

Abstract
Mortalin [also known as heat shock protein family A (Hsp70) member 9 (HSPA9) or glucose-regulated protein 75 (GRP75)] is a mitochondrial molecular chaperone that is often upregulated and mislocalized in tumors with abnormal activation of the kinases MEK and ERK. Here, we found that mortalin depletion was lethal selectively to tumor and immortalized normal cells expressing the mutant kinase B-RafV600E or the chimeric protein ΔRaf-1:ER, and that MEK-ERK–sensitive regulation of the peptide-binding domain in mortalin was critical to life/death decisions in these cells. Proteomics screening identified adenine nucleotide translocase 3 (ANT3) as a previously unknown mortalin client and life/death effector. Mechanistically, increased MEK-ERK signaling activity and mortalin function converge in opposition on the regulation of mitochondrial permeability. Specifically, whereas MEK-ERK activity increased mitochondrial permeability by promoting the interaction between ANT3 and the peptidyl-prolyl isomerase cyclophilin D (CypD), mortalin decreased mitochondrial permeability by inhibiting this interaction. As such, mortalin depletion increased mitochondrial permeability in MEK-ERK–deregulated cells, to the level triggering cell death. Moreover, chemical inhibitors of mortalin effectively suppressed the proliferation of B-RafV600E tumor cells in vitro and in vivo, including their B-Raf inhibitor-resistant progenies. This specific relationship between mortalin and deregulated MEK-ERK pathway activity suggest that mortalin has potential as a selective therapeutic target.
INTRODUCTION
Deregulated activity of the mitogen-activated protein kinase (MAPK) kinase–extracellular signal-regulated protein kinase (MEK/ERK) pathway, mainly caused by mutations in BRAF, is one of the most frequently detected oncogenic alterations and is a key therapeutic target. Indeed, development of Food and Drug Administration (FDA)-approved inhibitors targeting this pathway has advanced the therapy of BRAF-mutant (B-RafV600E/K/D) tumors (1–3). Nevertheless, most tumors that are initially responsive to these inhibitors develop resistance, leading to tumor relapse and patient death. Moreover, these drugs have shown limited efficacy against certain BRAF-mutant tumors due to intrinsic resistance (1, 3, 4). Therefore, additional therapeutic strategies are required to effectively treat BRAF-mutant tumors.
Mitochondria possess a powerful cell death machinery that is activated upon permeabilization of outer or inner mitochondrial membranes (5, 6). For example, whereas permeabilization of the outer membrane leads to the release of proapoptotic factors from the intermembrane space, formation of the mitochondrial membrane-spanning permeability transition pore (MPTP) can lead to metabolic catastrophe (6, 7), although molecular composition and mechanisms underlying the pore are not yet clearly understood (8–11). Mitochondria-originated cell death is mediated via various mitochondrial channels and regulators, including adenine nucleotide translocase (ANT), voltage-dependent anion channel (VDAC), mitochondrial Ca2+ uniporter (MCU), and cyclophilin D (CypD), and is often driven by metabolic stresses, such as deregulated mitochondrial redox or Ca2+ flux (5, 6). There is a growing interest in exploiting these processes for tumor therapy, because oncogenic transformation requires metabolic reprogramming, which inevitably increases the chance for various mitochondrial stresses in tumor cells (5, 12). It is conceivable, therefore, that malignant tumor cells have developed a protective mechanism (or mechanisms) in this context, the identification of which may lead to novel therapeutic strategies.
Mortalin [also known as heat shock protein family A (Hsp70) member 9 (HSPA9), glucose-regulated protein 75 (GRP75), or 74-kDa peptide-binding protein (PBP74)] is a mitochondrial chaperone that is often increased in expression and mislocalized in tumor cells (13–15). Mortalin regulates multiple aspects of tumorigenesis, including tumor cell proliferation and survival, stemness, epithelial-mesenchymal transition, and angiogenesis (16–20). We have previously demonstrated that mortalin facilitates tumor cell proliferation and survival by modulating MEK/ERK activity (21–23) and mitochondrial bioenergetics (24). In this study, we evaluated the potential of mortalin as a therapeutic target in MEK/ERK-deregulated tumors and investigated the underlying molecular mechanisms thereof.
RESULTS
Mortalin depletion selectively induces cell death in B-RafV600E-expressing cells
Mortalin levels are upregulated in melanoma 20, and its upregulation is correlated with poor patient survival, as suggested by our meta-analysis of different RNAseq datasets (fig. S1). Because RNA interference of mortalin expression can effectively suppress the proliferation of B-RafV600E tumor cells (21, 22), we sought to determine whether there is a specific relation between mortalin and B-RafV600E dictating these effects by examining the combinatory effects of B-RafV600E expression and mortalin depletion in IMR90E1A (immortalized normal human diploid fibroblasts) and MEL-ST (immortalized human melanocytes).
As determined by trypan blue exclusion assays, mortalin knockdown robustly suppressed the viability of IMR90E1A and MEL-ST cells expressing B-RafV600E, but not the viability of wild type B-Raf (B-RafWT)-expressing cells (Fig. 1, A and B). In these cells, concurrent B-RafV600E expression, but not B-RafWT expression, and mortalin knockdown substantially increased the cleavage of caspase 9, lamin A, and poly-(ADP-ribose)-polymerase (PARP), key surrogate markers for caspase-dependent apoptosis (25), along with p21CIP1 or p16INK4a expression (Fig. 1A; fig. S2A), although HMGB1 release, a necrosis marker, was not detected (fig. S2B). Mortalin knockdown with B-RafV600E expression substantially increased annexin V/propidium iodide co-staining and sub-G0/G1 phase population in IMR90E1A cells (Fig. 1C, D; fig. S3, A to C), indicating the onset of cell death and growth arrest. Indeed, the pan-caspase inhibitor Z-VAD(Ome)-FMK substantially rescued B-RafV600E expressing IMR90E1A and MEL-ST cells (fig. S4A), and the human B-RafV600E melanoma lines A375 and SK-MEL-28 cells from mortalin depletion-induced death (fig. S4, B and C). Of note, pretreatment of IMR90E1A cells with vemurafenib (PLX4032, B-RafV600E inhibitor), selumetinib (AZD6244, MEK1/2 inhibitor), or SCH772984 (ERK1/2 inhibitor) substantially attenuated cell viability loss (Fig. 1E) and lamin A cleavage induced by mortalin knockdown with B-RafV600E expression (Fig. 1, F to H), suggesting that MEK/ERK activity is necessary for the induction of cell death. Intriguingly, constitutively active AKTE40K did not elicit similar lethal effects when combined with mortalin knockdown, supporting the specificity of these effects (fig. S5, A to C).
Figure 1. Mortalin depletion selectively suppresses B-RafV600E-expressing cells.
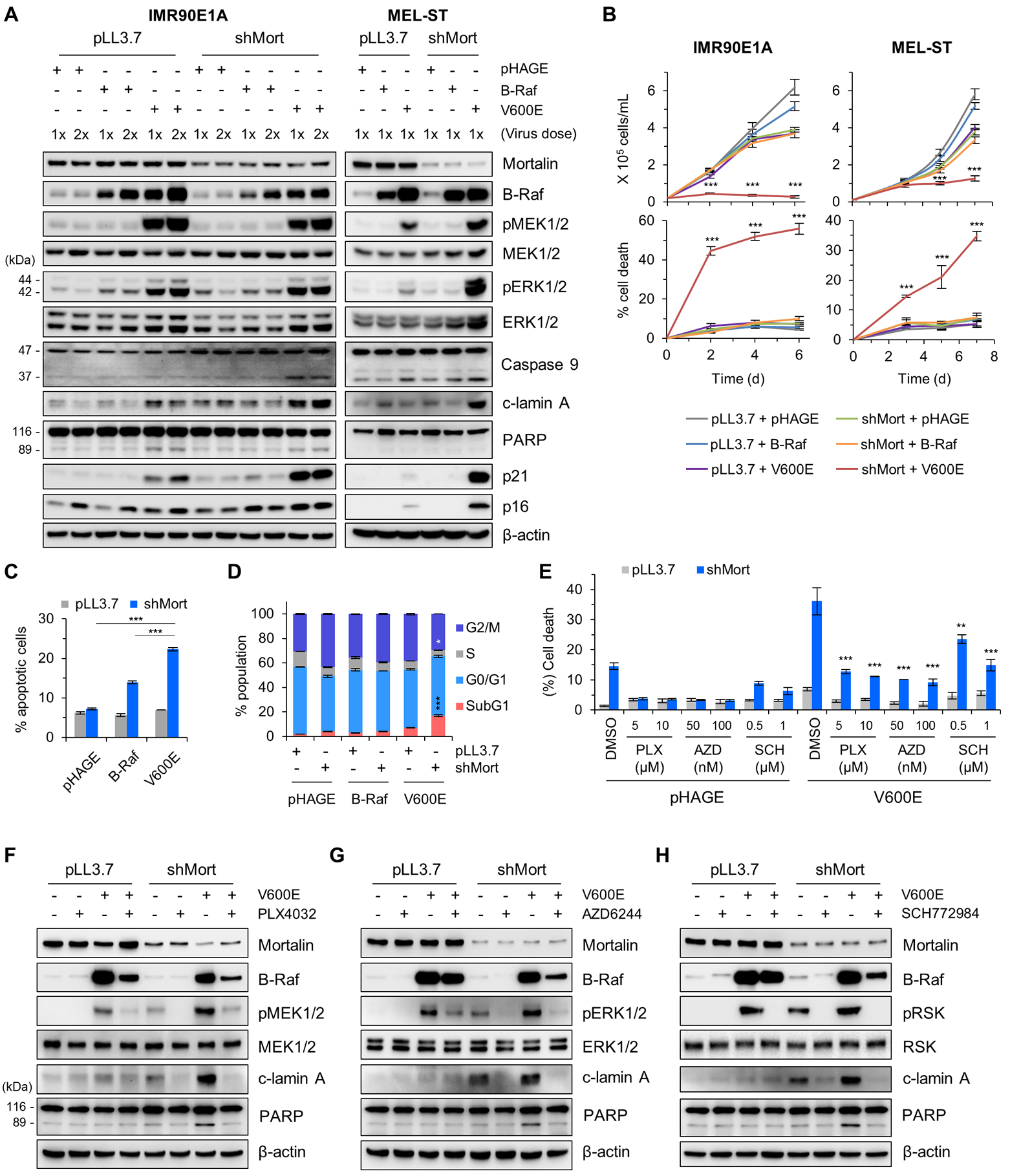
(A) Western blotting of total cell lysates from IMR90E1A and MEL-ST cells co-infected with pLL3.7 virus expressing shRNA targeting mortalin (shMort) and pHAGE expressing wild-type B-Raf or B-RafV600E (V600E), lysed at 3 days after infection. pMEK1/2, phosphorylated MEK1/2; pERK1/2, phosphorylated ERK1/2; c-lamin A, cleaved lamin A. β-actin was the control for equal protein loading. Densitometry of lamin A and caspase 9 cleavage is presented in fig. S2A. (B) Cell proliferation (top) and cell death (bottom) in cultures described in (A), monitored by trypan blue exclusion assays. (C) Apoptosis of IMR90E1A cells described in (A), assessed 2 days after infection by Annexin V/propidium iodide staining and flow cytometry. (D) Cell cycle analysis of IMR90E1A cells described in (A), assessed 3 days after infection. Infection efficiency and FACS histograms are presented in fig. S3. (E) IMR90E1A cells co-infected with pLL3.7-shMort and pHAGE-B-RafV600E were treated with indicated inhibitors for 2 days prior to TO-PRO-3 assays. p-values are relative to DMSO/V600E control. (F to H) Western blotting of total cell lysates harvested from the treated cells described in (E). Blots (A, F to H) are representative of two independent experiments; quantitative data (B to E) are mean ± SEM of three biological replicates. *p < 0.05, **p < 0.01, ***p < 0.001 by two-way ANOVA with Bonferroni post-tests.
Dysregulated mortalin-client interaction causes lethality in MEK/ERK-deregulated cells
Mortalin interacts with different clients and these interactions are regulated by its N-terminal ATPase and regulatory subdomains (26). Although mortalin has a mitochondrial targeting signal at its N-terminal end, it is also detected in different subcellular locations (27). To understand the molecular mechanism(s) by which mortalin regulates B-RafV600E tumor cell survival, we conducted a rescue experiment using different mortalin constructs (illustrated in Fig. 2A) in A375 engineered for doxycycline-inducible mortalin knockdown (A375-dox-shMort). We found that, whereas C-terminal HA-tagged mortalin expression effectively rescued A375-dox-shMort cells from doxycycline treatment, N-terminal HA-tagged mortalin did not but rather exacerbated doxycycline-induced cleavage of lamin A and PARP (Fig. 2, B and C, and fig. S6). Because the N-terminal, but not C-terminal, HA tag hindered mortalin localization to mitochondria (fig. S7), we suspected that abnormal enrichment of non-mitochondrial mortalin can be harmful to cells although mitochondrial mortalin is critical for cell survival. In subsequent truncation analyses, overexpression of the C-terminal peptide/client-binding domain (PBD), but not the ATPase domain (AD) or the subdomain 2 (SD2), also exacerbated mortalin depletion-induced effects in A375 cells (Fig. 2, B and C, and fig. S6). Notably, similar to mortalin depletion, PBD overexpression was sufficient to induce death in B-RafV600E melanoma cells, but not in immortalized non-tumor cells such as MEL-ST and HEK293T (Fig. 2D). However, PBD expression induced robust cell death upon B-RafV600E co-expression in IMR90E1A cells (Fig. 2, E and F) or upon ΔRaf-1:ER activation in LNCaP cells, a BRAF wild-type human prostate tumor line (fig. S8, A and B), highlighting its conditional lethal effects.
Figure 2. Dysregulated mortalin-PBD causes lethality in B-RafV600E-expressing cell.
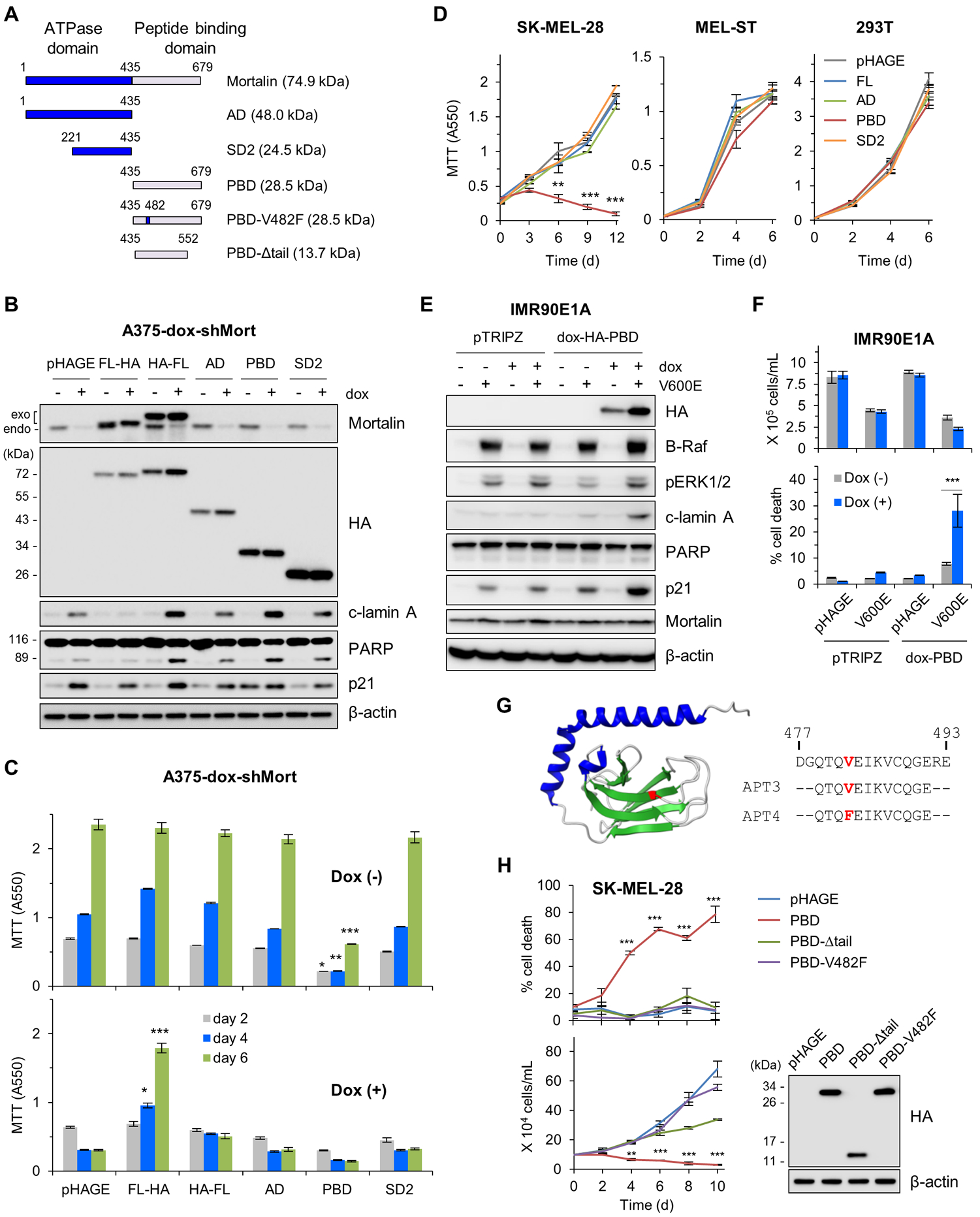
(A) Schematics of mortalin mutants used in this study. AD, ATPase domain; SD2, subdomain 2; PBD, peptide binding domain; V482F, Val482Phe; Δtail, tail deletion. (B and C) A375-dox-shMort cells infected with pHAGE expressing full-length mortalin (FL) or domain mutants were treated with 0.5 μg/ml doxycycline (dox) for 4 days prior to Western blotting of total cell lysates (B) and MTT assay (C). Exogenous and endogenous mortalin proteins are indicated. Densitometry of lamin A and PARP cleavage is presented in fig. S6. (D) MTT assay of cells expressing the indicated mortalin constructs. (E) Western blotting of total cell lysates from IMR90E1A -dox-PBD cells infected with pHAGE-B-RafV600E and treated with 0.5 μg/ml doxycycline for 3 days. pTRIPZ is the empty viral vector control for dox-HA-PBD. (F) Proliferation and death rates of cells in (E) were determined by trypan blue exclusion assays. (G) 3-D structure of mortalin-PBD (PDB:3N8E). Val482 in the substrate-binding cavity is highlighted in red in the structure and in synthetic decoy peptide aptamers (APT) used in this study. (H) Trypan blue exclusion assays of SK-MEL-28 cells expressing PBD mutants. Western blotting of total cell lysates (right panel) shows the expression levels of these constructs. Blots (B, E, and H) are representative of two independent experiments; quantitative data (C, D, F, and H) are mean ± SEM of three biological replicates. *p < 0.05, **p < 0.01, ***p < 0.001 by two-way ANOVA with Bonferroni post-tests.
HSP70-client interaction requires the central hydrophobic pocket in the client-binding cavity of PBD and the tail region that acts as “lid” for the pocket (illustrated in Fig. 2G). We and others previously reported that exchange of the conserved Val482 to Phe in this pocket (V482F, illustrated in red in Fig. 2G) impairs mortalin interaction with TP53, MEK1/2, and protein phosphatase1α (21, 28). For the bacterial HSP70 homologue, DnaK, this exchange substantially lowers client affinity without causing global structural changes or client specificity (29). We found that V482F exchange or deletion of the tail region abrogated the ability of PBD expression to induce cell death in B-RafV600E tumor cells (Fig. 2H). Of note, no similar effects were observed with corresponding domains of heat shock cognate 70 (Hsc70) (fig. S9, A to C), supporting the specificity of mortalin functions in B-RafV600E tumor cells. These data suggest that a dysregulated mortalin-client interaction can cause selective lethality in MEK/ERK-deregulated cells.
Proteomic analysis reveals mortalin interaction with ANT and VDAC
We then sought to identify the mortalin client(s) involved in its life/death regulation in MEK/ERK-deregulated cells by comparing the interactomes of full-length mortalin and the PBD in cell lines exhibiting different MEK/ERK activity, namely LNCaP-ΔRaf-1:ER and SK-MEL-28. As suggested by the greater number and intensity of bands visualized by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), PBD interacted more tightly and/or with more proteins than full-length mortalin or other domain mutants (Fig. 3A and fig. S10A). In contrast, AD and SD2 exhibited fewer protein interactions than did full-length mortalin or PBD in these cells. Proteins from the full-length mortalin and PBD fractions were identified by high mass accuracy mass spectrometry (data file S1 and table S1). High confidence proteins (<1% false-discovery rate) were then cataloged based on the overlap between those found with full-length mortalin and PBD, between cells with ΔRaf-1:ER activation and those without (Fig. 3A and data file S2), and between cell lines (fig. S10B). This screening identified multiple proteins whose interaction with mortalin was previously unknown. Subsequent analyses using Ingenuity Pathway Analysis (IPA) and Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) revealed that many proteins in the mortalin interactomes are involved in functions related to protein synthesis, DNA damage repair, post-transcriptional regulation, and cell death and survival, with the latter being the majority (Fig. 3, B to D; fig. S10, C to F; and data file S3). Notably, the interaction of full-length mortalin with proteins in the cell death and survival category increased in response to ΔRaf-1:ER activation, whereas that of the PBD was constitutive (Fig. 3, B and C). In this functional category, our STRING analyses consistently visualized a cluster indicating mitochondria-originated cell death based upon the presence of ANT2, ANT3, and VDAC (Fig. 3D and fig. S10, D and E).
Figure 3. Proteomic analysis identifies MPTP components in mortalin interactome.
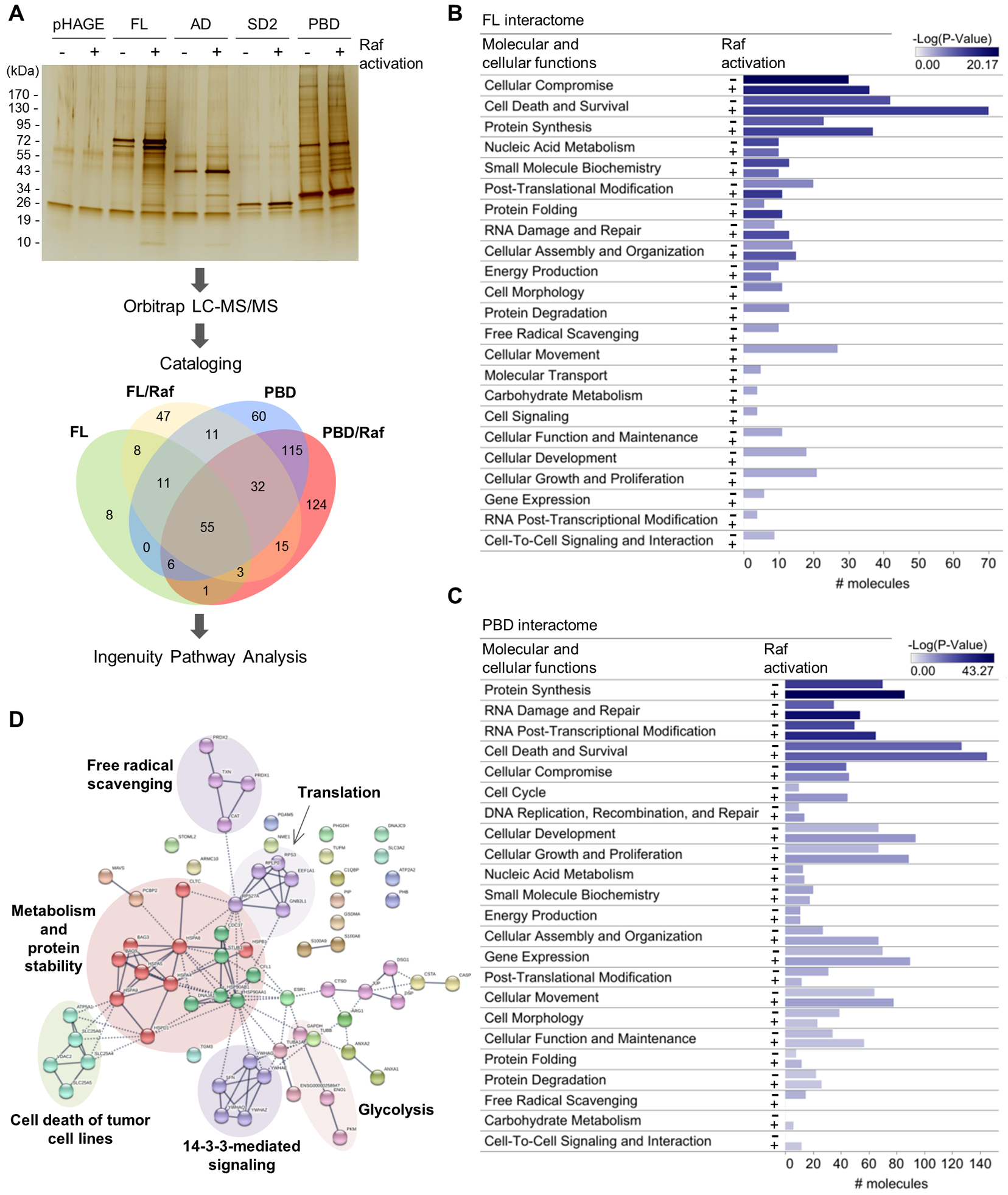
(A) Schematic of the proteomic analysis. Immunoprecipitates of the HA-tagged mortalin domain mutants from LNCaP-Raf:ER cells, after or without 4-hydroxytamoxifen (4-HT; 1 μM) treatment for 48 hours, were separated and visualized by SDS-PAGE and silver staining. Protein bands were excised, digested, and analyzed by LC-MS/MS using the instrument settings and search parameters in table S1. Identified proteins are cataloged in data file S1. Proteins in each sector of the Venn diagram are listed in data file S2. The screening in SK-MEL-28 is presented in fig. S10, A and B. Silver-stained SDS-PAGE image is representative of two independent samples. (B and C) Functional classification by IPA of the proteins identified from the interactomes of full-length mortalin (B) and PBD (C) reveal proteins in cell death and survival networks as a major constituent in the interactomes. # molecules indicate the number of different proteins in each functional category. IPA summary is shown in Comparison with SK-MEL-28 data is presented in fig. S10C. (D) STRING analysis of the cell death and survival protein networks of full-length mortalin interactome in Raf:ER-activated LNCaP cells. Edge intensity indicates the confidence of association between nodes. Nodes were clustered using MCL algorithm. Inter-cluster edges are represented by dashed-lines. Noted is the presence of ANT and VDAC in the “cell death of tumor cell lines” cluster in this network. Additional analysis is shown in fig. S10, D to F.
Mortalin directly interacts with ANT through a canonical chaperone-client interaction mechanism
Humans have three highly homologous ANT isoforms (ANT1/2/3) and, less homologous ANT4 (7). ANT can form a high affinity complex with VDAC at the mitochondrial intermembrane junction, which then can interact with different metabolic enzymes or the gatekeeper of MPTP, CypD (30) (illustrated in Fig. 4A). Consistent with our proteomics data, our co-immunoprecipitation (co-IP) analysis indicated that mortalin interacts with ANT1, ANT2, ANT3, and VDAC, but not with CypD (Fig. 4B and fig. S11A). This co-IP fraction also contained anti-apoptotic proteins BCL-xL and MCL-1, but not BCL-2 (Fig. 4B), which are suppressers of MPTP-mediated cell death (6). These proteins interacted more strongly with the PBD than with full-length mortalin (Fig. 4B), while their interactions with PBD were impaired by V482F exchange in the hydrophobic pocket of PBD (Fig. 4C and fig. S11B). Moreover, a short synthetic peptide (≤12 amino acids) designed from the β-strand containing Val482 (APT3, illustrated in Fig. 2G), inhibited the interaction of PBD with these proteins, whereas V482F exchange in APT3 (designated APT4) abrogated the decoying effect of this peptide (Fig. 4C).
Figure 4. Mortalin directly interacts with ANT via a canonical chaperone-client interaction mechanism.
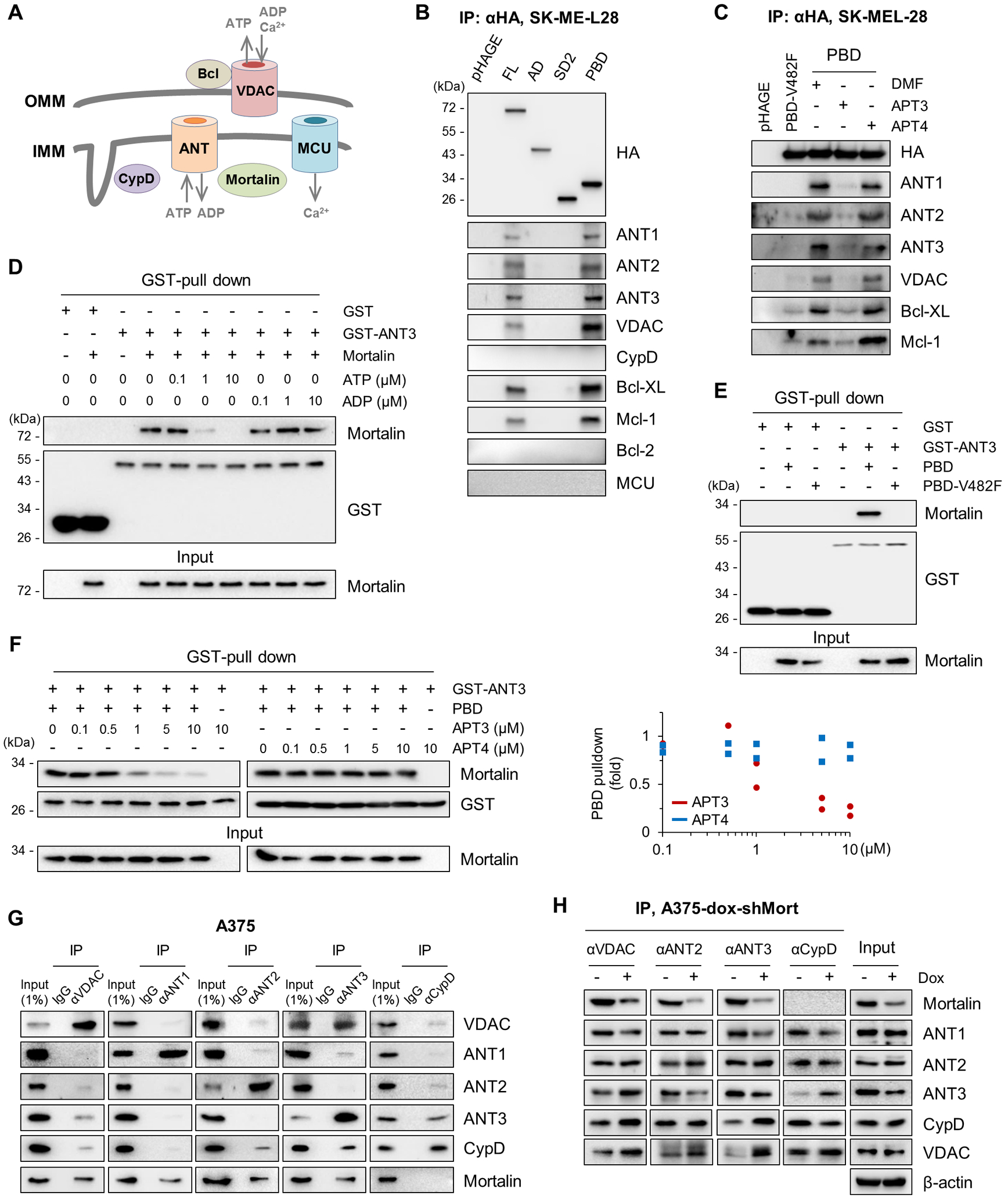
(A) Illustration of mitochondrial channels and regulators pertinent to this study. OMM, outer mitochondrial membrane; IMM, inner mitochondrial membrane. (B) Immunoprecipitation (IP) and Western analysis of the pulldown fractions of HA-tagged mortalin domain mutants from SK-MEL-28 total cell lysates. Inputs shown in fig. S11A. (C) IP-Western analysis of the pulldown fractions of HA-tagged PBD from total lysates of SK-MEL-28 cells incubated with 4 μM decoy peptides for 4 hours. Dimethylformamide (DMF) is the vehicle control. Inputs shown in fig. S11B. (D) In vitro binding assay to assess the effects of adenosine nucleotides on mortalin-ANT3 interaction. 0.5 μM recombinant GST-ANT3 was incubated for 60 min with 0.5 μM untagged recombinant mortalin in the presence of ATP or ADP before GST pulldown using Glutathione-Sepharose 4B and Western blotting. Recombinant GST was used as the control. (E) In vitro binding assay evaluating PBD-ANT3 interaction and the significance of Val482 in the client binding pocket. 0.5 μM GST-ANT3 was incubated for 60 min with 0.5 μM untagged recombinant PBD or PBD-V482F before GST pulldown using Glutathione-Sepharose 4B and Western blotting. (F) In vitro binding assay to assess the effects of APT3 and APT4 (left panel) on PBD-ANT3 interaction. PBD pulldown efficiency was determined by normalizing densitometry signals of PBD for GST signals (right panel) from two independent experiments. PBD was detected by the mortalin C-terminal–specific antibody. (G) IP-Western analysis of total lysates of A375 cells. Rabbit normal IgG was used as the control. Co-IP efficiency was determined by densitometry of signals from two independent experiments (fig. S12A). (H) IP-Western analysis of total lysates of A375-dox-shMort cells treated with 0.5 μg/ml doxycycline (dox) for 4 days. Co-IP efficiency was determined by densitometry of signals from two independent experiments (fig. S12B). Blots are representative of two (B, C, E-H) or three (D) independent experiments.
N-terminal ATPase-mediated ATP hydrolysis drives client release from the PBD of HSP70 chaperones (26). Because the mortalin-ANT interaction was previously unknown, we determined whether mortalin directly interacts with ANT. Indeed, recombinant mortalin and ANT3 interacted in our in vitro binding assays, and the addition of ATP, but not ADP, substantially decreased this interaction (Fig. 4D). The PBD-ANT3 interaction in vitro was also consistently impaired by V482F exchange (Fig. 4E) and was inhibited in a dose-dependent manner by APT3 but not by APT4 (Fig. 4F). These data are consistent with the canonical regulation, suggesting that ANT is a mortalin client. Notably, ANT3 interacted with VDAC more strongly than with ANT1 and ANT2 in our co-IP analyses of endogenous proteins in A375 and SK-MEL-28 cells (Fig. 4G and fig. S12A), and mortalin knockdown increased the interaction of ANT3, but not of ANT1 and ANT2, with CypD and VDAC in these cells (Fig. 4H and fig. S12B). Conversely, overexpression of the C-terminal HA-tagged mortalin decreased the interaction of ANT3 with CypD in the mitochondria (fig S13). Therefore, we investigated ANT3 in subsequent functional analyses.
ANT3, CypD, and MCU mediates the lethality caused by concurrent B-RafV600E expression and mortalin depletion
ANT3 knockdown, using two short-hairpin RNA (shRNA) constructs, markedly attenuated cell death and the cleavage of lamin A induced by B-RafV600E expression and mortalin depletion in IMR90E1A cells (Fig. 5A) and by mortalin depletion in SK-MEL-28 and A375 cells (Fig. 5B and fig. S14A). Conversely, ANT3 overexpression exacerbated cell death and the cleavage of lamin A and PARP induced by mortalin depletion in ΔRaf-1:ER-activated HEK293 cells (Fig. 5C). Of note, ANT3 overexpression increased ANT3 levels mainly in mitochondria (fig. S15A) and was, by itself, sufficient to suppress SK-MEL-28 and SK-MEL-1 cell survival (fig. S15, B and C). CypD knockdown also substantially attenuated cell death and the cleavage of lamin A induced by mortalin depletion in SK-MEL-28 and A375 cells (Fig. 5D and fig. S14B). MPTP opening can be caused by Ca2+ overflow from the endoplasmic reticulum (ER) to the mitochondria through the VDAC/MCU complex (6). Similar to ANT3 and CypD knockdown, MCU knockdown substantially attenuated mortalin depletion-induced lethality in these B-RafV600E melanoma cells (Fig. 5E and fig. S14C) and in B-RafV600E-expressing IMR90E1A cells (fig. S16, A and B). Of note, MCU did not interact with mortalin in our IP analysis (Fig. 4B), suggesting that mortalin does not directly regulate MCU.
Figure 5. ANT3, CypD, and MCU mediate cell death caused by concurrent B-RafV600E expression and mortalin depletion.
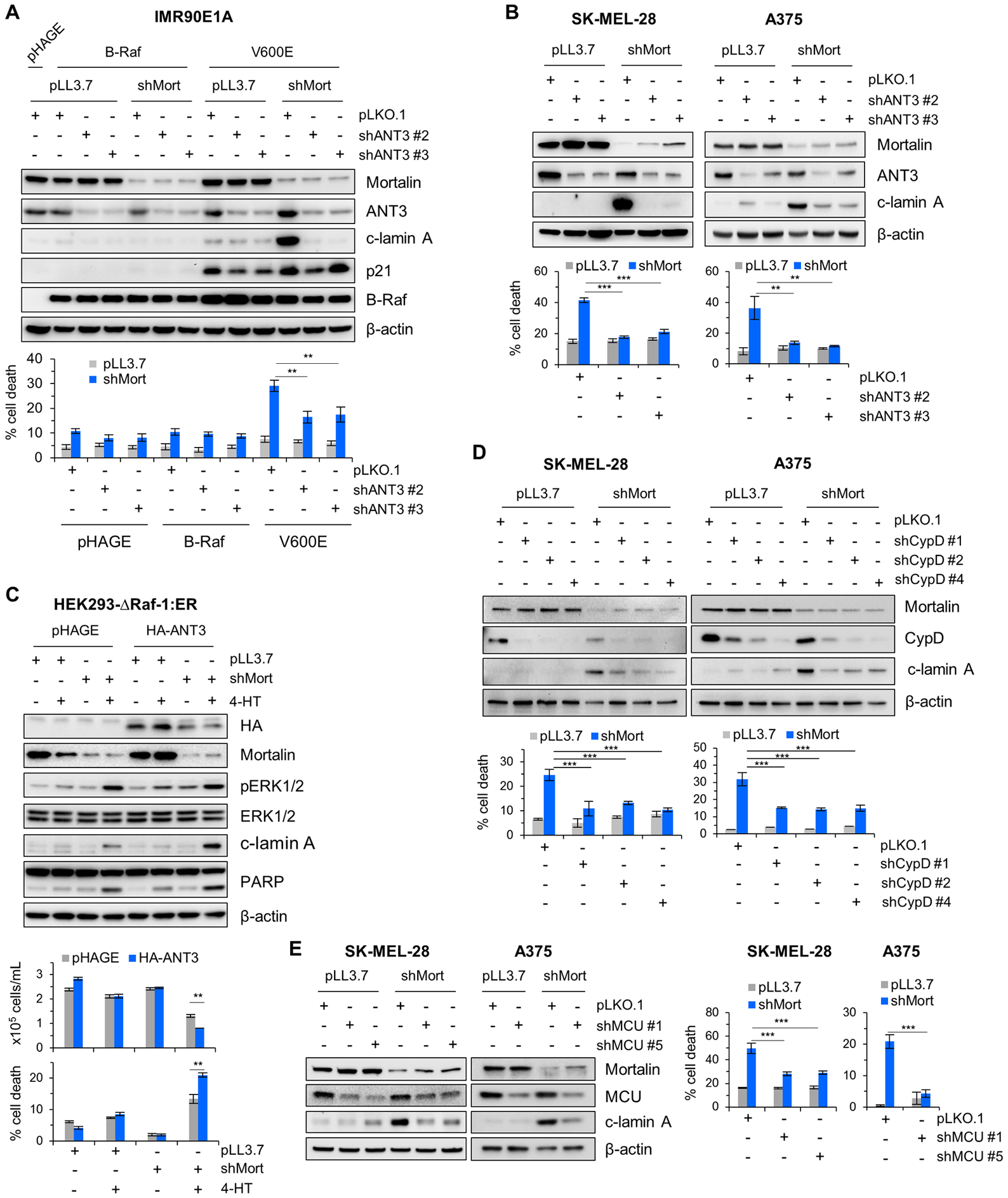
(A) IMR90E1A cells expressing B-Raf or B-RafV600E were co-infected for 4 days with pLL3.7-shMort and pLKO.1-shANT3 viruses (shANT3#2 and shANT3#3 targeting different ANT3 mRNA regions) prior to Western blotting of total cell lysates (top) and TO-PRO-3 assays (bottom). (B) SK-MEL-28 and A375 cells were co-infected for 4 days with pLL3.7-shMort and pLKO.1-shANT3 viruses prior to Western blotting of total cell lysates (top) and TO-PRO-3 assays (bottom). Densitometry of lamin A cleavage is presented in fig. S14A. (C) HEK293Δ-Raf-1:ER cells co-infected with pHAGE-ANT3 and pLL3.7-shMort viruses were treated with 1 μM 4-HT for 2 days prior to Western blotting of total cell lysates (top) and trypan blue exclusion assays (bottom). (D) SK-MEL-28 and A375 cells were co-infected for 4 days with pLL3.7-shMort and pLKO.1-shCypD viruses prior to Western blotting of total cell lysates (top) and TO-PRO-3 assays (bottom). Densitometry of lamin A cleavage is presented in fig. S14B. (E) SK-MEL-28 and A375 cells were co-infected for 4 days with pLL3.7-shMort and pLKO.1-shMCU viruses prior to Western blotting of total cell lysates (left) and TO-PRO-3 assays (right). Densitometry of lamin A cleavage is presented in fig. S14C. Blots (A to E) are representative of two independent experiments; quantitative data in each panel are mean ± SEM of three biological replicates. *p < 0.05, **p < 0.01, ***p < 0.001 by two-way ANOVA with Bonferroni post-tests.
Consistent with these “loss- or gain-of-function” analyses, the ANT inhibitor, bongkrekic acid (31); cyclosporin A, which inhibits the isomerase activity of CypD and displaces it from MPTP (32); the MCU inhibitors KB-R7943 (33) and ruthenium red (34); and nelfinavir, which inhibits ANT (35) and mitochondrial Ca2+ influx (36), each rescued B-RafV600E-expressing IMR90E1A and melanoma cells from mortalin knockdown in a dose-dependent manner (fig. S17, A to C). These data demonstrate the involvement of ANT3, CypD, and MCU in the lethality induced upon concurrent MEK/ERK deregulation and mortalin depletion.
Mortalin signaling and deregulated MEK/ERK activity converge on the ANT3-CypD interaction and mitochondrial permeability transition in an opposing manner
We sought to understand the mechanism by which ANT3 mediates B-RafV600E cell death upon mortalin depletion. Our co-IP experiments revealed that ANT3 interaction with CypD was very sensitive to mortalin and MEK/ERK activity although its interaction with VDAC was much less sensitive to these signals. In A375 cells, selumetinib treatment or ERK2 knockdown abolished basal as well as mortalin knockdown-induced ANT3-CypD interactions without affecting the ANT3-VDAC interaction as substantially (Fig. 6A and fig. S18A). Conversely, in HEK293 cells, ΔRaf-1:ER activation increased basal as well as mortalin knockdown-induced ANT-CypD interaction, with the effects of concurrent ΔRaf-1:ER activation and mortalin depletion being most prominent (Fig. 6B and fig. S18B). These data suggest that deregulated MEK/ERK activity upregulates ANT-CypD interaction, whereas mortalin counteracts this upregulation.
Figure 6. Mortalin and Raf-MEK-ERK signaling regulate the ANT3-CypD interaction and mitochondrial permeability transition in an opposing manner.
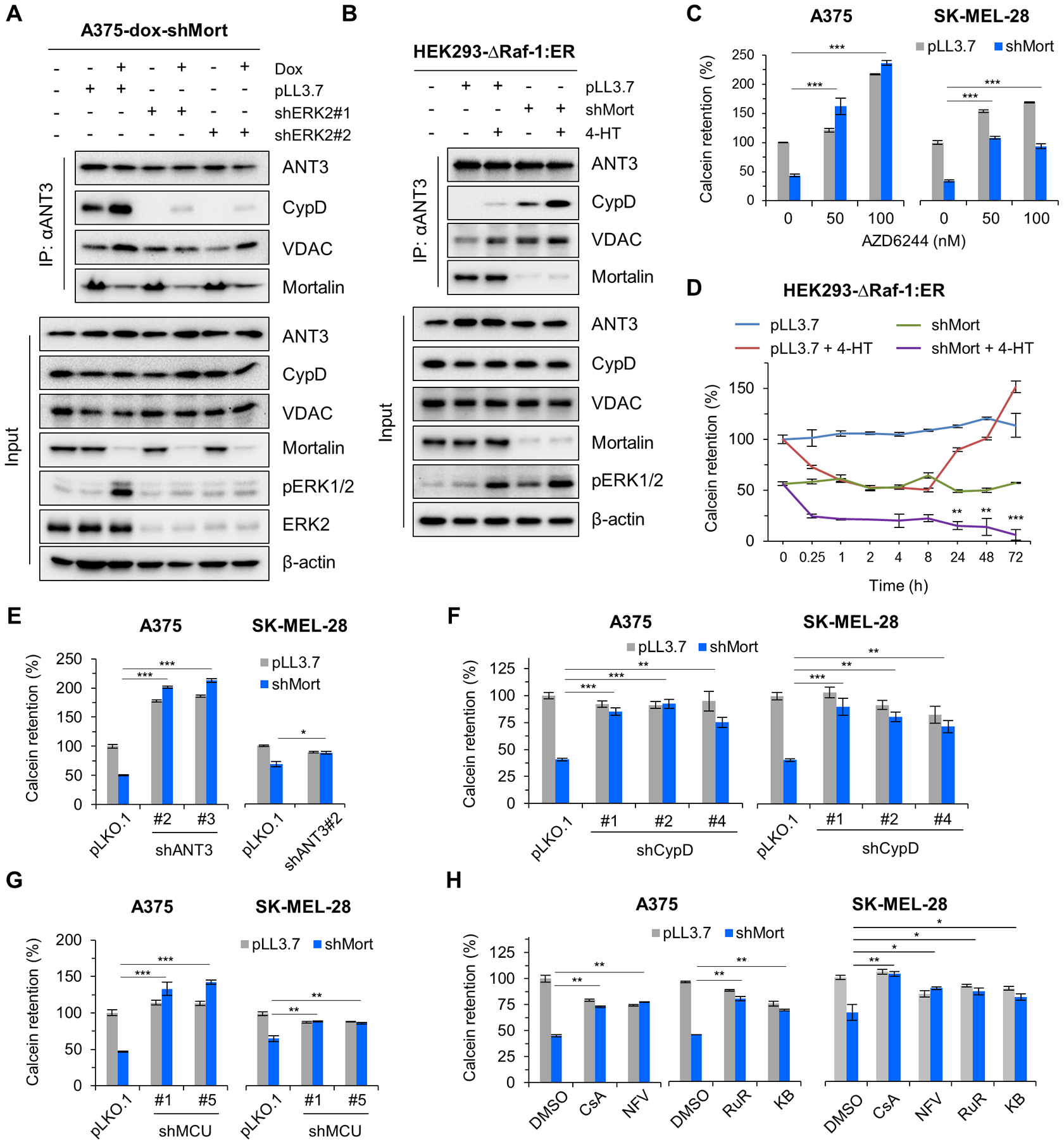
(A) A375-dox-shMort cells infected with pLL3.7-shERK2 viruses (shERK2#1 and shERK2#2 targeting different ERK2 mRNA regions) were treated with 0.5 μg/ml doxycycline for 4 days prior to IP-Western analysis of total lysates. Densitometry determining co-IP efficiency and similar effects of selumetinib are shown in fig. S18A. (B) HEK293Δ-Raf-1:ER cells depleted of mortalin were treated with 1 μM 4-HT for 2 days prior to IP-Western analysis of total lysates. Densitometry determining co-IP efficiency and selumetinib effects are shown in fig. S18B. (C) A375 (left) and SK-MEL-28 (right) cells were infected with pLL3.7-shMort (4 days) and treated with selumetinib (2 days) prior to Calcein labelling. Similar effects of ERK2 knockdown are shown in fig. S19A. FACS histograms are shown in fig. S19B. (D) Calcein retention assay of HEK293Δ-Raf-1:ER cells depleted of mortalin and treated with 1 μM 4-HT for the indicated time. FACS histograms are shown in fig. S19C. (E to H) Calcein retention assays of A375 and SK-MEL-28 cells depleted of mortalin (for 4 days) in combination with 4 days’ ANT3 knockdown (E), 4 days’ CypD knockdown (F), 4 days’ MCU knockdown (G), or 3 days’ treatment with MPTP inhibitor (H). CsA, 5 μM cyclosporine A; NFV, 5 μM Nelfinavir; RuR, 4 μM ruthenium red; KB, 4 μM KB-R7943; DMSO, vehicle control. FACS histograms are shown in fig. S19, D to G. Blots (A and B) are representative of two independent experiments; quantitative data (C to H) are mean ± SEM of three biological replicates. *p < 0.05, **p < 0.01, ***p < 0.001 by two-way ANOVA with Bonferroni post-tests.
Because the ANT3-CypD interaction has been implicated in mitochondrial permeability transition (9, 37, 38), we determined whether mortalin and MEK/ERK activity affect mitochondrial permeability in an opposite manner by conducting the calcein-acetyoxymethyl release assay (39), also known as “calcein labelling”, and mitochondrial membrane potential (Δψm) measurement. Consistent with their effects on ANT-CypD interaction, mortalin depletion substantially decreased mitochondrial calcein retention and Δψm in A375 and SK-MEL-28 cells, whereas selumetinib and ERK2 knockdown substantially abrogated mortalin depletion-induced decreases in calcein retention (Fig. 6C and fig. S19A, B) and Δψm (fig. S20, A and B) in these cells. Moreover, our time-course analysis in HEK293 cells revealed that mitochondrial calcein retention rapidly decreased upon ΔRaf-1:ER activation (~1 hour) but gradually returned to the basal level when ΔRaf-1:ER activation was prolonged (~72 hours), for which mortalin was necessary (Fig. 6D and fig. S19C). These data are consistent with the effects of mortalin and MEK/ERK activity on ANT-CypD interaction, suggesting that mitochondrial permeability can be deregulated by aberrant MEK/ERK activity in the absence of mortalin.
We confirmed the involvement of ANT3, CypD, and MCU in this mitochondrial permeability regulation using RNA interference and chemical inhibitors. Similar to the effects of selumetinib or ERK2 knockdown above, knockdown of ANT3, CypD, or MCU abolished mortalin knockdown-induced decreases in mitochondrial calcein retention (Fig. 6, E to G; and fig. S19, D to F) and Δψm (fig. S20, C to E) in A375 and SK-MEL-28 cells. Cyclosporin A, nelfinavir, ruthenium red, and KB-R7943 also consistently inhibited mortalin knockdown-induced decreases in mitochondrial calcein retention (Fig. 6H; fig. S19G) and Δψm (fig. S20F) in these cells. Moreover, these inhibitors inhibited mortalin depletion-induced release of death signals, cytochrome C and apoptosis-inducing factor (40), from mitochondria in the B-RafV600E melanoma lines, A375, SK-MEL-28, and SK-MEL-1 (fig. S21). These data suggest that perturbed mitochondrial permeability transition is a mechanism underlying the lethality induced by concurrent MEK/ERK deregulation and mortalin depletion.
Advanced MKT-077 derivatives phenocopy mortalin depletion and effectively suppress B-RafV600E melanoma cells, including their B-Raf inhibitor-resistant progenies
Observing the selective toxicity of mortalin knockdown in B-RafV600E-expressing cells, we sought for a “proof-of-principle demonstration” of mortalin targeting in B-RafV600E tumors. MKT-077, a water soluble benzothiazole-rhodacyanine (41), binds to a negatively charged pocket close to the nucleotide binding site of HSP70 in the ADP state, which is well conserved in human HSP70 paralogs (42). Being a Δψm-sensitive delocalized lipophilic cation, MKT-077 mainly partitions into mitochondria (43), and its interaction with mortalin in cells was demonstrated by affinity purification, although it also interacted with HSC70 (HSPA8) and F-actin (44, 45). Recently, a series of MKT-077 derivatives, such as JG-98 and JG-231, have been developed with improved affinity and selectivity for the HSP70 binding pocket (46). Although these inhibitors may display pan-activity for multiple HSP70 family members, (i) their lipophilic cationic property, (ii) different subcellular distribution of HSP70 chaperones, and (iii) mortalin dependency of BRAF tumor cell provided a rationale for evaluating them in this proof-of-concept study.
Similar to mortalin depletion or PBD overexpression, treatment with JG-98 induced robust cell death along with augmented p21CIP1 expression in IMR90E1A cells expressing B-RafV600E but not B-RafWT (Fig. 7A; fig. S22A). JG-98 also showed greater potency than previous MKT-077 derivatives in SK-MEL-28 cells (fig. S22B). Of note, mortalin overexpression conferred SK-MEL-28 and A375 cells partially resistant to JG-98 and JG-231, a JG-98 analog with improved pharmacokinetics and bioavailability (46), whereas mortalin knockdown sensitized these cells to these inhibitors (fig. S23, A and B). Moreover, selumetinib substantially rescued A375 cells from JG-98- or JG-231-induced death (fig. S24, A and B). These data suggest that the cellular activity of JG-98 and JG-231 occurs through mortalin in this system and that the cytotoxicity of these molecules, consistent with mortalin depletion, is also MEK/ERK-dependent. Notably, because mortalin depletion effectively suppressed cell viability of vemurafenib-resistant B-RafV600E melanoma lines A375PLX/R and Colo-829PLX/R (fig. S25, A to C), we evaluated JG-98 and its advanced analogs in these cells to find that all these compounds effectively suppressed viability in drug-resistant cells at IC50 values similar to those determined in parental cells (Fig. 7B; fig. S25D). We then evaluated JG-231 for in vivo efficacy. Indeed, when administered intraperitoneally, JG-231 effectively suppressed the growth of A375, A375-PLX/R, and Colo829 xenografts in athymic nude mice (Fig. 7, C and D) without causing significant body weight loss (Fig. 7E). Consistent with mortalin depletion effects, JG231-treated tumors exhibited increased lamin A cleavage and p21CIP1 expression (Fig. 7F and fig. S26). Moreover, JG231-treated tumors also exhibited increased MEK1/2 phosphorylation (Fig. 7F and fig. S26); this effect is consistent with the effects of JG-98 and JG-231 inducing ERK1/2 phosphorylation in vitro (fig. S24B) and the ability of mortalin to modulate MEK/ERK activity by regulating cellular levels of phosphorylated MEK1/2 via protein phosphatase 1α (21), without directly interacting with ERK1/2 (fig. S27). These data suggest the feasibility of exploiting mortalin to selectively suppress B-RafV600E tumors, including those that acquired B-Raf inhibitor resistance.
Figure 7. The effects of mortalin inhibitors in B-RafV600E-expressing IMR90E1A and vemurafenib -naïve and -resistant B-RafV600E tumor cells.
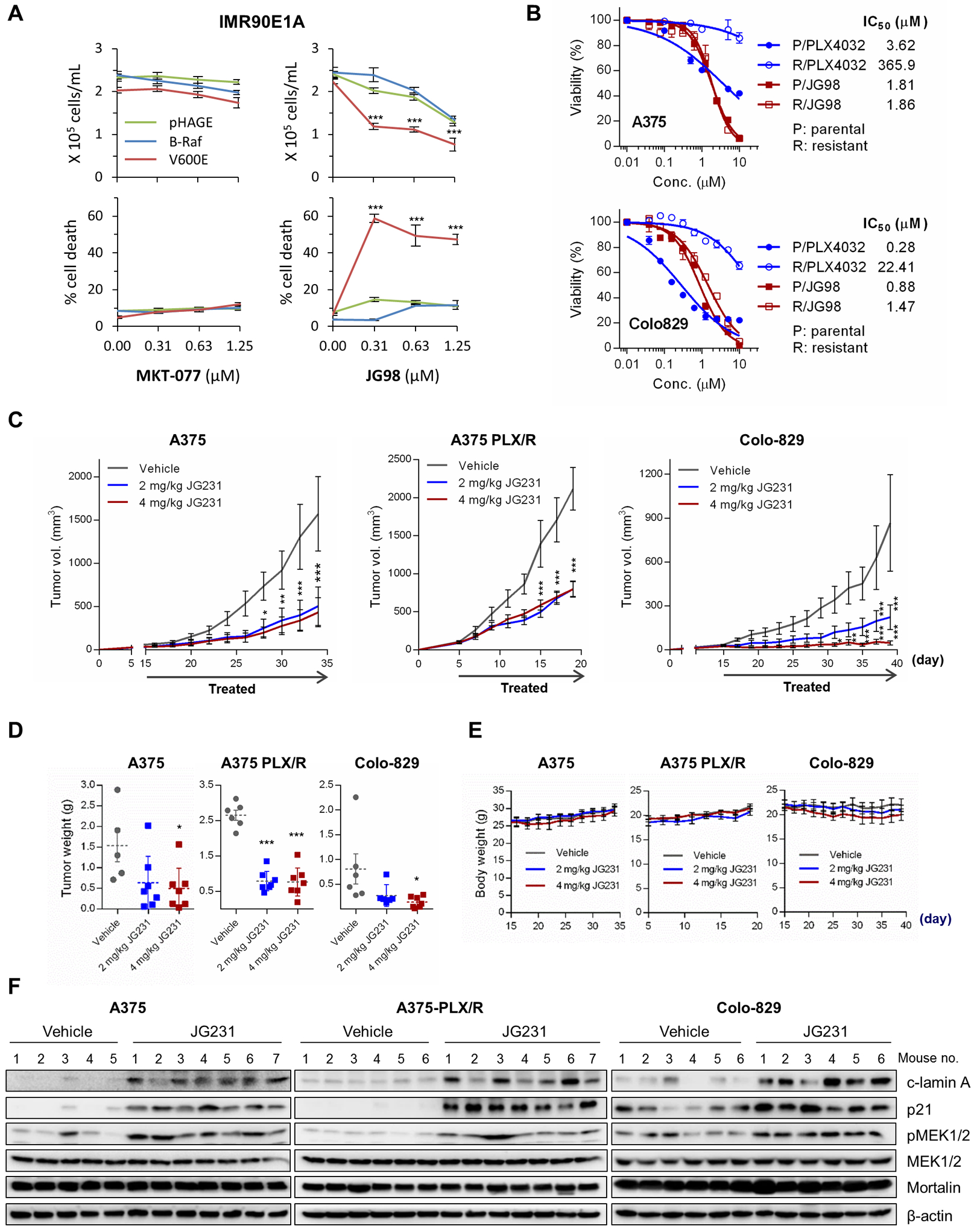
(A) Cell proliferation (top) and cell death (bottom) in IMR90E1A cells infected with pHAGE virus expressing wild-type B-Raf or B-RafV600E (V600E) and treated with MKT-077 or JG-98 for 2 days, monitored by trypan blue exclusion assays. Effects on day 1 and those of other MKT-077 derivatives are shown in fig. S22A. (B) IC50 analysis in vemurafenib/PLX4032-naïve and -resistant B-RafV600E tumor cells treated with JG-98 for 2 days. Cell viability was determined by MTT assay. 95% Confidence Intervals for P/JG98 vs. R/JG98 were 1.683 to 1.949 vs. 1.629 to 2.215 (A375) and 0.782 to 0.992 vs. 1.273 to 1.688 (Colo829), respectively. Results for other JG-98 analogs are shown in fig. S25D. (C to F) The effects of JG-231 on vemurafenib-naïve and -resistant B-RafV600E tumor xenografts in mice. Athymic mice bearing tumor xenografts were treated with one of two doses of JG-231. Drugs dissolved in 200 μl vehicle were administered intraperitoneally every other day for the indicated treatment period. The control group was treated with the vehicle only. Changes in tumor sizes (C), tumor weights (D), mouse body weight (E), and tumor protein abundances (by Western blotting; F) were assessed. Horizontal dotted line in (D) indicates mean. Densitometry of lamin A cleavage, MEK1/2 phosphorylation, and p21CIP in (F) is presented in fig. S26. Data in (A and B) are mean ± SEM of three biological replicates. Data in (C to E) are mean ± SEM from a single cohort of 5 to 7 mice per treatment group (A375, 5 mice for vehicle, 7 mice for each JG231 group; A375 PLX/R, 6 mice for vehicle, 7 mice for each JG231 group; Colo-829, 6 mice for vehicle, 6 mice for 2 mg/kg JG231, 7 mice for 4 mg/kg JG231). Each mouse developed one tumor. *p < 0.05, **p < 0.01, ***p < 0.001 in A, C, and E by two-way ANOVA with Bonferroni post-tests, and in D by one-way ANOVA with Dunnett post-tests.
DISCUSSION
Our data demonstrate that MEK/ERK deregulation puts cells at risk of cell death associated with altered mitochondrial permeability, but that mortalin counteracts this risk. Mechanistically, the effects of MEK/ERK and mortalin converge on ANT-CypD interaction in an opposing manner, whereby ANT-CypD interaction can be sustained upon MEK/ERK deregulation without the balancing mortalin effects. A failure in this regulation can trigger death signals and, as such, mortalin is a guardian of MEK/ERK-deregulated tumor cells against this risk (illustrated in Fig. 8). Of note, it appears that this risk is inherent in B-RafV600E tumor cells (not only vemurafenib-naïve but also vemurafenib-resistant) and can be exploited by targeting mortalin. Exploiting cytolethal risks associated with mitochondrial channels during metabolic reprogramming has been proposed as a promising cancer therapeutic strategy (5, 6). Given this, we propose mortalin as a candidate target for implementing this strategy in MEK/ERK-dependent tumors.
Figure 8. Graphic illustration summarizing the study’s findings.
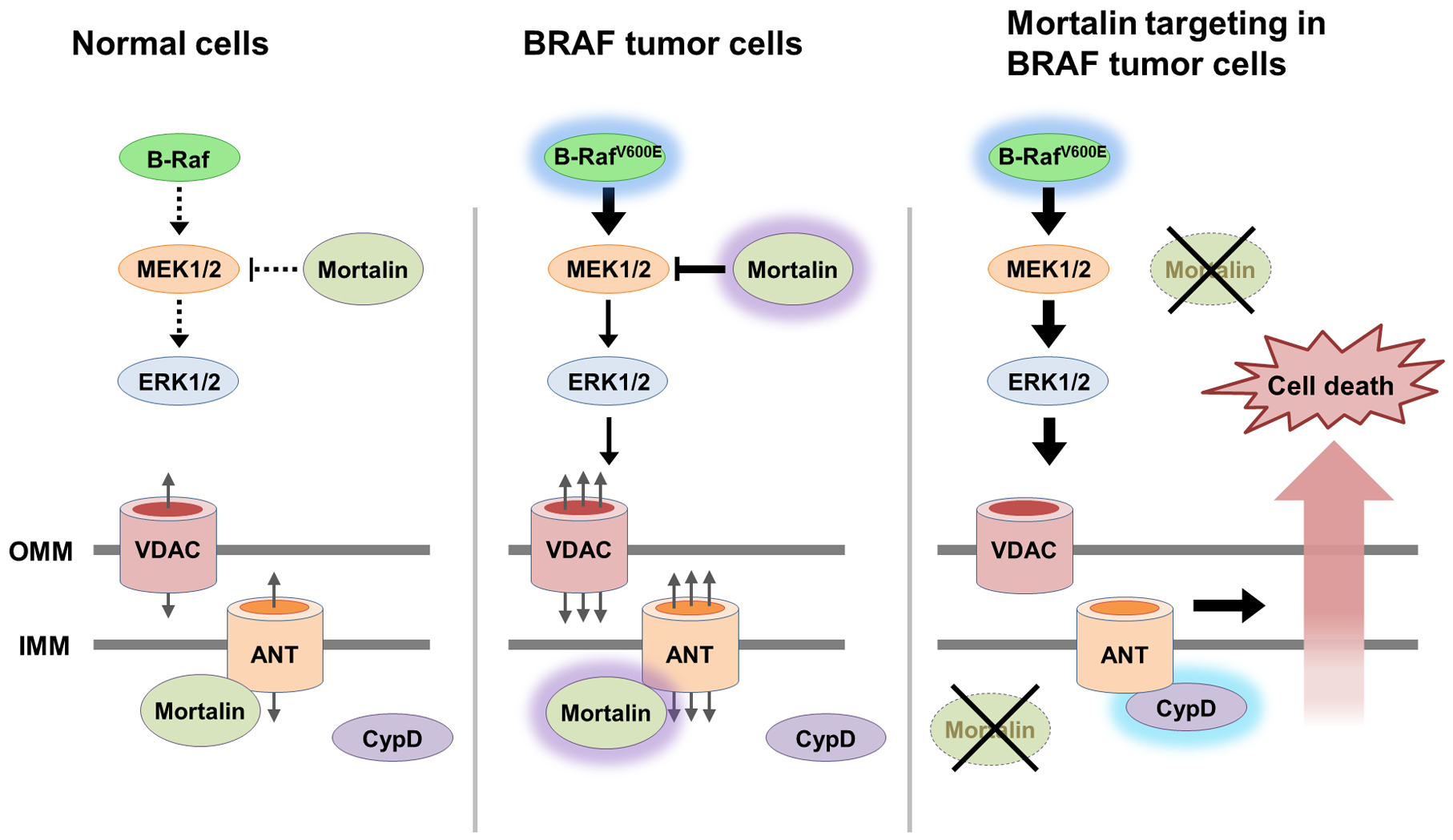
ANT is a mitochondrial channel that maintains cellular bioenergetics in collaboration with VDAC. However, ANT can also interact with CypD, the gatekeeper of MPTP, and cause cell death by perturbing mitochondrial membrane permeability. Deregulated MEK/ERK activity in BRAF-mutant tumor cells increases this lethal risk, but mortalin counteracts it by inhibiting ANT-CypD interaction and by modulating MEK1/2 activity. These processes may be exploited to selectively suppress BRAF-mutant tumor cells.
Increasing evidence suggests that molecular chaperones can facilitate tumorigenesis by altering the stability or activity of different molecular switches and effectors in diverse cellular processes (47). The composition of a chaperone interactome is sensitive to stress and is quite heterogeneous in cancer cells (48). While different chaperone interaction networks have been described (49), mortalin interaction networks have not been described previously for mammalian cells. Our data show that the composition of mortalin interactome is diverse, sensitive to Raf/MEK/ERK activity, and important for BRAF tumor cell survival. Our data suggest that ANT3 is a novel client for mortalin and critical for BRAF tumor cell survival. ANT plays essential roles in cellular bioenergetics by mediating ATP/ADP exchange across the mitochondrial inner membrane (7). For example, ANT usually exports ATP generated by respiration from mitochondrial matrix to cytosol to drive cellular processes whereas certain ANT isoforms, such as ANT2 in aerobic glycolytic tumor cells, imports ATP generated by glycolysis into mitochondrial matrix to sustain mitochondrial membrane potential (Δψm) and homeostasis (7). If these processes are impaired, ANT can promote cell death by complexing with VDAC and CypD (31, 50). Of note, mitochondrial bioenergetics is reprogramed to facilitate BRAF melanoma cell maintenance and B-Raf inhibitor resistance (51–53). Given these premises and our observed BRAF tumor cell sensitivity to mortalin-mediated ANT regulation, we speculate that (i) deregulated MEK/ERK activity upregulates bioenergetics at the risk of cell death associated with ANT dysregulation and (ii) mortalin upregulation is critical for protecting tumor cells from this risk.
Our data suggest that mortalin inhibits ANT-CypD interaction without directly interacting with CypD. Although CypD is generally accepted as the gatekeeper of MPTP, exact molecular composition of this channel is not clearly understood and there is no single model that universally addresses this phenomenon. For example, it has been proposed that mitochondrial permeability transition can occur by (at least) two CypD-dependent mechanisms, one involving ATP synthase and the other involving ANT (9, 10), while a recent report suggested that CypD is not necessary for calcium-induced MPTP activity (11). As such, although our data consistently implicate the ANT-CypD complex in the lethality caused by mortalin depletion in MEK/ERK-deregulated cells, the exact mechanism by which this complex mediates mitochondrial permeability transition in these cells remains to be elucidated.
Our data suggest that mortalin regulates the physical interactions between ANT, VDAC, and CypD via a coordinated process between its N-terminal regulatory domain and PBD, which is sensitive to MEK/ERK activity. We speculate that mortalin senses MEK/ERK activity through its N-terminal regulatory domain and, in turn, regulates PBD-client interaction; because mortalin neither directly interacts with ERK1/2 nor contains their substrate signature, this regulation would be indirect. Without the N-terminal domain, PBD-ANT3 interaction was sustained, which might have inhibited normal ANT3 function, eventually causing cell death. Of note, the HSP70 chaperone-client interaction is regulated via the communication between the subdomain 2 and different co-chaperones (26). As such, it is conceivable that a MEK/ERK-sensitive mortalin co-chaperone exists and regulates mortalin interaction with ANT3.
Our meta-analysis of different RNAseq datasets suggests that mortalin upregulation in melanoma is correlated with poor patient survival. Recent literature also suggests mortalin upregulation as a prognostic marker for poor patient survival in different tumors exhibiting aberrant MEK/ERK activity (54–56). Our data may therefore account for the significance of mortalin upregulation in these tumors. Intriguingly, the mortalin-MEK/ERK relationship appears quite specific because mortalin depletion did not induce any lethal effects when combined with constitutively active AKTE40K in immortalized normal fibroblasts. In support of this specificity, we detected MEK1/2, but not AKT, in mortalin interactomes while we previously detected mortalin in MEK1/2 interactomes (22). One should note that mortalin can modulate MEK1/2 activity by regulating the access of protein phosphatase 1α to MEK1/2 (21). This ability of mortalin has significance for oncogenic MEK/ERK signaling because sustained high MEK/ERK activity can induce cell death and growth arrest (57–59). Taken together, our findings propose that mortalin facilitates BRAF tumor cell survival and proliferation by modulating the degree of MEK/ERK activity and by preventing the risk of MEK/ERK-associated mitochondrial permeability dysregulation. Importantly, most tumors acquire resistance to anti-B-Raf/MEK/ERK therapy via a mechanism that reactivates the pathway (1, 3, 4). This MEK/ERK dependency of therapy-resistant tumors and the intriguing mortalin dependency of MEK/ERK-deregulated cells demonstrated in this study suggest a rationale for considering mortalin as a target to overcome B-Raf inhibitor resistance. In conclusion, our findings strongly demonstrate the significance of mortalin in MEK/ERK-deregulated cells and provide in-depth mechanistic insights into its potential as a therapeutic target for BRAF-mutant tumors.
MATERIALS AND METHODS
Cell lines
IMR90E1A, a gift from Yuri Lazebnik (Cold Spring Harbor Laboratory), were maintained in Dulbecco’s minimal essential medium (DMEM; Invitrogen) supplemented with 10% fetal bovine serum (FBS), 1% sodium pyruvate, and 1% nonessential amino acids. IMR90E1A-dox-HA-PBD and IMR90E1A-pTRIPZ were generated by stably infecting IMR90E1A with lentiviral pTRIPZ doxycycline-inducible HA-PBD and empty virus, respectively. The immortalized human melanocyte MEL-ST cells, a gift from Robert Weinberg (60), were maintained in DMEM supplemented with 5% bovine growth serum (HyClone). LNCaP-ΔRaf-1:ER and HEK293-ΔRaf-1:ER were maintained in 10% FBS-supplemented phenol red-free RPMI1640 (Invitrogen) or minimal essential medium (MEM, Invitrogen), respectively. Generation of LNCaP-ΔRaf-1:ER and HEK293-ΔRaf-1:ER were previously described (57, 59). ΔRaf-1:ER is the CR3 catalytic domain of Raf-1 fused to the hormone binding domain of the estrogen receptor (61) and was activated with 1 μM 4-hydroxytamoxifen (Sigma-Aldrich). SK-MEL-1 (ATCC), and SK-MEL-28 (ATCC) were maintained in MEM supplemented with 10% FBS, 1% sodium pyruvate, and 1% nonessential amino acids. A375 (ATCC), were cultured in DMEM supplemented with 10% FBS. A375-dox-shMort and SK-MEL-28-dox-shMort were generated by stably infecting A375 and SK-MEL-28 with lentiviral pTRIPZ doxycycline-inducible microRNA-adapted shRNA targeting human mortalin (Open Biosystems, V3THS_362249). Colo-829 was maintained in RPMI1640 supplemented with 10% FBS, 1% sodium pyruvate, and 1% nonessential amino acids. PLX4032-resistant (PLX/R) A375 and Colo-829 progenies and their parental cells were gifts from Richard Marais (62).
Xenograft experiments
Two million cells in 200 μL Hank’s balanced salt solution were subcutaneously inoculated into the rear flanks of 5-week-old female athymic nude (nu/nu) mice (The Jackson laboratory). Once palpable, tumors were measured using Vernier calipers every other day. Tumor volumes were calculated using the formula: length×width×height×0.5236. When tumor volumes reached about 50 mm3, mice bearing each cell line were sorted into three groups to achieve equal distribution of tumor size in all the treatment groups. Group 1 intraperitoneally received the vehicle (1:1.8:0.4:6.8 mixture of DMSO, Kolliphor RH40, dextrose, and HEPES) while groups 2 and 3 received JG231 (2 and 4 mg/kg body weight/dose, respectively). A375, A375-PLX/R, and Colo-829 received 9, 7, and 12 doses, respectively. At the end of the experiment, animals were euthanized by CO2 asphyxiation. All animal studies were performed according to the protocols approved by the Institutional Animal Care and Use Committee at Medical College of Wisconsin, Milwaukee, WI, USA.
Plasmids, RNA interference, and recombinant viruses
Primers used for cloning and mutagenesis are listed in table S2. Generation of pHAGE-BRAFV600E was previously reported (63). pHAGE-BRAFWT was generated by mutagenesis of pHAGE-BRAFV600E. Construction of pHAGE expression systems for N-terminally HA-tagged mortalin, ATPase domain, subdomain 2, peptide-binding domain, PBD-Δtail, and PBD-V482F was previously described (21, 22). C-terminally HA-tagged mortalin-encoding gene was generated by PCR and ligated into the NotI/XhoI sites of pHAGE. To construct pTRIPZ-dox-PBD, HA-tagged PBD-encoding gene were generated by PCR and obtained PCR product was used to replace the turboRFP in the AgeI/ClaI sites of pTRIPZ-dox. Generation of pHAGE-HA-HSC70 was previously described (22). HSC70 domain mutants in pHAGE were generated by PCR amplification of genes encoding ATPase domain (a.a. 1–391) and peptide-binding domain (a.a. 392–679), and by replacing HSPA8 in the NheI/BamHI sites of pHAGE-HA-HSC70 with obtained PCR products. Generation of pGEX-6-PBD and pGEX-6-PBD-V482F was previously described (21). pHAGE-HA-ANT3 was generated by PCR amplifying human ANT3 and by replacing HSPA9 in the NheI/BamHI sites of pHAGE-HA-Mort with obtained PCR product. pHAGE-AKT1-WT, pHAGE-AKT1-E40K, pLL3.7-shERK2, and pLL3.7-shMort constructs were previously described (22, 57, 64). pLKO.1-shANT3 (TRCN0000045014 and TRCN0000045015), pLKO.1-shCypD (TRCN0000232681, TRCN0000232682 and TRCN0000232684), and pLKO.1-shMCU (TRCN0000133861 and TRCN0000416434) constructs were purchased from Sigma-Aldrich. Lentivirus was generated and used as previously described (57).
Immunoprecipitation, immunoblotting, and In Vitro binding assays
For immunoprecipitation, cell lysates in 50 mM Tris (pH 7.5)/150 mM NaCl/1% NP-40 containing protease inhibitor (4-(2-aminoethyl)benzenesulfonyl fluoride, pepstatin A, E-64, bestatin, leupeptin, and aprotinin) and phosphatase inhibitor (cantharidin, calyculin A, and (–)-p-bromolevamisole oxalate) cocktails (Sigma-Aldrich) were immunoprecipitated using specific antibodies and Protein A/G Plus-agarose beads (Santa Cruz Biotechnology). For immunoblotting, equal amount of proteins in cell lysates in 62.5 mM Tris (pH 6.8)/2% SDS containing the protease and phosphatase inhibitor cocktails were resolved by SDS-PAGE, transferred to polyvinylidene difluoride membranes (Bio-Rad), and blocked with 5% nonfat dry milk or BSA in 0.1 M Tris (pH 7.5)/0.9% NaCl/0.05% Tween 20 prior to blotting. Antibodies and their dilutions are listed in table S3. For In vitro binding assay, 0.5 μM recombinant proteins were incubated at room temperature in 50 mM Tris (pH 7.4)/100 mM KCl/100 mM NaCl/5 mM MgCl2/5% glycerol/10 mM β-mercaptoethanol for 30 min and were subject to pulldown by 50 μL Glutathione-Sepharose 4B (GE Healthcare) for 1 hour at 4°C with gentle agitation. GST (Sigma-Aldrich) was used as the control. Chemiluminescence signals of immunoblots were visualized by SuperSignal West Pico and Femto chemiluminescence kits (Thermo Fisher Scientific), captured by ChemiDoc XRS+ (Bio-Rad), and analyzed by Image Lab software (Bio-Rad) for densitometry.
Chemicals
Chemicals, peptides, and other reagents are listed in table S4.
Recombinant proteins
BL21(DE3) transformed with pGEX-6P expressing GST-tagged mortalin-PBD, or PBD-V482F was grown in Luria-Bertani medium at 37°C for 16 hours. Protein expression was induced with 1 mM isopropyl-β-D-thiogalactopyranoside for 16 hours at 16°C. Cell pellets were collected by centrifugation, lysed in 50 mM Tris (pH 7.5)/150 mM NaCl/0.5% Triton X-100/1 mM EDTA/1 mM dithiothreitol/1 mM phenylmethylsulfonyl fluoride, and sonicated. Supernatants were loaded onto 5 mL GSTrap column (GE Healthcare), washed with 50 mM Tris (pH 7.5)/500 mM NaCl/0.5% Triton X-100/1 mM EDTA, and eluted by 0–100% gradients of 50 mM Tris (pH 7.5)/50 mM NaCl/0.5% Triton X-100/1 mM EDTA/10 mM DTT/50 mM reduced glutathione/10 % glycerol using AKTA FPLC (GE Healthcare). Elutes were dialyzed (8 kDa cutoff) in 20 mM Tris (pH 7.5)/100 mM NaCl/0.05% Triton X-100/5 mM β-mercaptoethanol at 4°C for 16 hours prior to PreScission protease (GE Healthcare) treatment. Proteolysis fractions were separated by glutathione-agarose column (Gold Biotechnology, St. Louis, MO) and the flow-through was concentrated by 30 kDa cutoff centricon (EMD Millipore). Purity of recombinant proteins was determined by SDS-PAGE using Stain-free fluorescent gel (Bio-Rad). Full length recombinant mortalin (65) was obtained from Abdussalam Azem (Tel Aviv Univ).
Cell viability, cycle, and death analyses
Cell viability was determined by trypan blue exclusion assay (Invitrogen) or by flow cytometry of cells stained with the fluorescent DNA intercalator TO-PRO-3 (Invitrogen). The colorimetric 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyltetrazolium bromide (MTT) assay was performed as previously described (59). For cell cycle and death analyses, flow cytometry of cells stained with annexin V and propidium iodide (Invitrogen) was performed using the Guava EasyCyte flowcytometry system (MilliporeSigma), as previously described (66). Data were analyzed by FCS EXPRESS software (De Novo Software).
Calcein retention assay and mitochondrial membrane potential measurement
MPTP opening and mitochondrial membrane potential were analyzed using MitoProbe Transition Pore Assay kit (Thermo Fisher Scientific) and tetramethylrhodamine, methyl ester (TMRM, Thermo Fisher Scientific), respectively, according to the manufacturer’s instruction. Calcein fluorescence retaining in cells after cobalt quenching and the fluorescence in TMRM-stained cells were determined by the Guava EasyCyte flowcytometry system (MilliporeSigma) and FCS EXPRESS software (De Novo Software).
Isolation of mortalin interactome and mass spectrometry
Lysates of cells infected with pHAGE virus expressing HA-tagged mortalin constructs were subject to immunoprecipitation using agarose beads conjugated with anti-HA antibody (Thermo Fisher Scientific) and, subsequently, to SDS-PAGE, as we previously described (22).
Silver-stained gel bands were excised, destained, reduced, and tryptic-digested. Peptides were extracted from these gel pieces using trifluoroacetic acid and acetonitrile, dried under vacuum prior to MS analyses using an Eksigent nanoLC in line with an LTQ Orbitrap Velos (Thermo Fisher Scientific). Two technical replicate injections of 2 μL in succession were performed using instrument settings outlined in table S1. MS-grade solvents and reagents were used. MS data were analyzed using Proteome Discoverer 2.2 (Thermo Fisher Scientific) according to details in table S1. All files for separate bands from a single lane (meaning the same sample) were searched together to produce a single output for each lane.
Analysis of proteomics data
Ingenuity Pathway Analysis software (https://www.qiagenbioinformatics.com/products/ingenuity-pathway-analysis/) was used to catalog proteins in different functional categories. Statistical significance (p value) was determined with 5% False Discovery Rate cutoff, as instructed by the supplier (http://qiagen.force.com/KnowledgeBase/KnowledgeIPAPage#). Molecular and cellular functions for each category are defined at http://qiagen.force.com/KnowledgeBase/KnowledgeIPAPage?id=kA1D0000000PIokKAG. Proteins in each category were then analyzed for their functional/physical interactions with the cutoff of confidence score 0.700 using STRING v11.0 software (67). Identified interaction clusters were visualized using the Markov Cluster (MCL) algorithm, inflation parameter = 3.0 (68). The overlap between interactomes was determined by jvenn (69).
Quantification and statistical analysis
All graphs represent the mean ± the standard error of the mean (SEM) of biological replicates. Statistical significance was determined by one-way ANOVA with Dunnett post-tests or two-way ANOVA with Bonferroni post-tests using Prism (GraphPad Software). IC50 and Confidence Intervals were determined by Prism. Gene expression and patient survival datasets from Jönsson study (70), Bhardwaj study (71), and TCGA-SKCM study (72) were analyzed by R2 Genomics Analysis and Visualization Platform (http://r2.amc.nl). p values of the Kaplan-Meier curves were determined by the log-rank test at R2.
Supplementary Material
Acknowledgements:
We thank Drs. Yuri Lazebnik (CSHL), Robert Weinberg (MIT), Richard Marais (Univ. Manchester), Richard Mulligan (Harvard Univ.), and Abdussalam Azem (Tel Aviv Univ.) for gifting us the IMR90E1A cells, MEL-ST cells, PLX4032-resistant melanoma cells, pHAGE, and recombinant mortalin, respectively. We also thank former Park laboratory members for technical support, and Drs. Albert Girotti (MCW) and Barry Nelkin (Johns Hopkins Univ.) for critical review of this manuscript.
Funding: This work was supported by NIH/National Cancer Institute grant (R01CA138441) to J.P. Development of HSP70 modulators was funded by NIH R01NS059690 to J.G.
Footnotes
This manuscript has been accepted for publication in Science Signaling. This version has not undergone final editing. Please refer to the complete version of record at https://urldefense.com/v3/__http://www.sciencesignaling.org/__;!!H8mHWRdzp34!sUHHuTeVklu0FDBRgc8ajouh4Yydci0l4jmmHRNWh1GGaAwqfAgnexb-Ac3eQTM$. The manuscript may not be reproduced or used in any manner that does not fall within the fair use provisions of the Copyright Act without the prior, written permission of AAAS.
Competing interests: J.G is an inventor on a US patent application that includes HSP70 modulators. All other authors declare that they have no competing interests.
Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials. JG series HSP70 modulators are available from J.G. under a material transfer agreement with UCSF. Mass spectrometry raw data (.RAW files) generated by this study are deposited in the MassIVE database (https://massive.ucsd.edu/ProteoSAFe/static/massive.jsp) and will be open to public upon publication of this report. Accession number, MassIVE: MSV000083228_reviewer; Password, parklabMCW.
REFERENCES AND NOTES
- 1.Karoulia Z, Gavathiotis E, Poulikakos PI, New perspectives for targeting RAF kinase in human cancer. Nat Rev Cancer 17, 676–691 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roskoski R Jr., Allosteric MEK1/2 inhibitors including cobimetanib and trametinib in the treatment of cutaneous melanomas. Pharmacol Res 117, 20–31 (2017). [DOI] [PubMed] [Google Scholar]
- 3.Kidger AM, Sipthorp J, Cook SJ, ERK1/2 inhibitors: New weapons to inhibit the RAS-regulated RAF-MEK1/2-ERK1/2 pathway. Pharmacol Ther 187, 45–60 (2018). [DOI] [PubMed] [Google Scholar]
- 4.Sale MJ, Cook SJ, Intrinsic and acquired resistance to MEK1/2 inhibitors in cancer. Biochem Soc Trans 42, 776–783 (2014). [DOI] [PubMed] [Google Scholar]
- 5.Leanza L, Zoratti M, Gulbins E, Szabo I, Mitochondrial ion channels as oncological targets. Oncogene 33, 5569–5581 (2014). [DOI] [PubMed] [Google Scholar]
- 6.Kroemer G, Galluzzi L, Brenner C, Mitochondrial membrane permeabilization in cell death. Physiol Rev 87, 99–163 (2007). [DOI] [PubMed] [Google Scholar]
- 7.Chevrollier A, Loiseau D, Reynier P, Stepien G, Adenine nucleotide translocase 2 is a key mitochondrial protein in cancer metabolism. Biochim Biophys Acta 1807, 562–567 (2011). [DOI] [PubMed] [Google Scholar]
- 8.Bonora M, Pinton P, A New Current for the Mitochondrial Permeability Transition. Trends Biochem Sci 44, 559–561 (2019). [DOI] [PubMed] [Google Scholar]
- 9.Neginskaya MA et al. , ATP Synthase C-Subunit-Deficient Mitochondria Have a Small Cyclosporine A-Sensitive Channel, but Lack the Permeability Transition Pore. Cell Rep 26, 11–17 e12 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.He J et al. , Persistence of the mitochondrial permeability transition in the absence of subunit c of human ATP synthase. Proc Natl Acad Sci U S A 114, 3409–3414 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hurst S, Baggett A, Csordas G, Sheu SS, SPG7 targets the m-AAA protease complex to process MCU for uniporter assembly, Ca(2+) influx, and regulation of mitochondrial permeability transition pore opening. J Biol Chem 294, 10807–10818 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Green DR, Galluzzi L, Kroemer G, Cell biology. Metabolic control of cell death. Science 345, 1250256 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Daugaard M, Rohde M, Jaattela M, The heat shock protein 70 family: Highly homologous proteins with overlapping and distinct functions. FEBS Lett 581, 3702–3710 (2007). [DOI] [PubMed] [Google Scholar]
- 14.Lee AS, Glucose-regulated proteins in cancer: molecular mechanisms and therapeutic potential. Nat Rev Cancer 14, 263–276 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kaul SC, Deocaris CC, Wadhwa R, Three faces of mortalin: a housekeeper, guardian and killer. Exp Gerontol 42, 263–274 (2007). [DOI] [PubMed] [Google Scholar]
- 16.Na Y et al. , Stress chaperone mortalin contributes to epithelial-mesenchymal transition and cancer metastasis. Cancer Res 76, 2754–2765 (2016). [DOI] [PubMed] [Google Scholar]
- 17.Yun CO et al. , Relevance of mortalin to cancer cell stemness and cancer therapy. Sci Rep 7, 42016 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wadhwa R et al. , Upregulation of mortalin/mthsp70/Grp75 contributes to human carcinogenesis. Int J Cancer 118, 2973–2980 (2006). [DOI] [PubMed] [Google Scholar]
- 19.Rozenberg P et al. , Elevated levels of mitochondrial mortalin and cytosolic HSP70 in blood as risk factors in patients with colorectal cancer. Int J Cancer 133, 514–518 (2013). [DOI] [PubMed] [Google Scholar]
- 20.Chen J et al. , Overexpression of Mortalin in hepatocellular carcinoma and its relationship with angiogenesis and epithelial to mesenchymal transition. Int J Oncol 44, 247–255 (2014). [DOI] [PubMed] [Google Scholar]
- 21.Wu PK, Hong SK, Park JI, Steady-State Levels of Phosphorylated Mitogen-Activated Protein Kinase Kinase 1/2 Determined by Mortalin/HSPA9 and Protein Phosphatase 1 Alpha in KRAS and BRAF Tumor Cells. Mol Cell Biol 37, e00061–00017 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu PK et al. , A Mortalin/HSPA9-Mediated Switch in Tumor-Suppressive Signaling of Raf/MEK/Extracellular Signal-Regulated Kinase. Mol Cell Biol 33, 4051–4067 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Karkhanis M, Park JI, Sp1 regulates Raf/MEK/ERK-induced p21 transcription in TP53-mutated cancer cells. Cell Signal 27, 479–486 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Starenki D, Hong SK, Lloyd RV, Park JI, Mortalin (GRP75/HSPA9) upregulation promotes survival and proliferation of medullary thyroid carcinoma cells. Oncogene 34, 4624–4634 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rosen A, Casciola-Rosen L, Macromolecular substrates for the ICE-like proteases during apoptosis. J Cell Biochem 64, 50–54 (1997). [DOI] [PubMed] [Google Scholar]
- 26.Kampinga HH, Craig EA, The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat Rev Mol Cell Biol 11, 579–592 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ryu J et al. , Identification and functional characterization of nuclear mortalin in human carcinogenesis. J Biol Chem 289, 24832–24844 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Iosefson O, Azem A, Reconstitution of the mitochondrial Hsp70 (mortalin)-p53 interaction using purified proteins--identification of additional interacting regions. FEBS Lett 584, 1080–1084 (2010). [DOI] [PubMed] [Google Scholar]
- 29.Laufen T et al. , Mechanism of regulation of hsp70 chaperones by DnaJ cochaperones. Proc Natl Acad Sci U S A 96, 5452–5457 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Crompton M, Barksby E, Johnson N, Capano M, Mitochondrial intermembrane junctional complexes and their involvement in cell death. Biochimie 84, 143–152 (2002). [DOI] [PubMed] [Google Scholar]
- 31.Halestrap AP, Brenner C, The adenine nucleotide translocase: a central component of the mitochondrial permeability transition pore and key player in cell death. Curr Med Chem 10, 1507–1525 (2003). [DOI] [PubMed] [Google Scholar]
- 32.Halestrap AP, Connern CP, Griffiths EJ, Kerr PM, Cyclosporin A binding to mitochondrial cyclophilin inhibits the permeability transition pore and protects hearts from ischaemia/reperfusion injury. Mol Cell Biochem 174, 167–172 (1997). [PubMed] [Google Scholar]
- 33.Santo-Domingo J et al. , The plasma membrane Na+/Ca2+ exchange inhibitor KB-R7943 is also a potent inhibitor of the mitochondrial Ca2+ uniporter. Br J Pharmacol 151, 647–654 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Broekemeier KM, Krebsbach RJ, Pfeiffer DR, Inhibition of the mitochondrial Ca2+ uniporter by pure and impure ruthenium red. Mol Cell Biochem 139, 33–40 (1994). [DOI] [PubMed] [Google Scholar]
- 35.Weaver JG et al. , Inhibition of adenine nucleotide translocator pore function and protection against apoptosis in vivo by an HIV protease inhibitor. J Clin Invest 115, 1828–1838 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Utkina-Sosunova IV et al. , Nelfinavir inhibits intra-mitochondrial calcium influx and protects brain against hypoxic-ischemic injury in neonatal mice. PLoS One 8, e62448 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Halestrap AP, Davidson AM, Inhibition of Ca2(+)-induced large-amplitude swelling of liver and heart mitochondria by cyclosporin is probably caused by the inhibitor binding to mitochondrial-matrix peptidyl-prolyl cis-trans isomerase and preventing it interacting with the adenine nucleotide translocase. The Biochemical journal 268, 153–160 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brustovetsky N, Klingenberg M, Mitochondrial ADP/ATP carrier can be reversibly converted into a large channel by Ca2+. Biochemistry 35, 8483–8488 (1996). [DOI] [PubMed] [Google Scholar]
- 39.Vaseva AV et al. , p53 opens the mitochondrial permeability transition pore to trigger necrosis. Cell 149, 1536–1548 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu X, Kim CN, Yang J, Jemmerson R, Wang X, Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell 86, 147–157 (1996). [DOI] [PubMed] [Google Scholar]
- 41.Koya K et al. , MKT-077, a novel rhodacyanine dye in clinical trials, exhibits anticarcinoma activity in preclinical studies based on selective mitochondrial accumulation. Cancer Res 56, 538–543 (1996). [PubMed] [Google Scholar]
- 42.Rousaki A et al. , Allosteric drugs: the interaction of antitumor compound MKT-077 with human Hsp70 chaperones. J Mol Biol 411, 614–632 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Modica-Napolitano JS, Aprille JR, Delocalized lipophilic cations selectively target the mitochondria of carcinoma cells. Adv Drug Deliv Rev 49, 63–70 (2001). [DOI] [PubMed] [Google Scholar]
- 44.Wadhwa R et al. , Selective toxicity of MKT-077 to cancer cells is mediated by its binding to the hsp70 family protein mot-2 and reactivation of p53 function. Cancer Res 60, 6818–6821 (2000). [PubMed] [Google Scholar]
- 45.Maruta H, Tikoo A, Shakri R, Shishido T, The anti-RAS cancer drug MKT-077 is an F-actin cross-linker. Ann N Y Acad Sci 886, 283–284 (1999). [DOI] [PubMed] [Google Scholar]
- 46.Shao H et al. , Exploration of Benzothiazole Rhodacyanines as Allosteric Inhibitors of Protein-Protein Interactions with Heat Shock Protein 70 (Hsp70). J Med Chem 61, 6163–6177 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Khalil AA, Kabapy NF, Deraz SF, Smith C, Heat shock proteins in oncology: Diagnostic biomarkers or therapeutic targets? Biochim Biophys Acta 1816, 89–104 (2011). [DOI] [PubMed] [Google Scholar]
- 48.Wang T et al. , Chaperome heterogeneity and its implications for cancer study and treatment. J Biol Chem 294, 2162–2179 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rizzolo K, Houry WA, Multiple functionalities of molecular chaperones revealed through systematic mapping of their interaction networks. J Biol Chem 294, 2142–2150 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Halestrap AP, Woodfield KY, Connern CP, Oxidative stress, thiol reagents, and membrane potential modulate the mitochondrial permeability transition by affecting nucleotide binding to the adenine nucleotide translocase. J Biol Chem 272, 3346–3354 (1997). [DOI] [PubMed] [Google Scholar]
- 51.Vazquez F et al. , PGC1alpha Expression Defines a Subset of Human Melanoma Tumors with Increased Mitochondrial Capacity and Resistance to Oxidative Stress. Cancer Cell 23, 287–301 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Haq R, Fisher DE, Widlund HR, Molecular pathways: BRAF induces bioenergetic adaptation by attenuating oxidative phosphorylation. Clin Cancer Res 20, 2257–2263 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Haq R et al. , Oncogenic BRAF regulates oxidative metabolism via PGC1alpha and MITF. Cancer Cell 23, 302–315 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sun J et al. , Mortalin overexpression predicts poor prognosis in early stage of non-small cell lung cancer. Tumour Biol 39, 1010428317695918 (2017). [DOI] [PubMed] [Google Scholar]
- 55.Xu M et al. , Mortalin is a distinct bio-marker and prognostic factor in serous ovarian carcinoma. Gene 696, 63–71 (2019). [DOI] [PubMed] [Google Scholar]
- 56.Cui X et al. , Mortalin expression in pancreatic cancer and its clinical and prognostic significance. Hum Pathol 64, 171–178 (2017). [DOI] [PubMed] [Google Scholar]
- 57.Hong SK, Yoon S, Moelling C, Arthan D, Park JI, Noncatalytic function of ERK1/2 can promote Raf/MEK/ERK-mediated growth arrest signaling. J Biol Chem 284, 33006–33018 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wu PK, Hong SK, Yoon SH, Park JI, Active ERK2 is sufficient to mediate growth arrest and differentiation signaling. FEBS J 282, 1017–1030 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hong SK, Wu PK, Park JI, A cellular threshold for active ERK1/2 levels determines Raf/MEK/ERK-mediated growth arrest versus death responses. Cell Signal 42, 11–20 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gupta PB et al. , The melanocyte differentiation program predisposes to metastasis after neoplastic transformation. Nat Genet 37, 1047–1054 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Samuels ML, Weber MJ, Bishop JM, McMahon M, Conditional transformation of cells and rapid activation of the mitogen-activated protein kinase cascade by an estradiol-dependent human raf-1 protein kinase. Mol Cell Biol 13, 6241–6252 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Girotti MR et al. , Inhibiting EGF receptor or SRC family kinase signaling overcomes BRAF inhibitor resistance in melanoma. Cancer Discov 3, 158–167 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hong SK, Kim JH, Lin MF, Park JI, The Raf/MEK/extracellular signal-regulated kinase 1/2 pathway can mediate growth inhibitory and differentiation signaling via androgen receptor downregulation in prostate cancer cells. Exp Cell Res 317, 2671–2682 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hong SK, Jeong JH, Chan AM, Park JI, AKT upregulates B-Raf Ser445 phosphorylation and ERK1/2 activation in prostate cancer cells in response to androgen depletion. Exp Cell Res 319, 1732–1743 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Iosefson O, Sharon S, Goloubinoff P, Azem A, Reactivation of protein aggregates by mortalin and Tid1--the human mitochondrial Hsp70 chaperone system. Cell Stress Chaperones 17, 57–66 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Starenki D, Park JI, Mitochondria-targeted nitroxide, Mito-CP, suppresses medullary thyroid carcinoma cell survival in vitro and in vivo. J Clin Endocrinol Metab 98, 1529–1540 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Szklarczyk D et al. , STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res 43, D447–452 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Brohee S, van Helden J, Evaluation of clustering algorithms for protein-protein interaction networks. BMC Bioinformatics 7, 488 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bardou P, Mariette J, Escudie F, Djemiel C, Klopp C, jvenn: an interactive Venn diagram viewer. BMC Bioinformatics 15, 293 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cirenajwis H et al. , Molecular stratification of metastatic melanoma using gene expression profiling: Prediction of survival outcome and benefit from molecular targeted therapy. Oncotarget 6, 12297–12309 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bogunovic D et al. , Immune profile and mitotic index of metastatic melanoma lesions enhance clinical staging in predicting patient survival. Proc Natl Acad Sci U S A 106, 20429–20434 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cancer Genome Atlas N, Genomic Classification of Cutaneous Melanoma. Cell 161, 1681–1696 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.