

Abstract
We have previously shown that activation of (pro)renin receptor (PRR) induces epithelial Na+ channel (ENaC) activity in cultured collecting duct cells. Here, we examined the role of soluble PRR (sPRR), the cleavage product of PRR in ENaC regulation, and further tested its relevance to aldosterone signaling. In cultured mpkCCD cells, administration of recombinant histidine-tagged sPRR (sPRR-His) at 10 nM within minutes induced a significant and transient increase in the amiloride-sensitive short-circuit current as assessed using the Ussing chamber technique. The acute ENaC activation was blocked by the NADPH oxidase 1/4 inhibitor GKT137892 and siRNA against Nox4 but not the β-catenin inhibitor ICG-001. In primary rat inner medullary collecting duct cells, administration of sPRR-His at 10 nM for 24 h induced protein expression of the α-subunit but not β- or γ-subunits of ENaC, in parallel with upregulation of mRNA expression as well as promoter activity of the α-subunit. The transcriptional activation of α-ENaC was dependent on β-catenin signaling. Consistent results obtained by epithelial volt ohmmeter measurement of equivalent current and Ussing chamber determination of short-circuit current showed that aldosterone-induced transepithelial Na+ transport was inhibited by the PRR decoy inhibitor PRO20 and PF-429242, an inhibitor of sPRR-generating enzyme site-1 protease, and the response was restored by the addition of sPRR-His. Medium sPRR was elevated by aldosterone and inhibited by PF-429242. Taken together, these results demonstrate that sPRR induces two phases of ENaC activation via distinct mechanisms and functions as a mediator of the natriferic action of aldosterone.
Keywords: collecting duct, epithelial Na+ channel activation, (pro)renin receptor, site-1 protease
The kidney plays a pivotal role in maintenance of extracellular fluid volume and blood pressure in mammals largely through fine tuning urinary Na+ excretion that matches Na+ intake (8). Aldosterone (Aldo) is the principal regulator of transepithelial Na+ transport, which is central to the maintenance of extracellular fluid volume and blood pressure in mammals (23, 30). As a steroid hormone, Aldo diffuses into the cell and binds the intracellular mineralocorticoid receptor, which then traffics to the nucleus to induce a complex transcriptional response (32). Despite intensive investigation, the detailed signaling mechanism responsible for Aldo-induced epithelial Na+ channel (ENaC) activation is still incompletely understood. A well-known mediator of Aldo action is serum- and glucocorticoid-regulated kinase 1 (Sgk1) (13). Sgk1 phosphorylates the ubiquitin-protein ligase neural precursor cell expressed developmentally downregulated protein (Nedd)4-2, a negative regulator of ENaC, thereby preventing the internalization and degradation of ENaC (1, 9, 12). However, the quite modest or inconsistent salt-wasting phenotype in whole body or renal-specific Sgk1 knockout mice was unmatched with the predicted role of Sgk1 in the regulation of renal function (5). In particular, these studies revealed no or modest changes in ENaC activity or expression after Sgk1 deletion (5). The suboptimal results from these studies may leave room for other mediators to be considered.
The (pro)renin receptor (PRR) is a 350-amino acid transmembrane receptor that binds both prorenin and renin with high affinity in the nanomolar range (22). Renin bound to PRR exhibits three- to fivefold increased renin activity (22). Furthermore, when PRR binds prorenin, the latter undergoes conformational changes to unfold the inhibitory prosegment of prorenin, leading to nonproteolytic activation (22). In vivo evidence is also available to shown that PRR regulates the activity of both local (24, 33) and circulating (29) renin activity. A large number of in vitro and in vivo studies have revealed that PPR functions as a potential regulator of ENaC activity. Interestingly, PRR seems to specially stimulate the expression of the α-subunt but not β- or γ-subunits of ENaC.
A soluble form of PRR (sPRR) is generated by protease-mediated cleavage (22). Recent evidence from two independent groups (4, 21) consistently demonstrates that site-1 protease (S1P) serves as a predominant source of sPRR following the controversial reports on the involvement of furin (3) and ADAM19 (43). In 2016, for the first time, we reported that recombinant sPRR exerted an important biological function in the upregulation of aquaporin-2 (AQP2) expression and enhancement of urine concentrating capability (18). Subsequently, we showed that sPRR promoted white-to-brown conversion in adipocytes of lithium-loaded mice (39). More recently, Gatineau et al. (7) reported that sPRR increased blood pressure in high fat-fed C57/BL6 mice. So far, to the best of our knowledge, there is no prior report on the direct role of sPRR in the regulation of transepithelial Na+ transport in general and in the collecting duct (CD) in particular.
METHODS
Animals.
Male Sprague-Dawley rats were from Charles River Laboratories (Wilmington, MA). All animals were cage housed and maintained in a temperature-controlled room with a 12:12-h light-dark cycle, with free access to tap water and standard rat chow for 14 days. Animal protocols were approved by the Animal Care and Use Committee of the University of Utah.
Materials.
Reagents used for cell culture were obtained from Life Technologies (Grand Island, NY). Histidine-tagged sPPR (sPRR-His) was a recombinant sPRR that included sPRR (residues 17–274) and an 8-histidine tag. Its production and purification have been described in a previous study (18). The antibody against the NH2-terminal domain in the sPRR sequence (termed “anti-PRR-N antibody”) was a gift from Dr. Yumei Feng (University of Nevada), and its neutralizing property was characterized by our previous studies (18, 35). The NADPH oxidase (Nox)1/4 inhibitor GKT137892 was a gift from Genkyotex. The FZD8 inhibitor OMP54F03 was provided by OncoMed Pharmaceuticals via Dr. John Lewicki. The following reagents were purchased from the commercial sources: ICG-001 (catalog no. HY14428, MedChemExpress, Monmouth Junction, NJ), PF429242 (Sigma-Aldrich, St. Louis, MO), Nox4 siRNA (catalog no. 184261, Invitrogen, Carlsbad, CA), FZD8 siRNA (catalog no. 157622, Invitrogen, Carlsbad, CA), PRR siRNA (catalog no. 81899, Invitrogen), S1P siRNA (catalog no. 74099, Invitrogen), and Aldo (Sigma-Aldrich).
Electrophysiological measurements of transepithelial transport.
Electrophysiology experiments were performed on immortalized mpkCCD cells after the cell monolayers reached confluence. Transepithelial Na+ transport was recorded using an epithelial volt ohmmeter (EVOM; World Precision Instruments) (17) or the Ussing chamber device (Physiologic Instruments, San Diego, CA) (31), as previously described. Transepithelial voltage (Vte) and resistance (Rte) across cell monolayers were measured with EVOM. Transepithelial current was calculated according to Ohm’s law, Ite = Vte/Rte, and normalized by the surface area of the insert. In performing Ussing chamber technique, voltage was clamped at zero using a VCC600 voltage-clamp apparatus (Physiologic Instruments), and short-circuit current (Isc) was then recorded using Ag-AgCl electrode in agar brides. Positive Isc reflects the active transport of cation (Na+) from the apical side to the basolateral side of media or transport of anion (Cl−) from the basolateral side to the apical side of media. For both measurements, the amiloride-sensitive component was taken as an index of ENaC activity. To examine the role of sPRR in the regulation of ENaC activity and its underlying mechanisms, cells were pretreated with an inhibitor (anti-PRR-N antibody, PRO20, GKT137892, ICG-001, and OMP54F03) or transfected with siRNA against Nox4 or FZD8 siRNA or their corresponding scrambled siRNA and then treated with sPRR-His in an acute (10 min) or chronic (24 h) setting. To test the role of PRR/sPRR in Aldo action, cells were pretreated with PRO20, PF-429242, or PF-429242 + sPRR-His and then treated with Aldo. Changes in transepithelial Na+ transport were monitored at various time points over 24 h using EVOM and further validated by Ussing chamber measurements of ENaC activity at a single time point of 24 h.
Native sPRR extraction.
Mouse urine samples were collected and centrifuged at 4,000 revolutions/min at 4°C for 10 min. The supernatant was carefully collected and applied to protein centrifugal filter devices (catalog nos. UFC903008 and UFC901008, Millipore, Burlington, MA) to harvest proteins between 10 and 30 kDa. sPRR-enriched protein samples were subjected to immunoprecipitation using a kit (catalog no. BSP002, Bio Basic, Markham, ON, Canada). Anti-PRR-N antibody was cross-linked with the magnetic beads (catalog no. 88805, Pierce, Appleton, WI) and then incubated for 30 min with urine protein samples. The beads were collected, washed, and eluted. The precipitated proteins termed as m-sPRR were stored in −80°C for subsequent experiments.
Primary cell culture experiments.
Primary rat inner medullary collecting duct (IMCD) cells were prepared from Sprague-Dawley rats as previously described (37) and grown to confluence in a six-well plate. Cells were pretreated for 1 h with an inhibitor (ICG-001 or OMP54F03) and treated for 24 h with sPRR-His or native sPRR. Cells were harvested with TRIzol for RNA extraction or RIPA buffer for protein extraction.
siRNA transfection.
Nox4 siRNA and FZD8 siRNA oligonucleotides were purchased from Invitrogen (catalog no. 4390771). At 50–70% confluence, mpkCCD cells were transfected with siRNA or nontargeting control siRNA using Lipofectamine RNAiMAX Transfection Reagent (catalog no. 13778-030, Invitrogen).
Measurement of reactive oxygen species.
Electron paramagnetic resonance (EPR) spectroscopy was performed with N-tert-butyl-α-phenylnitrone, a commonly used free radical spin trap, as previously described (17). After incubation with sPRR-His for 1 min at 37°C, N-tert-butyl-α-phenylnitrone was added to mpkCCD cells in the final concentration of 50 mM, and samples were extracted with toluene. After 10-min centrifugation at 4,000 revolutions/min, the toluene layer was collected as the measurement sample. Before the measurement, samples were deoxygenated by bubbling nitrogen gas for at least 15 min.
Measurement of H2O2.
H2O2 was measured using the ROS-Glo H2O2 Assay Kit (catalog no. G8820, Promega) according to the manufacturer’s instructions. The value was normalized by protein content.
Immunoblot analysis.
Renal tissues were lyzed and subsequently sonicated. Protein concentrations were determined using Coomassie reagent. Forty micrograms of protein for each sample were denatured in boiling water, separated by SDS-PAGE gels, and then transferred onto nitrocellulose membranes. Blots were blocked 1 h with 5% nonfat dry milk in Tris-buffered saline followed by incubation overnight with primary antibody. After a wash with Tris-buffered saline, blots were incubated with goat anti-rabbit/mouse/goat horseradish peroxidase-conjugated secondary antibody and visualized using ECL. The blots were quantitated using Imagepro-plus. The primary antibodies were as follows: rabbit anti-α-ENaC (catalog no. SPC403, Stressmarq), rabbit anti-β-ENaC (catalog no. SPC404, Stressmarq), and rabbit anti-γ-ENaC (catalog no. SPC405, Stressmarq).
Quantitative RT-PCR.
Total RNA isolation and reverse transcription were performed as previously described (34). Oligonucleotides were designed using Primer3 software (available at http://bioinfo.ut.ee/primer3-0.4.0/). Primers were as follows: for α-ENaC, 5′-GCGACAACAATCCCCAAG-3′ (sense) and 5′-TGAAGCGACAGGTGAAGATG-3′ (antisense); for β-ENaC, 5′-AAGCACCTGTAATGCCCAAG-3′ (sense) and 5′-ATAGCCCATCCCCACCAG-3′ (antisense); for γ-ENaC, 5′-CGAAGAAACTGGTGGGATTT-3′ (sense) and 5′-GATGGTGGAAAAGCGTGAAG-3′ (antisense); and for GAPDH, 5′-GTCTTCACTACCATGGAGAAGG-3′ (sense) and 5′-TCATGGATGACCTTGGCCAG-3′ (antisense).
Preparation of luciferase constructs.
Genomic DNA was extracted from rat tail using a tissue DNA kit (D3396-01, Promega). A 2,000-bp fragment of the 5′ flanking region (−2,000 ~ −1) of the α-ENaC gene (GenBank Accession No. NM_031548) was amplified from rat genomic DNA by PCR using the primers 5′-CGAGCTCTTACGCGTGCTAGC-3′ and 5′-GCTTACTTAGATCGCAGATCTTCTCCCTTTCTGTGCCAGGCT-3′ and subcloned to the pGL3-Basic reporter vector (Promega) using NheI and BgIII restriction sites. The construct was termed as “pGL3-α-ENaC” and validated by endonuclease analyzing and sequencing.
Luciferase assay.
mpkCCD cells were transfected with pGL3-α-ENaC plasmid or empty vector using HiPerFect Transfection Reagent (catalog no. 301702, Qiagen). Upon confluence, all cells were starved for 12 h, and transfected cells were then treated for 24 h with sPRR-His (10 nM). The vehicle-treated group served as a control. Luciferase activity was measured using a luciferase assay system (Promega), and luminescence was detected using an illuminometer (BMG FLUOstar OPTIMA).
Enzyme immunoassay.
Cell medium sPRR was determined by using the commercially available enzyme immunoassay kit (catalog no. JP27782, IBL, Toronto, ON, Canada) according to the manufacturer’s instructions.
Statistical analysis.
Data are expressed as means ± SE. All data points were included in the statistical analyses. Sample sizes were determined on the basis of similar previous studies or pilot experiments. Statistical analysis was performed using ANOVA with Bonferroni’s test for multiple comparisons or by paired or unpaired Student’s t tests for two comparisons. P values of <0.05 were considered statistically significant.
RESULTS
sPRR-His acutely induces ENaC activity via Nox4.
PRR is abundantly expressed in the CD, where its activation stimulates reabsorption of both Na+ and water via ENaC and AQP2, respectively, thereby promoting volume expression and hypertension. We have previously shown that activation of PRR by prorenin stimulated ENaC activity in cultured mpkCCD cells via Nox4-derived H2O2 (17). Further motivated by the recent observation that a recombinant histidine-tagged sPPR, termed sPRR-His, induced AQP2 expression in CD cells and elicited an antinatriuretic response (18), we suspected that sPRR might exert an effect on ENaC. Confluent monolayers of mpkCCD cells grown on Transwells were exposed to vehicle or 10 nM sPRR-His for 10 min. Ussing chamber technique was performed to monitor amiloride-sensitive Isc, an index of ENaC activity at 0, 2, 5, and 10 min. After the addition of 10 nM sPRR-His, ENaC activity was rapidly elevated, reaching the plateau within ~2 min and returning toward baseline within ~10 min. Addition of a PPR-neutralizing anti-PRR-N antibody almost completely abolished the action of sPRR-His, confirming its specificity (Fig. 1A). In light of the documented role of Nox4 in mediating PRR regulation of ENaC activity (17), we wondered if the same mediator may be responsible for the action of sPRR. Indeed, the Nox1/4 inhibitor GKT137892 completely blocked sPRR-His-induced ENaC activity (Fig. 1B). Subsequently, we validated this result using siRNA against Nox4. Immunoblot analysis revealed effective knockdown of Nox4 protein expression after transfection of Nox4 siRNA (Fig. 1C). siRNA-mediated Nox4 silencing significantly attenuated sPRR-induced ENaC activity compared with the scrambled siRNA control (Fig. 1D). Reactive oxygen species (ROS) were measured using EPR spectroscopy for total ROS and an ELISA kit for H202. Both indexes of ROS were elevated after sPRR-His. These results support an acute stimulatory effect of sPRR-His on ENaC activity via activation of Nox4.
Fig. 1.

Histidine-tagged soluble (pro)renin receptor (sPRR-His) acutely induced epithelial Na+ channel (ENaC) activity via NADPH oxidase (Nox) 4. A: mpkCCD cells were grown on Snapwells and treated with vehicle, 10 nM sPRR-His alone, or sPRR-His in combination with neutralizing antibody against the NH2-terminal domain in the sPRR sequence (termed “anti-PRR-N antibody”) for 30 min. Amiloride-sensitive short-circuit current (Isc) was determined at the indicated time points using the Ussing chamber technique. B: mpkCCD cells grown on Snapwells were pretreated with the Nox1/4 inhibitor GKT137892 (GKT) at 1 μM for 1 h and then treated with 10 nM sPRR-His. Amiloride-sensitive Isc was recorded over 10 min. C: cells were transfected with scrambled siRNA or Nox4 siRNA followed by immunoblot analysis of Nox4 protein abundance. D: amiloride-sensitive Isc was determined in cells transfected with scrambled siRNA or Nox4 siRNA. E and F: cells were treated for 1 min with 10 nM sPRR or vehicle. Total reactive oxygen species (ROS) electron paramagnetic resonance (EPR) signal and H2O2 were determined using EPR spectroscopy and an ELISA kit, respectively. The relative light unit (RLU) was normalized by protein abundance. Data are expressed as means ± SE; n = 4–5 per group for A, B, and D–F and n = 3 per group for C. *P < 0.05 vs. the control (CTR) group; #P < 0.05 vs. sPRR-His; $P < 0.05 vs. scrambled siRNA; &P < 0.05 vs. sPRR-His + scrambled siRNA.
sPRR-His chronically induces α-ENaC expression via β-catenin signaling.
Primary rat IMCD cells were used for gene expression experiments because of the ease of detecting ENaC protein expression and its sensitivity to stressors. In particular, ENaC antibodies worked in an optimal condition in rats versus mice. Accordingly, confluent primary cells were exposed to vehicle or 10 nM sPRR-His for 24 h followed by immunoblot analysis of various subunits of ENaC. The protein abundance of α-ENaC was elevated by sPRR-His, as shown by the representative immunoblot (Fig. 2A, top) and summary of densitometry data (Fig. 2A, bottom). In contrast, the protein abundance of β- or γ-ENaC was unaltered (Fig. 2A). Besides recombinant sPRR-His, we also used native sPRR isolated from mouse urine, termed m-sPRR. Like recombinant sPRR-His, m-sPRR exerted a similar stimulatory effect on α-ENaC protein expression (Fig. 2B). We have previously shown that sPRR-His stimulates AQP2 expression in cultured CD cells via FZD8-dependent β-catenin signaling (35). We wondered if the similar signaling pathway may be operative during stimulation of α-ENaC expression by sPRR-His. Indeed, inhibition of β-catenin signaling by ICG-001 (Fig. 3A) and FZD8 decoy inhibitor OMP54F03 (Fig. 3B) both effectively blocked sPRR-His-induced upregulation of α-ENaC protein expression. In agreement with this finding, quantitative RT-PCR showed that renal mRNA expression of α-ENaC was elevated by sPRR-His, which was blunted by ICG-001 (Fig. 3C) or OMP54F03 (Fig. 3D). To understand the transcriptional mechanism of this phenomenon, we generated an expression vector (pGL3-α-ENaC) that expressed luciferase under a ~2-kb 5′ flanking region of α-ENaC. Luciferase assay showed that pGL3-α-ENaC exhibited a twofold increase in baseline activity compared with the empty vector (Fig. 3E). In response to sPRR-His treatment, α-ENaC promoter exhibited a 2.5-fold increase in the activity, which was significantly blocked by either ICG-001 or OMP54F03 (Fig. 3E).
Fig. 2.
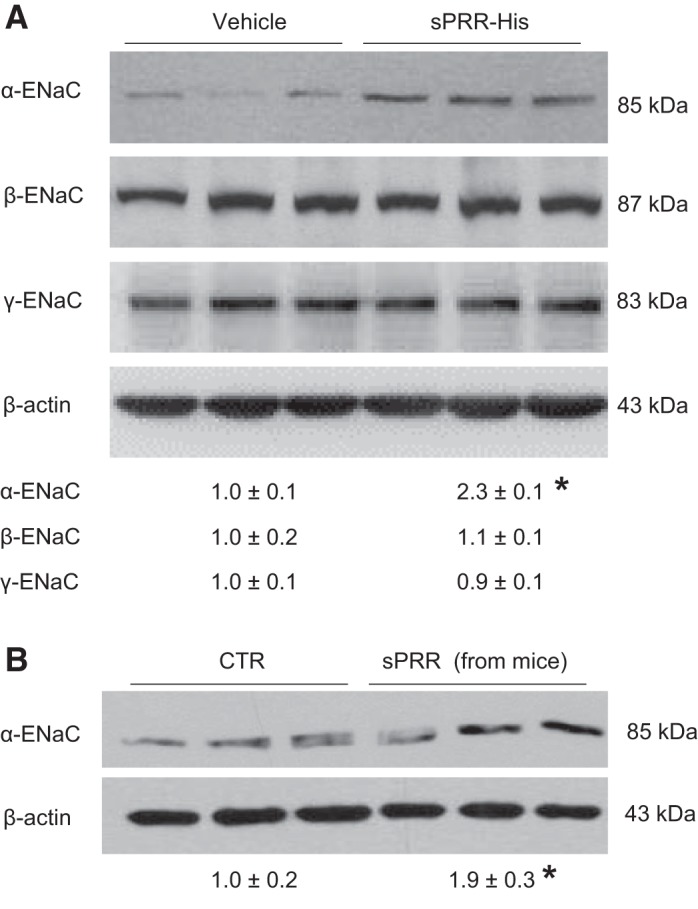
Recombinant and native soluble (pro)renin receptor (sPRR) stimulated protein expression of α-epithelial Na+ channel (ENaC) in primary rat inner medullary collecting duct (IMCD) cells. A: primary rat IMCD cells were exposed to histidine-tagged sPRR (sPRR-His) at 10 nM for 24 h. Protein abundances of the three subunits of ENaC were evaluated by immunoblot analysis. B: native sPRR was prepared from mouse urine samples using immunoprecipitation (termed as m-sPRR) and added to primary rat IMCD cells for 24 h. Protein abundance of α-ENaC protein was evaluated by immunoblot analysis. Data are means ± SE; n = 4–5 per group for A and B. *P < 0.05 vs. vehicle.
Fig. 3.

Analysis of the role of β-catenin pathway in the regulation of α-epithelial Na+ channel (ENaC) expression in response to histidine-tagged soluble (pro)renin receptor (sPRR-His) treatment. Primary rat inner medullary collecting duct cells were pretreated with the β-catenin inhibitor ICG-001 (ICG) at 10 μM or OMP54F03 (OMP) at 10 μM for 1 h and then treated with 10 nM sPRR-His for 24 h. α-ENaC protein and mRNA were determined by immunoblot analysis (A and B) and quantitative RT-PCR (C and D), respectively. A: effect of ICG on increased α-ENaC protein expression induced by sPRR-His. B: effect of OMP on increased α-ENaC protein expression induced by sPRR-His. C: effect of ICG on increased α-ENaC mRNA induced by sPRR-His. D: effect of OMP on increased α-ENaC mRNA induced by sPRR-His. mpkCCD cells were transfected with a construct expressing luciferase under the control of a flanking region of α-ENaC or empty plasmid (E). The transfected cells were pretreated with ICG at 10 μM or OMP at 10 μM for 1 h and then treated with vehicle or 10 nM sPRR-His for 24 h. Cells were lyzed and assayed for luciferase activity. Data are means ± SE; n = 4–5 per group for A–D and n = 9–10 per group for E. *P < 0.05 vs. control (CTR); #P < 0.05 vs. sPRR-His treatment.
The chronic regulation of α-ENaC expression as discussed above would predict the corresponding change in ENaC activity. Therefore, we measured Isc using Ussing chamber technique. As expected, 24-h exposure to sPRR-His treatment consistently upregulated ENaC activity, which was blocked by ICG-001 (Fig. 4A), OMP54F03 (Fig. 4B), or FZD8 siRNA (Fig. 4). In contrast, ICG-001 had no effect on the acute upregulation of ENaC activity by sPRR-His. These results support the concept that FZD8-dependent β-catenin signaling mediates the chronic but not acute upregulation of ENaC by sPRR-His.
Fig. 4.

β-Catenin signaling-dependent transcriptional mechanism mediated increased expression of α-epithelial Na+ channel (ENaC) by histidine-tagged soluble (pro)renin receptor (sPRR-His). Confluent mpkCCD cells grown on Snapwells were pretreated with ICG-001 (ICG) or OMP54F03 (OMP) or transfected with FZD8 siRNA and then treated with 10 nM sPRR-His for 24 h. Amiloride-sensitive short-circuit current was determined at the end of the experiment using the Ussing chamber technique. For acute experiments, cells were treated with vehicle or 10 nM sPRR-His alone or in combination of ICG. ENaC activity was determined at the indicated time points over 10 min. A: effect of ICG on sPRR-His-induced chronic elevation of ENaC activity. B: effect of OMP on sPRR-His-induced chronic elevation of ENaC activity. C: effect of silencing FZD8 on sPRR-His-induced chronic elevation of ENaC activity. D: effect of ICG on sPRR-His-induced acute elevation of ENaC activity. Data are means ± SE; n = 4–5 per group for A–E. *P < 0.05 vs. control (CTR).
S1P-derived sPRR mediates Aldo-induced ENaC activation.
Given the novel role of sPRR in the regulation of ENaC expression/activity, we were motivated to test the hypothesis that PRR/sPRR might serve as a potential mediator of Aldo signaling in mpkCCD cells. To this end, we used two different techniques to determine transepithelial Na+ transport. EVOM was initially used to monitor the time-dependent changes of transepithelial Na+ transport over 24 h. The result was subsequently validated by Ussing chamber measurement of Isc at the last time point.
In response to Aldo treatment, the equilibrium current (Ieq) increased to a plateau within 6 h and maintained at this level during the remaining time period (Fig. 5A). In sharp contrast, Aldo completely failed to elevate Ieq in the presence of PRO20 (Fig. 5A). PRO20 has been extensively characterized as an effective and specific inhibitor of PRR (14, 15, 18). The recent identification of S1P as the predominant PRR cleavage enzyme (4, 21) offers a unique opportunity to examine the contribution of endogenous sPRR to Aldo-induced ENaC activation. Inhibition of S1P by a specific S1P inhibitor, PF-429242, almost completely prevented the increase in the Ieq response to Aldo (Fig. 5B). To validate the involvement of sPRR, a rescue experiment was conducted by addition of sPRR-His to the PF-429242-treated group. Aldo-induced Ieq was largely restored in the PF-429242 + sPRR-His-treated group (Fig. 5B). In a separate experiment, mpkCCD cells were treated with the same protocols, but amiloride-sensitive Isc was determined using Ussing chamber technique. In agreement with the Ieq data, Aldo-induced amiloride-sensitive Isc was blunted by PF-429242 and restored by the addition of sPRR-His (Fig. 5C). By ELISA, medium sPRR concentration was elevated by Aldo, which was blocked by PF-429242 (Fig. 5D). Aldo-induced amiloride-sensitive Isc was also blunted by transfecting mpkCCD cells with siRNA against PRR (Fig. 5E) or S1P (Fig. 5F). These results validated the effect of PRO20 and PF-429242 on Aldo-induced amiloride-sensitive Isc.
Fig. 5.

Site-1 protease (S1P)-derived soluble (pro)renin receptor (sPRR) mediated aldosterone (Aldo)-induced epithelial Na+ channel (ENaC) activation. Confluent mpkCCD cells grown on Transwells or Snapwells were pretreated with 10 μM PF-429242 (PF) or 1.5 μM PRO20 for 1 h and then treated with 1 μM Aldo and 10 nM histidine-tagged sPRR (sPRR-His). Transepithelial Na+ transport was recorded using an epithelial volt ohmmeter (A and B) and the Ussing chamber technique (C). Medium sPRR was measured using ELISA. A: time course of equilibrium current (Ieq) changes in control (CTR), Aldo, and Aldo + PRO20 groups over 24 h. *P < 0.05 vs. CTR; #P < 0.05 vs. Aldo. B: time courses of Ieq changes in Aldo, Aldo + PF, Aldo + PF + sPRR-His, and PF groups. *P < 0.05 vs. the Aldo group; #P < 0.05 vs. the Aldo + PF + sPRR-His group; &P < 0.05 vs. PF. C: amiloride-sensitive current in CTR, Aldo, Aldo + PF, and Aldo + PF + sPRR-His groups at 12 h. D: medium sPRR content in CTR, Aldo, and Aldo + PF groups. E: effect of Aldo on ENaC activity in the presence or absence of PRR siRNA. F: effect of Aldo on ENaC activity in the presence or absence of S1P siRNA. Data are means ± SE; n = 4–5 per group for A–F.
DISCUSSION
Abundant evidence from multiple groups has established PRR as an important player in renal control of Na+ and water homeostasis and blood pressure as well as other physiological processes (27, 40–42, 44). After elucidation of the role of sPRR in the regulation of AQP2 expression and urine concentrating capability (18), we have begun to understand the biology of sPRR under various conditions. For example, sPRR infusion promotes conversion of white-to-brown adipocytes in lithium-treated mice (39). More recently, sPRR infusion elevates blood pressure in an obese mouse background (7). The present study represents the first report, to our knowledge, that sPRR functions as a direct regulator of ENaC in CD cells.
A significant number of previous reports from our group and others have documented PRR as an important regulator of ENaC (17, 24–26). In this regard, activation of PRR by prorenin, but not renin, induced ENaC activity in cultured mpkCCD cells (17). CD-specific deletion of PRR in mice attenuates ANG II-induced renal medullary expression of α-ENaC but not β- or γ-ENaC (24). In agreement with this finding, in vivo administration of PRR shRNA in rats reduced expressions of PRR throughout the kidney but only suppressed renal medullary but not cortical α-ENaC expression (25). However, to our knowledge, there is no prior study to show that the ENaC regulatory role of PRR can be ascribed to the generation of its soluble product sPRR. The present study extensively characterized the ENaC regulatory role of sPRR in cultured CD cells. In these cells, sPRR-His induced a rapid and transient upregulation of ENaC activity followed by a chronic stimulation of expression of the α-subunit but not β- or γ-subunits of ENaC. Indeed, among the three subunits, the synthesis of α-ENaC is a limiting factor in the assembly of the ENaC complex (20). Furthermore, salt restriction or Aldo infusion in rats selectively induces renal protein expression of α-ENaC (19).
On the basis of our results, it seems reasonable to speculate that much of PRR’s functions may be mediated by the generation of sPRR. In support of this notion, systemic pharmacological inhibition or renal-specific deletion of S1P, a recently identified sPRR-generating enzyme (4, 21), induced polyuria accompanied with downregulation of renal AQP2 expression (37), which largely recapitulates the phenotype in mice with conditional PRR deletion in the CD (35) or the whole nephron (28).
β-Catenin signaling plays a pivotal role in regulation of embryogenesis as well as tissue homeostasis (2, 11). Our previous study showed that sPRR directly interacted with FZD8 to activate β-catenin signaling, resulting in the upregulation of AQP2 expression in cultured CD cells (37). In the present study, the same β-catenin signaling mediated upregulation of α-ENaC expression by sPRR-His. Therefore, sPRR via β-catenin signaling targets both AQP2 and α-ENaC so that it can effectively coordinate the regulation of Na+ and water reabsorption in the CD. It is interesting to note that β-catenin signaling is shown to regulate various components of the intrarenal renin-angiotensin system, including PRR during renal injury (10, 16). It is likely that sPRR provides a potential link between PRR and β-catenin signaling in the kidney.
Besides the chronic regulation of α-ENaC expression, sPRR induces a rapid activation of ENaC activity, which occurs within minutes. Interestingly, the acute ENaC regulation by sPRR is dependent on Nox4 but not β-catenin signaling. We have previously shown that activation of PRR by prorenin induces a rapid increase of ENaC activity via Nox4-derived H2O2 in cultured mpkCCD cells (17). Given the similar timeframe of ENaC activation by sPRR and prorenin (within minutes), it seems reasonable to speculate that Nox4-derived H2O2 may similarly mediate the acute action of sPRR. Indeed, inhibition of Nox4 via siRNA or a Nox1/4 inhibitor effectively blocked sPRR-His-induced acute ENaC activation. The mechanism of how sPRR activates Nox4 is unknown and will warrant further investigation. In addition, it seems reasonable to speculate that the action of sPRR may be dependent on its physiological ligand prorenin. To verify its dependence on prorenin, the action of sPRR should be tested in renin-deficient cells.
We examined a potential role of sPRR as a mediator of Aldo-induced ENaC activity. By the use of EVOM, transepithelial Na+ transport induced by Aldo was almost completely blocked by PRO20 and PF-429242. The addition of sPRR fully reversed the inhibitory effect of PF-429242. In addition, the release of sPRR was increased in response to Aldo treatment. These results suggested that S1P-derived sPRR may function as a nonredundant mediator of Aldo-induced ENaC activation. It still remains elusive as to how Aldo induces sPRR production. Could this be the consequence of increased protease-mediated cleavage or the increased level of the substrate PRR? For the former case, could Aldo enhance the activity or expression of S1P? An obstacle exists in addressing this question at the present time because of the lack of a validated assay for S1P activity or a specific antibody for protein expression analysis.
The present study has a number of limitations. In particular, there is no in vivo evidence for sPRR mediation of Na+-retaining action of Aldo. Future studies are needed to test the effect of S1P inhibition on the Na+-retaining and prohypertensive action of Aldo. S1P can be inhibited using PF-429242 or making S1P knockout mice in a conventional or conditional manner. It is also interesting to test the possible interaction between sPRR and Sgk1 during Aldo signaling in the CD cells.
In summary, the present study, for the first time, reports that sPRR is capable of acutely activating ENaC activity and chronically stimulating α-ENaC expression in the cultured CD cells. There appear to be distinct mechanisms of this phenomenon with Nox4-derived ROS mediating the acute stimulation of ENaC activity but a β-catenin-dependent transcriptional mechanism accounting for the chronic upregulation of α-ENaC expression induced by sPRR-His. Furthermore, S1P-derived sPRR plays a key role in mediating Aldo-induced ENaC activation. It is likely that defining an sPRR-dependent pathway will open up a new area for a better understanding of the molecular basis of renal handling of Na+ balance and blood pressure, particularly the Na+-retaining action of Aldo in the distal nephron.
GRANTS
This work was supported by National Institutes of Health Grants HL-139689, DK-104072, and HL-135851 and a VA Merit Review from the Department of Veterans Affairs. T. Yang is a research career scientist in the Department of Veterans Affairs.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
T.Y., F.W, and R.L. conceived and designed research; F.W., R.L., K.P., X. Liu, C.X., X. Lu, S.S., and T.Y. performed experiments; F.W., R.L., K.P., X. Liu, and T.Y. analyzed data; F.W., R.L., and T.Y. interpreted results of experiments; F.W., R.L., and T.Y. prepared figures; F.W., R.L., and T.Y. drafted manuscript; F.W., R.L., and T.Y. edited and revised manuscript; T.Y. approved final version of manuscript.
REFERENCES
- 1.Abriel H, Loffing J, Rebhun JF, Pratt JH, Schild L, Horisberger JD, Rotin D, Staub O. Defective regulation of the epithelial Na+ channel by Nedd4 in Liddle’s syndrome. J Clin Invest 103: 667–673, 1999. doi: 10.1172/JCI5713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Clevers H, Nusse R. Wnt/β-catenin signaling and disease. Cell 149: 1192–1205, 2012. doi: 10.1016/j.cell.2012.05.012. [DOI] [PubMed] [Google Scholar]
- 3.Cousin C, Bracquart D, Contrepas A, Corvol P, Muller L, Nguyen G. Soluble form of the (pro)renin receptor generated by intracellular cleavage by furin is secreted in plasma. Hypertension 53: 1077–1082, 2009. doi: 10.1161/HYPERTENSIONAHA.108.127258. [DOI] [PubMed] [Google Scholar]
- 4.Fang H, Xu C, Lu A, Zou CJ, Xie S, Chen Y, Zhou L, Liu M, Wang L, Wang W, Yang T. (Pro)renin receptor mediates albumin-induced cellular responses: role of site-1 protease-derived soluble (pro)renin receptor in renal epithelial cells. Am J Physiol Cell Physiol 313: C632–C643, 2017. doi: 10.1152/ajpcell.00006.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Faresse N, Lagnaz D, Debonneville A, Ismailji A, Maillard M, Fejes-Toth G, Náray-Fejes-Tóth A, Staub O. Inducible kidney-specific Sgk1 knockout mice show a salt-losing phenotype. Am J Physiol Renal Physiol 302: F977–F985, 2012. doi: 10.1152/ajprenal.00535.2011. [DOI] [PubMed] [Google Scholar]
- 7.Gatineau E, Gong MC, Yiannikouris F. Soluble prorenin receptor increases blood pressure in high fat-fed male mice. Hypertension 74: 1014–1020, 2019. doi: 10.1161/HYPERTENSIONAHA.119.12906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hall JE. Control of sodium excretion by angiotensin II: intrarenal mechanisms and blood pressure regulation. Am J Physiol 250: R960–R972, 1986. doi: 10.1152/ajpregu.1986.250.6.R960. [DOI] [PubMed] [Google Scholar]
- 9.Harvey KF, Dinudom A, Cook DI, Kumar S. The Nedd4-like protein KIAA0439 is a potential regulator of the epithelial sodium channel. J Biol Chem 276: 8597–8601, 2001. doi: 10.1074/jbc.C000906200. [DOI] [PubMed] [Google Scholar]
- 10.He W, Dai C, Li Y, Zeng G, Monga SP, Liu Y. Wnt/beta-catenin signaling promotes renal interstitial fibrosis. J Am Soc Nephrol 20: 765–776, 2009. doi: 10.1681/ASN.2008060566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang SM, Mishina YM, Liu S, Cheung A, Stegmeier F, Michaud GA, Charlat O, Wiellette E, Zhang Y, Wiessner S, Hild M, Shi X, Wilson CJ, Mickanin C, Myer V, Fazal A, Tomlinson R, Serluca F, Shao W, Cheng H, Shultz M, Rau C, Schirle M, Schlegl J, Ghidelli S, Fawell S, Lu C, Curtis D, Kirschner MW, Lengauer C, Finan PM, Tallarico JA, Bouwmeester T, Porter JA, Bauer A, Cong F. Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature 461: 614–620, 2009. doi: 10.1038/nature08356. [DOI] [PubMed] [Google Scholar]
- 12.Kamynina E, Debonneville C, Bens M, Vandewalle A, Staub O. A novel mouse Nedd4 protein suppresses the activity of the epithelial Na+ channel. FASEB J 15: 204–214, 2001. doi: 10.1096/fj.00-0191com. [DOI] [PubMed] [Google Scholar]
- 13.Lang F, Böhmer C, Palmada M, Seebohm G, Strutz-Seebohm N, Vallon V. (Patho)physiological significance of the serum- and glucocorticoid-inducible kinase isoforms. Physiol Rev 86: 1151–1178, 2006. doi: 10.1152/physrev.00050.2005. [DOI] [PubMed] [Google Scholar]
- 14.Li W, Peng H, Cao T, Sato R, McDaniels SJ, Kobori H, Navar LG, Feng Y. Brain-targeted (pro)renin receptor knockdown attenuates angiotensin II-dependent hypertension. Hypertension 59: 1188–1194, 2012. doi: 10.1161/HYPERTENSIONAHA.111.190108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li W, Sullivan MN, Zhang S, Worker CJ, Xiong Z, Speth RC, Feng Y. Intracerebroventricular infusion of the (pro)renin receptor antagonist PRO20 attenuates deoxycorticosterone acetate-salt-induced hypertension. Hypertension 65: 352–361, 2015. doi: 10.1161/HYPERTENSIONAHA.114.04458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li Z, Zhou L, Wang Y, Miao J, Hong X, Hou FF, Liu Y. (Pro)renin receptor is an amplifier of Wnt/β-catenin signaling in kidney injury and fibrosis. J Am Soc Nephrol 28: 2393–2408, 2017. doi: 10.1681/ASN.2016070811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lu X, Wang F, Liu M, Yang KT, Nau A, Kohan DE, Reese V, Richardson RS, Yang T. Activation of ENaC in collecting duct cells by prorenin and its receptor PRR: involvement of Nox4-derived hydrogen peroxide. Am J Physiol Renal Physiol 310: F1243–F1250, 2016. doi: 10.1152/ajprenal.00492.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lu X, Wang F, Xu C, Soodvilai S, Peng K, Su J, Zhao L, Yang KT, Feng Y, Zhou SF, Gustafsson JA, Yang T. Soluble (pro)renin receptor via β-catenin enhances urine concentration capability as a target of liver X receptor. Proc Natl Acad Sci USA 113: E1898–E1906, 2016. doi: 10.1073/pnas.1602397113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Masilamani S, Kim GH, Mitchell C, Wade JB, Knepper MA. Aldosterone-mediated regulation of ENaC alpha, beta, and gamma subunit proteins in rat kidney. J Clin Invest 104: R19–R23, 1999. doi: 10.1172/JCI7840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.May A, Puoti A, Gaeggeler HP, Horisberger JD, Rossier BC. Early effect of aldosterone on the rate of synthesis of the epithelial sodium channel alpha subunit in A6 renal cells. J Am Soc Nephrol 8: 1813–1822, 1997. [DOI] [PubMed] [Google Scholar]
- 21.Nakagawa T, Suzuki-Nakagawa C, Watanabe A, Asami E, Matsumoto M, Nakano M, Ebihara A, Uddin MN, Suzuki F. Site-1 protease is required for the generation of soluble (pro)renin receptor. J Biochem 161: 369–379, 2017. doi: 10.1093/jb/mvw080. [DOI] [PubMed] [Google Scholar]
- 22.Nguyen G, Delarue F, Burcklé C, Bouzhir L, Giller T, Sraer JD. Pivotal role of the renin/prorenin receptor in angiotensin II production and cellular responses to renin. J Clin Invest 109: 1417–1427, 2002. doi: 10.1172/JCI0214276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nishimoto M, Fujita T. Renal mechanisms of salt-sensitive hypertension: contribution of two steroid receptor-associated pathways. Am J Physiol Renal Physiol 308: F377–F387, 2015. doi: 10.1152/ajprenal.00477.2013. [DOI] [PubMed] [Google Scholar]
- 24.Peng K, Lu X, Wang F, Nau A, Chen R, Zhou SF, Yang T. Collecting duct (pro)renin receptor targets ENaC to mediate angiotensin II-induced hypertension. Am J Physiol Renal Physiol 312: F245–F253, 2017. doi: 10.1152/ajprenal.00178.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Quadri S, Siragy HM. (Pro)renin receptor contributes to regulation of renal epithelial sodium channel. J Hypertens 34: 486–494, 2016. doi: 10.1097/HJH.0000000000000825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Quadri SS, Culver S, Ramkumar N, Kohan DE, Siragy HM. (Pro)renin receptor mediates obesity-induced antinatriuresis and elevated blood pressure via upregulation of the renal epithelial sodium channel. PLoS One 13: e0202419, 2018. doi: 10.1371/journal.pone.0202419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ramkumar N, Kohan DE. The (pro)renin receptor: an emerging player in hypertension and metabolic syndrome. Kidney Int 95: 1041–1052, 2019. doi: 10.1016/j.kint.2018.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ramkumar N, Stuart D, Calquin M, Quadri S, Wang S, Van Hoek AN, Siragy HM, Ichihara A, Kohan DE. Nephron-specific deletion of the prorenin receptor causes a urine concentration defect. Am J Physiol Renal Physiol 309: F48–F56, 2015. doi: 10.1152/ajprenal.00126.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Riquier-Brison ADM, Sipos A, Prókai Á, Vargas SL, Toma L, Meer EJ, Villanueva KG, Chen JCM, Gyarmati G, Yih C, Tang E, Nadim B, Pendekanti S, Garrelds IM, Nguyen G, Danser AHJ, Peti-Peterdi J. The macula densa prorenin receptor is essential in renin release and blood pressure control. Am J Physiol Renal Physiol 315: F521–F534, 2018. doi: 10.1152/ajprenal.00029.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rossier BC, Baker ME, Studer RA. Epithelial sodium transport and its control by aldosterone: the story of our internal environment revisited. Physiol Rev 95: 297–340, 2015. doi: 10.1152/physrev.00011.2014. [DOI] [PubMed] [Google Scholar]
- 31.Soodvilai S, Jia Z, Yang T. Hydrogen peroxide stimulates chloride secretion in primary inner medullary collecting duct cells via mPGES-1-derived PGE2. Am J Physiol Renal Physiol 293: F1571–F1576, 2007. doi: 10.1152/ajprenal.00132.2007. [DOI] [PubMed] [Google Scholar]
- 32.Verrey F, Fakitsas P, Adam G, Staub O. Early transcriptional control of ENaC (de)ubiquitylation by aldosterone. Kidney Int 73: 691–696, 2008. doi: 10.1038/sj.ki.5002737. [DOI] [PubMed] [Google Scholar]
- 33.Wang F, Lu X, Liu M, Feng Y, Zhou SF, Yang T. Renal medullary (pro)renin receptor contributes to angiotensin II-induced hypertension in rats via activation of the local renin-angiotensin system. BMC Med 13: 278, 2015. doi: 10.1186/s12916-015-0514-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang F, Lu X, Peng K, Du Y, Zhou SF, Zhang A, Yang T. Prostaglandin E-prostanoid4 receptor mediates angiotensin II-induced (pro)renin receptor expression in the rat renal medulla. Hypertension 64: 369–377, 2014. doi: 10.1161/HYPERTENSIONAHA.114.03654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang F, Lu X, Peng K, Fang H, Zhou L, Su J, Nau A, Yang KT, Ichihara A, Lu A, Zhou SF, Yang T. Antidiuretic action of collecting duct (pro)renin receptor downstream of vasopressin and PGE2 receptor EP4. J Am Soc Nephrol 27: 3022–3034, 2016. doi: 10.1681/ASN.2015050592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang F, Xu C, Luo R, Peng K, Ramkumar N, Xie S, Lu X, Zhao L, Zuo CJ, Kohan DE, Yang T. Site-1 protease-derived soluble (pro)renin receptor targets vasopressin receptor 2 to enhance urine concentrating capability. JCI Insight 4: e124174, 2019. doi: 10.1172/jci.insight.124174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang KT, Wang F, Lu X, Peng K, Yang T, David Symons J. The soluble (pro)renin receptor does not influence lithium-induced diabetes insipidus but does provoke beiging of white adipose tissue in mice. Physiol Rep 5: e13410, 2017. doi: 10.14814/phy2.13410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang T. Crosstalk between (pro)renin receptor and COX-2 in the renal medulla during angiotensin II-induced hypertension. Curr Opin Pharmacol 21: 89–94, 2015. doi: 10.1016/j.coph.2014.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang T. Unraveling the physiology of (pro)renin receptor in the distal nephron. Hypertension 69: 564–574, 2017. doi: 10.1161/HYPERTENSIONAHA.116.08318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang T, Xu C. Physiology and pathophysiology of the intrarenal renin-angiotensin system: an update. J Am Soc Nephrol 28: 1040–1049, 2017. doi: 10.1681/ASN.2016070734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yoshikawa A, Aizaki Y, Kusano K, Kishi F, Susumu T, Iida S, Ishiura S, Nishimura S, Shichiri M, Senbonmatsu T. The (pro)renin receptor is cleaved by ADAM19 in the Golgi leading to its secretion into extracellular space. Hypertens Res 34: 599–605, 2011. doi: 10.1038/hr.2010.284. [DOI] [PubMed] [Google Scholar]
- 44.Zhu Q, Yang T. Enzymatic sources and physio-pathological functions of soluble (pro)renin receptor. Curr Opin Nephrol Hypertens 27: 77–82, 2018. doi: 10.1097/MNH.0000000000000396. [DOI] [PMC free article] [PubMed] [Google Scholar]