

Abstract
In the murine venous thrombosis model induced by ligation of the inferior vena cava (IVCL), genetic deficiency of heme oxygenase-1 (HO-1) increases clot size. This study examined whether induction of HO-1 or administration of its products reduces thrombosis. Venous HO-1 upregulation by gene delivery reduced clot size, as did products of HO activity, biliverdin, and carbon monoxide. Induction of HO-1 by hemin reduced clot formation, clot size, and upregulation of plasminogen activator inhibitor-1 (PAI-1) that occurs in the IVCL model, while leaving urokinase plasminogen activator (uPA) and tissue plasminogen activator (tPA) expression unaltered. The reductive effect of hemin on clot size required HO activity. The IVCL model exhibited relatively high concentrations of heme that peaked just before maximum clot size, then declined as clot size decreased. Administration of hemin decreased heme concentration in the IVCL model. HO-2 mRNA was induced twofold in the IVCL model (vs. 40-fold HO-1 induction), but clot size was not increased in HO-2−/− mice compared with HO-2+/+ mice. Hemopexin, the major heme-binding protein, was induced in the IVCL model, and clot size was increased in hemopexin−/− mice compared with hemopexin+/+ mice. We conclude that in the IVCL model, the heme-degrading protein HO-1 and HO products inhibit thrombus formation, as does the heme-binding protein, hemopexin. The reductive effects of hemin administration require HO activity and are mediated, in part, by reducing PAI-1 upregulation in the IVCL model. We speculate that HO-1, HO, and hemopexin reduce clot size by restraining the increase in clot concentration of heme (now recognized as a procoagulant) that otherwise occurs.
NEW & NOTEWORTHY This study provides conclusive evidence that two proteins, one heme-degrading and the other heme-binding, inhibit clot formation. This may serve as a new therapeutic strategy in preventing and treating venous thromboembolic disease.
Keywords: bile pigments, carbon monoxide, heme oxygenase-1, hemopexin, murine model, venous thromboembolic disease
INTRODUCTION
Exceeded only by myocardial infarction and cerebrovascular disease, venous thromboembolic (VTE) disease is a major contributor to mortality arising from cardiovascular disease (3, 25, 28, 67). Fatality resulting from VTE disease rises remarkably with aging, as does the incidence of VTE disease (3), findings that speak to an ever-increasing significance of VTE disease in light of the recognized aging of the population of the United States. VTE disease may be primary and unprovoked, for example, as occurring in aged or obese subjects, or may be secondary and provoked by diverse conditions such as an acute illness, inactivity, and the postsurgical state (25, 67). In recent years concerted efforts have been consistently directed to the prevention of VTE disease, especially in the acute care setting, and yet the incidence of VTE remains essentially unchanged (3). In view of these considerations and the increasing understanding of the pathobiology of thrombogenesis (38, 41, 65, 66), considerable attention is directed to new strategies that can either prevent the initiation and propagation of VTE disease, hasten the resolution of venous thrombus once formed, or minimize the risk of embolic disease. In this regard, the armamentaria for inhibiting VTE disease now include, among others, novel anticoagulants (DOACs) and new agents that inhibit platelet activation (45, 67).
Among the experimental approaches widely used in the study of the mechanism of VTE disease and therapeutic strategies is a murine model involving the ligation of the inferior vena cava (IVC) below the origin of the renal veins (18, 48, 73). In this IVCL model, clotting promptly occurs following ligation of the IVC with the clot size peaking by day 2 after ligation; thereafter, endogenous thrombolytic mechanisms lead to progressive decrease in clot size such that the clot size is ~50% smaller by day 10 after IVCL. The clot size in this model is conventionally assessed by clot weight, clot length, and the clot weight-to-length ratio.
Our prior studies demonstrate that there is marked induction of heme oxygenase-1 (HO-1) in the IVCL model and that mice genetically deficient in HO-1, while exhibiting comparable clot size as HO-1+/+ mice at day 2, demonstrate a progressive increase in clot size by day 10; in contrast, HO-1+/+ mice displayed steady diminution in clot size by day 10 (73). HO is the rate-limiting, heme-degrading enzyme that produces the bile pigment biliverdin and carbon monoxide; biliverdin, generated by HO activity, is converted to bilirubin via biliverdin reductase (1, 11, 21, 49). There are two proteins that possess HO activity, the inducible (HO-1) and the constitutive (HO-2) isoforms. HO-1 exerts protective effects in diverse diseases, including, and as relevant to present studies, models of vascular injury (1, 11, 21, 49). For example, as shown in our prior studies, HO-1 is protective in such murine models as mineralocorticoid-induced systemic hypertension (51), the arteriovenous fistula (31), the partial carotid ligation model (36), and sickle cell vasculopathy (8, 10, 53).
The present studies extend our prior studies that examined the effect of HO-1 deficiency in the IVCL model by addressing in this model the complementary, more therapeutically relevant question, namely, whether the induction of HO-1 or the provision of its products reduces clot formation. In addressing this question emphasis was placed on a clinically translatable way of inducing of HO, namely, administration of hemin, as a brand of hemin (Panhematin) is currently approved for use in clinical conditions such as acute intermittent porphyria.
MATERIALS AND METHODS
Wild-type mice and genetically altered mice.
All experiments were performed in accordance with the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health and were approved by the Institutional Animal Care and Use Committee of Mayo Clinic.
The present study involved, primarily, male C57BL6/J mice from Jackson Laboratory (Bar Harbor, ME) which were 10–15 wk of age. For some experiments, female, age-matched HO-2+/+ and HO-2−/− mice (5 to 6 mo of age) were employed as in our previous studies (20, 34, 36). In additional studies, female mice (12–14 mo of age) with targeted disruption of the hemopexin gene, as previously described (13, 71), and age- and sex-matched hemopexin+/+ mice were also used. Separate colonies of HO-2+/+ and HO-2−/− mice, as well as hemopexin+/+ and hemopexin−/− mice, were generated and maintained by the mating of heterozygous males and females.
The IVC ligation model of thrombosis in mice.
The IVC ligation (IVCL) model was created as described in our prior study (73) and as originally described (48). The procedure was performed on a temperature-regulated table under pentobarbital sodium anesthesia (50 mg/kg ip) and with postsurgical buprenorphine analgesia. Through a midline incision, the bowel was gently retracted, and the aorta and IVC were carefully isolated and separated by blunt dissection. The IVC was then ligated just below the renal veins with 6-0 silk suture, and side branches of the IVC were ligated. Sham operation for control mice included midline incision and retraction of the bowel but with no IVC or branch ligation. At time points ranging from 1 to 10 days after ligation, the thrombus-containing IVC was carefully removed by cutting the IVC proximally, just below the ligature, and distally, at the lower end of the thrombus. For control mice the IVC segment distal to the renal veins and proximal to the bifurcation was harvested. The harvested IVC and contained clot (the IVCL model) were assessed for thrombus weight and length; for those samples of the IVCL model in which no clot was visible, clot weight, length, and weight-to-length ratio were evaluated as zero. Studies of gene expression and heme concentration in the IVCL model were also undertaken with the sham-ligated IVC serving as a control.
Studies of the effect of HO-1 upregulation in the IVCL model.
To upregulate HO-1, hemin (Cat. No. H651-9, Frontier Scientific, Logan, UT) was administered by intraperitoneal injection (25 mg/kg); the control group received saline vehicle. C57BL6/J mice were treated with doses of hemin or saline vehicle at 2 days before and 1 day before IVCL or sham surgery, again immediately after surgery, and then daily until 1 day before harvest. At 1, 2, and 3 days after ligation or sham surgery, the IVCL model for ligated mice and the IVC for control mice were harvested, as described above, for histological and morphometric assessment of wall thickness (31, 73), measurement of heme concentration, and for analysis of gene expression (31, 73).
In additional studies employing this protocol for hemin administration, concomitant administration of a specific inhibitor of HO activity, tin protoporphyrin (SnPP, 30 mg/kg, Frontier Scientific), the day before and the day of ligation surgery was used to determine the role of HO activity in mediating the effect of hemin in the IVCL model. For these studies the IVCL model was harvested at 1 day after ligation for the determination of thrombus weight and length.
The protocol for administering hemin was established in preliminary studies in which C57BL6/J mice were treated with hemin or saline, dosed as described above, and euthanized 6 h after the third dose; veins were harvested for the assessment of gene and protein expression as well as HO activity. In other studies, the effect of upregulation of HO-1 by an adeno-associated viral serotype 9 (AAV9) vector on the IVCL model was examined. For these studies, the AAV9 vector [AAV9(HO-1)] and a negative control vector [AAV9(revHO-1)], containing a reverse orientation of the HO-1 transgene, were constructed and produced as described in our previous studies (29, 35); our prior studies demonstrate this method of gene delivery leads to robust upregulation of HO-1 protein expression and markedly increased HO activity in central veins in mice (29, 35). AAV9(HO-1) vector or control AAV9(revHO-1) vector was intravenously administered (1 × 1011 vg/mouse) 3 wk before creating the IVCL model. Two days after IVCL, the IVCL model was harvested for assessment of thrombus weight and length.
Studies of the effect of administration of products of HO activity on the IVCL model.
As a method of delivering the product of HO activity, carbon monoxide (CO), a CO-releasing molecule [CORM-3; tricarbonylchloro(glycinato) ruthenium (II); Sigma-Aldrich, St. Louis, MO] was administered to mice as in our previous studies (35, 36). CORM-3 (40 mg/kg ip) was administered 1 day before IVCL; additional doses were administered on the day of surgery and 1 day thereafter. Inactivated CORM-3 (iCORM-3), prepared as described in our previous study, was injected as a control (35, 36). In other studies, biliverdin (50 µmol/kg ip, Cat. No. B655-9, Frontier Scientific) or saline vehicle was administered 1 day before IVCL, immediately after surgery, and 1 day thereafter (75). In each of these studies, the IVCL model was harvested for assessment of thrombus weight and length 2 days after IVCL.
Effect of genetic deficiencies of HO-2 and hemopexin on the IVCL model.
The effects of the deficiencies of HO-2 and hemopexin on the IVCL model were examined in separate studies. In these studies, HO-2+/+ and HO-2−/− mice were subjected to IVCL, and 2 and 10 days later the IVC and contained clot were harvested for assessment of thrombus weight and length. Similar studies were performed using hemopexin+/+ and hemopexin−/− mice, with the IVC and contained clot harvested 2 days after IVCL.
mRNA expression by quantitative real-time reverse transcriptase-PCR.
Gene expression in the mouse IVCL model was assessed by quantitative real-time RT-PCR employing a two-step method as in our previous study (73). These studies of gene expression were performed in the ligated IVC and contained clot unless otherwise stated. Briefly, the TRIzol method (Invitrogen, Carlsbad, CA) was used to extract total RNA, which was further purified with an RNeasy Mini Kit (Qiagen, Valencia, CA). Reverse transcription was performed with a Transcriptor First Strand cDNA Synthesis Kit (Roche Applied Science, Indianapolis, IN), and the resulting cDNA was used in quantitative real-time PCR analysis. Probes and primers used for quantification were obtained as assay sets for each target mRNA (TaqMan Gene Expression Assays, Applied Biosystems) and relative quantitation was performed on an ABI Prism 7900HT (Applied Biosystems, Foster City, CA) employing standard curves constructed for each target and housekeeping gene consisting of serial 10-fold dilutions of cDNA stocks. Expression of 18S rRNA was used for normalization of the expression of each target gene.
Western blot analysis.
Western blot analysis of HO-1 and hemopexin protein was performed as in our previous studies (64, 73). Primary antibodies against HO-1 (Cat. No. ADI-SPA-895; Enzo Life Sciences, Farmingdale, NY), hemopexin (Cat. No. ab133415; Abcam, Cambridge, MA), and β-actin (Cat. No. 612656; BD Biosciences, San Jose, CA) were employed in 18-h incubations at 4°C. Peroxidase-conjugated secondary antibodies were then used, and an enhanced chemiluminescence method was used for band visualization.
Measurement of heme oxygenase activity.
As performed in our previous studies, HO activity was quantitated by the measurement of the rate of formation of bilirubin from hemin (50, 53). Activity was normalized to the lysate protein content as assessed by the Lowry method.
Measurement of heme concentration in the IVCL model.
Heme concentration in the IVCL model was measured by the pyridine-chromogen method as previously employed by our laboratory (52, 54). The ligated IVC and contained clot were homogenized and centrifuged at 500 g for 3 min, and heme concentration was then measured in the supernatant.
Statistics.
Data are expressed as means ± SD and considered statistically significant for P < 0.05. All graphs display individual data values for all measurements as dot-whisker plots. The Student's t-test was used for parametric data, and the Mann-Whitney U-test was employed for nonparametric data. The incidence of clot formation was assessed using Fisher's exact test.
RESULTS
Upregulation of HO-1/HO by gene delivery reduces clot formation.
Our prior studies demonstrate that systemic delivery of AAV9 (HO-1) markedly induces HO-1 protein (greater than 3-fold) and increases HO activity approximately threefold in veins (35); additionally, we demonstrated that such induction of HO-1 is protective against neointimal hyperplasia and venous injury in a murine model of an arteriovenous fistula (35). We thus used this previously validated method for inducing HO-1 to determine whether such upregulation of HO-1 reduces clot formation. As demonstrated in Fig. 1, clot weight, clot length, and the clot weight-to-length ratio were all significantly reduced by the administration of AAV9(HO-1) as compared with AAV9(revHO-1) at day 2 after IVCL. AAV9(HO-1) as compared with AAV9(revHO-1) did not reduce clot number at day 2 after IVCL.
Fig. 1.

Effect of adeno-associated viral serotype 9 (AAV9)-mediated HO-1 upregulation on clot size in the inferior vena cava ligation (IVCL) model. Clot weight (A), clot length (B), and the clot weight-to-length ratio (C) in negative control vector [AAV9(revHO-1)]-treated (n = 9) and AAV9 vector [AAV9(HO-1)]-treated (n = 10) mice were determined at 2 days following IVCL. *P < 0.05 vs. AAV9(revHO-1)-treated group for that index.
Products of HO activity reduce clot formation.
Carbon monoxide (CO) is one of the main products of HO activity. We thus examined in this model whether the administration of CO, as delivered by a CO-releasing molecule (CORM), reduces clot formation. The administration of the CO-releasing molecule (CORM-3), as compared with control iCORM-3 (which does not release CO), reduced clot weight, clot length, and clot weight-to-length ratio at day 2 after IVCL (Fig. 2). CORM-3, as compared with control iCORM-3, did not reduce clot number at day 2 after IVCL.
Fig. 2.

Effect of carbon monoxide delivered via carbon monoxide-releasing molecule 3 (CORM-3) on clot size in the inferior vena cava ligation (IVCL) model. Clot weight (A), clot length (B), and the clot weight-to-length ratio (C) in inactivated CORM-3 (iCORM-3)-treated (n = 10) and CORM-3-treated (n = 9) mice were determined at 2 days following IVCL. *P < 0.05 vs. iCORM-3-treated group for that index.
We also determined whether the other main HO product, biliverdin, exerted an inhibitory effect on clot formation. As shown in Fig. 3, biliverdin reduced clot weight, clot length, and weight-to-length ratio on day 2 after IVCL. Biliverdin did not reduce clot number at day 2 after IVCL.
Fig. 3.

Effect of biliverdin on clot size in the inferior vena cava ligation (IVCL) model. Clot weight (A), clot length (B), and the clot weight-to-length ratio (C) in saline-treated (n = 10) and biliverdin-treated (n = 9) mice were determined at 2 days following IVCL. *P < 0.05 vs. saline-treated group for that index.
Upregulation of HO-1/HO by hemin reduces clot formation.
In preliminary studies we established a protocol for administering hemin that effectively and robustly induced HO-1. As shown in Fig. 4, 6 h after the administration of the third dose of hemin in mice with an intact IVC (and not subjected to IVCL), veins exhibited marked induction of HO-1 mRNA and protein and increased HO activity. With this protocol, the administration of hemin significantly decreased clot weight, clot length, and clot weight-to-length ratio on days 1 and 2 after IVCL, and decreased clot weight-to-length ratio on day 3 after IVCL (Fig. 5). Interestingly, while saline-treated mice exhibited on visual inspection clot formation at the time of harvest on all 3 days after IVCL, no visible clot was macroscopically observed at day 1 in 4 out of 7 hemin-treated mice with IVCL (P = 0.07 vs. saline-treated mice with IVCL on day 1), and at day 3, no visible clot was seen in 5 out of 12 hemin-treated mice with IVCL (P = 0.04 vs. saline-treated mice with IVCL on day 3).
Fig. 4.
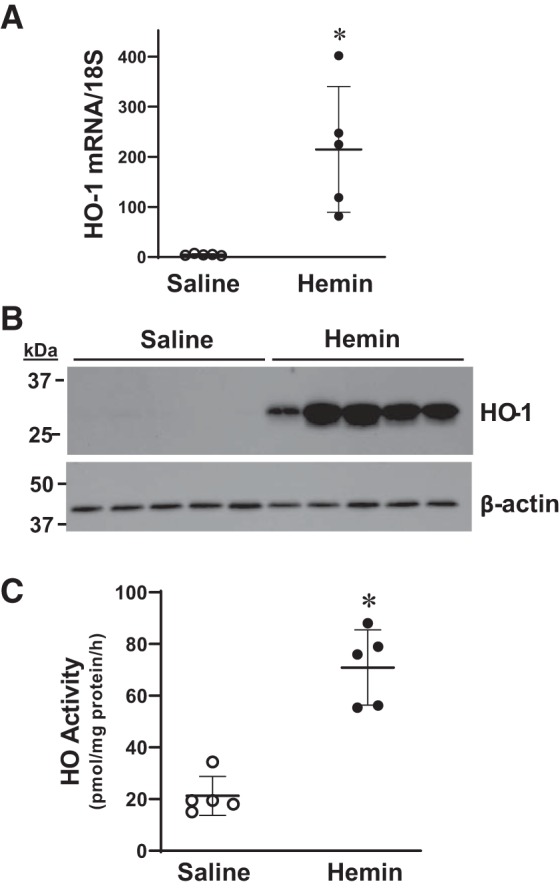
Heme oxygenase-1 (HO-1) expression and HO activity in intact veins in mice administered hemin. HO-1 mRNA expression in jugular veins was measured by quantitative real-time RT-PCR (A), HO-1 protein expression in thoracic inferior vena cava (IVC) was assessed by Western blot analysis (B), and HO activity was determined in the abdominal IVC (C) in mice treated with saline or hemin daily for 3 days, but without IVC ligation. Assessments were made 6 h after the 3rd dose of saline or hemin. *P < 0.05 vs. saline-treated group; n = 5 in each group.
Fig. 5.

Effect of hemin on clot size in the inferior vena cava ligation (IVCL) model. Clot weight (A), clot length (B), and the clot weight-to-length ratio (C) were determined in saline-treated and hemin-treated mice at days 1, 2, and 3 following IVCL. n = 8, 6, and 10 in each group for days 1, 2, and 3, respectively, in saline-treated mice, and n = 7, 7, and 12 in each group for days 1, 2, and 3, respectively, in hemin-treated mice. *P < 0.05 vs. saline-treated group at that day.
Histologic assessment of the IVC wall demonstrated that the IVC exhibited, as previously described, thickening and inflammation of the venous wall in the control, saline-treated IVCL model; such changes were reduced in hemin-treated mice with IVCL. Morphometric studies demonstrated that the wall thickness of the ligated IVC was reduced on days 2 and 3 after IVCL in hemin-treated as compared with saline-treated mice (Fig. 6A). Shown in Fig. 6, B and C, are representative sections at day 3 after IVCL, depicting foci of neutrophil-enriched inflammation involving the venous endothelium in saline-treated mice (Fig. 6B) and less prominent focal inflammation in hemin-treated mice (Fig. 6C).
Fig. 6.
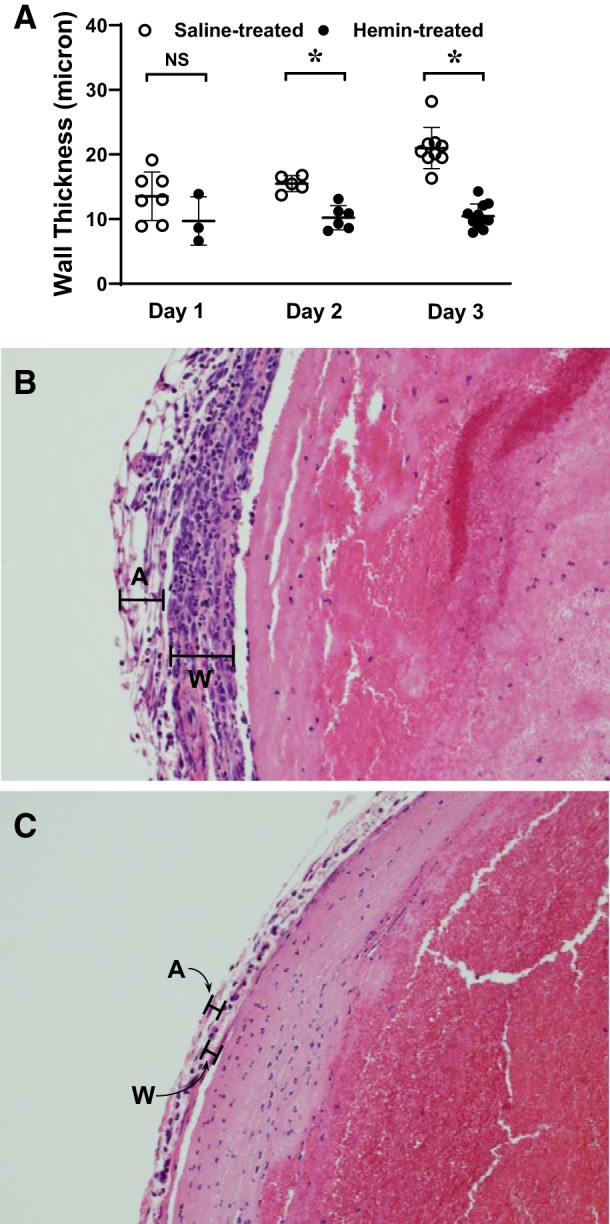
Effect of hemin on venous wall thickness in the inferior vena cava ligation (IVCL) model. Venous wall thickness in the IVCL model in histologic samples available after tissue preparation and histological processing was determined morphometrically in saline-treated and hemin-treated mice at days 1, 2, and 3 following IVCL (A). n = 7, 5, and 9 in each group for days 1, 2, and 3, respectively, in saline-treated mice, and n = 3, 6, and 11 in each group for days 1, 2, and 3, respectively, in hemin-treated mice. *P < 0.05 vs. saline-treated group at that day. Representative histological views (×200) of the IVCL model in saline-treated (B) and hemin-treated (C) mice at day 3 after IVCL are shown. In B and C, the wall (W) and adventitia (A) are demarcated by black lines. The IVCL in hemin-treated mice displays reduced wall thickness and smaller and fewer foci of neutrophil-enriched inflammation.
As shown previously, the IVCL is characterized by markedly increased expression of proinflammatory genes. We thus questioned whether hemin altered the expression of such genes in the IVCL model at day 2. As demonstrated in Fig. 7, the administration of hemin decreased expression of critical genes in this model [keratinocyte chemoattractant (KC), interleukin-6 (IL-6), monocyte chemoattractant protein-1 (MCP-1) and inducible nitric oxide synthase (iNOS)], along with other proinflammatory genes as listed in Table 1. These data demonstrate that hemin suppressed expression of genes that are strong chemoattractants for leukocytes, and among the genes prominently suppressed by hemin was KC, the murine homologue for human IL-8, which is a potent chemoattractant for neutrophils. Neutrophils, either through neutrophil extracellular trap (NET)-dependent or NET-independent processes, are incriminated in thrombus formation (38, 63, 66). The observed reductive effects of hemin on KC expression (and that of other genes) and on neutrophil infiltration suggest the possibility that the beneficial effect of hemin may be mediated, in part, by suppressing neutrophil infiltration.
Fig. 7.
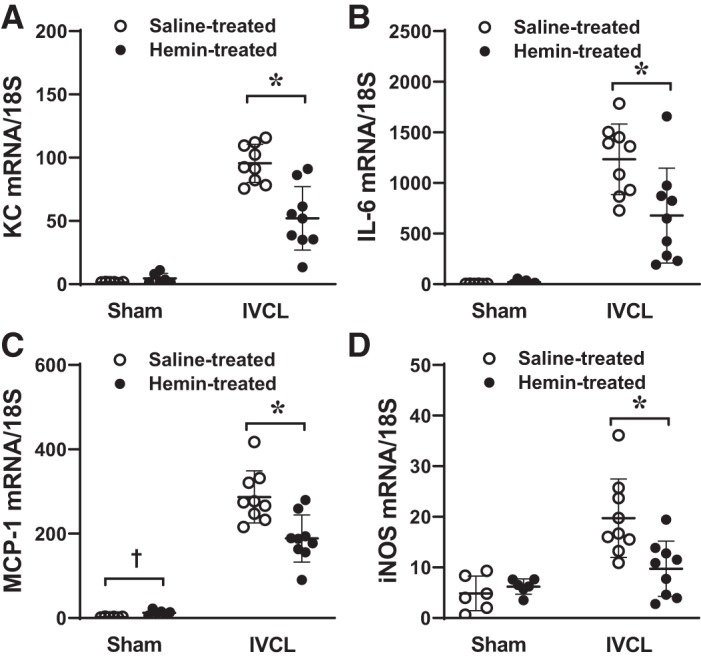
Effect of hemin on the expression of inflammation-related genes in the inferior vena cava ligation (IVCL) model. Expression of keratinocyte chemoattractant (KC; A), interleukin-6 (IL-6; B), monocyte chemoattractant protein-1 (MCP-1; C) and inducible nitric oxide synthase (iNOS; D) was determined by quantitative real-time RT-PCR following IVCL or sham operation in saline-treated or hemin-treated mice and undertaken at day 2 after IVCL. n = 6 for each sham group, n = 9 for each IVCL group. *P < 0.05 vs. saline-treated IVCL. †P < 0.05 vs. saline-treated sham.
Table 1.
Gene expression in the IVCL model (clot and IVC wall) at day 2 after IVCL
| Gene | Sham + Saline |
Sham + Hemin |
IVCL + Saline |
IVCL + Hemin |
|---|---|---|---|---|
| SELP | 7.9 ± 1.6 | 11.1 ± 2.0† | 38.4 ± 9.3 | 27.4 ± 7.9* |
| TGFβ1 | 2.0 ± 0.3 | 2.6 ± 0.5† | 9.8 ± 2.1 | 6.9 ± 2.2* |
| TNFα | 7.0 ± 3.3 | 9.8 ± 4.4 | 194.6 ± 59.9 | 135.8 ± 57.3* |
| MMP-9 | 1.2 ± 0.3 | 3.0 ± 1.5† | 16.6 ± 5.0 | 10.4 ± 5.8* |
| PDGFa | 1.6 ± 0.2 | 1.5 ± 0.2 | 3.1 ± 0.4 | 2.5 ± 0.6* |
| PDGFb | 3.6 ± 1.5 | 4.8 ± 0.6 | 8.6 ± 1.3 | 6.7 ± 1.7* |
| FN1 | 5.3 ± 1.1 | 5.2 ± 1.0 | 18.4 ± 2.5 | 12.4 ± 4.1* |
| VEGFa | 4.7 ± 3.7 | 7.7 ± 2.9 | 6.8 ± 1.3 | 4.1 ± 1.6* |
Values are means ± SD; n = 6 for each sham and n = 9 for each inferior vena cava (IVC) ligation (IVCL) group. Values are the result of relative quantification performed against a standard curve constructed for each mRNA target, normalized for expression of 18S rRNA, and expressed in arbitrary units. SELP, selectin P; TGF, transforming growth factor; TNF, tumor necrosis factor; MMP, matrix metalloproteinase; PDGF, platelet-derived growth factor; FN, fibronectin; VEGF, vascular endothelial growth factor.
P < 0.05 vs. IVCL + saline;
P < 0.05 vs. sham + saline.
Hemin attenuates induction of plasminogen activator inhibitor-1 in the IVCL model.
To pursue a mechanism that may underlie the reductive effect of hemin on clot formation in the IVCL model, we examined the effect of hemin on expression of plasminogen activator inhibitor-1 (PAI-1) as well as the countervailing molecules for PAI-1, namely, urokinase plasminogen activator (uPA) and tissue plasminogen activator (tPA); in models of venous thrombosis, the balance of procoagulant species (PAI-1) and relevant anticoagulant species (uPA and tPA) is a significant determinant of clot formation. As shown in Fig. 8, PAI-1, uPA, and tPA were all induced in the IVCL model. Hemin significantly reduced expression of PAI-1 but did not influence the expression of uPA and tPA (Fig. 8). Accordingly, from these data we calculated the relevant procoagulant-to-anticoagulant ratios, demonstrating that hemin significantly reduced the elevated ratios for PAI-1 to uPA (51.9 ± 9.2 vs. 40.0 ± 12.8, saline vs. hemin, respectively, P < 0.05) and for PAI-1 to tPA (10.8 ± 1.9 vs. 7.7 ± 3.2, saline vs. hemin, respectively, P < 0.05) that occur in the IVCL model. As PAI-1 is known to significantly contribute to clot formation in the IVCL model (6), the reductive effect of hemin on PAI-1 upregulation provides at least one molecular mechanism, accounting for the reduction in clot formation and size observed with the administration of hemin.
Fig. 8.

Effect of hemin on the expression of plasminogen activator inhibitor-1 (PAI-1), urokinase plasminogen activator (uPA), and tissue plasminogen activator (tPA) mRNA in the inferior vena cava ligation (IVCL) model. Expression of PAI-1 (A), uPA (B), and tPA (C) was determined by quantitative real time RT-PCR following IVCL or sham operation in saline-treated or hemin-treated mice and undertaken at day 2 after IVCL. n = 6 for each sham group; n = 9 for each IVCL group. *P < 0.05 vs. saline-treated IVCL.
Reductive effect of hemin on clot formation is dependent on HO activity.
As shown in Fig. 4, hemin robustly induces HO-1 mRNA and HO activity. To determine whether the reductive effect of hemin on clot size was dependent on HO activity, we determined clot size in additional groups of mice subjected to the IVCL model and which were administered hemin alone or hemin along with tin protoporphyrin, a competitive inhibitor of HO activity; this competitive inhibitor of HO activity is widely used in disease models to probe whether HO activity is functionally significant. As shown in Fig. 9, inhibiting HO activity while concomitantly administering hemin led to significantly larger clots as compared with clots in mice treated with hemin alone. Thus, the reductive effect of hemin on clot size requires HO activity.
Fig. 9.

Role of heme oxygenase (HO) activity in mediating the effect of hemin on clot size in the inferior vena cava ligation (IVCL) model. Clot weight (A), clot length (B), and the clot weight-to-length ratio (C) were determined in hemin-treated (n = 8) and hemin + SnPP (tin protoporphyrin)-treated (n = 10) mice at day 1 following IVCL. SnPP is a competitive inhibitor of HO activity. *P < 0.05 vs. hemin-treated group.
Concentration of heme is increased in the IVCL model.
We assessed the concentration of heme in the IVCL model for two reasons. First, inducing the major heme-degrading enzyme by two different strategies, AAV9(HO-1) and hemin, significantly decreased clot size. Second, recent constructs of clotting assign a pathogenic role for heme in the initiation of clot formation; in this construct, hemoglobin from trapped, damaged erythrocytes undergo oxidative denaturation and releases free heme (12, 40, 41, 66). Our findings in this model support this construct: as shown in Fig. 10A, concentrations of heme in the IVCL model are markedly increased at days 1, 2, and 3 after IVCL and decrease thereafter and are back to baseline by day 10.
Fig. 10.

Heme concentration in the inferior vena cava (IVC) ligation (IVCL) model. A: heme concentration was measured by the pyridine-chromogen method and normalized for lysate protein content at day 1, 2, 3, and 10 following IVCL. n = 6 in each group for studies at day 1, 2, 3, and 10; n = 5 in the day 0 (IVC only control) group. *P < 0.05 vs. day 0. B: heme concentration was determined by pyridine-chromogen method following IVCL or sham operation in saline-treated or hemin-treated mice and undertaken at day 2 after IVCL. n = 5 for each sham group; n = 9 for each IVCL group. *P < 0.05 vs. saline-treated IVCL.
Administration of hemin reduces concentration of heme in the IVCL model.
Since hemin reduced clot size through an HO-dependent mechanism, we next questioned whether heme concentration in the IVCL model would be reduced when hemin is administered. As shown in Fig. 10B, the prior administration of hemin diminished heme concentration in the IVCL model, thereby reflecting the robust induction of HO activity by hemin, with attendant heme degradation. Interestingly, in the sham-treated mice (without IVCL), heme content in the intact IVC (sham-ligated) in mice treated with repeated doses of hemin was entirely comparable with that seen in the saline-treated sham-ligated mice. Thus, the induction of HO in mice repeatedly treated with hemin is sufficiently robust to entirely prevent any elevation in heme content in the control IVC (sham ligated), despite the repeated administration of hemin.
Hemopexin inhibits clot formation in the IVCL model.
Since heme concentration is markedly increased in the IVCL model and inducing the heme-degrading enzyme HO by two different strategies reduced clot formation, we questioned whether a heme-binding protein would also reduce clot formation. The major heme-binding protein is hemopexin. In the IVCL model, hemopexin mRNA and protein are both significantly induced in the IVC wall itself; in these studies, the clot was removed from the resected IVCL model such that hemopexin expression was examined only in the IVC wall in the absence of the attached clot (Fig. 11). Hemopexin mRNA expression was also markedly induced in the combined ligated IVC and contained clot (data not shown).
Fig. 11.
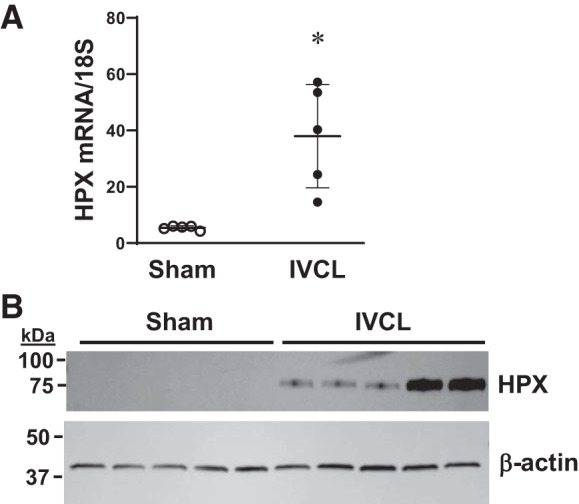
Expression of hemopexin (HPX) in the inferior vena cava (IVC) wall in the IVC ligation (IVCL) model. HPX mRNA was measured by quantitative real-time RT-PCR (A) and HPX protein was assessed by Western blot analysis (B) in the IVC wall (without contained clot) of mice 2 days following IVCL or sham operation. n = 5 in each group for the mRNA measurements in A. *P < 0.05 vs. sham group.
To determine the functional significance of hemopexin in this model, we imposed the IVCL model in hemopexin+/+ and hemopexin−/− mice. The genetic deficiency of hemopexin augmented clot size as evidenced by more than a 30% increase in the clot weight-to-length ratio at day 2 after IVCL (Fig. 12).
Fig. 12.

Effect of genetic deficiency of hemopexin (HPX) on clot size in the inferior vena cava ligation (IVCL) model. Clot weight (A), clot length (B), and the clot weight-to-length ratio (C) were determined in HPX+/+ mice and HPX−/− mice at day 2 following IVCL. n = 7 and n = 8 in HPX+/+ mice and HPX−/− groups, respectively. *P < 0.05 vs. HPX+/+ group for that index.
Role of HO-2 on clot formation in the IVCL model.
Heme-degrading HO activity is possessed not only by the inducible HO-1 isoform but also the constitutive HO-2 isoform. We thus questioned whether the constitutive HO isoform, HO-2, may inhibit either clot formation or promote clot resolution. In this model, HO-2 mRNA, a constitutive gene that is rarely induced in diseased models, quite surprisingly, is significantly induced at approximately twofold (Fig. 13A), and as shown for comparison in the same IVCL specimens, the induction of HO-1 mRNA in IVCL is markedly greater at 40-fold (Fig. 13B). To examine the functional significance of HO-2, we imposed the IVCL model in HO-2+/+ and HO-2−/− mice. However, neither at day 2 nor at day 10 did the deficiency of HO-2 significantly increased clot weight, clot length, or clot weight-to-length ratio (Fig. 14). The lack of a functional role for HO-2 likely reflects much less HO activity from HO-2 compared with HO-1, given the markedly lower relative HO-2 mRNA induction (2-fold vs. 40-fold, HO-2 vs. HO-1).
Fig. 13.
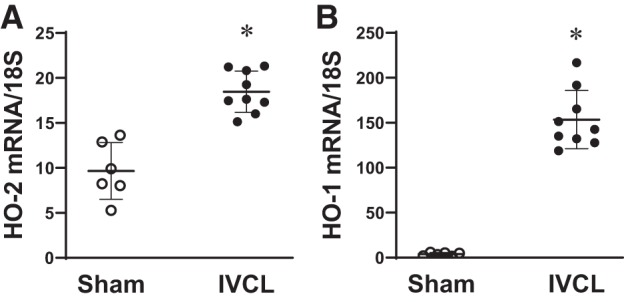
Expression of heme oxygenase-2 (HO-2) and heme oxygenase-1 (HO-1) mRNA in the inferior vena cava (IVC) ligation (IVCL) model. Expression of HO-2 (A) and HO-1 (B) mRNA in the IVCL model (IVC and contained clot) was determined in mice at 2 days after IVCL or sham surgery. n = 6 for each sham group; n = 9 for each IVCL group. *P < 0.05 vs. sham group.
Fig. 14.

Effect of genetic deficiency of heme oxygenase-2 (HO-2) on clot size in the inferior vena cava ligation (IVCL) model. Clot weight (A), clot length (B), and the clot weight-to-length ratio (C) were determined in HO-2+/+ mice and HO-2−/− mice at 2 and 10 days following IVCL. n = 5 in each group.
DISCUSSION
Our prior work demonstrated that genetic HO-1 deficiency promoted clot formation in the IVCL model (73). The current studies addressed whether the induction of HO-1 or the provision of its products reduces clot formation. Induction of HO-1 by viral gene delivery reduced clot size, as did products of HO activity, CO and biliverdin. Induction of HO-1 by hemin reduced clot size and number, and IVC wall thickness and inflammation. The clinical relevance of our findings is at least threefold. First, the protective effects of hemin we observed offer a therapeutic strategy for reducing thrombus formation in high-risk clinical settings; brands of hemin such as Panhematin are already approved for use in human diseases such as porphyria. Second, CO-releasing molecules (CORMs) are currently undergoing clinical trials in human diseases. Third, individuals with long GT repeats, as compared with short GT repeats, in the human HO-1 gene promoter are prone to VTE disease (7, 47); notably, long GT repeats, as compared with short GT repeats, in the HO-1 gene promoter lead to less HO-1 inducibility and less HO activity (19).
The protective effects of heme-degrading and heme-binding proteins along with increased heme content in the IVCL model are consistent with the recent concept that heme contributes to clot formation. Red blood cells (RBCs) are now regarded as an active participant, rather than a passive bystander, in clot formation, and among the mechanisms underpinning such participation is the release of hemoglobin from trapped and injured RBCs in the developing clot (12, 40, 41, 66). Plasma hemoglobin, no longer surrounded by the reducing RBC milieu, is oxidatively destabilized, releasing its heme. Supporting this thesis are the clinical findings indicating release of hemoglobin from RBCs during acute coronary syndromes (17), increased plasma bilirubin levels (which correlate with the thrombus burden) following myocardial infarction (27), and increased plasma heme levels when thrombosis occurs after cardiopulmonary bypass (70).
Free heme in plasma is thrombogenic via several mechanisms. First, in sickle cell disease, heme activates the endothelium and causes the degranulation of Weibel-Palade bodies that contain thrombogenic constituents (9, 11, 33); heme also activates NF-κB which elicits a thrombogenic and proinflammatory phenotype (9, 11, 33). Second, heme induces tissue factor (69) that is a trigger for the extrinsic coagulation cascade. Third, heme activates the alternative complement pathway, producing the thrombogenic component C3a (22, 46). Fourth, heme causes cross-linking of fibrinogen (37). In the IVCL model the thrombogenicity of heme is supported by the following findings: 1) levels of heme peaked just before maximum clot size (day 2), with heme levels and clot size declining thereafter; 2) inducing the heme-degrading enzyme HO-1 by gene delivery or hemin decreased clot size; and 3) deficiency of the heme-binding protein, hemopexin, increased clot size.
The effects of hemin in the IVCL model illustrate the Janus-faced nature of heme; the countenance presented depends upon the administered dose of heme, plasma/tissue levels of heme, and the time point at which such effects are assessed (11, 72). In addition to its procoagulant and proinflammatory effects, heme is prooxidant and proapoptotic (26). However, heme concomitantly upregulates countervailing systems such as HO-1: heme promotes HO-1 mRNA expression by inducing its activating transcription factor, Nrf2 (2); heme also binds to Bach1, the suppressive transcription factor for HO-1, thereby derepressing HO-1 gene expression (30). Induction of HO-1 not only degrades and reduces levels of heme but also produces bile pigments, which are antioxidant and anti-inflammatory, and CO, which, in low doses, is vasorelaxant, anti-inflammatory, and antiapoptotic (1, 5, 11, 21, 49). Thus, rapid degradation of heme by HO-1 induction concomitantly generates products that counteract the adverse effects of any heme that remains undegraded. The efficacy of HO-1 induction and the generation of HO products as antithrombotic responses reside on the following: first, this system removes heme, a thrombogenic species; second, this system produces antioxidant and anti-inflammatory species, oxidant stress and inflammation being potent drivers of thromobogenesis; and third, both CO and bile pigments are antithrombotic (see Figs. 2 and 3 and discussion).
The increase in venous HO activity induced by AAV9(HO-1) in our prior studies (35) and by hemin in the present studies were comparable at approximately threefold. There appeared to be a stronger effect of hemin as compared with AAV9(HO-1), CO, and biliverdin on clot size. We suggest the reason why hemin may be more effective is that hemin administration not only generates HO activity but also provides a ready and copious source of substrate for induced HO activity; this ensures not only rapid removal of heme but also copious and rapid generation of products which are both antithrombotic and anti-inflammatory. In contrast, HO-1/HO, when induced by AAV9(HO-1), is not provided with substrate as is HO-1/HO when induced by hemin. HO-1, when induced by AAV9(HO-1), relies on much lower amounts of heme substrate, as is available from the quite low ambient levels of cellular heme that exist or arise from destabilized intracellular heme proteins; generation of antithrombotic HO products (CO and biliverdin) is thus strikingly lower.
Our studies focused on hemin as an inducer of HO-1 and its beneficial effects as an antithrombotic strategy because of the potential for clinical translation. As a potential mechanism underlying the reductive effects of hemin, we examined PAI-1 expression because of the following considerations: 1) PAI-1 exerts potent prothrombotic effects (32, 76), 2) our present studies showed marked induction of PAI-1 in the IVCL model, and 3) prior studies demonstrated that genetic deficiency of PAI-1 reduced clot formation in the murine IVCL model (6). PAI-1 is upregulated in the IVCL model, at least in part, by oxidative stress (heme being a contributor to oxidant stress) and inflammatory processes present in the early stages of clot formation. In the present studies, hemin administration significantly reduced the observed upregulation of PAI-1, thereby providing at least one molecular mechanism for the beneficial effects of hemin. Additionally, prior studies demonstrate that upregulation of HO-1 decreases PAI-1 expression in other vascular models (15), while HO-1 deficiency, as shown in our prior studies in the arteriovenous fistula, upregulates PAI-1 (31). Finally, both carbon monoxide and bile pigments reduce PAI-1 expression in models of tissue injury (15, 23, 42, 44, 68); in humans, higher levels of bilirubin are associated with decreased serum levels of PAI-1 (16). We thus suggest that hemin reduces clot formation, at least in part, through HO-dependent suppression of PAI-1 upregulation that occurs in this model, and extrapolating from the known reductive effects of CO and bile pigments on PAI-1 expression, we speculate that suppression of PAI-1 may also underlie the beneficial effects of these products of HO activity.
There is a bidirectional relationship between clotting and inflammation, with clotting creating an inflammatory milieu and inflammation creating a prothrombotic milieu. Because hemin suppressed the upregulation of KC (murine homologue of IL-8) and MCP-1, among other proinflammatory species, and decreased neutrophilic infiltration in the clot, these effects of hemin would tend to be antithrombotic. As such they would act in concert with the suppressive effects of hemin on the upregulation of PAI-1 that occurs in the IVCL model.
Induction of HO-1 and increased levels of carboxyhemoglobin (an index of CO production) occur in conditions with a risk for thrombosis (43, 56–62). Based largely on in vitro studies, it has been inferred that such induction of HO-1/CO promotes thrombosis. Such interpretations need to be tempered because they involve extrapolation of in vitro findings, and our current studies in vivo attest to the antithrombotic effect of HO-1, CORM-3, and biliverdin. We suggest that the HO-1/CO/biliverdin system is acting as a countervailing antithrombotic process when induced, rather than a prothrombotic one.
Prior studies in the IVCL model demonstrating that activated protein C diminishes clot size, in part, through HO-1 (24) are consistent with our findings. HO-1 induction is antithrombotic in other models of venous (39) and arterial thrombosis (74); additionally, CO reduces arterial thrombosis after aortic transplantation in HO-1−/− mice (14). The present findings are also relevant to our prior studies in which HO-1 induction inhibits vasoocclusion in sickle cell disease (8, 10) and prolongs the patency of the dialysis arteriovenous fistula (31). As procoagulant processes contribute to both vasoocclusion and fistula failure, the beneficial effects of HO-1 in these conditions may reflect the antithrombotic actions of HO-1.
The construct articulated in this study (the counterbalancing between the effects of heme on one hand and those of HO-1 and hemopexin on the other) may be germane to other diseases that exhibit a procoagulant milieu, increased plasma heme levels, and HO-1 induction. Two such conditions are preeclampsia and paroxysmal nocturnal hemoglobinuria (PNH). In preeclampsia, HO-1 and hemopexin may exert a protective role (4), as does HO-1 in PNH (55).
In summary, in the IVCL model we provide the first demonstration of elevated concentrations of heme and the antithrombotic effects of the major heme-degrading protein HO-1 and its products. Our studies support the thesis that oxidative denaturation of hemoglobin, leaked from RBCs, and the attendant release of heme contribute to thrombogenesis. Our studies also provide the first evidence in vivo for the antithrombotic effect of hemopexin. It would be of interest to examine this prothrombotic/antithrombotic construct in other models, in particular, models of venous thrombosis occurring in the setting of venous stenosis. Our findings suggest therapeutic strategies not only for VTE diseases but also for diverse diseases driven in part by thrombogenic processes, including, among others, acute coronary syndromes, sickle cell disease, dialysis fistula failure, and thrombotic microangiopathies.
GRANTS
These studies were supported by National Institutes of Health Grants DK-47060 and DK-119167 (to K. A. Nath and J. P. Grande), HL-114567 (to J. D. Belcher and G. M. Vercellotti), HL-136348 (to V. D. Garovic), and R01-NS-095205 (to R. F. Regan).
DISCLOSURES
G. M. Vercellotti and J. D. Belcher receive research funding from CSL Behring and Mitobridge/Astellas. Dr. Regan has received research funding from CSL Behring.
AUTHOR CONTRIBUTIONS
K.A.N. and A.J.C. conceived and designed research; A.J.C., M.L.H., and M.C.N. performed experiments; K.A.N., J.P.G., A.J.C., and M.C.N. analyzed data; K.A.N., J.P.G., J.D.B., V.D.G., A.J.C., M.L.H., M.A.B., M.C.N., R.F.R., and G.M.V. interpreted results of experiments; K.A.N. and A.J.C. prepared figures; K.A.N. and A.J.C. drafted manuscript; K.A.N., J.P.G., J.D.B., V.D.G., A.J.C., M.L.H., M.A.B., M.C.N., R.F.R., and G.M.V. edited and revised manuscript; K.A.N., J.P.G., J.D.B., V.D.G., A.J.C., M.L.H., M.A.B., M.C.N., R.F.R., and G.M.V. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Allan W. Ackerman for technical assistance in these studies.
REFERENCES
- 1.Agarwal A, Bolisetty S. Adaptive responses to tissue injury: role of heme oxygenase-1. Trans Am Clin Climatol Assoc 124: 111–122, 2013. [PMC free article] [PubMed] [Google Scholar]
- 2.Alam J, Killeen E, Gong P, Naquin R, Hu B, Stewart D, Ingelfinger JR, Nath KA. Heme activates the heme oxygenase-1 gene in renal epithelial cells by stabilizing Nrf2. Am J Physiol Renal Physiol 284: F743–F752, 2003. doi: 10.1152/ajprenal.00376.2002. [DOI] [PubMed] [Google Scholar]
- 3.Alotaibi GS, Wu C, Senthilselvan A, McMurtry MS. Secular trends in incidence and mortality of acute venous thromboembolism: the AB-VTE population-based study. Am J Med 129: 819–825, 2016. [DOI] [PubMed] [Google Scholar]
- 4.Anderson UD, Jälmby M, Faas MM, Hansson SR. The hemoglobin degradation pathway in patients with preeclampsia - Fetal hemoglobin, heme, heme oxygenase-1 and hemopexin - Potential diagnostic biomarkers? Pregnancy Hypertens 14: 273–278, 2018. doi: 10.1016/j.preghy.2018.02.005. [DOI] [PubMed] [Google Scholar]
- 5.Ayer A, Zarjou A, Agarwal A, Stocker R. Heme oxygenases in cardiovascular health and disease. Physiol Rev 96: 1449–1508, 2016. doi: 10.1152/physrev.00003.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baldwin JF, Sood V, Elfline MA, Luke CE, Dewyer NA, Diaz JA, Myers DD, Wakefield T, Henke PK. The role of urokinase plasminogen activator and plasmin activator inhibitor-1 on vein wall remodeling in experimental deep vein thrombosis. J Vasc Surg 56: 1089–1097, 2012. doi: 10.1016/j.jvs.2012.02.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bean CJ, Boulet SL, Ellingsen D, Trau H, Ghaji N, Hooper WC, Austin H. Increased risk of venous thromboembolism is associated with genetic variation in heme oxygenase-1 in Blacks. Thromb Res 130: 942–947, 2012. doi: 10.1016/j.thromres.2012.08.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Belcher JD, Chen C, Nguyen J, Abdulla F, Zhang P, Nguyen H, Nguyen P, Killeen T, Miescher SM, Brinkman N, Nath KA, Steer CJ, Vercellotti GM. Haptoglobin and hemopexin inhibit vaso-occlusion and inflammation in murine sickle cell disease: Role of heme oxygenase-1 induction. PLoS One 13: e0196455, 2018. doi: 10.1371/journal.pone.0196455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Belcher JD, Chen C, Nguyen J, Milbauer L, Abdulla F, Alayash AI, Smith A, Nath KA, Hebbel RP, Vercellotti GM. Heme triggers TLR4 signaling leading to endothelial cell activation and vaso-occlusion in murine sickle cell disease. Blood 123: 377–390, 2014. doi: 10.1182/blood-2013-04-495887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Belcher JD, Mahaseth H, Welch TE, Otterbein LE, Hebbel RP, Vercellotti GM. Heme oxygenase-1 is a modulator of inflammation and vaso-occlusion in transgenic sickle mice. J Clin Invest 116: 808–816, 2006. doi: 10.1172/JCI26857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Belcher JD, Nath KA, Vercellotti GM. Vasculotoxic and proinflammatory effects of plasma heme: cell signaling and cytoprotective responses. ISRN Oxidative Med 2013: 831596, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Byrnes JR, Wolberg AS. Red blood cells in thrombosis. Blood 130: 1795–1799, 2017. doi: 10.1182/blood-2017-03-745349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen-Roetling J, Liu W, Regan RF. Hemopexin decreases hemin accumulation and catabolism by neural cells. Neurochem Int 60: 488–494, 2012. doi: 10.1016/j.neuint.2012.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen B, Guo L, Fan C, Bolisetty S, Joseph R, Wright MM, Agarwal A, George JF. Carbon monoxide rescues heme oxygenase-1-deficient mice from arterial thrombosis in allogeneic aortic transplantation. Am J Pathol 175: 422–429, 2009. doi: 10.2353/ajpath.2009.081033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen YH, Tsai HL, Chiang MT, Chau LY. Carbon monoxide-induced early thrombolysis contributes to heme oxygenase-1-mediated inhibition of neointimal growth after vascular injury in hypercholesterolemic mice. J Biomed Sci 13: 721–730, 2006. doi: 10.1007/s11373-006-9093-7. [DOI] [PubMed] [Google Scholar]
- 16.Cho HS, Lee SW, Kim ES, Shin J, Moon SD, Han JH, Cha BY. Serum bilirubin levels are inversely associated with PAI-1 and fibrinogen in Korean subjects. Atherosclerosis 244: 204–210, 2016. doi: 10.1016/j.atherosclerosis.2015.11.008. [DOI] [PubMed] [Google Scholar]
- 17.Cubedo J, Suades R, Padro T, Martin-Yuste V, Sabate-Tenas M, Cinca J, Sans-Rosello J, Sionis A, Badimon L. Erythrocyte-heme proteins and STEMI: implications in prognosis. Thromb Haemost 117: 1970–1980, 2017. doi: 10.1160/TH17-05-0314. [DOI] [PubMed] [Google Scholar]
- 18.Diaz JA, Obi AT, Myers DD Jr, Wrobleski SK, Henke PK, Mackman N, Wakefield TW. Critical review of mouse models of venous thrombosis. Arterioscler Thromb Vasc Biol 32: 556–562, 2012. doi: 10.1161/ATVBAHA.111.244608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Exner M, Minar E, Wagner O, Schillinger M. The role of heme oxygenase-1 promoter polymorphisms in human disease. Free Radic Biol Med 37: 1097–1104, 2004. doi: 10.1016/j.freeradbiomed.2004.07.008. [DOI] [PubMed] [Google Scholar]
- 20.Farrugia G, Lei S, Lin X, Miller SM, Nath KA, Ferris CD, Levitt M, Szurszewski JH. A major role for carbon monoxide as an endogenous hyperpolarizing factor in the gastrointestinal tract. Proc Natl Acad Sci USA 100: 8567–8570, 2003. doi: 10.1073/pnas.1431233100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fredenburgh LE, Merz AA, Cheng S. Haeme oxygenase signalling pathway: implications for cardiovascular disease. Eur Heart J 36: 1512–1518, 2015. doi: 10.1093/eurheartj/ehv114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Frimat M, Tabarin F, Dimitrov JD, Poitou C, Halbwachs-Mecarelli L, Fremeaux-Bacchi V, Roumenina LT. Complement activation by heme as a secondary hit for atypical hemolytic uremic syndrome. Blood 122: 282–292, 2013. doi: 10.1182/blood-2013-03-489245. [DOI] [PubMed] [Google Scholar]
- 23.Fujita T, Toda K, Karimova A, Yan SF, Naka Y, Yet SF, Pinsky DJ. Paradoxical rescue from ischemic lung injury by inhaled carbon monoxide driven by derepression of fibrinolysis. Nat Med 7: 598–604, 2001. doi: 10.1038/87929. [DOI] [PubMed] [Google Scholar]
- 24.Gabre J, Chabasse C, Cao C, Mukhopadhyay S, Siefert S, Bi Y, Netzel-Arnett S, Sarkar R, Zhang L. Activated protein C accelerates venous thrombus resolution through heme oxygenase-1 induction. J Thromb Haemost 12: 93–102, 2014. doi: 10.1111/jth.12424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goldhaber SZ, Bounameaux H. Pulmonary embolism and deep vein thrombosis. Lancet 379: 1835–1846, 2012. doi: 10.1016/S0140-6736(11)61904-1. [DOI] [PubMed] [Google Scholar]
- 26.Gonzalez-Michaca L, Farrugia G, Croatt AJ, Alam J, Nath KA. Heme: a determinant of life and death in renal tubular epithelial cells. Am J Physiol Renal Physiol 286: F370–F377, 2004. doi: 10.1152/ajprenal.00300.2003. [DOI] [PubMed] [Google Scholar]
- 27.Hamur H, Duman H, Bakirci EM, Kucuksu Z, Demirelli S, Kalkan K, Degirmenci H. Bilirubin levels and thrombus burden in patients with ST-segment elevation myocardial infarction. Angiology 67: 565–570, 2016. doi: 10.1177/0003319715603899. [DOI] [PubMed] [Google Scholar]
- 28.Heit JA. The epidemiology of venous thromboembolism in the community. Arterioscler Thromb Vasc Biol 28: 370–372, 2008. doi: 10.1161/ATVBAHA.108.162545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hillestad ML, Guenzel AJ, Nath KA, Barry MA. A vector-host system to fingerprint virus tropism. Hum Gene Ther 23: 1116–1126, 2012. doi: 10.1089/hum.2011.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Igarashi K, Watanabe-Matsui M. Wearing red for signaling: the heme-bach axis in heme metabolism, oxidative stress response and iron immunology. Tohoku J Exp Med 232: 229–253, 2014. doi: 10.1620/tjem.232.229. [DOI] [PubMed] [Google Scholar]
- 31.Juncos JP, Tracz MJ, Croatt AJ, Grande JP, Ackerman AW, Katusic ZS, Nath KA. Genetic deficiency of heme oxygenase-1 impairs functionality and form of an arteriovenous fistula in the mouse. Kidney Int 74: 47–51, 2008. doi: 10.1038/ki.2008.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jung RG, Simard T, Labinaz A, Ramirez FD, Di Santo P, Motazedian P, Rochman R, Gaudet C, Faraz MA, Beanlands RS, Hibbert B. Role of plasminogen activator inhibitor-1 in coronary pathophysiology. Thromb Res 164: 54–62, 2018. doi: 10.1016/j.thromres.2018.02.135. [DOI] [PubMed] [Google Scholar]
- 33.Kanakiriya SK, Croatt AJ, Haggard JJ, Ingelfinger JR, Tang SS, Alam J, Nath KA. Heme: a novel inducer of MCP-1 through HO-dependent and HO-independent mechanisms. Am J Physiol Renal Physiol 284: F546–F554, 2003. doi: 10.1152/ajprenal.00298.2002. [DOI] [PubMed] [Google Scholar]
- 34.Kang L, Grande JP, Farrugia G, Croatt AJ, Katusic ZS, Nath KA. Functioning of an arteriovenous fistula requires heme oxygenase-2. Am J Physiol Renal Physiol 305: F545–F552, 2013. doi: 10.1152/ajprenal.00234.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kang L, Grande JP, Hillestad ML, Croatt AJ, Barry MA, Katusic ZS, Nath KA. A new model of an arteriovenous fistula in chronic kidney disease in the mouse: beneficial effects of upregulated heme oxygenase-1. Am J Physiol Renal Physiol 310: F466–F476, 2016. doi: 10.1152/ajprenal.00288.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kang L, Hillestad ML, Grande JP, Croatt AJ, Barry MA, Farrugia G, Katusic ZS, Nath KA. Induction and functional significance of the heme oxygenase system in pathological shear stress in vivo. Am J Physiol Heart Circ Physiol 308: H1402–H1413, 2015. doi: 10.1152/ajpheart.00882.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ke Z, Huang Q. Haem-assisted dityrosine-cross-linking of fibrinogen under non-thermal plasma exposure: one important mechanism of facilitated blood coagulation. Sci Rep 6: 26982, 2016. doi: 10.1038/srep26982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Key NS. Bench to bedside: new developments in our understanding of the pathophysiology of thrombosis. J Thromb Thrombolysis 35: 342–345, 2013. doi: 10.1007/s11239-013-0898-8. [DOI] [PubMed] [Google Scholar]
- 39.Lindenblatt N, Bordel R, Schareck W, Menger MD, Vollmar B. Vascular heme oxygenase-1 induction suppresses microvascular thrombus formation in vivo. Arterioscler Thromb Vasc Biol 24: 601–606, 2004. doi: 10.1161/01.ATV.0000118279.74056.8a. [DOI] [PubMed] [Google Scholar]
- 40.Litvinov RI, Weisel JW. Role of red blood cells in haemostasis and thrombosis. ISBT Sci Ser 12: 176–183, 2017. doi: 10.1111/voxs.12331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mackman N. New insights into the mechanisms of venous thrombosis. J Clin Invest 122: 2331–2336, 2012. doi: 10.1172/JCI60229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Maruyama K, Morishita E, Yuno T, Sekiya A, Asakura H, Ohtake S, Yachie A. Carbon monoxide (CO)-releasing molecule-derived CO regulates tissue factor and plasminogen activator inhibitor type 1 in human endothelial cells. Thromb Res 130: e188–e193, 2012. doi: 10.1016/j.thromres.2012.07.002. [DOI] [PubMed] [Google Scholar]
- 43.Matika RW, Nielsen VG, Steinbrenner EB, Sussman AN, Madhrira M. Hemodialysis patients have plasmatic hypercoagulability and decreased fibrinolytic vulnerability: role of carbon monoxide. ASAIO J 60: 716–721, 2014. doi: 10.1097/MAT.0000000000000144. [DOI] [PubMed] [Google Scholar]
- 44.Matsumoto H, Ishikawa K, Itabe H, Maruyama Y. Carbon monoxide and bilirubin from heme oxygenase-1 suppresses reactive oxygen species generation and plasminogen activator inhibitor-1 induction. Mol Cell Biochem 291: 21–28, 2006. doi: 10.1007/s11010-006-9190-y. [DOI] [PubMed] [Google Scholar]
- 45.McFadyen JD, Schaff M, Peter K. Current and future antiplatelet therapies: emphasis on preserving haemostasis. Nat Rev Cardiol 15: 181–191, 2018. doi: 10.1038/nrcardio.2017.206. [DOI] [PubMed] [Google Scholar]
- 46.Merle NS, Grunenwald A, Rajaratnam H, Gnemmi V, Frimat M, Figueres ML, Knockaert S, Bouzekri S, Charue D, Noe R, Robe-Rybkine T, Le-Hoang M, Brinkman N, Gentinetta T, Edler M, Petrillo S, Tolosano E, Miescher S, Le Jeune S, Houillier P, Chauvet S, Rabant M, Dimitrov JD, Fremeaux-Bacchi V, Blanc-Brude OP, Roumenina LT. Intravascular hemolysis activates complement via cell-free heme and heme-loaded microvesicles. JCI Insight 3: e96910, 2018. doi: 10.1172/jci.insight.96910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mustafa S, Weltermann A, Fritsche R, Marsik C, Wagner O, Kyrle PA, Eichinger S. Genetic variation in heme oxygenase 1 (HMOX1) and the risk of recurrent venous thromboembolism. J Vasc Surg 47: 566–570, 2008. doi: 10.1016/j.jvs.2007.09.060. [DOI] [PubMed] [Google Scholar]
- 48.Myers D Jr, Farris D, Hawley A, Wrobleski S, Chapman A, Stoolman L, Knibbs R, Strieter R, Wakefield T. Selectins influence thrombosis in a mouse model of experimental deep venous thrombosis. J Surg Res 108: 212–221, 2002. doi: 10.1006/jsre.2002.6552. [DOI] [PubMed] [Google Scholar]
- 49.Nath KA. Heme oxygenase-1: a provenance for cytoprotective pathways in the kidney and other tissues. Kidney Int 70: 432–443, 2006. doi: 10.1038/sj.ki.5001565. [DOI] [PubMed] [Google Scholar]
- 50.Nath KA, Balla G, Vercellotti GM, Balla J, Jacob HS, Levitt MD, Rosenberg ME. Induction of heme oxygenase is a rapid, protective response in rhabdomyolysis in the rat. J Clin Invest 90: 267–270, 1992. doi: 10.1172/JCI115847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nath KA, d’Uscio LV, Juncos JP, Croatt AJ, Manriquez MC, Pittock ST, Katusic ZS. An analysis of the DOCA-salt model of hypertension in HO-1−/− mice and the Gunn rat. Am J Physiol Heart Circ Physiol 293: H333–H342, 2007. doi: 10.1152/ajpheart.00870.2006. [DOI] [PubMed] [Google Scholar]
- 52.Nath KA, Grande JP, Croatt AJ, Likely S, Hebbel RP, Enright H. Intracellular targets in heme protein-induced renal injury. Kidney Int 53: 100–111, 1998. doi: 10.1046/j.1523-1755.1998.00731.x. [DOI] [PubMed] [Google Scholar]
- 53.Nath KA, Grande JP, Haggard JJ, Croatt AJ, Katusic ZS, Solovey A, Hebbel RP. Oxidative stress and induction of heme oxygenase-1 in the kidney in sickle cell disease. Am J Pathol 158: 893–903, 2001. doi: 10.1016/S0002-9440(10)64037-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nath KA, Haggard JJ, Croatt AJ, Grande JP, Poss KD, Alam J. The indispensability of heme oxygenase-1 in protecting against acute heme protein-induced toxicity in vivo. Am J Pathol 156: 1527–1535, 2000. doi: 10.1016/S0002-9440(10)65024-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nath KA, Vercellotti GM, Grande JP, Miyoshi H, Paya CV, Manivel JC, Haggard JJ, Croatt AJ, Payne WD, Alam J. Heme protein-induced chronic renal inflammation: suppressive effect of induced heme oxygenase-1. Kidney Int 59: 106–117, 2001. doi: 10.1046/j.1523-1755.2001.00471.x. [DOI] [PubMed] [Google Scholar]
- 56.Nielsen VG, Galvani CA, Boyle PK, Steinbrenner EB, Matika RW. Bariatric patients have plasmatic hypercoagulability and systemic upregulation of heme oxygenase activity. Blood Coagul Fibrinolysis 26: 200–204, 2015. doi: 10.1097/MBC.0000000000000194. [DOI] [PubMed] [Google Scholar]
- 57.Nielsen VG, Garol BD, Zelman EA, Guerrero MA. Hemeoxygenase-1 mediated hypercoagulability in a patient with thyroid cancer. Blood Coagul Fibrinolysis 24: 663–665, 2013. doi: 10.1097/MBC.0b013e328363ab86. [DOI] [PubMed] [Google Scholar]
- 58.Nielsen VG, Garza JI. Comparison of the effects of CORM-2, CORM-3 and CORM-A1 on coagulation in human plasma. Blood Coagul Fibrinolysis 25: 801–805, 2014. doi: 10.1097/MBC.0000000000000146. [DOI] [PubMed] [Google Scholar]
- 59.Nielsen VG, Kirklin JK, George JF. Carbon monoxide releasing molecule-2 increases the velocity of thrombus growth and strength in human plasma. Blood Coagul Fibrinolysis 20: 377–380, 2009. doi: 10.1097/MBC.0b013e32832ca3a3. [DOI] [PubMed] [Google Scholar]
- 60.Nielsen VG, Ley ML, Waer AL, Alger PW, Matika RW, Steinbrenner EB. Plasmatic hypercoagulation in patients with breast cancer: role of heme oxygenase-1. Blood Coagul Fibrinolysis 24: 809–813, 2013. doi: 10.1097/MBC.0b013e3283658b00. [DOI] [PubMed] [Google Scholar]
- 61.Nielsen VG, Nfonsam VN, Matika RW, Ong ES, Jie T, Warneke JA, Steinbrenner EB. Colon and pancreas tumors enhance coagulation: role of hemeoxygenase-1. Blood Coagul Fibrinolysis 25: 435–438, 2014. doi: 10.1097/MBC.0000000000000075. [DOI] [PubMed] [Google Scholar]
- 62.Nielsen VG, Sobieski MA 2nd, Slaughter MS. Left ventricular assist device-associated carbon monoxide and iron-enhanced hypercoagulation: Impact of concurrent disease. ASAIO J 61: 417–423, 2015. doi: 10.1097/MAT.0000000000000210. [DOI] [PubMed] [Google Scholar]
- 63.Noubouossie DF, Reeves BN, Strahl BD, Key NS. Neutrophils: back in the thrombosis spotlight. Blood 133: 2186–2197, 2019. doi: 10.1182/blood-2018-10-862243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pittock ST, Norby SM, Grande JP, Croatt AJ, Bren GD, Badley AD, Caplice NM, Griffin MD, Nath KA. MCP-1 is up-regulated in unstressed and stressed HO-1 knockout mice: Pathophysiologic correlates. Kidney Int 68: 611–622, 2005. doi: 10.1111/j.1523-1755.2005.00439.x. [DOI] [PubMed] [Google Scholar]
- 65.Preston RJS, O’Sullivan JM, O’Donnell JS. Advances in understanding the molecular mechanisms of venous thrombosis. Br J Haematol 186: 13–23, 2019. doi: 10.1111/bjh.15869. [DOI] [PubMed] [Google Scholar]
- 66.Saha P, Humphries J, Modarai B, Mattock K, Waltham M, Evans CE, Ahmad A, Patel AS, Premaratne S, Lyons OT, Smith A. Leukocytes and the natural history of deep vein thrombosis: current concepts and future directions. Arterioscler Thromb Vasc Biol 31: 506–512, 2011. doi: 10.1161/ATVBAHA.110.213405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Serhal M, Barnes GD. Venous thromboembolism: A clinician update. Vasc Med 24: 122–131, 2019. doi: 10.1177/1358863X18821159. [DOI] [PubMed] [Google Scholar]
- 68.Soni H, Jain M, Mehta AA. Investigation into the mechanism(s) of antithrombotic effects of carbon monoxide releasing molecule-3 (CORM-3). Thromb Res 127: 551–559, 2011. doi: 10.1016/j.thromres.2011.02.009. [DOI] [PubMed] [Google Scholar]
- 69.Sparkenbaugh EM, Chantrathammachart P, Wang S, Jonas W, Kirchhofer D, Gailani D, Gruber A, Kasthuri R, Key NS, Mackman N, Pawlinski R. Excess of heme induces tissue factor-dependent activation of coagulation in mice. Haematologica 100: 308–314, 2015. doi: 10.3324/haematol.2014.114728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Stolla M, Henrichs K, Cholette JM, Pietropaoli AP, Phipps RP, Spinelli SL, Blumberg N. Haem is associated with thrombosis in neonates and infants undergoing cardiac surgery for congenital heart disease. Vox Sang 113: 72–75, 2018. doi: 10.1111/vox.12606. [DOI] [PubMed] [Google Scholar]
- 71.Tolosano E, Hirsch E, Patrucco E, Camaschella C, Navone R, Silengo L, Altruda F. Defective recovery and severe renal damage after acute hemolysis in hemopexin-deficient mice. Blood 94: 3906–3914, 1999. doi: 10.1182/blood.V94.11.3906. [DOI] [PubMed] [Google Scholar]
- 72.Tracz MJ, Alam J, Nath KA. Physiology and pathophysiology of heme: implications for kidney disease. J Am Soc Nephrol 18: 414–420, 2007. doi: 10.1681/ASN.2006080894. [DOI] [PubMed] [Google Scholar]
- 73.Tracz MJ, Juncos JP, Grande JP, Croatt AJ, Ackerman AW, Katusic ZS, Nath KA. Induction of heme oxygenase-1 is a beneficial response in a murine model of venous thrombosis. Am J Pathol 173: 1882–1890, 2008. doi: 10.2353/ajpath.2008.080556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.True AL, Olive M, Boehm M, San H, Westrick RJ, Raghavachari N, Xu X, Lynn EG, Sack MN, Munson PJ, Gladwin MT, Nabel EG. Heme oxygenase-1 deficiency accelerates formation of arterial thrombosis through oxidative damage to the endothelium, which is rescued by inhaled carbon monoxide. Circ Res 101: 893–901, 2007. doi: 10.1161/CIRCRESAHA.107.158998. [DOI] [PubMed] [Google Scholar]
- 75.Vitali SH, Mitsialis SA, Liang OD, Liu X, Fernandez-Gonzalez A, Christou H, Wu X, McGowan FX, Kourembanas S. Divergent cardiopulmonary actions of heme oxygenase enzymatic products in chronic hypoxia. PLoS One 4: e5978, 2009. doi: 10.1371/journal.pone.0005978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wang M, Hao H, Leeper NJ, Zhu L; Early Career Committee . Thrombotic Regulation From the Endothelial Cell Perspectives. Arterioscler Thromb Vasc Biol 38: e90–e95, 2018. doi: 10.1161/ATVBAHA.118.310367. [DOI] [PMC free article] [PubMed] [Google Scholar]