

Abstract
Introduction
Vasopressors are a commonly used treatment in beta-blocker poisoning despite evidence they may be ineffective or harmful. The primary objective of the present study is to use previously collected data from two prior studies (high-dose insulin (HDI) versus vasopressin + epinephrine and a placebo-controlled HDI study) to compare survival between vasopressin + epinephrine and placebo. Secondary outcomes included a comparison with HDI as well as comparisons with hemodynamic parameters, including mean arterial pressure (MAP), cardiac output (CO), heart rate (HR), and systemic vascular resistance (SVR).
Methods
Cardiogenic shock was induced in healthy pigs with a bolus of 0.5 mg/kg of intravenous propranolol followed by an infusion of 0.25 mg/kg/minute until the point of toxicity, defined as (0.75 × initial HR × initial MAP), at which point the infusion was reduced to 0.125 mg/kg/minute for 240 (vasopressin + epinephrine or HDI) or 360 minutes (placebo) or until death.
Results
Survival was significantly lower in pigs receiving vasopressin + epinephrine (0%, 0/5) than in pigs receiving placebo (50%, 2/4) (p < 0.01). Survival was significantly higher with HDI compared with both groups (100%, 5/5) (p < 0.01). All vasopressin + epinephrine pigs died within 100 minutes after reaching toxicity. Over the course of the resuscitation, we observed a statistically significant steady decrease in CO and HR in the vasopressin + epinephrine group compared with placebo (p < 0.01). In contrast, we observed a statistically significant change in MAP and SVR that followed a parabolic arc, with MAP and SVR rising significantly initially in the vasopressin + epinephrine group then rapidly falling until death (p < 0.01).
Conclusions
Mortality was higher with vasopressors compared with placebo in this porcine model of propranolol poisoning. Further studies are warranted to define the optimal timing and role of vasopressors in beta-blocker poisoning.
Keywords: Vasopressors, Propranolol, Beta-blockers, Poisoning, Overdose
Introduction
Beta-blocker poisoning is common [1]. In 2017, US poison centers reported 26,225 exposures to beta-blockers, including 1,042 with a moderate outcome, 109 with a major outcome, and 18 deaths [2]. Beta-blockers are even more toxic when combined with other medications; they were contributory to 118 deaths reported to US poison centers in 2017 [2]. Morbidity and mortality are multifactorial in nature, but occur primarily due to cardiogenic shock [3]. Multiple therapies, including glucagon, high-dose insulin (HDI), and vasopressors [4–6], have been proposed as ideal agents to treat beta-blocker poisoning; however, there is no consensus first-line agent after basic supportive measures, such as preload augmentation and calcium supplementation, have failed.
Vasopressors are a standard treatment for hemodynamic shock in general [7, 8], given their ability to increase cardiac output and systemic vascular resistance (SVR). The use of beta-agonists, in particular, has appeal in beta-blocker poisoning as high doses of a receptor agonist to overcome receptor blockade is a common treatment strategy in poisoning. Data for the use of vasopressors in beta-blocker poisoning, however, are scant. A recent systematic review [9] of the use of vasopressors for poison-induced cardiogenic shock found evidence to support the use of vasopressors in beta-blocker poisoning was limited to 51 case reports and 7 animal studies [6, 10–15] and identified no human studies. Not only are data scant, but also results are mixed. Of these 7 studies, 6 evaluated mortality, and in 5 of these 6 studies, vasopressors were associated with statistically significantly lower survival rates [10–14]. Furthermore, of these 7 studies, only 4 contained a placebo arm [12–15], and vasopressors commonly did not outperform placebo.
Though vasopressors remain a standard treatment for shock, they are also associated with adverse outcomes. In addition to causing tissue ischemia and dysrhythmias, the rise in SVR associated with vasopressors may contribute to an overall decrease in cardiac output [16]. If shock is truly cardiogenic, this increase in afterload and subsequent decrease in cardiac output may further worsen the patient’s shock state. As such, it is possible in a poison-induced cardiogenic shock state, such as that from beta-blockers, vasopressors (or vasopressors combined with inotropes) may actually be harmful. Indeed, there is some evidence to suggest this [11, 12, 16]. The combination of dopamine with either isoproterenol or glucagon significantly reduced survival time compared with placebo in a rat model of propranolol poisoning [13, 14].
Our group has used a porcine model to evaluate various therapies for propranolol poisoning. In one study, we compared HDI with a combination of vasopressin and epinephrine infusions; that study did not include a placebo arm [6]. In another study, we performed a randomized, placebo-controlled trial of 3 unique doses of HDI [3]. These two studies were conducted by the same primary group of investigators utilizing the same animal model of toxicity, including instrumentation, monitoring, laboratory space, and laboratory personnel. Upon completion of these two studies, we had an opportunity to compare the use of vasopressors (specifically vasopressin and epinephrine) with placebo in a porcine model of propranolol poisoning, an important evaluation not previously done. Therefore, the primary objective of the present study is to use these existing data in a post hoc fashion to determine if the use of vasopressin + epinephrine is associated with higher mortality than placebo. Secondary outcomes include a mortality comparison with HDI as well as comparisons with various hemodynamic parameters among vasopressors, placebo, and HDI.
Methods
Study Setting and Animal Preparation
These studies were approved by the Scientific Review and Animal Care and Use Committees within the HealthPartners Institutional Review Board. Experiments were performed in the HealthPartners Animal Care Facility, a secure animal care facility licensed by the United States Department of Agriculture. The laboratory was accredited by the American Association for Accreditation of Laboratory Animal Care.
Healthy Yorkshire pigs were sedated with tiletamine and zolazepam to facilitate instrumentation. Thiopental sodium (2.5%) was administered while a tracheostomy was performed, and anesthesia was maintained throughout the study protocol with a combination of isoflurane and 30% nitrous oxide. Anesthesia was titrated by monitoring of reflexes to minimize cardiovascular depressant effects of the anesthetics. Pigs were mechanically ventilated, typically at a rate of 10 breaths/minute with an FiO2 of 30%. A cutdown technique was used to access the right internal jugular vein, and a Swan-Ganz catheter was inserted into the pulmonary artery. Femoral cutdowns were used to place arterial and central venous groin lines. Electrocardiogram leads were placed for cardiac monitoring. Body temperature was maintained at 37–38° using active warming as needed, and a suprapubic urinary catheter was placed.
Experimental Design
We observed a stabilization period of 30 minutes after instrumentation before inducing toxicity. All propranolol doses were administered intravenously (IV). A bolus of 0.5 mg/kg propranolol was administered, followed by an infusion of 0.25 mg/kg/minute until the point of toxicity was reached, which was defined as a 25% reduction in the product of the initial MAP × HR. This point of toxicity was previously used in other large animal poisoning studies [12, 17]. Upon reaching the point of toxicity, the propranolol infusion was halved to 0.125 mg/kg/minute, and each animal received a 20 mL/kg bolus of normal saline. Glucose concentrations were measured in all subjects at baseline, at point of toxicity, and every 10 minutes thereafter. Dextrose was administered as needed (a 25 g bolus of 50% dextrose was administered for a blood glucose < 60 mg/dL; a 50 g bolus was administered for a blood glucose < 40 mg/dL). Dextrose infusions were used and started at 12.5 g/hour; if a dextrose bolus was required in addition to the dextrose bolus, the infusion was increased by 12.5 g/hour. Resuscitation continued until death or 4 hours in the vasopressin + epinephrine and HDI groups, or until death or 6 hours in the placebo group. This difference in resuscitation duration was the only substantial difference between the two studies. Any living pigs at the end of the resuscitation protocols were euthanized with IV sodium pentobarbital. Data from 14 pigs were used in the present study: 4 pigs receiving saline placebo from Cole et al. and 5 pigs each receiving either vasopressin + epinephrine or HDI from Holger et al. Animal instrumentation and induction of toxicity are displayed in Fig. 1.
Fig. 1.

Animal instrumentation and induction of toxicity.
Interventions
Vasopressin and epinephrine were chosen to represent vasopressor therapy for multiple reasons. First, we found vasopressin tended toward superiority compared to the more traditional therapy of glucagon in a similar porcine model of propranolol poisoning [6]. Epinephrine was chosen because it offered both a way to synergistically maximize vasopressor effects via α1-agonism while simultaneously providing a high-affinity beta-agonist to directly compete with propranolol. Last, at the time of the investigation, there was reason to believe that a combination of vasopressin and epinephrine was more effective than epinephrine alone in cardiopulmonary resuscitation [18, 19].
For the 5 pigs receiving vasopressin + epinephrine, infusions of vasopressin dosed at 0.0028 units/kg/minute along and epinephrine at 10 mcg/kg/minute were started at the point of toxicity. The vasopressin infusion was increased by 0.0028 units/kg/minute every 10 minutes until the baseline HR × MAP product normalized or a maximum infusion of 0.014 units/kg/minute was reached. This vasopressin infusion was based upon a human equivalency infusion of a titration between 0.17 and 0.84 units/kg/hour [6]. The epinephrine infusion was increased by 10 mcg/kg/minute until the HR × MAP product returned to baseline or a maximum dose of 50 mcg/kg/minute. For the 5 pigs receiving HDI, HDI was initiated at 2 units/kg/hour at the point of toxicity and increased every 10 minutes until the MAP × HR product returned to baseline or a maximum of 10 units/kg/hour dose was reached. Pigs in the placebo group received normal saline at a rate commensurate with similar infusions of either vasopressors or HDI.
Measurements
Baseline measurements were taken before the induction of toxicity on each animal. Continuous cardiac output (CO) was measured using thermodilution technique. Pulse oximetry (SpO2), heart rate (HR), systolic blood pressure (SBP), mean arterial pressure (MAP), SVR, and arterial pH were monitored and recorded every 10 minutes. Glucose monitoring was described as above; results were recorded every 10 minutes. Potassium concentrations were measured and recorded at baseline, at the point of toxicity, and every hour thereafter.
Outcomes
The primary outcome of the current study was mortality between groups treated with vasopressors or placebo. Secondary outcomes included difference in mortality between HDI and the other two groups, as well as differences in CO, HR, MAP, and SVR among all three groups.
Analysis
Data analysis was performed using SAS 9.1. Kruskal-Wallis and one-way ANOVA tests were used as appropriate to evaluate baseline characteristics, which were described using means and standard errors. The Kaplan-Meier method was used for the analysis. CO was evaluated using a linear mixed-effects regression model. Like our previous work [3, 6, 11, 20], this model featured CO as a function of time, quadratic time, a time by CO interaction, and a random effect for each subject to account for correlation between measurements taken from the same identical animal. Similar regression models were used for HR, MAP, and SVR. The use of this dose-by-time interaction model for hemodynamic parameters allowed us to make several observations on each animal to determine if differences in hemodynamic parameters occurred between arms over time and not just at any one given point. Multiple imputation was used to correct for truncation of data in animals that died, assuming that after death, unobserved measurements of cardiovascular variables were proportional to the mean values for that subject, as well as the values of that particular variable observed in other subjects to account for imputation error [21]. In addition to the assumptions implicit in all ANOVA and linear regression models, two additional assumptions were made in the analysis of CO. First, we assumed a linear change in CO over time and did not fit any more complicated models involving exponential, logarithmic, or polynomial coefficients. Second, we imputed values for the vasopressin + epinephrine group when the recorded value for cardiac output was “< 1,” by assuming that the decline from the last recorded value of CO until the time of death would be roughly linear, with a standard error proportionate to the expected value. Assumptions for HR were similar to the ones used in the CO analysis: that the change in HR was linear over time and that the conditions of regression held. We assumed that MAP was a quadratic function of time (rather than linear).
Results
A total of 14 pigs were included in the final analysis. There was no difference in baseline characteristics (weight, CO, cardiac index (CI), or heart rate) between the groups at the point of toxicity with the exceptions of mean MAP, SVR, and time to point of toxicity which were all lower (or shorter) in the placebo group than either the HDI or the vasopressin + epinephrine groups. Table 1 includes baseline characteristics at the point of toxicity for all groups, as well as times to maximum infusions of both vasopressin + epinephrine and HDI. Metabolic parameters, including potassium measurements, were reported in the previous manuscripts.
Table 1.
Comparisons between treatment arms at point of toxicity and time to maximum infusions for interventions.
| Variable | Vasopressin + epinephrine | High-dose insulin | Placebo | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Mean | SEM | Range | Mean | SEM | Range | Mean | SEM | Range | p value | |
| Weight (kg) | 29.4 | 2.28 | 22.4–35.8 | 32.6 | 3.66 | 22–38.6 | 39.6 | 2.94 | 31.8–44.4 | 0.083 |
| Time to toxicity (minute) | 45.0 | 8.94 | 20–60 | 49.0 | 4.00 | 40–60 | 13.8 | 1.25 | 10–15 | 0.014 |
| Cardiac output (L/minute) | 2.55 | 0.24 | 2.1–3.1 | 3.00 | 0.27 | 2.3–3.8 | 2.80 | 0.30 | 2.2–3.5 | 0.525 |
| Cardiac index (L/minute/m2) | 3.77 | 0.73 | 3.5–4.6 | 4.14 | 0.63 | 3.9–5.5 | 3.44 | 0.76 | 3.6–5.1 | 0.418 |
| Heart rate (beats/minute) | 80.2 | 1.65 | 67–84 | 77.8 | 3.38 | 72–90 | 68.7 | 3.77 | 64–80 | 0.073 |
| SVR (dynes × s/cm5) | 2046 | 149 | 1496–2400 | 1787 | 165 | 1357–2330 | 1263 | 117 | 955–1418 | 0.013 |
| Mean arterial pressure (mmHg) at time of toxicity | 75.3 | 2.36 | 49–82 | 73.8 | 4.94 | 65–92 | 49.0 | 2.42 | 45–56 | 0.001 |
| Time to maximal infusion rate for therapy (minute)* | 40 | 0 | 40–40 | 40 | 6.32 | 20–60 | NA | NA | NA | |
Italicized values represent a significant difference between groups
*All pigs in the vasopressin + epinephrine reached maximal dosing. All but one pig in the HDI group reached maximal dosing; this unique pig reached a steady state at 6 U/kg/hour at 20 minutes and required no additional up-titration of HDI
Survival
Two pigs survived in the placebo group (2/4, 50%), no pigs survived in the vasopressin + epinephrine group (0/5, 0%), and all pigs survived in the HDI group (5/5, 100%). The two deaths in the placebo group occurred at 220 and 270 minutes into resuscitation. All pigs in the vasopressin + epinephrine group died in less than 100 minutes. Regarding the primary outcome, there was a statistically significant difference in survival favoring the placebo group over the vasopressin + epinephrine group (p < 0.01). HDI was superior to both placebo and vasopressin + epinephrine in terms of mortality (p < 0.01). Figure 2 depicts the Kaplan-Meier survival curve for the three groups.
Fig. 2.

Survival rates post resuscitation protocol.
Cardiovascular Parameters
Cardiac Output
While CO was not significantly different in any of the three groups at time of toxicity, the interaction effects of the treatment groups and time upon CO were significant. As calculated from the linear mixed-effects regression fit to these data, the predicted change for the insulin treatment is a gain of 0.0113 L/minute2 over the 240 minutes of study time. Thus, over a 240-minute resuscitation, the mean increase in CO for HDI-treated pigs can be calculated using the following formula:
(dose/time interaction for CO [0.0113 L/min2] × 240 minutes) = 2.71 L/minute.
The dose/time interaction for HDI was significantly higher (p < 0.0001) than the change in CO for the placebo group, which was a loss of cardiac output of − 0.0119 L/minute2 over the 240 minutes of study time. Thus, the loss of CO over a 240-minute resuscitation can be calculated as follows:
(dose/time interaction for CO [− 0.0119 L/min2] × 240 minutes) = − 2.86 L/min.
In turn, the placebo arm’s predicted change in CO over time was significantly greater (p < 0.0001) than that of the vasopressin + epinephrine group, which was − 0.0352 L/minute2. Thus, the loss of CO over a 240-minute resuscitation can be calculated as follows:
(dose/time interaction for CO [− 0.0352 L/min2] × 240 minutes) = − 8.45 L/minute.
CO is displayed over time for all three groups in Fig. 3.
Fig. 3.
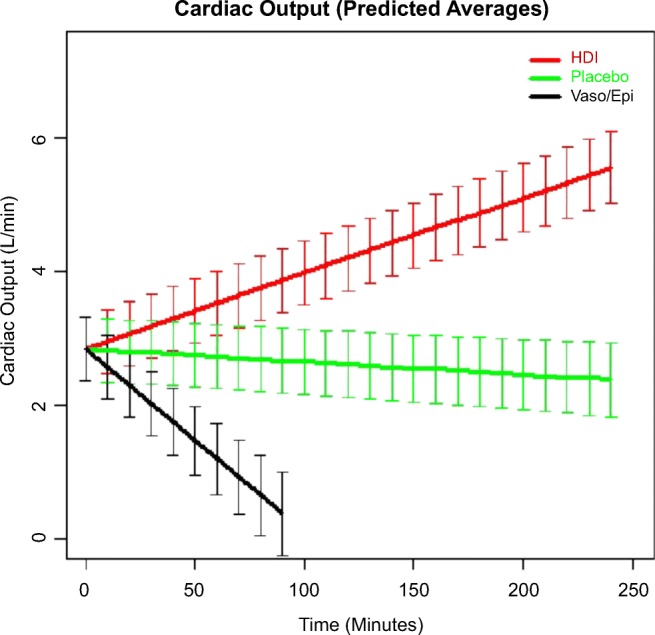
Cardiac output (predicted average).
Heart Rate
Heart rates at time of toxicity were significantly lower in the placebo group compared with the insulin group (p < 0.001). Because of this, the linear mixed-effects regression used to model the change in HR over time did not attempt to force a common intercept term at t = 0. The predicted model for the insulin arm showed subjects’ HR starting at 87 beats/minute and increasing by 0.099 beats/minute every minute, such that by the end of the study, it would be 110.7 beats/minute. This is significantly different from the placebo group (p < 0.0001), whose initial heart rate is 69 beats/minute and experiences a projected decline that is quite close to zero. The placebo arm had significantly higher HR over time than did the vasopressin + epinephrine arm (p < 0.0001), which started at 86 beats/minute and declined at − 0.501 beats/minute until all subjects died in the interval between 90 and 100 minutes. Heart rate over time is displayed for all three groups in Fig. 4.
Fig. 4.

Heart rate over time.
Mean Arterial Pressure
MAP at time of toxicity was significantly lower in the saline arm than in the other two arms. Due to this, we did not attempt to force a common intercept at t = 0 in linear mixed-effects regression. While the change in MAP over time was not significantly different in placebo and HDI arms (p = 0.1975, p = 0.1076 for linear and quadratic treatment by time interaction terms, respectively), the change in MAP projected for the vasopressin + epinephrine arm was a parabolic arc following the formula MAP = 78 + 1.83 × (time) − 0.03 × (time2). The respective p values for the linear and quadratic interaction terms with time for vasopressin + epinephrine were < 0.0001 and < 0.0001. MAP over time for all three groups is displayed in Fig. 5.
Fig. 5.
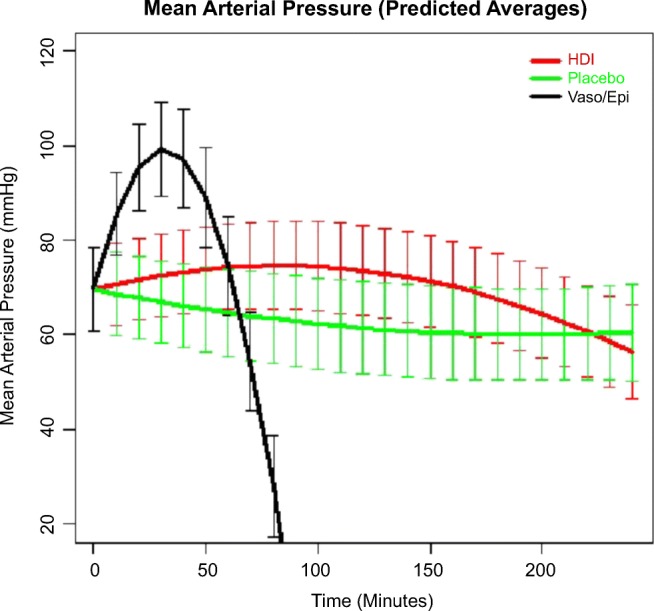
Mean arterial pressure (predicted averages).
Systemic Vascular Resistance
SVR showed a statistically significant difference between placebo and HDI arms, both at baseline and in terms of interaction effects (p < 0.001). SVR was higher in both the HDI and vasopressin + epinephrine groups initially, peaking at 30 minutes, then fell to 0 in all pigs as they died. Meanwhile, placebo and insulin groups did not experience a significantly different change in SVR over time. Both the placebo and HDI groups experience a shallow decline in SVR that was not statistically different from zero. SVR over time for all three groups is displayed in Fig. 6.
Fig. 6.

Systemic vascular resistance (predicted average).
Discussion
In this study of a lethal porcine model of propranolol poisoning, we found the combination of vasopressin and epinephrine, compared with placebo, to be associated with decreased survival. HDI was associated with increased survival compared with both groups. Hemodynamic parameters were also more favorable for placebo than vasopressin + epinephrine. Cardiac output and heart rate were both significantly higher in the placebo group than in the vasopressin + epinephrine group when accounting for the entire study protocol, and HDI was associated with both increased heart rate and cardiac output compared with both placebo and vasopressin + epinephrine. Systemic vascular resistance and mean arterial pressure were initially higher in the vasopressin + epinephrine group compared with placebo or HDI, but experienced a parabolic arc as the animals died, resulting ultimately in significantly lower readings.
Existing literature evaluating vasopressors and inotropes for beta-blocker poisoning is sparse. In addition to the four studies evaluating vasopressors and inotropes in a placebo-controlled manner identified by Skoog et al., we found an additional 7 studies by conducting our own literature search [22–28] (see summary in Table 2). This was performed by searching PubMed and MEDLINE using the search terms used by Skoog et al. restricted to any controlled or comparative study dating back to 1990. We supplemented this search by the review of the references in the controlled trials previously identified by Skoog et al. Three groups studied only inotropes, with two (Love et al. and Sato et al.) studying glucagon, amrinone, milrinone, and combination therapies in canine models [15, 22–24] and one group studying dobutamine and levosimendan in a porcine model [25]. The Love and Sato groups evaluated only hemodynamic parameters and not mortality and found all therapies to be superior to placebo with subtle differences in each therapy to increase either heart rate (glucagon) or MAP (phosphodiesterase inhibitors). Leppikangas et al. found no difference between placebo and dobutamine but did note a substantial mortality advantage with levosimendan [25]. Another group studied levosimendan in a rat model, comparing it with placebo and epinephrine, and found improved survival in metoprolol poisoning [27] but not propranolol [28]. Currently, levosimendan is not approved for use in the USA. Toet and colleagues conducted two studies using a rat model of propranolol poisoning evaluating isoproterenol, dopamine, and glucagon [13, 14]. There was no difference in time to survival between arms (including placebo), with the exception of the isoproterenol + dopamine arm, in which survival time was significantly decreased compared with all other groups [13]. In their second study, which utilized their previous rat model as well as a rabbit model, they evaluated glucagon and dopamine and found that no intervention improved survival compared with placebo in either model. In addition, in the rat model, glucagon + dopamine was associated with decreased survival compared with placebo, as was glucagon monotherapy in the rabbit model [14]. Lastly, Kerns et al. compared saline placebo with glucagon, with epinephrine, and with HDI in a lethal canine model [12]. Survival was significantly greater with HDI over the other arms. No placebo animals survived; one of six canines treated with epinephrine survived.
Table 2.
Summary of comparative effectiveness animal studies evaluating vasopressors and inotropes in beta-blocker poisoning.
| Reference | Model** | Treatment arms | Results |
|---|---|---|---|
| Love et al. [15] | Canine |
Placebo (n = 6) Glucagon (n = 6) Amrinone (n = 6) |
Amrinone and glucagon effective in reversing depressed ventricular pressures, cardiac output, and stroke volume compared with placebo, only glucagon improved heart rate |
| Love et al. [22] | Canine | Amrinone + glucagon (n = 6)* | Addition of amrinone to glucagon had a detrimental effect on MAP |
| Sato et al. [23] | Canine |
Placebo (n = 3) Glucagon (n = 5) Milrinone (n = 5) |
Milrinone and glucagon both superior to placebo, hemodynamic parameters favor milrinone |
| Sato et al. [24] | Canine |
Placebo (n = 5) Glucagon (n = 5) Milrinone (n = 5) Glucagon + milrinone (n = 5) |
No evaluation of survival. All arms improved over placebo; combination therapy resulted in undesired tachycardia |
| Toet et al. [13] | Rat |
Placebo (n = 10) Isoproterenol (n = 10) Isoproterenol + glucagon (n = 10) Isoproterenol + dopamine (n = 10) |
Survival not improved over placebo in any arm, but reduced in the isoproterenol + dopamine group |
| Toet et al. [14] | Rat and rabbit |
Placebo (n = 10 rats, 8 rabbits) Glucagon (n = 10 rats, 8 rabbits) Dopamine (n = 10 rats, 8 rabbits) Dopamine + glucagon (n = 10 rats, 8 rabbits) |
Survival not improved over placebo in any arm, but reduced in dopamine + glucagon arm in rat study |
| Kerns et al. [12] | Canine |
Placebo (n = 6) Epinephrine (n = 6) Glucagon (n = 6) HDI (n = 6) |
Survival as follows: placebo (0/6), epinephrine (1/6), glucagon (4/6), and HDI (6/6) |
| Holger et al. [6] | Porcine |
Vasopressin (n = 8) Glucagon (n = 8) |
No difference between arms |
| Uechi et al. [26] | Canine (oral carvedilol) |
Placebo (n = 6) Dobutamine (n = 6) Dopamine (n = 6) Milrinone (n = 6) |
Mortality not evaluated, hemodynamic parameters favor milrinone |
| Holger et al. [11] | Porcine |
Vasopressin + epinephrine (n = 5) HDI (n = 5) |
Significantly lower survival with vasopressors |
| Leppikangas et al. [25] | Porcine |
Placebo (n = 6) Dobutamine (n = 9) Levosimendan (n = 9) |
No difference in survival between placebo and dobutamine, all pigs receiving levosimendan lived |
| Kalam and Graudins [27] | Rat (IV metoprolol) |
Placebo (n = 10) Epinephrine (n = 10) Levosimendan (n = 20) |
Improved survival with levosimendan over control but not epinephrine |
| Kalam and Graudins [28] | Rat |
Placebo (n = 10) Epinephrine (n = 10) Levosimendan (n = 50) |
No difference in survival between any groups |
| Katzung et al. [10] | Porcine |
HDI (n = 5) HDI + norepinephrine (n = 5) Norepinephrine + epinephrine (n = 5) |
Significantly reduced survival with vasopressors |
*Results compared with data from Love et al. [15]
**All models in table involve IV propranolol, except as noted
As noted in the review by Skoog et al., evidence examining the use of vasopressors for beta-blocker poisoning is perplexing. Animal studies generally show no improvement over placebo or poor outcomes, whereas human case reports, subject to publication bias as they may be [29, 30], are generally associated with positive outcomes. Our data align with previous studies demonstrating vasopressors, in this case vasopressin and epinephrine, are associated with worse outcomes than placebo. Clinical experience with severe beta-blocker poisoning, however, clearly demonstrates that multiple therapies are frequently needed to maintain perfusion [5, 31, 32]. Therefore, the most pertinent question is likely not “if” vasopressors should be used in beta-blocker poisoning but “when.” Recent data from our laboratory may shed some light on these questions. In propranolol poisoned pigs, Katzung et al. found the combination of norepinephrine and epinephrine to be associated with rapid death and that HDI was superior to the combination of these two vasopressors [10]. However, survival with HDI alone was not universal. In this model, when norepinephrine was added to HDI, not only did survival increase, but brain perfusion, as measured by cerebral oxygen tension [33], improved as well. Given multiple animal models have demonstrated that HDI is superior to vasopressors in beta-blocker poisoning [10–12] and that vasopressors may be worse than placebo in this context, the available data suggest severe poisoning should first be treated with HDI, with vasopressors added as adjuncts if HDI fails to produce adequate organ perfusion as it appears that HDI and vasopressors may be synergistic [10, 20]. Data from human patients poisoned with beta-blockers would help clarify these matters; however, a randomized trial in humans is unlikely to be feasible in the near future.
Limitations
Our study has several limitations. First, this is an animal model only of propranolol poisoning. We chose a porcine model to mitigate this as much as possible, given the many similarities between porcine and human hearts [34]; however, porcine is clearly not a perfect model, and unappreciated differences between humans and pigs may have introduced bias and limited generalizability.
Second, this study was not planned a priori. Rather, it was an analysis of previously collected data. This was done not only for feasibility and cost reasons, but also to prevent unnecessarily sacrificing animals. This methodology has been used by other researchers on this topic [15, 22]. The lack of a priori planning and the spaced data collection between arms could have introduced bias. It is possible this existed as there were some differences in the baseline characteristics between groups. Time to toxicity was shorter, and MAP and SVR were lower in the placebo group than in the vasopressin + epinephrine group. We also followed placebo pigs for up to 360 minutes, whereas HDI and vasopressin + epinephrine pigs were only followed up to 240 minutes. These differences, however, would bias the groups toward the vasopressin + epinephrine group having healthier pigs and better outcomes, and yet the vasopressin + epinephrine group performed worst overall. For instance, one of the placebo pigs died at 270 minutes—had we followed placebo pigs for only 240 minutes, the placebo arm would have had an even better survival profile. As the placebo group performed better despite having less optimal baseline characteristics and a longer observation time, we believe any differences that may have been present between the groups did not substantially bias our results against vasopressors.
Third, our model included only propranolol poisoning, and yet as both large studies of patients with beta-blocker poisoning [1, 5, 35] and numerous case reports [36–38] demonstrate, co-ingestion with other medications that cause vasodilatory shock is common. Therefore, even if our results translate to human beta-blocker poisoning, they may not apply if a drug that causes profound vasodilatory shock is also co-ingested. In addition, as demonstrated by Kalam and Graudins with metoprolol and propranolol, different beta-blockers themselves may respond differently to different therapies [27, 28]. Propranolol is somewhat unique among beta-blockers in that it causes sodium channel blockade. We did not measure QRS duration formally in this study, though anecdotally, we observed QRS duration on continuous ECG monitoring did not become visibly wide until death was imminent. Nevertheless, the fact that HDI proved to be a superior therapy may have implications for poisoning from other drugs that cause cardiogenic shock via sodium channel blockade, such as flecainide [39–41]. Further study is needed to determine if HDI has a role in such poisonings.
Last, our results may only apply to the medications we studied and not to other inotropes or vasopressors. For instance, we did not study glucagon, a commonly recommended first-line drug for beta-blocker poisoning [42]. We chose to study vasopressin rather than glucagon because in our previous work, we found no significant difference between vasopressin and glucagon [6]. In addition, it may be that a single unopposed vasopressor is not harmful in beta-blocker poisoning. Similar to our results, in many of the animal studies where vasopressors were found to be harmful, worse outcomes occurred when the vasopressor was used in combination with another inotrope or a second vasopressor [10, 11, 13, 14]. It is possible a single vasopressor, particularly if used at a modest dose, may not be harmful in beta-blocker poisoning.
Conclusions
This study demonstrated an increased mortality with the combination of vasopressin and epinephrine when compared with placebo in a porcine model of propranolol poisoning. Further studies are warranted to define the role and optimal timing of the use of vasopressors in beta-blocker poisoning.
Acknowledgments
The authors would like to recognize Dr. Joel Holger for his valued research mentorship. We would like to thank Kate Faltesek and Alex Adams as well; without their laboratory expertise, the execution of these studies would not have been possible. Finally, with admiration and sadness, we wish to thank and recognize Dr. Kristin Engebretsen. Our colleague, teacher, mentor, collaborator, and friend passed away during the processing of this study.
Sources of Funding
This project was funded by a grant from the American Academy of Clinical Toxicology and by the HealthPartners Research Foundation.
Compliance with Ethical Standards
Conflicts of Interest
None
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Lauterbach M. Clinical toxicology of beta-blocker overdose in adults. Basic Clin Pharmacol Toxicol. 2019;125(2):178–186. doi: 10.1111/bcpt.13231. [DOI] [PubMed] [Google Scholar]
- 2.Gummin DD, Mowry JB, Spyker DA, Brooks DE, Osterthaler KM, Banner W. 2017 annual report of the American Association of Poison Control Centers’ National Poison Data System (NPDS): 35th annual report. Clin Toxicol. 2018;56(12):1213–1415. doi: 10.1080/15563650.2018.1533727. [DOI] [PubMed] [Google Scholar]
- 3.Cole JB, Stellpflug SJ, Ellsworth H, Anderson CP, Adams AB, Engebretsen KM, Holger JS. A blinded, randomized, controlled trial of three doses of high-dose insulin in poison-induced cardiogenic shock. Clin Toxicol. 2013;51(4):201–207. doi: 10.3109/15563650.2013.770152. [DOI] [PubMed] [Google Scholar]
- 4.Taboulet P, Cariou A, Berdeaux A, Bismuth C. Pathophysiology and management of self-poisoning with beta-blockers. J Toxicol Clin Toxicol. 1993;31(4):531–551. doi: 10.3109/15563659309025759. [DOI] [PubMed] [Google Scholar]
- 5.Cole JB, Arens AM, Laes JR, Klein LR, Bangh SA, Olives TD. High dose insulin for beta-blocker and calcium channel-blocker poisoning. Am J Emerg Med. 2018;36(10):1817–1824. doi: 10.1016/j.ajem.2018.02.004. [DOI] [PubMed] [Google Scholar]
- 6.Holger JS, Engebretsen KM, Obetz CL, Kleven TL, Harris CR. A comparison of vasopressin and glucagon in beta-blocker induced toxicity. Clin Toxicol. 2006;44(1):45–51. doi: 10.1080/15563650500394795. [DOI] [PubMed] [Google Scholar]
- 7.Levy B, Buzon J, Kimmoun A. Inotropes and vasopressors use in cardiogenic shock: when, which and how much? Curr Opin Crit Care. 2019;25(4):384–390. doi: 10.1097/MCC.0000000000000632. [DOI] [PubMed] [Google Scholar]
- 8.Knotzer H, Poidinger B, Kleinsasser A. Pharmacologic agents for the treatment of vasodilatory shock. Curr Pharm Des. 2019;25(19):2133–2139. doi: 10.2174/1381612825666190704101907. [DOI] [PubMed] [Google Scholar]
- 9.Skoog CA, Engebretsen KM. Are vasopressors useful in toxin-induced cardiogenic shock? Clin Toxicol. 2017;55(4):285–304. doi: 10.1080/15563650.2017.1284329. [DOI] [PubMed] [Google Scholar]
- 10.Katzung KG, Leroy JM, Boley SP, Stellpflug SJ, Holger JS, Engebretsen KM. A randomized controlled study comparing high-dose insulin to vasopressors or combination therapy in a porcine model of refractory propranolol-induced cardiogenic shock. Clin Toxicol. 2019;57(11):1073–1079. doi: 10.1080/15563650.2019.1580372. [DOI] [PubMed] [Google Scholar]
- 11.Holger JS, Engebretsen KM, Fritzlar SJ, Patten LC, Harris CR, Flottemesch TJ. Insulin versus vasopressin and epinephrine to treat beta-blocker toxicity. Clin Toxicol. 2007;45(4):396–401. doi: 10.1080/15563650701285412. [DOI] [PubMed] [Google Scholar]
- 12.Kerns W, 2nd, Schroeder D, Williams C, Tomaszewski C, Raymond R. Insulin improves survival in a canine model of acute beta-blocker toxicity. Ann Emerg Med. 1997;29(6):748–757. doi: 10.1016/s0196-0644(97)70196-3. [DOI] [PubMed] [Google Scholar]
- 13.Toet AE, te Biesebeek JD, Vleeming W, Wemer J, Meulenbelt J, de Wildt DJ. Reduced survival after isoprenaline/dopamine in d,l-propranolol intoxicated rats. Hum Exp Toxicol. 1996;15(2):120–128. doi: 10.1177/096032719601500204. [DOI] [PubMed] [Google Scholar]
- 14.Toet AE, Wemer J, Vleeming W, te Biesebeek JD, Meulenbelt J, de Wildt DJ. Experimental study of the detrimental effect of dopamine/glucagon combination in d,l-propranolol intoxication. Hum Exp Toxicol. 1996;15(5):411–421. doi: 10.1177/096032719601500509. [DOI] [PubMed] [Google Scholar]
- 15.Love JN, Leasure JA, Mundt DJ, Janz TG. A comparison of amrinone and glucagon therapy for cardiovascular depression associated with propranolol toxicity in a canine model. J Toxicol Clin Toxicol. 1992;30(3):399–412. doi: 10.3109/15563659209021555. [DOI] [PubMed] [Google Scholar]
- 16.Holger JS, Stellpflug SJ, Cole JB, Harris CR, Engebretsen KM. High-dose insulin: a consecutive case series in toxin-induced cardiogenic shock. Clin Toxicol. 2011;49(7):653–658. doi: 10.3109/15563650.2011.593522. [DOI] [PubMed] [Google Scholar]
- 17.Kline JA, Tomaszewski CA, Schroeder JD, Raymond RM. Insulin is a superior antidote for cardiovascular toxicity induced by verapamil in the anesthetized canine. J Pharmacol Exp Ther. 1993;267(2):744–750. [PubMed] [Google Scholar]
- 18.Stadlbauer KH, Wagner-Berger HG, Wenzel V, Voelckel WG, Krismer AC, Klima G, et al. Survival with full neurologic recovery after prolonged cardiopulmonary resuscitation with a combination of vasopressin and epinephrine in pigs. Anesth Analg. 2003;96(6):1743–1749. doi: 10.1213/01.ANE.0000066017.66951.7F. [DOI] [PubMed] [Google Scholar]
- 19.Wenzel V, Krismer AC, Arntz HR, Sitter H, Stadlbauer KH, Lindner KH, et al. A comparison of vasopressin and epinephrine for out-of-hospital cardiopulmonary resuscitation. N Engl J Med. 2004;350(2):105–113. doi: 10.1056/NEJMoa025431. [DOI] [PubMed] [Google Scholar]
- 20.Engebretsen KM, Morgan MW, Stellpflug SJ, Cole JB, Anderson CP, Holger JS. Addition of phenylephrine to high-dose insulin in dihydropyridine overdose does not improve outcome. Clin Toxicol. 2010;48(8):806–812. doi: 10.3109/15563650.2010.521753. [DOI] [PubMed] [Google Scholar]
- 21.Rubin DB. Multiple imputation for survey nonresponse. New York: Wiley; 1987. [Google Scholar]
- 22.Love JN, Leasure JA, Mundt DJ. A comparison of combined amrinone and glucagon therapy to glucagon alone for cardiovascular depression associated with propranolol toxicity in a canine model. Am J Emerg Med. 1993;11(4):360–363. doi: 10.1016/0735-6757(93)90168-b. [DOI] [PubMed] [Google Scholar]
- 23.Sato S, Tsuji MH, Okubo N, Naito H. Milrinone versus glucagon: comparative hemodynamic effects in canine propranolol poisoning. J Toxicol Clin Toxicol. 1994;32(3):277–289. doi: 10.3109/15563659409017960. [DOI] [PubMed] [Google Scholar]
- 24.Sato S, Tsuji MH, Okubo N, Nishimoto C, Naito H. Combined use of glucagon and milrinone may not be preferable for severe propranolol poisoning in the canine model. J Toxicol Clin Toxicol. 1995;33(4):337–342. doi: 10.3109/15563659509028919. [DOI] [PubMed] [Google Scholar]
- 25.Leppikangas H, Ruokonen E, Rutanen J, Kiviniemi V, Lindgren L, Kurola J. Levosimendan as a rescue drug in experimental propranolol-induced myocardial depression: a randomized study. Ann Emerg Med. 2009;54(6):811–7.e1–3. doi: 10.1016/j.annemergmed.2009.06.512. [DOI] [PubMed] [Google Scholar]
- 26.Uechi M, Hori Y, Fujimoto K, Ebisawa T, Yamano S, Maekawa S. Cardiovascular effects of a phosphodiesterase III inhibitor in the presence of carvedilol in dogs. J Vet Med Sci. 2006;68(6):549–553. doi: 10.1292/jvms.68.549. [DOI] [PubMed] [Google Scholar]
- 27.Kalam Y, Graudins A. Levosimendan infusion improves cardiac output but not blood pressure in a rodent model of severe metoprolol toxicity. Hum Exp Toxicol. 2012;31(9):955–963. doi: 10.1177/0960327111433182. [DOI] [PubMed] [Google Scholar]
- 28.Kalam Y, Graudins A. Levosimendan does not improve cardiac output or blood pressure in a rodent model of propranolol toxicity when administered using various dosing regimens. Int J Toxicol. 2012;31(2):166–174. doi: 10.1177/1091581811435366. [DOI] [PubMed] [Google Scholar]
- 29.Cole JB, Stellpflug SJ, Engebretsen KM. Asystole immediately following intravenous fat emulsion for overdose. J Med Toxicol. 2014;10(3):307–310. doi: 10.1007/s13181-014-0382-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bradley PJ. Guidelines to authors publishing a case report: the need for quality improvement. AME Case Rep. 2018;2:10. doi: 10.21037/acr.2018.04.02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang GS, Levitan R, Wiegand TJ, Lowry J, Schult RF, Yin S, Toxicology Investigators Consortium Extracorporeal membrane oxygenation (ECMO) for severe toxicological exposures: review of the Toxicology Investigators Consortium (ToxIC) J Med Toxicol. 2016;12(1):95–99. doi: 10.1007/s13181-015-0486-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lewis J, Zarate M, Tran S, Albertson T. The recommendation and use of extracorporeal membrane oxygenation (ECMO) in cases reported to the California Poison Control System. J Med Toxicol. 2019;15(3):169–177. doi: 10.1007/s13181-019-00704-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Orozco BS, Engebretsen KM, Holger JS, Stellpflug SJ. A swine model of severe propranolol toxicity permitting direct measurement of brain tissue oxygenation. J Med Toxicol. 2019;15(3):178–183. doi: 10.1007/s13181-019-00707-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Camacho P, Fan H, Liu Z, He J-Q. Large mammalian animal models of heart disease. J Cardiovasc Dev Dis [Internet]. 2016;3(4). Available from:). 10.3390/jcdd3040030. [DOI] [PMC free article] [PubMed]
- 35.Truitt CA, Brooks DE, Dommer P, LoVecchio F. Outcomes of unintentional beta-blocker or calcium channel blocker overdoses: a retrospective review of poison center data. J Med Toxicol. 2012;8(2):135–139. doi: 10.1007/s13181-011-0209-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Heise CW, Beutler D, Bosak A, Orme G, Loli A, Graeme K. Massive atenolol, lisinopril, and chlorthalidone overdose treated with endoscopic decontamination, hemodialysis, impella percutaneous left ventricular assist device, and ECMO. J Med Toxicol. 2015;11(1):110–114. doi: 10.1007/s13181-014-0419-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Laes JR, Williams DM, Cole JB. Improvement in hemodynamics after methylene blue administration in drug-induced vasodilatory shock: a case report. J Med Toxicol. 2015;11(4):460–463. doi: 10.1007/s13181-015-0500-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stellpflug SJ, Fritzlar SJ, Cole JB, Engebretsen KM, Holger JS. Cardiotoxic overdose treated with intravenous fat emulsion and high-dose insulin in the setting of hypertrophic cardiomyopathy. J Med Toxicol. 2011;7(2):151–153. doi: 10.1007/s13181-010-0133-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ellsworth H, Stellpflug SJ, Cole JB, Dolan JA, Harris CR. A life-threatening flecainide overdose treated with intravenous fat emulsion. Pacing Clin Electrophysiol. 2013;36(3):e87–e89. doi: 10.1111/j.1540-8159.2012.03485.x. [DOI] [PubMed] [Google Scholar]
- 40.Bruccoleri RE, Burns MM. A literature review of the use of sodium bicarbonate for the treatment of QRS widening. J Med Toxicol. 2016;12(1):121–129. doi: 10.1007/s13181-015-0483-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brumfield E, KRL B, Kabrhel C. Life-threatening flecainide overdose treated with intralipid and extracorporeal membrane oxygenation. Am J Emerg Med. 2015;33(12):1840.e3–1840.e5. doi: 10.1016/j.ajem.2015.04.012. [DOI] [PubMed] [Google Scholar]
- 42.Bailey B. Glucagon in beta-blocker and calcium channel blocker overdoses: a systematic review. J Toxicol Clin Toxicol. 2003;41(5):595–602. doi: 10.1081/clt-120023761. [DOI] [PubMed] [Google Scholar]