

Abstract
Excitation-contraction (EC) coupling is the coordinated process by which an action potential triggers cardiac myocyte contraction. EC coupling is initiated in dyads where the junctional sarcoplasmic reticulum (jSR) is in tight proximity to the sarcolemma of cardiac myocytes. Existing models of EC coupling critically depend on dyad stability to ensure the fidelity and strength of EC coupling, where even small variations in ryanodine receptor channel and voltage-gated calcium channel-α 1.2 subunit separation dramatically alter EC coupling. However, dyadic motility has never been studied. Here, we developed a novel strategy to track specific jSR units in dissociated adult ventricular myocytes using photoactivatable fluorescent proteins. We found that the jSR is not static. Instead, we observed dynamic formation and dissolution of multiple dyadic junctions regulated by the microtubule-associated molecular motors kinesin-1 and dynein. Our data support a model where reproducibility of EC coupling results from the activation of a temporally averaged number of SR Ca2+ release units forming and dissolving SR-sarcolemmal junctions. These findings challenge the long-held view that the jSR is an immobile structure and provide insights into the mechanisms underlying its motility.
Keywords: cardiomyocytes, dyadic plasticity, EC coupling, junctional sarcoplasmic reticulum, live imaging
INTRODUCTION
Cardiac contractions are triggered by action potentials (AP). The chain of events that couple an AP to contraction is collectively known as excitation-contraction (EC) coupling. Multiple factors determine the strength of contraction, including Ca2+ influx, sarcoplasmic reticulum (SR) Ca2+ release, and the sensitivity of contractile proteins to cytosolic Ca2+.
The coefficient of variation of the AP-evoked intracellular Ca2+ concentration ([Ca2+]i) transient in an adult ventricular myocyte is ~0.1 (21), suggesting high EC coupling reproducibility. To understand mechanisms underlying [Ca2+]i transient reproducibility, it is pertinent to review key events during EC coupling. Activation of sarcolemmal voltage-gated calcium channel-α 1.2 subunit (Cav1.2 channel) during the AP causes a small influx of Ca2+ that activates ryanodine receptors (RyRs) in the nearby junctional SR (jSR). The small distance and volume separating Cav1.2 and RyR channels allow even brief openings of Cav1.2 channels to increase local [Ca2+]i high enough to activate nearby RyRs. Thus the fidelity of local EC coupling critically depends on dyad structural stability and hence the separation between RyR and Cav1.2 channels.
The jSR is a complex structure with several proteins contributing to its architecture and function. Junctophilin-2 (JPH2) is anchored to the jSR in its COOH terminus and contacts the sarcolemma through lipid-interacting motifs in its NH2 terminus (7, 15). JPH2 is hypothesized to provide a molecular bridge between the jSR and the transverse tubules (T-tubules; Refs. 1, 19). The Ca2+-binding protein calsequestrin is anchored to the jSR membrane by triadin (TRD) and junctin (23). TRD and junctin knockout mice show changes in jSR architecture (8, 23). This is important as even small changes in dyadic size and structure can perturb EC coupling by altering peak [Ca2+] levels and Ca2+ decay rates in the dyad (3, 9, 17).
There is growing evidence that the dyad is not a stable structure; T-tubule-SR junctions show dynamic regulation, particularly during disease states (21). Multiple studies show that changes in T-tubule sarcolemma-SR junctions likely contribute to Ca2+ instability during pathology. In heart failure, physical uncoupling of the SR and T-tubules (23), T-tubule disorganization (11), and remodeled T-tubules moving away from Z-lines (18) underlie loss of local control and Ca2+ instability. Similarly, EC uncoupling and SR network fracturing occur during postmyocardial infarction (22).
However, compared with T-tubule structural studies, jSR plasticity has received little attention. This can be attributed to the ease at which the T-tubule membrane can be visualized and the ineffectiveness of fluorescent labeling of the jSR (10, 21). In this study, we developed a novel strategy to monitor the stability of the T-tubule sarcolemma-SR junction in freshly isolated adult ventricular myocytes. Our data indicate that the jSR is a dynamic structure. This supports a novel paradigm shift that departs from the long-standing model of Ca2+ signaling modulated by a static dyad. The dynamic jSR creates another layer of Ca2+ signaling regulation; moreover, this regulation could exhibit exquisite local precision.
METHODS
Isolation of mouse ventricular myocytes.
Male mice (C57BL/6J, 6–8 wk old) were euthanized with a lethal dose of sodium pentobarbital administered intraperitoneally as approved by the institutional animal care and use committee. Ventricular myocytes were isolated using a Langendorff perfusion apparatus as previously described (5). The isolated ventricular myocytes were kept either at room temperature (22°C) or at 37°C in Tyrode solution 0.5–5 h after isolation.
Viral expression system.
Mice were anesthetized by isoflurane and injected retro-orbitally with adeno-associated viral vector 9 (AAV9) encoding the following: TRD tagged with photoactivatable green fluorescent protein (AAV9-TRD-paGFP); a SR/endoplasmic reticulum (ER) retention signal tagged with red fluorescent protein (AAV9-SR-RFP); an shRNA disruption expression of dynein tagged with RFP (AAV9-Dnchc1-shRNA-RFP); and a dominant-negative version of the kinesin-1 tagged with RFP (AAV9-Kif5b-DN-RFP). To allow robust expression of fluorescent proteins and shRNA knockdown, experiments were performed ≥3 wk after virus injection.
jSR, T-tubule, and [Ca2+]i imaging.
To label jSR, paGFP was activated by brief illumination with 405-nm light. To label T-tubules, 1-(3-sulfonatopropyl)-8-vinylpyridium betaine (di-8-ANEPPS) was loaded into myocytes as previously described (6). To visualize and acquire paGFP and di-8-ANEPPS fluorescent signals, we used a laser-scanning Olympus FV1000 confocal microscope equipped with an Olympus ×60 oil-immersion lens. Images were acquired at 1-Hz frequency with the zero-drift compensation module engaged, continuously correcting for drifts and thus maintaining a stable focal plane throughout the duration of the experiment. For recordings of TRD-paGFP, each cell was scanned axially and recorded at a plane where the predominant number of TRDs could be seen and tracked. Myocytes were corrected for photobleaching using the Histogram Matching feature of Photocorrection in ImageJ. Cells were analyzed blind to experimental condition via name randomization. Whole cell [Ca2+]i transients were measured in myocytes loaded with fluo 4-AM.
Statistics.
Data are presented as coefficients of variation (COVs) or means ± SE. Two-sample comparisons of normally distributed data were made using a Student’s t test. A Kruskal–Wallis test was used to compare nonparametric data sets. “N” represents number of mice used, and “C” and “S” represent the number of cells and sites analyzed, respectively.
RESULTS
Expression of TRD-paGFP allows the visualization of the jSR in ventricular myocytes.
We designed two AAV9s for specific infection of adult ventricular myocytes: one to express TRD tagged with a photoactivatable GFP (AAV9-TRD-paGFP) and the other a constitutively fluorescent RFP tag with a retention signal for the SR lumen (SR-RFP; Fig. 1A). TRD-paGFP fluorescence is restricted to the set of proteins expressed at the time of photoactivation and is, therefore, proportional to the number of junctional proteins expressed and their relative position within the imaging focal plane.
Fig. 1.

Triadin-tagged photoactivatable green fluorescent protein (TRD-paGFP) is expressed in the junctional sarcoplasmic reticulum (jSR) of ventricular myocytes. A: illustration depicting the location of TRD-paGFP on the luminal aspect of jSR. B: 1-(3-sulfonatopropyl)-8-vinylpyridium betaine (di-8-ANEPPS) labeling of the surface sarcolemma and transverse tubules (T-tubules) in an isolated ventricular myocyte. White box highlights a representative area where high-power images are acquired. C: higher-magnification imaging of the di-8-ANEPPS-labeled ventricular myocyte shows labeled T-tubules. After local photoactivation (circular region) of TRD-paGFP with UV light, GFP fluorescence appears along the T-tubules, representing the location of the jSR. D: SR/endoplasmic reticulum retention signal-tagged red fluorescent protein (SR-RFP) fluorescence is closely opposed to TRD-paGFP after photoactivation. Cav1.2 channel, voltage-gated calcium channel-α 1.2 subunit; MT, microtubule; RyR, ryanodine receptor; BINI, bridging integrator 1.
Isolated ventricular myocytes expressing TRD-paGFP were exposed to the red-shifted potentiometric fluorescent indicator di-8-ANEPPS (Di-8). Di-8 labeling identifies the sarcolemma and the T-tubular membrane in ventricular myocytes (Fig. 1B). On photoactivation, we observed TRD-paGFP fluorescence in close proximity to Di-8-labeled T-tubules (Fig. 1C). We analyzed the physical interaction of these signals by calculating a Pearson colocalization coefficient of 0.95 (C = 10, N = 5). In parallel experiments, myocytes expressing both TRD-paGFP and SR-RFP were photoactivated to validate targeting of TRD-paGFP to the SR. As previously described (21), SR-RFP is broadly expressed in the lumen of the SR, showing that this organelle forms a vast network that extends throughout the cell. On photoactivation, TRD-paGFP fluorescence is observed at opposing ends of SR-expressing SR-RFP (Fig. 1D). Together, these data indicate that TRD-paGFP is expressed and properly targeted to the jSR of ventricular myocytes.
jSR has multiple modalities of mobility.
To assess jSR mobility at T-tubules, we temporally measured and tracked TRD-paGFP and Di-8 signals after photoactivation. Confocal images were acquired in zero-drift compensation mode to ensure stable imaging of the same focal plane. Supplemental Video S1 (supplemental material is available at https://doi.org/10.6084/m9.figshare.11396283.v1) shows examples of stable and unstable jSR segments after photoactivation of TRD-paGFP. During analysis, we measured the averaged fluorescence intensity in 0.2-µm-diameter regions of interest within the area of the cell where TRD-paGFP was photoactivated. While averaging TRD-paGFP intensity, we also determined the time course of Di-8 fluorescence in each of the sites where TRD-paGFP was activated. The coefficient of variation (COV = SD ÷ mean) of all Di-8-labeled sites analyzed had a normal, unimodal distribution (0.021 ± 0.008, C = 14, S = 132, N = 8; Fig. 2A). This suggests that T-tubules are stable and that the focal plane did not change during the time course of imaging. In sharp contrast, analysis of TRD-paGFP fluorescent signal COV from these same sites reveals a larger degree of variation in intensities over time, both within and between cells. TRD-paGFP COVs demonstrate a multimodal distribution of fluorescent signals (Fig. 2A). These distributions could be fit with the sum of four Gaussian functions with centers at 0.06, 0.16, 0.24, and 0.36 (Fig. 2A). On the basis of this analysis, we classified jSR sites as stable and unstable. For a jSR to be considered stable, it must have a COV that is within the mean ± 3 SDs (SD = 0.02) from the lowest TRD-paGFP COV average. Sites with a COV >0.12 were considered to be unstable jSR sites. Figure 2, B–G, shows representative time courses of TRD-paGFP and Di-8 signals from different imaging sites with variable COV values. In Fig. 2B, TRD-paGFP and Di-8 fluorescence signals were stable throughout imaging. Indeed, the COVs of this site were 0.06 and 0.07 for TRD-paGFP and Di-8, respectively. By contrast, Fig. 2, C and D, shows two representative sites displaying TRD-paGFP COVs of 0.18 and 0.25. These relatively unstable sites showed frequent changes in TRD-paGFP but not Di-8 fluorescence. Some sites demonstrated much greater COV values, suggesting a higher degree of jSR mobility (Fig. 2, E–G).
Fig. 2.

Junctional sarcoplasmic reticulum (jSR) is mobile at the cardiac dyad. A: histogram shows the frequency of transverse tubule (TT) and jSR sites (n = 132) and their respective coefficient of variance (COV). TT sites show lower COV values indicating that they are static. jSR sites, on the other hand, are both static and mobile and can be fit by 4 Gaussian functions. Calculated threshold for a jSR to be mobile (static mean + 3 SDs) is noted on the histogram as a dotted line. B–G: fluorescent intensity plots (arbitrary units, A.U.) of TT fluorescence (red trace) and triadin-tagged photoactivatable green fluorescent protein (TRD-paGFP) fluorescence (green trace) as a function of time. Blue lines at the beginning of each trace denotes period of photoactivation of TRD-paGFP. Each representative plot demonstrates individual jSR and TT tracking and fluorescent measurements with variable jSR COVs.
Because newly synthesized TRD does not fluoresce before activation with UV light, fluorescent intensity fluctuations likely represent changes in the localization of TRD-paGFP proteins previously activated in the experiment. For example, partial changes in TRD-paGFP fluorescence (<100%) likely reflect positional changes of the jSR within the focal plane. We speculate that complete loss and recovery of TRD-paGFP fluorescence is produced by the movement of the jSR in and out the focal plane.
In addition to measuring jSR mobility, we discerned dwell times of jSR at T-tubules (i.e., time Di-8 and TRD-paGFP fluorescent signals overlap). By default, stable TRD-paGFP/jSR sites had membrane dwell times >15 min, or the duration of our experiments. The membrane dwell times of unstable TRD-paGFP varied from 20 to 780 s, with an average of 257 ± 17.1 s (C = 14, N = 8).
We determined three distinct patterns of jSR biogenesis and motility. Figure 3B shows exemplary segments of jSR that 1) split to become two distinct segments, 2) retract or withdraw from the T-tubule, or 3) emerge to approach the T-tubule. Images in Fig. 3A demonstrate a TRD-paGFP signal that splits into two distinct segments. Split events represent the minority (15%) of jSR mobility, whereas jSR retraction (57%) and emergence (28%) encompass the majority of movement signatures.
Fig. 3.

Junctional sarcoplasmic reticulum (jSR) displays different modalities for mobility. A, top: 1-(3-sulfonatopropyl)-8-vinylpyridium betaine (di-8-ANEPPS)-labeled transverse tubules (T-tubules, TT) in a ventricular myocyte. White box indicates zoomed region. Middle: after photoactivation with UV light, triadin-tagged photoactivatable green fluorescent protein (TRD-paGFP) fluorescence is localized to T-tubules. Bottom: during time-lapse imaging, TRD-paGFP fluorescence changes and TT fluorescence is static. B: representative images of jSR mobility. In the example retraction images, a region of TRD-paGFP fluorescence disappears from the focal plane, whereas TT fluorescence in the same region remains. During emergence events, distinct regions within the focal plane increase in fluorescence over time. Split events represent the minority of jSR mobility and are characterized as an area of TRD fluorescence once a single unit is divided into 2 distinct regions over time.
Dynein and kinesin-1 regulate jSR motility and decrease the variance and amplitude of the AP-evoked [Ca2+]i transient.
Work from our laboratory (21) and others (14) implicates the molecular motors dynein and kinesin-1 in SR/ER motility. Dynein drives the processive movement of numerous intracellular cargos toward the minus end of microtubules (2). Kinesin-1 moves unidirectionally toward the plus end of microtubules (12). However, the role of these proteins in jSR mobility is unknown. We packaged motorless kinesin-1 into viral vectors (AAV9-Kif5b-DN-RFP) for live cell imaging. We also generated AAV9-Dnchc1-shRNA-RFP, which expresses an shRNA directed against Dnchc1, encoding dynein. We confirmed successful expression of these viruses by assaying for RFP fluorescence. Expression of this virus reduced dynein mRNA expression by 62%. We found that in myocytes expressing Kif5b-DN-RFP (C = 8, S = 93, N = 5), the median (0.12) and range (0.65) of the COV values of jSR in these cells were lower than those from control myocytes (median = 0.69, range 1.77; C = 10, S = 166, N = 4; Fig. 4A). Similarly, Dnchc1-shRNA-RFP-expressing myocytes had a lower median and smaller range of jSR COVs (median = 0.13, range = 0.48; C = 9, S = 100, N = 5) than myocytes expressing wild-type Dnchc1 (median = 0.72, range = 0.1.45; C = 9, S = 148, N = 5). These data suggest that dynein and kinesin-1 regulate jSR mobility in ventricular myocytes.
Fig. 4.
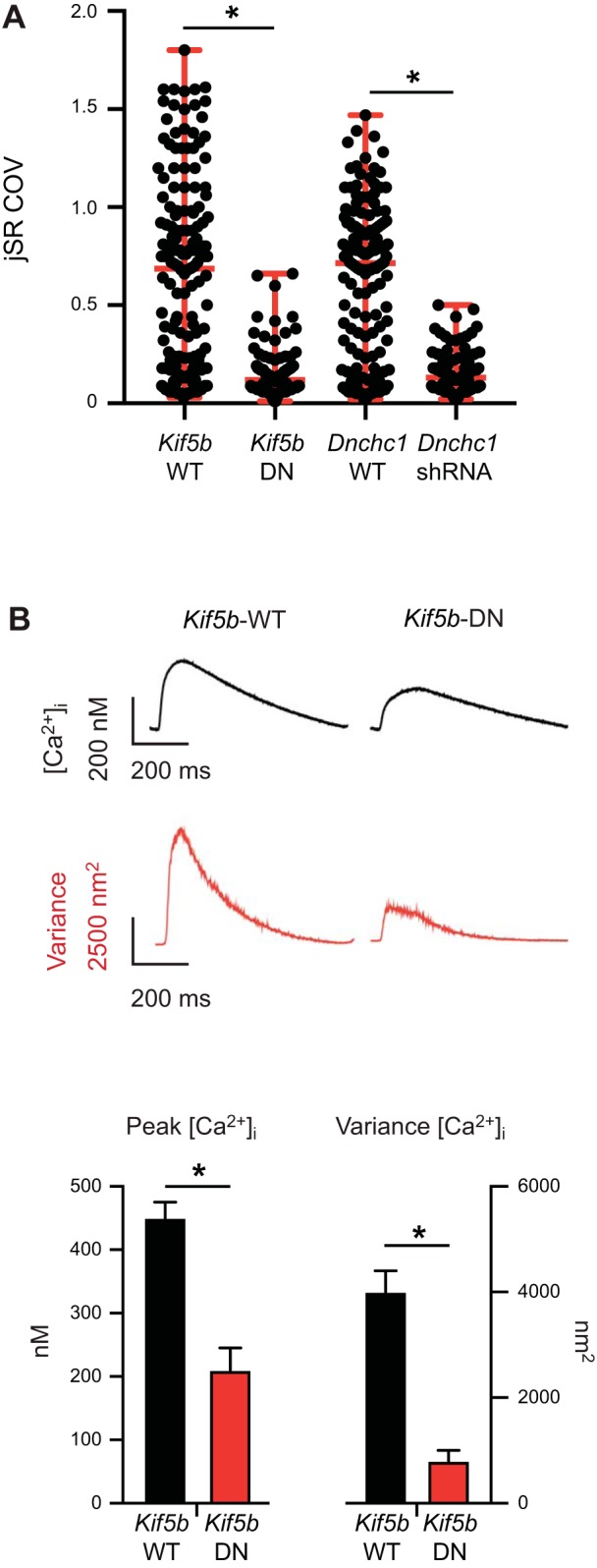
Dynein and kinesin-1 regulate junctional sarcoplasmic reticulum (jSR) mobility. A: scatter plot of the coefficient of variance (COV) of individual jSR sites from cells expressing Dnchc1-shRNA [number of sites (S) = 100] or Kif5b-DN (S = 93) vs. their respective wild-type (WT) controls (Kif5b-WT: S = 166; Dnchc1-WT: S = 148). Horizontal red lines in the plots mark the median and range of the data in each group. *P < 0.05, Kruskal–Wallis test. B: intracellular Ca2+ concentration ([Ca2+]i) transients recorded from fluo 4-AM-loaded ventricular myocytes have a higher amplitude and variance in Kif5b-WT vs. Kif5b-DN myocytes. Data are presented as means ± SE. *P < 0.05, Student’s unpaired t test. DN, dominant-negative.
Finally, we investigated the consequences of disrupting Kif5b expression on EC coupling. To do this, we loaded ventricular myocytes expressing wild-type Kif5b-WT and Kif5b-DN with the fluorescent Ca2+ indicator fluo 4-AM and evoked whole cell [Ca2+]i transients via field stimulation (1 Hz). During analysis, we aligned and averaged 20 successive [Ca2+]i transients. Figure 4B shows a representative averaged [Ca2+]i transient from Kif5b-WT (C = 7, N = 5)- and Kif5b-DN (C = 6, N = 4)-expressing myocytes. Notably, expression of Kif5b-DN decreased the amplitude and the variance of AP-evoked [Ca2+]i in ventricular myocytes (Fig. 4B). These data suggest that kinesin-1-dependent movement of the jSR contributes to the fidelity of the [Ca2+]i transients in ventricular myocytes to elicit EC coupling.
DISCUSSION
This study is the first of its kind to visualize and characterize jSR movement in real time. We have made multiple novel observations relating to cardiac SR dynamics. First, the jSR is dynamic and moves in several modalities. Second, this bidirectional mobility is dependent on the molecular motors, kinesin-1 and dynein. Third, jSR mobility contributes to beat-to-beat variability of the AP-evoked [Ca2+]i in ventricular myocytes. These findings oppose the traditional dogma that the jSR is static to maximize the reproducibility of myocyte response to an AP. Instead, we see a complex system of potential regulation, where jSR segments locally move in and out of the T-tubules. Thus, whereas the reproducibility of the heartbeat under steady-state conditions is perpetuated by local SR Ca2+ release from a similar number of couplons in the myocyte, the spatial dwell time of many dyads could be transient.
An intriguing observation in our study is that jSR mobility is multimodal. Interestingly, we find that retraction events are almost twice as common compared with emergence events. However, the number of functional couplons activated during an AP does not change under steady-state conditions, implying that numbers of jSR emergence and retraction events should be matched (21). How can we reconcile these seemingly contradictory observations? We speculate that although we detected more retractions than emergence events, the average number of functional couplons remains the same under steady-state conditions because the retracting jSR could be replaced by new, nonfluorescent jSR. This could be either a result of a nonphotoactivated TRD-paGFP (i.e., initially located outside of the field of activation) or by a jSR that only expressed wild-type TRD. Alternatively, retracting jSR with a photoactivated TRD-paGFP could be moving to a different region of the cell, one that is outside of the area being imaged.
Several studies provide insights into the potential mechanisms underlying the mobility of the jSR and hence stability of the dyad. JPH2 is necessary for stabilizing the plasmalemma and jSR by providing a structural bridge between membranes. Cardiac-specific disruption of JPH2 expression reduces the number of junctional membranes and increases dyadic size (20). JPH2 can undergo Ca2+-dependent proteolysis under conditions of high [Ca2+]i in the dyad (13), compromising the coupling between the jSR and plasmalemma. In the context of our findings, we speculate that static jSR segments are stabilized by bound JPH2, whereas mobile jSR, rapidly moving in and out of T-tubules, lacks JPH2. Accordingly, jSR mobility could be related to the number of JPH2 in each dyad.
A second determinant of jSR stability could be the lifetime of Cav1.2 channel clusters in the dyad. Cav1.2 clusters are formed by a stochastic self-assembly mechanism in the sarcolemma of ventricular myocytes (16). In developing cardiomyocytes, the nucleocytoplasmic adaptor protein BIN1 serves as a delivery site for Cav1.2 channels, thereby facilitating the clustering of Cav1.2 channels (4). Furthermore, BIN1 serves as an anchor point for the jSR, promoting stable dyads and enhancing EC coupling (4). Thus the formation of Cav1.2 clusters in the T-tubular membrane may be the first step in the formation of a functional dyad. Interestingly, the averaged membrane dwell time of mobile jSR that we measured here is similar to that of Cav1.2 channels (16). Thus it is intriguing to hypothesize that all elements, including junctophilin, Cav1.2 channels, and BIN1, must be present for a functional, stable dyad to form and that the lifetime of a dyad may be coupled to the lifetime of the associated Cav1.2 cluster. Further investigations will be required to test this hypothesis.
Another interesting finding in our study is that decreasing jSR mobility by downregulating Kif5b expression was associated with a decrease in the variance but also a decrease in the amplitude of the [Ca2+]i transient. Thus jSR mobility may be a determinant of beat-to-beat variability of the [Ca2+]i transient. At present, however, the mechanisms by which loss of Kif5b decreases SR Ca2+ release are unclear and should be the subject of a future study.
Finally, it is important to note that due to the diffraction-limited resolution of our microscope, we are unable to resolve jSR movement below approximately 250 nm in the lateral axis and 500–600 nm in the axial axis. Therefore, our measurement for jSR mobility or jSR dwell times could be lower-level estimates of dyad stability.
In conclusion, our findings suggest a novel mechanism of EC coupling regulation. These studies suggest that jSR mobility may provide another layer of regulation for EC coupling. Future studies are necessary to define the precise extent of jSR mobility, investigate regulatory mechanisms, and understand whether jSR mobility is disrupted during pathology.
GRANTS
This work was funded by National Heart, Lung, and Blood Institute Grant 5-R01-HL-085686-14.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
B.M.D. and L.F.S. conceived and designed research; B.M.D., C.Y., and A.d.l.M. performed experiments; B.M.D., A.d.l.M., and L.F.S. analyzed data; B.M.D. and L.F.S. interpreted results of experiments; B.M.D., C.Y., and L.F.S. prepared figures; L.F.S. drafted manuscript; N.G. and L.F.S. edited and revised manuscript; B.M.D., A.d.l.M., N.G., and L.F.S. approved final version of manuscript.
REFERENCES
- 1.Beavers DL, Landstrom AP, Chiang DY, Wehrens XH. Emerging roles of junctophilin-2 in the heart and implications for cardiac diseases. Cardiovasc Res 103: 198–205, 2014. doi: 10.1093/cvr/cvu151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bhabha G, Johnson GT, Schroeder CM, Vale RD. How dynein moves along microtubules. Trends Biochem Sci 41: 94–105, 2016. doi: 10.1016/j.tibs.2015.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cannell MB, Soeller C. Numerical analysis of ryanodine receptor activation by L-type channel activity in the cardiac muscle diad. Biophys J 73: 112–122, 1997. doi: 10.1016/S0006-3495(97)78052-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.De La Mata A, Tajada S, O’Dwyer S, Matsumoto C, Dixon RE, Hariharan N, Moreno CM, Santana LF. BIN1 induces the formation of T-tubules and adult-like Ca2+ release units in developing cardiomyocytes. Stem Cells 37: 54–64, 2019. doi: 10.1002/stem.2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Drum BM, Dixon RE, Yuan C, Cheng EP, Santana LF. Cellular mechanisms of ventricular arrhythmias in a mouse model of Timothy syndrome (long QT syndrome 8). J Mol Cell Cardiol 66: 63–71, 2014. doi: 10.1016/j.yjmcc.2013.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Drum BM, Yuan C, Li L, Liu Q, Wordeman L, Santana LF. Oxidative stress decreases microtubule growth and stability in ventricular myocytes. J Mol Cell Cardiol 93: 32–43, 2016. doi: 10.1016/j.yjmcc.2016.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Garbino A, Wehrens XH. Emerging role of junctophilin-2 as a regulator of calcium handling in the heart. Acta Pharmacol Sin 31: 1019–1021, 2010. doi: 10.1038/aps.2010.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Glover L, Quinn S, Ryan M, Pette D, Ohlendieck K. Supramolecular calsequestrin complex. Eur J Biochem 269: 4607–4616, 2002. doi: 10.1046/j.1432-1033.2002.03160.x. [DOI] [PubMed] [Google Scholar]
- 9.Gómez AM, Valdivia HH, Cheng H, Lederer MR, Santana LF, Cannell MB, McCune SA, Altschuld RA, Lederer WJ. Defective excitation-contraction coupling in experimental cardiac hypertrophy and heart failure. Science 276: 800–806, 1997. doi: 10.1126/science.276.5313.800. [DOI] [PubMed] [Google Scholar]
- 10.Jones PP, MacQuaide N, Louch WE. Dyadic plasticity in cardiomyocytes. Front Physiol 9: 1773, 2018. doi: 10.3389/fphys.2018.01773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Louch WE, Hake J, Mørk HK, Hougen K, Skrbic B, Ursu D, Tønnessen T, Sjaastad I, Sejersted OM. Slow Ca2+ sparks de-synchronize Ca2+ release in failing cardiomyocytes: evidence for altered configuration of Ca2+ release units? J Mol Cell Cardiol 58: 41–52, 2013. doi: 10.1016/j.yjmcc.2013.01.014. [DOI] [PubMed] [Google Scholar]
- 12.Lu H, Ali MY, Bookwalter CS, Warshaw DM, Trybus KM. Diffusive movement of processive kinesin-1 on microtubules. Traffic 10: 1429–1438, 2009. doi: 10.1111/j.1600-0854.2009.00964.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Murphy RM, Dutka TL, Horvath D, Bell JR, Delbridge LM, Lamb GD. Ca2+-dependent proteolysis of junctophilin-1 and junctophilin-2 in skeletal and cardiac muscle. J Physiol 591: 719–729, 2013. doi: 10.1113/jphysiol.2012.243279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nath S, Bananis E, Sarkar S, Stockert RJ, Sperry AO, Murray JW, Wolkoff AW. Kif5B and Kifc1 interact and are required for motility and fission of early endocytic vesicles in mouse liver. Mol Biol Cell 18: 1839–1849, 2007. doi: 10.1091/mbc.e06-06-0524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pritchard HAT, Griffin CS, Yamasaki E, Thakore P, Lane C, Greenstein AS, Earley S. Nanoscale coupling of junctophilin-2 and ryanodine receptors regulates vascular smooth muscle cell contractility. Proc Natl Acad Sci USA 116: 21874–21881, 2019. doi: 10.1073/pnas.1911304116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sato D, Hernández-Hernández G, Matsumoto C, Tajada S, Moreno CM, Dixon RE, O’Dwyer S, Navedo MF, Trimmer JS, Clancy CE, Binder MD, Santana LF. A stochastic model of ion channel cluster formation in the plasma membrane. J Gen Physiol 151: 1116–1134, 2019. doi: 10.1085/jgp.201912327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Soeller C, Cannell MB. Numerical simulation of local calcium movements during L-type calcium channel gating in the cardiac diad. Biophys J 73: 97–111, 1997. doi: 10.1016/S0006-3495(97)78051-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Song LS, Sobie EA, McCulle S, Lederer WJ, Balke CW, Cheng H. Orphaned ryanodine receptors in the failing heart. Proc Natl Acad Sci USA 103: 4305–4310, 2006. doi: 10.1073/pnas.0509324103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Takeshima H, Komazaki S, Nishi M, Iino M, Kangawa K. Junctophilins: a novel family of junctional membrane complex proteins. Mol Cell 6: 11–22, 2000. doi: 10.1016/s1097-2765(00)00003-4. [DOI] [PubMed] [Google Scholar]
- 20.van Oort RJ, Garbino A, Wang W, Dixit SS, Landstrom AP, Gaur N, De Almeida AC, Skapura DG, Rudy Y, Burns AR, Ackerman MJ, Wehrens XH. Disrupted junctional membrane complexes and hyperactive ryanodine receptors after acute junctophilin knockdown in mice. Circulation 123: 979–988, 2011. doi: 10.1161/CIRCULATIONAHA.110.006437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vega AL, Yuan C, Votaw VS, Santana LF. Dynamic changes in sarcoplasmic reticulum structure in ventricular myocytes. J Biomed Biotechnol 2011: 382586, 2011. doi: 10.1155/2011/382586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wagner E, Lauterbach MA, Kohl T, Westphal V, Williams GS, Steinbrecher JH, Streich JH, Korff B, Tuan HT, Hagen B, Luther S, Hasenfuss G, Parlitz U, Jafri MS, Hell SW, Lederer WJ, Lehnart SE. Stimulated emission depletion live-cell super-resolution imaging shows proliferative remodeling of T-tubule membrane structures after myocardial infarction. Circ Res 111: 402–414, 2012. doi: 10.1161/CIRCRESAHA.112.274530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang L, Kelley J, Schmeisser G, Kobayashi YM, Jones LR. Complex formation between junctin, triadin, calsequestrin, and the ryanodine receptor. Proteins of the cardiac junctional sarcoplasmic reticulum membrane. J Biol Chem 272: 23389–23397, 1997. doi: 10.1074/jbc.272.37.23389. [DOI] [PubMed] [Google Scholar]