

Iron homeostasis plays a key role at the host-pathogen interface during the process of infection. Bacterial growth restriction resulting from host-imposed iron starvation (nutritional immunity) highlights the importance of iron import during pathogenesis. Conversely, bacterial iron efflux pumps function as virulence factors in several systems. The requirement for iron efflux in pathogens such as Listeria monocytogenes, Streptococcus pyogenes, and Mycobacterium tuberculosis suggests that both import and efflux are needed for cells to successfully navigate rapidly changing levels of iron availability in the host. Here, we provide insight into how iron efflux genes are controlled, an aspect of bacterial iron homeostasis relevant to infectious disease processes.
KEYWORDS: Bacillus subtilis, Fur, Listeria monocytogenes, P-type ATPase, PerR, iron, peroxide stress, regulation
ABSTRACT
The physiological relevance of bacterial iron efflux has only recently been appreciated. The Bacillus subtilis P1B4-type ATPase PfeT (peroxide-induced ferrous efflux transporter) was one of the first iron efflux pumps to be characterized, and cells lacking pfeT accumulate high levels of intracellular iron. The pfeT promoter region has binding sites for both PerR, a peroxide-sensing Fur-family metalloregulator, and the ferric uptake repressor Fur. Both Fur and PerR bind DNA with Fe(II) as a cofactor. While reaction of PerR-Fe(II) with peroxide can account for the induction of pfeT under oxidative stress, binding of Fur-Fe(II) would be expected to lead to repression, which is inconsistent with the known role of PfeT as an iron efflux protein. Here, we show that expression of pfeT is repressed by PerR, as anticipated, and induced by Fur in response to Fe(II). Activation by Fur is mediated both by antagonism of the PerR repressor and by direct transcriptional activation, as confirmed using in vitro transcription assays. A similar mechanism of regulation can explain the iron induction of the Listeria monocytogenes PfeT ortholog and virulence factor, FrvA. Mutational studies support a model in which Fur activation involves regions both upstream and downstream of the pfeT promoter, and Fur and PerR have overlapping recognition of a shared regulatory element in this complex promoter region. This work demonstrates that B. subtilis Fur can function as an iron-dependent activator of transcription.
IMPORTANCE Iron homeostasis plays a key role at the host-pathogen interface during the process of infection. Bacterial growth restriction resulting from host-imposed iron starvation (nutritional immunity) highlights the importance of iron import during pathogenesis. Conversely, bacterial iron efflux pumps function as virulence factors in several systems. The requirement for iron efflux in pathogens such as Listeria monocytogenes, Streptococcus pyogenes, and Mycobacterium tuberculosis suggests that both import and efflux are needed for cells to successfully navigate rapidly changing levels of iron availability in the host. Here, we provide insight into how iron efflux genes are controlled, an aspect of bacterial iron homeostasis relevant to infectious disease processes.
INTRODUCTION
Iron is an essential micronutrient across all domains of life, including bacteria. It is used as a cofactor by enzymes involved in a wide array of metabolic processes, including the tricarboxylic acid (TCA) cycle, respiration, and DNA precursor (deoxynucleoside triphosphate [dNTP]) synthesis (1). Although iron is abundant in nature, its bioavailability is limited in oxic environments because of its facile oxidation from its soluble Fe(II) (ferrous) form to the Fe(III) (ferric) form, which readily forms insoluble hydrates. As a result, microorganisms have evolved a variety of high-affinity import mechanisms to acquire iron under iron-depleted conditions. The production of Fe(III)-binding siderophores is one of the most widespread adaptations to iron limitation in microorganisms, including Bacteria and many fungi (2).
Although the adaptive responses to iron limitation are widely appreciated, the role of iron efflux proteins has received comparatively little attention (3). Previously, we described the P1B4-type ATPase PfeT (formerly ZosA) as an iron efflux pump in the model Gram-positive bacterium Bacillus subtilis (4). PfeT is the first member of the P1B4-type ATPases implicated in iron efflux and among the first bacterial iron efflux systems to be discovered (4–8). Cells that lack pfeT accumulate high levels of intracellular iron and are therefore more sensitive to iron overload. Although the PfeT ATPase is specifically activated by ferrous iron, it has a relatively low apparent binding affinity (520 ± 120 μM) in vitro, consistent with a function limited to relieving iron overload (4). Orthologs of PfeT have been identified in other Gram-positive bacteria, many of which have been determined to act as virulence factors (9, to ,12). These findings create a conundrum since it is now clear that both iron deprivation and iron intoxication are physiologically relevant stresses that can limit the success of bacterial pathogens (3, 13). Presumably, these stresses occur at different stages or in different contexts during the infection process.
Oxidative stress and iron homeostasis are interlinked in light of the fact that ferrous iron acts as a catalyst for harmful Fenton reactions in which hydrogen peroxide (H2O2) is reduced with the generation of highly reactive hydroxyl radicals (14). To ameliorate the toxicity of H2O2, bacteria routinely deploy catalases and peroxidases to prevent endogenously produced intracellular H2O2 from accumulating. These enzymes work in concert with other proteins that can reduce the availability and reactivity of intracellular ferrous iron, often by sequestration. For example, in B. subtilis the Fur family peroxide-sensing metalloregulator PerR controls expression of two peroxide detoxification enzymes, catalase (KatA) and alkyl hydroperoxide reductase (AhpCF), as well as the Dps-like miniferritin MrgA that can sequester iron (15, to ,18). The ferrous iron efflux pump PfeT is also regulated by PerR and is derepressed under conditions of oxidative stress (17, 19).
Prior studies of the PerR regulon in Bacillus subtilis have laid the groundwork for our understanding of how bacteria respond to oxidative stress through PerR (17, 20). Genes under the control of this metalloregulator contain one or more PerR operator sites defined by their characteristic consensus sequence (21). Upon exposure to H2O2, PerR-Fe(II) metal-catalyzed oxidation (MCO) of iron-coordinated histidines at the Fe(II)-binding site triggers a conformational change that causes PerR to become unbound from the DNA, thus derepressing transcription of the regulon (22, 23).
As a member of the PerR regulon, pfeT has a well-conserved PerR operator site (here designated PerR I [PI]) within its promoter region (Fig. 1A and B). Through DNase I footprinting assays and in vivo experiments, it was shown that PerR binds to DNA regions overlapping the PI operator site (24). Although PfeT helps alleviate peroxide stress, it plays a secondary role compared to that of the enzymatic detoxification pathways and likely works in concert with MrgA to help reduce the labile iron pool in the cell (4). PfeT plays a more prominent role in survival under conditions of iron overload. Although pfeT was originally identified as under the control of PerR (19), a reassessment of its promoter region revealed two additional regulator binding sites shown to interact with Fur in vitro (24). One site is located upstream of the PI site (designated Fur II [FII]), and a second begins 65 bp downstream of the transcriptional start site (Fur III [FIII]) (Fig. 1A and B).
FIG 1.

Promoter region of pfeT. (A) Sequence logo of the Fur and PerR consensus operator sites. A total of 23 Fur and 11 Per sites was used to generate the sequence logos with WebLogo. The list of sequences is found in Tables S3 and S4 in the supplemental material. (B) The sequence of the pfeT promoter region contains one conserved PerR box (gray), named PerR I (PI), which is adjacent to the −35 region. Two Fur operator sites (bold), named Fur II (FII) and Fur III (FIII), are positioned both upstream and downstream of the Per site. FII is separated by 8 bp from the PerR box, while FIII is more than 55 bp downstream of the transcriptional start site. The transcriptional start site is denoted in bold. The −10 and −35 RNAP binding sites are in italics. Vertical lines (|) indicate conserved bases, and asterisks indicate mutated bases in the constructs containing operator site mutations. (C) Model for the pfeT control region (i), illustrating transcriptional repression by PerR (PDB accession number 3F8N) binding at sites PI and FII (ii) and induction by iron mediated by Fur (represented by the homolog Streptomyces coelicolor Zur; PDB code 3MWM) binding at FII (to antagonize PerR repression and as a direct transcription activator) and/or FIII (as a transcription activator) (iii).
Here, we have investigated the interplay between the PerR and Fur metalloregulators in the regulation of the pfeT gene. Our genetic and biochemical results lead to a model in which PerR forms a repression complex that responds to elevated H2O2 to derepress pfeT expression. In parallel, Fur senses elevated cytosolic iron and both antagonizes PerR repression and directly activates pfeT transcription. These results reveal complexities of operator site overlap and regulator interaction in this complex control region.
RESULTS
Molecular model for the regulation of pfeT.
The pfeT regulatory region contains three operator sites defined by sequence similarity to the PerR and Fur consensus binding sites (Fig. 1A and B). The results presented here culminate in a model, presented at the outset for clarity, in which PerR forms a repression complex involving cooperative binding to both sites PI and FII (Fig. 1C), consistent with the extended region of PerR binding noted in prior footprinting studies (24). When cytosolic iron levels are elevated, Fur replaces PerR at site FII, thereby disrupting the repression complex, and Fur protein bound to site FII and/or FIII contributes to activation of pfeT expression (Fig. 1C). This model results from a combination of in vivo and in vitro studies to explore the role of Fur and PerR in pfeT regulation.
Expression of pfeT is induced in response to excess iron.
To quantify the induction of pfeT in response to high iron levels, we fused the pfeT promoter region to a lacZ reporter. Cells grown in lysogeny broth (LB) medium showed a low basal level of expression of about 1 to 2 Miller units. When grown in LB medium amended with Fe(II), pfeT was induced (Fig. 2A). This induction was not observed in LB amended with Zn, Mn, or Co salts at concentrations known to induce genes responsive to these metals (25–27), indicating that pfeT induction is iron specific.
FIG 2.
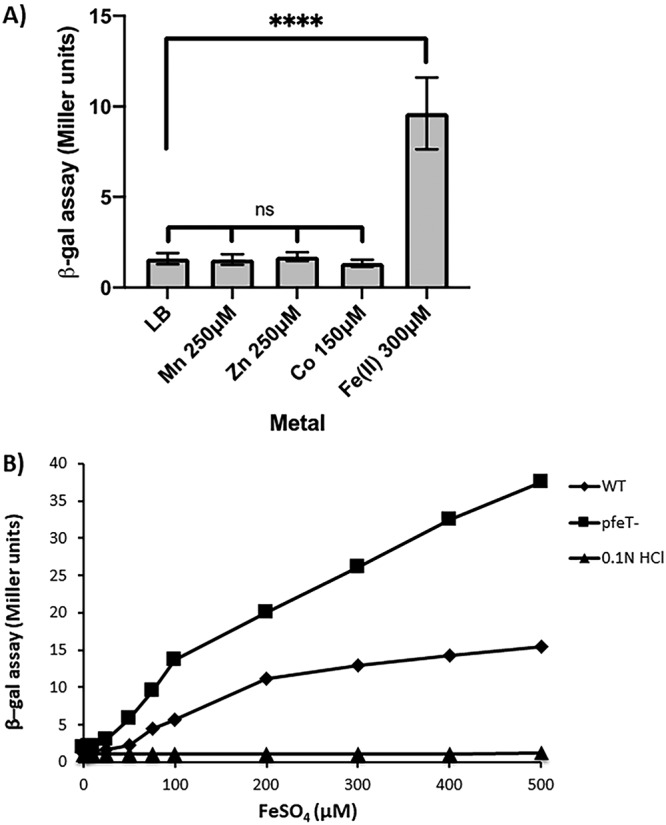
β-Galactosidase assays showing that induction of the pfeT gene is specific to iron. (A) WT cells containing a pfeT-lacZ transcriptional fusion were grown in LB medium amended with different metals, each at a specific concentration, to an OD600 of 0.4. β-Galactosidase (β-Gal) activity was measured. Data represent means ± standard deviations for three or more biological replicates. Multipronged significance bars indicate statistical significance for a given condition in reference to results with LB medium, and significance was calculated using a one-way analysis of variance (ns, no statistical significance). Significance between results for the iron and normal LB conditions was calculated using an unpaired Student's t test (****, P < 0.0001). (B) β-Galactosidase assays to monitor pfeT-lacZ expression in LB medium amended with different concentrations of iron. WT cells show increasing levels of induction as a function of iron. Induction was even more sensitive to added iron when measured in pfeT::kan cells. As a control, the strain was also grown in LB medium containing the equivalent volume of 0.1 N HCl for each particular iron concentration. Results shown are representative of three biological replicates.
Cells grown over a range of iron concentrations in LB medium displayed an increase in pfeT induction as a function of added iron. We confirmed that this induction was dependent on the concentration of added FeSO4 (dissolved in 0.1 N HCl) and was not observed in cells treated with 0.1 N HCl alone (Fig. 2B). Since cells lacking pfeT are unable to export iron and have higher intracellular iron concentrations even with nontoxic amounts of added iron (4), we anticipated that induction of the pfeT-lacZ reporter would be more sensitive to Fe(II) in cells lacking PfeT. As expected, induction levels were increased in a pfeT null mutant compared to levels in the wild type (WT) (Fig. 2B). Thus, the pfeT promoter region provides a sensitive bioreporter for sensing intracellular, bioavailable iron.
Fur and PerR differentially contribute to pfeT expression in vivo.
In order to understand how PerR and Fur contribute to the expression of pfeT, we measured expression levels in perR, fur, and perR fur null backgrounds in the presence or absence of added iron. As expected, in WT cells pfeT is iron induced, whereas expression is constitutive in the perR null strain (Fig. 3). This confirms that PerR functions as a repressor (19). However, even in the perR null mutant, the addition of iron led to a modest, but reproducible, increase in pfeT-lacZ expression (Fig. 3). This suggests that gene induction in response to iron may be mediated by Fur. Consistent with this notion, in a perR fur double mutant, transcription levels were somewhat lower, and no increase was observed upon iron addition (Fig. 3). Studies of iron induction in the perR null mutant strain are challenging to interpret since this mutant is itself altered in iron homeostasis (28). Specifically, derepression of catalase and Fur in the perR null mutant combine to impose an iron restriction on growth (28).
FIG 3.
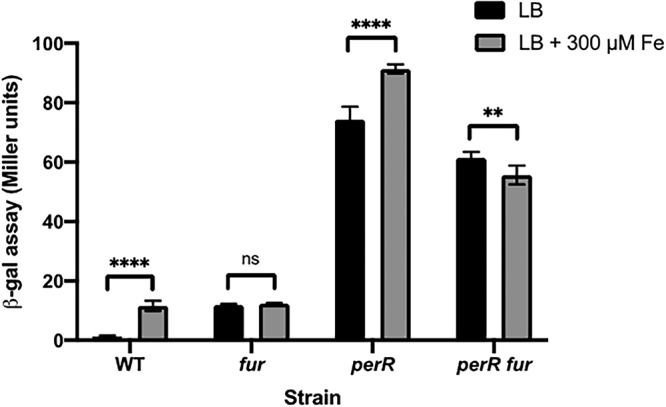
β-Galactosidase assay of pfeT in different strain backgrounds. The pfeT-lacZ transcriptional fusion was monitored in WT, fur::kan, perR::tet, and fur::kan perR::tet strain backgrounds. Cells were grown in LB medium containing either no added iron or 300 μM iron (a concentration known to induce pfeT) to an OD600 of 0.4. Data represent means ± standard deviations for at least three biological replicates. Significance was calculated using a two-way analysis of variance test with Šidák correction (**, P < 0.01; ****, P < 0.0001; ns, no statistical significance).
Unlike expression in the perR null strain, in the fur null mutant expression of pfeT is repressed and no longer iron responsive (Fig. 3). We note that repression is not as strong as in WT cells. We suggest that this is due to an increased level of bioavailable iron in the fur mutant strain. The efficacy of PerR repression is known to be highly sensitive to the balance between Mn(II) and Fe(II) since PerR bound to Mn(II) as a corepressor is less susceptible to oxidative inactivation during aerobic growth (22, 29–31). Indeed, supplementation of LB medium with 25 μM Mn(II) reduces the expression of a pfeT-lacZ fusion in medium with 500 μM iron by severalfold (from 17 ± 2.3 to 3.6 ± 0.6 Miller units). In this model, Fur activates pfeT expression, but the absence of Fur alters metal physiology to reduce the efficiency of PerR repression. Alternatively, these results are consistent with a model in which apo-Fur is a repressor of pfeT, and iron binding leads to dissociation. While this alternative model can account for the modest derepression seen in the fur null mutant, it is inconsistent with the decrease in expression in the perR fur double mutant compared to expression in the perR mutant (Fig. 3). To distinguish between these and other possible models, we next sought to define the role of each of the three protein binding sites in regulation.
Functional analysis of the pfeT promoter region through operator site dissection in vivo.
We made point mutations of each regulator binding site (Fig. 1B, PI*, FII*, and FIII*) and used a pfeT promoter region-lacZ fusion to measure promoter activity in LB and LB amended with iron (Fig. 4A). Consistent with the reported role of PerR as a repressor, mutation of the PerR-binding site (PI*) led to derepression of pfeT (Fig. 4A). As we also observed in the strain lacking PerR (Fig. 3), we still noted an iron-dependent increase in pfeT expression in this strain.
FIG 4.
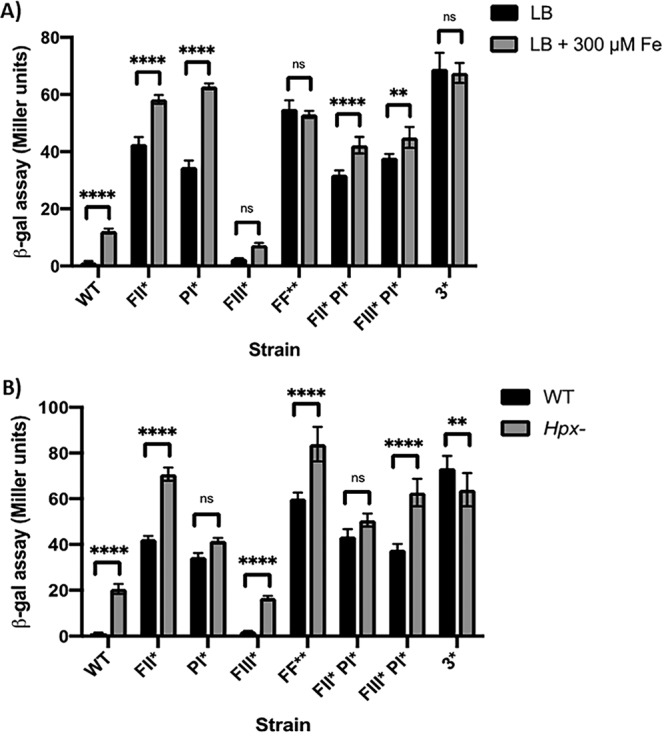
Promoter dissection of pfeT. (A) β-Galactosidase assay to monitor iron induction of pfeT-lacZ in cells grown in LB medium or in LB plus 300 μM Fe. Data represent means ± standard deviations for at least three biological replicates. Significance was calculated using a two-way analysis of variance test with Šidák correction (**, P < 0.01; ****, P < 0.0001; ns, no statistical significance). (B) Comparison of β-galactosidase activities of the promoter constructs shown in panel A measured in the WT and an Hpx− background grown in LB medium. Data represent means ± standard deviations for three or more biological replicates. Significance was calculated using a two-way analysis of variance test with Šidák correction (**, P < 0.01; ****, P < 0.0001; ns, no statistical significance).
Interpretation of the effects of mutations in the two Fur binding sites was not straightforward. For a simple activator protein binding site, one would expect a noninducible phenotype as seen in a fur null mutant (Fig. 3). However, the FII* mutation led to constitutive expression, similar to the result with PI* (Fig. 3 and 4A). We can rationalize this effect by proposing that PerR binds to FII, and this binding is important for formation of a PerR repression complex. In this model (Fig. 1C), binding of PerR is cooperative, and mutation of either FII (FII*) or PI (PI*) leads to derepression.
In both single FII* and FIII* mutant strains, there was still a statistically significant increase in expression in response to iron (∼1.5- and 3.5-fold, respectively) (Fig. 4A). In contrast, a strain with mutations in both Fur binding sites (FF**) no longer responded to iron. This supports the idea that Fur is the physiologically relevant iron sensor, that Fur is an activator of transcription, and that Fur activation can occur with Fur bound at either FII or FIII (Fig. 1C).
In addition to its induction by high iron (Fig. 2), pfeT has previously been shown to be induced by H2O2 (17, 19). We decided to measure the contribution of each regulator binding site in the context of H2O2 induction by measuring pfeT-lacZ expression in a strain lacking the two major hydroperoxidases, KatA and AhpCF (Hpx−). As observed also for comparable Hpx− mutant strains in Escherichia coli (32), the absence of H2O2 detoxification enzymes leads to an increase in endogenous levels of H2O2 sufficient to derepress the cognate stress response (Fig. 4B). In a strain with a mutation in the PerR binding site (PI*) pfeT is derepressed, and there is no further increase in the Hpx− mutant background, consistent with the idea that PerR repression requires PI. In contrast, most strains with mutations in the Fur binding sites (FII* or FIII* or FF**) still display some increase in gene expression when transferred to the Hpx−background unless the PI box is also mutated (Fig. 4, strain 3*). The one exception is the FIII* PI* mutant, which still responds to elevated H2O2 in the absence of a PI site (Fig. 4B). This might reflect sensitivity of Fur bound at FII to H2O2 inactivation or possibly an ability of PerR to bind to the FII region in this strain even in the absence of PI.
Together, the analysis of iron and H2O2 induction of strains bearing operator site mutations illustrates the complementary roles of the PerR and Fur regulatory proteins. When both Fur binding sites are mutated (FII* FIII*), gene expression is elevated, but no iron induction is observed (Fig. 4A), whereas in an Hpx− background there is still a small but significant response to increased H2O2 (Fig. 4B). Conversely, the PI* mutation that disrupts the PerR repression complex leads to a loss of H2O2 responsiveness (Fig. 4B), but the cells retain iron induction (Fig. 4A).
Operator site specificity in vitro.
To test the model for PerR repression and Fur activation that emerged from these gene expression studies (Fig. 1C), we measured the binding of PerR and Fur to the pfeT regulatory region using electrophoretic mobility shift assays (EMSAs). Both regulators were previously found to bind to this region (24), as confirmed here, and we have used our operator site mutations to test the role of each consensus site in complex formation. For these studies, both Fur and PerR were metallated with Mn(II), known from prior studies to activate DNA binding and to allow in vitro binding studies to be conducted aerobically (24, 33).
Fur binds to the WT promoter region (WT PpfeT) with high affinity (dissociation constant [Kd] of ∼20 nM) (Fig. 5A). Fur also bound with high affinity (Kd of ∼20 nM) with the PI*pfeT fragment. Given that the PI site is PerR specific, according to the in vivo data (Fig. 4) and prior results (19, 24), this was not surprising. Fur still bound with high affinity to promoter fragments retaining at least one Fur binding site (FII* PpfeT and FIII* PpfeT), but binding was abolished when both were mutated (FF*). These data suggest that FII and FIII bind Fur independently and with high affinity and that either site can mediate iron-dependent activation (Fig. 4A and 5B). However, when both FII and FIII were inactivated (FF*), Fur no longer bound to the promoter region (Fig. 5B), and iron induction was no longer observed (Fig. 4A).
FIG 5.
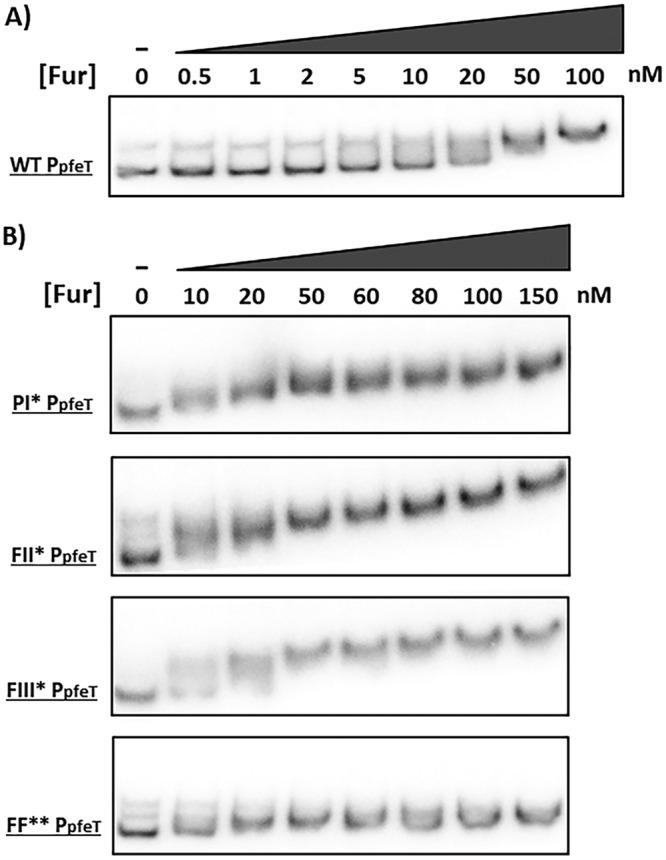
Electrophoretic mobility shift assays (EMSAs) of the pfeT promoter region (268 bp) with Fur. (A) EMSA of the WT pfeT promoter region showing Fur binding. (B) EMSAs of pfeT PI*, FII*, FIII*, and FF** promoter region mutants (Fig. 1B) showing the effect on Fur binding. All EMSAs were performed a total of three times with very similar results, and a representative result is shown.
Next, we tested PerR binding to these same promoter region variants. The WT PpfeT bound PerR with high affinity (Kd of ∼50 nM) (Fig. 6A). As expected, mutating the PI site compromised the ability of PerR to bind to DNA. Moreover, the FII* promoter variant also had a greatly reduced affinity for PerR (Fig. 6). These results suggest that both the PI and FII sites interact with PerR and that there is positive cooperativity in binding. This inference is consistent with prior results from DNase I footprinting that document that PerR binds to an extended region on the pfeT promoter spanning both the PI and FII sites (24). These results support a model in which PerR binding to the PI and FII sites contributes to the formation of a stable repression complex (Fig. 1C).
FIG 6.
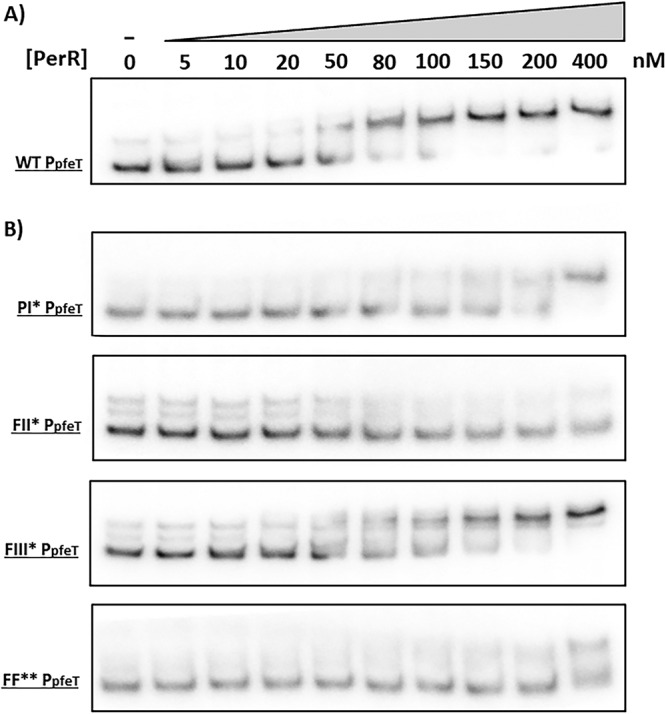
Electrophoretic mobility shift assays (EMSAs) of the pfeT promoter region (268 bp) with PerR. (A) EMSA of the WT pfeT promoter region showing PerR binding. (B) EMSAs of pfeT PI*, FII*, FIII*, and FF** promoter region mutants (Fig. 1B) showing the effect on PerR binding. All EMSAs were performed a total of three times with very similar results, and a representative result is shown.
Collectively, the Fur EMSA data (Fig. 5) and the mutational results (Fig. 4A) suggest that the FII site is promiscuous. That is, although this site displays high similarity to the Fur operator consensus sequence (Fig. 1A), it seems to be able to bind both Fur and PerR in vitro (Fig. 5B and 6B), with a switch in operator occupancy accompanying the iron-dependent induction of pfeT (Fig. 1C).
Fur is a direct activator of pfeT in vitro.
The emerging model for pfeT regulation postulates that Fur can function both to disrupt the PerR repression complex (by competition for FII) and also to directly activate transcription (Fig. 1C). We therefore performed in vitro transcription assays to test this model. In the absence of both regulators, pfeT is transcribed at a basal level (Fig. 7A, lane 1), consistent with the high expression noted in the perR fur null mutant (Fig. 3). Upon addition of PerR, pfeT transcription was almost completely abolished (Fig. 7A, lane 2), consistent with the role of PerR as a repressor. Conversely, when Fur was added, transcription was activated beyond the level obtained with RNA polymerase (RNAP) alone (Fig. 7A, lane 3 versus lane 1), consistent with Fur as an activator of transcription.
FIG 7.
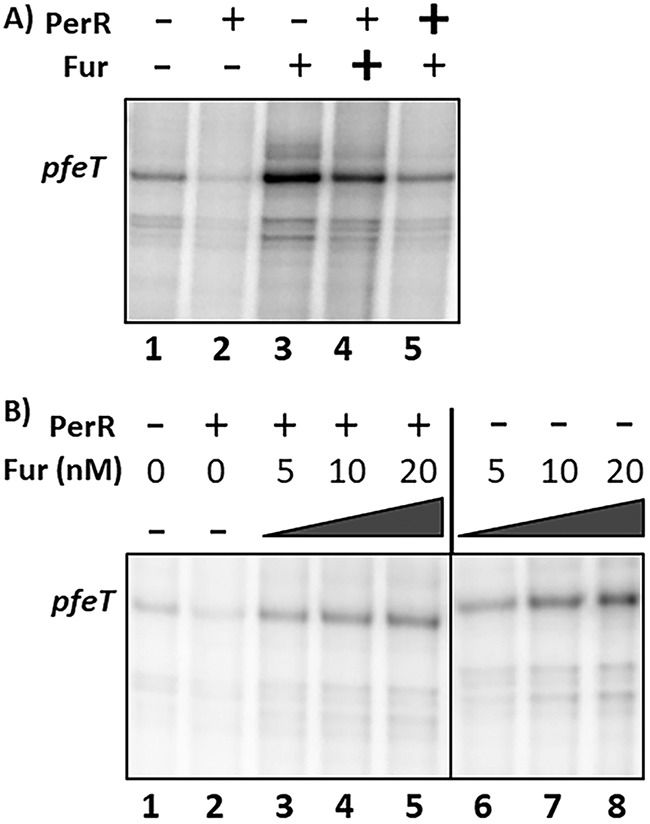
In vitro transcription of pfeT. (A) Polyacrylamide gel electrophoresis (PAGE) was used to visualize radiolabeled RNA (runoff transcripts) from the pfeT promoter region. The pfeT transcript is labeled and is 171 bp long. Manganese was present in the reaction mixture at 10 μM, and RNAP holoenzyme was present at 100 nM. PerR and Fur regulators were both added to a final concentration of 50 nM each. Plus signs indicate presence of a component, minus signs indicate absence, and bold plus signs indicate that this regulator was added 5 min prior to the second regulator, with an additional 5-min equilibration prior to initiation of the reaction. Band quantification values were determined using ImageJ. Values for lanes 1 through 5 normalized to the value for lane 1 (set to 100) are 27, 294, 191, and 110, respectively. (B) PAGE analysis of runoff transcripts illustrating activation by Fur in the presence or absence of PerR. Manganese was present in the reaction mixture at 10 μM, and RNAP holoenzyme was present at 100 nM. PerR regulator was added to a final concentration of 30 nM to each reaction mixture. Fur was added at the indicated concentrations to each reaction mixture. The vertical line distinguishes the two sets of reaction mixtures, one in which PerR was included (after Fur) and one in which PerR was not included. Band quantification values were determined using ImageJ. Values for lanes 1 through 8 are 100, 52, 179, 217, 290, 230, 370, and 439, respectively, and are normalized with respect to values for lane 1.
To test the hypothesis that Fur may act to antagonize the PerR repression complex, we explored the effect of the order of addition of PerR and Fur. When Fur was incubated with the DNA for 5 min prior to the addition of PerR (lane 3), transcript levels were 3-fold higher than those with RNAP alone (lane 1). When PerR was added before addition of Fur (lane 5), transcription was elevated compared to the level of the PerR-repressed state (lane 2) but not as high as that when Fur was added first. Thus, with only 5 min allowed for regulator competition, the system had not reached equilibrium. This is consistent with the expected low off-rate for dissociation of PerR-Mn(II) from FII, which may be a prerequisite for Fur binding at this site to preclude formation of the PerR repression complex.
To further assess the effects of Fur on pfeT activation, we carried out a Fur titration in the presence or absence of PerR (Fig. 7B). As expected, pfeT was expressed at a low level in the absence of both metalloregulators and highly repressed when only PerR was added to the reaction mixture (Fig. 7B, lanes 1 and 2). We confirmed that pfeT expression increases as a function of Fur concentration with and without PerR. Transcript bands were stronger in the Fur-only reactions even at 20 nM (Fig. 7B, lane 8) than in the reaction mixtures containing PerR. This is indicative of the fact that Fur can act as an antirepressor in the presence of PerR as well as a direct activator (Fig. 7A). This is in alignment with our in vivo data wherein we also saw PerR-dependent repression and Fur activation (Fig. 3).
Fur activation of pfeT orthologs.
A homolog of PfeT, the Fur-regulated virulence protein FrvA, has been previously identified in Listeria monocytogenes (9). These two efflux pumps share 54% protein identity (see Fig. S2 in the supplemental material) although FrvA displays a greater affinity for Fe(II), as judged by the amount required for activation of ATPase activity (9). The similarity between these two efflux pumps extends to the architecture of their promoter regions (Fig. 8 and Fig. S1). They both possess a PerR box and multiple Fur binding sites across the promoter region. In fact, frvA also has a predicted Fur box adjacent to a PerR binding site (which itself overlaps the −10 region) and another Fur binding site beyond the transcriptional start site (Fig. 9A and Fig. S1B). Previous studies supported the idea that Fur is a transcriptional activator of FrvA (9, 34) although other studies suggested that it might function as a repressor (10, 35).
FIG 8.

Promoter structure of pfeT orthologs. Schematic showing the different regulatory regions controlling pfeT orthologs in L. monocytogenes (frvA), Streptococcus pyogenes (pmtA), and Mycobacterium tuberculosis (ctpD). Arrows outlined in white indicate genes that are virulence factors.
FIG 9.

Promoter analysis of the Listeria monocytogenes ortholog, frvA. (A) Promoter region of frvA reveals a similar structure to that seen for pfeT. A conserved PerR box (gray) overlaps the −10 region (italics) of the RNAP binding site. Like pfeT, frvA has two Fur operator sites. One is located upstream of the PerR box and is adjacent to the −35 region, and the other is downstream of the transcriptional start site (in black). (B) PAGE of runoff RNA transcripts showing PerR repression and Fur activation of frvA. The frvA transcript is 182 bp long. Manganese was present in the reaction mixture at 10 μM, and RNAP holoenzyme was present at 100 nM. B. subtilis PerR was added to a final concentration of 30 nM to each reaction mixture. B. subtilis Fur was added at the indicated concentrations to each reaction mixture. The vertical line distinguishes the two sets of reaction mixtures, one in which PerR was added (after a 5-min Fur incubation) and one in which PerR was not added. Band quantification values were determined using ImageJ. Values for lanes 1 through 6 are 100, 25, 49, 498, 291, and 500, respectively, and are normalized with respect to the value for lane 1. All lanes were from a single PAGE experiment, with the line indicating the deletion from the image of intervening lanes for clarity.
Here, we used in vitro transcription assays to test the effects of PerR and Fur on transcription of the L. monocytogenes frvA gene. Since the B. subtilis and L. monocytogenes regulators and RNAP are highly similar in sequence, we used purified B. subtilis proteins in this study. In the absence of Fur and PerR, basal levels of frvA transcription were observed (Fig. 9B, lane 1). Upon addition of PerR to the reaction mixture, this basal level of transcription was completely abolished (lane 2). Addition of Fur to the reaction mixture increased transcript levels in a protein concentration-dependent manner (Fig. 9B, lanes 3 to 6), as observed for pfeT (Fig. 7B).
DISCUSSION
Bacterial iron homeostasis has been an active area of research for many years (2). In many environments, iron is a limiting nutrient, and bacteria devote considerable resources to obtaining iron. Given this focus on iron import, a role for iron efflux was initially dismissed as unlikely to be physiologically important (36). PfeT was the first P1B4-type ATPase to be characterized as an Fe(II) efflux pump (3, 4). Homologous proteins that also serve as Fe(II) efflux systems play important roles in pathogenesis (9–12) (Fig. 8). However, the mechanisms by which these transporters are transcriptionally regulated are still poorly understood and seem to vary across bacterial types, as is evident by the differences in their promoter architecture (24, 35, 37, 38).
Bacterial Fur proteins function as Fe(II)-dependent repressors to control genes required during iron depletion (39). Previous work in B. subtilis has mapped the Fur regulon, which includes ∼60 genes that are repressed under iron-replete conditions (40, 41). In addition, Fur indirectly activates the expression of some genes in response to Fe(II) as mediated by the Fur-repressed small RNA, FsrA (42–44). Our work here has shown that Fur also functions as a direct transcription activator of pfeT under high-iron conditions (Fig. 7A) and that this mode of regulation is conserved for the L. monocytogenes ortholog frvA (Fig. 9B). Fur has been previously shown to function as an activator for selected genes in Gram-negative bacteria such as Escherichia coli (45), Salmonella enterica (46), and Neisseria meningitidis (47). Past studies in Gram-positive bacteria such as Staphylococcus aureus (48) and L. monocytogenes (9, 34) have suggested that Fur might also function as a transcriptional activator in these systems but have never shown it directly.
Our new understanding of pfeT and frvA transcription will provide insight into how and when iron efflux is activated during infection. Host-imposed iron limitation, by sequestration and through efflux pumps that deplete the phagocytic vacuole of iron, is countered by the induction of high-affinity bacterial iron uptake systems (49–51). Transient iron overload, due to the expression of high-affinity iron uptake systems and the transition to a comparatively iron-replete environment, may need to be counterbalanced by efflux pumps that serve as a relief valve to help adjust cytosolic iron levels. When this occurs during infection is not well understood, but we have previously suggested that it might be correlated with escape of L. monocytogenes from the phagocytic vacuole into the host cytosol (9).
The precise way in which Fur activates gene transcription remains unclear and will require further attention. Our mutational analysis of the different operator sites of the pfeT promoter region suggests that either FII or FIII can support gene induction (Fig. 4A and 5). Inactivation of both of these sites prevents Fur binding and therefore gene activation. Multiple Fur binding sites are also present in the frvA promoter region (see Fig. S1B in the supplemental material). In B. subtilis, MntR-mediated activation of the genes encoding the manganese efflux pumps MneP and MneS also involves multiple operator sites upstream of the transcriptional start site. In this case, all of these multiple sites appear to be required for activation (25). Operator sites located downstream of the transcriptional start site are typically involved in gene repression. However, in some cases downstream sites may play a positive role in gene expression. For example, the E. coli virulence regulator Rns, as well as related AraC family members, can activate gene expression from downstream sites (52).
Our results here provide an example of operator site promiscuity in which a single site can interact with either PerR or Fur. Previous work has defined the binding specificity of Fur family proteins (Fur, PerR, and Zur) at their cognate operator sites (24, 39). In this prior work, it was shown that introduction of two point mutations into the mrgA PerR box to match the Fur consensus site resulted in a promoter region (mrgAFur) that was bound by both Fur and PerR in vitro. Moreover, high-level expression of an mrgAFur-lacZ fusion was observed in a double fur perR mutant but not in the single mutants (24). The FII site here matches the operator site introduced in the mrgAFur construct and appears to represent the first naturally occurring example of a site recognized by both regulators.
Our work shows that the PerR and Fur regulons are interlinked. Formation of a PerR repression complex involves cooperative binding of PerR to both the PI and FII sites. This extended DNA binding behavior is reminiscent of what has been previously observed for E. coli Fur (53, 54). Activation is mediated by Fur binding to either FII or FIII (Fig. 1C). These results extend our understanding of the regulation of iron efflux genes and thereby set the stage for a new understanding of host-pathogen interactions.
MATERIALS AND METHODS
Bacterial strains, phage, plasmids, and growth conditions.
Bacillus subtilis strains derived from CU1065 (WT) were grown on lysogeny broth (LB) medium (10 g/liter casein digest peptone, 5 g/liter yeast extract, 5 g/liter NaCl) at 37°C with vigorous shaking. Ferrous iron (FeSO4·7H2O) was titrated into the medium as indicated in the figure legends from a freshly made 100 mM iron stock solution dissolved in 0.1 N HCl. Ampicillin (Amp; 100 μg ml−1) was used to select E. coli transformants. Erythromycin (Ery; 1 μg ml−1) and lincomycin (Linc; 25 μg ml−1; for testing macrolide-lincosamide-streptogramin B [MLS] resistance), spectinomycin (Spec; 100 μg ml−1), kanamycin (Kan; 10 μg ml−1), and neomycin (Neo; 10 μg ml−1) were used for the selection of various B. subtilis strains.
Mutant strain construction.
Null mutant strains were obtained from the BKE collection (a B. subtilis 168 gene knockout library) available from the Bacillus Genetic Stock Center (BGSC). Each BKE strain contains an erythromycin resistance cassette inserted into the gene in the B. subtilis 168 genome. Mutations were transformed into CU1065 for this study. The pDR244 plasmid (from BGSC) was used to excise the BKE erythromycin cassette to generate a markerless deletion of the target gene (55). Isolation of B. subtilis chromosomal DNA, transformation, and specialized SPβ transduction were performed as described previously (56). Strains were confirmed by PCR.
Reporter strain construction.
For monitoring gene expression, we generated promoter region-cat-lacZ operon fusions integrated into the SPβ prophage as previously described (57). Promoter regions were cloned into pJPM122, which integrates in single copy into the chromosome of strain ZB307A. This strain (SPβ c2Δ2::Tn917::pBSK10Δ6) carries a plasmid (pPSK10Δ6) with a promoterless lacZ gene inserted into a Tn917 transposon inside a temperature-sensitive derivative of the SPβ prophage, designated SPβ c2Δ2 (57). Integration of pJPM122 derivatives into strain ZB307A generates promoter region-cat-lacZ operon fusions that can be mobilized by specialized transduction after induction of phage by a temperature shift to 50°C for 15 min. The cat-lacZ operon fusions generated in strain ZB307A were moved to different backgrounds by SPβ transduction and selection for MLS and neomycin resistance. All strains were confirmed by PCR. All SPβ prophages containing the reporter fusions were stable under the experimental conditions tested (including high iron and elevated peroxide production in Hpx− cells) as judged by a lack of visible cell lysis and by phage titers. In contrast, heat stress led to cell lysis and SPβ phage induction.
Site-directed mutagenesis of the pfeT promoter.
PCR primers containing the desired changes within the PI, FII, and FIII boxes were used to amplify the pfeT promoter region from CU1065 WT chromosomal DNA. Final fragments were restriction digested with high-fidelity HindIII and BamHI enzymes (NEB) and cloned into the pJPM122 vector (57). Plasmids containing the desired fragments were transformed into competent DH5α E. coli and selected with ampicillin. All constructs were verified by DNA sequencing prior to transfer into strain ZB307A.
β-Galactosidase assays.
Cells containing the pfeT promoter-lacZ fusions were grown in LB amended with different concentrations of FeSO4·7H2O to an optical density at 600 nm (OD600) of ∼0.4. Cells were then harvested, and β-galactosidase assays were performed as described previously (58), except that cells were lysed for 30 min at 37°C with 0.1 mg ml−1 lysozyme instead of chloroform.
EMSAs.
PCR fragments (268 bp) containing the wild-type and mutant promoter regions of pfeT (WT, PI*, FII*, FIII*, and FF**) were purified (PCR purification kit; Qiagen) and labeled with [γ-32P]ATP using T4 polynucleotide kinase. After labeling, a G10 column (NucAwayTM spin columns; Invitrogen) was used to remove the unincorporated [γ-32P]ATP, and the radioactivity of each probe was quantified by a scintillation counter. The γ-32P-labeled promoter fragments were incubated with either Fur or PerR in 1× binding buffer at room temperature for 15 min. The 1× binding buffer used for Fur reactions contained 10 mM Tris-HCl (pH 8.0), 50 mM NaCl, 5% glycerol, 1 mM dithiothreitol (DTT), 50 μg/ml bovine serum albumin (BSA), 10 μM MnCl2, and 2 μg/ml salmon testes DNA. The 1× binding buffer used for PerR binding reactions was as previously described (22). Fur and PerR proteins were purified as previously described (31, 33). Samples were loaded on a 5% polyacrylamide gel and run in 40 mM Tris-acetate buffer (no EDTA; pH 8.0). The gel was dried and exposed to a phosphorimager screen overnight and scanned using a Typhoon FLA 7000 system.
Multiple-round in vitro transcription.
In vitro transcription was performed as previously described (59). Briefly, σA-saturated RNAP holoenzyme was reconstituted by mixing purified RNAP with purified σA (1:5 molar ratio) in transcription buffer (10 mM Tris [pH 8.0], 10 mM MgCl2, 1 mM DTT, 10 mM potassium glutamate, 10 μg ml−1 acetylated BSA) and incubated on ice for 15 min. A promoter DNA fragment (10 nM) and 50 nM PerR and/or Fur in transcription buffer were mixed and incubated for 10 min at room temperature. Both PerR and Fur were incubated with Mn(II) on ice for 15 min to ensure full metallation before addition to the reaction mixture. For the transcription reaction, an ∼400-bp PCR product was amplified from CU1065 B. subtilis chromosomal DNA to give a DNA fragment that yielded a 171-nucleotide (nt) transcript. For the L. monocytogenes ortholog, a 478-bp PCR fragment was amplified from WT L. monocytogenes chromosomal DNA to give a DNA fragment that yielded a 182-nt transcript. Next, RNAP was added, and the reaction mixtures were incubated for 10 min at 37°C. Transcription was initiated by adding 0.5 mM each GTP, CTP, and ATP, 0.1 mM UTP, and 2.5 mCi of [α-32P]UTP. After a 10-min incubation, the reaction products were ethanol precipitated in the presence of 0.3 M sodium acetate (pH 5.2) and 1 ml of GlycoBlue (15 mg ml−1; Ambion). The RNA pellet was washed with 70% cold ethanol, dried, and dissolved in formamide loading dye and separated on a 6% denaturing polyacrylamide sequencing gel (UreaGEL). A single-stranded DNA ladder was made by generating [γ-32P]ATP-labeled PCR fragments of 100 bp, 200 bp, and 300 bp in size from a pET17b plasmid. The fragments were pooled, dissolved in formamide loading dye, and boiled for 5 min at 90°C. The gel was dried and exposed to a phosphorimager screen overnight and scanned by a Typhoon FLA 7000 system. These experiments were repeated at least twice. Band quantification and normalization were performed using ImageJ.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by an NIH MIRA grant (award number R35GM122461 to J.D.H.).
We thank Xiaojuan Huang for purification of RNAP, Ahmed Gaballa for technical assistance, and the entire Helmann Lab for their feedback and support.
This work is dedicated in loving memory of A.P.-B.’s grandmother and scientist, Nelly Lafuente Indo. Gracias por tus enseñanzas que siguen vivas en tu memoria.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Andrews SC, Robinson AK, Rodríguez-Quiñones F. 2003. Bacterial iron homeostasis. FEMS Microbiol Rev 27:215–237. doi: 10.1016/S0168-6445(03)00055-X. [DOI] [PubMed] [Google Scholar]
- 2.Cornelis P, Andrews SC (ed). 2010. Iron uptake and homeostasis in microorganisms. Caister Academic Press, Norfolk, United Kingdom. [Google Scholar]
- 3.Pi H, Helmann JD. 2017. Ferrous iron efflux systems in bacteria. Metallomics 9:840–851. doi: 10.1039/c7mt00112f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guan G, Pinochet-Barros A, Gaballa A, Patel SJ, Argüello JM, Helmann JD. 2015. PfeT, a P1B4 -type ATPase, effluxes ferrous iron and protects Bacillus subtilis against iron intoxication. Mol Microbiol 98:787–803. doi: 10.1111/mmi.13158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bhubhanil S, Chamsing J, Sittipo P, Chaoprasid P, Sukchawalit R, Mongkolsuk S. 2014. Roles of Agrobacterium tumefaciens membrane-bound ferritin (MbfA) in iron transport and resistance to iron under acidic conditions. Microbiology 160:863–871. doi: 10.1099/mic.0.076802-0. [DOI] [PubMed] [Google Scholar]
- 6.Sankari S, O'Brian MR. 2014. A bacterial iron exporter for maintenance of iron homeostasis. J Biol Chem 289:16498–16507. doi: 10.1074/jbc.M114.571562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Frawley ER, Crouch ML, Bingham-Ramos LK, Robbins HF, Wang W, Wright GD, Fang FC. 2013. Iron and citrate export by a major facilitator superfamily pump regulates metabolism and stress resistance in Salmonella Typhimurium. Proc Natl Acad Sci U S A 110:12054–12059. doi: 10.1073/pnas.1218274110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Frawley ER, Fang FC. 2014. The ins and outs of bacterial iron metabolism. Mol Microbiol 93:609–616. doi: 10.1111/mmi.12709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pi H, Patel SJ, Argüello JM, Helmann JD. 2016. The Listeria monocytogenes Fur-regulated virulence protein FrvA is an Fe(II) efflux P1B4-type ATPase. Mol Microbiol 100:1066–1079. doi: 10.1111/mmi.13368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McLaughlin HP, Xiao Q, Rea RB, Pi H, Casey PG, Darby T, Charbit A, Sleator RD, Joyce SA, Cowart RE, Hill C, Klebba PE, Gahan CG. 2012. A putative P-type ATPase required for virulence and resistance to haem toxicity in Listeria monocytogenes. PLoS One 7:e30928. doi: 10.1371/journal.pone.0030928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Patel SJ, Lewis BE, Long JE, Nambi S, Sassetti CM, Stemmler TL, Arguello JM. 2016. Fine-tuning of substrate affinity leads to alternative roles of Mycobacterium tuberculosis Fe2+-ATPases. J Biol Chem 291:11529–11539. doi: 10.1074/jbc.M116.718239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.VanderWal AR, Makthal N, Pinochet-Barros A, Helmann JD, Olsen RJ, Kumaraswami M. 2017. Iron efflux by PmtA is critical for oxidative stress resistance and contributes significantly to group A Streptococcus virulence. Infect Immun 85:e00091-17. doi: 10.1128/IAI.00091-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Silva-Gomes S, Vale-Costa S, Appelberg R, Gomes MS. 2013. Iron in intracellular infection: to provide or to deprive? Front Cell Infect Microbiol 3:96. doi: 10.3389/fcimb.2013.00096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Imlay JA. 2013. The molecular mechanisms and physiological consequences of oxidative stress: lessons from a model bacterium. Nat Rev Microbiol 11:443–454. doi: 10.1038/nrmicro3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen L, Keramati L, Helmann JD. 1995. Coordinate regulation of Bacillus subtilis peroxide stress genes by hydrogen peroxide and metal ions. Proc Natl Acad Sci U S A 92:8190–8194. doi: 10.1073/pnas.92.18.8190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Faulkner MJ, Helmann JD. 2011. Peroxide stress elicits adaptive changes in bacterial metal ion homeostasis. Antioxid Redox Signal 15:175–189. doi: 10.1089/ars.2010.3682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Helmann JD, Wu MF, Gaballa A, Kobel PA, Morshedi MM, Fawcett P, Paddon C. 2003. The global transcriptional response of Bacillus subtilis to peroxide stress is coordinated by three transcription factors. J Bacteriol 185:243–253. doi: 10.1128/jb.185.1.243-253.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen L, Helmann JD. 1995. Bacillus subtilis MrgA is a Dps(PexB) homologue: evidence for metalloregulation of an oxidative-stress gene. Mol Microbiol 18:295–300. doi: 10.1111/j.1365-2958.1995.mmi_18020295.x. [DOI] [PubMed] [Google Scholar]
- 19.Gaballa A, Helmann JD. 2002. A peroxide-induced zinc uptake system plays an important role in protection against oxidative stress in Bacillus subtilis. Mol Microbiol 45:997–1005. doi: 10.1046/j.1365-2958.2002.03068.x. [DOI] [PubMed] [Google Scholar]
- 20.Helmann JD. 2014. Specificity of metal sensing: iron and manganese homeostasis in Bacillus subtilis. J Biol Chem 289:28112–28120. doi: 10.1074/jbc.R114.587071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Herbig AF, Helmann JD. 2001. Roles of metal ions and hydrogen peroxide in modulating the interaction of the Bacillus subtilis PerR peroxide regulon repressor with operator DNA. Mol Microbiol 41:849–859. doi: 10.1046/j.1365-2958.2001.02543.x. [DOI] [PubMed] [Google Scholar]
- 22.Lee JW, Helmann JD. 2006. The PerR transcription factor senses H2O2 by metal-catalysed histidine oxidation. Nature 440:363–367. doi: 10.1038/nature04537. [DOI] [PubMed] [Google Scholar]
- 23.Jacquamet L, Traore DA, Ferrer JL, Proux O, Testemale D, Hazemann JL, Nazarenko E, El Ghazouani A, Caux-Thang C, Duarte V, Latour JM. 2009. Structural characterization of the active form of PerR: insights into the metal-induced activation of PerR and Fur proteins for DNA binding. Mol Microbiol 73:20–31. doi: 10.1111/j.1365-2958.2009.06753.x. [DOI] [PubMed] [Google Scholar]
- 24.Fuangthong M, Helmann JD. 2003. Recognition of DNA by three ferric uptake regulator (Fur) homologs in Bacillus subtilis. J Bacteriol 185:6348–6357. doi: 10.1128/jb.185.21.6348-6357.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang X, Shin JH, Pinochet-Barros A, Su TT, Helmann JD. 2017. Bacillus subtilis MntR coordinates the transcriptional regulation of manganese uptake and efflux systems. Mol Microbiol 103:253–268. doi: 10.1111/mmi.13554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ma Z, Chandrangsu P, Helmann TC, Romsang A, Gaballa A, Helmann JD. 2014. Bacillithiol is a major buffer of the labile zinc pool in Bacillus subtilis. Mol Microbiol 94:756–770. doi: 10.1111/mmi.12794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moore CM, Gaballa A, Hui M, Ye RW, Helmann JD. 2005. Genetic and physiological responses of Bacillus subtilis to metal ion stress. Mol Microbiol 57:27–40. doi: 10.1111/j.1365-2958.2005.04642.x. [DOI] [PubMed] [Google Scholar]
- 28.Faulkner MJ, Ma Z, Fuangthong M, Helmann JD. 2012. Derepression of the Bacillus subtilis PerR peroxide stress response leads to iron deficiency. J Bacteriol 194:1226–1235. doi: 10.1128/JB.06566-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fuangthong M, Herbig AF, Bsat N, Helmann JD. 2002. Regulation of the Bacillus subtilis fur and perR genes by PerR: not all members of the PerR regulon are peroxide inducible. J Bacteriol 184:3276–3286. doi: 10.1128/jb.184.12.3276-3286.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ji CJ, Kim JH, Won YB, Lee YE, Choi TW, Ju SY, Youn H, Helmann JD, Lee JW. 2015. Staphylococcus aureus PerR is a hypersensitive hydrogen peroxide sensor using iron-mediated histidine oxidation. J Biol Chem 290:20374–20386. doi: 10.1074/jbc.M115.664961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ma Z, Lee JW, Helmann JD. 2011. Identification of altered function alleles that affect Bacillus subtilis PerR metal ion selectivity. Nucleic Acids Res 39:5036–5044. doi: 10.1093/nar/gkr095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Park S, You X, Imlay JA. 2005. Substantial DNA damage from submicromolar intracellular hydrogen peroxide detected in Hpx− mutants of Escherichia coli. Proc Natl Acad Sci U S A 102:9317–9322. doi: 10.1073/pnas.0502051102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ma Z, Faulkner MJ, Helmann JD. 2012. Origins of specificity and cross-talk in metal ion sensing by Bacillus subtilis Fur. Mol Microbiol 86:1144–1155. doi: 10.1111/mmi.12049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ledala N, Sengupta M, Muthaiyan A, Wilkinson BJ, Jayaswal RK. 2010. Transcriptomic response of Listeria monocytogenes to iron limitation and Fur mutation. Appl Environ Microbiol 76:406–416. doi: 10.1128/AEM.01389-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rea R, Hill C, Gahan CG. 2005. Listeria monocytogenes PerR mutants display a small-colony phenotype, increased sensitivity to hydrogen peroxide, and significantly reduced murine virulence. Appl Environ Microbiol 71:8314–8322. doi: 10.1128/AEM.71.12.8314-8322.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Neilands JB. 1991. A brief history of iron metabolism. Biol Met 4:1–6. doi: 10.1007/bf01135550. [DOI] [PubMed] [Google Scholar]
- 37.Brenot A, King KY, Caparon MG. 2005. The PerR regulon in peroxide resistance and virulence of Streptococcus pyogenes. Mol Microbiol 55:221–234. doi: 10.1111/j.1365-2958.2004.04370.x. [DOI] [PubMed] [Google Scholar]
- 38.Raimunda D, Long JE, Padilla-Benavides T, Sassetti CM, Argüello JM. 2014. Differential roles for the Co2+/Ni2+ transporting ATPases, CtpD and CtpJ, in Mycobacterium tuberculosis virulence. Mol Microbiol 91:185–197. doi: 10.1111/mmi.12454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee JW, Helmann JD. 2007. Functional specialization within the Fur family of metalloregulators. Biometals 20:485–499. doi: 10.1007/s10534-006-9070-7. [DOI] [PubMed] [Google Scholar]
- 40.Baichoo N, Wang T, Ye R, Helmann JD. 2002. Global analysis of the Bacillus subtilis Fur regulon and the iron starvation stimulon. Mol Microbiol 45:1613–1629. doi: 10.1046/j.1365-2958.2002.03113.x. [DOI] [PubMed] [Google Scholar]
- 41.Pi H, Helmann JD. 2018. Genome-wide characterization of the Fur regulatory network reveals a link between catechol degradation and bacillibactin metabolism in Bacillus subtilis. mBio 9:e01451-18. doi: 10.1128/mBio.01451-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gaballa A, Antelmann H, Aguilar C, Khakh SK, Song KB, Smaldone GT, Helmann JD. 2008. The Bacillus subtilis iron-sparing response is mediated by a Fur-regulated small RNA and three small, basic proteins. Proc Natl Acad Sci U S A 105:11927–11932. doi: 10.1073/pnas.0711752105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Smaldone GT, Antelmann H, Gaballa A, Helmann JD. 2012. The FsrA sRNA and FbpB protein mediate the iron-dependent induction of the Bacillus subtilis lutABC iron-sulfur-containing oxidases. J Bacteriol 194:2586–2593. doi: 10.1128/JB.05567-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Smaldone GT, Revelles O, Gaballa A, Sauer U, Antelmann H, Helmann JD. 2012. A global investigation of the Bacillus subtilis iron-sparing response identifies major changes in metabolism. J Bacteriol 194:2594–2605. doi: 10.1128/JB.05990-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Seo SW, Kim D, Latif H, O'Brien EJ, Szubin R, Palsson BO. 2014. Deciphering Fur transcriptional regulatory network highlights its complex role beyond iron metabolism in Escherichia coli. Nat Commun 5:4910. doi: 10.1038/ncomms5910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Teixidó L, Carrasco B, Alonso JC, Barbé J, Campoy S. 2011. Fur activates the expression of Salmonella enterica pathogenicity island 1 by directly interacting with the hilD operator in vivo and in vitro. PLoS One 6:e19711. doi: 10.1371/journal.pone.0019711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Delany I, Rappuoli R, Scarlato V. 2004. Fur functions as an activator and as a repressor of putative virulence genes in Neisseria meningitidis. Mol Microbiol 52:1081–1090. doi: 10.1111/j.1365-2958.2004.04030.x. [DOI] [PubMed] [Google Scholar]
- 48.Horsburgh MJ, Ingham E, Foster SJ. 2001. In Staphylococcus aureus, Fur is an interactive regulator with PerR, contributes to virulence, and is necessary for oxidative stress resistance through positive regulation of catalase and iron homeostasis. J Bacteriol 183:468–475. doi: 10.1128/JB.183.2.468-475.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gunshin H, Mackenzie B, Berger UV, Gunshin Y, Romero MF, Boron WF, Nussberger S, Gollan JL, Hediger MA. 1997. Cloning and characterization of a mammalian proton-coupled metal-ion transporter. Nature 388:482–488. doi: 10.1038/41343. [DOI] [PubMed] [Google Scholar]
- 50.Knutson MD, Oukka M, Koss LM, Aydemir F, Wessling-Resnick M. 2005. Iron release from macrophages after erythrophagocytosis is up-regulated by ferroportin 1 overexpression and down-regulated by hepcidin. Proc Natl Acad Sci U S A 102:1324–1328. doi: 10.1073/pnas.0409409102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Palmer LD, Skaar EP. 2016. Transition metals and virulence in bacteria. Annu Rev Genet 50:67–91. doi: 10.1146/annurev-genet-120215-035146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Munson GP, Holcomb LG, Scott JR. 2001. Novel group of virulence activators within the AraC family that are not restricted to upstream binding sites. Infect Immun 69:186–193. doi: 10.1128/IAI.69.1.186-193.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Escolar L, Pérez-Martín J, de Lorenzo V. 1998. Binding of the Fur (ferric uptake regulator) repressor of Escherichia coli to arrays of the GATAAT sequence. J Mol Biol 283:537–547. doi: 10.1006/jmbi.1998.2119. [DOI] [PubMed] [Google Scholar]
- 54.Escolar L, Pérez-Martín J, de Lorenzo V. 1999. Opening the iron box: transcriptional metalloregulation by the Fur protein. J Bacteriol 181:6223–6229. doi: 10.1128/JB.181.20.6223-6229.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Koo BM, Kritikos G, Farelli JD, Todor H, Tong K, Kimsey H, Wapinski I, Galardini M, Cabal A, Peters JM, Hachmann AB, Rudner DZ, Allen KN, Typas A, Gross CA. 2017. Construction and analysis of two genome-scale deletion libraries for Bacillus subtilis. Cell Syst 4:291–305.e7. doi: 10.1016/j.cels.2016.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Harwood CR, Cutting SM. 1990. Molecular biological methods for Bacillus. Wiley, Chichester, United Kingdom. [Google Scholar]
- 57.Slack FJ, Mueller JP, Sonenshein AL. 1993. Mutations that relieve nutritional repression of the Bacillus subtilis dipeptide permease operon. J Bacteriol 175:4605–4614. doi: 10.1128/jb.175.15.4605-4614.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Miller JH. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 59.Gaballa A, MacLellan S, Helmann JD. 2012. Transcription activation by the siderophore sensor Btr is mediated by ligand-dependent stimulation of promoter clearance. Nucleic Acids Res 40:3585–3595. doi: 10.1093/nar/gkr1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.