

Abstract
The association of an electron-rich substrate with an electron-accepting molecule can generate a new molecular aggregate in the ground state, called an electron donor–acceptor (EDA) complex. Even when the two precursors do not absorb visible light, the resulting EDA complex often does. In 1952, Mulliken proposed a quantum-mechanical theory to rationalize the formation of such colored EDA complexes. However, and besides a few pioneering studies in the 20th century, it is only in the past few years that the EDA complex photochemistry has been recognized as a powerful strategy for expanding the potential of visible-light-driven radical synthetic chemistry. Here, we explain why this photochemical synthetic approach was overlooked for so long. We critically discuss the historical context, scientific reasons, serendipitous observations, and landmark discoveries that were essential for progress in the field. We also outline future directions and identify the key advances that are needed to fully exploit the potential of the EDA complex photochemistry.
Introduction
Chemists have long been fascinated by the use of visible light to trigger chemical processes.1 Besides offering a sustainable way to synthesize molecules,2 photochemistry has the potential to unlock reaction manifolds that are unavailable to conventional thermal pathways. This is because the chemical reactivity of electronically excited molecules differs fundamentally from that in the ground state.3 Despite its intrinsic potential, synthetic photochemistry was for a long time a specialized area with limited practical applications, mastered by only a few chemists. In recent years, this situation has changed dramatically and many photochemical methods have been developed, greatly expanding the synthetic toolbox of modern chemists. Progress within the field has mainly been spurred by photoredox catalysis.4 This strategy relies on the use of colored photocatalysts that harvest the energy of visible light to activate readily available bench-stable substrates and to generate reactive radicals5 under very mild reaction conditions.6
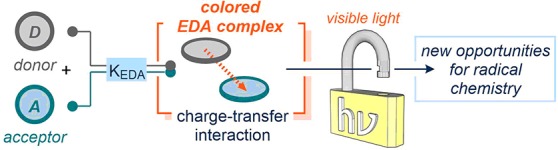
Recently, the synthetic community has recognized the potential of a photochemical approach that intrinsically differs from photoredox catalysis, since it does not rely on the use of an exogenous photoredox catalyst. This strategy exploits the association of an electron acceptor substrate A and a donor molecule D (Lewis acids and bases, respectively) to bring about the formation of a new molecular aggregation in the ground state, called an electron donor–acceptor (EDA) complex7 (Figure 1a). The two components A and D may not absorb visible light themselves, but the resulting EDA complex does. Light excitation then triggers an intramolecular single-electron-transfer (SET) event that can generate radical intermediates under mild conditions.8 The photophysics of EDA complexes have been extensively studied since the 1950s.7−10 In contrast, until very recently, they found only limited application in chemical synthesis. In the past few years, however, the EDA complex photochemistry has attracted the interest of a growing number of chemists, providing fresh opportunities in synthetic chemistry.11
Figure 1.

(a) Classical EDA complex theory and the factors that complicate synthetic applications. (b) A general strategy to make the EDA complex synthetically productive. KEDA: association constant for the formation of the EDA complex; kSET, kBET, kP: kinetic constants; ΨGS: wave function associated with the ground state; ΨES: wave function associated with the excited state; SET: single-electron transfer; LG: leaving group.
With this Perspective, we will critically assess the synthetic potential of EDA complex photochemistry. We will provide a possible rationale for why this photochemical approach was overlooked so long, and highlight the accomplishments that were crucial for developing the existing tools. In addition to charting the ideas, challenges, and milestone reactions that were essential for progress in the field, we will discuss future directions and identify the key advances that are needed to fully exploit the potential of the EDA complex photochemistry.
Background and Pioneering Synthetic Applications
Chemists are familiar with the appearance of strong color on combining two colorless organic compounds. The observation that iodine forms different colored solutions in different solvents prompted Hildebrand to investigate this phenomenon in a series of studies, which covered a time span of about 40 years.12 Eventually, spectroscopic investigations showed that benzene and mesitylene form 1:1 complexes of considerable stability with iodine.12b In 1952, this series of studies, among others,13 inspired Robert Mulliken to propose a quantum-mechanical theory to rationalize the formation of these complexes.10 According to the Mulliken charge-transfer theory, the association of an electron-rich substrate (a donor D with a low ionization potential, IP) with an electron-accepting molecule (an acceptor A having a high electronic affinity, EA)14 can elicit the formation of a new complex in the ground state, the EDA complex (Figure 1a, KEDA being the association constant for the complex formation). The physical properties of an EDA complex differ from those of the separated substrates. This is because new molecular orbitals are formed from the electronic coupling of the D and A frontier orbitals (HOMO/LUMO). This new chemical entity is characterized by the appearance of a new absorption band, the charge-transfer band (hνCT), associated with an ΨGS → ΨES electronic transition (Ψ is the wave function, associated with ground and excited states). In many cases, the energy of this transition lies within the visible range. Upon excitation of the EDA complex (orange box in Figure 1a), the ΨES is populated, which translates in an intracomplex transfer of an electron from D to A to generate a radical ion pair characterized by a net charge separation (light blue box). This complex may ultimately furnish reactive radicals. The EDA complex photochemistry may therefore offer the possibility of using visible light to activate substances that would not normally absorb in the visible spectrum.
Initial research efforts focused on the photophysical characterization of EDA complexes.8,9 For example, since the energetic gap of the electronic transition is proportional to the electron affinity of A and the ionization potential of D, cyclic voltammetry measurements14 were used to assess the feasibility of a donor and acceptor pair to undergo EDA complex formation.15 In contrast, EDA complex photochemistry initially found limited application in chemical synthesis.16 This is probably due to the difficulties of avoiding an unproductive back electron transfer (BET) from the radical ion pair, which restores the ground-state EDA complex (Figure 1a). If other possible processes leading to reactive radicals and eventually to the products are kinetically not competitive with the BET, then the photoactivation of the EDA complex will be synthetically unproductive.
To overcome this limitation and transform EDA complex activation into a productive synthetic approach, one strategy relies on the presence of a suitable leaving group (LG) within the radical anion partner ([D+•,A–•] in Figure 1b), which can trigger an irreversible fragmentation event rapid enough to compete with the BET. This can productively render two reactive radical intermediates, which can initiate synthetically useful transformations (Figure 1b). The viability of this approach was demonstrated by seminal contributions in 1970s. For example, Cantacuzene,17 Bunnett,18 Russell,19 Kornblum,20 Kochi,21 and others22 showed that the EDA complex photochemistry could trigger synthetically useful radical processes under mild reaction conditions (Figure 2). Specifically, Cantacuzene and Bunnett investigated the participation of enamines and enolates (structure 1) as donors in charge-transfer interactions with aryl and perfluoroalkyl iodides of type 2, to form an EDA aggregate (EDA-1, Figure 2a).17,18 Following the mechanistic pattern depicted in Figure 1b, the native iodide functionality, embedded within the acceptor core of 2, served as a suitable leaving group to foster the fragmentation of the radical anion, preventing an unproductive BET. The net process was a photochemical alkylation/arylation of carbonyl compounds via an electron transfer substitution reaction, which could not be achieved under thermal activation. Russell expanded this substitution protocol to other halogen native moieties, demonstrating that electron-poor benzyl chlorides 4 could participate in EDA complex formation with stoichiometric donor enamines (Figure 2b).19 Kornblum used akin acceptors 4 in combination with nitrogen-centered nucleophiles 3 to form a photoactive EDA-2 that could trigger displacement of the chloride.20,23 Kochi’s laboratory identified electron-rich arenes of type 5 as effective donors in EDA complex activation (Figure 2c).21a Charge-transfer interaction with tetranitromethane 6 produced the photoactive EDA-3, which promoted the nitration of the electron-rich aromatic ring. Kochi also demonstrated that alkyl stannanes 7 could engage tetracyanoethylene (TCNE) 8(24) in the formation of a photoactive EDA-4.21b In contrast with previous examples, where the leaving group was a native functionality within the acceptor core, here the fragmenting group (i.e., the metal center) favoring radical formation is embedded within the donor structure. Excitation of EDA-4 and intracomplex SET, followed by fragmentation of the radical cation, generated a metal cation and an alkyl radical, which were both embedded in the core of the final product.
Figure 2.

Seminal examples reporting the use of EDA complex photochemistry for synthetic applications. RF: perfluoroalkyl residue.
These early examples demonstrated the potential of the EDA complex photochemistry as a radical generation strategy useful for synthetic transformations. However, there was no emphasis on the real potential benefits for chemical synthesis, probably because synthetic photochemistry was at the time considered a specialized area requiring specific experimental expertise. Overall, these early studies were viewed more as unique chemical reactions than as integral parts of a larger research field. It was not until 2013 that the photoactivity of EDA complexes was recognized as an independent field of synthetic research, which could provide an overarching and powerful strategy in visible-light-driven radical synthesis.
Synthetic Renaissance of EDA Complex Photochemistry
In 2013, independent efforts by Chatani25 and our laboratory26 revisited and reintroduced EDA complex photochemistry as a useful radical generation strategy for chemical synthesis (Figure 3). To better contextualize these studies and provide the historical context that motivated them, it is important to appreciate how deeply the advent of photoredox catalysis, in 2008,4,27 attracted the interest of the synthetic community. Photoredox catalysis provides access under very mild conditions to open-shell species, whose unique reactivity allows transformations that are not accessible through polar pathways. This created new opportunities to apply radical chemistry in synthesis. The 2013 studies on EDA complex photochemistry were developed in the context of a photoredox system, and arose from the serendipitous observation (linked to control experiments) that an exogenous photoredox catalyst was not needed.28
Figure 3.

(a) Visible-light-induced C2-arylation of pyrroles in the absence of a photocatalyst: EDA-5 formed upon association of two stoichiometric substrates. (b) Enantioselective catalytic α-alkylation of aldehydes enabled by irradiation of an enamine-based EDA complex: EDA-6 formed upon association of a transient catalytic chiral intermediate III with substrate 13. LED: light-emitting diode; CFL: compact fluorescent lamp; EWG: electron-withdrawing group; MTBE: methyl tert-butyl ether; TMS: trimethylsilyl. The filled gray circle represents a bulky substituent on the chiral organic catalyst.
During the development of a photoredox protocol for the arylation of heteroaromatics 9 with iodonium salts 10 (Figure 3a), Chatani and co-workers observed that, when using electron-rich pyrroles 9a as substrates, the corresponding product 11a could also be obtained in the absence of the exogenous iridium photoredox catalyst.25 The photoredox catalyst was used to generate aryl radicals via SET reduction of 10. With pyrroles 9a, however, simple visible light illumination of the substrates was enough to trigger the radical process. The researchers rationalized this unusual reactivity with the formation of a yellow-orange complex EDA-5 (Figure 3a), generated in solution upon association of substrates 9a and 10. Optical absorption spectroscopic studies confirmed the appearance of a new absorption band in the visible region (the charge-transfer band hνCT). Irradiation of the colored EDA complex (EDA-5), followed by irreversible extrusion of aryl iodide, generated the radical intermediates I and II, which furnished the C2-arylated product 11a.29
Concomitantly, prompted by the interest in asymmetric organocatalysis,30 our laboratory had been investigating the direct α-alkylation of aldehydes 12 with electron-deficient alkyl bromides 13, including benzyl and phenacyl bromides, catalyzed by the chiral amine 14 (Figure 3b).26 Previous studies27a on similar reactivity established the need for a photoredox catalyst to generate radicals via reductive cleavage of the alkyl bromide 13. However, a control experiment revealed that, for specific substrates 13, the reaction could efficiently proceed in a stereoselective fashion without an external photoredox catalyst.26 The chemistry did not proceed at all without light illumination, and evidence was collected supporting a radical manifold. Mechanistic studies revealed the ability of the electron-rich chiral enamines III, generated upon condensation of the amine catalyst 14 with aldehyde 12, to trigger the formation of visible-light-absorbing EDA complexes (EDA-6) with electron-deficient dinitrobenzyl and phenacyl bromides 13. An intracomplex SET, induced by irradiation of EDA-6, afforded the chiral radical ion pair IV. By facilitating an irreversible cleavage of the carbon–halogen bond, the bromide within IV avoided an unproductive BET securing access to the reactive open-shell intermediate V. Optical absorption spectroscopic studies confirmed the formation of the enamine-based complex EDA-6, which could absorb in visible frequency regions where the individual components (enamine III and bromide 13) could not. Quantum yield measurements31 established that the reaction proceeded through a self-propagating radical chain mechanism.32 This implies that the photochemical activity of the enamine-based EDA-6 served as an initiation to sustain a chain process. The propagation manifold relied on the ability of the α-aminoalkyl radical (not shown in Figure 3b), emerging from the trap of radical V from the ground-state chiral enamine III, to regenerate V upon SET reduction of organic bromides 13. This study demonstrated that transiently generated catalytic intermediates, such as chiral enamines, can engage in the formation of photoactive EDA complexes and trigger asymmetric radical processes that are not achievable with ground-state organocatalysis. Broadly speaking, this study demonstrated that the synthetic potential of organocatalysis can be enhanced when combined with photochemical reactivity to unlock reaction pathways inaccessible via thermal activation.33
Collectively, the reports highlighted in Figure 3 showcased the synthetic potential of the EDA photochemistry and revived interest in this radical generation strategy. The synthetic community has since developed a variety of synthetically useful photochemical procedures. This Perspective, instead of providing an exhaustive list of reactions, critically describes developments since 2013, charting the ideas and advances that were crucial in developing the photochemical synthetic tools. We categorize the selected examples in two classes, based on whether the intermediates involved in EDA complex formation are present in stoichiometric (in analogy to the chemistry reported in Figure 3a) or catalytic amounts (in analogy to Figure 3b). EDA complexes can also be thermally activated to trigger synthetic transformations.34 This ground-state EDA complex reactivity falls outside of the scope of this Perspective.
Photoactivity of Stoichiometric EDA Complexes
Direct Coupling between Donor and Acceptor Substrates
The most straightforward synthetic application of the EDA complex activation strategy is based on the light-driven coupling of two substrates, the donor and the acceptor (Figure 4a). The viability of the resulting radical processes is strictly dependent on the intrinsic electronic properties of the two partners, which should be prone to forming a photoactive EDA complex. The structural moieties of the substrates, which are responsible for EDA complex formation, would both eventually end up in the core of the final product. Critical for reactivity is the presence of a suitable leaving group (blue circle in Figure 4a), which is generally a native functionality (e.g., halides) adorning the structure of one of the substrates. As explained above (Figure 1b), this leaving group is essential to securing, upon photoinduced SET, an irreversible fragmentation that productively affords open-shell intermediates, responsible for the formation of the product. The net process is a selective coupling reaction, although these photochemical processes are often based on radical chain propagation manifolds and not on radical coupling events.32
Figure 4.

(a) General strategy for the coupling of electron-rich (donor) and electron-poor (acceptor) stoichiometric substrates via EDA complex activation. (b) Photochemical C2-alkylation of indoles and the X-ray structure of the photoactive complex EDA-7, formed upon association of 3-methylindole and 2,4-dinitrobenzyl bromide. (c) Photochemical C(sp2)–C(sp2) coupling between aniline derivatives 19 and bromothiophenes 20.
This strategy has been used to promote carbon–carbon bond-forming processes. Generally, electron-rich aromatic compounds have served as donors, while electron-poor alkyl halides have been used as acceptors. The halides act as suitable leaving groups. Along these lines, a photochemical strategy was developed for the direct alkylation of 3-substituted indoles 16 with electron-accepting benzyl 13 and phenacyl bromides 17 (Figure 4b).35
The most significant result was the successful isolation and full characterization by X-ray single-crystal spectroscopic analysis24 of a visible-light-absorbing EDA complex (EDA-7), whose photochemical activity triggered the alkylation process. Remarkably, the latter analysis established that the average interplanar distance between the 3-methylindole and the 2,4-dinitrobenzyl bromide fragments (3.33 Å) is significantly lower than the van der Waals separation for aromatic molecules (3.40 Å),36 which is consonant with intermolecular binding forces being at work in the solid state. Irradiation of EDA-7 by a compact fluorescence lamp (CFL) bulb induced the formation of the radical ion pair VI, which evolved into the radicals VII and VIII upon extrusion of the bromide anion. The low quantum yield of the process (Φ = 0.2) indicated that a radical combination could be responsible for delivering the C2-alkylated indole 18. Similar photochemical C–C bond-forming processes have been developed, using an array of electron-rich aromatics.37 For example, König and co-workers used a photoactive EDA complex (EDA-8), formed upon aggregation of aniline derivatives 19 and electron-poor bromothiophenes 20, to forge a C(sp2)–C(sp2) bond within product 21 (Figure 4c).37a
α-Ketoacids of type 22 have also been reported to be suitable donors for productive EDA complex formation with different acceptors, including imines38 and alkyl boronic acids.39 In particular, the latter strategy enabled the 1,2-radical addition to the carbonyl system of 22 (Figure 5). The radical addition to a carbonyl compound, in particular ketones, is a difficult process. This is because it is generally hampered by the strong tendency of the resulting alkoxyl radical to undergo β-fragmentation, which makes the process reversible.40 The EDA complex photochemistry provided an effective strategy to overcome this limitation, highlighting its potential applicability to difficult synthetic problems. Specifically, the chemistry is triggered by the boron complex formation between α-ketoacids 22 and alkyl boronic acids 23, which act as Lewis acids. The resulting complex can be represented as either the Lewis acid–base pair EDA-9a or the boron anhydride EDA-9b (Figure 5). This boron complex EDA-9 was confirmed to have a 1:1 composition in the substrates and to absorb in the visible region. Irradiation furnished alkyl radicals IX, which could add on the activated carbonyl of another molecule of the boron anhydride EDA-9b. Interception of the oxygen-centered radical within the resulting intermediate X by the vicinal boron atom prevented an unproductive β-scission, while feeding a radical chain manifold by regenerating the alkyl radical IX. Hydrolysis of the boracycle XI, followed by telescoped esterification, delivered lactate products 24. Here, the complex EDA-9 served as the radical precursor and activated the carbonyl to facilitate the alkyl radical addition.
Figure 5.

1,2-Radical addition to carbonyl compounds driven by light-irradiation of EDA-9.
An alternative strategy for productive EDA complex formation is to generate stoichiometric transient highly electron-rich intermediates from stable weakly polarized substrates. Our laboratory used this strategy for the photochemical perfluoroalkylation of arenes (Figure 6a).41 The presence of a base unmasked an electron-rich enolate, which was generated in situ upon facile deprotonation of α-cyano arylacetates 25, which bear a highly acidic proton. The enolate formed a colored EDA complex (EDA-10) upon association with electron poor perfluoroalkyl iodides (RFI, where RF indicates the perfluoroalkyl fragment). The photoactivity of EDA-10 afforded electrophilic perfluoroalkyl radicals (RF·), which could be intercepted by the aryl moiety of substrate 25 via a homolytic aromatic substitution (HAS) pathway. Quantum yield determination (Φ = 3.8, λ = 400 nm) established a radical chain mechanism as the main reaction pathway, implying that the EDA complex photoactivity served as an initiation step.
Figure 6.
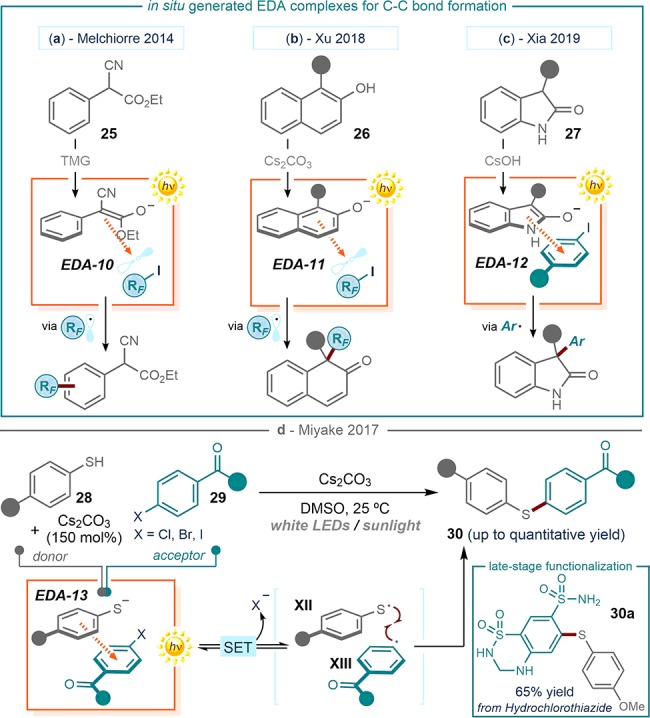
Survey of photoactive EDA complexes, formed upon in situ generation of the donor counterpart from weakly polarized substrates, and their use for the construction of C–C (a–c) and C–S bonds (d), TMG: 1,1,3,3-tetramethylguanidine; RF: perfluoroalkyl residue.
Since perfluoroalkyl iodides are particularly prone to EDA complex formation, they have been extensively used as acceptors.42 One recent representative example is a photochemical EDA complex strategy for the dearomative functionalization of naphthols 26 (Figure 6b).42a Treating 26 with cesium carbonate secured the formation of the naphtholate anion, which acted as the donor for the formation of EDA-11. The donor ability of enolates, generated in situ upon deprotonation of oxindoles 27, was also used to trigger the formation of aryl radicals by means of the photoactivity of the colored EDA-12, formed by association with aryl iodides (Figure 6c).42b This approach was used for the C3-arylation of oxindoles 27.
All the protocols discussed above deal with the formation of novel C–C bonds. The photoactivity of stoichiometric EDA complexes could also be used to design efficient carbon-heteroatom coupling procedures. For carbon–sulfur bond-forming methods,43 one often requires forcing experimental conditions or specialized catalytic systems, mostly relying on the use of transition metals.44 Miyake recently developed a metal-free alternative for the formation of the C–S bond in aryl thioethers 30 (Figure 6d).43a
This reactivity is based on the charge-transfer interaction between an electron-rich thiolate, formed upon deprotonation of aryl thiol 28, and an electron-poor aryl halide 29. Irradiation by white light emitting-diodes (LEDs) of the resulting EDA complex (EDA-13) generated radicals XII and XIII, which delivered products 30 upon radical coupling. The mild experimental conditions of this photochemical process secured an excellent functional group tolerance, as demonstrated by the late-stage functionalization of diuretic remedy hydrochlorothiazide (structure 30a in Figure 6d).
Eluding Structural Constrictions: Using Sacrificial Donors and Redox Auxiliaries
The methods highlighted above enable the coupling of two substrates, which are also involved in the EDA complex formation. Therefore, the diversity of the reaction products is somehow restricted by the need to select highly polarized reagents with donor and acceptor properties, which eventually end up in the product scaffold. One strategy to evade this limitation is to use sacrificial donor compounds that elicit EDA complex formation by aggregation with electron-poor substrates (Figure 7a). Upon light-induced radical formation, the resulting open-shell intermediate is intercepted by an external substrate, which serves as a trap. This approach has an enhanced synthetic versatility since the radical trap does not need specific electronic properties to elicit EDA complex formation. For example, stoichiometric amounts of secondary amines 31 have been used as sacrificial donors to activate perfluoroalkyl iodides (RFI 32) via EDA complex formation (EDA-14, Figure 7b).45 Visible-light irradiation of EDA-14 generates perfluoroalkyl radicals (RF·,XIV), which add on isocyanide 33, acting as an external radical trap. The ensuing intermediate XV triggers a cyclization to afford radical XVI, which abstracts an iodine from RFI to form the quinoxaline product 34 while propagating a radical chain via regeneration of RF·.
Figure 7.

(a) General strategy for radical formation based on the use of a stoichiometric sacrificial donor to drive EDA complex formation. The structure of the radical trap, which is not involved in the radical formation process, ends up in the final product. (b) Photochemical generation of perfluoroalkyl radicals for the synthesis of quinoxalines; RF: perfluoroalkyl residue.
Another limitation of the EDA complex-based synthetic strategies discussed so far is that one substrate must be both electronically biased and bear a fragmenting functionality. This is necessary to form an EDA aggregate and trigger the fragmentation needed for radical formation. Generally, simple and easily available substrates adorned with native functionalities (mostly halides within acceptors) were used for this purpose. The use of native fragmenting groups is advantageous in terms of the availability of the reagents, but it requires the EDA complex formation to be elicited exclusively by the electronic properties of the substrate’s main core. This means that only highly polarized radicals can be generated. For example, in the previous contribution reported in Figure 7, it is the electron-poor nature of the perfluoralkyl fragment within RFI that secures the formation of the EDA complex, while the iodide is a mere fragmenting group. The resulting perfluoroalkyl radical is therefore electronically biased (highly electrophilic). In an alternative strategy, which proved useful to expand the synthetic potential of EDA complex photochemistry, a reaction partner is decorated with a purposely installed activating group, which serves as both redox-auxiliary (RA, blue circle in Figure 8a) and leaving group. The substrate’s main core does not need to be electronically biased here, since the EDA complex formation is facilitated by the electronic properties of the redox-auxiliary/fragmenting group. The radical emerging from the excitation of the EDA complex is therefore electronically unbiased.
Figure 8.

(a) General representation of the use of a redox auxiliary that drives both the formation of an EDA complex and, upon photoexcitation and fragmentation, the generation of an electronically unbiased radical. (b) Photochemical generation of nitrogen-centered radicals by the installation of an appropriately functionalized dinitro-substituted auxiliary on the O-aryl oximes 35. RA: redox auxiliary; CHD: cyclohexadiene; HAT: hydrogen atom transfer.
This strategy was elegantly exploited by Daniele Leonori to generate nitrogen-centered radicals (NCRs) using appropriately functionalized dinitro-substituted O-aryl oximes 35 as bench-stable precursors (Figure 8b).46 The electron-poor dinitro aryl moiety on the oxime substrate served as a redox tag to elicit the formation of an EDA complex (EDA-15) upon aggregation with electron-rich triethylamine, as supported by UV–vis analyses. Upon photoinduced SET, EDA-15 delivered the ion pair XVII. Here, the reduced electron auxiliary acted as a leaving group, extruding the stable phenoxide 38. The resulting iminyl radical underwent a 5-exo-trig cyclization to give the C-centered radical XVIII.47 The latter can either abstract a hydrogen atom from the cyclohexadiene additive, or be oxidized by 38 to deliver cyclic imines 36 and 37, respectively. This iminyl radical generation from dinitro-substituted aryl oximes was also used to synthesize imidazoles,48a cyclic sulfonimides,48b phenanthridines, and quinolines.48c Further studies highlighted the ability of potassium carbonate to participate as donor in similar EDA complexes to form amidyl radicals.48d
The main advantage of installing suitable redox auxiliaries is the possibility of generating unbiased radicals, not bearing any stabilizing/activating functionality. Often, the redox auxiliary is easily installed on readily available substrates. Chen’s group implemented a protocol for generating alkyl radicals from alcohol precursors, simply by adorning the native hydroxyl moiety of the substrate with a N-phthalimide fragment (Figure 9a).49a The aromatic ring of the resulting N-(alkoxy)phthalimide 39 was electron-poor, which elicited a productive π–π interaction with the electron-rich Hantzsch ester 40, leading to the EDA complex EDA-16. Light-mediated SET generated the phthalimide anion 43, the oxygen-centered radical XIX, and the radical cation XX. Extrusion of formaldehyde from XIX formed the alkyl radical XXI, which added to an external trap, namely allyl phenyl sulfone 41, through a Giese-type addition to ultimately afford products 42.
Figure 9.

(a) N-phthalimide as a redox auxiliary that activates alcohols and forms an EDA complex (EDA-16) with Hantzsch ester: generation of alkyl radicals XXII. (b) Pyridinium salts as redox auxiliaries for the activation of primary amines and the generation of alkyl radicals.
In a similar approach developed by Varinder Aggarwal,50 the electron-accepting properties of pyridinium salts,51 which can be easily prepared from amines, served as redox auxiliaries for EDA complex formation. Pyridinium derivatives 44 formed EDA-17 with Hantzsch ester 40 to generate alkyl radicals and promote a Giese addition to electron-poor olefins 45 (Figure 9b). The versatility of this radical generation method has been used to design other photochemical processes, including hydrodeamination, alkynylation, alkenylation, allylation, thioetherification,50 and thioesterification52 reactions. Glorius found that indoles can also aggregate with pyridinium derivatives to form EDA complexes, whose photoactivity can trigger C2-functionalization of indoles.53
As highlighted by the previous examples (Figure 9), redox auxiliaries modulate the redox properties of a substrate and, upon aggregation with a sacrificial donor (the Hantzsch ester 40), enable the photochemical generation of electronically unbiased radicals. The use of external radical traps allows for a wide diversification of products. But the redox-auxiliary-based strategy is also useful for implementing coupling processes, where both partners of the EDA complex provide fragments to the final products of the photochemical process (Figure 10a). In this context, Aggarwal54 demonstrated that redox-active N-(acyloxy)phthalimides 47a,54a Katritizky N-alkylpyridinium salts 47b,54b,54d and thionocarbonates 47c(54c) can act as both suitable acceptors and radical precursors for photochemical borylation processes (Figure 10b). These protocols convert readily available carboxylic acid, amine, and alcohol derivatives into valuable boronic esters. Crucial for reactivity was the in situ formation of an aggregate between bis(catecholato)diboron 48 and the amide-based solvent (N,N-dimethylacetamide, DMA), which can act as an effective donor for EDA complex formation with N-(acyloxy)phthalimides 47a and N-alkylpyridinium salts 47b. The photoactivity of the resulting colored complexes (EDA-18a and EDA-18b) triggered the radical borylation process. Conversely, for thionocarbonates 47c, additional triethylamine was required to increase the electron-donating character of 48, leading to the complex EDA-18c. In all these examples, the formation of visible-light-absorbing EDA-18 was ascertained by UV–vis absorption studies, showing the appearance of the charge-transfer band. Visible-light excitation of EDA-18 triggers an intracomplex SET-forming radical cation XXII, along with the open-shell intermediate XXI, formed upon extrusion of the redox auxiliary. XXI is trapped by a second 48·DMA aggregate to furnish the desired boronic ester product 49 after ligand exchange. The trapping event delivers a strongly reducing boron-centered radical XXIII, which is responsible for propagating a radical chain pattern by SET reduction of substrate 47. The high quantum yield measured for the borylation process with 47b (Φ = 7) is congruent with this mechanistic scenario. The mild reaction conditions of the protocol provided for the efficient borylation of primary, secondary, and specific tertiary radical precursors, with high functional group tolerance.
Figure 10.

(a) Schematic representation of coupling reactions exploiting redox auxiliaries for EDA complex activation. (b) Photochemical borylation of carboxylic acid, amine and alcohol derivatives; RA: redox auxiliary.
The EDA complex activation strategy is generally characterized by mild operational conditions and a high functional group tolerance, which makes it potentially suitable for the functionalization of biologically relevant macromolecules. A first demonstration of this potential comes from the Ragains laboratory, which developed a photochemical O-glycosylation of thioglycosides 51 (Figure 11).55
Figure 11.

Visible-light promoted O-glycosylation reported by Ragains and co-workers.55 OTf: triflate; PMP: p-methoxyphenyl.
Here, to foster the formation of a charge-transfer interaction with Umemoto’s reagent 50,56 thioglycoside 51 is adorned with an electron-rich p-methoxy styrene moiety. This leads to a π–π stacking interaction between the styrene and the S-trifluoromethyldibenzothiophenium cation to form EDA-19, as indicated by spectroscopic and computational studies. This aggregate can absorb light in the visible region, promoting the formation of the distonic radical cation XXIV and the trifluoromethyl radical XXV, upon fragmentation of the dibenzothiophene 53, which acted as a redox auxiliary and a leaving group. Intramolecular attack of the nucleophilic sulfur followed by intermolecular trap of the trifluoromethyl radical XXV generates the sulfonium intermediate 54. The presence of an external oxygen-centered nucleophile (an alcohol) displaced the sulfur-based leaving group, furnishing the O-glycosylated product 52.
Photoactivity of Catalytic EDA Complexes
The efficiency and scope of the photochemical processes was considerably improved by adorning substrates with well-tailored activating groups that could elicit EDA interactions and facilitate both redox processes and radical formation. But this approach still required the use of stoichiometric substrates that can form an EDA complex. An important advance was to implement the EDA complex activation strategy within a catalytic regime. This requires a catalyst to activate one of the substrates, which is weakly polarized in its native form. The ensuing generation of a transient catalytic intermediate, characterized by a greatly enhanced polarization, could then trigger the formation of a photoactive EDA aggregate (Figure 12a). This approach provided opportunities to expand the efficiency of the EDA complex photochemistry, while implementing asymmetric radical processes when using a chiral catalyst.
Figure 12.

(a) Moving the EDA complex activation strategy into a catalytic regime: in situ generation of a catalytic intermediate acting as a donor. (b) Photo-organocatalytic enantioselective perfluoroalkylation of β-ketoesters 55 driven by the photochemical activity of the chiral enolate-based EDA complex EDA-20. PTC: phase transfer catalyst.
Organocatalysis and Asymmetric Photochemical Processes
Organocatalysis proved effective for developing catalytic asymmetric reactions driven by the photoactivity of EDA complexes.33 The organocatalytic mechanisms of substrate activation and induction, which had been so successful in promoting ionic processes in the thermal regime with high enantioselectivity, could also be used for the photoactivation of substrates and to control the stereochemical outcome of the ensuing radical process (Figure 12a). As an early example, our laboratory (Figure 3b) used transiently generated catalytic enamines as donors for EDA complex formation.26 Here, the key reactivity aspect is that the chiral organocatalyst can activate weakly polarized substrates (such as aldehydes or ketones57) that would normally not be suitable donors to elicit a charge-transfer interaction with an acceptor. This is because the enamine, resulting from amine catalyst condensation with the carbonyl substrate, has a greatly enhanced donor ability, which makes it prone to EDA complex formation. Irradiation with visible light started a radical chain manifold, where the ground-state chiral enamine could stereoselectively intercept the photochemically generated open-shell intermediate.
This strategy was expanded to include other chiral organocatalytic intermediates and thus to develop enantioselective processes that are not achievable with the ground-state chemistry of organocatalysis. For example, the electronic similarities with enamines suggested the use of chiral enolates of type XXVI, generated in situ under phase transfer (PTC) conditions58 by deprotonation of cyclic β-ketoesters 55 (Figure 12b).59 Perfluoroalkyl iodides 32 were selected as electron-accepting substrates. The chiral enolate XXVI was sufficiently electron-rich to interact with the σ* of RFI and promote the formation of a colored EDA complex (EDA-20). Visible-light irradiation induced an SET which triggered the formation of the perfluoroalkyl radical (RF·) XIV via the reductive cleavage of the C–I bond. Since RF· is an electrophilic radical, it was intercepted by the ground-state chiral enolate XXVI to furnish the enantioenriched ketoester products 57 bearing a perfluoroalkyl- or a trifluoromethyl-containing quaternary stereocenter.59
Ryan Gilmour developed a complementary strategy (Figure 13),60 exploiting the electron-poor character of another classical organocatalytic intermediate: the iminium ion.61 This strongly electrophilic intermediate XXVII is generated from weakly polarized α,β-unsaturated aldehydes 58 upon activation by a chiral amine catalyst. The ground-state reactivity of chiral iminium ions has found wide application in the stereoselective β-functionalization of enals with nucleophilic compounds.62 Here, the electronic nature of the transient catalytic iminium ion was used to trigger the formation of a photoactive EDA complex with a donor substrate (Figure 13b). The use of α-keto acids 22 as donor substrates was crucial for reaction development: (i) acting as acids, they facilitated the condensation of the aminocatalysts 59 with enals 58, leading to the iminium ions XXVII; (ii) along with electrostatic interactions, their electron-rich nature secured the formation of the EDA complex with the electron-poor organocatalytic intermediate (EDA-21); and (iii) they acted as latent acyl radicals. Indeed, the excitation of the EDA complex with UV light at 402 nm triggered an SET event, which induced rapid decarboxylation prior to radical–radical combination between the 5-π β-enaminyl intermediate XXVIII and the acyl radical XXIX. The overall process provided access to 1,4-dicarbonyl compounds 60 by means of an acyl radical conjugate addition, namely a formal radical Stetter reaction (Figure 13b).60 Quantum yield determination (Φ = 0.01) and computational analysis suggested that a closed radical catalytic cycle was operative. Although an asymmetric variant of this process could not be implemented, these studies established the possibility of electron-poor iminium ions serving as acceptors in the formation of intermolecular EDA complexes. Another peculiarity of this process was that, in contrast to many of the previous examples, a chain propagation mechanism was not operative. This implied that the photoactivity of the EDA complex was not limited to promoting an initiation step, but rather was iteratively driving every catalytic cycle.
Figure 13.

(a) Moving the EDA complex activation strategy into a catalytic regime: in situ generation of a catalytic intermediate acting as an acceptor. (b) Photocatalytic radical Stetter reaction driven by the photochemical activity of the iminium ion-based EDA complex EDA-21.
As the examples in this Perspective show, the synthetic methods triggered by the photoactivity of EDA complexes generally rely on the excitation of intermolecular aggregates formed upon association of two substrates/intermediates. Our laboratory recently demonstrated that photon-absorbing intramolecular EDA complexes can also promote synthetically useful processes (Figure 14).63 Similarly to the chemistry discussed above (cf. Figure 13b), this approach uses the electron-poor character of catalytically generated iminium ions. Here, however, we used a chiral amine catalyst adorned with an electron-rich carbazole moiety64 (amine 63 in Figure 14b). Upon condensation with cyclic enone 61, this catalyst generated chiral iminium ions that showed a broad absorption band in the visible region. This optical property arises from an intramolecular charge transfer π–π interaction between the electron-rich carbazole fragment and the electron-deficient iminium double bond: for example, aliphatic iminium ions typically can only absorb in the UV region (below 400 nm). The formation of the intramolecular EDA complex EDA-22 was confirmed by X-ray crystallographic analysis, which showed how the interatomic separation between the carbazole nitrogen and the sp2 α-carbon of the iminium ion (3.10 Å) was significantly shorter than the van der Waals distance. Excitation of the intramolecular EDA-22 at 420 nm triggered an SET event from the carbazole to the iminium ion, furnishing the chiral radical intermediate XXX. The long-lived carbazole radical cation in XXX then acted as an effective oxidant to generate a radical from an easily oxidizable electron-rich alkyl silane 62. The resulting radical was then stereoselectively intercepted by the ground-state electron-poor iminium ion. The overall process, which proceeded by virtue of a radical chain propagation manifold, enabled radical conjugate additions to β-substituted cyclic enones to form synthetically valuable quaternary carbon stereocenters65 with high stereocontrol using visible light irradiation. Besides the synthetic implications, this study63 demonstrates that the photoactivity of visible-light-absorbing intramolecular EDA complexes can be used to generate radicals under mild conditions.66
Figure 14.

(a) Photoactivity of an intramolecular EDA complex; a catalyst adorned with a donor unit activates a weakly polarized substrate to generate an electron-poor intermediate, which is prone to intramolecular EDA complex formation with the catalyst fragment. (b) Enantioselective catalytic radical conjugate addition driven by the excitation of an intramolecular EDA complex.
Toward a General Catalytic Radical Generation Strategy
The catalytic strategies discussed so far are all based on the exploitation of organocatalytic intermediates that are directly involved in the photochemical radical formation and the trapping of the ensuing open-shell intermediates. Eventually, a portion of the structure of the catalytic intermediate is embedded in the core of the final products. These systems therefore require judiciously chosen catalysts and reagents, which lowers the substrate generality and the scope of the reactions. A more flexible and effective catalytic system for EDA complex photochemistry would require these processes, namely the photochemical generation of radicals and the trapping event, to be decoupled. This would require the use of a catalyst to exclusively generate radicals. In the general strategy depicted in Figure 15a, an electron-rich catalyst would trigger EDA complex formation upon aggregation with an electron-poor substrate. Photoinduced SET would then lead to radicals, which could be intercepted by an external trap to form a product. The essential step would be an effective catalyst turnover through SET reduction of the catalyst radical cation, arising from the photoactivity of the progenitor EDA complex.
Figure 15.

(a) General strategy for catalysis in EDA complex photochemistry: a donor catalyst that can photochemically generate radicals and then be turned over. (b) Photocatalytic radical alkylations mediated by the catalytic combination of triphenylphosphine and sodium iodide; TFA: trifluoroacetic acid.
Recent studies have demonstrated the feasibility of this catalytic approach. Shang and Fu reported a combination of easily available and inexpensive catalysts, namely triphenylphosphine (Ph3P) and sodium iodide (NaI), which, despite not absorbing in the visible spectrum individually, can promote synthetically useful reactions under blue light irradiation (Figure 15b).67 Specifically, these catalysts could mediate the formation of radicals from redox-active esters 47a since they could trigger the formation of a photoactive three-component EDA complex (EDA-23 in Figure 15b). A light-induced intracomplex SET from iodide to the substrate phthalimide moiety in 47a produced the catalyst radical cation XXXI and, upon CO2 extrusion, the open-shell intermediate XXI. Radicals XXI are then intercepted by acid-activated heteroarenes 65 in a Minisci manifold. The crucial step of this mechanism is the catalyst turnover: the radical cation XXXII, generated upon C–C bond formation, is reduced by the Ph3P–I· intermediate XXXI, which was proposed to be a persistent radical.68 This SET event delivers the alkylated heteroaromatic product 66 while turning over the Ph3P/NaI catalytic system. Quantum yield determination (Φ = 0.15) is consonant with a closed radical catalytic cycle being operational. The triphenylphosphine plays a key role in this catalytic machinery. It is crucial for facilitating, upon association with iodine, the intermolecular EDA complex formation (EDA-23) and stabilizing the iodine radical as a Ph3P–I· intermediate. The supposed persistency of the latter radical intermediate secured an effective catalyst turnover through SET reduction. The same NaI/PPh3 catalytic system was then used to promote the formation of radicals via photoinduced SET reduction of other radical precursors, including hypervalent iodine reagent 67 and pyridinium salts 44. The ensuing photochemically generated radicals were later intercepted by suitable electron-rich radical traps. However, since the quantum yield of these processes was not measured, a radical chain manifold (which would not require an effective catalyst turnover) could not be excluded.
Bosque and Bach expanded the concept of using a catalytic electron donor species to trigger visible-light-mediated radical reactions via EDA complex formation. They demonstrated that 3-acetoxyquinuclidine (q-OAc, 69) could be used in a catalytic fashion (Figure 16).69 Combination with electron-poor tetrachlorophthalimide ester 68 affords the colored complex EDA-24. Blue-light irradiation triggers an intracomplex SET from the catalyst (q-OAc) to the tetrachlorophthalimide moiety, leading to decarboxylation and formation of the α-amino radical XXXIII. The latter intermediate is then oxidized by the catalyst radical cation (q-OAc+•, XXXIV): this step turns the catalyst over and affords iminium ion XXXV, which is trapped by the previously liberated tetrachlorophthalimide anion 43, delivering the final product 70. The low value of the quantum yield (Φ = 0.02) is consonant with a closed catalytic cycle with no radical propagation chain being operative. Overall, q-OAc 69 triggers a redox-neutral pathway, since it acts first as a donor for an intracomplex SET within the EDA complex, and then can get back the electron from intermediate XXXIII. The crucial aspect for catalysis here relies on the rigid, geometrically constrained structure of the catalyst’s quinuclidine core, which prevents a possible degradation path proceeding through α-deprotonation70 of the radical cation q-OAc+•, XXXIV.
Figure 16.

Use of 3-acetoxyquinuclidine as an external electron-donor catalyst for visible-light-mediated radical processes via EDA complex formation; q-OAC: 3-acetoxyquinuclidine; BOC: tert-butyloxycarbonyl.
Sami Lakhdar has recently reported a different, interesting catalytic approach for EDA complex photochemistry.71 In contrast to the examples in Figures 15 and 16, here the catalyst does not directly activate substrates toward radical formation (Figure 17a). Instead, the donor catalyst forms an EDA complex with an electron-poor additive. The resulting photoactivity affords an open-shell intermediate (A· in Figure 17a) that is eventually responsible to generate radicals (R·), which participate in the process leading to the final products. This means that neither partners of the photoactive EDA complex (the catalyst and the additive) end up in the product’s structure. This catalytic strategy was used to photochemically generate hydrogen atom transfer (HAT) agents, which could then promote radical cascade reactions upon activation of diphenylphosphine oxide 72 (Figure 17b).71 Specifically, the ground-state association between eosin Y (73), present in catalytic amounts, and pyridinium salts 74, used as additives, formed EDA-25.72 The formation of this photoactive aggregate was confirmed by both UV–vis and X-ray spectroscopic analyses. Upon irradiation of EDA-25, photoinduced SET afforded the oxidized form of 73 (73+•) along with ethoxy radical XXXVI, generated upon reductive fragmentation of the pyridinium salts 74. The ethoxy radical XXXVI, due to its propensity for hydrogen abstraction, became the real promoter for radical formation. Acting as a HAT agent, it activated diphenylphosphine oxide 72 to form the phosphorus-centered radical XXXVII. This intermediate then started a radical cascade sequence: addition to acetylene 71 generated the C(sp2)-centered radical XXXVIII, which triggered a cyclization leading to cyclohexadienyl intermediate XXXIX. The latter intermediate transferred an electron to the oxidized catalyst 73+• to deliver, after deprotonation, product 75 while closing the catalytic cycle. The low quantum yield value (Φ = 0.19) was consonant with this photochemical mechanism.
Figure 17.

(a) A different strategy for catalysis in for catalysis in EDA complex photochemistry: a donor catalyst that aggregates with an additive to form a radical promoter (A·), which is responsible to generate reactive radicals: here, none of the EDA partners is incorporated in the product’s structure. (b) Photochemical synthesis of benzo[b]phosphole oxides triggered by HAT agent XXXVI, generated upon light excitation of EDA-25; HAT: hydrogen atom transfer.
The reported catalytic systems have shown potential for expanding the synthetic applicability of EDA complex photochemistry. Further applications are expected, for example, driven by the identification of more effective and general catalyst turnover events or by the use of catalytic electron acceptors.
EDA Complex Photochemistry and Asymmetric Enzymatic Catalysis
The utility of the EDA complex photochemistry in stereoselective catalytic radical processes can be expanded to include biocatalysis.73 Recent advances highlighted the ability of some enzymes, dependent on photoactive cofactors,74 to alter their native reactivity upon light excitation and catalyze completely different processes than those for which they evolved. This strategy holds great potential, given that charge-transfer interactions between substrates can be facilitated by the spatial proximity secured by the enzyme active sites. In addition, the functions of a natural enzyme can be opportunely enhanced and tuned by directed evolution.75 As a general approach (Figure 18a), specific electron-rich cofactors can serve as donors in EDA complex formation with electron-poor substrates, which are brought in close proximity upon selective binding within the enzyme active site (symbolized as a light blue oval). The SET event triggered by the direct excitation of the EDA complex delivers an open-shell radical intermediate (R· in green circle), which is still bound to the active site. The chiral environment provided by the enzyme then secures a high stereocontrol over the ensuing radical process. Substrate exchange and regeneration of the cofactor (from deactivated gray to active purple circle) re-establish the catalytic activity of the enzyme.
Figure 18.

(a) Schematic representation of the synergistic use of enzymatic catalysis and EDA complex activation for the development of asymmetric processes. (b) Photobiocatalytic enantioselective radical dehalogenation of α-bromolactones. (c) Photochemical flavoenzymes-catalyzed stereoselective radical cyclization of olefin-tethered α-chloroamides; SET: single-electron transfer; LED: Light-emitting diode; HAT: hydrogen atom transfer; LKADH: Lactobacillus kefiri alcohol dehydrogenase; NADP(H): nicotinamide adenine dinucleotide phosphate; GDH-105: glucose dehydrogenase-105.
This strategy was successfully applied by Todd Hyster, who demonstrated that the natural reactivity of nicotinamide-dependent ketoreductases (KREDs) can be altered upon light excitation of the photoresponsive NADH/NADPH cofactor, which is bound into the enzyme active site (Figure 18b).76 The native reactivity of these enzymes, which is based on classical polar mechanisms, enables the stereoselective reduction of ketone substrates.77 The ground-state carbonyl reductase activity depends on the enzymes’ ability to bring the carbonyl compound and the cofactor in close proximity through noncovalent weak interactions. The NADH cofactor can then stereoselectively deliver a hydride (H–). However, the close proximity within the active site was also found to elicit the formation of a photoactive EDA complex between the NADP(H) cofactor 79 and electron-poor α-bromolactones 76 (EDA-26). These substrates can bind in the active site of KREDs but are not primed to carbonyl reduction. Photoinduced SET under blue light illumination triggers the mesolytic cleavage of the C–Br bond, leading to the prochiral α-carboxyl radical XLI. The tendency of the cofactor radical cation XL, emerging from the SET, to act as a good hydrogen atom (H·) donor for XLI drives the formation of the reduced chiral product 78. Mechanistic insights indicated that the last HAT step, which happens in the chiral environment provided by the enzyme, is the enantio-determining event, since the binding of the racemic bromolactones 76 from the KRED enzyme is unselective. The turnover of the cofactor is obtained by reduction of the deactivated NADP+ intermediate 77 by either i-PrOH, taking advantage of the native dehydrogenase activity of the enzyme, or a glucose dehydrogenase (GDH-105) coenzyme. The net reaction of this photochemical process is the enantioselective dehalogenation of racemic α-bromo lactones 76.
Building upon these findings, the Hyster laboratory expanded this photobiocatalytic strategy to the use of flavoenzymes (flavin-dependent “ene”-reductases). The strategy was used to implement the synthetically elusive stereoselective radical hydroalkylation of alkenes (Figure 18c).78 Here, the flavin hydroquinone cofactor 82 and the alkene-tethered α-chloroamide 80 are responsible for the formation of the EDA complex within the active site of the ene-reductase enzyme (EDA-27). Excitation by cyan LEDs (497 nm) triggers both the SET and chloride fragmentation events, which generate the radical intermediates XLII and XLIII. Radical cyclization from XLII and an ensuing HAT from XLI forge two novel bonds within the chiral lactam product 81 in a stereodefined fashion. Remarkably, the highly structured chiral environment of the flavoenzyme’s active site enables exquisite control of absolute and relative stereoselectivity of the process. These methods illustrate how using light to develop enzymes with new catalytic functions holds great potential for the design of stereoselective radical-mediated biocatalytic reactions.
Conclusions and Future Outlook
Over the past few years, the photochemistry of EDA complexes has provided fresh opportunities in synthetic radical chemistry. We have outlined here the evolution of this strategy from the simple coupling of specialized, electronically biased substrates to the development of more general platforms providing products with a wider structural diversity. More sophisticated variants have shown the potential of this chemistry in asymmetric catalytic strategies, including within biological systems. Overall, the resulting methods provide new synthetic frameworks to successfully tackle some major challenges in radical reactivity, which traditional methodologies have not been able to address. But major developments are probably still to come.
Novel synthetic developments are expected to arise from the identification of other substrates that can engage in the formation of productive EDA complexes. In particular, the installation of redox auxiliaries within substrates has greatly expanded the potential of this strategy. However, the structural diversity of the auxiliaries is still very limited, thus offering the possibility for further developments. For example, the redox auxiliaries identified to date all have electron-accepting properties. A future goal for the continued expansion of the field will be to design redox-active scaffolds capable of acting as donors in charge-transfer interactions, which could open up complementary reaction manifolds.
The EDA complex activation has been successfully used in asymmetric processes when coupled with organocatalytic strategies. This approach has been limited to a few organocatalytic mechanisms of induction and substrate activation, namely aminocatalysis and phase-transfer catalysis. We foresee that other organocatalytic strategies, including N-heterocyclic carbene79 or hydrogen-bonding catalysis,80 could be useful to activate inactive substrates and turn them into potential chiral donors or acceptors, unlocking novel radical enantioselective processes. Along the same lines, another force for innovation may be the use of chiral Lewis acids to foster EDA complex formation and control the stereochemical outcome of the ensuing radical process, thus providing new mechanisms for stereocontrolled bond-formation. Finally, since the EDA complex activation strategy and the ensuing radical reactivity proceed under mild conditions while exhibiting high functional group tolerance, we expect great strides in the development of novel visible light-driven processes for the late-stage derivatization of advanced biologically relevant intermediates and macromolecules, including proteins (i.e., bioconjugation).81
Given the many innovative reactivity concepts identified in the past few years, and their impact on the field of radical relativity and synthetic photochemistry, EDA complex activation has a bright future.
Acknowledgments
We thank MINECO (CTQ2016-75520-P) and the European Research Council (ERC 681840 - CATA-LUX) for financial support. D.M. thanks H2020-MSCA-ITN-2016 (722591-PHOTOTRAIN) for a predoctoral fellowship. G.E.M.C. thanks the Marie Skłodowska-Curie Actions for a postdoctoral fellowship (H2020-MSCA-IF-2017 795793).
Author Contributions
# G.E.M.C. and D.M. contributed equally.
The authors declare no competing financial interest.
References
- a Ciamician G. The Photochemistry of the Future. Science 1912, 36, 385–394. 10.1126/science.36.926.385. [DOI] [PubMed] [Google Scholar]; b Ciamician G. Actions Chimiques de la Lumière. Bull. Soc. Chim. Fr. 1908, 3–4, i-xxvii. [Google Scholar]; c Paternò E. Sintesi in Chimica Organica per Mezzo della Luce. Nota I. Introduzione. Gazz. Chim. Ital. 1914, 44, 31. [Google Scholar]
- a Handbook of Synthetic Photochemistry; Albini A., Fagnoni M., Eds.; Wiley-VCH, 2010. [Google Scholar]; b Schultz D. M.; Yoon T. P. Solar Synthesis: Prospects in Visible Light Photocatalysis. Science 2014, 343, 1239176. 10.1126/science.1239176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Photochemistry and Photophysics; Balzani V., Ceroni P., Juris A., Eds.; Wiley-VCH, 2014. [Google Scholar]; b Modern Molecular Photochemistry of Organic Molecules; Turro N. J., Ramamurthy V., Scaiano J. C., Eds.; University Science Books: Sausalito, CA, 2010. [Google Scholar]
- a Shaw M. H.; Twilton J.; MacMillan D. W. C. Photoredox Catalysis in Organic Chemistry. J. Org. Chem. 2016, 81, 6898–6926. 10.1021/acs.joc.6b01449. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Romero N.; Nicewicz D. Organic Photoredox Catalysis. Chem. Rev. 2016, 116, 10075–10166. 10.1021/acs.chemrev.6b00057. [DOI] [PubMed] [Google Scholar]; c Marzo L.; Pagire S. K.; Reiser O.; König B. Visible-Light Photocatalysis: Does It Make a Difference in Organic Synthesis?. Angew. Chem., Int. Ed. 2018, 57, 10034–10072. 10.1002/anie.201709766. [DOI] [PubMed] [Google Scholar]; d Buzzetti L.; Crisenza G. E. M.; Melchiorre P. Mechanistic Studies in Photocatalysis. Angew. Chem., Int. Ed. 2019, 58, 3730–3747. 10.1002/anie.201809984. [DOI] [PubMed] [Google Scholar]
- a Encyclopedia of Radicals in Chemistry, Biology and Materials; Chatgilialoglu C., Studer A., Eds.; John Wiley & Sons, 2012. [Google Scholar]; b Yan M.; Lo J. C.; Edwards J. T.; Baran P. S. Radicals: Reactive Intermediates with Translational Potential. J. Am. Chem. Soc. 2016, 138, 12692–12714. 10.1021/jacs.6b08856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Photoredox processes have also been adopted in an industrial setting; for selected examples, see:; a DiRocco D. A.; Dykstra K.; Krska S.; Vachal P.; Conway D. V.; Tudge M. Late-Stage Functionalization of Biologically Active Heterocycles Through Photoredox Catalysis. Angew. Chem., Int. Ed. 2014, 53, 4802–4806. 10.1002/anie.201402023. [DOI] [PubMed] [Google Scholar]; b ElMarrouni A.; Ritts C. B.; Balsells J. Silyl-mediated Photoredox-catalyzed Giese Reaction: Addition of Non-Activated Alkyl Bromides. Chem. Sci. 2018, 9, 6639–6646. 10.1039/C8SC02253D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster R. Electron Donor-Acceptor Complexes. J. Phys. Chem. 1980, 84, 2135–2141. 10.1021/j100454a006. [DOI] [Google Scholar]
- Rosokha S. V.; Kochi J. K. Fresh Look at Electron-Transfer Mechanisms via the Donor/Acceptor Bindings in the Critical Encounter Complex. Acc. Chem. Res. 2008, 41, 641–653. 10.1021/ar700256a. [DOI] [PubMed] [Google Scholar]
- a Hilinski E. F.; Masnovi J. M.; Amatore C.; Kochi J. K.; Rentzepis P. M. Charge-Transfer Excitation of Electron Donor-Acceptor Complexes. Direct Observation of Ion Pairs by Time-Resolved (Picosecond) Spectroscopy. J. Am. Chem. Soc. 1983, 105, 6167–6168. 10.1021/ja00357a042. [DOI] [Google Scholar]; b Singh J. O.; Anunziata J. D.; Silber J. J. n-π Electron Donor-Acceptor Complexes. II. Aliphatic Amines with Dinitrobenzenes. Can. J. Chem. 1985, 63, 903–907. 10.1139/v85-150. [DOI] [Google Scholar]
- a Mulliken R. S. Structures of Complexes Formed by Halogen Molecules with Aromatic and with Oxygenated Solvents. J. Am. Chem. Soc. 1950, 72, 600–608. 10.1021/ja01157a151. [DOI] [Google Scholar]; b Mulliken R. S. Molecular Compounds and their Spectra. II. J. Am. Chem. Soc. 1952, 74, 811–824. 10.1021/ja01123a067. [DOI] [Google Scholar]; c Mulliken R. S. Molecular Compounds and their Spectra. III. The Interaction of Electron Donors and Acceptors. J. Phys. Chem. 1952, 56, 801–822. 10.1021/j150499a001. [DOI] [Google Scholar]
- a Lima C. G. S.; Lima T. d. M.; Duarte M.; Jurberg I. D.; Paixão M. W. Organic Synthesis Enabled by Light-Irradiation of EDA Complexes: Theoretical Background and Synthetic Applications. ACS Catal. 2016, 6, 1389–1407. 10.1021/acscatal.5b02386. [DOI] [Google Scholar]; b Yuan Y.-Q.; Majumder S.; Yang M.-H.; Guo S.-R. Recent Advances in Catalyst-free Photochemical Reactions via Electron-Donor Acceptor (EDA) Complex Process. Tetrahedron Lett. 2020, 61, 151506. 10.1016/j.tetlet.2019.151506. [DOI] [Google Scholar]
- a Hildebrand J. H.; Glascock B. L. The Color of Iodine Solutions. J. Am. Chem. Soc. 1909, 31, 26–31. 10.1021/ja01931a005. [DOI] [Google Scholar]; b Benesi H. A.; Hildebrand J. H. A Spectrophotometric Investigation of the Interaction of Iodine with Aromatic Hydrocarbons. J. Am. Chem. Soc. 1949, 71, 2703–2707. 10.1021/ja01176a030. [DOI] [Google Scholar]
- a Lewis G. N. Acids and Bases. J. Franklin Inst. 1938, 226, 293–313. 10.1016/S0016-0032(38)91691-6. [DOI] [Google Scholar]; b Winstein S.; Lucas H. J. The Coordination of Silver Ion with Unsaturated Compounds. J. Am. Chem. Soc. 1938, 60, 836–847. 10.1021/ja01271a021. [DOI] [Google Scholar]; c Dewar M. J. S. Mechanism of the Benzidine and Related Rearrangementes. Nature 1945, 156, 784. 10.1038/156784a0. [DOI] [Google Scholar]
- The electron-donor property of an electron-rich molecule is indicated by the magnitude of its ionization potential (IP in the gas phase) or its oxidation potential (E°ox in solution). Conversely, a compound is a viable acceptor in measure with the magnitude of its electron affinity (EA in the gas phase) or its reduction potential (E°red in solution).
- Zon M. A.; Fernández H.; Sereno L.; Silber J. J. Electrochemical Study of the Stability of the Electron-donor-acceptor (EDA) Complex Between N,N,N′,N′-Tetramethyl-p-phenylendiamine and m-Dinitrobenzene in Acetonitrile. Electrochim. Acta 1987, 32, 71–77. 10.1016/0013-4686(87)87011-1. [DOI] [Google Scholar]
- Rathore R.; Kochi J. K. Donor/Acceptor Organizations and the Electron-Transfer Paradigm for Organic Reactivity. Adv. Phys. Org. Chem. 2000, 35, 193–318. 10.1016/S0065-3160(00)35014-6. [DOI] [Google Scholar]
- Cantacuzène D.; Wakselman C.; Dorme R. Condensation of Perfluoroalkyl Iodides with Unsaturated Nitrogen Compounds. J. Chem. Soc., Perkin Trans. 1 1977, 1365–1371. 10.1039/P19770001365. [DOI] [Google Scholar]
- Bunnett J. F. Aromatic Substitution by the SRN1 Mechanism. Acc. Chem. Res. 1978, 11, 413–420. 10.1021/ar50131a003. [DOI] [Google Scholar]
- Russell G. A.; Wang K. Homolytic Alkylation of Enamines by Electrophilic Radicals. J. Org. Chem. 1991, 56, 3475–3479. 10.1021/jo00011a007. [DOI] [Google Scholar]
- Wade P. A.; Morrison H. A.; Kornblum N. The Effect of Light on Electron-Transfer Substitution at a Saturated Carbon Atom. J. Org. Chem. 1987, 52, 3102–3107. 10.1021/jo00390a026. [DOI] [Google Scholar]
- a Sankararaman S.; Haney W. A.; Kochi J. K. Annihilation of Aromatic Cation Radicals by Ion-Pair and Radical-Pair Collapse. Unusual Solvent and Salt Effects in the Competition for Aromatic Substitution. J. Am. Chem. Soc. 1987, 109, 7824–7838. 10.1021/ja00259a035. [DOI] [Google Scholar]; b Fukuzumi S.; Mochida K.; Kochi J. K. A Unified Mechanism for Thermal and Photochemical Activation of Charge-Transfer Processes with Organometals. Steric Effects in the Insertion of Tetracyanoethylene. J. Am. Chem. Soc. 1979, 101, 5961–5972. 10.1021/ja00514a016. [DOI] [Google Scholar]
- a Gotoh T.; Padias A. B.; Hall J. H. K. An Electron Donor-Acceptor Complex and Thermal Triplex as Intermediates in the Cycloaddition Reaction of N-Vinylcarbazole with Dimethyl 2,2-Dicyanoethylene-1,1-dicarboxylate. J. Am. Chem. Soc. 1991, 113, 1308–1312. 10.1021/ja00004a035. [DOI] [Google Scholar]; b Fox M. A.; Younathan J.; Fryxell G. E. Photoinitiation of the SRN1 Reaction by Excitation of Charge-Transfer Complexes. J. Org. Chem. 1983, 48, 3109–3112. 10.1021/jo00166a038. [DOI] [Google Scholar]
- An analogous charge-transfer interaction between nitrogen nucleophiles and nitroarenes was also documented by Silber; see ref (9b).
- TCNE 8 and derivatives have been extensively used in EDA complex formation, and also for synthetic applications. For a recent example, see:; Berionni G.; Bertelle P.-A.; Marrot J.; Goumont R. X-ray Structure of a CT Complex Relevant to Diels-Alder Reactivity of Anthracenes. J. Am. Chem. Soc. 2009, 131, 18224–18225. 10.1021/ja908747j. [DOI] [PubMed] [Google Scholar]; This study also detailed the characterization of an EDA complex via X-ray spectroscopic analysis.
- Tobisu M.; Furukawa T.; Chatani N. Visible Light-mediated Direct Arylation of Arenes and Heteroarenes Using Diaryliodonium Salts in the Presence and Absence of a Photocatalyst. Chem. Lett. 2013, 42, 1203–1205. 10.1246/cl.130547. [DOI] [Google Scholar]
- Arceo E.; Jurberg I. D.; Álvarez-Fernández A.; Melchiorre P. Photochemical activity of a key donor-acceptor complex can drive stereoselective catalytic α-alkylation of aldehydes. Nat. Chem. 2013, 5, 750–756. 10.1038/nchem.1727. [DOI] [PubMed] [Google Scholar]
- a Nicewicz D. A.; MacMillan D. W. C. Merging Photoredox Catalysis with Organocatalysis: The Direct Asymmetric Alkylation of Aldehydes. Science 2008, 322, 77–80. 10.1126/science.1161976. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Ischay M. A.; Anzovino M. E.; Du J.; Yoon T. P. Efficient Visible light Photocatalysis of [2 + 2] Enone Cycloadditions. J. Am. Chem. Soc. 2008, 130, 12886–12887. 10.1021/ja805387f. [DOI] [PubMed] [Google Scholar]; c Narayanam J. M. R.; Tucker J. W.; Stephenson C. R. J. Electron-Transfer Photoredox Catalysis: Development of a Tin-free Reductive Dehalogenation Reaction. J. Am. Chem. Soc. 2009, 131, 8756–8757. 10.1021/ja9033582. [DOI] [PubMed] [Google Scholar]
- This demonstrates the importance of control experiments in overriding experimenter bias and preconceived ideas, e.g., the idea that a photoredox catalyst was necessary to generate a radical via SET and drive the processes depicted in Figure 3.
- An alternative C–C bond-forming event would be the trap of the aryl radical II from the closed-shell pyrrole 9a. This path would be part of a radical chain propagation manifold.
- a MacMillan D. W. C. The advent and development of organocatalysis. Nature 2008, 455, 304–308. 10.1038/nature07367. [DOI] [PubMed] [Google Scholar]; b Melchiorre P.; Marigo M.; Carlone A.; Bartoli G. Asymmetric Aminocatalysis - Gold Rush in Organic Chemistry. Angew. Chem., Int. Ed. 2008, 47, 6138–6171. 10.1002/anie.200705523. [DOI] [PubMed] [Google Scholar]
- Bahamonde A.; Melchiorre P. Mechanism of the Stereoselective α-Alkylation of Aldehydes Driven by the Photochemical Activity of Enamines. J. Am. Chem. Soc. 2016, 138, 8019–8030. 10.1021/jacs.6b04871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studer A.; Curran D. P. Catalysis of Radical Reactions: a Radical Chemistry Perspective. Angew. Chem., Int. Ed. 2016, 55, 58–102. 10.1002/anie.201505090. [DOI] [PubMed] [Google Scholar]
- Silvi M.; Melchiorre P. Enhancing the potential of enantioselective organocatalysis with light. Nature 2018, 554, 41–49. 10.1038/nature25175. [DOI] [PubMed] [Google Scholar]
- For selected examples, see:; a Dohi T.; Ito M.; Yamaoka N.; Morimoto K.; Fujioka H.; Kita Y. Unusual ipso Substitution of Diaryliodonium Bromides Initiated by a Single-Electron-Transfer Oxidizing Process. Angew. Chem., Int. Ed. 2010, 49, 3334–3337. 10.1002/anie.200907281. [DOI] [PubMed] [Google Scholar]; b Cheng Y.; Yuan X.; Ma J.; Yu S. Direct Aromatic C-H Trifluoromethylation via an Electron-Donor-Acceptor Complex. Chem. - Eur. J. 2015, 21, 8355–8359. 10.1002/chem.201500896. [DOI] [PubMed] [Google Scholar]; c Cheng Y.; Yu S. Hydrotrifluoromethylation of Unactivated Alkenes and Alkynes Enabled by an Electron-Donor-Acceptor Complex of Togni’s Reagent with a Tertiary Amine. Org. Lett. 2016, 18, 2962–2965. 10.1021/acs.orglett.6b01301. [DOI] [PubMed] [Google Scholar]; d Zhou S.; Song T.; Chen H.; Liu Z.; Shen H.; Li C. Catalytic Radical Trifluoromethylalkynylation of Unactivated Alkenes. Org. Lett. 2017, 19, 698–701. 10.1021/acs.orglett.6b03870. [DOI] [PubMed] [Google Scholar]; e Shirke R. P.; Ramasastry S. V. Organocatalytic β-Azidation of Enones Initiated by an Electron Donor-Acceptor Complex. Org. Lett. 2017, 19, 5482–5485. 10.1021/acs.orglett.7b02861. [DOI] [PubMed] [Google Scholar]
- Kandukuri S. R.; Bahamonde A.; Chatterjee I.; Jurberg I. D.; Escudero-Adán E. C.; Melchiorre P. X-Ray Characterization of an Electron Donor-Acceptor Complex Drives the Photochemical Alkylation of Indoles. Angew. Chem., Int. Ed. 2015, 54, 1485–1489. 10.1002/anie.201409529. [DOI] [PubMed] [Google Scholar]
- Alvarez S. A Cartography of the van der Waals territories. Dalton Trans. 2013, 42, 8617–8636. 10.1039/c3dt50599e. [DOI] [PubMed] [Google Scholar]
- a Marzo L.; Wang S.; König B. Visible-Light-Mediated Radical Arylation of Anilines with Acceptor-Substituted (Hetero)aryl Halides. Org. Lett. 2017, 19, 5976–5979. 10.1021/acs.orglett.7b03001. [DOI] [PubMed] [Google Scholar]; b Hsu C.-W.; Sundén H. α-Aminoalkyl Radical Addition to Maleimides via Electron Donor-Acceptor Complexes. Org. Lett. 2018, 20, 2051–2054. 10.1021/acs.orglett.8b00597. [DOI] [PubMed] [Google Scholar]
- Zhang H.-H.; Yu S. Visible-Light-Induced Radical Acylation of Imines with α-Ketoacids Enabled by Electron-Donor-Acceptor Complexes. Org. Lett. 2019, 21, 3711–3715. 10.1021/acs.orglett.9b01169. [DOI] [PubMed] [Google Scholar]
- Xie S.; Li D.; Huang H.; Zhang F.; Chen Y. Intermolecular Radical Addition to Ketoacids Enabled by Boron Activation. J. Am. Chem. Soc. 2019, 141, 16237–16242. 10.1021/jacs.9b09099. [DOI] [PubMed] [Google Scholar]
- a Salamone M.; Bietti M. Reaction Pathways of Alkoxyl Radicals. The Role of Solvent Effects on C-C Bond Fragmentation and Hydrogen Atom Transfer Reactions. Synlett 2014, 25, 1803–1816. 10.1055/s-0033-1341280. [DOI] [Google Scholar]; b Hartung J.; Gottwald T.; Špehar K. Selectivity in the Chemistry of Oxygen-Centered Radicals - The Formation of Carbon-Oxygen Bonds. Synthesis 2002, 1469–1498. 10.1055/s-2002-33335. [DOI] [Google Scholar]
- Nappi M.; Bergonzini G.; Melchiorre P. Metal-Free Photochemical Aromatic Perfluoroalkylation of α-Cyano Arylacetates. Angew. Chem., Int. Ed. 2014, 53, 4921–4925. 10.1002/anie.201402008. [DOI] [PubMed] [Google Scholar]
- a Guo Q.; Wang M.; Liu H.; Wang R.; Xu Z. Visible-Light-Promoted Dearomative Fluoroalkylation of β-Naphthols through Intermolecular Charge Transfer. Angew. Chem., Int. Ed. 2018, 57, 4747–4751. 10.1002/anie.201800767. [DOI] [PubMed] [Google Scholar]; b Liang K.; Li N.; Zhang Y.; Li T.; Xia C. Transition-metal-free α-arylation of oxindoles via visible-light-promoted electron transfer. Chem. Sci. 2019, 10, 3049–3053. 10.1039/C8SC05170D. [DOI] [PMC free article] [PubMed] [Google Scholar]; For other recent examples, see:; c Tang X.; Studer A. α-Perfluoroalkyl-β-alkynylation of alkenes via radical alkynyl migration. Chem. Sci. 2017, 8, 6888–6892. 10.1039/C7SC02175E. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Wang Y.; Wang J.; Li G.-X.; He G.; Chen G. Halogen-Bond-Promoted Photoactivation of Perfluoroalkyl Iodides: A Photochemical Protocol for Perfluoroalkylation Reactions. Org. Lett. 2017, 19, 1442–1445. 10.1021/acs.orglett.7b00375. [DOI] [PubMed] [Google Scholar]; e Tang X.; Studer A. Alkene 1,2-Difunctionalization by Radical Alkenyl Migration. Angew. Chem., Int. Ed. 2018, 57, 814–817. 10.1002/anie.201710397. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Liu Y.; Chen X.-L.; Sun K.; Li X.-Y.; Zeng F.-L.; Liu X.-C.; Qu L.-B.; Zhao Y.-F.; Yu B. Visible-Light Induced Radical Perfluoroalkylation/Cyclization Strategy To Access 2-Perfluoroalkylbenzothiazoles/Benzoselenazoles by EDA Complex. Org. Lett. 2019, 21, 4019–4024. 10.1021/acs.orglett.9b01175. [DOI] [PubMed] [Google Scholar]; g Rosso C.; Williams J. D.; Filippini G.; Prato M.; Kappe C. O. Visible-Light-Mediated Iodoperfluoroalkylation of Alkenes in Flow and Its Application to the Synthesis of a Key Fulvestrant Intermediate. Org. Lett. 2019, 21, 5341–5345. 10.1021/acs.orglett.9b01992. [DOI] [PubMed] [Google Scholar]
- a Liu B.; Lim C.-H.; Miyake G. M. Visible-Light-Promoted C-S Cross Coupling via Intermolecular Charge Transfer. J. Am. Chem. Soc. 2017, 139, 13616–13619. 10.1021/jacs.7b07390. [DOI] [PMC free article] [PubMed] [Google Scholar]; For related studies, see:; b Liu B.; Lim C.-H.; Miyake G. M. Light-Driven Intermolecular Charge Transfer Induced Reactivity of Ethynylbenziodoxol(on)e and Phenols. J. Am. Chem. Soc. 2018, 140, 12829–12835. 10.1021/jacs.8b05870. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Li G.; Yan Q.; Gan Z.; Li Q.; Dou X.; Yang D. Photocatalyst-Free Visible-Light-Promoted C(sp2)-S Coupling: A Strategy for the Preparation of S-Aryl Dithiocarbamates. Org. Lett. 2019, 21, 7938–7942. 10.1021/acs.orglett.9b02921. [DOI] [PubMed] [Google Scholar]
- a Kwong F. Y.; Buchwald S. L. A general, Efficient and Inexpensive Catalyst System for the Coupling of Aryl Iodides and Thiols. Org. Lett. 2002, 4, 3517–3520. 10.1021/ol0266673. [DOI] [PubMed] [Google Scholar]; b Murata M.; Buchwald S. L. A general and efficient method for the palladium-catalyzed cross-coupling of thiols and secondary phosphines. Tetrahedron 2004, 60, 7397–7403. 10.1016/j.tet.2004.05.044. [DOI] [Google Scholar]
- Sun X.; Wang W.; Li Y.; Ma J.; Yu S. Halogen-Bond-Promoted Double Radical Isocyanide Insertion under Visible-Light Irradiation: Synthesis of 2-Fluoroalkylated Quinoxalines. Org. Lett. 2016, 18, 4638–4641. 10.1021/acs.orglett.6b02271. [DOI] [PubMed] [Google Scholar]
- Davies J.; Booth S. G.; Essafi S.; Dryfe R. A. W.; Leonori D. Visible-Light-Mediated Generation of Nitrogen-Centered Radicals: Metal-Free Hydroimination and Iminohydroxylation Cyclization Reactions. Angew. Chem., Int. Ed. 2015, 54, 14017–14021. 10.1002/anie.201507641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Forrester A. R.; Gill M.; Sadd J. S.; Thomson R. H. Iminyls. Part 2. Intramolecular Aromatic Substitution by Iminyls. A New Route to Phenanthridines and Quinolines. J. Chem. Soc., Perkin Trans. 1 1979, 612–615. 10.1039/p19790000612. [DOI] [Google Scholar]; b Mikami T.; Narasaka K. Photochemical Transformation of γ,δ-Unsaturated Ketone O-(p-Cyanophenyl)oximes to 3,4-Dihydro-2H-pyrrole Derivatives. Chem. Lett. 2000, 29, 338–339. 10.1246/cl.2000.338. [DOI] [Google Scholar]
- a Li J.; Zhang P.; Jiang M.; Yang H.; Zhao Y.; Fu H. Visible Light as a Sole Requirement for Intramolecular C(sp3)-H Imination. Org. Lett. 2017, 19, 1994–1997. 10.1021/acs.orglett.7b00533. [DOI] [PubMed] [Google Scholar]; b Li Y.; Mao R.; Wu J. N-Radical Initiated Aminosulfonylation of Unactivated C(sp3)-H Bond through Insertion of Sulfur Dioxide. Org. Lett. 2017, 19, 4472–4475. 10.1021/acs.orglett.7b02010. [DOI] [PubMed] [Google Scholar]; c Sun J.; He Y.; An X.-D.; Zhang X.; Yu L.; Yu S. Visible-light-induced iminyl radical formation via electron-donor-acceptor complexes: a photocatalyst-free approach to phenanthridines and quinolines. Org. Chem. Front. 2018, 5, 977–981. 10.1039/C7QO00992E. [DOI] [Google Scholar]; d Buglioni L.; Mastandrea M. M.; Frontera A.; Pericàs M. A. Anion-π Interactions in Light-Induced Reactions: Role in the Amidation of (Hetero)aromatic Systems with Activated N-Aryloxyamides. Chem. - Eur. J. 2019, 25, 11785–11790. 10.1002/chem.201903055. [DOI] [PubMed] [Google Scholar]
- a Zhang J.; Li Y.; Xu R.; Chen Y. Donor-Acceptor Complex Enables Alkoxyl Radical Generation for Metal-Free C(sp3)- C(sp3) Cleavage and Allylation/Alkenylation. Angew. Chem., Int. Ed. 2017, 56, 12619–12623. 10.1002/anie.201707171. [DOI] [PubMed] [Google Scholar]; b Li Y.; Zhang J.; Li D.; Chen Y. Metal-Free C(sp3)-H Allylation via Aryl Carboxyl Radicals Enabled by Donor-Acceptor Complex. Org. Lett. 2018, 20, 3296–3299. 10.1021/acs.orglett.8b01172. [DOI] [PubMed] [Google Scholar]
- Wu J.; Grant P. S.; Li X.; Noble A.; Aggarwal V. K. Catalyst-Free Deaminative Functionalizations of Primary Amines by Photoinduced Single-Electron Transfer. Angew. Chem., Int. Ed. 2019, 58, 5697–5701. 10.1002/anie.201814452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Katritzky A. R.; de Ville G. Z.; Patel R. C. The SRN2 Mechanism of Nucleophilic Substitution. Tetrahedron Lett. 1980, 21, 1723–1726. 10.1016/S0040-4039(00)77820-8. [DOI] [Google Scholar]; b Katritzky A. R.; de Ville G. Z.; Patel R. C. Carbon-Alkylation of Simple Nitronate Anions by N-Substituted Pyridiniums. Tetrahedron 1981, 37, 25–30. 10.1016/0040-4020(81)85037-5. [DOI] [Google Scholar]; c Katritzky A. R.; Chen J.-L.; Marson C. M.; Maia A.; Kashmiri M. A. The Non-Chain Radicaloid C-Alkylation of Nitronate Anions: Further Evidence for the Mechanism. Tetrahedron 1986, 42, 101–108. 10.1016/S0040-4020(01)87407-X. [DOI] [Google Scholar]
- Yang M.; Cao T.; Xu T.; Liao S. Visible-Light-Induced Deaminative Thioesterification of Amino Acid Derived Katritzky Salts via Electron Donor-Acceptor Complex Formation. Org. Lett. 2019, 21, 8673–8678. 10.1021/acs.orglett.9b03284. [DOI] [PubMed] [Google Scholar]
- James M. J.; Strieth-Kalthoff F.; Sandfort F.; Klauck F. J. R.; Wagener F.; Glorius F. Visible-Light-Mediated Charge Transfer Enables C-C Bond Formation with Traceless Acceptor Groups. Chem. - Eur. J. 2019, 25, 8240–8244. 10.1002/chem.201901397. [DOI] [PubMed] [Google Scholar]
- a Fawcett A.; Pradeilles J.; Wang Y.; Mutsuga T.; Myers E. L.; Aggarwal V. K. Photoinduced decarboxylative borylation of carboxylic acids. Science 2017, 357, 283–286. 10.1126/science.aan3679. [DOI] [PubMed] [Google Scholar]; b Wu J.; He L.; Noble A.; Aggarwal V. K. Photoinduced Deaminative Borylation of Alkylamines. J. Am. Chem. Soc. 2018, 140, 10700–10704. 10.1021/jacs.8b07103. [DOI] [PubMed] [Google Scholar]; c Wu J.; Bär R. M.; Guo L.; Noble A.; Aggarwal V. K. Photoinduced Deoxygenative Borylations of Aliphatic Alcohols. Angew. Chem., Int. Ed. 2019, 58, 18830–18834. For a related study, see: 10.1002/anie.201910051. [DOI] [PubMed] [Google Scholar]; d Sandfort F.; Strieth-Kalthoff F.; Klauck F. J. R.; James M. J.; Glorius F. Deaminative Borylation of Aliphatic Amines Enabled by Visible Light Excitation of an Electron Donor-Acceptor Complex. Chem. - Eur. J. 2018, 24, 17210–17214. 10.1002/chem.201804246. [DOI] [PubMed] [Google Scholar]
- Spell M. L.; Deveaux K.; Bresnahan C. G.; Bernard B. L.; Sheffield W.; Kumar R.; Ragains J. R. A Visible-Light-Promoted O-Glycosylation with a Thioglycoside Donor. Angew. Chem., Int. Ed. 2016, 55, 6515–6519. 10.1002/anie.201601566. [DOI] [PubMed] [Google Scholar]
- For additional examples using Umemoto’s reagent in EDA complex activation, see:; a Liu Y.-Y.; Yu X.-Y.; Chen J.-R.; Qiao M.-M.; Qi X.; Shi D.-Q.; Xiao W.-J. Visible-Light-Driven Aza-ortho-quinone Methide Generation for the Synthesis of Indoles in a Multicomponent Reaction. Angew. Chem., Int. Ed. 2017, 56, 9527–9531. 10.1002/anie.201704690. [DOI] [PubMed] [Google Scholar]; b Zhu M.; Zhou K.; Zhang X.; You S.-L. Visible-Light-Promoted Cascade Alkene Trifluoromethylation and Dearomatization of Indole Derivatives via Intermolecular Charge Transfer. Org. Lett. 2018, 20, 4379–4383. 10.1021/acs.orglett.8b01899. [DOI] [PubMed] [Google Scholar]
- Arceo E.; Bahamonde A.; Bergonzini G.; Melchiorre P. Enantioselective direct α-alkylation of cyclic ketones by means of photo-organocatalysis. Chem. Sci. 2014, 5, 2438–2442. 10.1039/c4sc00315b. [DOI] [Google Scholar]
- Shirakawa S.; Maruoka K. Recent Developments in Asymmetric Phase-Transfer Reactions. Angew. Chem., Int. Ed. 2013, 52, 4312–4348. 10.1002/anie.201206835. [DOI] [PubMed] [Google Scholar]
- Woźniak Ł.; Murphy J. J.; Melchiorre P. Photo-organocatalytic Enantioselective Perfluoroalkylation of β-Ketoesters. J. Am. Chem. Soc. 2015, 137, 5678–5681. 10.1021/jacs.5b03243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morack T.; Mück-Lichtenfeld C.; Gilmour R. Bioinspired Radical Stetter Reaction: Radical Umpolung Enabled by Ion-Pair Photocatalysis. Angew. Chem., Int. Ed. 2019, 58, 1208–1212. 10.1002/anie.201809601. [DOI] [PubMed] [Google Scholar]
- Ahrendt K. A.; Borths C. J.; MacMillan D. W. C. New Strategies for Organic Synthesis: the First Highly Enantioselective Organocatalytic Diels-Alder Reaction. J. Am. Chem. Soc. 2000, 122, 4243–4244. 10.1021/ja000092s. [DOI] [Google Scholar]
- Lelais G.; MacMillan D. W. C. Modern strategies in organic catalysis: the advent and development of iminium activation. Aldrichimica Acta 2006, 39, 79–87. [Google Scholar]
- Cao Z.-Y.; Ghosh T.; Melchiorre P. Enantioselective radical conjugate additions driven by a photoactive intramolecular iminium-ion-based EDA complex. Nat. Commun. 2018, 9, 3274. 10.1038/s41467-018-05375-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Murphy J. J.; Bastida D.; Paria S.; Fagnoni M.; Melchiorre P. Asymmetric catalytic formation of quaternary carbons by iminium ion trapping of radicals. Nature 2016, 532, 218–222. 10.1038/nature17438. [DOI] [PubMed] [Google Scholar]; b Bahamonde A.; Murphy J. J.; Savarese M.; Bremond E.; Cavalli A.; Melchiorre P. Studies on the Enantioselective Iminium Ion Trapping of Radicals Triggered by an Electron-Relay Mechanism. J. Am. Chem. Soc. 2017, 139, 4559–4567. 10.1021/jacs.7b01446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quasdorf K. W.; Overman L. E. Catalytic enantioselective synthesis of quaternary carbon stereocentres. Nature 2014, 516, 181–191. 10.1038/nature14007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For an additional application of an intramolecular EDA complex, see:; Ho H. E.; Pagano A.; Rossi-Ashton J. A.; Donald J. R.; Epton R. G.; Churchill J. C.; James M. J.; O’Brien P.; Taylor R. J. K.; Unsworth W. P. Visible-light-induced intramolecular charge transfer in the radical spirocyclisation of indole-tethered ynones. Chem. Sci. 2020, 11, 1353. 10.1039/C9SC05311E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu M.-C.; Shang R.; Zhao B.; Wang B.; Fu Y. Photocatalytic decarboxylative alkylations mediated by triphenylphosphine and sodium iodide. Science 2019, 363, 1429–1434. 10.1126/science.aav3200. [DOI] [PubMed] [Google Scholar]
- Symons M. C. R.; Petersen R. L. Halide ion capture by radicals. Electron spin resonance spectra of R3P·—hal and R2S·—hal σ* radicals. J. Chem. Soc., Faraday Trans. 2 1979, 75, 210–219. 10.1039/F29797500210. [DOI] [Google Scholar]
- Bosque I.; Bach T. 3-Acetoxyquinuclidine as Catalyst in Electron Donor-Acceptor Complex-Mediated Reactions Triggered by Visible Light. ACS Catal. 2019, 9, 9103–9109. 10.1021/acscatal.9b01039. [DOI] [Google Scholar]
- Griller D.; Howard J. A.; Marriott P. R.; Scaiano J. C. Absolute Rate Constants for the Reactions of Tert-butoxyl, Tert-butylperoxyl, and Benzophenone Triplet with Amines: the Importance of a Stereoelectronic Effect. J. Am. Chem. Soc. 1981, 103, 619–623. 10.1021/ja00393a020. [DOI] [Google Scholar]
- Quint V.; Morlet-Savary F.; Lohier J.-F.; Lalevée J.; Gaumont A.-C.; Lakhdar S. Metal-Free, Visible Light-Photocatalyzed Synthesis of Benzo[b]phosphole Oxides: Synthetic and Mechanistic Investigations. J. Am. Chem. Soc. 2016, 138, 7436–7441. 10.1021/jacs.6b04069. [DOI] [PubMed] [Google Scholar]
- a Willner I.; Eichen Y.; Rabinovitz M.; Hoffman R.; Cohen S. Structure and Thermodynamic and Kinetic Properties of Eosin-Bipyridinium Complexes. J. Am. Chem. Soc. 1992, 114, 637–644. 10.1021/ja00028a033. [DOI] [Google Scholar]; b Willner I.; Marx S.; Eichen Y. Photoswitchable Association of an Azobenzene-Bipyridinium Diad to Eosin: Photostimulated “On-Off” Guest Binding. Angew. Chem., Int. Ed. Engl. 1992, 31, 1243–1244. 10.1002/anie.199212431. [DOI] [Google Scholar]
- Prier C. K.; Arnold F. H. Chemomimetic Biocatalysis: Exploiting the Synthetic Potential of Cofactor-Dependent Enzymes to Create New Catalysts. J. Am. Chem. Soc. 2015, 137, 13992–14006. 10.1021/jacs.5b09348. [DOI] [PubMed] [Google Scholar]
- a Maciá-Agulló J. A.; Corma A.; Garcia H. Photobiocatalysis: The Power of Combining Photocatalysis and Enzymes. Chem. - Eur. J. 2015, 21, 10940–10959. 10.1002/chem.201406437. [DOI] [PubMed] [Google Scholar]; b Schmermund L.; Jurkaš V.; Özgen F. F.; Barone G. D.; Büchsenschütz H. C.; Winkler C. K.; Schmidt S.; Kourist R.; Kroutil W. Photo-Biocatalysis: Biotransformations in the Presence of Light. ACS Catal. 2019, 9, 4115–4144. 10.1021/acscatal.9b00656. [DOI] [Google Scholar]
- a Arnold F. H. Directed Evolution: Bringing New Chemistry to Life. Angew. Chem., Int. Ed. 2018, 57, 4143–4148. 10.1002/anie.201708408. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Hyster T. K.; Ward T. R. Genetic Optimization of Metalloenzymes: Enhancing Enzymes for Non-Natural Reactions. Angew. Chem., Int. Ed. 2016, 55, 7344–7357. 10.1002/anie.201508816. [DOI] [PubMed] [Google Scholar]
- Emmanuel M. A.; Greenberg N. R.; Oblinsky D. G.; Hyster T. K. Accessing non-natural reactivity by irradiating nicotinamide-dependent enzymes with light. Nature 2016, 540, 414–417. 10.1038/nature20569. [DOI] [PubMed] [Google Scholar]
- Huisman G. W.; Liang J.; Krebber A. Practical chiral alcohol manufacture using ketoreductases. Curr. Opin. Chem. Biol. 2010, 14, 122–129. 10.1016/j.cbpa.2009.12.003. [DOI] [PubMed] [Google Scholar]
- Biegasiewicz K. F.; Cooper S. J.; Gao X.; Oblinsky D. G.; Kim J. H.; Garfinkle S. E.; Joyce L. A.; Sandoval B. A.; Scholes G. D.; Hyster T. K. Photoexcitation of flavoenzymes enables a stereoselective radical cyclization. Science 2019, 364, 1166–1169. 10.1126/science.aaw1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flanigan D. M.; Romanov-Michailidis F.; White N. A.; Rovis T. Organocatalytic Reactions Enabled by N-Heterocyclic Carbenes. Chem. Rev. 2015, 115, 9307–9387. 10.1021/acs.chemrev.5b00060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Taylor M. S.; Jacobsen E. N. Asymmetric Catalysis by Chiral Hydrogen-bond Donors. Angew. Chem., Int. Ed. 2006, 45, 1520–1543. 10.1002/anie.200503132. [DOI] [PubMed] [Google Scholar]; b Zhang Z.; Schreiner P. (Thio)urea organocatalysis - What can be learnt from anion recognition?. Chem. Soc. Rev. 2009, 38, 1187–1198. 10.1039/b801793j. [DOI] [PubMed] [Google Scholar]
- Bottecchia C.; Noël T. Photocatalytic Modification of Amino acids, Peptides, and Proteins. Chem. - Eur. J. 2019, 25, 26–42. 10.1002/chem.201803074. [DOI] [PMC free article] [PubMed] [Google Scholar]