

Abstract
Sepsis-induced diaphragm dysfunction contributes to respiratory failure and mortality in critical illness. There are no treatments for this form of diaphragm weakness. Studies show that sepsis-induced muscle dysfunction is triggered by enhanced mitochondrial free radical generation. We tested the hypothesis that SS31, a mitochondrially targeted antioxidant, would attenuate sepsis-induced diaphragm dysfunction. Four groups of mice were studied: 1) sham-operated controls, 2) sham-operated+SS31 (10 mg·kg−1·day−1), 3) cecal ligation puncture (CLP), and 4) CLP+SS31. Forty-eight hours postoperatively, diaphragm strips with attached phrenic nerves were isolated, and the following were assessed: muscle-field-stimulated force-frequency curves, nerve-stimulated force-frequency curves, and muscle fatigue. We also measured calpain activity, 20S proteasomal activity, myosin heavy chain (MHC) levels, mitochondrial function, and aconitase activity, an index of mitochondrial superoxide generation. Sepsis markedly reduced diaphragm force generation; SS31 prevented these decrements. Diaphragm-specific force generation averaged 30.2 ± 1.4, 9.4 ± 1.8, 25.5 ± 2.3, and 27.9 ± 0.6 N/cm2 for sham, CLP, sham+SS31, and CLP+SS31 groups (P < 0.001). Similarly, with phrenic nerve stimulation, CLP depressed diaphragm force generation, effects prevented by SS31. During endurance trials, force was significantly reduced with CLP, and SS31 prevented these reductions (P < 0.001). Sepsis also increased diaphragm calpain activity, increased 20S proteasomal activity, decreased MHC levels, reduced mitochondrial function (state 3 rates and ATP generation), and reduced aconitase activity; SS31 prevented each of these sepsis-induced alterations (P ≤ 0.017 for all indices). SS31 prevents sepsis-induced diaphragm dysfunction, preserving force generation, endurance, and mitochondrial function. Compounds with similar mechanisms of action may be useful therapeutically to preserve diaphragm function in patients who are septic and critically ill.
NEW & NOTEWORTHY Sepsis-induced diaphragm dysfunction is a major contributor to mortality and morbidity in patients with critical illness in intensive care units. Currently, there is no proven pharmacological treatment for this problem. This study provides the novel finding that administration of SS31, a mitochondrially targeted antioxidant, preserves diaphragm myosin heavy chain content and mitochondrial function, thereby preventing diaphragm weakness and fatigue in sepsis.
Keywords: diaphragm fatigue, diaphragm force generation, neuromuscular transmission, sepsis, SS31
INTRODUCTION
Recent studies reveal that patients in mechanically ventilated, medical intensive care units develop profound diaphragm muscle weakness (11, 18, 20, 41, 47, 49). Diaphragm weakness is associated with prolonged duration of mechanical ventilation in this patient population (41, 47), and diaphragm weakness also correlates with mortality. Mortality rates are significantly higher when profound diaphragm weakness is present, and patients with stronger diaphragm muscles have far lower rates of death (41, 43, 47). Several factors are thought to increase the risk for developing diaphragm weakness in critical illness, including use of mechanical ventilation per se (i.e., ventilator-induced diaphragm dysfunction, VIDD; Refs. 23, 28, 30), the combined use of steroids and neuromuscular blocking agents (1), and electrolyte imbalances (e.g., hypophosphatemia; Ref. 2). Sepsis, however, is a major risk factor for diaphragm weakness (13, 27, 41, 43, 47), with patients with sepsis having an average level of diaphragm force generation that is less than half that of patients without sepsis (41). Although these previous studies have better defined the causes and consequences of diaphragm weakness, this prior work has failed to identify a proven therapy to prevent or reverse weakness in patients who are critically ill.
Animal studies suggest that sepsis-induced diaphragm dysfunction may be linked to alterations in mitochondrial free radical generation (6, 8, 37). This previous work has shown that sepsis activates upstream processes (e.g., cytokine-induced muscle neutral sphingomyelinase activation), which lead to increased mitochondrial free radical generation (38). Mitochondrial generation of superoxide and other reactive oxygen species (ROS), in turn, are linked to activation of proteolytic pathways (i.e., calpain and the proteasome; Refs. 33, 40). It has also been suggested that mitochondrial ROS may damage components of the mitochondrial electron transport chain, leading to reductions in the capacity of mitochondria to achieve high state 3 respiration rates (9). In theory, increased proteolytic activity could lead to loss of muscle proteins, engendering weakness, and loss of mitochondrial electron transport chain constituents could lead to reduced ATP generation, producing increased fatigability (9, 45).
Based on this prior work, administration of agents that either scavenge or prevent generation of mitochondrial ROS should theoretically be capable of preventing sepsis-induced muscle dysfunction. SS31 (d-Arg-Dmt-Lys-Phe-NH2; Dmt, 2′,6′-dimethyltyrosine) is a novel cell-permeable antioxidant that achieves high mitochondrial concentrations (3, 15, 48). The purpose of the present study, therefore, was to test the hypothesis that administration of SS31 would ameliorate sepsis-induced diaphragm weakness and fatigability. Sepsis was induced using the cecal ligation puncture (CLP) model (38). Four groups of mice were studied: sham-operated vehicle-treated controls, CLP vehicle-treated, sham-operated SS31-treated, and CLP SS31-treated. At 48 h after induction of sepsis (or sham operation), animals were euthanized, and diaphragms were excised to permit assessment of diaphragm function, mass, proteolytic enzyme activity, myosin heavy chain content, and mitochondrial function. Results from the four groups were compared to determine whether SS31 administration attenuates sepsis-induced diaphragm weakness and fatigue.
METHODS
Experimental protocols.
All experiments were approved by the University of Kentucky Institutional Animal Care and Use Committee. Male CD1 mice (25–30 g) were obtained from Charles River (Wilmington, MA). We studied four groups (n = 5–6 mice per group) including: 1) sham-operated animals treated with vehicle (saline 0.2 mL ip), 2) cecal ligation puncture (CLP)-operated animals treated with vehicle, 3) sham-operated animals treated with SS31 (10 mg·kg−1·day−1 ip in saline), and 4) CLP animals treated with SS31 (10 mg·kg−1·day−1 ip in saline, injected 2–5 min after surgery). Forty-eight hours after sham or CLP surgery, animals were euthanized and costal diaphragm muscle strips with intact phrenic nerves were dissected for in vitro determinations of diaphragm force generation. The remaining costal diaphragm was frozen, stored at −80°C, and subsequently used to determine diaphragm protein levels, proteolytic enzyme activities, and myosin heavy chain levels. The SS31 dosage chosen for this study was selected because it is similar to that used in previous animal studies (10).
Cecal ligation puncture-induced sepsis.
Induction anesthesia was achieved with 2–4% isoflurane delivered via an anesthetic gas vaporizer. After achieving a steady plane of anesthesia, the surgical area was prepped using sterile technique and a 1-cm incision was made in the abdomen. The cecum was identified, and a portion was ligated (1.0 cm). The ligated cecum was punctured through-and-through with an 18-gauge sterile needle, and a small amount of feces was expressed through the hole. The abdominal musculature was approximated and sutured, followed by closure of the skin with surgical staples. For sham surgeries, the abdomen was opened and closed without cecal ligation or puncture. All animals were resuscitated with 60 mL/kg saline administered subcutaneously following the procedure and then immediately treated with analgesics (buprenorphine 0.05 mg/kg subcutaneously) and every 12 h thereafter until the time of euthanasia. Animals were euthanized at 48 h after surgery using an intraperitoneal overdose of pentobarbital (150 mg/kg).
Assessment of diaphragm strength, neuromuscular transmission, and fatigue.
For diaphragm force measurements, diaphragm strips with attached phrenic nerves were dissected from the left midcostal diaphragm and mounted in water-jacketed glass organ baths containing a physiological solution without curare at 25°C as previously described (7, 37–39, 45). One end of each strip was tied to the base of the organ bath and the other to a force transducer. The intact phrenic nerve was placed in a cuff electrode, and platinum field stimulation electrodes were placed around the muscle. Supramaximal currents from a constant current amplifier connected to a Grass S48 stimulator (West Warwick, RI) were delivered using phrenic nerve and muscle field electrodes. Supramaximal muscle field stimulation currents were in the 0.36–0.54 mA·s range. After an equilibration period (15 min), muscle length was adjusted to Lo, defined as the length at which twitch muscle force generation was maximal. Lo for diaphragm strips ranged from 0.9 to 1.1 cm.
To determine muscle force-frequency relationships, diaphragm muscles were first stimulated via the phrenic nerve with trains of 1-, 20-, and 50-Hz stimuli (train duration 800 ms, 30 s between adjacent trains). Muscles were then stimulated via direct muscle field stimulation with trains of 1, 10, 20, 50, 100, 150, and 200 Hz (train duration 800 ms, 30 s between adjacent trains). Force was recorded with a BIOPAC MP150 transducer data acquisition system (BIOPAC Systems, Goleta, CA).
To assess neuromuscular transmission, we employed the technique described by Greising et al. (16). For this measurement, the diaphragm was made to repetitively contract in response to phrenic nerve stimulation (1 train of phrenic nerve stimuli per second) with intermittent superimposition of field muscle stimulation (1 train of field muscle stimulation every 10 s). The rates of decline of force in response to nerve (NF) and field muscle (MF) stimulation were determined using the formula transmission failure index = 100 × (NF − MF)/(1 − MF), as previously described (16, 17). After a 15-min rest, diaphragm fatigability in response to supramaximal muscle field stimulation (trains of 330 ms, 40-Hz stimulation, at a rate of 1 train per second over 2 min) was assessed.
Muscle cross-sectional area was calculated as muscle strip weight divided by muscle density (1.06) and muscle length; muscle-specific force was calculated as force divided by cross-sectional area. The total weight of the costal diaphragm was determined. Diaphragm tissues not used for force determinations were frozen, stored at −80°C, and subsequently used for assays.
Calpain activity assay.
Calpain activity was assessed as previously described in detail (40, 44, 46). Briefly, diaphragm muscle homogenates (100 μg of protein) and a calpain-specific fluorogenic substrate [succinyl-Leu-Leu-Val-Tyr-7-amino-4-methylcoumarin (Suc-LLVY-AMC)] were added to assay buffer. Duplicate determinations were made with muscle homogenate, assay buffer, Suc-LLVY-AMC, and a highly specific calpain inhibitor (0.1 mg/mL calpain inhibitor III). Immediately after substrate was added, baseline fluorescent measurements of AMC (excitation frequency of 360 nm and an emission frequency of 460 nm) were performed using a spectrofluorophotometer (Molecular Devices, San Jose, CA). Measurements were repeated after 0.5 h of incubation at 30°C. The change in AMC fluorescence from the initial reading to the final reading was calculated to obtain the raw increase in fluorescence; the increase in reading for the calpain inhibitor III duplicate was subtracted from the raw reading to determine the calpain activity for an individual sample. AMC standards were used to calibrate fluorescent measurements of calpain activity.
20S proteasome subunit activity assay.
Proteasome activity of diaphragm homogenates was measured using a commercially available kit (20S proteasome subunit activity kit; Calbiochem, San Diego, CA) as previously reported (39, 44). The manufacturer’s protocol was followed, and AMC standards were used to calibrate fluorescent measurements of proteasomal activity.
Protein determination by Western blotting.
Western blotting was employed to measure myosin heavy chain (MHC) protein levels. Briefly, muscles were homogenized in buffer, and protein levels were determined using the Bradford assay (Bio-Rad, Hercules, CA). The V3 Western Workflow system and ChemiDoc Touch Imaging System were used to perform Western blotting and analyses (Bio-Rad). Samples were diluted with an equal volume of 2× Laemmli loading buffer, and equal amounts of protein were loaded onto 7.5% Mini-PROTEAN TGX Stain-Free gels (Bio-Rad). To ensure proper loading conditions for assessment of changes in MHC content between the different experimental groups, a series of studies were performed to ensure that conditions were in the linear range of detection. MHC levels were probed with a MHC antibody (sc-20641; 1:50,000; Santa Cruz Biotechnology, Dallas, TX). MHC levels were normalized to the total protein loaded for each lane using Image Lab 5.2.1 software (Bio-Rad).
Mitochondrial function and aconitase activity.
For this assessment, diaphragm mitochondrial homogenates, prepared as previously described (42), were mixed with mitochondrial assay buffer and placed in a fiber-optic fluorescence monitoring system (Instech, Plymouth Meeting, PA). We then measured rates of oxygen consumption (states 3 and 4), respiratory control ratio (RCR), ADP-to-O ratios, and ATP production as previously described (42). Oxygen consumption rates were normalized to homogenate protein content. We also determined the aconitase activity of mitochondrial homogenates. Since aconitase is only present in mitochondria in skeletal muscle and mitochondrial superoxide generation reacts with and inactivates this enzyme (21), reductions in aconitase activity are an index of increased mitochondrial superoxide generation. Aconitase was measured using a commercially available kit according to the manufacturer’s instructions (Abcam, Cambridge, MA).
Statistical analysis.
ANOVA was used to compare variables (e.g., force) across groups of animals treated with different agents, with post hoc testing (Tukey) to determine differences between groups.
RESULTS
Diaphragm force generation.
We found that cecal ligation puncture (CLP)-induced sepsis elicited a large reduction in the force-generating capacity of the diaphragm, as evidenced by measurement of the force-frequency relationship of excised diaphragm muscle strips in response to direct supramaximal electrical field stimulation (Fig. 1). For example, the specific diaphragm force generated in response to 200-Hz muscle stimulation averaged 30.2 ± 1.4 N/cm2 for sham controls and was only 9.4 ± 1.8 N/cm2 for the CLP group (P < 0.001 for this comparison and for all other stimulation frequencies tested). Administration of SS31 ablated this sepsis-induced reduction in muscle force generation, with force generation in response to 200-Hz muscle stimulation for diaphragms from CLP+SS31-treated animals averaging 25.5 ± 2.3 N/cm2, a value similar to sham controls and significantly higher than the CLP group (P < 0.001 for this latter comparison). Similarly, force in response to muscle stimulation at all other frequencies tested (1–150 Hz) was also significantly higher for CLP+SS31 animals than for the CLP group (P < 0.001 for each comparison). Diaphragm force generation for sham+SS31-treated animals was similar to that for the sham controls (not significant, NS).
Fig. 1.
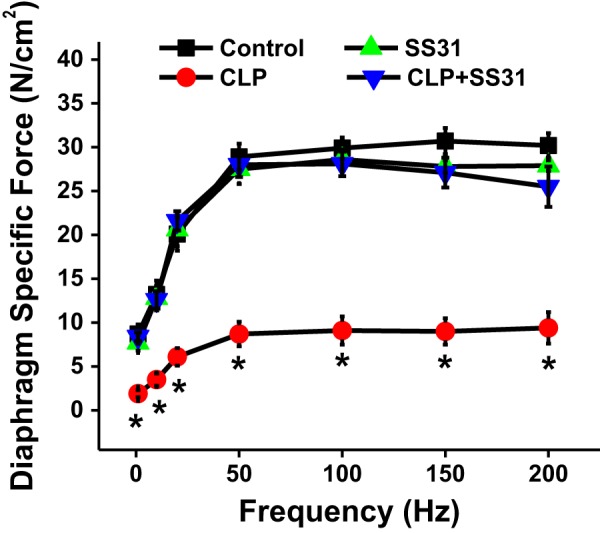
Diaphragm-specific force generation (i.e., force per cross-sectional area) as a function of direct muscle field stimulation frequency. Group mean data ±1 SE are shown for sham controls (black) and cecal ligation puncture (CLP) sepsis (red)-, sham+SS31 (green)-, and CLP+SS31 (blue)-treated groups (n = 5–6 mice per group). CLP-induced sepsis produced a large reduction in diaphragm force generation at all stimulation frequencies compared with values for sham controls (P < 0.001 for comparison at each stimulation frequency). SS31 administration prevented this sepsis-induced reduction in diaphragm force, with diaphragm-specific force at each stimulation frequency for the CLP+SS31-treated group higher than the value for the CLP group (P < 0.001 for each comparison) and similar to the value for sham controls and sham+SS31 (not significant for comparison at each stimulation frequency). *Statistical significance.
A similar pattern for the effects of CLP sepsis and SS31 on diaphragm force generation was observed when diaphragm muscle-specific force generation was assessed in response to supramaximal phrenic nerve stimulation (Fig. 2). Specifically, CLP sepsis resulted in a marked reduction in the diaphragm force generated in response to all frequencies of phrenic nerve stimulation tested (P < 0.01 at 1 Hz, P < 0.001 for 20 and 50 Hz) and administration of SS31 to CLP animals preserved diaphragm force, with diaphragm-specific force generation achieved at all frequencies of phrenic nerve stimulation markedly higher for the CLP+SS31 group compared with the CLP group (P < 0.02). Diaphragm force generation in response to phrenic nerve stimulation was similar between sham controls and sham+SS31 groups (NS for each stimulation frequency).
Fig. 2.
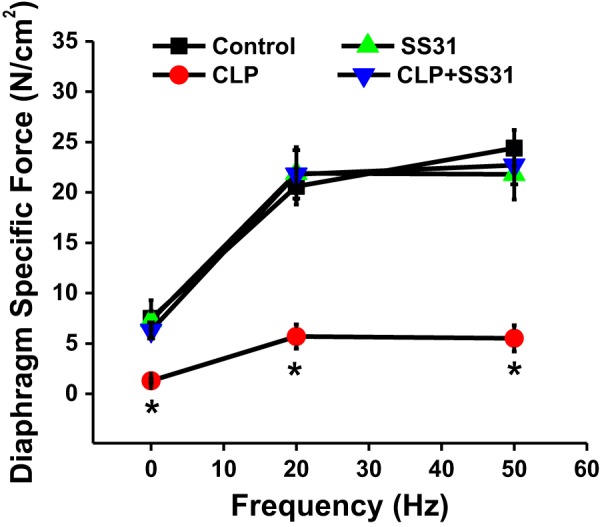
Diaphragm-specific force generation in response to phrenic nerve stimulation as a function of phrenic nerve stimulation frequency. Group mean data ±1 SE are shown for sham controls (black) and cecal ligation puncture (CLP) sepsis (red)-, sham+SS31 (green)-, and CLP+SS31 (blue)-treated groups (n = 5–6 mice per group). Phrenic nerve-stimulated diaphragm force generation was similar to that observed in response to direct muscle stimulation, with diaphragm force generation in response to sepsis markedly reduced compared with sham controls (P < 0.01 at 1 Hz, P < 0.001 at 20 and 50 Hz for comparison of these 2 groups). SS31 prevented sepsis-induced reductions in diaphragm force assessed in response to phrenic nerve stimulation, with force for CLP+SS31 significantly higher than that for CLP for each stimulation frequency (P < 0.02 at 1 Hz, P < 0.001 at 20 and 50 Hz). Phrenic nerve-stimulated diaphragm force generation for sham+SS31 was similar to that for sham controls (not significant). *Statistical significance.
Neuromuscular transmission.
Previous reports indicate that sepsis can sometimes alter neuromuscular transmission. To assess neuromuscular transmission in the phrenic nerve-diaphragm preparation, we employed the technique described by Greising et al. (16). For this assessment, the diaphragm was repetitively made to contract in response to phrenic nerve stimulation (1 train of phrenic stimuli per second) with intermittent superimposition of direct muscle field stimulation (1 train of field muscle stimulation every 10 s; Fig. 3A). We then calculated the relative rates of decline of force in response to nerve and field muscle stimulation using the formula for transmission failure index (TFI) as described by Greising et al. (16). We found that the TFI was not significantly different between sham controls and the CLP sepsis animals as shown in Fig. 3B. In addition, administration of SS31 to sham-operated or CLP animals did not significantly change the TFI (TFI similar across the 4 experimental groups; NS for each comparison).
Fig. 3.
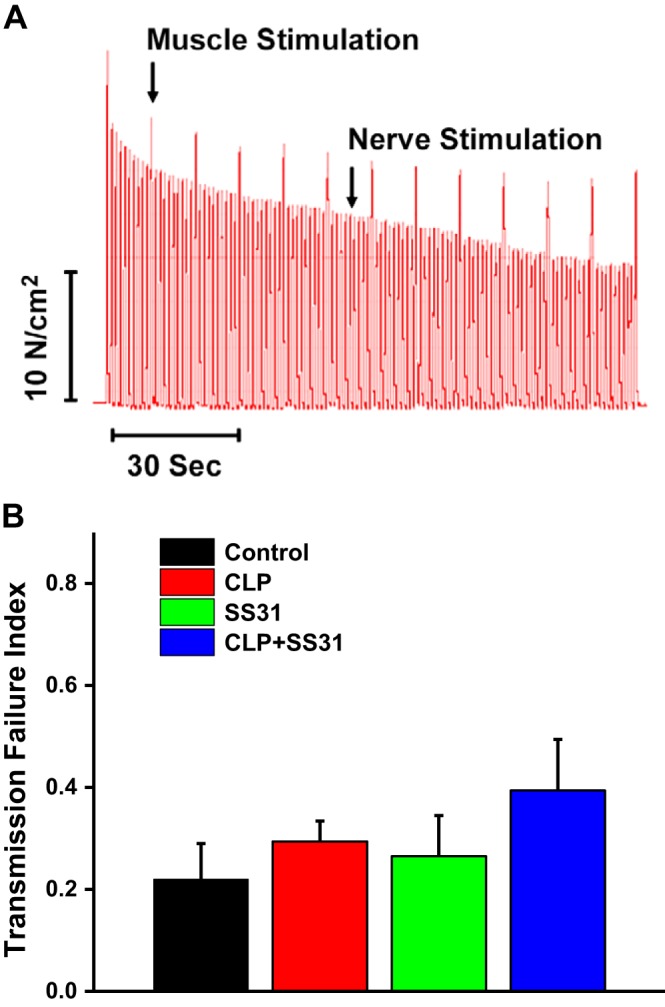
Neuromuscular transmission failure during a repetitive contraction trial for diaphragms taken from the 4 experimental groups performed according to the method of Greising et al. (16). As shown in A, there was a proportionately greater fall, over time, observed for force generated in response to phrenic nerve stimulation compared with direct muscle stimulation in this representative experiment. Similar results were observed for all diaphragm muscles taken from the 4 experimental groups. Group mean data ±1 SE for the transmission failure index (B) are shown for sham controls (black) and cecal ligation puncture (CLP) sepsis (red)-, sham+SS31 (green)-, and CLP+SS31 (blue)-treated groups. Neuromuscular transmission failure index was not statistically different between the 4 groups (n = 5–6 mice per group), suggesting that under the conditions tested in the current study, CLP sepsis did not alter diaphragm neuromuscular transmission.
Diaphragm fatigue.
To assess diaphragm fatigability, we repetitively stimulated diaphragm muscle strips using supramaximal muscle field stimulation for 2 min, as shown in Fig. 4 (stimulation with trains of 330 ms, 40-Hz stimulation, at a rate of 1 train per second over 2 min). Representative force-time curves for individual muscles taken from each of the 4 experimental groups are shown in Fig. 4A, whereas mean force-time curves for the 4 experimental groups are shown in Fig. 4B. Force-time curves were similar for sham, sham+SS31-treated, and CLP+SS31-treated groups (NS for comparison at each time point for these 3 groups). In contrast, specific force was severely reduced both initially and at all subsequent time points for the CLP group (P < 0.001 for comparison of force for this group compared with the other 3 experimental groups at baseline and at all subsequent time points).
Fig. 4.

Repetitive contraction trial of diaphragm muscle strips in response to direct muscle field stimulation was also performed for all diaphragm samples. A shows force-time curves from representative experiments for an animal from each experimental group. B displays group mean data ±1 SE for the force-time curves for sham controls (black) and cecal ligation puncture (CLP) sepsis (red), sham+SS31 (green), and CLP+SS31 (blue) groups (n = 5–6 mice per group). At all time points, the diaphragm-specific force generated by diaphragms taken from the CLP group was markedly lower than the force generated by muscles from the other 3 experimental groups (P < 0.001 for comparison). *Statistical significance.
To further measure the capacity of diaphragm strips to maintain force generation over time during repetitive muscle stimulation trials, we calculated the force-time integrals for the 4 experimental groups; these data are presented in Fig. 5A. The force-time integral for CLP animals was only 30% of the value of this index for sham controls (P < 0.001). We also found that SS31 administration dramatically improved the force-time integral, with this index for the CLP+SS31-treated group averaging 250% of the level observed for CLP group (P < 0.001). The force-time integral for the sham+SS31 group was similar to that for sham controls (NS).
Fig. 5.
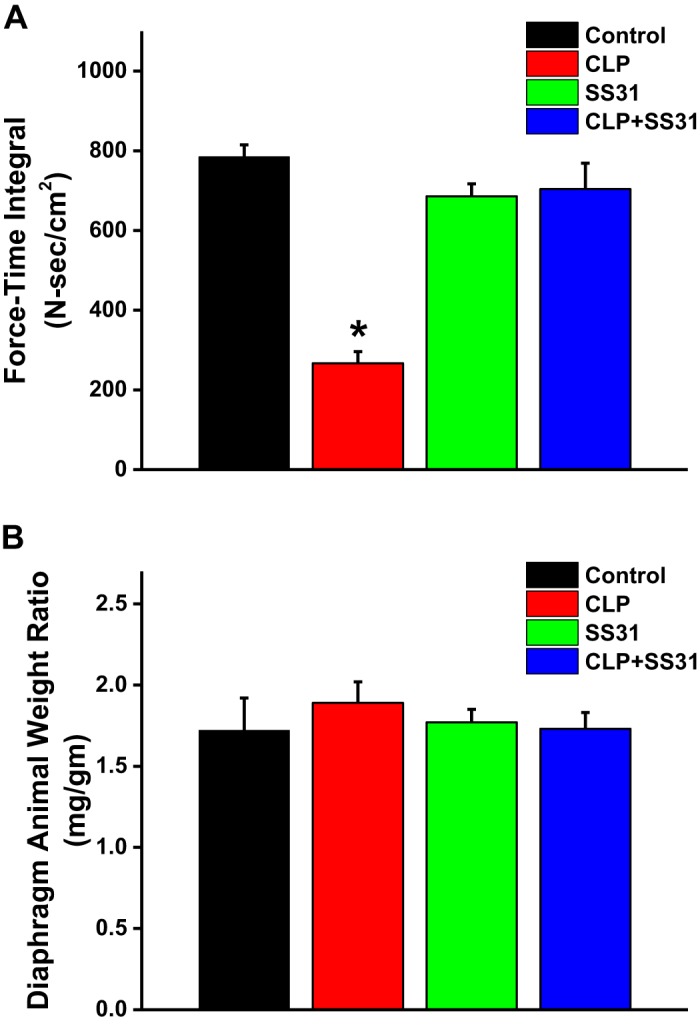
Force-time integrals for diaphragm fatigue curves for the 4 experimental groups in A. Ability of the diaphragm to maintain force generation over time during a fatiguing, repetitive direct muscle stimulation trial was substantially lower for muscles taken from septic animals [i.e., the cecal ligation puncture (CLP) group] than for sham controls (P < 0.001 for comparison of these 2 groups). SS31 prevented this sepsis-induced reduction in force maintenance, with the force-time integral for the CLP+SS31-treated group similar to the sham controls and significantly higher than the value for the CLP septic animals (P < 0.001 for this latter comparison). B shows group mean data ±1 SE for the diaphragm weight-to-animal weight ratios for sham controls (black) and CLP sepsis (red), sham+SS31 (green), and CLP+SS31 (blue) groups. This ratio was similar for the 4 groups (not significant for all comparisons). These data indicate that loss of specific diaphragm force in response to CLP-induced sepsis in the current study was not associated with a generalized loss of diaphragm muscle mass under the conditions tested. *Statistical significance; n = 5–6 mice per group.
Diaphragm muscle mass.
Sepsis can theoretically induce reductions in muscle force generation both by reducing the muscle force generated per unit muscle mass (i.e., reducing specific force generation) and by reducing total muscle mass (i.e., by inducing muscle atrophy). To assess the impact of sepsis on diaphragm mass, we normalized diaphragm mass, at the time of animal euthanasia, to total animal weight. As demonstrated in Fig. 5B, sepsis did not elicit a significant loss in diaphragm mass by the time point studied in the current protocol (i.e., 48 h after CLP surgery), with diaphragm mass-to-animal weight ratios similar for sham, CLP, sham+SS31, and CLP+SS31 groups (NS for each comparison).
Proteolytic enzyme activity.
To assess the effects of CLP sepsis on diaphragm proteolytic activity, we measured calpain and 20S proteasome activity on diaphragm homogenates by determining the rate of cleavage of fluorescent substrates in vitro with and without addition of specific calpain and proteasomal enzyme inhibitors. CLP sepsis elicited large increases in both diaphragm calpain (Fig. 6A) and 20S proteasomal (Fig. 6B) enzyme activity. On average, sepsis increased calpain activity threefold (P < 0.02 for comparison of sham and CLP groups) and increased 20S proteasomal activity more than twofold (P = 0.013 for comparison of sham and CLP). SS31 treatment substantially reduced both calpain and 20S proteasomal activities. Specifically, calpain activity for diaphragm homogenates from CLP+SS31-treated animals was only 28% of the level observed for the CLP group (P < 0.02 for this comparison). In addition, diaphragm homogenate 20S proteasome activity for CLP+SS31-treated animals was only 34% of the level observed for the CLP group (P < 0.05 for this comparison). Calpain and 20S proteasome activity levels were similar for sham and sham+SS31-treated groups (NS).
Fig. 6.
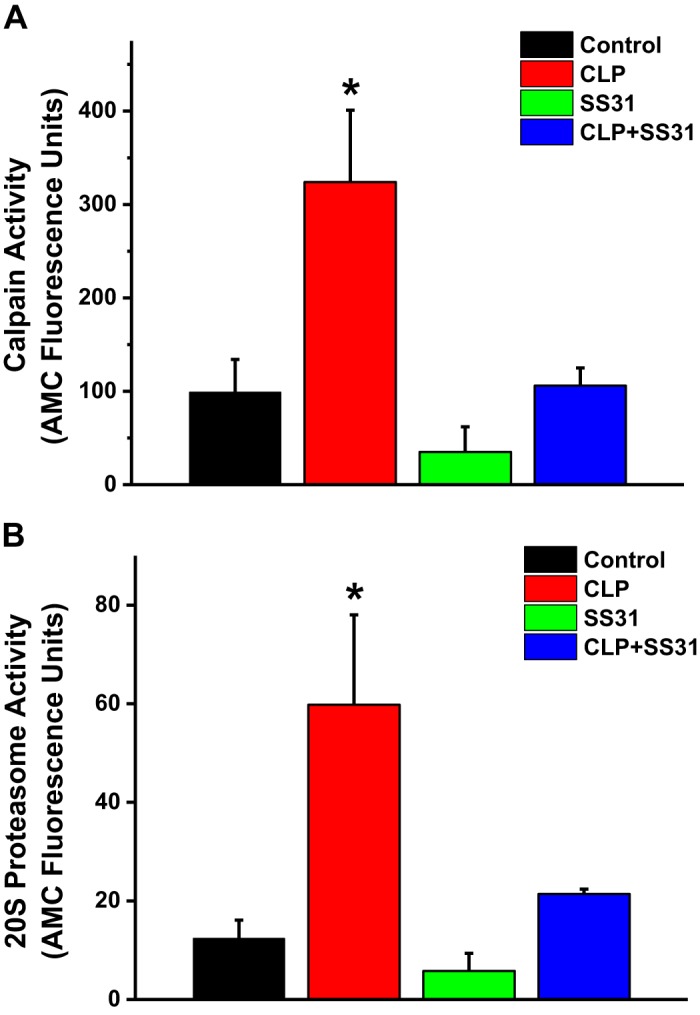
A indicates group mean data ±1 SE for diaphragm calpain activity in sham controls (black) and cecal ligation puncture (CLP) sepsis (red), sham+SS31 (green), and CLP+SS31 (blue) groups. Calpain activity significantly increased with CLP sepsis compared with controls (P < 0.02), and administration of SS31 to septic animals reduced calpain activity to control levels [P < 0.02 for comparison of CLP and CLP+SS31 groups and not significant (NS) for comparison of CLP+SS31 and sham controls]. Calpain activity for sham+SS31 animals was similar to controls (NS). B displays group mean data ±1 SE for diaphragm 20S proteasome activity for sham controls (black) and CLP sepsis (red), sham+SS31 (green), and CLP+SS31 (blue) groups. Proteasome activity was increased in CLP sepsis compared with sham controls (P = 0.013 for this comparison), and SS31 administration to CLP sepsis animals prevented increases in proteasome activity (NS for comparison of CLP+SS31 and sham controls). *Statistical significance; n = 5 mice per group. AMC, amino-4-methylcoumarin.
Diaphragm myosin heavy chain concentrations.
Sepsis elicited a significant reduction in diaphragm myosin heavy chain (MHC) protein levels, with MHC levels reduced by 38% for diaphragm samples from the CLP group compared with sham controls (P = 0.015; Fig. 7). SS31 treatment significantly attenuated sepsis-induced reductions in MHC protein levels, with MHC levels for the CLP+SS31-treated group significantly higher than levels for CLP animals (P < 0.03). MHC levels for sham and sham+SS31 animals were similar (NS).
Fig. 7.
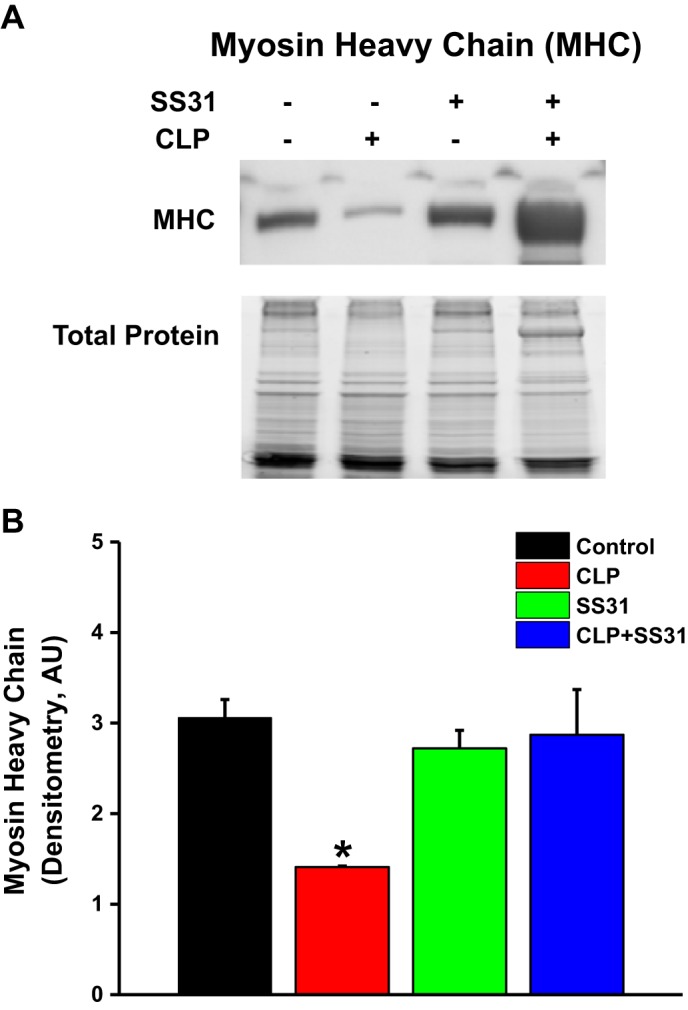
Diaphragm myosin heavy chain protein levels for the 4 experimental groups. A shows a representative Western blot in samples from individual diaphragm muscle homogenates taken from (left to right) a sham control and cecal ligation puncture (CLP) sepsis, sham+SS31, and CLP+SS31 animals. Myosin heavy chain band intensity was reduced for the CLP sepsis animal. CLP+SS31 band intensity was similar to the sham and sham+SS31-treated animal. Similar pattern was observed for comparisons between all diaphragm samples for the 4 groups, with group mean densitometry (±1 SE) for MHC bands presented in B. MHC content was significantly lower for diaphragms from CLP sepsis (red) compared with the sham controls (black; P = 0.015 for this comparison). SS31 administration prevented MHC chain depletion, with MHC levels for the CLP+SS31 group (blue) similar to values for the sham controls and the sham+SS31 group (not significant for these comparisons). *Statistical significance; n = 4 mice per group. AU, arbitrary units.
Diaphragm mitochondrial function and aconitase activity.
We measured oxygen consumption rates of diaphragm mitochondrial suspensions prepared from the four experimental groups to assess mitochondrial function. ADP-stimulated oxygen consumption rate (state 3 respiration) was significantly reduced for mitochondria from the CLP group, and SS31 administration to CLP animals prevented this reduction (Fig. 8A; P = 0.008 for comparison of the CLP group to the other 3 groups). We also found that CLP sepsis reduced the mitochondrial respiratory control ratio (RCR) and ATP generation rate, and SS31 blocked both of these sepsis-induced changes (Fig. 8, B and C; P = 0.002 for comparison of the RCR in the CLP group with the other 3 experimental groups, P = 0.051 for comparison of the ATP generation rate in the CLP group with the other 3 experimental groups). We also measured aconitase activity as an index of mitochondrial superoxide generation. Diaphragm mitochondrial aconitase activity decreased following CLP sepsis, consistent with sepsis-induced increases in mitochondrial superoxide generation, and SS31 administration prevented this increase (Fig. 8D; P = 0.024 for comparison of the CLP group to the other 3 experimental groups).
Fig. 8.

Mitochondrial function was assessed on diaphragm mitochondrial homogenates by measuring ADP-stimulated respiration (state 3 oxygen consumption rates; A) and the respiratory control ratio (RCR; B) and calculating ATP production rates (C). We found that cecal ligation puncture (CLP) induced significant reductions in mitochondrial ADP-stimulated oxygen consumption (state 3 respiration; P = 0.002 compared with control) in the RCR (P = 0.002 compared with control) and in ATP production rates (P = 0.031 compared with control). SS31 ablated each of these CLP-induced alterations in mitochondrial function, with these parameters for the CLP+SS31 group higher than values for the CLP group (P = 0.008 for ADP-stimulated respiration, P = 0.002 for RCR, and P = 0.051 for ATP production rates). We also measured diaphragm aconitase activity, an index of mitochondrial free radical generation (greater free radical generation induces reductions in aconitase activity), and found that CLP reduced aconitase levels and SS31 blocked this response (D; P = 0.024 for each of these comparisons). *Statistical significance; n = 3–4 mice per group.
DISCUSSION
Recent reports indicate that diaphragm weakness is a common clinical problem in patients with critical illness who are mechanically ventilated (11, 13, 18, 20, 27, 41, 47, 49), with an average diaphragm force generation for this patient population that is only 23% of the value observed in healthy, normal subjects (41). Importantly, diaphragm weakness is associated with poor outcomes, with patients who are weaker requiring a longer time to wean from mechanical ventilation (41, 47). In addition, there is a strong correlation between weakness and mortality, with weakness representing a greater risk factor for death than the level of lung function, the sequential organ failure assessment (SOFA) score, age, or the degree of chronic comorbidities (41, 47).
There are several factors that are thought to contribute to the development of skeletal muscle weakness in patients who are critically ill, including the use of mechanical ventilation (31), the simultaneous use of steroids and neuromuscular blocking agents (1), hyperglycemia (5), and electrolyte abnormalities (2). In addition, multiple reports indicate that the presence of sepsis and other severe infections are a major risk factor for the development of skeletal muscle weakness in general and diaphragm weakness in particular (13, 27, 41). In keeping with this concept, diaphragm strength has been reported to decrease as much as 80% within 48 h in animal models of sepsis (e.g., cecal ligation puncture-induced peritonitis, endotoxin administration, and induction of pneumonia; Refs. 12, 34, 51). Similarly, recent studies examining diaphragm strength in patients in medical intensive care units found that patients with severe, active infections had a level of diaphragm strength that was less than half that of patients without evidence of infection (41).
The cellular mechanisms by which infections induce muscle weakness have been recently studied in animal models. This work has shown that infection is associated with a loss of force generation by the contractile proteins, with a marked downward shift of the force-pCa curve measured for single diaphragm muscle fibers taken from septic animals compared with curves for muscle fibers from healthy, control animals (35, 36, 46). This loss in contractile protein function is thought to be the result of reductions in levels of critical contractile proteins (e.g., myosin heavy chain) and due to chemical modification of proteins (45).
The data obtained from the present study are largely consistent with these previous observations. We found that CLP sepsis induced large reductions in diaphragm strength. We also assessed diaphragm fatigability by measuring diaphragm force generation over time during repetitive contractions over 2 min. CLP sepsis produced a marked downward shift in the diaphragm force-time relationship and dramatically reduced the total force-time integral of this curve; these findings are consistent with an effect of sepsis to increase diaphragm fatigability. We also found that CLP sepsis elicited significant increases in the activity of two proteolytic pathway indices for diaphragm homogenates, i.e., diaphragm calpain activity and diaphragm 20S proteasome activity. Both of these enzymes can induce cleavage of selected skeletal muscle proteins, including myosin heavy chain (31, 52). In keeping with this possibility, we also found that CLP sepsis was associated with a significant reduction in diaphragm levels of myosin heavy chain protein.
Most importantly, the current study found that administration of a novel mitochondrially targeted antioxidant, SS31, prevented CLP sepsis-induced reductions in diaphragm muscle strength. Treatment with SS31 also reduced the effect of CLP sepsis to increase diaphragm fatigability, maintaining force generation over time during a repetitive contraction trial. Administration of SS31 also blocked sepsis-induced increases in calpain and 20S proteasome activity and prevented CLP sepsis-induced reductions in MHC levels.
These findings that SS31 administration both reduces proteolytic pathway activity and increases MHC contractile protein levels are consistent with the concept that sepsis-induced reductions in muscle strength are due, in large part, to the downstream effects of excessive mitochondrial free radical generation (4–6, 8). According to this theory, sepsis increases mitochondrial superoxide generation, which, in turn, activates the calpain and proteasome proteolytic systems (6, 8, 14). This engenders a loss of critical contractile proteins (e.g., MHC; Ref. 45) and produces a downshift in the contractile protein force-pCa relationship (35), decreasing force generation during contraction. Consistent with this theory, the present study also found that CLP sepsis induced a reduction in diaphragm mitochondrial aconitase activity, an index of heightened superoxide generation. We also found that SS31 prevented sepsis-induced reductions in aconitase activity, consistent with an effect of SS31 to prevent and/or scavenge mitochondrial superoxide production. We found that CLP sepsis reduced diaphragm mitochondrial function, decreasing ADP-stimulated respiration rate (state 3) and reducing mitochondrial ATP generation; SS31 prevented both effects of sepsis.
These latter findings raise the issue as to the exact mechanism by which SS31 exerted its effects on the diaphragm in the present study. The primary process by which SS31 is thought to exert its effects is via localization to the mitochondrial inner membrane where this compound scavenges superoxide anions and their products (hydrogen peroxide, peroxynitrite, and hydroxyl radicals; Refs. 22, 24). In keeping with this concept, SS31 has been shown, in previous studies, to reduce mitochondrial reactive oxygen species in epithelial, endothelial, and neuronal cells (3, 10, 50). By this mechanism, SS31 may have directly reduced sepsis-induced diaphragm damage in the current study. SS31 has, however, additional effects to reduce cytokine levels, inflammatory signaling pathways, mitochondrial dynamics (fission and fusion), and apoptotic pathway activation (24, 50). It is also possible, therefore, that these additional actions of SS31 may have contributed to its protective effects on the diaphragm.
Previous work has suggested that sepsis can also induce reductions in neural transmission (26) as well as inducing a myopathy (8), with reductions in neural propagation of motor action potentials, alterations in muscle neuromuscular junction transmission, and reductions in sarcolemmal propagation of action potentials (26). To provide an assessment of diaphragm neuromuscular transmission in our sepsis model, we used the technique described by Greising et al. (16). We found that CLP sepsis did not increase transmission failure compared with responses observed for muscles from nonseptic control animals. We also found that SS31 treatment did not alter the transmission failure index in either control or septic animals. Our results suggest that neuromuscular transmission failure is not a cause of diaphragm weakness or enhanced fatigability in the CLP model of sepsis under the conditions tested in the present experiment.
As mentioned earlier, the use of mechanical ventilators per se has been previously shown to induce a significant degree of diaphragm weakness in patients, a phenomenon has been termed ventilator-induced diaphragm dysfunction (VIDD; Refs. 23, 28, 30, 31). One previous report found that SS31 administration is capable of attenuating the development of diaphragm weakness due to VIDD in an animal model (29), suggesting that VIDD, as well as sepsis-induced diaphragm dysfunction, may both be causally related to excessive skeletal muscle generation of free radicals. Both conditions are known to be associated with significant loss of diaphragm force generation as well as activation of calpain and proteasomal proteolytic enzyme systems (8, 19, 37–40, 45), suggesting that both clinical entities may share some pathophysiological features.
The present work is the first to examine the ability of SS31 to alter sepsis-induced diaphragm dysfunction. This raises the question as to whether this agent may also reduce sepsis-induced dysfunction of other organs. Only a single recent report has studied this possibility. Specifically, Liu et al. (24) found that administration of SS31 prevented morphological changes in the heart in response to administration of LPS (endotoxin). Of interest, these authors found that SS31 also reduced cardiac inflammatory responses to LPS, suppressing cardiac mRNA levels of IL-6, IL-1β, and TNF-α (24).
The results of the current study suggest it is theoretically possible that SS31 or other agents that either inhibit mitochondrial superoxide production or scavenge these radicals to block their downstream effects may provide a therapy to prevent the development of diaphragm weakness in patients with sepsis. Of note, two other mitochondrially targeted antioxidants, SkQ1 and MitoQ (25, 32), have recently been tested in human clinical trials with success (32), albeit for purposes that do not involve treatment of patients who are critically ill. In theory, further testing of such agents and translation to use in critical illness may ultimately prove to be a viable treatment to improve diaphragm strength. We speculate that improvements in diaphragm strength, in turn, may also reduce the duration of mechanical ventilation, attenuate morbidity, and decrease the mortality in this population.
GRANTS
G. S. Supinski is supported by Grants R01-HL-113494 and R01-HL-141356 from the National Heart, Lung, and Blood Institute of the National Institutes of Health and 5I01BX002132 from the Department of Veterans Affairs. L. A. P. Callahan is supported by Grants R01-HL-112085 and R01-HL-141356 from the National Heart, Lung, and Blood Institute of the National Institutes of Health. E. A. Schroder is supported by Grant R01-HL-141356 from the National Heart, Lung, and Blood Institute of the National Institutes of Health.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
G.S.S. conceived and designed research; G.S.S., L.W., and E.A.S. performed experiments; G.S.S. and L.A.P.C. analyzed data; G.S.S., E.A.S., and L.A.P.C. interpreted results of experiments; L.A.P.C. prepared figures; G.S.S. drafted manuscript; G.S.S., E.A.S., and L.A.P.C. edited and revised manuscript; G.S.S., L.W., E.A.S., and L.A.P.C. approved final version of manuscript.
REFERENCES
- 1.Annane D. What is the evidence for harm of neuromuscular blockade and corticosteroid use in the intensive care unit? Semin Respir Crit Care Med 37: 51–56, 2016. doi: 10.1055/s-0035-1570355. [DOI] [PubMed] [Google Scholar]
- 2.Aubier M, Murciano D, Lecocguic Y, Viires N, Jacquens Y, Squara P, Pariente R. Effect of hypophosphatemia on diaphragmatic contractility in patients with acute respiratory failure. N Engl J Med 313: 420–424, 1985. doi: 10.1056/NEJM198508153130705. [DOI] [PubMed] [Google Scholar]
- 3.Cai M, Li J, Lin S, Chen X, Huang J, Jiang X, Yang L, Luo Y. Mitochondria-targeted antioxidant peptide SS31 protects cultured human lens epithelial cells against oxidative stress. Curr Eye Res 40: 822–829, 2015. doi: 10.3109/02713683.2014.959607. [DOI] [PubMed] [Google Scholar]
- 4.Callahan LA, Nethery D, Stofan D, DiMarco A, Supinski G. Free radical-induced contractile protein dysfunction in endotoxin-induced sepsis. Am J Respir Cell Mol Biol 24: 210–217, 2001. doi: 10.1165/ajrcmb.24.2.4075. [DOI] [PubMed] [Google Scholar]
- 5.Callahan LA, She ZW, Nosek TM. Superoxide, hydroxyl radical, and hydrogen peroxide effects on single-diaphragm fiber contractile apparatus. J Appl Physiol (1985) 90: 45–54, 2001. doi: 10.1152/jappl.2001.90.1.45. [DOI] [PubMed] [Google Scholar]
- 6.Callahan LA, Stofan DA, Szweda LI, Nethery DE, Supinski GS. Free radicals alter maximal diaphragmatic mitochondrial oxygen consumption in endotoxin-induced sepsis. Free Radic Biol Med 30: 129–138, 2001. doi: 10.1016/S0891-5849(00)00454-8. [DOI] [PubMed] [Google Scholar]
- 7.Callahan LA, Supinski GS. Hyperglycemia-induced diaphragm weakness is mediated by oxidative stress. Crit Care 18: R88, 2014. doi: 10.1186/cc13855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Callahan LA, Supinski GS. Sepsis-induced myopathy. Crit Care Med 37, Suppl: S354–S367, 2009. doi: 10.1097/CCM.0b013e3181b6e439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Callahan LA, Supinski GS. Sepsis induces diaphragm electron transport chain dysfunction and protein depletion. Am J Respir Crit Care Med 172: 861–868, 2005. doi: 10.1164/rccm.200410-1344OC. [DOI] [PubMed] [Google Scholar]
- 10.Cho S, Szeto HH, Kim E, Kim H, Tolhurst AT, Pinto JT. A novel cell-permeable antioxidant peptide, SS31, attenuates ischemic brain injury by down-regulating CD36. J Biol Chem 282: 4634–4642, 2007. doi: 10.1074/jbc.M609388200. [DOI] [PubMed] [Google Scholar]
- 11.Demoule A, Jung B, Prodanovic H, Molinari N, Chanques G, Coirault C, Matecki S, Duguet A, Similowski T, Jaber S. Diaphragm dysfunction on admission to the intensive care unit. Prevalence, risk factors, and prognostic impact–a prospective study. Am J Respir Crit Care Med 188: 213–219, 2013. doi: 10.1164/rccm.201209-1668OC. [DOI] [PubMed] [Google Scholar]
- 12.Divangahi M, Matecki S, Dudley RW, Tuck SA, Bao W, Radzioch D, Comtois AS, Petrof BJ. Preferential diaphragmatic weakness during sustained Pseudomonas aeruginosa lung infection. Am J Respir Crit Care Med 169: 679–686, 2004. doi: 10.1164/rccm.200307-949OC. [DOI] [PubMed] [Google Scholar]
- 13.Dres M, Goligher EC, Heunks LMA, Brochard LJ. Critical illness-associated diaphragm weakness. Intensive Care Med 43: 1441–1452, 2017. doi: 10.1007/s00134-017-4928-4. [DOI] [PubMed] [Google Scholar]
- 14.Furukawa K, Kikusato M, Kamizono T, Toyomizu M. Time-course changes in muscle protein degradation in heat-stressed chickens: possible involvement of corticosterone and mitochondrial reactive oxygen species generation in induction of the ubiquitin-proteasome system. Gen Comp Endocrinol 228: 105–110, 2016. doi: 10.1016/j.ygcen.2016.02.007. [DOI] [PubMed] [Google Scholar]
- 15.Gilliam LA, Moylan JS, Patterson EW, Smith JD, Wilson AS, Rabbani Z, Reid MB. Doxorubicin acts via mitochondrial ROS to stimulate catabolism in C2C12 myotubes. Am J Physiol Cell Physiol 302: C195–C202, 2012. doi: 10.1152/ajpcell.00217.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Greising SM, Ermilov LG, Sieck GC, Mantilla CB. Ageing and neurotrophic signalling effects on diaphragm neuromuscular function. J Physiol 593: 431–440, 2015. doi: 10.1113/jphysiol.2014.282244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Greising SM, Vasdev AK, Zhan WZ, Sieck GC, Mantilla CB. Chronic TrkB agonist treatment in old age does not mitigate diaphragm neuromuscular dysfunction. Physiol Rep 5: e13103, 2017. doi: 10.14814/phy2.13103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hermans G, Agten A, Testelmans D, Decramer M, Gayan-Ramirez G. Increased duration of mechanical ventilation is associated with decreased diaphragmatic force: a prospective observational study. Crit Care 14: R127, 2010. doi: 10.1186/cc9094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hooijman PE, Beishuizen A, Witt CC, de Waard MC, Girbes AR, Spoelstra-de Man AM, Niessen HW, Manders E, van Hees HW, van den Brom CE, Silderhuis V, Lawlor MW, Labeit S, Stienen GJ, Hartemink KJ, Paul MA, Heunks LM, Ottenheijm CA. Diaphragm muscle fiber weakness and ubiquitin-proteasome activation in critically ill patients. Am J Respir Crit Care Med 191: 1126–1138, 2015. doi: 10.1164/rccm.201412-2214OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Laghi F, Cattapan SE, Jubran A, Parthasarathy S, Warshawsky P, Choi YS, Tobin MJ. Is weaning failure caused by low-frequency fatigue of the diaphragm? Am J Respir Crit Care Med 167: 120–127, 2003. doi: 10.1164/rccm.200210-1246OC. [DOI] [PubMed] [Google Scholar]
- 21.Larsen FJ, Schiffer TA, Ørtenblad N, Zinner C, Morales-Alamo D, Willis SJ, Calbet JA, Holmberg HC, Boushel R. High-intensity sprint training inhibits mitochondrial respiration through aconitase inactivation. FASEB J 30: 417–427, 2016. doi: 10.1096/fj.15-276857. [DOI] [PubMed] [Google Scholar]
- 22.Lee FY, Shao PL, Wallace CG, Chua S, Sung PH, Ko SF, Chai HT, Chung SY, Chen KH, Lu HI, Chen YL, Huang TH, Sheu JJ, Yip HK. Combined therapy with SS31 and mitochondria mitigates myocardial ischemia-reperfusion injury in rats. Int J Mol Sci 19: 2782, 2018. doi: 10.3390/ijms19092782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Levine S, Nguyen T, Taylor N, Friscia ME, Budak MT, Rothenberg P, Zhu J, Sachdeva R, Sonnad S, Kaiser LR, Rubinstein NA, Powers SK, Shrager JB. Rapid disuse atrophy of diaphragm fibers in mechanically ventilated humans. N Engl J Med 358: 1327–1335, 2008. doi: 10.1056/NEJMoa070447. [DOI] [PubMed] [Google Scholar]
- 24.Liu Y, Yang W, Sun X, Xie L, Yang Y, Sang M, Jiao R. SS31 ameliorates sepsis-induced heart injury by inhibiting oxidative stress and inflammation. Inflammation 42: 2170–2180, 2019. doi: 10.1007/s10753-019-01081-3. [DOI] [PubMed] [Google Scholar]
- 25.Manskikh VN, Gancharova OS, Nikiforova AI, Krasilshchikova MS, Shabalina IG, Egorov MV, Karger EM, Milanovsky GE, Galkin II, Skulachev VP, Zinovkin RA. Age-associated murine cardiac lesions are attenuated by the mitochondria-targeted antioxidant SkQ1. Histol Histopathol 30: 353–360, 2015. doi: 10.14670/HH-30.353. [DOI] [PubMed] [Google Scholar]
- 26.Nayci A, Atis S, Comelekoglu U, Ozge A, Ogenler O, Coskun B, Zorludemir S. Sepsis induces early phrenic nerve neuropathy in rats. Eur Respir J 26: 686–692, 2005. doi: 10.1183/09031936.05.0111004. [DOI] [PubMed] [Google Scholar]
- 27.Petrof BJ. Diaphragm weakness in the critically ill: basic mechanisms reveal therapeutic opportunities. Chest 154: 1395–1403, 2018. doi: 10.1016/j.chest.2018.08.1028. [DOI] [PubMed] [Google Scholar]
- 28.Petrof BJ, Hussain SN. Ventilator-induced diaphragmatic dysfunction: what have we learned? Curr Opin Crit Care 22: 67–72, 2016. doi: 10.1097/MCC.0000000000000272. [DOI] [PubMed] [Google Scholar]
- 29.Powers SK, Hudson MB, Nelson WB, Talbert EE, Min K, Szeto HH, Kavazis AN, Smuder AJ. Mitochondria-targeted antioxidants protect against mechanical ventilation-induced diaphragm weakness. Crit Care Med 39: 1749–1759, 2011. doi: 10.1097/CCM.0b013e3182190b62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Powers SK, Shanely RA, Coombes JS, Koesterer TJ, McKenzie M, Van Gammeren D, Cicale M, Dodd SL. Mechanical ventilation results in progressive contractile dysfunction in the diaphragm. J Appl Physiol (1985) 92: 1851–1858, 2002. doi: 10.1152/japplphysiol.00881.2001. [DOI] [PubMed] [Google Scholar]
- 31.Powers SK, Wiggs MP, Sollanek KJ, Smuder AJ. Ventilator-induced diaphragm dysfunction: cause and effect. Am J Physiol Regul Integr Comp Physiol 305: R464–R477, 2013. doi: 10.1152/ajpregu.00231.2013. [DOI] [PubMed] [Google Scholar]
- 32.Rossman MJ, Santos-Parker JR, Steward CAC, Bispham NZ, Cuevas LM, Rosenberg HL, Woodward KA, Chonchol M, Gioscia-Ryan RA, Murphy MP, Seals DR. Chronic supplementation with a mitochondrial antioxidant (MitoQ) improves vascular function in healthy older adults. Hypertension 71: 1056–1063, 2018. doi: 10.1161/HYPERTENSIONAHA.117.10787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stana F, Vujovic M, Mayaki D, Leduc-Gaudet JP, Leblanc P, Huck L, Hussain SNA. Differential regulation of the autophagy and proteasome pathways in skeletal muscles in sepsis. Crit Care Med 45: e971–e979, 2017. doi: 10.1097/CCM.0000000000002520. [DOI] [PubMed] [Google Scholar]
- 34.Supinski G, Nethery D, DiMarco A. Effect of free radical scavengers on endotoxin-induced respiratory muscle dysfunction. Am Rev Respir Dis 148: 1318–1324, 1993. doi: 10.1164/ajrccm/148.5.1318. [DOI] [PubMed] [Google Scholar]
- 35.Supinski G, Nethery D, Nosek TM, Callahan LA, Stofan D, DiMarco A. Endotoxin administration alters the force vs. pCa relationship of skeletal muscle fibers. Am J Physiol Regul Integr Comp Physiol 278: R891–R896, 2000. doi: 10.1152/ajpregu.2000.278.4.R891. [DOI] [PubMed] [Google Scholar]
- 36.Supinski G, Stofan D, Callahan LA, Nethery D, Nosek TM, DiMarco A. Peroxynitrite induces contractile dysfunction and lipid peroxidation in the diaphragm. J Appl Physiol (1985) 87: 783–791, 1999. doi: 10.1152/jappl.1999.87.2.783. [DOI] [PubMed] [Google Scholar]
- 37.Supinski GS, Alimov AP, Wang L, Song XH, Callahan LA. Calcium-dependent phospholipase A2 modulates infection-induced diaphragm dysfunction. Am J Physiol Lung Cell Mol Physiol 310: L975–L984, 2016. doi: 10.1152/ajplung.00312.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Supinski GS, Alimov AP, Wang L, Song XH, Callahan LA. Neutral sphingomyelinase 2 is required for cytokine-induced skeletal muscle calpain activation. Am J Physiol Lung Cell Mol Physiol 309: L614–L624, 2015. doi: 10.1152/ajplung.00141.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Supinski GS, Callahan LA. β-Hydroxy-β-methylbutyrate (HMB) prevents sepsis-induced diaphragm dysfunction in mice. Respir Physiol Neurobiol 196: 63–68, 2014. doi: 10.1016/j.resp.2014.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Supinski GS, Callahan LA. Calpain activation contributes to endotoxin-induced diaphragmatic dysfunction. Am J Respir Cell Mol Biol 42: 80–87, 2010. doi: 10.1165/rcmb.2008-0275OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Supinski GS, Callahan LA. Diaphragm weakness in mechanically ventilated critically ill patients. Crit Care 17: R120, 2013. doi: 10.1186/cc12792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Supinski GS, Callahan LA. Hemin prevents cardiac and diaphragm mitochondrial dysfunction in sepsis. Free Radic Biol Med 40: 127–137, 2006. doi: 10.1016/j.freeradbiomed.2005.09.025. [DOI] [PubMed] [Google Scholar]
- 43.Supinski GS, Morris PE, Dhar S, Callahan LA. Diaphragm dysfunction in critical illness. Chest 153: 1040–1051, 2018. doi: 10.1016/j.chest.2017.08.1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Supinski GS, Vanags J, Callahan LA. Eicosapentaenoic acid preserves diaphragm force generation following endotoxin administration. Crit Care 14: R35, 2010. doi: 10.1186/cc8913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Supinski GS, Wang L, Song XH, Moylan JS, Callahan LA. Muscle-specific calpastatin overexpression prevents diaphragm weakness in cecal ligation puncture-induced sepsis. J Appl Physiol (1985) 117: 921–929, 2014. doi: 10.1152/japplphysiol.00975.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Supinski GS, Wang W, Callahan LA. Caspase and calpain activation both contribute to sepsis-induced diaphragmatic weakness. J Appl Physiol (1985) 107: 1389–1396, 2009. doi: 10.1152/japplphysiol.00341.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Supinski GS, Westgate P, Callahan LA. Correlation of maximal inspiratory pressure to transdiaphragmatic twitch pressure in intensive care unit patients. Crit Care 20: 77, 2016. doi: 10.1186/s13054-016-1247-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Szeto HH. Mitochondria-targeted peptide antioxidants: novel neuroprotective agents. AAPS J 8: E521–E531, 2006. doi: 10.1208/aapsj080362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Watson AC, Hughes PD, Louise Harris M, Hart N, Ware RJ, Wendon J, Green M, Moxham J. Measurement of twitch transdiaphragmatic, esophageal, and endotracheal tube pressure with bilateral anterolateral magnetic phrenic nerve stimulation in patients in the intensive care unit. Crit Care Med 29: 1325–1331, 2001. doi: 10.1097/00003246-200107000-00005. [DOI] [PubMed] [Google Scholar]
- 50.Yang SK, Li AM, Han YC, Peng CH, Song N, Yang M, Zhan M, Zeng LF, Song PA, Zhang W, Tang SQ, Zhang H. Mitochondria-targeted peptide SS31 attenuates renal tubulointerstitial injury via inhibiting mitochondrial fission in diabetic mice. Oxid Med Cell Longev 2019: 2346580, 2019. doi: 10.1155/2019/2346580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang HX, Du JM, Ding ZN, Zhu XY, Jiang L, Liu YJ. Hydrogen sulfide prevents diaphragm weakness in cecal ligation puncture-induced sepsis by preservation of mitochondrial function. Am J Transl Res 9: 3270–3281, 2017. [PMC free article] [PubMed] [Google Scholar]
- 52.Zhu X, van Hees HWH, Heunks L, Wang F, Shao L, Huang J, Shi L, Ma S. The role of calpains in ventilator-induced diaphragm atrophy. Intensive Care Med Exp 5: 14, 2017. doi: 10.1186/s40635-017-0127-4. [DOI] [PMC free article] [PubMed] [Google Scholar]