by the United States Food and Drug Administration for treatment of patients with deleterious or suspected deleterious germline breast cancer susceptibility genes (BRCA)‐mutated human epidermal growth factor receptor 2 (HER2)‐negative locally advanced/metastatic breast cancer4 based on results from the pivotal Phase 3 EMBRACA study, which showed statistically significant and clinically meaningful improvement in progression‐free survival (PFS) vs physician's choice of chemotherapy.5 Treatment with talazoparib was generally safe and well tolerated. The most common adverse events (AEs) were cytopenia, fatigue and nausea. Grade 3–4 AEs were primarily haematological and occurred in 55% of patients on talazoparib; only 1.4% of these patients permanently discontinued treatment due to a haematological AE.5
The recommended dose of talazoparib is the maximum tolerated dose (MTD) of 1 mg once daily (QD) as determined in a Phase 1 dose escalation study.6 Pharmacokinetic (PK) analysis of talazoparib 1 mg QD in patients with advanced tumours showed that plasma talazoparib exposure is dose proportional in the dose range of 0.025 mg to 2 mg QD, suggesting linear PK.6, 7 Talazoparib is rapidly absorbed with a median time to first occurrence of maximum observed plasma concentration (Tmax) ranging from approximately 1.0 to 2.0 h post dose.6 Talazoparib undergoes minimal hepatic metabolism, and renal excretion of unchanged talazoparib is the major elimination pathway.6, 8 Based on urinary excretion data following administration of a single 1 mg oral dose of9 C‐talazoparib, the absolute bioavailability is at least 54.6% with fraction absorbed of at least 68.7%.4 An exposure–response analysis suggested that the higher talazoparib exposure seen in the EMBRACA study was associated with longer PFS. This finding further supports the use of the MTD of 1 mg QD as the recommended dose for talazoparib as single‐agent therapy in the treatment of solid tumours to provide the highest tolerable exposure that will lead to the best PFS outcomes.10
At therapeutic exposures, talazoparib does not markedly induce or inhibit any enzymes or transporters.4 Therefore, it is unlikely that talazoparib will demonstrate clinically significant drug enzyme or transporter induction‐ or inhibition‐based drug–drug interactions when coadministered with corresponding substrates. In vitro, talazoparib was shown to be a substrate for P‐glycoprotein (P‐gp) and breast cancer resistant protein transporters.4 Therefore, plasma talazoparib concentrations may increase or decrease when coadministered with P‐gp or breast cancer resistance protein inhibitors or inducers, respectively. Population PK analysis using data pooled from multiple clinical trials indicated that potent P‐gp inhibitors increased the relative bioavailability of talazoparib by 45%.11 The objectives of this study were to evaluate the effect of multiple doses of itraconazole as a P‐gp inhibitor or rifampicin as a P‐gp inducer on the PK and safety of a single dose of talazoparib in patients with advanced solid tumours.
2. METHODS
2.1. Study design and participants
This trial was an open‐label, 2‐armed, fixed‐sequence drug–drug interaction Phase 1 study in patients with advanced solid tumours for the investigation of the effect of P‐gp inhibition and induction on the PK of talazoparib (EudraCT number: 2016–001813‐26). A total of 36 patients with advanced solid tumours were enrolled in this study according to the inclusion criteria.
The study was conducted in accordance with the principles of the Declaration of Helsinki12 in place at the time of study conduct and in compliance with the International Council for Harmonisation (ICH) E6 Guideline for Good Clinical Practice (GCP) (Committee for Proprietary Medicinal Products [CPMP] guideline CPMP/ICH/135/95)13 and the European Union Clinical Trial Directive (EU CTD): Directive 2001/20/EC.14 Details of the Independent Ethics Committees for this study are provided in the Supplemental Materials. All patients provided written informed consent before enrolment. The protocol, its amendments, and the consent forms were reviewed and approved by the independent ethics committees.
2.2. Inclusion criteria
For both arms, eligible patients were aged ≥18 years. For Arm A, eligible patients were aged <65 years; there was no upper age limit for Arm B. Patients had to provide informed consent, have a histologically confirmed advanced solid tumour judged by the investigator as not appropriate for standard therapy and Eastern Cooperative Oncology Group (ECOG) performance status ≤2 at screening and time of enrolment. Female patients of childbearing potential and fertile males had to agree to use a highly effective birth control method.
Patients were excluded if they had previous treatment with any other prescription or nonprescription drugs or supplements with potential P‐gp interaction within 7 days or 5 half‐lives (t½), whichever was longer, before Day 1; had major surgery within 8 weeks prior to screening or received any investigational or anticancer drug within 28 days before screening; had an estimated glomerular filtration rate ≤50 mL/min/1.73‐m2 by the Modification of Diet in Renal Disease equation at screening and Day −1 or had positive tests for human immunodeficiency virus, hepatitis B or hepatitis C.
2.3. Treatment selection and timing of doses
Patients were assigned to Arm A (n = 19) or Arm B (n = 17) of the study based on meeting the respective eligibility criteria. Patients in Arm A received 0.5 mg talazoparib, which is 50% of the recommended dose, for daily administration. For Arm B, 1 mg of talazoparib was administered. In Period 1, talazoparib was administered without the interacting drugs the morning of Day 1 under fasted conditions (8 h before until 2 h after dosing, consumption of water was allowed), followed by a 14‐day wash‐out period (Days 2–15). In Period 2, talazoparib was administered in combination with the interacting drugs the morning of Day 23 (Arm A) and of Day 25 (Arm B) under fasted conditions (8 h before until 2 h after dosing) (Figure 1). The administration of itraconazole and rifampicin was supervised by the investigator on days when coadministration with talazoparib occurred. On all other days, the patients self‐administered these drugs and recorded the dose and time and date of intake in a diary.
Figure 1.
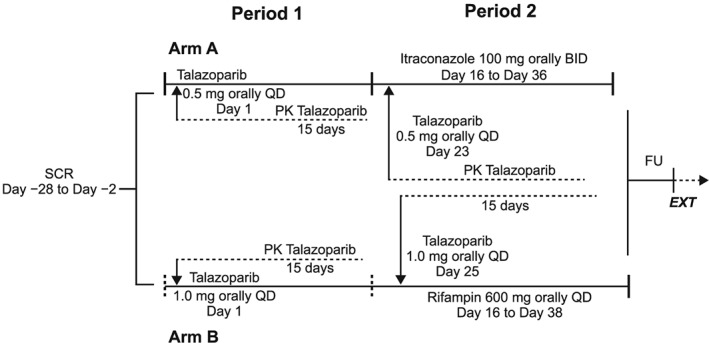
Study design and patient assignment. BID, twice a day; EXT, extension study MDV3800–13/C3441010; FU, follow‐up; QD, once daily; PK, pharmacokinetics; SCR, screening
The doses of itraconazole and rifampicin and their administration with respect to meals were selected following the dosing recommendations for their use as antifungal and antimicrobial agents in their respective summaries of product characteristics in order to provide optimal absorption and systemic exposures of each interacting agent.9, 15 In Period 2, on non‐talazoparib dosing days, itraconazole 100 mg was administered twice daily, once in the morning and once in the evening (approximately 12 h apart), from Day 16 to Day 36, immediately after meals (breakfast and dinner, respectively). Rifampicin 600 mg was administered once daily in the morning from Day 16 to Day 38 in Period 2, at least 30 min before breakfast. On talazoparib dosing days, itraconazole 100 mg or rifampicin 600 mg was taken 30 min before talazoparib under fasted conditions.
2.4. Study assessments
Serial blood samples were collected for PK analysis at predetermined times after talazoparib administration, up to 336 h (14 days). Blood samples for PK analysis were drawn at the following selected time‐points during each treatment period: pre‐dose and 0.5, 1, 2, 4, 8, 12, 24, 48, 72, 96, 120, 168, 216, 264 and 336 h after talazoparib dosing in both arms. In both arms and periods, patients were confined from 1 day prior to talazoparib administration until 3 days after talazoparib administration.
2.5. Bioanalytical methods
Plasma samples were analysed for talazoparib concentrations at Alliance Pharma (Malvern, PA, USA) using a validated, sensitive and specific high‐performance liquid chromatography tandem mass spectrometry, as described previously.11 Additional details on assay performance included in Supplemental Materials.
2.6. Sample size considerations
Eighteen patients were to be enrolled in each arm to allow for approximately 15% of early discontinuation or loss to follow‐up, for a target number of 15 evaluable patients in each arm. Using an analysis of variance (ANOVA) model that includes treatment as a fixed factor and subject as a random effect with intrasubject variability of 42% (CV%) as estimated from population PK analysis,11 with 15 evaluable patients, and a true ratio of 100%, the equivalence limits of 90% confidence interval (CI) were estimated to be 77% and 130%.
2.7. PK analyses
PK parameters were estimated from talazoparib plasma concentrations using non‐compartmental analysis for the PK population, which was defined as all patients who received at least 1 dose of talazoparib and had at least 1 reportable talazoparib plasma concentration. Statistical analyses were performed to assess the effect of multiple doses of itraconazole and rifampicin on the PK of talazoparib using itraconazole or rifampicin in combination with talazoparib as Test and the treatment with talazoparib alone as Reference. A mixed effects model that included treatment as a fixed factor and patients as a random effect was fitted to the log‐transformed PK parameters (for area under the plasma concentration–time profile from time 0 extrapolated to infinity [AUCinf], area under the plasma concentration time profile from time 0 to the time of last quantifiable concentration [AUClast] and maximum observed plasma concentration [Cmax]). A point estimate and the corresponding 90% CI for the difference between least squares means of Test and Reference treatment (Test – Reference) was calculated. The antilogarithm of this value was calculated to obtain the point estimate and the 90% CI for the ratio (Test/Reference) of the geometric means on the untransformed scale.
2.8. Safety analyses
All safety analyses were performed using the safety population, defined as all patients who had received at least 1 dose of talazoparib. Safety and tolerability were evaluated using summaries of AEs, physical examinations (including weight), vital signs assessments, 12‐lead electrocardiograms and clinical laboratory evaluations. AEs were coded to preferred term and system organ class using the Medical Dictionary for Regulatory Activities version 19.1 and classified by severity using the National Cancer Institute Common Terminology Criteria for Adverse Events version 4.03. AEs were collected following the first dose of study drug (Day 1) until completion of the follow‐up visit (20 ± 3 days after the last dose of study drug). Serious AEs were collected starting at informed consent signing until completion of the follow‐up visit.
Treatment‐emergent AEs (TEAEs), laboratory values and vital signs following the first administration of talazoparib for both arms were summarised by the last treatment received, i.e. talazoparib alone treatment period, itraconazole/rifampicin treatment period or talazoparib + itraconazole/rifampicin treatment period.
3. RESULTS
3.1. Patient demographics and disposition
A total of 36 patients with advanced solid tumours were assigned to Arm A (n = 19) or Arm B (n = 17). The majority of patients were female (n = 17, 89.5% and n = 13, 76.5% of patients for Arms A and B, respectively). The median age was 56 years (range 20–63 years) for Arm A and 69 years (range 41–76 years) for Arm B. All patients were white. Patient demographics are shown in Table 1. In Arm A, 19 patients were treated in Period 1 and 15 patients were treated in Period 2, with 17 patients (89.5%) completing treatment Period 1 and 14 patients (73.7%) completing both treatment Periods 1 and 2 (Table S1). In total, 5 patients (26.3%) in Arm A discontinued treatment due to AEs. No patients discontinued treatment due to study‐drug related AEs. In Arm B, 17 patients (100%) were treated and completed Period 1, 15 patients (88.2%) were treated in Period 2, 15 patients (88.2%) completed both treatment periods and 2 patients (11.8%) discontinued treatment due to AEs, 1 (5.9%) of which was study‐drug related (Table S1).
Table 1.
Summary of demographics and baseline characteristics
| Talazoparib + itraconazole (n = 19) | Talazoparib + rifampicin (n = 17) | |
|---|---|---|
| Age, ya | 56 (20–63) | 69 (41–76) |
|
Sex, n (%) Male Female |
2 (10.5) 17 (89.5) |
4 (23.5) 13 (76.5) |
| Weight, kga | 69.0 (48.0–89.0) | 67.2 (46.0–98.0) |
| Height, cma | 162.0 (150–174) | 163.0 (144–180) |
| BMI, kg/m2a | 25.4 (19.2–34.2) | 25.2 (18.0–39.0) |
BMI, body mass index
Data expressed as median (range).
3.2. PK
3.2.1. Arm A (Itraconazole)
Median talazoparib plasma concentrations were higher when talazoparib was administered in the presence of multiple oral doses of itraconazole than when administered alone (Figure 2). The median Tmax was similar for the 2 periods (1 h for talazoparib and 1.02 h for talazoparib + itraconazole; Table 2). The mean estimated t½ was slightly higher when talazoparib was administered with itraconazole compared to talazoparib alone (Table 2). The adjusted geometric mean talazoparib AUCinf and Cmax values following coadministration of itraconazole were approximately 56 and 40% higher, respectively, compared to when talazoparib was administered alone (Table 3, Figure S2). Variability of talazoparib AUCinf and Cmax geometric means was similar when talazoparib was administered with itraconazole compared to talazoparib alone.
Figure 2.
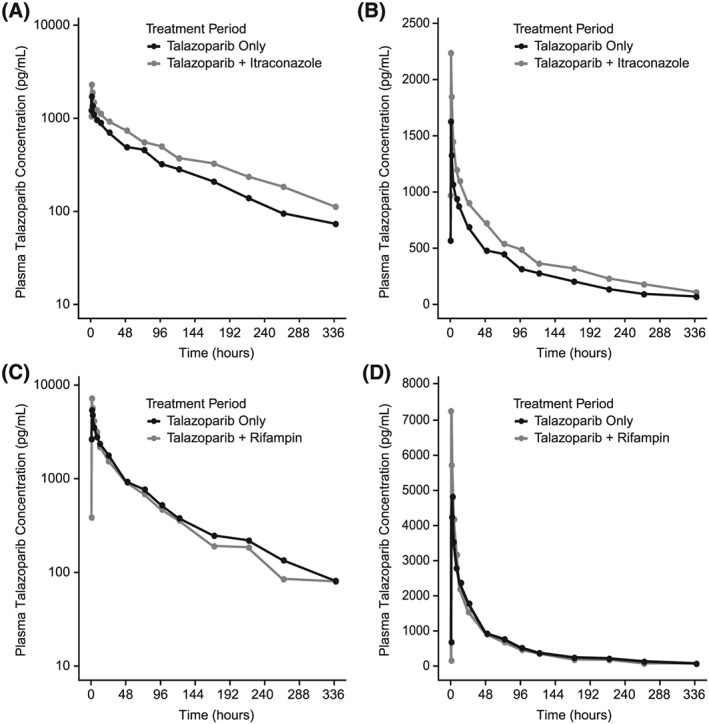
Median plasma talazoparib concentration–time profile following a single dose of talazoparib alone and with multiple doses of itraconazole (semilogarithmic, A; linear, B) or following a single dose of talazoparib with multiple doses of rifampicin (semilogarithmic, C; linear, D). Concentration values below the lower limit of quantification were set to zero. Only pharmacokinetic samples collected within the 10% of the scheduled time point are included
Table 2.
Summary of plasma talazoparib pharmacokinetic (PK) parameters without and with multiple doses of itraconazole (arm a) and rifampicin (arm B)
| Arm A | Arm B | |||
|---|---|---|---|---|
| PK parameter | Talazoparib alone (0.5 mg) | Talazoparib (0.5 mg) + itraconazole | Talazoparib alone (1.0 mg) | Talazoparib (1.0 mg) + rifampicin |
| Cmax (pg/mL) |
19 2092.0 (50.0) |
15 2936.8 (56.0) |
17 6007.0 (53.0) |
15 8336.8 (71.0) |
| Tmax (h) |
19 1.0 (0.5–24.1) |
15 1.0 (0.5–4.0) |
17 1.0 (0.5–24.0) |
15 1.0 (0.5–8.0) |
| AUClast (h*pg/mL) |
19 98 532.3 (38.0) |
15 145 944.6 (38.0) |
17 196 631.3 (32.0) |
15 196 100.7 (33.0) |
| AUCinf (h*pg/mL) |
18 109 762.1 (42.0) |
12 151 919.6 (36.0) |
17 209 521.6 (34.0) |
12 194 307.7 (36.0) |
| CL/F (L/h) |
18 4.6 (42.0) |
12 3.3 (36.0) |
17 4.8 (34.0) |
12 5.2 (36.0) |
| Vz/F (L) |
18 644.81 (42.0) |
12 552.01 (27.0) |
17 623.9 (30.0) |
12 588.1 (33.0) |
| T1/2 (h) |
18 101.26 (26.3) |
12 118.47 (23.6) |
17 92.1 (17.7) |
12 80.6 (16.5) |
AUCinf, area under the plasma concentration–time profile from time 0 extrapolated to infinity; AUClast, area under the plasma concentration–time profile from time 0 to the time of last quantifiable concentration; CL/F, apparent clearance; Cmax, maximum observed plasma concentration; CV, coefficient of variation; SD, standard deviation; t½, half‐life; Tmax, time to first occurrence of maximum observed plasma concentration; Vz/F, apparent volume of distribution
Geometric mean (CV%) is presented for all PK parameters with the exception of Tmax (median, range) and T1/2 (arithmetic mean and SD).
Table 3.
Statistical summary of analysis of effect of itraconazole on talazoparib primary pharmacokinetic (PK) parameters
| PK parameters | Talazoparib + itraconazole (test) | Talazoparib alone (reference) | Geometric mean ratio (test/reference), % (90% CI) |
|---|---|---|---|
| AUCinf (h pg/mL) | 171,489.1 | 109,762.1 | 156.2 (137.6, 177.4) |
| AUClast (h pg/mL) | 148,495.3 | 98,532.3 | 150.7 (136.5, 166.4) |
| Cmax (pg/mL) | 2927.2 | 2092.0 | 139.9 (113.3, 172.9) |
AUCinf, area under the plasma concentration–time profile from time 0 extrapolated to infinity; AUClast, area under the plasma concentration time profile from time 0 to the time of last quantifiable concentration; CI, confidence interval; Cmax, maximum observed plasma concentration
3.2.2. Arm B (rifampicin)
The median talazoparib Tmax was similar, i.e. 1 h, for both periods (Table 2). The mean talazoparib t½ was slightly shorter when administered with rifampicin (80.6 h) compared to when administered alone (92.1 h). The adjusted geometric means for talazoparib AUCinf values were similar when talazoparib was administered with rifampicin compared with when talazoparib was administered alone, while the adjusted geometric mean for talazoparib Cmax was approximately 37% higher when talazoparib was administered with rifampicin compared to when talazoparib was administered alone (Table 4 , Figures 2C, 2D and S2). Variability of talazoparib Cmax geometric means was higher when talazoparib was administered with rifampicin compared to its administration alone (Table 4).
Table 4.
Statistical summary of analysis of effect of rifampicin on talazoparib primary pharmacokinetic (PK) parameters
| PK parameters | Talazoparib + rifampicin (test) |
Talazoparib alone (reference) |
Geometric mean ratio (test/reference), % (90% CI) |
|---|---|---|---|
| AUCinf (h pg/mL) | 213 789.1 | 209 521.6 | 102.0 (94.0, 110.7) |
| AUClast (h pg/mL) | 207 181.8 | 196 631.3 | 105.4 (98.0, 113.2) |
| Cmax (pg/mL) | 8207.0 | 6007.0 | 136.6 (103.2, 180.9) |
AUCinf, area under the plasma concentration–time profile from time 0 extrapolated to infinity; AUClast, area under the plasma concentration time profile from time 0 to the time of last quantifiable concentration; CI, confidence interval; Cmax, maximum observed plasma concentration
4. SAFETY
Talazoparib was generally well tolerated with no new safety signals identified for either talazoparib administered alone or together with itraconazole or rifampicin. In both arms, the majority of TEAEs were reported within the system organ class of gastrointestinal disorders, and most TEAEs were considered by the investigator to be not related to the study drugs and of mild or moderate severity. More details on TEAEs, serious AEs and laboratory values/vital signs/electrocardiograms are presented in Supplemental Materials and Table S2.
5. DISCUSSION
Coadministration of multiple doses of itraconazole increased both talazoparib Cmax by 40% and overall exposure by 56%. Population PK analysis using data from first‐in‐human studies, as well as from Phase 2 and 3 clinical studies, showed that coadministration of talazoparib with potent P‐gp inhibitors (i.e. those that result in ≥ 2‐fold increase in the exposure of an in vivo probe P‐gp substrate), increased the relative bioavailability of talazoparib by 44.7%.4 The results from Arm A of this study are consistent with population PK analysis.11
In the Phase 2 ABRAZO study and the Phase 3 EMBRACA study, the most common Grade ≥3 AEs were haematological.5, 16 Exposure–safety analyses based on pooled data from the ABRAZO and EMBRACA studies showed that higher talazoparib exposure is also associated with a higher risk for Grade ≥ 3 anaemia and thrombocytopenia.17 As these haematological AEs correlate with talazoparib exposure levels, lowering the exposure by dosing interruption or dose reduction may lead to a lower probability of having these AEs. Exposure‐efficacy analysis showed that higher talazoparib exposure in the EMBRACA study was associated with longer PFS in patients with germline BRCA‐mutated, HER2‐negative locally advanced or metastatic breast cancer,10 which justifies the use of talazoparib MTD (1 mg QD) as the recommended talazoparib dose in the single‐agent setting to provide the highest tolerable exposure that will lead to the best PFS outcomes. Findings from exposure–safety and efficacy analysis suggest that when talazoparib concentration is increased under certain conditions, such as when talazoparib is coadministered with potent P‐gp inhibitors, dose reduction is an effective approach to manage AEs, while overcorrection should be avoided in order to maximize efficacy.
Based on data from the population PK analysis and data from the itraconazole arm of the current trial and exposure‐safety analysis, coadministration of talazoparib with potent P‐gp inhibitors should be avoided.4, 18, 19 If a potent P‐gp inhibitor must be coadministered, the talazoparib dose should be reduced from 1 to 0.75 mg QD to account for the increase in talazoparib exposure.6 Further dose reduction when talazoparib is coadministered with potent P‐gp inhibitors would lead to lower exposure than that following 1 mg talazoparib administered alone, which is not recommended as suggested by exposure–efficacy analysis.10 Patients experiencing haematological AEs can be managed via dose interruption, dose reduction and standard supportive care.
While itraconazole increased talazoparib talazoparib AUCinf and Cmax, only a small increase in mean talazoparib t1/2 was observed with itraconazole administration. This observation suggests that itraconazole predominantly increased talazoparib bioavailability with a minor effect on elimination. However, this observation cannot be generalised to all P‐gp inhibitors as several P‐gp inhibitors are known to inhibit P‐gp in the renal tubules and could therefore alter drug elimination of P‐gp substrates that are predominantly renally eliminated as unchanged drug (e.g. digoxin). For example, known P‐gp inhibitors including amiodarone, quinidine, propafenone and verapamil reduced digoxin renal clearance.20, 21, 22 Therefore, the potential of a significantly longer talazoparib t1/2 when coadministered with other P‐gp inhibitors cannot be excluded. Consequently, upon discontinuation of the potent P‐gp inhibitor, the talazoparib dose should be maintained at the reduced dose for 3–5 half‐lives of the P‐gp inhibitor, after which the talazoparib dose should be increased to the dose used prior to the initiation of the P‐gp inhibitor.
The effect of P‐gp inducers on talazoparib exposure has not been previously studied. The results of Arm B of this study show that coadministration of rifampicin increased talazoparib Cmax by approximately 37% and did not affect talazoparib overall exposure (AUCinf). The increase in talazoparib Cmax with rifampicin coadministration might be explained by rifampicin's inhibition effect on P‐gp in the enterocytes during the absorption phase of talazoparib23 as rifampicin was administered 30 min before talazoparib in Period 2. Therefore, the potential for rifampicin‐mediated P‐gp inhibition during the initial talazoparib absorption phase cannot be excluded.24 However, rifampicin effect on P‐gp induction in the renal proximal tubules is expected to be sustained by daily rifampicin dosing throughout Period 2. The net result could be the lack of change in talazoparib AUC with this rifampicin administration schedule. The similar exposure observed when talazoparib was administered alone and with rifampicin (adjusted geometric mean ratio 102.0% with talazoparib alone as reference) and the similar talazoparib terminal half‐life when administered alone and with rifampicin (arithmetic mean ± standard deviation of 92.1 ± 17.7 and 80.6 ± 16.5 h for talazoparib administered alone and with rifampicin, respectively) suggested that the effect of rifampicin administration 30 min before talazoparib on talazoparib exposure, in particular on talazoaprib elimination, was limited. It is also worth highlighting that the variability of talazoparib Cmax was higher with rifampicin administration (71%) compared to talazoparib alone (53%), reflecting variability in the effect of rifampicin on talazoparib absorption. The limited impact of P‐gp induction on talazoparib elimination is consistent with findings from several studies investigating the effect of P‐gp inducers on the PK of P‐gp substrates that showed that other known pregnane‐X receptor agonists resulted in a mild to moderate reduction of exposure of P‐gp substrates (ranged from ~19 to 42%).25, 26, 27, 28
The design of both arms was sufficient to evaluate the effect of P‐gp inhibition and induction on talazoparib PK. First, the choice of perpetrators (itraconazole and rifampicin) was acceptable as P‐gp modulators because itraconazole and rifampicin modulate both CYP3A and P‐gp and talazoparib is not a CYP3A substrate. Second, a 0.5 mg talazoparib dose used in Arm A provided a 2‐fold safety margin relative to the MTD and the recommended dose to account for potential increase in talazoparib exposure when coadministered with itraconazole. In Arm B, the recommended dose of 1 mg talazoparib was used because it was expected that rifampicin could reduce the exposure of talazoparib. Third, itraconazole and rifampicin were dosed for adequate duration prior to the second dose of talazoparib to enable maximum P‐gp modulation prior to administration of talazoparib in Period 2. Administration of itraconazole and rifampicin was continued until the completion of PK samplings in order to maintain maximal P‐gp modulation throughout this period. Finally, a Phase 1 food effect study showed that food reduced talazoparib Cmax but had no effect on AUCinf. 7 To avoid any potential confounding food effects in this study, talazoparib was administered under fasting conditions. According to their respective summaries of product characteristics,9, 15 itraconazole should be administered immediately after a meal and rifampicin should be administered at least 30 min before a meal in order to achieve optimum intestinal absorption. Here, the meal plan followed for itraconazole and rifampicin was consistent with their respective summaries of product characteristics with the exception of the talazoparib dosing days where itraconazole and rifampicin were administered under fasting conditions.
In summary, coadministration of itraconazole with talazoparib increased talazoparib plasma exposure compared to talazoparib administered alone, consistent with P‐gp inhibition by itraconazole. A reduced talazoparib dose is recommended if coadministration of potent P‐gp inhibitors is unavoidable. The similar talazoparib exposure observed when talazoparib was administered alone and with rifampicin suggests that the effect of rifampicin administration 30 min before talazoparib on talazoparib exposure is limited. The safety profile of talazoparib coadministered with multiple doses of itraconazole or multiple doses of rifampicin showed good tolerability and was overall consistent with that observed in previous studies where talazoparib was administered as a single‐dose monotherapy.
COMPETING INTERESTS
M.E., C.‐H.C., A.P., H.S. and D.W. are employees of Pfizer and receive stock and stock options as part of their employment.
CONTRIBUTORS
All authors contributed to the design of the study and/or assisted with the data analysis/interpretation of the data. All authors assisted in the preparation of the manuscript, reviewed the manuscript and provided their approval for submission. All authors agree to be accountable for all aspects of the work presented.
Supporting information
Table S1: Summary of patient disposition
Table S2: Summary of treatment‐emergent adverse events (TEAEs)
Figure S1: Individual, median and geometric mean plasma talazoparib AUCinf and AUClast values (A) and Cmax values (B) following a single dose of talazoparib alone and with multiple oral doses of itraconazole. Box plot provides median and 25%/75% quartiles with whiskers to the last point within 1.5 ×x interquartile range. Geometric means are shown as triangles. AUCinf, area under the plasma concentration‐–time profile from time 0 extrapolated to infinity; AUClast, area under the plasma concentration time profile from time 0 to the time of last quantifiable concentration; Cmax, maximum observed plasma concentration.
Figure S2: Individual, median and geometric mean plasma talazoparib AUCinf and AUClast values (A) and Cmax values (B) following a single dose of talazoparib alone and with multiple oral doses of rifampinrifampicin. Box plot provides median and 25%/75% quartiles with whiskers to the last point within 1.5×x interquartile range. Geometric means are shown as triangles. AUCinf, area under the plasma concentration‐–time profile from time 0 extrapolated to infinity; AUClast, area under the plasma concentration time profile from time 0 to the time of last quantifiable concentration; Cmax, maximum observed plasma concentration
ACKNOWLEDGEMENTS
This study was sponsored by Medivation, which was acquired by Pfizer in September 2016.
Editorial and medical writing support was provided by Chantel Cadwell, PhD, and Mary Kacillas of Ashfield Healthcare Communications (Middletown, CT, USA) and was funded by Pfizer.
Elmeliegy M, Láng I, Smolyarchuk EA, et al. Evaluation of the effect of P‐glycoprotein inhibition and induction on talazoparib disposition in patients with advanced solid tumours. Br J Clin Pharmacol. 2020;86:771–778. 10.1111/bcp.14178
Trial register and clinical trial registration number: NCT03077607
PI statement: The authors confirm that the principal investigator for the study on which this paper is based is Elena A. Smolyarchuk and that she had direct clinical responsibility for patients.
DATA AVAILABILITY STATEMENT
Upon request, and subject to certain criteria, conditions and exceptions (see https://www.pfizer.com/science/clinical-trials/trial-data-and-results for more information), Pfizer will provide access to individual de‐identified participant data from Pfizer‐sponsored global interventional clinical studies conducted for medicines, vaccines and medical devices (i) for indications that have been approved in the US and/or EU or (ii) in programmes that have been terminated (i.e. development for all indications has been discontinued). Pfizer will also consider requests for the protocol, data dictionary and statistical analysis plan. Data may be requested from Pfizer trials 24 months after study completion. The de‐identified participant data will be made available to researchers whose proposals meet the research criteria and other conditions, and for which an exception does not apply, via a secure portal. To gain access, data requestors must enter into a data access agreement with Pfizer.
REFERENCES
- 1. Lord CJ, Ashworth A. PARP inhibitors: synthetic lethality in the clinic. Science. 2017;355(6330):1152‐1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bitler BG, Watson ZL, Wheeler LJ, Behbakht K. PARP inhibitors: clinical utility and possibilities of overcoming resistance. Gynecol Oncol. 2017;147(3):695‐704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Murai J, Feng Y, Yu GK, et al. Resistance to PARP inhibitors by SLFN11 inactivation can be overcome by ATR inhibition. Oncotarget. 2016;7(47):76534‐76550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Talzenna (talazoparib) [prescribing information]. Pfizer. NY, New York. 2018.
- 5. Litton JK, Rugo HS, Ettl J, et al. Talazoparib in patients with advanced breast cancer and a germline BRCA mutation. N Engl J Med. 2018;379(8):753‐763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. de Bono J, Ramanathan RK, Mina L, et al. Phase I, dose‐escalation, two‐part trial of the PARP inhibitor talazoparib in patients with advanced germline BRCA1/2 mutations and selected sporadic cancers. Cancer Discov. 2017;7(6):620‐629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. FDA. Multi‐disciplinary Review and Evaluation NDA 211651 TALZENNA (Talazoparib). https://www.accessdata.fda.gov/drugsatfda_docs/nda/2018/211651Orig1s000MultidisciplineR.pdf. Accessed April 4, 2019.
- 8. Yu Y, Chung CH, Plotka A, et al. A phase 1 mass balance study of (14) C‐labeled talazoparib in patients with advanced solid tumors. J Clin Pharmacol. 2019;59(9):1195‐1203. [DOI] [PubMed] [Google Scholar]
- 9. http://Medicines.org.uk. Rifadin 300mg Capsules (SmPC). 2018. https://www.medicines.org.uk/emc/product/6384/smpc. Accessed April 4, 2019.
- 10. Yu Y, Elmeliegy M, Litton JK, et al. Exposure‐efficacy progression‐free survival (PFS) analyses of breast cancer patients with germline BRCA1/2 mutations receiving talazoparib in the phase III EMBRACA trial. ESMO 2018 congress, Munich, Germany. Ann Onc. 2018;29:90‐121. [Google Scholar]
- 11. Yu Y, Durairaj C, Shi H, Wang DD. Population pharmacokinetics of talazoparib in patients with advanced cancer. J Clin Pharmacol. 2019. Sep;6:1‐12. [DOI] [PubMed] [Google Scholar]
- 12.WMA Declaration of Helsinki (18th WMA General Assembly 1964), revised at 64th
- 13. International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. ICH Harmonised Tripartite Guideline, E6: Guideline for Good Clinical Practice (CPMP/ICH/135/95). January 1997.
- 14. Directive 2001/20/EC of the European Parliament and of the Council of 4 April 2001 on the approximation of the laws, regulations and administrative provisions of the Member States relating to the implementation of good clinical practice in the conduct of clinical trials on medicinal products for human use. [PubMed]
- 15. http://Medicines.org.uk. Itraconazole 100mg Capsules, hard (SmPC). 2017. https://www.medicines.org.uk/emc/product/5914/smpc. April 4, 2019.
- 16. Turner NC, Telli ML, Rugo HS, et al. Final results of a phase 2 study of talazoparib (TALA) following platinum or multiple cytotoxic regimens in advanced breast cancer patients (pts) with germline BRCA1/2 mutations (ABRAZO). J Clin Oncol. 2017;35(15_suppl):1007‐1007.
- 17. Elmeliegy M, Yu Y, Litton JK, et al. Exposure‐safety analyses in breast cancer patients with germline BRCA1/2 mutations receiving talazoparib (TALA) in EMBRACA and ABRAZO trials. In: ESMO 2018 congress, Munich. Germany: Ann Onc. 2018;29:90‐121. [Google Scholar]
- 18. School of Pharmacy University of Washington . Drug Interaction Database Program. https://www.druginteractioninfo.org/. Accessed April 4, 2019.
- 19. FDA . Drug development and drug interactions: table of substrates, inhibitors and inducers. 2017. https://www.fda.gov/drugs/developmentapprovalprocess/developmentresources/druginteractionslabeling/ucm093664.htm. Accessed April 4, 2019.
- 20. Nademanee K, Piwonka RW, Singh BN, Hershman JM. Amiodarone and thyroid function. Prog Cardiovasc Dis. 1989;31(6):427‐437. [DOI] [PubMed] [Google Scholar]
- 21. Su SF, Huang JD. Inhibition of the intestinal digoxin absorption and exsorption by quinidine. Drug Metab Dispos. 1996;24(2):142‐147. [PubMed] [Google Scholar]
- 22. Calvo MV, Martin‐Suarez A, Martin Luengo C, Avila C, Cascon M, Dominguez‐Gil HA. Interaction between digoxin and propafenone. Ther Drug Monit. 1989;11(1):10‐15. [DOI] [PubMed] [Google Scholar]
- 23. Reitman ML, Chu X, Cai X, et al. Rifampin's acute inhibitory and chronic inductive drug interactions: experimental and model‐based approaches to drug‐drug interaction trial design. Clin Pharmacol Ther. 2011;89(2):234‐242. [DOI] [PubMed] [Google Scholar]
- 24. Kirby BJ, Collier AC, Kharasch ED, Whittington D, Thummel KE, Unadkat JD. Complex drug interactions of the HIV protease inhibitors 3: effect of simultaneous or staggered dosing of digoxin and ritonavir, nelfinavir, rifampicin, or bupropion. Drug Metab Dispos. 2012;40(3):610‐616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Juan H, Wan‐Hua Y, Juan S, Xiao‐Lei L, Wen‐Xing P. P‐gp induction by curcumin: an effective antidotal pathway. J Bioequiv Availab. 2013;5:236‐241. [Google Scholar]
- 26. Lutz JD, Kirby BJ, Wang L, et al. Cytochrome P450 3A induction predicts P‐glycoprotein induction; part 2: prediction of decreased substrate exposure after rifabutin or carbamazepine. Clin Pharmacol Ther. 2018;104(6):1191‐1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rameis H. On the interaction between phenytoin and digoxin. Eur J Clin Pharmacol. 1985;29(1):49‐53. [DOI] [PubMed] [Google Scholar]
- 28. Yamada S, Yasui‐Furukori N, Akamine Y, Kaneko S, Uno T. Effects of the P‐glycoprotein inducer carbamazepine on fexofenadine pharmacokinetics. Ther Drug Monit. 2009;31(6):764‐768. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: Summary of patient disposition
Table S2: Summary of treatment‐emergent adverse events (TEAEs)
Figure S1: Individual, median and geometric mean plasma talazoparib AUCinf and AUClast values (A) and Cmax values (B) following a single dose of talazoparib alone and with multiple oral doses of itraconazole. Box plot provides median and 25%/75% quartiles with whiskers to the last point within 1.5 ×x interquartile range. Geometric means are shown as triangles. AUCinf, area under the plasma concentration‐–time profile from time 0 extrapolated to infinity; AUClast, area under the plasma concentration time profile from time 0 to the time of last quantifiable concentration; Cmax, maximum observed plasma concentration.
Figure S2: Individual, median and geometric mean plasma talazoparib AUCinf and AUClast values (A) and Cmax values (B) following a single dose of talazoparib alone and with multiple oral doses of rifampinrifampicin. Box plot provides median and 25%/75% quartiles with whiskers to the last point within 1.5×x interquartile range. Geometric means are shown as triangles. AUCinf, area under the plasma concentration‐–time profile from time 0 extrapolated to infinity; AUClast, area under the plasma concentration time profile from time 0 to the time of last quantifiable concentration; Cmax, maximum observed plasma concentration
Data Availability Statement
Upon request, and subject to certain criteria, conditions and exceptions (see https://www.pfizer.com/science/clinical-trials/trial-data-and-results for more information), Pfizer will provide access to individual de‐identified participant data from Pfizer‐sponsored global interventional clinical studies conducted for medicines, vaccines and medical devices (i) for indications that have been approved in the US and/or EU or (ii) in programmes that have been terminated (i.e. development for all indications has been discontinued). Pfizer will also consider requests for the protocol, data dictionary and statistical analysis plan. Data may be requested from Pfizer trials 24 months after study completion. The de‐identified participant data will be made available to researchers whose proposals meet the research criteria and other conditions, and for which an exception does not apply, via a secure portal. To gain access, data requestors must enter into a data access agreement with Pfizer.