

Abstract
Biallelic variants in the EYS gene are a major cause of autosomal recessive inherited retinal disease (IRD), with a high prevalence in the Asian population. The purpose of this study was to identify pathogenic EYS variants, to determine the clinical/genetic spectrum of EYS-associated retinal disease (EYS-RD), and to discover disease-associated variants with relatively high allele frequency (1%-10%) in a nationwide Japanese cohort. Sixty-six affected subjects from 61 families with biallelic or multiple pathogenic/disease-associated EYS variants were ascertained by whole-exome sequencing. Three phenotype groups were identified in EYS-RD: retinitis pigmentosa (RP; 85.94%), cone-rod dystrophy (CORD; 10.94%), and Leber congenital amaurosis (LCA; 3.12%). Twenty-six pathogenic/disease-associated EYS variants were identified, including seven novel variants. The two most prevalent variants, p.(Gly843Glu) and p.(Thr2465Ser) were found in 26 and twelve families (42.6%, 19.7%), respectively, for which the allele frequency (AF) in the Japanese population was 2.2% and 3.0%, respectively. These results expand the phenotypic and genotypic spectrum of EYS-RD, accounting for a high proportion of EYS-RD both in autosomal recessive RP (23.4%) and autosomal recessive CORD (9.9%) in the Japanese population. The presence of EYS variants with relatively high AF highlights the importance of considering the pathogenicity of non-rare variants in relatively prevalent Mendelian disorders.
Subject terms: Disease genetics, Genetic counselling
Introduction
Inherited retinal disease (IRD) is one of the major causes of blindness in children and the working population in developed countries1. The prevalence of IRD is high among Mendelian disorders, with 1 in 3,500–4,000 individuals thought to be affected (Genetic Home References; https://ghr.nlm.nih.gov/condition)1–3.
In 2008, variants in the eyes shut homologue (EYS) gene (OMIM: 612424) were first identified as disease-causing for autosomal recessive retinitis pigmentosa (ARRP) by Abd El–Aziz et al. and Collins et al., independently4,5. Since the discovery, over 270 disease-associated variants have been reported according to the Human Gene Mutation Database (HGMD; 2018.4 version, https://portal.biobase-international.com)6.
EYS (NM_001142800.1) spans approximately 2 Mb of chr6q12 and encodes a protein with 3144 amino acids that contains 27 epidermal growth factor-like (EGF) domains and five Laminin G-like domains4. EYS is an orthologue of Drosophila’s spacemaker (spam), which has an essential role in the morphogenesis of photoreceptors4,5,7. Recent studies have shown that the absence of EYS causes disruptions of the photoreceptor structure and leads to cone-rod dystrophy (CORD) in zebrafish8,9. However, the exact molecular mechanism has not been clarified due to the limited resources of animal models, as the EYS gene is lacking in several rodent species4,9,10.
Several phenotypes have been associated with pathogenic EYS variants, such as RP and CORD; thus, “EYS-associated retinal disease (EYS-RD)” can be used as an accurate description for this disease, in consideration of the phenotypic spectrum5,11–13.
EYS-RD is currently thought to be one of the most prevalent IRDs in Asian and European populations13–19. Barragan et al. reported that EYS-RD accounted for 15.9% of ARRP in the Spanish population17, and Hosono et al. and Arai et al. reported a high prevalence (18%-23%) in Japanese cohorts of RP patients15,20. However, the pathogenicity of the EYS variants and the accurate prevalence of EYS-RD, as well as what underlies this high prevalence of EYS-RD, are still uncertain due to the lack of large cohort studies.
Recently, disease-associated variants with relatively high allele frequency (AF; >1% in the general population) have been identified in ABCA4-associated retinal disease, representing one of the most prevalent IRDs21–24. A survey of variants with relatively high AF has become increasingly important to obtain an accurate genetic diagnosis in the era of next-generation sequencing. This is because high-throughput genetic screening technologies with automated variant filtering remove variants with a population frequency above the selected cut-off23,24. The analysis of variants with relatively high AF is anticipated to be more relevant for relatively prevalent Mendelian disorders such as IRD, especially in an isolated population such as the Japanese population, given the potential presence of founder variants.
The purpose of this study was to determine pathogenic/disease-associated EYS variants utilizing an analysis of variants with relatively high AF and to clarify the clinical and genetic spectrum of EYS-RD in a large nationwide Japanese cohort. The AF of EYS variants in affected subjects and in the general population were also investigated to estimate the AF-based prevalence of EYS-RD.
Results
Participants, demographics and visual acuity
In total, 66 affected subjects from 61 families with EYS-RD were included in this study. Co-segregation analysis was performed within 20 families. Thirty-six family members from 61 families underwent clinical examination and whole-exome sequencing. The pedigree charts showing the clinical and genetic status of each affected and unaffected subject are presented in Supplementary Fig. S1. The phenotypic and genotypic features of the affected subjects are summarized in Supplementary Table S1.
The detailed clinical data are presented in Supplementary Table S2. All 66 affected subjects were Japanese, and there were 36 females and 30 males. The median age of 66 affected subjects was 46.0 years at examinations (range, 11.0–84.0), with a median age of onset of 21.0 (range, 1.0–65.0). The median best-corrected visual acuity (BCVA) was 0.1 logarithm of minimum angle of resolution (LogMAR) unit (range, −0.18–1.8). There were three eyes with hand motion, two with light perception, and one with non-light perception. Retinal images of four representative cases caused by prevalent variants in the EYS gene are presented in Fig. 1.
Figure 1.
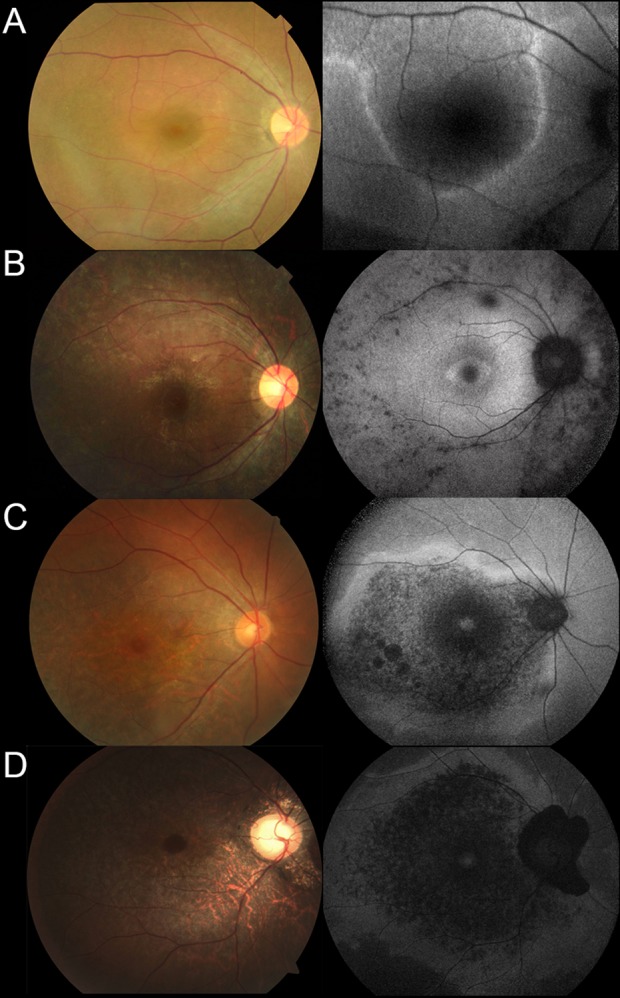
Fundus and autofluorescence findings of four representative cases with EYS-associated retinal disease (EYS-RD). (A) A 43-year-old female (2-III:2) diagnosed with retinitis pigmentosa (RP) harbouring homozygous variants (c.[2528 G > A];[2528 G > A], p.[(Gly843Glu)];[(Gly843Glu)]) in the EYS gene, showing retinal atrophic changes along the vessel arcade. (B) A 17-year-old male (22-II:2) diagnosed with RP harbouring homozygous variants (c.[4957dupA];[4957dupA], p.[(Ser1653Lysfs*2)];[(Ser1653Lysfs*2)]) in the EYS gene, showing retinal atrophic changes outside the vessel arcade. (C) A 50-year-old female (11-II:1) diagnosed with cone-rod dystrophy (CORD) harbouring two pairs of homozygous variants (c.[2528 G > A;c.7394 C > G];[2528 G > A;c.7394 C > G], p.[(Gly843Glu);(Thr2465Ser)];[(Gly843Glu);(Thr2465Ser)]) with relatively high allele frequency (AF) in the EYS gene, showing retinal atrophic changes within the vessel arcade. (D) A 39-year-old male (23-II:1) diagnosed with CORD harbouring compound heterozygous variants (c.[4957dupA];[8805 C > A], p.[(Ser1653Lysfs*2)];[(Tyr2935*)]) in the EYS gene, showing retinal atrophic changes within the vessel arcade.
Phenotype subgroups
The described clinical diagnosis was RP in 52 families (57 affected subjects), CORD in seven families (seven affected subjects), and early-onset retinitis pigmentosa or Leber congenital amaurosis (EORP/LCA) in two families (two affected subjects). The median age of onset and the median age at examination of 57 affected subjects with RP were 20.0 (range 1.0–65.0) and 46.5 (range 17.0–84.0), respectively. The median age of onset and the median age at examination of seven affected subjects with CORD were 40.0 (range 29.0–43.0) and 46.0 (range 39.0–76.0), respectively. In three families, different clinical diagnoses were described for the affected subjects within the family (Families 2, 3 and 28). Candidate variants were identified in the CACNA1F gene (OMIM; 300110), the TOPORS gene (OMIM; 609507), the RIMS1 gene (OMIM; 606629), the DRAM2 gene (OMIM; 613360), and the RP1L1 gene (OMIM; 608581) gene, respectively. (Supplementary Table S1, Supplementary Figs. S1 and S2).
EYS variants and in silico molecular genetic analysis
Whole-exome sequencing was performed for all the genomic DNA samples of 102 subjects from 61 families. The mean depth (±standard deviation) was 81.87 ± 25.15× and the mean coverage for the targeted regions was 96.03 ± 3.02% with a depth higher than 15×. The mean depth/coverage for the detected EYS variants in this study is listed in Table 1. No copy number variants associated with the disease were identified.
Table 1.
Summary of genetic analyses for 26 EYS variants.
| No. | Variant | Variant type | Family count (%*) | AC* | AF* | Mean read depth | Coverage (≥15 reads) | AF in general databases | ACMG classification | Reference | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HGVD | iJGVD 3.5k | gnomAD EA | gnomAD Total | ||||||||||
| 1 | c.2528 G > A, p.(Gly843Glu) | Missense | 26 (42.6%) | 32 | 26.23% | 37.24 | 99.87% | 2.25% | 1.7000% | 0.0391% | 0.0026% | LP | 25 |
| 2 | c.4957dupA, p.(Ser1653Lysfs*2) | Frameshift | 22 (36.1%) | 29 | 23.77% | 38.27 | 100.00% | 0.2084% | 0.0000% | 0.0098% | 0.0007% | P | 15,20,25,32 |
| 3 | c.8805 C > A, p.(Tyr2935*) | Nonsense | 14 (23.0%) | 17 | 13.93% | 37.11 | 100.00% | 0.2901% | 0.1700% | 0.0293% | 0.0020% | P | 15,20,25 |
| 4 | c.7394 C > G, p.(Thr2465Ser) | Missense | 12 (19.7%) | 15 | 12.30% | 36.70 | 99.74% | 3.0468% | 2.9000% | 0.1465% | 0.0126% | US | Novel†20 |
| 5 | c.6557 G > A, p.(Gly2186Glu) | Missense | 7 (11.5%) | 7 | 5.74% | 35.09 | 99.15% | 0.0000% | 0.0000% | 0.0497% | 0.0035% | LP | 14,20,32 |
| 6 | c.6563 T > C, p.(Ile2188Thr) | Missense | 5 (8.2%) | 6 | 4.92% | 32.15 | 97.12% | 0.0000% | 0.1000% | NA | NA | US | 15 |
| 7 | c.1211dupA, p.(Asn404Lysfs*3) | Frameshift | 3 (4.9%) | 3 | 2.46% | 48.45 | 99.80% | 0.0000% | 0.0000% | 0.0000% | 0.0016% | P | 25,33 |
| 8 | c.632 G > A, p.(Cys211Tyr) | Missense | 2 (3.3%) | 2 | 1.64% | 35.97 | 99.41% | 0.0000% | 0.0300% | NA | NA | LP | 15 |
| 9 | c.7665_7666del, p.(Tyr2555*) | Nonsense | 2 (3.3%) | 2 | 1.64% | 33.26 | 93.72% | 0.0000% | 0.0000% | NA | NA | P | 25 |
| 10 | c.5644 + 5 G > A | Splicing | 1 (1.6%) | 1 | 0.82% | 31.63 | 93.85% | 0.0000% | 0.0000% | NA | NA | P | 34 |
| 11 | c.3809 T > G, p.(Val1270Gly) | Missense | 1 (1.6%) | 1 | 0.82% | 35.88 | 99.93% | 0.4570% | 0.0455% | 0.0492% | 0.0033% | US | 25 |
| 12 | c.5027 C > G, p.(Ser1676*) | Nonsense | 1 (1.6%) | 1 | 0.82% | 35.37 | 98.50% | 0.0000% | 0.0000% | 0.0000% | 0.0000% | P | This study |
| 13 | c.7002 C > A, p.(Cys2334*) | Nonsense | 1 (1.6%) | 1 | 0.82% | 27.08 | 90.26% | 0.0000% | 0.0100% | NA | NA | P | 35 |
| 14 | c.6714del, p.(Ile2239Serfs*17) | Frameshift | 1 (1.6%) | 1 | 0.82% | 27.97 | 91.69% | 0.0000% | 0.0000% | 0.0098% | 0.0039% | P | 5,17,27,35 |
| 15 | c.1485_1493delinsCGAAAAG, p.(Val495Glufs*13) | Frameshift | 1 (1.6%) | 1 | 0.82% | 33.67 | 99.22% | 0.0000% | 0.0000% | NA | NA | P | 25 |
| 16 | c.137 C > T, p.(Thr46Ile) | Missense | 1 (1.6%) | 1 | 0.82% | 38.23 | 99.28% | 0.0000% | 0.0000% | NA | NA | US | This study |
| 17 | c.9186_9187del, p.(Asn3062Lysfs*9) | Frameshift | 1 (1.6%) | 1 | 0.82% | 39.32 | 99.61% | 0.0000% | 0.0000% | 0.0000% | 0.0013% | P | 36 |
| 18 | c.8608 A > T, p.(Asn2870Tyr) | Missense | 1 (1.6%) | 1 | 0.82% | 48.93 | 100.00% | 0.0000% | 0.0000% | NA | NA | US | This study |
| 19 | c.141 A > T, p.(Glu47Asp) | Missense | 1 (1.6%) | 1 | 0.82% | 38.97 | 100.00% | 0.0000% | 0.0100% | NA | NA | US | 15,35 |
| 20 | c.4022del, p.(Ser1341Phefs*11) | Frameshift | 1 (1.6%) | 1 | 0.82% | 43.99 | 100.00% | 0.0000% | 0.0000% | NA | NA | P | 35 |
| 21 | c.8278 C > T, p.(Arg2760Cys) | Missense | 1 (1.6%) | 1 | 0.82% | 39.08 | 99.80% | 0.0000% | 0.0000% | 0.0000% | 0.0000% | US | This study |
| 22 | c.8516dupA, p.(Asn2839Lysfs*2) | Frameshift | 1 (1.6%) | 1 | 0.82% | 24.03 | 87.22% | 0.0000% | 0.0000% | NA | NA | P | This study |
| 23 | c.7609 G > A, p.(Ala2537Thr) | Missense | 1 (1.6%) | 1 | 0.82% | 33.56 | 98.63% | 0.5013% | 0.2300% | 0.2678% | 0.0266% | US | 36,37 |
| 24 | c.2000G > A, p.(Arg667His) | Missense | 1 (1.6%) | 1 | 0.82% | 35.18 | 99.87% | 0.0000% | 0.0327% | 0.0000% | 0.0295% | US | 38 |
| 25 | c.7919 G > A, p.(Trp2640*) | Nonsense | 1 (1.6%) | 1 | 0.82% | 41.19 | 100.00% | 0.0000% | 0.0600% | 0.0000% | 0.0027% | P | 4,17,39 |
| 26 | c.7392dupT, p.(Thr2465Tyrfs*12) | Frameshift | 1 (1.6%) | 1 | 0.82% | 41.27 | 100.00% | 0.0000% | 0.0000% | NA | NA | P | This study |
AC = allele count; AF = allele frequency; EA = East Asian; ACMG = American College of Medical Genetics and Genomics; P = pathogenic; LP = likely pathogenic; US = uncertain significance; HGVD = Human Genetic Variation Database (http://www.genome.med.kyoto-u.ac.jp/SnpDB/; accessed on July 1, 2017); iJGVD 3.5k=Integrative Japanese Genome Variation 3.5k (https://ijgvd.megabank.tohoku.ac.jp/download_3.5kjpn/; accessed on July 1, 2017); gnomAD=the Genome Aggregation Database (http://gnomad.broadinstitute.org/; accessed on 1st of August, 2018).
*Number is this cohort; †: a variant listed in the cited reference, but not associated with the disease.
Twenty-six EYS variants were identified in total. Six variants have never been reported, and one variant (c.7394 C > G, p.(Thr2465Ser)) has never been associated with the specific phenotype of RP/CORD/LCA. The detected variants are widely distributed in the EYS gene (Fig. 2). There were twelve missense variants, eight frameshift variants, five nonsense variants, and one variant with splice site alteration. The genetic results are summarized in Table 1 and Supplementary Table S1.
Figure 2.

EYS variants detected in a Japanese cohort with inherited retinal disease (IRD). (A) A schematic genetic and protein structure of EYS and the location of the detected variants in this study. The EYS gene (ENST00000503581.5) contains 43 exons. Exons 4 to 43 encode a 3144-amino acid protein containing 27 epidermal growth factor-like domains (highlighted with diagonal lines) and five laminin G-like domains (highlighted with horizontal lines) as well as one N-terminal signal peptide (highlighted with grey). Truncating variants (nonsense, frameshift, and splice site alteration) are shown in red, and missense variants are shown in black. Novel variants identified in this study are underlined. (B) Distribution of the types of the detected variants. In total, 26 variants were identified, including twelve missense variants (46%), eight frameshift variants (31%), five nonsense variants (19%) and one splicing site alteration variant (4%). (C) Allele frequency (AF) of the detected variants in this EYS-RD cohort and in the general population presented in public databases. Top: The AF in this EYS affected cohort. c.2528 G > A (p.(Gly843Glu)), c.4957dupA (p.(Ser1653Lysfs*2)), c.8805 C > A (p.(Tyr2935*)) and c.7394 C > G (p.(Thr2465Ser)) are the four most prevalent variants, with AFs of 26.23%, 23.77%, 13.93%, and 12.30%, respectively. Bottom: The AF in the general population in the two public databases: the Genome Aggregation Database (gnomAD; a database for the ethnic and the total general population) and the Human Genetic Variation Database (HGVD; a database for the Japanese general population). The AF of the four most prevalent variants provided by the HGVD/gnomAD East Asian/gnomAD total databases was 2.25%/0.04%/0.00%, 0.21%/0.01%/0.00%, 0.29%/0.03%/0.00%, and 3.05%/0.15%/0.01%, respectively, for c.2528 G > A (p.(Gly843Glu)), c.4957dupA (p.(Ser1653Lysfs*2)), c.8805 C > A (p.(Tyr2935*)) and c.7394 C > G (p.(Thr2465Ser)).
Detailed results of the in silico analyses of 26 variants are presented in Supplementary Table S3. Thirteen variants were classified as pathogenic, four were likely pathogenic, and nine were variants of uncertain significance (VUS). Eight VUS were found with likely pathogenic/pathogenic variants or variants previously reported as likely pathogenic elsewhere. One variant (c.8608 A > T, p.(Asn2870Tyr)) was found with the recurrent relatively high AF variant (c.7394 C > G, p.(Thr2465Ser)).
The AF of all 26 variants detected in this affected cohort and in the general population is presented in Table 1 and Fig. 2. The AF of EYS variants ranged from 0.00% to 3.05% in the Human Genetic Variation Database (HGVD), 0.00% to 0.27% in the East Asian in the Genome Aggregation Database (gnomAD), and 0.00% to 0.03% in total in the gnomAD database. The four most prevalent variants were c.2528 G > A (p.(Gly843Glu)), c.4957dupA (p.(Ser1653Lysfs*2)), c.8805 C > A (p.(Tyr2935*)), and c.7394 C > G (p.(Thr2465Ser)), with an AF in this affected EYS cohort of 26.23% (32/122), 23.77% (29/122), 13.93% (17/122), and 12.30% (15/122), respectively. The AF of these four most prevalent variants in the HGVD/gnomAD East Asian/gnomAD total was 2.25%/0.04%/0.00%, 0.21%/0.01%/0.00%, 0.29%/0.03%/0.00%, and 3.05%/0.15%/0.01%, respectively. A considerably higher AF in the Japanese population was observed in these four variants than in the other populations. There were two more variants with higher AF ( > 0.45%) in the general Japanese population: c.3809 T > G (p.(Val1270Gly)) and c.7609 G > A (p.(Ala2537Thr)), with the AF provided by the HGVD/gnomAD East Asian/gnomAD total being 0.46%/0.05%/0.00% and 0.50%/0.27%/0.03%, respectively.
Discussion
The genetic spectrum of EYS-RD is illustrated in a well-characterized large Japanese cohort of 61 families. The identification of variants with relatively high AF confirmed by the co-segregation analysis in multiple families helped to clarify the high proportion of EYS-RD in the IRDs of the Japanese population; 23.4% of AR or sporadic RP (52/222) and 9.9% of AR or sporadic CORD (7/71).
Two EYS variants with allele frequencies higher than 1%
Two variants with relatively high AF (>1% in the general population) were confirmed in our cohort: p.(Gly843Glu) and p.(Thr2465Ser). All the subjects harbouring these variants in a homozygous or compound heterozygous status in the Japan Eye Genetics Consortium (JEGC; http://www.jegc.org/) study cohort of 1302 subjects from 729 families demonstrated retinal dystrophy, which supports the disease causation/association of these two variants.
The variant p.(Gly843Glu) was first described by Iwanami et al. in 2012. Five subjects with this variant found with the other proven truncating variants, such as p.(Ser1653Lysfs*2) and p.(Ser2428*), were presented in this report25. In our study, there were five families homozygous for this variant, showing various phenotypic findings in the spectrum of RP. In the HGVD database, there is one subject homozygous for this variant out of 1207 subjects (1/1207, 0.08%) with no registered diseases on the records for whom no further ophthalmic information is available26. Given the variable disease onset and phenotype associated with this variant, it is still uncertain whether this subject will develop visual defects in the future. The AF of this variant in our molecularly proven ARRP cohort of 112 families (32/224; 14.3%) was significantly higher than that in the general Japanese population (53/2361, 2.25%; HGVD) calculated with Fisher’s exact test (P < 0.001), as implied by the previous studies in the Japanese population19,25. Moreover, the AF in the general Japanese population was approximately 50/1000 times higher than that in the East Asian/total population of gnomAD. The pathogenicity of this variant is not fully proven; however, a founder effect in the Japanese population should be considered to explain this most prevalent disease-associated allele.
The other variant with relatively high AF (p.(Thr2465Ser)) was first described by Hosono et al. in 2012 as a possible non-pathogenic variant with the allele frequency of affected (8/200; 4.0%) and normal subjects (2/192; 1.0%)20. In our cohort, twelve families harboured this variant and no other candidate variants in any other known retinal disease-associated gene. Three of these twelve families had proven biallelic EYS variants confirmed by the co-segregation analysis. It is of note that five alleles of this variant were associated with CORD. In addition, this variant was found in cis with the p.(Gly843Glu) variant in two families with an additional family harbouring three candidate unsegregated variants. The AF of this variant in our molecularly confirmed ARRP cohort (15/224; 6.70%) was higher than that of the general Japanese population (67/2203, 3.04%; HGVD) calculated with Fisher’s exact test, which reached a statistically significant value (P = 0.01). The AF in the general Japanese population was approximately 200/2000 times higher than those in the East Asian/total population, respectively. In the HGVD database, there are two subjects (2/1207, 0.17%) homozygous for this variant with no available ophthalmic information. The results of in silico analysis and comparison analysis between the AF of the affected cohort and the general population suggest some supporting evidence for the disease causation, and a founder effect in the Japanese population could also be considered for this prevalent allele that is possibly associated with IRDs.
The EYS gene and the high prevalence of IRDs in the Japanese population
Two prevalent truncating variants (p.(Ser1653Lysfs*2) and p.(Tyr2935*)) were also frequently found in our cohort. As previously described, these two variants have a higher AF in the Japanese population than in other populations15,19,20,25. Together with the other two variants with high AF (p.(Val1270Gly) and p.(Ala2537Thr); AF > 0.45%) in the Japanese general population, several frequent variants especially prevalent in the Japanese population were determined in this study.
The total value of AF of the total detected EYS variants was 6.75% in the general Japanese population. Given this number, the estimated prevalence of subjects at risk for EYS-RD in Japan should be higher than the current estimated value of 1 in 3500–4000 for RP. However, it is of note that the genetic risk does not perfectly correspond to the prevalence of the disease in the real world, as shown for the most prevalent ABCA4 variant (p.(Asn1868Ile) in the European population (AF > 6.7%)21.
Genotype-phenotype association of EYS-RD
Three phenotype groups were identified in our cohort: RP (85.9%), CORD (10.9%), and EORP/LCA (3.1%). There were only a few patients with EYS-CORD reported to date in the previous literature; however, our EYS-RD cohort provided the largest number of CORD patients associated with EYS variants5,11–13. Seven out of 45 molecularly proven cases of ARCORD in the JEGC cohort are caused by biallelic or putative biallelic EYS variants (7/45, 15.6%). This fact highlighted that EYS should be the major IRD gene in the Japanese population, with the a significantly higher prevalence than that in the European population5,16,17,27.
There was no clear genotype-phenotype association/correlation between RP/CORD/EORD due to the limited number of CORD and EORD/LCA cases. Although further detailed analysis is needed for accurate assessment, both of the two aforementioned variants with relatively high AF are associated with either RP or CORD; thus, the prediction of predominant functional failure (rod or cone) seems hard based on the genotype. It is noteworthy that even patients with the identical genotype presented with the contrasting clinical phenotypes (RP/CORD), which suggests the possible presence of modifiers outside of the EYS gene that contribute to the disease presentation.
There were seven families with multiple disorders (Supplementary Fig. S2) or non-AR inheritance in our cohort. Conclusive genetic diagnosis is still unavailable in four affected subjects with limited clinical information from three families (Families 5, 8 and 16). Whole-exome sequencing was not performed in three subjects from two families (Families 5 and 16). Given the presence of variants with a relatively high AF, such coincidence with the other EYS variants or other pathogenic variants in the non-EYS genes should be considered in the clinical/genetic diagnosis of IRD. For this reason, comprehensive gene screening is helpful to elucidate the cause of complicated phenotypes in such families.
Limitations of this study
There are limitations to this study. First, the molecular mechanisms of disease causation for most variants have not yet been known, and the clinical effect of the variants (e.g., functional loss by a single variant, acting as a modifier, complexing with missing disease-causing variants, and others) is poorly understood. Further functional analysis is needed to conclude the disease causation of each variant. Second, the identification of copy number variants is technically hard with the results of whole-exome sequencing; thus, the possible presence of copy number variants was not completely excluded in our study. As previously reported28,29, it is crucial to examine the structural variants in the EYS gene. Third, the AF data of general populations were not studied in the detail due to the limited data resources of ophthalmic findings and natural history, which should be valuable to assess the clinical effect of each variant in subjects at risk in the real world, especially in relatively prevalent IRDs with diverse onset and phenotype. Last, the identification of background ethnicity for each variant was not available in this study. Extensive genomic analysis with detailed haplotype information could delineate the ethnic specificity of EYS-RD.
In conclusion, the phenotypic and genotypic characteristics of EYS-RD were determined in this largest cohort of the Japanese population. The presence of variants with a relatively high AF in a specific population requires the survey of non-rare variants in consideration of founder effects, especially in relatively prevalent Mendelian disorders.
Methods
The protocol of this study adhered to the tenets of the Declaration of Helsinki and was approved by the ethics committee of the participating institutions from Japan; National Institute of Sensory Organs, National Hospital Organization Tokyo Medical Center (Reference; R18–029). A signed informed consent was obtained from all subjects.
Participants and clinical investigation
Patients with a clinical diagnosis of IRD and available genetic data were studied between 2008 and 2018 as part of the JEGC (http://www.jegc.org/)30. A total of 1302 subjects from 729 families, including 222 families with autosomal recessive or sporadic RP and 71 families with autosomal recessive or sporadic CORD, registered in the JEGC cohort were surveyed. Clinical information is available via the NISO online database, including medical history, family history, ethnicity, chief complaints of visual symptoms, the onset of disease, the best-corrected decimal visual acuity converted to the LogMAR unit, fundoscopy, fundus photography, autofluorescence imaging, and phenotypic categorization.
EYS variant detection
Genomic DNA was extracted from the peripheral blood of all affected subjects and the available unaffected family members with the Gentra Puregene Blood Kit (Qiagen, Tokyo, Japan). Whole-exome sequencing with targeted analysis of retinal disease-associated genes (RetNet; https://sph.uth.edu/retnet/home.htm; accessed on 1 January 2017) was performed on the affected subjects and unaffected family members according to previously published methods30. Briefly, paired-end sequence library construction and exome capturing were performed with the Agilent Bravo automated liquid-handling platform with SureSelect XT Human All Exon V3–5+UTRs kit (Agilent Technologies, Santa Clara, CA, USA). Enriched libraries were sequenced with the Illumina HiSeq. 2000/2500 sequencer (San Diego, CA, USA; read length 2×101 bp). Reads were aligned to the University of California, Santa Cruz (USCS; California, United States) human genome 19 reference sequence with Burrows-Wheeler Aligner software. Duplicated reads were removed by the Picard MarkDuplicates module, and mapped reads around insertion-deletion polymorphisms (INDELs) were realigned by the Genome Analysis Toolkit (GATK) Version 3.0. Base-quality scoring was recalibrated by the GATK. Mutation calling was performed by the GATK Unified Genotyper module. Called single-nucleotide variants (SNVs) and INDELs were annotated by the snpEff software (snpEff score; “high”, “moderate” or “low”). The XHMM (eXome-Hidden Markov Model; https://atgu.mgh.harvard.edu/xhmm/) tool was applied for the detection of copy number variations. The read depth and coverage of the targeted regions were also confirmed with Integrate Genome Viewer (IGV; http://software.broadinstitute.org/software/igv/). All called SNVs and INDELs of the RetNet genes were selected for further analysis. Variants with the read depths higher than 15× were selected for this study.
The identified variants with an allele frequency of less than 1% in the HGVD (http://www.genome.med.kyoto-u.ac.jp/SnpDB/; accessed on 1 July 2017), and Integrative Japanese Genome Variation (iJGVD 3.5k, https://ijgvd.megabank.tohoku.ac.jp/download_3.5kjpn/; accessed on 1 July 2017), which are two allele frequency databases specific for the Japanese population, were filtered. Only for the two autosomal recessive genes with high prevalence (EYS and ABCA4), the identified variants were filtered with an AF of less than 10.0% in the HGVD to avoid missing the pathogenic/disease-associated variants with a relatively high AF (1%-10%). Together with phenotypic features and inheritance, as well as co-segregation, disease-causing/disease-associated variants were determined from the called variants in the retinal disease-associated genes.
In silico molecular genetic analysis
Sequence variant nomenclature was performed according to the guidelines of the Human Genome Variation Society (HGVS; version 2.0; http://varnomen.hgvs.org/). The AF of all detected variants in the HGVD, iJGVD, 1000 Genome Project (http://www.internationalgenome.org/; accessed on 1 August 2018), and gnomAD (http://gnomad.broadinstitute.org/; accessed on 1 August 2018) databases was established. All variants were analysed using two general prediction programmes and three functional prediction programmes: MutationTaster (http://www.mutationtaster.org/; accessed on 1 August 2018), FATHMM (http://fathmm.biocompute.org.uk/9; accessed on 1 August 2018), SIFT (https://www.sift.co.uk/; accessed on 1 August 2018), PROVEAN (http://provean.jcvi.org/index.php; accessed on 1 August 2018), and PolyPhen 2 (http://genetics.bwh.harvard.edu/pph2/; accessed on 1 August 2018). Evolutional conservation scores were calculated from the UCSC database (https://genome.ucsc.edu/index.html; accessed on 1 August 2018). Variant classification was performed according to the guidelines of the American College of Medical Genetics and Genomics (ACMG) for all detected variants31.
Supplementary information
Supplementary Figure S1 and Supplementary Figure S2.
Acknowledgements
We thank the patients and their families for participation in this study. We are grateful to Dr. Kazuki Yamazawa and Dr. Satomi Inoue, National Institute of Sensory Organs, National Tokyo Medical Center, Japan for their help in clinical and genetic data analysis. We also thank all the collaborators of Japan Eye Genetics Consortium (URL: http://www.jegc.org/) for data collection. Kaoru Fujinami is supported by grants from Grant-in-Aid for Young Scientists (A) of the Ministry of Education, Culture, Sports, Science and Technology, Japan (16H06269); grants from Grant-in-Aid for Scientists to support international collaborative studies of the Ministry of Education, Culture, Sports, Science and Technology, Japan (16KK01930002); grants from the National Hospital Organization Network Research Fund, Japan (H30-NHO-Sensory Organs-03); grants from FOUNDATION FIGHTING BLINDNESS ALAN LATIES CAREER DEVELOPMENT PROGRAM (CF-CL-0416-0696-UCL); grants from Health Labour Sciences Research Grant, the Ministry of Health, Labour and Welfare, Japan (201711107 A); and grants from the Great Britain Sasakawa Foundation Butterfield Awards, UK. Yu Fujinami-Yokokawa is supported by grants from Grant-in-Aid for Young Scientists of the Ministry of Education, Culture, Sports, Science and Technology, Japan (18K16943). Gavin Arno is supported by a Fight for Sight (UK) early career investigator award, NIHR-BRC at Moorfields Eye Hospital and the UCL Institute of Ophthalmology, NIHR-BRC at Great Ormond Street Hospital and UCL Institute of Child Health, and Great Britain Sasakawa Foundation Butterfield Award, UK. Nikolas Pontikos is funded by the NIHR-BRC at Moorfields Eye Hospital and the UCL Institute of Ophthalmology. Toshihide Kurihara is supported by Tsubota Laboratory, Inc, Fuji Xerox Co., Ltd, Kirin Company, Ltd, Kowa Company, Ltd, Novartis Pharmaceuticals, Santen Pharmaceutical Co. Ltd, and ROHTO Pharmaceutical Co., Ltd. Takeshi Iwata is supported by the Japan Agency for Medical Research and Development (AMED) (18ek0109282h0002). Kazushige Tsunoda is supported by AMED; the Ministry of Health, Labor and Welfare, Japan (18ek0109282h0002); Grants-in-Aid for Scientific Research, Japan Society for the Promotion of Science, Japan (H26-26462674); grants from the National Hospital Organization Network Research Fund, Japan (H30-NHO-Sensory Organs-03) and Novartis Research Grant (2018). Role of the Funder/Sponsor: The funding sources had no role in the design and conduct of the study; the collection, management, analysis, and interpretation of the data; the preparation, review, and approval of the manuscript; or the decision to submit the manuscript for publication.
Author contributions
Each author meets the ICMJE criteria for authorship and agrees with all the contents of the manuscript. L.Y., K.F., T.I. and K.T. designed the research. All authors performed data acquisition and/or research execution. All authors analyzed and/or interpret data. L.Y., K.F., G.A., N.P., and K.T. prepared the manuscript. All authors reviewed the manuscript. K.F. (corresponding author) has full access to all the data in the present study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Competing interests
All authors have completed and submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Individual investigators who participate in the sponsored project(s) are not directly compensated by the sponsor but may receive salary or other support from the institution to support their effort on the project(s). Kaoru Fujinami is a paid consultant of Astellas Pharma Inc, Kubota Pharmaceutical Holdings Co., Ltd, Acucela Inc., Novartis AG., NightstaRx Limited, and Sanofi Genzyme. Kaoru Fujinami reports personal fees from Astellas Pharma Inc, personal fees from Kubota Pharmaceutical Holdings Co., Ltd., personal fees from Acucela Inc., personal fees from Novartis AG., personal fees from SANTEN Company Limited, personal fees from Foundation Fighting Blindness, personal fees from Foundation Fighting Blindness Clinical Research Institute, and personal fees from Japanese Ophthalmology Society, personal fees from Japan Retinitis Pigmentosa Society. The laboratory of Visual Physiology, Division for Vision Research, National Institute of Sensory Organs, National Hospital Organization, Tokyo Medical Center, Tokyo, Japan is supported by grants from Astellas Pharma Inc. (NCT03281005), outside the submitted work. Toshihide Kurihara reports personal fees from Novartis Pharmaceuticals Japan, and Bayer Yakuhin, Ltd., outside the submitted work.
Footnotes
A comprehensive list of consortium members appears at the end of the paper.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Lizhu Yang and Kaoru Fujinami.
Contributor Information
Kaoru Fujinami, Email: k.fujinami@ucl.ac.uk.
JEGC study group:
Toshihide Nishimura, Yoshihide Hayashizaki, Nobuhiro Shimozawa, Masayuki Horiguchi, Shuichi Yamamoto, Manami Kuze, Shigeki Machida, Yoshiaki Shimada, Makoto Nakamura, Takashi Fujikado, Yoshihiro Hotta, Masayo Takahashi, Kiyofumi Mochizuki, Akira Murakami, Hiroyuki Kondo, Susumu Ishida, Mitsuru Nakazawa, Tetsuhisa Hatase, Tatsuo Matsunaga, Akiko Maeda, Kosuke Noda, Atsuhiro Tanikawa, Syuji Yamamoto, Hiroyuki Yamamoto, Makoto Araie, Makoto Aihara, Toru Nakazawa, Tetsuju Sekiryu, Kenji Kashiwagi, Kenjiro Kosaki, Carninci Piero, Takeo Fukuchi, Atsushi Hayashi, Katsuhiro Hosono, Keisuke Mori, Kouji Tanaka, Koichi Furuya, Keiichirou Suzuki, Ryo Kohata, Yasuo Yanagi, Yuriko Minegishi, Daisuke Iejima, Akiko Suga, Brian P. Rossmiller, Yang Pan, Tomoko Oshima, Mao Nakayama, Megumi Yamamoto, Naoko Minematsu, Daisuke Mori, Yusuke Kijima, Kentaro Kurata, Norihiro Yamada, Masayoshi Itoh, Hideya Kawaji, and Yasuhiro Murakawa
Supplementary information
is available for this paper at 10.1038/s41598-020-62119-3.
References
- 1.Liew G, Michaelides M, Bunce C. A comparison of the causes of blindness certifications in England and Wales in working age adults (16-64 years), 1999-2000 with 2009-2010. BMJ Open. 2014;4:e004015. doi: 10.1136/bmjopen-2013-004015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. 2006;368:1795–1809. doi: 10.1016/S0140-6736(06)69740-7. [DOI] [PubMed] [Google Scholar]
- 3.Roosing S, et al. Causes and consequences of inherited cone disorders. Prog. Retin. Eye Res. 2014;42:1–26. doi: 10.1016/j.preteyeres.2014.05.001. [DOI] [PubMed] [Google Scholar]
- 4.Abd El-Aziz MM, et al. EYS, encoding an ortholog of Drosophila spacemaker, is mutated in autosomal recessive retinitis pigmentosa. Nat. Genet. 2008;40:1285–1287. doi: 10.1038/ng.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Collin RW, et al. Identification of a 2 Mb human ortholog of Drosophila eyes shut/spacemaker that is mutated in patients with retinitis pigmentosa. Am. J. Hum. Genet. 2008;83:594–603. doi: 10.1016/j.ajhg.2008.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Messchaert M, Haer-Wigman L, Khan MI, Cremers FPM, Collin RWJ. EYS mutation update: In silico assessment of 271 reported and 26 novel variants in patients with retinitis pigmentosa. Hum. Mutat. 2018;39:177–186. doi: 10.1002/humu.23371. [DOI] [PubMed] [Google Scholar]
- 7.Zelhof AC, Hardy RW, Becker A, Zuker CS. Transforming the architecture of compound eyes. Nature. 2006;443:696–699. doi: 10.1038/nature05128. [DOI] [PubMed] [Google Scholar]
- 8.Lu Z, et al. Ablation of EYS in zebrafish causes mislocalisation of outer segment proteins, F-actin disruption and cone-rod dystrophy. Sci. Rep. 2017;7:46098. doi: 10.1038/srep46098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Messchaert M, et al. Eyes shut homolog is important for the maintenance of photoreceptor morphology and visual function in zebrafish. PLoS One. 2018;13:e0200789. doi: 10.1371/journal.pone.0200789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Seko Y, et al. The manner of decay of genetically defective EYS gene transcripts in photoreceptor-directed fibroblasts derived from retinitis pigmentosa patients depends on the type of mutation. Stem Cell Res. Ther. 2018;9:279. doi: 10.1186/s13287-018-1016-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Katagiri S, et al. Autosomal recessive cone-rod dystrophy associated with compound heterozygous mutations in the EYS gene. Doc. Ophthalmol. 2014;128:211–217. doi: 10.1007/s10633-014-9435-0. [DOI] [PubMed] [Google Scholar]
- 12.Littink KW, et al. Homozygosity mapping in patients with cone-rod dystrophy: novel mutations and clinical characterizations. Invest. Ophthalmol. Vis. Sci. 2010;51:5943–5951. doi: 10.1167/iovs.10-5797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sengillo JD, et al. A Distinct Phenotype of Eyes Shut Homolog (EYS)-Retinitis Pigmentosa Is Associated With Variants Near the C-Terminus. Am. J. Ophthalmol. 2018;190:99–112. doi: 10.1016/j.ajo.2018.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abd El-Aziz MM, et al. Identification of novel mutations in the ortholog of Drosophila eyes shut gene (EYS) causing autosomal recessive retinitis pigmentosa. Invest. Ophthalmol. Vis. Sci. 2010;51:4266–4272. doi: 10.1167/iovs.09-5109. [DOI] [PubMed] [Google Scholar]
- 15.Arai Y, et al. Retinitis Pigmentosa with EYS Mutations Is the Most Prevalent Inherited Retinal Dystrophy in Japanese Populations. J. Ophthalmol. 2015;2015:819760. doi: 10.1155/2015/819760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Audo I, et al. EYS is a major gene for rod-cone dystrophies in France. Hum. Mutat. 2010;31:E1406–1435. doi: 10.1002/humu.21249. [DOI] [PubMed] [Google Scholar]
- 17.Barragan I, et al. Mutation spectrum of EYS in Spanish patients with autosomal recessive retinitis pigmentosa. Hum. Mutat. 2010;31:E1772–1800. doi: 10.1002/humu.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen X, et al. Targeted next-generation sequencing reveals novel EYS mutations in Chinese families with autosomal recessive retinitis pigmentosa. Sci. Rep. 2015;5:8927. doi: 10.1038/srep08927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maeda A, et al. Development of a molecular diagnostic test for Retinitis Pigmentosa in the Japanese population. Jpn. J. Ophthalmol. 2018;62:451–457. doi: 10.1007/s10384-018-0601-x. [DOI] [PubMed] [Google Scholar]
- 20.Hosono K, et al. Two novel mutations in the EYS gene are possible major causes of autosomal recessive retinitis pigmentosa in the Japanese population. PLoS One. 2012;7:e31036. doi: 10.1371/journal.pone.0031036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Allikmets R, Zernant J, Lee W. Penetrance of the ABCA4 p.Asn1868Ile Allele in Stargardt Disease. Invest. Ophthalmol. Vis. Sci. 2018;59:5564–5565. doi: 10.1167/iovs.18-25579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cremers FPM, Cornelis SS, Runhart EH, Astuti GDN. Author Response: Penetrance of the ABCA4 p.Asn1868Ile Allele in Stargardt Disease. Invest. Ophthalmol. Vis. Sci. 2018;59:5566–5568. doi: 10.1167/iovs.18-25944. [DOI] [PubMed] [Google Scholar]
- 23.Fujinami, K. et al. Detailed genetic characteristics of an international large cohort of patients with Stargardt disease: ProgStar study report 8. Br J Ophthalmol, 10.1136/bjophthalmol-2018-312064 (2018). [DOI] [PMC free article] [PubMed]
- 24.Zernant J, et al. Frequent hypomorphic alleles account for a significant fraction of ABCA4 disease and distinguish it from age-related macular degeneration. J. Med. Genet. 2017;54:404–412. doi: 10.1136/jmedgenet-2017-104540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Iwanami M, Oshikawa M, Nishida T, Nakadomari S, Kato S. High prevalence of mutations in the EYS gene in Japanese patients with autosomal recessive retinitis pigmentosa. Invest. Ophthalmol. Vis. Sci. 2012;53:1033–1040. doi: 10.1167/iovs.11-9048. [DOI] [PubMed] [Google Scholar]
- 26.Higasa K, et al. Human genetic variation database, a reference database of genetic variations in the Japanese population. J. Hum. Genet. 2016;61:547–553. doi: 10.1038/jhg.2016.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Littink, K. W. et al. Mutations in the EYS gene account for approximately 5% of autosomal recessive retinitis pigmentosa and cause a fairly homogeneous phenotype. Ophthalmology117, 2026–2033, 2033 e2021–2027, 10.1016/j.ophtha.2010.01.040 (2010). [DOI] [PubMed]
- 28.Pieras JI, et al. Copy-number variations in EYS: a significant event in the appearance of arRP. Invest. Ophthalmol. Vis. Sci. 2011;52:5625–5631. doi: 10.1167/iovs.11-7292. [DOI] [PubMed] [Google Scholar]
- 29.Perez-Carro R, et al. Panel-based NGS Reveals Novel Pathogenic Mutations in Autosomal Recessive Retinitis Pigmentosa. Sci. Rep. 2016;6:19531. doi: 10.1038/srep19531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fujinami K, et al. Novel RP1L1 Variants and Genotype-Photoreceptor Microstructural Phenotype Associations in Cohort of Japanese Patients With Occult Macular Dystrophy. Invest. Ophthalmol. Vis. Sci. 2016;57:4837–4846. doi: 10.1167/iovs.16-19670. [DOI] [PubMed] [Google Scholar]
- 31.Richards S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015;17:405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yoon CK, et al. The diagnostic application of targeted re-sequencing in Korean patients with retinitis pigmentosa. BMC Genomics. 2015;16:515. doi: 10.1186/s12864-015-1723-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bandah-Rozenfeld D, et al. Novel null mutations in the EYS gene are a frequent cause of autosomal recessive retinitis pigmentosa in the Israeli population. Invest. Ophthalmol. Vis. Sci. 2010;51:4387–4394. doi: 10.1167/iovs.09-4732. [DOI] [PubMed] [Google Scholar]
- 34.Huang H, et al. Systematic evaluation of a targeted gene capture sequencing panel for molecular diagnosis of retinitis pigmentosa. PLoS One. 2018;13:e0185237. doi: 10.1371/journal.pone.0185237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Katagiri S, et al. Whole exome analysis identifies frequent CNGA1 mutations in Japanese population with autosomal recessive retinitis pigmentosa. PLoS One. 2014;9:e108721. doi: 10.1371/journal.pone.0108721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang XF, et al. Genotype-phenotype correlation and mutation spectrum in a large cohort of patients with inherited retinal dystrophy revealed by next-generation sequencing. Genet. Med. 2015;17:271–278. doi: 10.1038/gim.2014.138. [DOI] [PubMed] [Google Scholar]
- 37.Xu Y, et al. Mutations of 60 known causative genes in 157 families with retinitis pigmentosa based on exome sequencing. Hum. Genet. 2014;133:1255–1271. doi: 10.1007/s00439-014-1460-2. [DOI] [PubMed] [Google Scholar]
- 38.Carss KJ, et al. Comprehensive Rare Variant Analysis via Whole-Genome Sequencing to Determine the Molecular Pathology of Inherited Retinal Disease. Am. J. Hum. Genet. 2017;100:75–90. doi: 10.1016/j.ajhg.2016.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Oishi M, et al. Comprehensive molecular diagnosis of a large cohort of Japanese retinitis pigmentosa and Usher syndrome patients by next-generation sequencing. Invest. Ophthalmol. Vis. Sci. 2014;55:7369–7375. doi: 10.1167/iovs.14-15458. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1 and Supplementary Figure S2.