

Abstract
Pediatric acute lymphoblastic leukemia (ALL) is defined by recurrent chromosomal aberrations including hyperdiploidy and chromosomal translocations. Many of these aberrations originate in utero and the cells transform in early childhood through acquired secondary mutations. In this review, we will discuss the most common prenatal lesions that can lead to childhood ALL, with a special emphasis on the most common translocation in childhood ALL, t(12;21), which results in the ETV6-RUNX1 gene fusion. The ETV6-RUNX1 fusion arises prenatally and at a 500-fold higher frequency than the corresponding ALL. Even though the findings regarding the frequency of ETV6-RUNX1 were originally challenged, newer studies have confirmed the higher frequency. The prenatal origin has also been proven for other gene fusions, including KMT2A, the translocations t(1;19) and t(9;22) leading to TCF3-PBX1 and BCR-ABL1, respectively, as well as high hyperdiploidy. For most of these aberrations, there is evidence for more frequent occurrence than the corresponding leukemia incidences. We will briefly discuss what is known about the cells of origin, the mechanisms of leukemic transformation through lack of immunosurveillance, and why only a part of the carriers develops ALL.
Keywords: Prenatal origin, ALL, Preleukemia, Fusion genes
Introduction
Acute lymphoblastic leukemia (ALL) is the most common leukemia subtype in children [1]. The vast majority of cases belong to the B cell precursor subtype, whereas roughly 15% carry T cell progenitor markers [2]. ALL is common in young children and incidence peaks at ages 2–5 years. This peak is absent from acute myelogenous leukemia (AML), which is more common in adults (Fig. 1a) [1]. Infant ALL with KMT2A (previously known as MLL) rearrangements being a possible exception, childhood ALL is caused by a combination of genetic susceptibility factors and subsequently acquired somatic mutations. These mutations often occur in genes that are critical for lymphoid development [4]. The genetic susceptibility factors are often recurrent nonrandom mutations, like translocations or hyperdiploidy. These factors correspond to the ALL group in which they appear and can be used to classify the ALL. Depending on the age of the patients, different mutations are predominant. For infants, KMT2A rearrangements are the most common aberration; in young children, high hyperdiploidy and the translocation t(12;21), causing ETV6-RUNX1 (TEL-AML1), are most common (Fig. 1b) [2]. Another common translocation is t(1;19), leading to TCF3-PBX1 (E2A-PBX1), which occurs in approximately 5% of childhood cases as well as in adult ALL [5]. The BCR-ABL1 fusion, as a result of a t(9;22), is relatively rare in childhood ALL but is the most common aberration in adult ALL (Fig. 1b) [5].
Fig. 1.
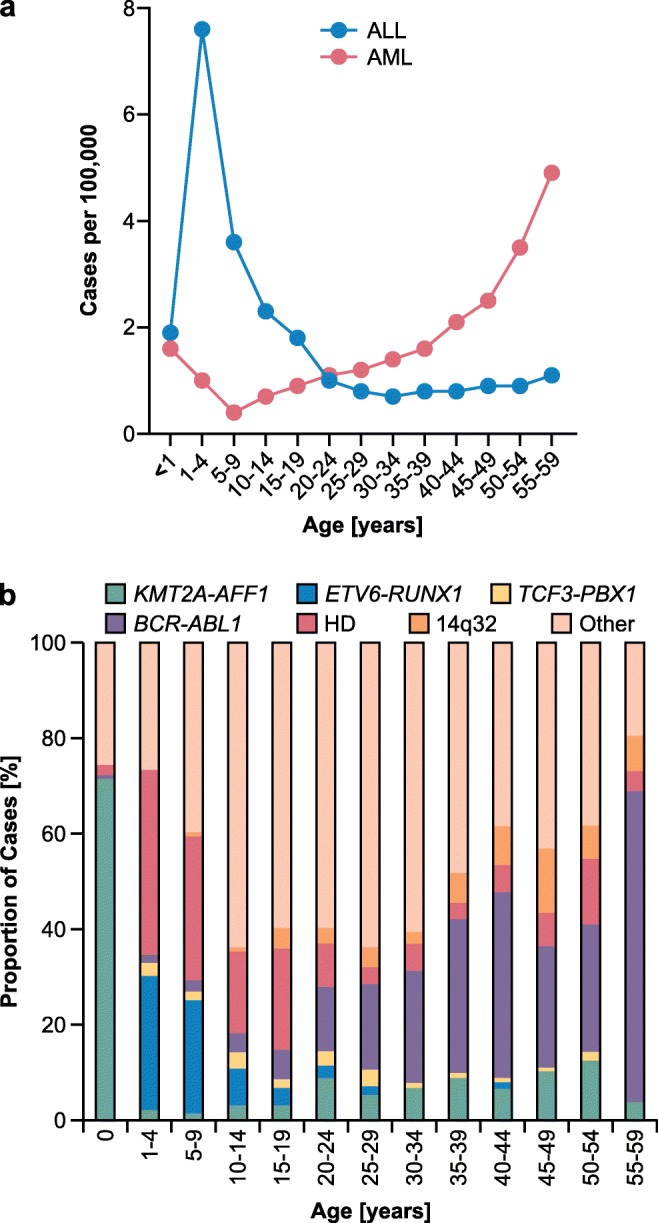
Age distribution and major subtypes of ALL. a Age distribution of ALL and AML in the USA from 1975 to 2016. Cases per 100,000 are shown. ALL has a clear peak at ages 1–4 and 5–9, whereas AML rates rise with age. b Major subtypes of ALL divided by age groups. The KMT2A-AFF1 fusion is very prevalent in infants, ETV6-RUNX1 and high hyperdiploidy (HD) dominate childhood ALL, and BCR-ABL1 is the most prevalent aberration in adults. Data for (a) taken from [1], data from (b) taken from [3]
As secondary alterations are needed for most subtypes, these first hits produce a preleukemic state. The secondary mutations only occur in a fraction of carriers [6–9]. There is convincing evidence that a significant percentage of these preleukemic lesions can arise prenatally and transform after postnatal secondary events occur [6–12] (Fig. 2). This evidence comes primarily from twin studies in which both twins had identical breakpoints, immunoglobulin heavy chain (IgH), or T cell receptor (TCR) rearrangements [10, 11, 14–16]. Additionally, by tracing the leukemia back to neonatal blood spots (Guthrie cards) of the twins, studies have been able to generate further proof of prenatal origin [17–19]. Here, the identical breakpoints were also present on the Guthrie cards. Since the blood is taken immediately after birth, a postnatal origin can be ruled out. The first hit occurs in the cell of origin, which differs from subtype to subtype. KMT2A rearrangements probably occur at the earliest state in CD34+ and CD19− cells [20], while other translocations seem to arise later in B cell development, although the exact cell is usually unknown. Infection [21] and delayed infection [22] have been discussed as possible causes for leukemic transformation for over a century. Furthermore, the mixing of populations has been postulated as a causal factor for leukemic transformation [23]. Recent studies have shown that exposure to infection can trigger the progression from preleukemia to ALL [24, 25]. A dysregulated immune response by activation of preleukemic B cells through memory T helper cells [26] and inactivity of NK cells have also been discussed [27–29].
Fig. 2.
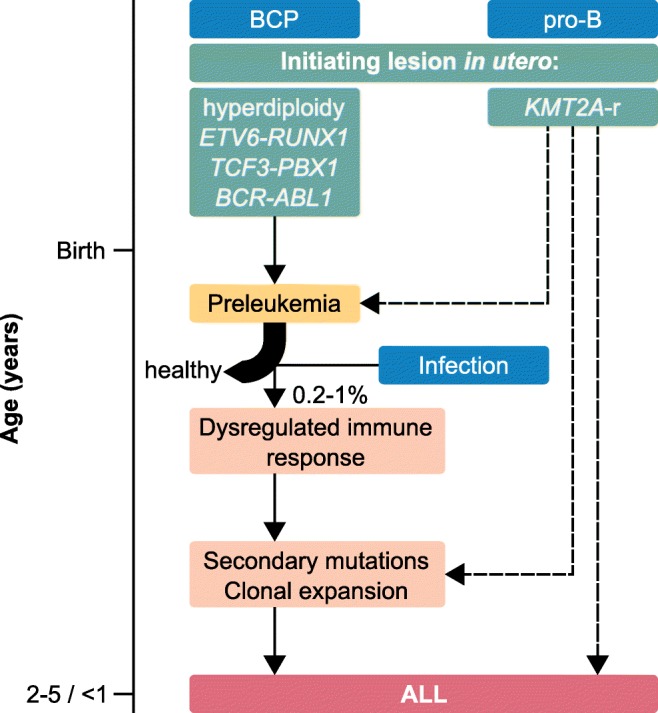
Timeline of ALL development. The initiating lesions (green) occur in utero and lead to a state of preleukemia after birth. Exposure to infection leads to a dysregulated immune response in a small fraction (about 0.2–1%) of preleukemic children. Most preleukemic children remain healthy [6, 7]. The children develop ALL by acquiring secondary mutations, eventually leading to clonal expansion. In the case of KMT2A rearrangements (KMT2A-r, right), it is not completely clear whether the KMT2A-r are sufficient for ALL development (right dashed arrow). It is also possible that the KMT2A-r directly trigger secondary mutations (central dashed arrow). One case of a healthy KMT2A-r carrier has been reported [13], leading to the possibility of a preleukemic state for those cases (left dashed arrow). The given median ages of ALL onset are 2–5 years of age for B cell precursor subtypes (BCP, left) and < 1 year of age for pro-B cell subtype (pro-B, right)
To date, the prenatal origin of ALL with KMT2A rearrangements [10], ETV6-RUNX1 [11], TCF3-PBX1 [9], BCR-ABL1 [8], and (high) hyperdiploidy [12] has been shown (Table 1).
Table 1.
Studies that identified prenatal ALL or preleukemia
| Subtype | Year | Study | Specimen | Positives (prevalence) | Study type | Method of detection | Population | Preleukemic cells |
|---|---|---|---|---|---|---|---|---|
| KMT2A-r | 1993 | Ford et al. [10] | Bone marrow, peripheral blood, testicles | 6/6 (100%) | LTS | Southern Blotting | N/S | |
| 1994 | Gill Super et al. [15] | Peripheral blood | 2/2 (100%) | LTS | Southern Blotting | N/S | ||
| 1997 | Gale et al. [17] | Guthrie cards | 3/3 (100%) | BT | PCR | N/S | ||
| 1998 | Uckun et al. [13] | Fetal bone marrow, fetal liver | 9/29 (31.03%) | PNS | nRT-PCR | N/S | ||
| 2000 | Yagi et al. [12] | Guthrie cards | 2/2 (100%) | BT | nPCR | N/S | ||
| 2000 | Fasching et al. [30] | Guthrie cards | 2/2 (100%) | BT | PCR | Austrian | ||
| 2002 | Taub et al. [31] | Guthrie cards | 1/1 (100%) | BT | PCR | US American | ||
| ETV6-RUNX1 | 1998 | Ford et al. [11] | Guthrie cards | 2/2 (100%) | LTS/BT | PCR | Dutch | |
| 1999 | Wiemels et al. [14, 18] | Guthrie cards | 8/11 (72.73%) | BT | LDI-PCR | British, Italian | ||
| 2001 | Maia et al. [19] | Guthrie cards | 3/3 (100%) | BT | PCR | N/S | ||
| 2001 | Eguchi-Ishimae et al. [32] | Cord blood | 1/67 (1.49%) | NBS | nRT-PCR | Japanese | ||
| 2002 | Mori et al. [6] | Frozen cord blood | 6/567 (1.06%) | NBS | nRT-PCR qRT-PCR FISH | British | 10−3 to 10−4 | |
| 2002 | Taub et al. [31] | Guthrie cards | 1/1 (100%) | BT | PCR | US American | ||
| 2006 | Burjanivova et al. [33] | Guthrie cards | 1/3 (33.33%) | BT | PCR | Czech | ||
| 2008 | Gruhn et al. [34] | Guthrie cards | 3/6 (50.00%) | BT | snPCR | German | 10−4 to 10−5 | |
| 2011 | Zuna et al. [35] | Cord blood | 5/253 (1.98%) | NBS | RT-PCR, qRT-PCR | Czech | ||
| 2012 | Olsen et al. [36] | Fresh cord blood | 3/1258 (0.24%) | NBS | qRT-PCR | Danish | < 10−4 | |
| 2014 | Škorvaga et al. [37] | Frozen cord blood | 8/200 (4.00%) | NBS | qRT-PCR | Slovak | ≤ 10−5 | |
| 2015 | Ornelles et al. [38] | Fresh cord blood | 5/210 (2.38%) | NBS | nRT-PCR | US American | ||
| 2017 | Kosik et al. [39] | Cord blood | 4/300 (1.33%) | NBS | qRT-PCR | Slovak | ≤ 10−5 | |
| 2018 | Schäfer et al. [7] | Frozen cord blood | 50/1000 (5.00%) | NBS | GIPFEL | Danish | 10−2 to 10−5 | |
| TCF3-PBX1 | 2002 | Wiemels et al. [40] | Guthrie cards | 2/15 (13.33%) | BT | PCR | US American | |
| 2002 | Taub et al. [31] | Guthrie cards | 1/1 (100%) | BT | PCR | US American | ||
| 2019 | Hein et al. [9] | Frozen cord blood | 2/340 (0.59%) | NBS | GIPFEL | Danish | 10−3 to 10−4 | |
| BCR-ABL1 | 2011 | Cazzaniga et al. [8] | Bone marrow, peripheral blood, Guthrie cards | 4/4 (100%) | LTS/BT | RT-PCR, qRT-PCR, FISH | Italian, British | |
| Hyperdiploid | 2000 | Yagi et al. [12] | Guthrie cards | 1/1 (100%) | BT | nPCR | N/S | |
| 2002 | Panzer-Grümeyer et al. [41] | Guthrie cards | 1/1 (100%) | BT | nPCR | Austrian | ||
| 2002 | Taub et al. [31] | Guthrie cards | 5/5 (100%) | BT | PCR | US American | ||
| 2003 | Maia et al. [16] | Bone marrow, peripheral blood | 2/2 (100%) | BT | nPCR | British | ||
| 2008 | Gruhn et al. [34] (including Taub et al. [31]) | Guthrie cards | 10/11 (90.91%) | BT | snPCR | German, US American | 10−4 | |
| Other | 2000 | Yagi et al. [12] | Guthrie cards | 1/4 (25.00%) | BT | nPCR | N/S | |
| 2002 | Taub et al. [31] | Guthrie cards | 4/9 (44.44%) | BT | PCR | US American | ||
| 2006 | Burjanivova et al. [33] | Guthrie cards | 2/9 (22.22%) | BT | PCR | Czech | ||
| 2008 | Gruhn et al. [34] | Guthrie cards | 11/20 (55.00%) | BT | snPCR | German | 10−3 to 10−5 |
The number of preleukemic cells is only given for studies that investigated preleukemia and determined its frequency. Frequencies for studies that used GIPFEL are from sorted CD19+ cells
KMT2A-r, cases with KMT2A rearrangements; other, not specified ALL cases; LTS, leukemic twin study; BT, backtracking study; PNS, prenatal screening study; NBS, newborn screening study; N/S, not specified
Prenatal subtypes
KMT2A rearrangements
The KMT2A gene has a myriad of fusion partners. For ALL, the most important one is surely AFF1, with MLLT3 (AF9) and MLLT1 (ENL) also being more frequent than others [5]. KMT2A translocations frequently occur in infants, and some newborns already show signs of full-blown leukemia [15]. Therefore, these translocations are a natural candidate for prenatal development. Among several studies that investigated the prenatal status of ALL in general and KMT2A-rearranged ALL in particular, were those that first traced ALL back to an in utero event [10, 15] and the first to trace it back to Guthrie cards [17]. Uckun et al. [13] showed actual in utero presence of the fusion gene in fetal tissue from abortions. This study also found one case of a healthy infant expressing the KMT2A-AFF1 fusion transcript. This suggests that KMT2A fusions are also present in healthy individuals and will not necessarily lead to overt leukemia. However, it is unknown whether this infant developed leukemia later on. Additionally, other studies [42, 43] failed to reproduce these findings, leaving the question of whether KMT2A translocations also occur more frequently than the corresponding leukemia at least in part unanswered. It is of note, though, that one should expect to find KMT2A fusions in fetal tissue, even if leukemia development was inevitable. For that to happen, the cohort size would simply have to be much larger than the 29 samples studied by Uckun et al. [13].
There are two possibilities that explain how the KMT2A fusions contribute to leukemia development and the short latency periods after birth: (1) The fusion itself is sufficient for leukemia onset. This would mean that leukemia development is inevitable and no healthy individuals carrying the fusions exist. (2) A secondary mutation is required, but is triggered by the fusion protein. This would also be in line with the short latency. If the fusions trigger the additional mutation, leukemia development might be inevitable, but it would allow for the theoretical possibility of healthy carriers (Fig. 2). As only one such case has been described [13], it is not possible to infer from the presence or absence of healthy carriers which model is actually at work.
ETV6-RUNX1
There is ample evidence that the translocation t(12;21) leading to the fusion of the transcription factors ETV6 and RUNX1 predominantly, maybe even always, arises in utero [11, 18]. This was first shown in a twin study in 1998 [11]. Here, both twins had exactly the same breakpoint, something that had never before been described for ETV6-RUNX1+ leukemia [11]. This supports a model in which the preleukemic clone arises in one twin and spreads to the other via the shared placenta. The secondary mutations in the twins differed, hinting at postnatal origin. Additionally, this and several other studies were able to trace back this leukemia type to Guthrie cards [11, 18, 19, 31, 33, 34].
The ETV6-RUNX1 fusion alone is not sufficient for leukemia development. For that, secondary postnatal mutations are necessary. Therefore, not without controversy regarding the frequency of the translocation, several studies have investigated the ETV6-RUNX1 fusion in healthy individuals, especially newborns. Initially identified in umbilical cord blood of one healthy newborn and the peripheral blood of 13 healthy children and adults [32], ETV6-RUNX1 was shown to be present in ~ 1% of newborns [6]. Several Danish studies later challenged these findings [36, 44–47], but newer reports confirmed the original results [7, 37–39].
There are several possible explanations for the contradicting results of these studies. The material used is one of the factors that can influence the outcome. All studies used umbilical cord blood (UCB) for the investigation of newborns. However, some studies used fresh UCB, handled within 24 h of blood draw, while others used frozen UCB or did not specify whether the material was fresh. Interestingly, all studies that identified no or very few ETV6-RUNX1+ cells in the UCB used fresh UCB [45, 46, 48] or in one case fresh embryonic liver [44]. Using freshly harvested cells has the advantage of accurately representing the neonatal hematopoietic environment. It does, however, require a great deal of time and money. It is unlikely that the different results are influenced by the use of fresh or stored UCB, because (1) one study by Ornelles et al. [38] used fresh UCB and identified 2.38% ETV6-RUNX1+ samples, and (2) storage has a negative effect on RNA, especially when RNA is released from dead cells [49], and therefore the studies using frozen UCB should have found fewer ETV6-RUNX1+ cells. Then again, it was shown that apoptotic signals can induce double-strand breaks in both ETV6 and RUNX1 and that this can lead to the ETV6-RUNX1 fusion [32]. Storage therefore could induce the translocation but probably at very low levels and in much fewer samples than reported by the studies using frozen UCB [6, 7, 37]. Also, if the freezing induced the ETV6-RUNX1 fusion, Ornelles et al. [38] should not have found any positive samples.
A more likely cause of the different results is the use of different detection methods. Most studies used nested reverse transcriptase PCR (nRT-PCR) or quantitative RT-PCR (qRT-PCR). The advantage of qRT-PCR is that it allows for quantification of the fusion transcript. The nRT-PCR may be more sensitive, as it uses a nested PCR setup, but it is not quantitative. Both methods are, like all RNA methods, vulnerable to contamination, the nRT-PCR approach even more so as it is an open-tube technique. However, contaminations in qRT-PCR can also lead to overestimation of prevalence. However, almost all studies regardless of results used qRT-PCR, and some used multiple techniques for validation. Mori et al. [6] used nRT-PCR and then qRT-PCR and FISH to validate their finding that ~ 1% carried the fusion. Lausten-Thomsen et al. [46] initially found 14 of 1417 (~ 1%) samples to be ETV6-RUNX1+ by qRT-PCR. After dot-blot validation, nine positives remained. It was only the second validation with RNA of flow-cytometric-sorted frozen UCB cells that led the authors to conclude that the results were falsely positive. Hence, the specific method used may positively or negatively impact the detection of the ETV6-RUNX1 fusion, in combination with the quality and quantity of the input material. Low-quality or -quantity input material might lead to false-negative results. Ultimately, all studies but one used RNA as basis for their analysis. DNA is more stable than RNA by a factor of 10,000 when stored frozen [49] and is thus the better choice for stored material. Furthermore, RNA produces the same fusion point for every breakpoint. This is advantageous for screening purposes but makes identification of contaminants impossible. Identical breakpoints on the DNA level have only been reported for identical twins [11]. Hence, a possible contamination is easy to detect. To date, we have conducted the only study identifying ETV6-RUNX1+ cells via DNA quantification [7]. We used the novel GIPFEL technique [50], allowing for the indirect identification of chromosomal translocations at the DNA level. In this study, we identified 5% of healthy newborns to be ETV6-RUNX1+. Additionally, we sequenced the chromosomal breakpoints of five positive samples.
One could argue that differences between populations might lead to different ETV6-RUNX1 frequencies in the healthy population. Population differences have been identified for some tumor entities, including ETV6-RUNX1+ and TCF3-PBX1+ leukemias, the latter of which is more frequent in Latin America [51, 52]. ETV6-RUNX1 is much less common in East Asians [53], Hispanics [54], and especially in Maori, where only 5.4% of pediatric ALL cases harbor this translocation [55]. Interestingly, the survival rates of ETV6-RUNX1+ Maori did not differ from those of other ethnicities, probably due to equal access to ALL treatment for all in New Zealand [55]. Except for the Japanese study by Eguchi-Ishimae et al. [32] and the US-American study by Ornelles et al. [38], all studies used primarily Caucasian European populations. Therefore, an influence of the population on the frequency of ETV6-RUNX1 is highly unlikely.
Notably, all studies that could not identify ETV6-RUNX1+ newborns were conducted with a Danish population. However, Olsen et al. [48] found 10 out of 2005 healthy adults to express ETV6-RUNX1 at low levels. That is statistically more than would be expected if the incidence were equal to the leukemia rate (t test, P = 0.0019). This implies that adults carry the fusion at a higher prevalence than the leukemia rate. Therefore, it is safe to assume that the same is also true for children, even though it is not a proof of prenatal origin. Furthermore, we also screened UCB samples from Denmark and we were able to identify ETV6-RUNX1 carriers [7]. Hence, it is highly unlikely that the differences between the studies are a result of using samples from the Danish population, especially as the leukemia incidence in Denmark does not differ from the incidences of other European countries [56].
The real discussion might not be whether the ETV6-RUNX1 fusion is present in healthy newborns but at what frequency. Originally, Mori et al. [6] reported frequencies of 10−4 to 10−3, but those frequencies were not confirmed by later studies [36, 37, 39, 46, 48]. The frequency in investigated adults was markedly lower, but that is in line with the reduced risk for ETV6-RUNX1+ leukemia in adults [48]. However, all studies confirming the presence of ETV6-RUNX1 in healthy newborns that looked at the frequency found it to be much lower [37, 39]. Lausten-Thomsen et al. [46] initially found ~ 1% of ETV6-RUNX1-positive samples with a frequency of ≤ 10−5, therefore this study is very important in challenging the proposed frequency of the preleukemic cells. We also tried to address this in our study [7], but the frequency we found can only be compared with the others under reserve. We used CD19+-sorted cells and had a bias, because not all PCR products are amplified in the same way. Therefore, these numbers should be considered an estimate. Furthermore, we used DNA instead of RNA, so this must be taken into account when comparing the studies. In our study, we also confirmed the presence of ETV6-RUNX1 by qRT-PCR in two cases [7]. The frequency was ~ 10−4, which would be more in line with the studies that found low frequencies.
TCF3-PBX1
The TCF3-PBX1 fusion is the product of a balanced or unbalanced t(1;19) translocation and is among the most frequent aberrations in childhood ALL. It is especially common in Latin America [51, 52] and among black children [57], where as many as 11.8% of childhood ALL cases carry this fusion.
Unlike the aforementioned translocations, TCF3-PBX1 has long been considered to only arise postnatally. Still, the fusion could be traced back to Guthrie cards by Wiemels et al. in two cases [40]. In both cases, only one segment of the blood spot was positive for the fusion and the fusion points showed signs of site specificity and of terminal deoxynucleotidyl transferase activity, and so TCF3-PBX1 was declared postnatal. The site specificity hints at aberrant V(D)J recombination. During fetal hematopoiesis, none or few nontemplate nucleotides are inserted, whereas this insertion is common in children and adults [58–60]. However, in another backtracking study, one TCF3-PBX1 patient could be traced back to the respective Guthrie card by screening for IgH rearrangements [31].
Following our success with GIPFEL and ETV6-RUNX1 [7], we also looked for TCF3-PBX1 in healthy newborns. In 2 of 340 (0.6%) cases, we were able to identify the fusion and also the exact fusion point [9]. The presence of the TCF3-PBX1 fusion in the UCB of newborns is definite proof of prenatal origin. It is, however, not clear if TCF3-PBX1+ newborns will remain healthy throughout their lifetime. In the ETV6-RUNX1 study [7], 50/1000 (5%) were translocation positive, which gave the study enough statistical power to conclude that most of them will never develop leukemia. For TCF3-PBX1, it is unlikely that both newborns will develop ALL but not impossible. Identifying the TCF3-PBX1 fusion in healthy newborns could prove that TCF3-PBX1 can arise prenatally but not that the frequency definitely exceeds the ALL incidence. The data from these studies paint a picture in which TCF3-PBX1 can arise prenatally but possibly also throughout an individual’s lifetime. Studies investigating the frequency of TCF3-PBX1+ ALL in children and adults found a slight decrease from 5% of ALL cases in children to 3% in adults [5]. Thus, either TCF3-PBX1 (1) always arises prenatally and can have a very long latency phase, or (2) it can also arise postnatally, explaining the mild decrease from childhood to adulthood.
BCR-ABL1
The BCR-ABL1 fusion is the product of a t(9;22) translocation, widely known as the Philadelphia chromosome, which was the first ever to be described [61]. The fusion of these genes can create three different proteins: p190, p210, and p230. Each of these differ in their BCR breakpoints, with m-BCR (minor) leading to p190, M-BCR (major) to p210, and μ-BCR (micro) to p230 [62, 63]. Classically, BCR-ABL1 is present in adult chronic myelogenous leukemia (CML), where 90–95% carry the Philadelphia chromosome. Of these patients, over 99% express the p210 isoform. However, the fusion is also present in ALL. In the adult form, 25% have a t(9;22) [5], with the majority also expressing the p210. In this entity, the p190 isoform is also prominently present. In pediatric ALL, BCR-ABL1 plays a minor role, with only 3% of cases being positive for this fusion [2]. It is of interest, though, that the p190 isoform is the predominant form in pediatric ALL, with 90% expressing this protein.
The p190 isoform was shown to arise prenatally in at least two pairs of monozygotic twins [8]. In both cases, both twins had the identical breakpoint, indicative of prenatal origin. Also, in one twin pair, the fusion could be traced back to the respective Guthrie cards. Interestingly, in one pair of twins, only one twin developed leukemia [8]. It is, of course, possible that the second twin developed ALL later in life, but it shows that secondary hits are necessary and that these hits are acquired postnatally. This also hints at the possibility that BCR-ABL1 may arise prenatally in children who will never develop ALL. Additionally, the specificity of the isoforms, regarding the resulting leukemia subtype, indicates that the p210 isoform probably arises postnatally, especially when one considers that it is typical for CML, which usually arises later in life.
IgH or TCR rearrangements
Leukemia can be traced back to birth using not only gene fusions but also rearrangements of the IgH or the TCR. Several studies have used this approach to investigate ALL cohorts [12, 16, 31, 33, 34, 41] (Table 1). Using IgH or TCR rearrangements has the advantage of enabling the study of leukemia entities without a defining gene fusion. In this way, hyperdiploid ALLs have been shown to arise prenatally [12, 16, 31, 34, 41]. Moreover, it is not surprising that the prenatal origin is not restricted to only ALL with fusion genes. In fact, prenatal origin has been shown for roughly two-thirds of childhood ALL subforms [2, 8–12]. One might argue that prenatal origin is not restricted to those subforms but can also occur in other subgroups, as it is also not restricted to ALL. Prenatal origin of childhood leukemia could also be shown for AML [6] but seems to be more common in ALL [33].
Mechanisms of transformation
Some details remain unknown, even after the origin of a particular ALL is proven to be prenatal, such as the cell of origin or the mechanisms by which this prenatal preleukemia turns into postnatal ALL.
The cell of origin depends on the underlying lesion. Even though shown in leukemic cases that were not traced back to birth, BCR-ABL1 fusions can have different cells of origin, depending on the isoform. The p210 form originates in hematopoietic stem cells, whereas the p190 form, typical for childhood ALL, originates in a B cell progenitor [64]. For ETV6-RUNX1, there is also evidence that the cell of origin is CD34+ and CD19+, hinting at a more differentiated lymphoid-committed precursor [64–66]. However, there is also evidence for the opposite. Experiments in zebrafish showed that ETV6-RUNX1 expression restricted to lymphoid cells was incapable of inducing leukemia, whereas expression in all linages led to ALL at a low level, comparable with the human situation [67]. Mice in which the ETV6-RUNX1 fusion was put under the control of the IgH promoter did not develop leukemia [68], whereas mice developed ALL when the entire bone marrow was transplanted [69], hinting at a cell of origin with an earlier developmental stage. TCF3-PBX1 fusions also seem to originate at later stages in a more differentiated, lymphoid-committed progenitor [70]. Translocations including the KMT2A gene, however, seem to originate earlier, in a CD34+, CD19− cell [20]. Importantly, it is generally difficult to determine the exact cell of origin for the ALL subtypes. The leukemic lesions can alter the cell of origin so that its properties change. It is therefore also possible that the preleukemic cell moves—at least in part—backwards in its evolution, so that it eventually shows surface markers and properties of a cell that is hierarchically upstream from the cell of origin. Similarly, it has been postulated that the initiating lesion, e.g., a fusion gene, does not arise in a committed cell but a hematopoietic stem/progenitor cell, which is then reprogrammed by this oncogene. This reprogramming then leads to epigenetic changes that are inherited by the daughter cells [71]. In this model, the oncogene might not even be needed in the actual tumor but would only act in the cell of origin in a hit and run manner [72].
Progression from preleukemia to overt leukemia might differ in the various prenatal subtypes. As mentioned in the KMT2A section, these translocations have a very short latency period and may inevitably lead to ALL development. However, one case of a healthy carrier has been reported [13]. For other translocations, healthy carriers have been described [6–9, 35], suggesting that the translocation itself is insufficient for ALL development. KMT2A fusions might be sufficient by themselves or actively trigger a secondary mutation, whereas ETV6-RUNX1, TCF3-PBX1, and BCR-ABL1 require independent secondary events for transformation. These secondary events often target genes that are important for immune cells in general or B cells in particular. The complete or partial deletion of IKFZ1 is a common secondary aberration in BCR-ABL1+ cases and confers a dismal prognosis [8]. Deletions of the second ETV6 allele have been reported in up to 70% of ETV6-RUNX1+ cases [73]. PAX5 and CDKN2A are deleted in 28% and 26% of cases, respectively [73]. These deletions in ETV6-RUNX1+ patients are predominantly caused by aberrant RAG1/2 activity [74]. The fact that only a part of the translocation carriers suffers secondary mutations and the resulting ALL hints at the involvement of environmental factors in acquiring said mutations. A natural candidate for a possible cause of acute leukemia is infection. This was proposed as early as 1917 [21]. Additional alterations to this hypothesis have been made, including delayed infection [22] and population mixing [23]. Recent studies have provided in vivo evidence by linking ALL in Pax5-heterozygous mice [24] and ETV6-RUNX1+ mice [25] to exposure to infection. The exact mechanisms of leukemic transformation through infection are not yet fully understood, but dysregulation of the immune system may play a major role. It has been proposed that memory T helper cells can support preleukemic B cell precursors in the bone marrow, and thus support ALL development [26].
Natural killer (NK) cells are also interesting candidates, as they not only play a role in distinguishing between self and foreign but also play an important role in cancer control [75]. One task of NK cells is it to kill aberrant cells. The interaction between killer immunoglobulin-like receptors (KIRs) on NK cells and human lymphocyte antigen (HLA) class I ligands regulates the activity of NK cells. Both, KIRs and HLA, are polymorphic. HLA-C alleles, which are considered to be the dominant KIR ligands, can be grouped as C1 and C2. The KIR haplotypes on chromosome 19q13.4 are A and B. The A-haplotype harbors more inhibitory KIRs, whereas the B-haplotype has more activating KIRs [28]. For both, there is a telomeric and a centromeric cluster. The presence of more activating KIRs seems to increase the risk of ALL development [29]. Furthermore, it has been shown that in patients with positive minimal residual disease, the activating KIR2DS1 receptor was more present, whereas the inhibitory receptors KIR2DL1 and KIR3DL1 were less frequent [28]. In general, a higher frequency of activating KIRs, which are mostly encoded by the B-haplotype, seems to confer a greater risk of developing ALL [29], with the risk coming mainly from the telomeric B-motifs cluster [28]. The elevated risk is due to the fact that a greater number of inhibitory receptors on NK cells leads to a stronger responsiveness and the absence of inhibitory receptors leaves the cells hyporesponsive [76, 77]. Furthermore, an increase in activating signals leads to a decrease in signal intensity over time [78, 79]. The use of the HLA-C allele also has an influence on susceptibility to ALL. The C2 epitope leads to an elevated risk of ALL and a late relapse, whereas C1 homozygosity has a protective effect [27]. The binding of HLA-C2 ligands to KIR2DS1 induces tolerance and renders the NK cells hyporesponsive [80, 81].
Taken together with the data from the prenatal lesions and the fact that these often only induce preleukemia, it is reasonable to assume that the use of HLA-C epitope and KIRs influences not only the likelihood of leukemic transformation (Fig. 3) but also the process itself. If the NK cells fail to clear the preleukemic or leukemic cells, the onset of ALL is more likely or inevitable, respectively. Therefore, among those children in whom the preleukemia progressed to overt leukemia, there should be a higher percentage of HLA-C2 and activating KIRs, whereas among those who are healthy, HLA-C1 and inhibitory KIRs should be predominant.
Fig. 3.
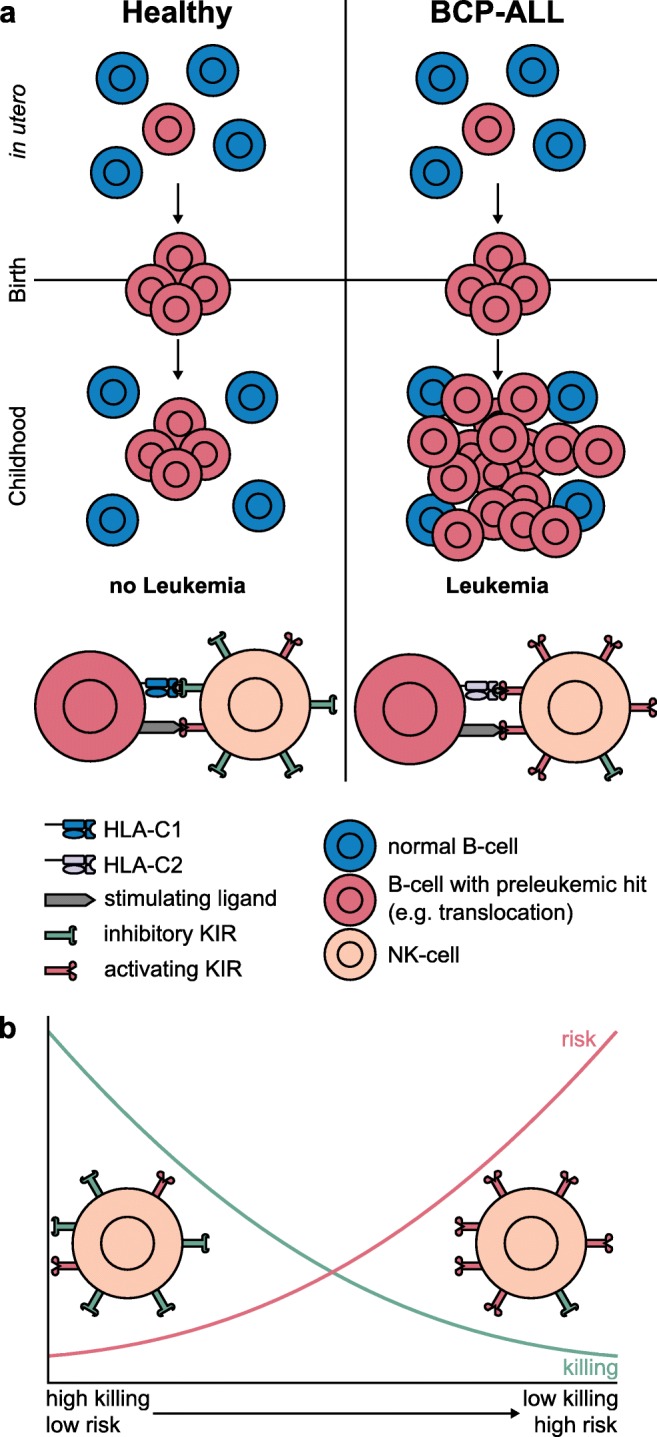
Model of the impact of NK cells on ALL development. a A prenatal preleukemic hit emerges in utero (red) and an expanded clone is present at birth. According to this model, interaction with NK cells plays a key role in leukemia development. HLA-C2 receptors (lavender) pose an elevated risk for ALL and can also interact with activating KIRs, e.g., KIR2DS1 [82]. An elevated number of activating KIRs (red), especially from the telomeric B cluster also pose a risk (right), whereas HLA-C1 (blue) and more inhibitory KIRs, i.e., the A-haplotype (green) seems to protect against ALL (left). b Scheme of NK cell killing efficiency and risk of developing ALL. NK cells with more inhibitory KIRs (green) have a higher killing efficiency and confer a lower risk of ALL; NK cells with more activating KIRs (red) have a lower killing efficiency and confer a higher risk of ALL
Conclusion
Most carriers of prenatal lesions will remain healthy throughout their lifetime. This emphasizes the need for a secondary mutation and possibly a dysregulated immune system. On the other hand, it opens up new possibilities for prevention and treatment. If environmental and genetic factors play a role in leukemic transformation, this offers opportunities for prevention and interference. For instance, if a prenatal lesion is diagnosed, one could also check for HLA-C epitopes and KIR haplotypes to assess the risk of ALL development. A better understanding of the mechanisms of progression from preleukemia to leukemia and why it only happens in a fraction of cases will not only help to treat the respective ALLs but ultimately help to prevent them from occurring in the first place.
Funding information
This work was supported by the German Federal Office for Radiation Protection (grants 36 14S 30034 and 36 18S 32275) and by an intramural grant (2019-03) from the Research Commission of the Medical Faculty of the Heinrich-Heine-University Düsseldorf.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Howlader N, Noone AM, Krapcho M, Miller D, Brest A, Yu M, et al. SEER cancer statistics review, 1975–2016. Bethesda, MD: National Cancer Institute; 2019. [Google Scholar]
- 2.Pui CH, Carroll WL, Meshinchi S, Arceci RJ. Biology, risk stratification, and therapy of pediatric acute leukemias: An update. Journal of Clinical Oncology. 2011;29(5):551–565. doi: 10.1200/jco.2010.30.7405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Harrison CJ. Cytogenetics of paediatric and adolescent acute lymphoblastic leukaemia. British Journal of Haematology. 2009;144(2):147–156. doi: 10.1111/j.1365-2141.2008.07417.x. [DOI] [PubMed] [Google Scholar]
- 4.Lim JY, Bhatia S, Robison LL, Yang JJ. Genomics of racial and ethnic disparities in childhood acute lymphoblastic leukemia. Cancer. 2014;120(7):955–962. doi: 10.1002/cncr.28531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pui CH, Relling MV, Downing JR. Acute lymphoblastic leukemia. The New England Journal of Medicine. 2004;350(15):1535–1548. doi: 10.1056/NEJMra023001. [DOI] [PubMed] [Google Scholar]
- 6.Mori H, Colman SM, Xiao Z, Ford AM, Healy LE, Donaldson C, Hows JM, Navarrete C, Greaves M. Chromosome translocations and covert leukemic clones are generated during normal fetal development. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(12):8242–8247. doi: 10.1073/pnas.112218799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schäfer D, Olsen M, Lähnemann D, Stanulla M, Slany R, Schmiegelow K, et al. Five percent of healthy newborns have an ETV6-RUNX1 fusion as revealed by DNA-based GIPFEL screening. Blood. 2018;131(7):821–826. doi: 10.1182/blood-2017-09-808402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cazzaniga G, van Delft FW, Lo Nigro L, Ford AM, Score J, Iacobucci I, et al. Developmental origins and impact of BCR-ABL1 fusion and IKZF1 deletions in monozygotic twins with Ph+ acute lymphoblastic leukemia. Blood. 2011;118(20):5559–5564. doi: 10.1182/blood-2011-07-366542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hein D, Dreisig K, Metzler M, Izraeli S, Schmiegelow K, Borkhardt A, Fischer U. The preleukemic TCF3-PBX1 gene fusion can be generated in utero and is present in ≈0.6% of healthy newborns. Blood. 2019;134(16):1355–1358. doi: 10.1182/blood.2019002215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ford AM, Ridge SA, Cabrera ME, Mahmoud H, Steel CM, Chan LC, Greaves M. In utero rearrangements in the trithorax-related oncogene in infant leukaemias. Nature. 1993;363(6427):358–360. doi: 10.1038/363358a0. [DOI] [PubMed] [Google Scholar]
- 11.Ford AM, Bennett CA, Price CM, Bruin MC, Van Wering ER, Greaves M. Fetal origins of the TEL-AML1 fusion gene in identical twins with leukemia. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(8):4584–4588. doi: 10.1073/pnas.95.8.4584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yagi T, Hibi S, Tabata Y, Kuriyama K, Teramura T, Hashida T, Shimizu Y, Takimoto T, Todo S, Sawada T, Imashuku S. Detection of clonotypic IGH and TCR rearrangements in the neonatal blood spots of infants and children with B-cell precursor acute lymphoblastic leukemia. Blood. 2000;96(1):264–268. doi: 10.1182/blood.V96.1.264. [DOI] [PubMed] [Google Scholar]
- 13.Uckun FM, Herman-Hatten K, Crotty ML, Sensel MG, Sather HN, Tuel-Ahlgren L, Sarquis MB, Bostrom B, Nachman JB, Steinherz PG, Gaynon PS, Heerema N. Clinical significance of MLL-AF4 fusion transcript expression in the absence of a cytogenetically detectable t(4;11)(q21;q23) chromosomal translocation. Blood. 1998;92(3):810–821. [PubMed] [Google Scholar]
- 14.Wiemels JL, Ford AM, Van Wering ER, Postma A, Greaves M. Protracted and variable latency of acute lymphoblastic leukemia after TEL-AML1 gene fusion in utero. Blood. 1999;94(3):1057–1062. doi: 10.1182/blood.V94.3.1057.415k10_1057_1062. [DOI] [PubMed] [Google Scholar]
- 15.Gill Super HJ, Rothberg PG, Kobayashi H, Freeman AI, Diaz MO, Rowley JD. Clonal, nonconstitutional rearrangements of the MLL gene in infant twins with acute lymphoblastic leukemia: In utero chromosome rearrangement of 11q23. Blood. 1994;83(3):641–644. doi: 10.1182/blood.V83.3.641.641. [DOI] [PubMed] [Google Scholar]
- 16.Maia AT, van der Velden VH, Harrison CJ, Szczepanski T, Williams MD, Griffiths MJ, et al. Prenatal origin of hyperdiploid acute lymphoblastic leukemia in identical twins. Leukemia. 2003;17(11):2202–2206. doi: 10.1038/sj.leu.2403101. [DOI] [PubMed] [Google Scholar]
- 17.Gale KB, Ford AM, Repp R, Borkhardt A, Keller C, Eden OB, et al. Backtracking leukemia to birth: Identification of clonotypic gene fusion sequences in neonatal blood spots. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(25):13950–13954. doi: 10.1073/pnas.94.25.13950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wiemels JL, Cazzaniga G, Daniotti M, Eden OB, Addison GM, Masera G, Saha V, Biondi A, Greaves MF. Prenatal origin of acute lymphoblastic leukaemia in children. Lancet. 1999;354(9189):1499–1503. doi: 10.1016/S0140-6736(99)09403-9. [DOI] [PubMed] [Google Scholar]
- 19.Maia AT, Ford AM, Jalali GR, Harrison CJ, Taylor GM, Eden OB, Greaves MF. Molecular tracking of leukemogenesis in a triplet pregnancy. Blood. 2001;98(2):478–482. doi: 10.1182/blood.V98.2.478. [DOI] [PubMed] [Google Scholar]
- 20.Hotfilder M, Rottgers S, Rosemann A, Schrauder A, Schrappe M, Pieters R, et al. Leukemic stem cells in childhood high-risk ALL/t(9;22) and t(4;11) are present in primitive lymphoid-restricted CD34+CD19- cells. Cancer Research. 2005;65(4):1442–1449. doi: 10.1158/0008-5472.Can-04-1356. [DOI] [PubMed] [Google Scholar]
- 21.Ward G. The infective theory of acute leukaemia. Br J Child Dis. 1917;14:10–20. [Google Scholar]
- 22.Greaves MF. Speculations on the cause of childhood acute lymphoblastic leukemia. Leukemia. 1988;2(2):120–125. [PubMed] [Google Scholar]
- 23.Kinlen LJ, Clarke K, Hudson C. Evidence from population mixing in British new towns 1946-85 of an infective basis for childhood leukaemia. Lancet. 1990;336(8715):577–582. doi: 10.1016/0140-6736(90)93389-7. [DOI] [PubMed] [Google Scholar]
- 24.Martin-Lorenzo A, Hauer J, Vicente-Duenas C, Auer F, Gonzalez-Herrero I, Garcia-Ramirez I, et al. Infection exposure is a causal factor in B-cell precursor acute lymphoblastic leukemia as a result of Pax5-inherited susceptibility. Cancer Discovery. 2015;5(12):1328–1343. doi: 10.1158/2159-8290.cd-15-0892. [DOI] [PubMed] [Google Scholar]
- 25.Rodríguez-Hernández G, Hauer J, Martín-Lorenzo A, Schäfer D, Bartenhagen C, García-Ramírez I, Auer F, González-Herrero I, Ruiz-Roca L, Gombert M, Okpanyi V, Fischer U, Chen C, Dugas M, Bhatia S, Linka RM, Garcia-Suquia M, Rascón-Trincado MV, Garcia-Sanchez A, Blanco O, García-Cenador MB, García-Criado FJ, Cobaleda C, Alonso-López D, de Las Rivas J, Müschen M, Vicente-Dueñas C, Sánchez-García I, Borkhardt A. Infection exposure promotes ETV6-RUNX1 precursor B-cell leukemia via impaired H3K4 Demethylases. Cancer Research. 2017;77(16):4365–4377. doi: 10.1158/0008-5472.can-17-0701. [DOI] [PubMed] [Google Scholar]
- 26.Bürgler S, Nadal D. Pediatric precursor B acute lymphoblastic leukemia: Are T helper cells the missing link in the infectious etiology theory? Mol Cell Pediatr. 2017;4(1):6. doi: 10.1186/s40348-017-0072-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Babor F, Manser AR, Fischer JC, Scherenschlich N, Enczmann J, Chazara O, Moffett A, Borkhardt A, Meisel R, Uhrberg M. KIR ligand C2 is associated with increased susceptibility to childhood ALL and confers an elevated risk for late relapse. Blood. 2014;124(14):2248–2251. doi: 10.1182/blood-2014-05-572065. [DOI] [PubMed] [Google Scholar]
- 28.Sullivan EM, Jeha S, Kang G, Cheng C, Rooney B, Holladay M, Bari R, Schell S, Tuggle M, Pui CH, Leung W. NK cell genotype and phenotype at diagnosis of acute lymphoblastic leukemia correlate with postinduction residual disease. Clinical Cancer Research. 2014;20(23):5986–5994. doi: 10.1158/1078-0432.Ccr-14-0479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Misra MK, Prakash S, Moulik NR, Kumar A, Agrawal S. Genetic associations of killer immunoglobulin like receptors and class I human leukocyte antigens on childhood acute lymphoblastic leukemia among north Indians. Human Immunology. 2016;77(1):41–46. doi: 10.1016/j.humimm.2015.10.009. [DOI] [PubMed] [Google Scholar]
- 30.Karin Fasching, Simon Panzer, Oskar A. Haas, Rolf Marschalek, Helmut Gadner, E. Renate Panzer-Grümayer, (2000) Presence of clone-specific antigen receptor gene rearrangements at birth indicates an in utero origin of diverse types of early childhood acute lymphoblastic leukemia. Blood 95 (8):2722-2724 [PubMed]
- 31.Taub JW, Konrad MA, Ge Y, Naber JM, Scott JS, Matherly LH, et al. High frequency of leukemic clones in newborn screening blood samples of children with B-precursor acute lymphoblastic leukemia. Blood. 2002;99(8):2992–2996. doi: 10.1182/blood.v99.8.2992. [DOI] [PubMed] [Google Scholar]
- 32.Eguchi-Ishimae M, Eguchi M, Ishii E, Miyazaki S, Ueda K, Kamada N, Mizutani S. Breakage and fusion of the TEL (ETV6) gene in immature B lymphocytes induced by apoptogenic signals. Blood. 2001;97(3):737–743. doi: 10.1182/blood.V97.3.737. [DOI] [PubMed] [Google Scholar]
- 33.Burjanivova T, Madzo J, Muzikova K, Meyer C, Schneider B, Votava F, Marschalek R, Stary J, Trka J, Zuna J. Prenatal origin of childhood AML occurs less frequently than in childhood ALL. BMC Cancer. 2006;6:100. doi: 10.1186/1471-2407-6-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gruhn B, Taub JW, Ge Y, Beck JF, Zell R, Hafer R, et al. Prenatal origin of childhood acute lymphoblastic leukemia, association with birth weight and hyperdiploidy. Leukemia. 2008;22(9):1692–1697. doi: 10.1038/leu.2008.152. [DOI] [PubMed] [Google Scholar]
- 35.Zuna J, Madzo J, Krejci O, Zemanova Z, Kalinova M, Muzikova K, Zapotocky M, Starkova J, Hrusak O, Horak J, Trka J. ETV6/RUNX1 (TEL/AML1) is a frequent prenatal first hit in childhood leukemia. Blood. 2011;117(1):368–369. doi: 10.1182/blood-2010-09-309070. [DOI] [PubMed] [Google Scholar]
- 36.Olsen M, Hjalgrim H, Melbye M, Madsen HO, Schmiegelow K. RT-PCR screening for ETV6-RUNX1-positive clones in cord blood from newborns in the Danish National Birth Cohort. Journal of Pediatric Hematology/Oncology. 2012;34(4):301–303. doi: 10.1097/MPH.0b013e3182332268. [DOI] [PubMed] [Google Scholar]
- 37.Skorvaga M, Nikitina E, Kubes M, Kosik P, Gajdosechova B, Leitnerova M, et al. Incidence of common preleukemic gene fusions in umbilical cord blood in Slovak population. PLoS One. 2014;9(3):e91116. doi: 10.1371/journal.pone.0091116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ornelles DA, Gooding LR, Garnett-Benson C. Neonatal infection with species C adenoviruses confirmed in viable cord blood lymphocytes. PLoS One. 2015;10(3):e0119256. doi: 10.1371/journal.pone.0119256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kosik P, Skorvaga M, Durdik M, Jakl L, Nikitina E, Markova E, et al. Low numbers of pre-leukemic fusion genes are frequently present in umbilical cord blood without affecting DNA damage response. Oncotarget. 2017;8(22):35824–35834. doi: 10.18632/oncotarget.16211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wiemels JL, Leonard BC, Wang Y, Segal MR, Hunger SP, Smith MT, Crouse V, Ma X, Buffler PA, Pine SR. Site-specific translocation and evidence of postnatal origin of the t(1;19) E2A-PBX1 fusion in childhood acute lymphoblastic leukemia. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(23):15101–15106. doi: 10.1073/pnas.222481199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Panzer-Grümayer ER, Fasching K, Panzer S, Hettinger K, Schmitt K, Stockler-Ipsiroglu S, et al. Nondisjunction of chromosomes leading to hyperdiploid childhood B-cell precursor acute lymphoblastic leukemia is an early event during leukemogenesis. Blood. 2002;100(1):347–349. doi: 10.1182/blood-2002-01-0144. [DOI] [PubMed] [Google Scholar]
- 42.Kim-Rouille MH, MacGregor A, Wiedemann LM, Greaves MF, Navarrete C. MLL-AF4 gene fusions in normal newborns. Blood. 1999;93(3):1107–1108. doi: 10.1182/blood.V93.3.1107. [DOI] [PubMed] [Google Scholar]
- 43.Trka, J., Zuna, J., Hrusak, O., Michalova, K., Muzikova, K., Kalinova, M., et al. (1999). No evidence for MLL/AF4 expression in normal cord blood samples. Blood, 93(3), 1106–1107; author reply 1108-1110. [PubMed]
- 44.Lausten-Thomsen U, Hjalgrim H, Marquart H, Lutterodt M, Petersen BL, Schmiegelow K. ETV6-RUNX1 transcript is not frequent in early human haematopoiesis. European Journal of Haematology. 2008;81(2):161–162. doi: 10.1111/j.1600-0609.2008.01091.x. [DOI] [PubMed] [Google Scholar]
- 45.Lausten-Thomsen U, Madsen HO, Vestergaard TR, Hjalgrim H, Lando A, Schmiegelow K. Increased risk of ALL among premature infants is not explained by increased prevalence of pre-leukemic cell clones. Blood Cells, Molecules & Diseases. 2010;44(3):188–190. doi: 10.1016/j.bcmd.2009.12.007. [DOI] [PubMed] [Google Scholar]
- 46.Lausten-Thomsen U, Madsen HO, Vestergaard TR, Hjalgrim H, Nersting J, Schmiegelow K. Prevalence of t(12;21)[ETV6-RUNX1]-positive cells in healthy neonates. Blood. 2011;117(1):186–189. doi: 10.1182/blood-2010-05-282764. [DOI] [PubMed] [Google Scholar]
- 47.Kusk MS, Lausten-Thomsen U, Andersen MK, Olsen M, Hjalgrim H, Schmiegelow K. False positivity of ETV6/RUNX1 detected by FISH in healthy newborns and adults. Pediatric Blood & Cancer. 2014;61(9):1704–1706. doi: 10.1002/pbc.25050. [DOI] [PubMed] [Google Scholar]
- 48.Olsen M, Madsen HO, Hjalgrim H, Gregers J, Rostgaard K, Schmiegelow K. Preleukemic TEL-AML1-positive clones at cell level of 10(-3) to 10(-4) do not persist into adulthood. Journal of Pediatric Hematology/Oncology. 2006;28(11):734–740. doi: 10.1097/01.mph.0000243652.33561.0f. [DOI] [PubMed] [Google Scholar]
- 49.Shibata Y, Malhotra A, Dutta A. Detection of DNA fusion junctions for BCR-ABL translocations by anchored ChromPET. Genome Medicine. 2010;2(9):70. doi: 10.1186/gm191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Füller E, Schäfer D, Fischer U, Krell PF, Stanulla M, Borkhardt A, et al. Genomic inverse PCR for exploration of ligated breakpoints (GIPFEL), a new method to detect translocations in leukemia. PLoS One. 2014;9(8):e104419. doi: 10.1371/journal.pone.0104419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jimenez-Morales S, Miranda-Peralta E, Saldana-Alvarez Y, Perez-Vera P, Paredes-Aguilera R, Rivera-Luna R, et al. BCR-ABL, ETV6-RUNX1 and E2A-PBX1: Prevalence of the most common acute lymphoblastic leukemia fusion genes in Mexican patients. Leukemia Research. 2008;32(10):1518–1522. doi: 10.1016/j.leukres.2008.03.021. [DOI] [PubMed] [Google Scholar]
- 52.Bekker-Mendez VC, Miranda-Peralta E, Nunez-Enriquez JC, Olarte-Carrillo I, Guerra-Castillo FX, Pompa-Mera EN, et al. Prevalence of gene rearrangements in Mexican children with acute lymphoblastic leukemia: A population study-report from the Mexican Interinstitutional Group for the identification of the causes of childhood leukemia. BioMed Research International. 2014;2014:210560. doi: 10.1155/2014/210560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liang DC, Shih LY, Yang CP, Hung IJ, Liu HC, Jaing TH, et al. Frequencies of ETV6-RUNX1 fusion and hyperdiploidy in pediatric acute lymphoblastic leukemia are lower in far east than west. Pediatric Blood & Cancer. 2010;55(3):430–433. doi: 10.1002/pbc.22628. [DOI] [PubMed] [Google Scholar]
- 54.Aldrich MC, Zhang L, Wiemels JL, Ma X, Loh ML, Metayer C, et al. Cytogenetics of Hispanic and White children with acute lymphoblastic leukemia in California. Cancer Epidemiology, Biomarkers & Prevention. 2006;15(3):578–581. doi: 10.1158/1055-9965.Epi-05-0833. [DOI] [PubMed] [Google Scholar]
- 55.Pettit, T., Cole, N., Leung, W., Ballantine, K., & Macfarlane, S. (2017). Analysis of common cytogenetic abnormalities in New Zealand pediatric ALL shows ethnically diverse carriage of ETV6-RUNX1, without a corresponding difference in survival. Pediatric Blood & Cancer, 64(12). 10.1002/pbc.26676. [DOI] [PubMed]
- 56.Ferlay J, Colombet M, Soerjomataram I, Dyba T, Randi G, Bettio M, Gavin A, Visser O, Bray F. Cancer incidence and mortality patterns in Europe: Estimates for 40 countries and 25 major cancers in 2018. European Journal of Cancer. 2018;103:356–387. doi: 10.1016/j.ejca.2018.07.005. [DOI] [PubMed] [Google Scholar]
- 57.Pui CH, Sandlund JT, Pei D, Rivera GK, Howard SC, Ribeiro RC, Rubnitz JE, Razzouk BI, Hudson MM, Cheng C, Raimondi SC, Behm FG, Downing JR, Relling MV, Evans WE. Results of therapy for acute lymphoblastic leukemia in black and white children. JAMA. 2003;290(15):2001–2007. doi: 10.1001/jama.290.15.2001. [DOI] [PubMed] [Google Scholar]
- 58.Sanz I. Multiple mechanisms participate in the generation of diversity of human H chain CDR3 regions. Journal of Immunology. 1991;147(5):1720–1729. [PubMed] [Google Scholar]
- 59.Shiokawa S, Mortari F, Lima JO, Nunez C, Bertrand FE, 3rd, Kirkham PM, et al. IgM heavy chain complementarity-determining region 3 diversity is constrained by genetic and somatic mechanisms until two months after birth. Journal of Immunology. 1999;162(10):6060–6070. [PubMed] [Google Scholar]
- 60.Zemlin M, Bauer K, Hummel M, Pfeiffer S, Devers S, Zemlin C, Stein H, Versmold HT. The diversity of rearranged immunoglobulin heavy chain variable region genes in peripheral blood B cells of preterm infants is restricted by short third complementarity-determining regions but not by limited gene segment usage. Blood. 2001;97(5):1511–1513. doi: 10.1182/blood.v97.5.1511. [DOI] [PubMed] [Google Scholar]
- 61.Nowell PC, Hungerford D. A minute chromosome in human chronic granulocytic leukemia. Science. 1960;132:1497–1501. [Google Scholar]
- 62.Kurzrock R, Shtalrid M, Romero P, Kloetzer WS, Talpas M, Trujillo JM, et al. A novel c-abl protein product in Philadelphia-positive acute lymphoblastic leukaemia. Nature. 1987;325(6105):631–635. doi: 10.1038/325631a0. [DOI] [PubMed] [Google Scholar]
- 63.Kurzrock R, Shtalrid M, Gutterman JU, Koller CA, Walters R, Trujillo JM, Talpaz M. Molecular analysis of chromosome 22 breakpoints in adult Philadelphia-positive acute lymphoblastic leukaemia. British Journal of Haematology. 1987;67(1):55–59. doi: 10.1111/j.1365-2141.1987.tb02296.x. [DOI] [PubMed] [Google Scholar]
- 64.Castor A, Nilsson L, Astrand-Grundstrom I, Buitenhuis M, Ramirez C, Anderson K, et al. Distinct patterns of hematopoietic stem cell involvement in acute lymphoblastic leukemia. Nature Medicine. 2005;11(6):630–637. doi: 10.1038/nm1253. [DOI] [PubMed] [Google Scholar]
- 65.Hotfilder M, Rottgers S, Rosemann A, Jurgens H, Harbott J, Vormoor J. Immature CD34+CD19- progenitor/stem cells in TEL/AML1-positive acute lymphoblastic leukemia are genetically and functionally normal. Blood. 2002;100(2):640–646. doi: 10.1182/blood.V100.2.640. [DOI] [PubMed] [Google Scholar]
- 66.Hong D, Gupta R, Ancliff P, Atzberger A, Brown J, Soneji S, Green J, Colman S, Piacibello W, Buckle V, Tsuzuki S, Greaves M, Enver T. Initiating and cancer-propagating cells in TEL-AML1-associated childhood leukemia. Science. 2008;319(5861):336–339. doi: 10.1126/science.1150648. [DOI] [PubMed] [Google Scholar]
- 67.Sabaawy HE, Azuma M, Embree LJ, Tsai HJ, Starost MF, Hickstein DD. TEL-AML1 transgenic zebrafish model of precursor B cell acute lymphoblastic leukemia. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(41):15166–15171. doi: 10.1073/pnas.0603349103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Andreasson P, Schwaller J, Anastasiadou E, Aster J, Gilliland DG. The expression of ETV6/CBFA2 (TEL/AML1) is not sufficient for the transformation of hematopoietic cell lines in vitro or the induction of hematologic disease in vivo. Cancer Genetics and Cytogenetics. 2001;130(2):93–104. doi: 10.1016/s0165-4608(01)00518-0. [DOI] [PubMed] [Google Scholar]
- 69.Bernardin F, Yang Y, Cleaves R, Zahurak M, Cheng L, Civin CI, Friedman AD. TEL-AML1, expressed from t(12;21) in human acute lymphocytic leukemia, induces acute leukemia in mice. Cancer Research. 2002;62(14):3904–3908. [PubMed] [Google Scholar]
- 70.Cobaleda C, Sanchez-Garcia I. B-cell acute lymphoblastic leukaemia: Towards understanding its cellular origin. Bioessays. 2009;31(6):600–609. doi: 10.1002/bies.200800234. [DOI] [PubMed] [Google Scholar]
- 71.Vicente-Duenas C, Romero-Camarero I, Cobaleda C, Sanchez-Garcia I. Function of oncogenes in cancer development: A changing paradigm. The EMBO Journal. 2013;32(11):1502–1513. doi: 10.1038/emboj.2013.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Perez-Caro M, Cobaleda C, Gonzalez-Herrero I, Vicente-Duenas C, Bermejo-Rodriguez C, Sanchez-Beato M, et al. Cancer induction by restriction of oncogene expression to the stem cell compartment. The EMBO Journal. 2009;28(1):8–20. doi: 10.1038/emboj.2008.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mullighan CG, Goorha S, Radtke I, Miller CB, Coustan-Smith E, Dalton JD, Girtman K, Mathew S, Ma J, Pounds SB, Su X, Pui CH, Relling MV, Evans WE, Shurtleff SA, Downing JR. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature. 2007;446(7137):758–764. doi: 10.1038/nature05690. [DOI] [PubMed] [Google Scholar]
- 74.Papaemmanuil E, Rapado I, Li Y, Potter NE, Wedge DC, Tubio J, et al. RAG-mediated recombination is the predominant driver of oncogenic rearrangement in ETV6-RUNX1 acute lymphoblastic leukemia. Nature Genetics. 2014;46(2):116–125. doi: 10.1038/ng.2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Imai K, Matsuyama S, Miyake S, Suga K, Nakachi K. Natural cytotoxic activity of peripheral-blood lymphocytes and cancer incidence: An 11-year follow-up study of a general population. Lancet. 2000;356(9244):1795–1799. doi: 10.1016/s0140-6736(00)03231-1. [DOI] [PubMed] [Google Scholar]
- 76.Yu J, Heller G, Chewning J, Kim S, Yokoyama WM, Hsu KC. Hierarchy of the human natural killer cell response is determined by class and quantity of inhibitory receptors for self-HLA-B and HLA-C ligands. Journal of Immunology. 2007;179(9):5977–5989. doi: 10.4049/jimmunol.179.9.5977. [DOI] [PubMed] [Google Scholar]
- 77.Joncker NT, Fernandez NC, Treiner E, Vivier E, Raulet DH. NK cell responsiveness is tuned commensurate with the number of inhibitory receptors for self-MHC class I: The rheostat model. Journal of Immunology. 2009;182(8):4572–4580. doi: 10.4049/jimmunol.0803900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Raulet DH, Vance RE. Self-tolerance of natural killer cells. Nature Reviews. Immunology. 2006;6(7):520–531. doi: 10.1038/nri1863. [DOI] [PubMed] [Google Scholar]
- 79.Joncker NT, Raulet DH. Regulation of NK cell responsiveness to achieve self-tolerance and maximal responses to diseased target cells. Immunological Reviews. 2008;224:85–97. doi: 10.1111/j.1600-065X.2008.00658.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chewning JH, Gudme CN, Hsu KC, Selvakumar A, Dupont B. KIR2DS1-positive NK cells mediate alloresponse against the C2 HLA-KIR ligand group in vitro. Journal of Immunology. 2007;179(2):854–868. doi: 10.4049/jimmunol.179.2.854. [DOI] [PubMed] [Google Scholar]
- 81.Pittari G, Liu XR, Selvakumar A, Zhao Z, Merino E, Huse M, et al. NK cell tolerance of self-specific activating receptor KIR2DS1 in individuals with cognate HLA-C2 ligand. Journal of Immunology. 2013;190(9):4650–4660. doi: 10.4049/jimmunol.1202120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Campbell KS, Purdy AK. Structure/function of human killer cell immunoglobulin-like receptors: Lessons from polymorphisms, evolution, crystal structures and mutations. Immunology. 2011;132(3):315–325. doi: 10.1111/j.1365-2567.2010.03398.x. [DOI] [PMC free article] [PubMed] [Google Scholar]