

Abstract
Desmoplastic small round-cell tumour is a very rare neoplasm, which usually arises from the abdominal or pelvic peritoneum of adolescents and young adults. Early diagnosis is difficult, because most tumours present with non-specific gastrointestinal symptoms after a long asymptomatic period. It is generally a very aggressive tumour, which grows rapidly with poor prognosis and an overall five-year survival rate of 15% despite multimodal treatment. Despite multiple treatment strategies, the management of desmoplastic small round-cell tumour still remains a clinical challenge and no consensus about a therapeutic protocol has been established. A 35-year-old man presented with mild abdominal pain, constipation and weight gain, and was eventually diagnosed with desmoplastic small round-cell tumour, which was shown to be limited to the abdomen. After incomplete debulking surgery, radiotherapy and chemotherapy, he developed multiple metastatic nodular foci in chest and the pleura and, unfortunately, he died due to disease progression.
Keywords: Desmoplastic small round-cell tumour, Abdominal peritoneum, Rare tumour, Prognosis, Adolescents, Young adults
Background
Desmoplastic small round-cell tumour (DSRCT) is a rare abdominal tumour with only, approximately 450 cases described in the literature since its first description in 1989 by Gerald et al.1 DSRCT typically affects adolescents and young adults, mostly 18–25 years old, with a fourfold male predominance.2 Most abdominal DSRCTs present with notoriously nonspecific symptoms such as abdominal distention, constipation and nausea. Patients are thus diagnosed at an advanced stage and they have a poor prognosis despite multimodal treatment approach.3 DSRCT is a sarcoma, belonging to the family of small round blue cell tumours of the paediatric population.4 It mainly originates from the abdominal and/or pelvic peritoneum with widespread intra-abdominal involvement with surface masses and nodules, and more rarely with invasion of extra peritoneal sites.5 The main metastatic site beyond the peritoneum is the lymphoid tissue.6 DSRCTs are characterised by a chromosomal translocation t(11:22) (p 13; q 12) that involves the EWSR1 and WT1 genes.7
Case history
A 35-year-old man presented with a three-month history of flatulence, mild abdominal pain, constipation and weight gain. Clinical examination showed ascites and laboratory tests revealed CA125 level of 1.786 u/ml. Computed tomography (CT) showed the presence of diffuse intraperitoneal implantations (the largest was 13.5 × 8cm) and ascitic fluid (Fig 1). Staging workup did not show distant metastases. A biopsy of the largest lesion revealed a DSRCT.
Figure 1.
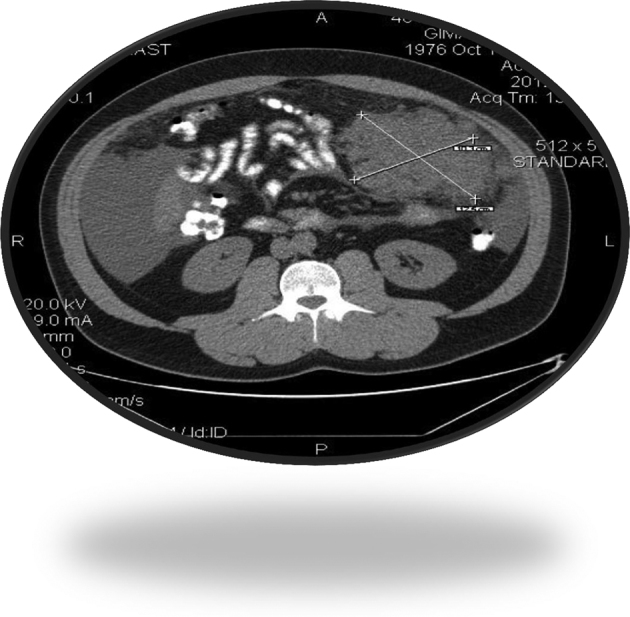
Computed tomography of the abdomen.
The patient underwent surgical removal of the tumour (not complete) with simultaneous implantation of radioactive implants in places where exclusion of the residual tumour was impossible, and administration of intraoperative hyperthermic chemotherapy using mitomycin. The resected tumour appeared macroscopically as a solid, large multilobulated white-grey mass on cross-section, distorted by cystic change and areas of necrosis (Fig 2). Histological examination of the tumour showed it to be composed of sharply demarcated nests of varying size with small round or oval cells embedded in a hypervascular desmoplastic stroma. The neoplastic cells were undifferentiated, had small hyperchromatic nuclei with inconspicuous nucleoli, scant amounts of eosinophilic cytoplasm and high mitotic index Ki-67 (Figures 3–4). Immunohistochemical analysis showed tumour cells positive for cytokeratins, WT-1, desmin and neuron-specific enolase (Figures 5–8).
Figure 2.
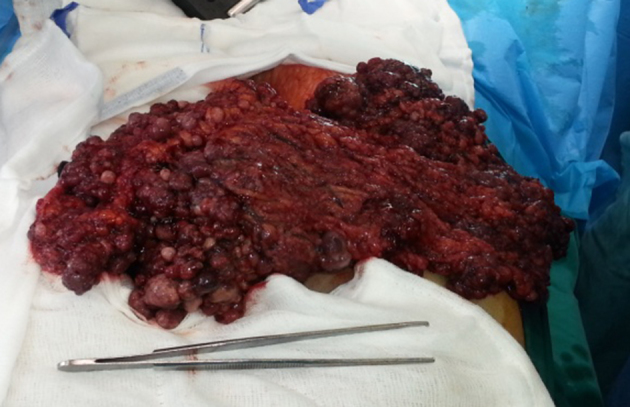
Macroscopic appearance of the desmoplastic small round cell tumour.
Figure 3.
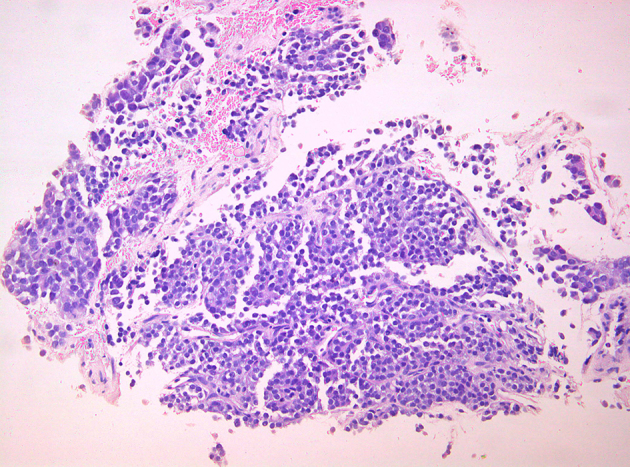
Nests and anastomosing cords of uniform appearance of small blue cells within desmoplastic stroma. Scant eosinophilic cytoplasm with prominent cell borders (H-Ex200).
Figure 4.
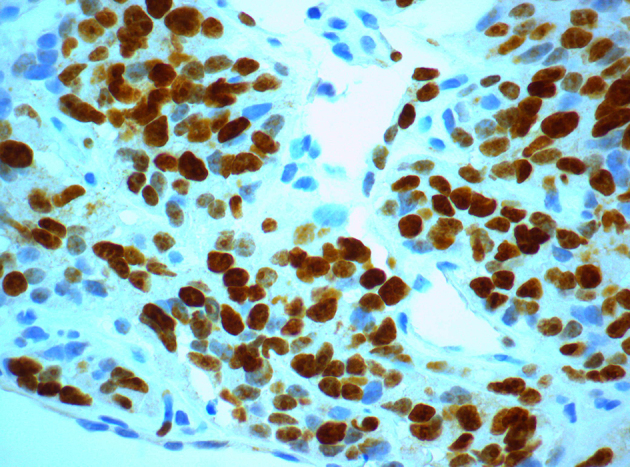
High mitotic index Ki-67 among neoplastic cells (Ki-67x600).
Figure 5.
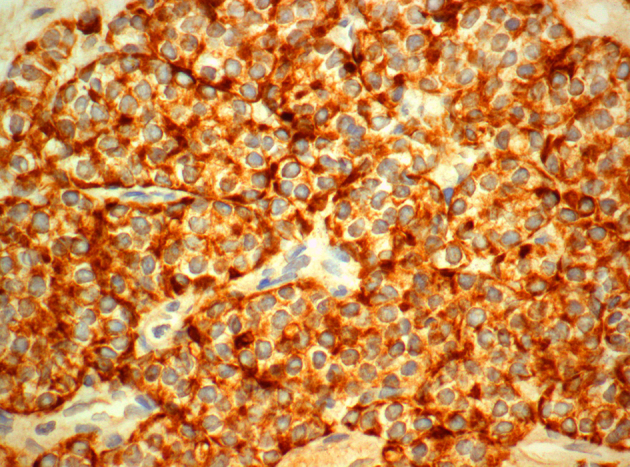
Strong immunopositivity of neoplastic cells for pankeratin (AE1/AE3x600).
Figure 6.
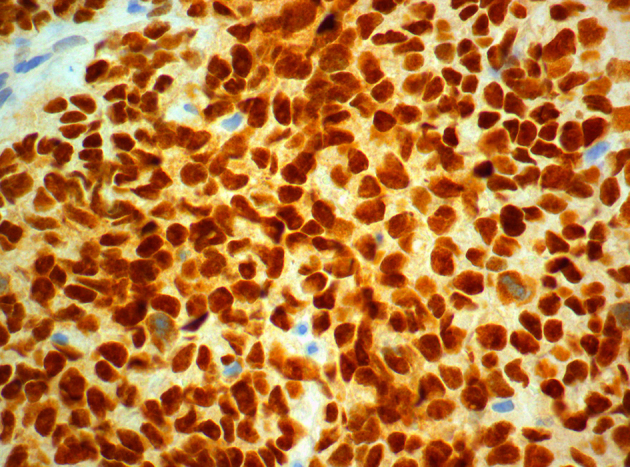
Strong, diffuse, nuclear positivity for WT-1 (WT-1x600).
Figure 7.
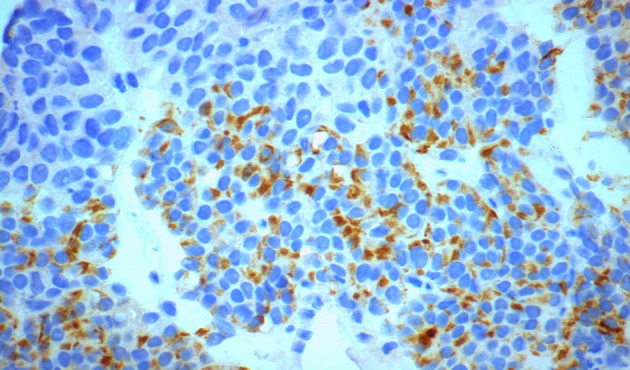
Positive cytoplasmic staining for desmin (desminx600).
Figure 8.
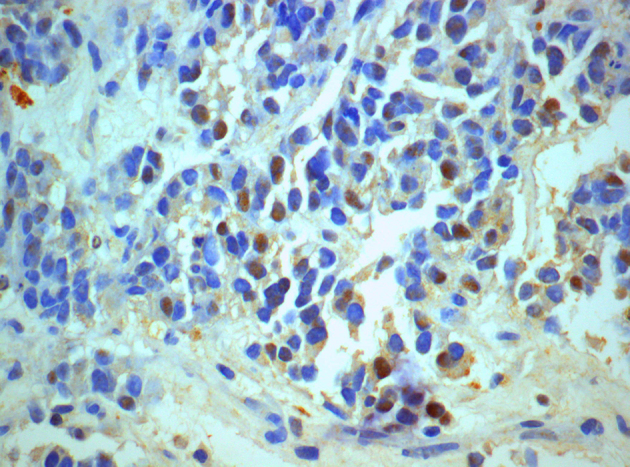
Many cells show positive cytoplasmic staining for neuron specific enolase NSE (NSEx600).
Subsequent CT revealed multiple nodular foci in the lung, probable pleural metastasis as well as pleural and ascitic fluid. The patient received a chemotherapy regimen with cyclophosphamide, adriamycin, vincristine, ifosfamide and etoposide. He developed pancytopenia, febrile neutropenia, left subclavian thrombosis and mucositis with dysphagia, which were successfully treated. He continued the chemotherapy schedule but, unfortunately, he died five months later from disease progression.
Discussion
It is speculated that the DSRCT probably arises from mesothelial cells explaining its widespread abdominal serosal involvement, mainly peritoneum as in this case, and occasionally in the pleura, lung, tunica vaginalis, sinus cavities and the posterior cranial fossa.8 It is also characterized by the presence of the specific reciprocal translocation t(11;22)(p13;q12), which is observed in more than 90% of all patients, regardless of the primary site.9 The identification of a unique cytogenetic abnormality t(11;22)(p13; q12) in this tumour has helped to establish DSRCT as a distinct clinicopathologic entity. The breakpoints involve the EWS gene on 22q12 and the WT1 on 11p13. WT1 is a tumour suppressor gene that encodes a transcription factor, which normally represses promoters that control expression of growth factors such as platelet-derived growth factor. This fusion product causes a loss of suppressing function of WT1 and an uncontrolled regulation of many growth factors from the EWS gene.9 The fusion transcript can be detected by RT-PCR or fluorescence in-situ hybridisation (FISH) using frozen or fixed tissue. Most commonly, this fusion involves exon 7 of EWS and exon 8 of WT1; rare variant fusions have been described.
The tumour is characterised by a polyphenotypic profile with expression of epithelial, mesenchymal, and neural markers. Virtually all tumours stain for epithelial markers, including cytokeratins and epithelial membrane antigen. Occasionally, immunostains for cytokeratin reveal a dot-like pattern of cytoplasmic immunoreactivity. Stains for cytokeratin (CK) 20 (positive in Merkel cell carcinoma) and CK5/6 (positive in malignant mesothelioma) are typically negative in DSRCT, although epithelial markers including MOC-31 and Ber-Ep4 are commonly expressed.2,7
Virtually all DSRCTs stain for vimentin, but perhaps the most useful diagnostic marker is desmin. Up to 90% of cases stain for this antigen, typically with a perinuclear dot-like pattern, a unique pattern of desmin immunoreactivity peculiar to DSRCT. Although often taken as evidence of myogenic differentiation, immunostains for nuclear myogenic regulatory proteins including MyoD1 and myogenin are negative. Rare lesions express muscle-specific or smooth muscle actin.2,9
A variety of neural antigens have also been detected in DSRCT, most commonly neuron-specific enolase and Leu-7. Finally, CD99 and NB-84 are markers that have been used in the differential diagnosis of small round cell neoplasms.7
The diagnosis is often delayed because most DSRCTs are found in the abdominal cavity and, consequently the majority of patients are asymptomatic for a significant period of time. Even when the first symptoms such as discomfort or distention, constipation or bowel obstruction, nausea or emesis occur, they are nonspecific.10 Our patient had only undefined abdominal symptoms at presentation, but he had also ascites, which occasionally can be the first clinical presentation in some patients, reflecting the large tumour burden. If present in extraperitoneal sites, DSRCT can cause specific symptoms, mostly due to expansion of and compression by the tumour. Hepatic parenchyma is the most common site of DSRCT extraperitoneal involvement, followed by lung, bone, splenic parenchyma, pleura and soft tissue.11 Weight loss may occur when there is tumour dissemination through lymphogenous and haematological spread.10 In our case, the patient gained weight due to ascites. Distant metastatic foci, mostly to the pleura and lungs, can be found at the time of diagnosis in 40% of cases or they may appear later, as in this patient.
Despite the absence of typical clinical characteristics, imaging methods can contribute to diagnosis of DSRCT. Bulky peritoneal soft tissue masses without an apparent primary organ origin, indicate possible DSRCT (12). Two CT subtype patterns are noted, and the most common imaging findings are extensively disseminated masses in the peritoneal cavity and/or mesentery with low attenuation and a modest uniform enhancement on contrast-enhanced CT (subtype 1, 9/12; 75%), as in our patient. In subtype 2 (3/12; 25%), the tumour is solitary with a bulky soft-tissue mass, localised in the retroperitoneum or retrovesical space. It manifests with heterogeneous enhancement, usually suggestive of central haemorrhage or necrosis.13 18F2-fluoro-2-deoxyglucose positron emission tomography combined with CT has been well used as a useful imaging modality for identifying primary tumours of unknown origin and metastases, revealing at the same time the metabolic activity of tumours.14
The differential diagnosis of DSRCT encompasses a wide range of neoplastic and inflammatory conditions, especially those presenting as multiple solid peritoneal masses. The majority of other small round-cell tumours such as metastatic neuroblastoma, Merkel cell carcinoma, malignant mesothelioma, Ewing’s sarcoma and lymphoma should be considered. Neoplasms including sarcomatoid carcinoma, spindle-cell sarcomas of various types, metastatic adenocarcinoma and malignant extra renal rhabdoid tumour may occasionally be entertained as diagnostic considerations. The presence of perinuclear dot-like immunostaining with desmin strongly suggests DSRCT.15 Desmoid fibromatosis, peritoneal tuberculosis, malignant peritoneal mesothelioma, fibrosing mesenteritis, tuberculosis, amyloidosis and Castleman disease are disorders whose infiltrative and invasive manifestations overlap with the appearance of DSRCT.1 The immunohistochemical expression of epithelial, mesenchymal and neural antigens, particularly the dot-like pattern of desmin staining, are useful for arriving at a diagnosis. In questionable cases, reverse transcription polymerase chain reaction or FISH analysis for evidence of a EWS–WT1 fusion is indicated.
Despite multiple therapy strategies, DSRCT treatment remains challenging, with no established therapeutic protocol at present.16 There is no level I evidence from clinical trials to support particular management strategies. Complete surgical excision may improve survival rates, mainly for non-metastatic DSRCTs. However, total resection is often impossible when disease has progressed. Debulking or cytoreductive type of surgery remains the main approach for DSRCT management, to offer a symptomatic relief, and it is performed before chemotherapy.6 Hyperthermic intraperitoneal chemotherapy (HIPEC) has been used in some institutions, as in our patient, where HIPEC with mitomycin, was combined with debulking surgery.17
Aggressive surgical resection combined with multi-agent adjuvant chemotherapy can ameliorate symptoms and may improve the management of advanced metastatic DSRCTs. Many chemotherapy combinations have been used in treating DSRCT, but most of them appeared to be partially effective.18 Radiotherapy is only recommended for salvage therapy. For patients with advanced disease palliative treatment is indicated. New promising approaches are emerging according the literature. Immunotherapy, bone marrow ablation, dose-intensive chemotherapy with autologous peripheral blood stem cell support, may have potential benefit for DSRCTs.19
To sum up, this is a characteristic case of desmoplastic small-cell carcinoma typically presented in the abdominal cavity of a young man as numerous abdominal peritoneal tumours with unidentifiable primary site. The histological appearance of this case is in complete accordance with the literature. The tumour cells are always small, having uniform round hyperchromatic nuclei, inconspicuous nucleoli and scanty to modest cytoplasm.20 The cells grow in nests and sheets separated by sclerotic fibrous stroma, showing a polyphenotypic differentiation, typically a mixture of epithelial, mesenchymal and neural features, confirmed with stain positive for desmin, cytokeratin, and S100, just as in our patient.20,21 High mitotic index Ki-67 is another key feature, but not so frequently reported in the literature. The unique feature in our patient was the remarkably high levels of CA125, which were more than twice as high as the maximum levels previously observed. Namely, our patient had CA125 levels of 1.786 u/ml on diagnosis, while expected levels are 22–735 u/ml.22
Conclusion
DSRCT is a rare type of neoplasm that is difficult to diagnose and manage. Establishing clinical management guidelines for this disease is very difficult. It is still considered a very aggressive neoplasm, with poor prognosis despite multimodal treatment.
References
- 1.Honore C, Amroun K, Vilcot L. et al. Abdominal desmoplastic small round cell tumor: multimodal treatment combining chemotherapy, surgery, and radiotherapy is the best option. Ann Surg Oncol 2015; : 1,073–1,079. [DOI] [PubMed] [Google Scholar]
- 2.Gerald WL, Ladanyi M, De Alava E. et al. Clinical, pathologic, and molecular spectrum of tumors associated with t(11;22)(p13;q12): desmoplastic small round-cell tumor and its variants. J Clin Oncol 1998; : 3,028–3,036. [DOI] [PubMed] [Google Scholar]
- 3.Tang Y, Song H, Bao Y, Zhi Y. Multimodal treatment of abdominal and pelvic desmoplastic smallround cell tumor with relative good prognosis Int J Surg 2015; : 1–6. [DOI] [PubMed] [Google Scholar]
- 4.Dorsey BV, Benjamin LE, Rauscher F. et al. Intra-abdominal desmoplastic small round cell tumour: expansion of the pathologic profile. Mod Pathol 1996; : 703–709. [PubMed] [Google Scholar]
- 5.Gerald WL, Miller HK, Battifora H. et al. Intra-abdominal desmoplastic small round-cell tumor: report of 19 cases of a distinctive type of high-grade polyphenotypic malignancy affecting young individuals, Am J Surg Pathol 1991; : 499–513. [PubMed] [Google Scholar]
- 6.Chan AS, MacNeill S, Thorner P. et al. VariantEWS-WT1 chimeric product in the desmoplastic small round cell tumor. Pediatr Dev Pathol 1999; : 188–192. [DOI] [PubMed] [Google Scholar]
- 7.Zhang PJ, Goldblum JR, Pawel BR. et al. Immunophenotype of desmoplastic small round cell tumors as detected in cases with EWS-WT1 gene fusion product. Mod Pathol 2003; : 229–235. [DOI] [PubMed] [Google Scholar]
- 8.Kushner BH, La Quaglia MP, Wollner N. et al. Desmoplastic small round-cell tumor: prolonged progression-free survival with aggressive multimodality therapy. J Clin Oncol 1996, : 1,526–1,531. [DOI] [PubMed] [Google Scholar]
- 9.Kim J, Lee JM, Branton PE, Pelletier J. Modulation of EWS/WT1 activity by the v-Src protein tyrosine kinase. FEBS Lett 2000; : 121–128. [DOI] [PubMed] [Google Scholar]
- 10.Zhang GM, Zhu Y, Gan HL, Ye DW. Testicular desmoplastic small round cell tumor: a case report and review of literature. World J Surg Oncol 2014; : 227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lettieri CK, Garcia-Filion P, Hingorani P. Incidence and outcomes of desmoplastic small roundcell tumor: results from the surveillance, epidemiology, and end results database. J Cancer Epidemiol 2014; : article ID 680126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pickhardt PJ, Fisher AJ, Balfe MD. Desmoplastic small round cell tumor of the abdomen: radiologic-histopathologic correlation. Radiology 1999; : 633–638. [DOI] [PubMed] [Google Scholar]
- 13.Thuret R, Renaudin K, Leclere J. et al. Uncommon malignancies: case 3: paratesticular desmoplastic small round-cell tumor. J Clin Oncol 2005; : 6,253–6,255. [DOI] [PubMed] [Google Scholar]
- 14.Tateishi U, Hosono A, Makimoto A. et al. Accuracy of 18Ffluorodeoxyglucose positron emission tomography/computed tomographyin staging of pediatric sarcomas. J Pediatr Hematol Oncol 2007; : 608–612. [DOI] [PubMed] [Google Scholar]
- 15.Iyer RS, Schaunaman G, Pruthi S, Finn LS. Imaging of pediatric desmoplastic small-round-cell tumor with pathologic correlation. Curr Probl Diagn Radiol 2013; : 26–32. [DOI] [PubMed] [Google Scholar]
- 16.Dufresne A, Cassier P, Couraud L. et al. Desmoplastic small round cell tumor: current management and recent findings. Sarcoma 2012; : 714986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Msika S, Gruden E, Sarnacki S. et al. Cytoreductive surgery associated to hyperthermic intraperitoneal chemoperfusion for desmoplastic round small cell tumor with peritoneal carcinomatosis in young patients. J Pediatr Surg 2010; : 1,617–1,621. [DOI] [PubMed] [Google Scholar]
- 18.Lippe P, Berardi R, Cappelletti C. et al. Desmoplastic small round cell tumour: a description of two cases and review of the literature. Oncology 2003; : 14–17. [DOI] [PubMed] [Google Scholar]
- 19.Bisogno G, Ferrari A, Rosolen A. et al. Sequential intensified chemotherapy with stem cell rescue for children and adolescents with desmoplastic small round-cell tumor. Bone Marrow Transplant 2010; : 907–911. [DOI] [PubMed] [Google Scholar]
- 20.Mora J, Modak S, Cheung NK. et al. Desmoplastic small round cell tumor 20 years after its discovery. Future Oncol 2015; : 1,071–1,081. [DOI] [PubMed] [Google Scholar]
- 21.Mandal PK, Adhikari A, De A, Mondal SK. Desmoplastic small round cell tumor: diagnostic dilemma and uncertain prognosis: Report of few cases. J Cancer Res Ther 2015; : 1,028. [DOI] [PubMed] [Google Scholar]
- 22.Fizazi K, Farhat F, Theodore C. et al. CA125 and neuron-specific enolase (NSE) as tumour markers for intra-abdominal desmoplastic small round-cell tumours. Br J Cancer 1997; : 76–78. [DOI] [PMC free article] [PubMed] [Google Scholar]