

Introduction
Neurons are unique cells because of their morphology and polarity, with the cell body extending two types of processes. These are the dendrites that primarily receive signals from other cells and the surrounding environment and the axons that are long processes connecting neurons to their target cells. The longest neuronal process, the axon, can extend up to a meter in length from the cell body, in the case of some motor and sensory neurons. One of the unique challenges of the neuron is the provision of materials to distant synapses, where communication with target cells takes place. Such provision relies on the trafficking of material to and from the synapse by a complex set of intracellular trafficking events collectively referred to as fast axonal transport (FAT) (reviewed in [6]). Microtubule motor-based FAT along axons is critical to the function and health of neurons, delivering organelles, vesicles, and other cellular materials that ultimately support communication with target cells. FAT is also necessary for returning damaged material to the cell body for recycling, and for delivering neurotrophic signals received at the axon terminal to the cell body, where they can affect gene transcription (reviewed in [67]). The importance of this process is highlighted by examples of neurodegeneration following its disruption. Mutations in the genes encoding proteins involved in transport along the axon (including molecular motors, cytoskeletal tracks, and adaptor proteins) cause several neurodevelopmental and neurodegenerative diseases (reviewed in [65, 79]). The tau protein is among those implicated in affecting FAT in a group of neurodegenerative diseases known as tauopathies (reviewed in [42, 45]). In this chapter, we will discuss the relationship between tau and FAT under normal conditions and how disruptions to this process may be a toxic mechanism in multiple neurodegenerative diseases.
Tau Protein and Disease
From its initial discovery in 1975, the tau protein was closely associated with microtubules and is somewhat enriched in axons [5, 105]. It is primarily expressed in the brain with higher levels of mRNA and protein in the cortex and hippocampus than white matter and the cerebellum [95]. The protein exists as six major isoforms in the adult human brain, generated through alternative mRNA splicing. The isoforms differ in their inclusion of 2, 1 or 0 of exons 2 and 3 in the N-terminus and their inclusion of exon 10 which determines whether the protein contains either 4 or 3 microtubule-binding repeat regions (+exon 10 = 4R tau, −exon 10 = 3R tau; Fig. 7.1a). The microtubule-binding regions in the C-terminal half of the protein are positively-charged, while the N-terminus is enriched in acidic amino acids leaving the protein isoforms with a relatively low net charge. Directly upstream of the microtubule-binding domains is a proline-rich region which includes several phosphorylation sites and PXXP motifs that may bind to SH3 domains (Fig. 7.1b). Other functional domains include a phosphatase-activating domain (PAD, discussed in greater detail below) at the extreme N-terminus [40]. Structural studies indicate that tau is a highly dynamic protein, capable of multiple conformations that may underlie its diverse role in multiple cellular functions. The protein contains regions of secondary structure and acquires global conformations such as the “paperclip”, which involves the N- and C-termini interacting with each other and the C-terminus interacting with the microtubule-binding repeat regions [38, 69].
Fig. 7.1.
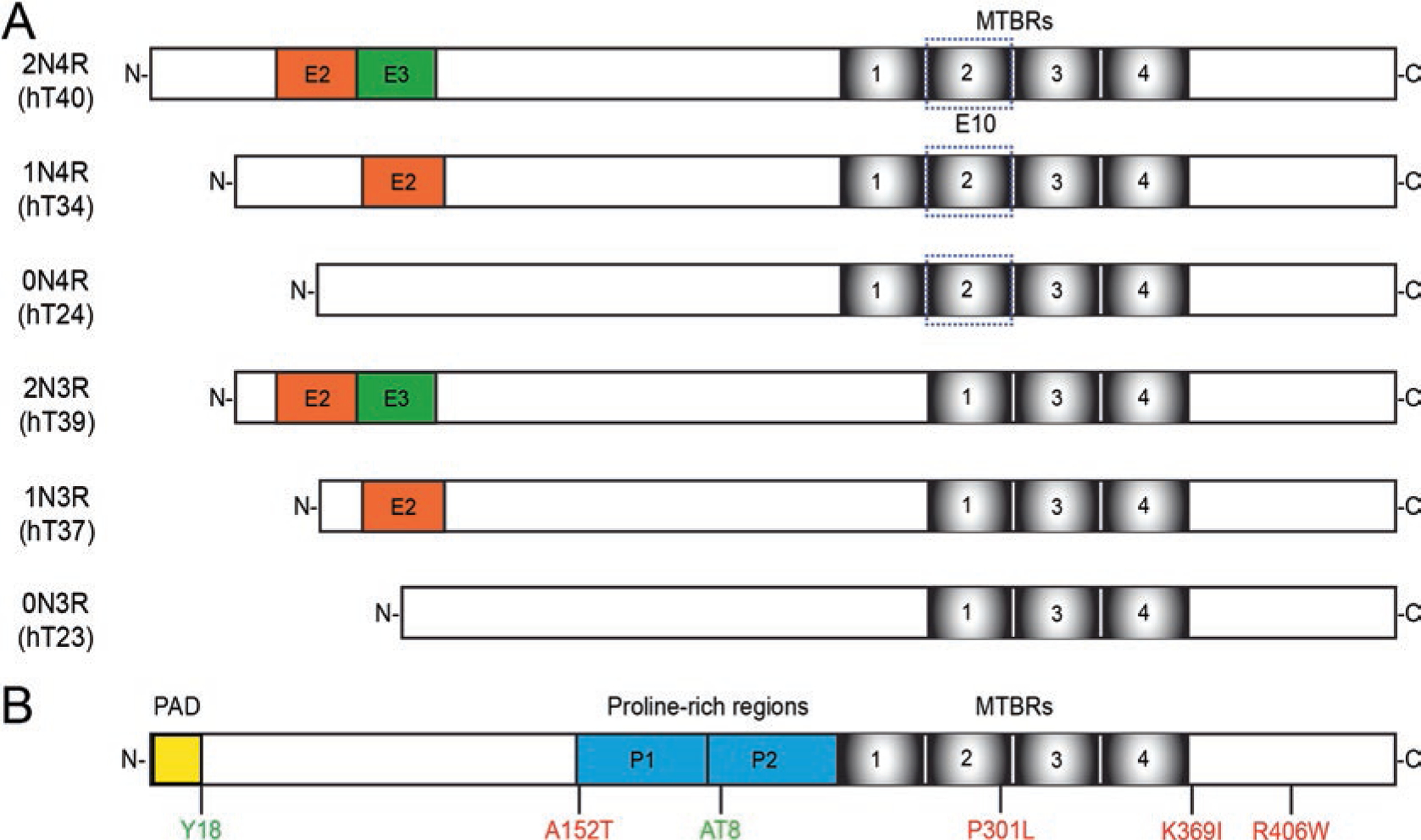
Tau isoforms and selected modifications. (a) In the adult human brain tau primarily exists as six isoforms generated through alternative mRNA splicing. The isoforms differ based on the inclusion of exons 2 and 3 in the N-terminus of the protein (2 N, 1 N, or 0 N) and exon 10. Exon 10 contains the second of four potential microtubule-binding repeat regions. Isoforms are referred to as 3R or 4R based on the number of repeat regions they contain. (b) Modifications to the protein can affect its function and induce dysfunction in disease. Some selected modifications discussed here include phosphorylation sites (green) at tyrosine 18 and the AT8 sites (serine 202 and threonine 205). FTDP-17 mutations can lead to inherited early-onset frontotemporal dementias (reviewed in [53]). Several of these mutations have been linked to FAT dysfunction including A152T, P301L, K369I, and R406W (red). Functional domains associated with transport include the phosphatase activating domain (yellow), a motif that is conformationally displayed in disease-related forms of tau and linked to changes in signaling pathways that regulate transport. The tau molecule contains many phosphorylation sites throughout the sequence, some of which are found in healthy tissues and others are associated with tau pathology. (Reviewed in [84])
Approximately a decade after the discovery of tau, a series of studies demonstrated that tau was the primary component of the hallmark tangle pathology in Alzheimer’s disease (AD) and that tau was heavily phosphorylated in these inclusions [29, 30, 107]. Interest in the tau protein has continued to grow as a central role in AD and several other neurodegenerative disorders became apparent. The discovery of inherited mutations within the tau gene that lead to early-onset frontotemporal dementias (FTDs) was the first line of evidence demonstrating that pathological tau is sufficient to cause neurodegenerative disease [35]. In these diseases, tau undergoes a number of pathological post-translational modifications (reviewed in [55, 84]) and forms a variety of morphologically different aggregates that range from small oligomers to much larger filamentous inclusions (reviewed in [46]). Pathological tau is highly modified compared to its normal state with some of the most prominent modifications including increased levels of phosphorylation, changes in overall conformation, and truncation of the protein, among many others [7, 15, 29, 31, 34]. These modified versions of tau accumulate in the somatodendritic and axonal compartments of neurons and often display impaired microtubule binding [8, 12, 28, 82], fueling the notion that reduced stability of the microtubule cytoskeleton represents a critical pathogenic event in tauopathies. However, almost 50 years after its discovery, the exact mechanisms of tau toxicity continue to be debated.
Some potential clues may be found by elucidating tau’s normal cellular functions as well as examining the neurodegenerative phenotypes associated with tauopathies. As mentioned above, tau was traditionally linked to microtubule-based functions involving stabilization and dynamics. However, the protein’s putative functions have expanded to include regulation of FAT, scaffolding for phosphotransferases, synaptic plasticity, neuronal activity, actin bundling, and mediating interactions between cellular components (reviewed in [45, 103]). These functions may be reliant on the local tau isoform composition as well as specific sets of regulatory post-translational modifications. Thus, many, if not all, of these potential functions of tau are likely affected in disease as the protein undergoes abnormal modification, misfolding and aggregation.
Axonal Transport
Projection neurons affected in tauopathies face a unique challenge in maintaining their health and functional abilities due to their cellular architecture. On one end of a neuron is the somatodendritic compartment, the primary location of protein synthesis and degradation, and on the other end are the synapses where inter-neuronal communication takes place. The transport of material in both directions is therefore vital to the cell and defects in this process contribute to a variety of neurodegenerative disorders (reviewed in [9]).
Transport in the axon occurs along the axonal microtubules [49] while actin microfilaments and neurofilaments have roles that are more structural; although microfilaments are important in short distance movements such as secretion and neurotransmitter release. Two major molecular motor families are responsible for bidirectional transport along microtubules in the axon, kinesins and dyneins (Fig. 7.2). The kinesin superfamily is organized into 14 families, from which 46 kinesins are expressed in the human brain (reviewed in [59]). Conventional kinesin, or kinesin-1, is the most abundant member of the kinesin superfamily and exists as a heterotetramer composed of two heavy chains (involved in ATP and microtubule binding and movement) and two light chains (involved in cargo binding) that produce plus-end directed movement of cargoes toward the axon terminal [20], a process known as anterograde fast axonal transport (aFAT) (reviewed in [67]). Other members of the kinesin superfamily, kinesin-2, kinesin-3 and kinesin-4 may also be involved in a subset of aFAT [75, 86], but the bulk of trafficking appears to be mediated by conventional kinesin [3, 10, 88]. Cytoplasmic dynein is a large multisubunit (two proteins each of a heavy chain, intermediate chain, light intermediate chain, and three light chains) motor complex responsible for minus-end directed movement, or retrograde FAT (rFAT) (reviewed in [76]). In contrast to the FAT of membrane-bound organelles, other cytoplasmic and cytoskeletal proteins move through slow axonal transport, a process that likely involves kinesin-1 and dynein but is not fully understood yet ([51, 98] and reviewed in [78]).
Fig. 7.2.
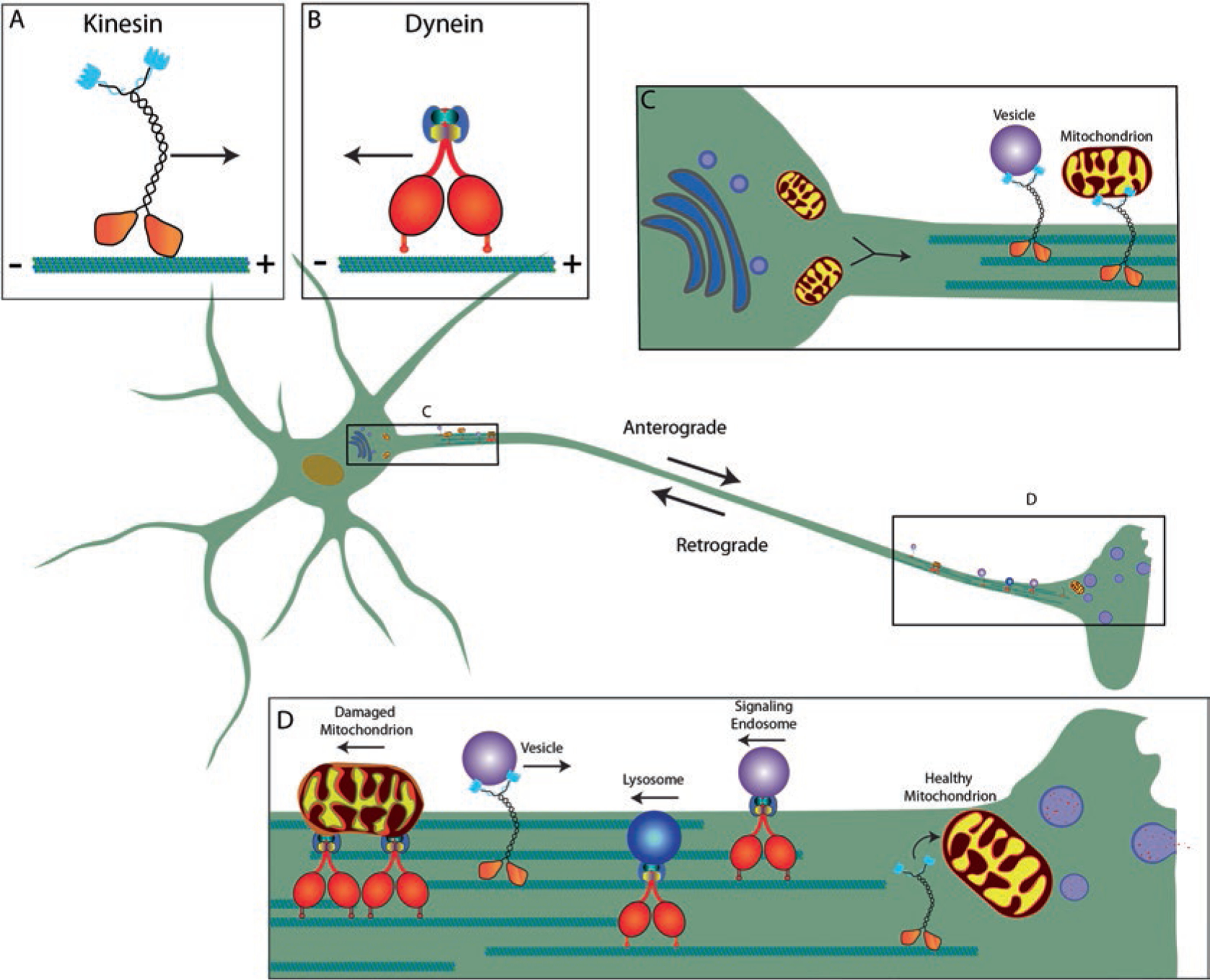
Neurons depend on robust microtubule-based transport in axons. A healthy, functional neuron is dependent on the molecular motor complexes kinesin (a) and dynein (b), whose roles are to transport material along microtubules in the plus- (anterograde) or minus-end (retrograde) direction, respectively. Materials synthesized in the soma (e.g., cytoskeletal components, mitochondria and membrane-bound organelles) rely on kinesin for their delivery to the correct axonal compartment (c). Kinesin-driven anterograde transport is necessary for the delivery of synaptic components, including mitochondria and vesicles, to the axon terminal where they will aid in cell signaling as well as delivery of channels to axon to support propagations of the action potential. Dynein-driven retrograde transport is necessary for the transport of signaling endosomes and material undergoing breakdown and recycling, like damaged mitochondria, multivesicular bodies and lysosomes (d) back to the neuronal soma
Normal FAT is regulated through a complicated system involving the composition of different motor protein complexes and regulatory post-translational modifications of the motor complexes (e.g. phosphorylation) (reviewed in [9, 67]). Several studies have suggested that variation in kinesin-1 motor protein subunits can lead to specificity in the cargoes being transported [20, 90, 92]. Phosphorylation is the best studied post-translational modification in the context of regulating multisubunit motor complexes. Studies in various experimental systems, most notably the isolated squid axoplasm preparation [44, 85], revealed that signaling pathways involving several kinases or phosphatases can tightly regulate FAT by inhibiting binding to microtubules or promoting cargo dissociation [19, 61, 62]. In fact, several regulatory kinase and phosphatase pathways for FAT are disrupted in tauopathies and other neurodegenerative diseases providing another potential link between FAT, loss of axonal connectivity, and neuronal degeneration (reviewed in [42, 65]).
Axonal Degeneration in Tauopathies
There are 27 different tauopathies described to date including Alzheimer’s disease (AD), chronic traumatic encephalopathy (CTE), Pick’s disease (PiD), progressive supranuclear palsy (PSP), frontotemporal dementia with parkinsonism linked to chromosome 17 (FTDP-17), and corticobasal degeneration (CBD) (reviewed in [53]). Tauopathies typically display pathological features consistent with a “dying back” pattern of neuronal degeneration. These include dystrophic axons and spheroids, as well as evidence of disrupted FAT (Fig. 7.3) (reviewed in [42, 45]). Accordingly, synaptic dysfunction and loss also occur very early in disease and synaptic loss correlates closely with cognitive deficits in AD and other tauopathies [21]. Studies using post-mortem human tissue samples suggest that tau inclusions appear as neuropil threads or dystrophic neurites early during the progressive accumulation of tau pathology in AD brains [25, 47]. Advanced brain imaging studies confirm degeneration of white matter containing axonal projections during the mild cognitive impairment stage, prior to onset of AD, and continued degeneration in other regions with disease progression. Tau pathology and degeneration are also prominent features of other tauopathies. In PSP, pathological tau is observed in neuropil threads and in subcortical white matter along with dystrophic axons and spheroids indicative of FAT disruptions and degeneration in the same regions [2, 33, 36, 70]. Tau-positive neuropil threads are also observed in CBD and closely associated with white matter degeneration detected by advanced imaging [36, 111]. Pick bodies are the defining feature of PiD but tau pathology can also be detected in axonal terminals along with neuritic threads and axonal spheroids in mossy fibers and cerebellar white matter [74]. Again, this is associated with degeneration of white matter regions that can be quite severe [108]. CTE brains present with neuropil threads and axonal atrophy within subcortical white matter and transported proteins accumulate in axons after traumatic brain injuries [48, 56, 99].
Fig. 7.3.
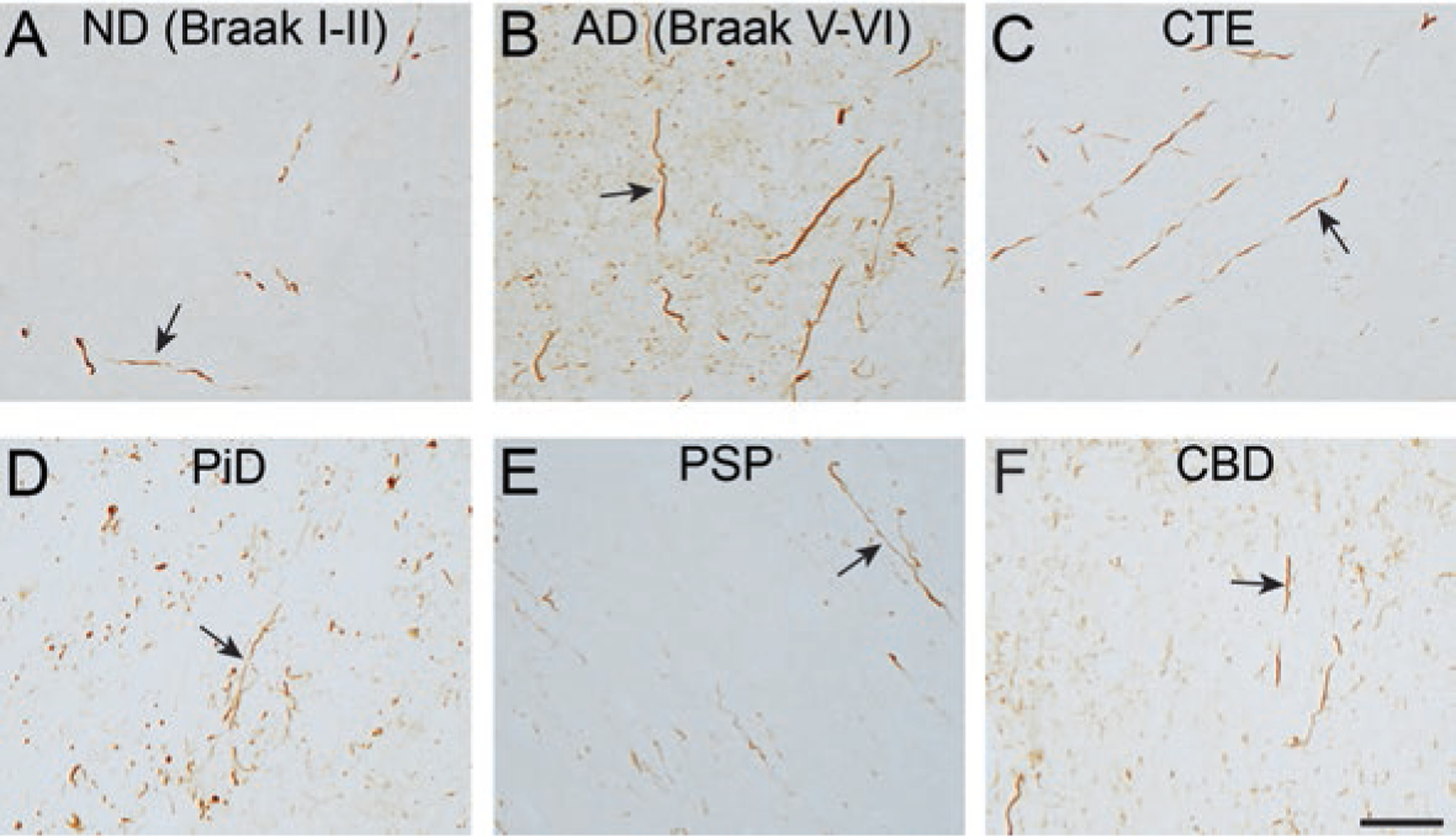
Dystrophic axons containing tau pathology is a prominent feature in multiple tauopathies. The TNT1 antibody detects exposure of the phosphatase activating domain (PAD). (a, b) TNT1 pathology-containing axons are observed in the subcortical white matter in non-demented aged patients with early stages of tau deposition (a; ND (Braak stage I-II)) and robust TNT1 axonal pathology is seen in severe Alzheimer’s disease brains (b; AD (Braak stage V-VI). C-F) Axonal tau pathology in the subcortical white matter displays PAD exposure (i.e. TNT1 reactivity) in chronic traumatic encephalopathy (c; CTE), Pick’s disease (d; PiD), progressive supranuclear palsy (e; PSP) and corticobasal degeneration (f; CBD) as well. Scale bar is 50 μm
Together, these and other studies demonstrate that the most common tauopathies are characterized by several features that point to a significant role for axonal dysfunction that may be caused by deficits in FAT. Pathological tau modifications and axonal degeneration are closely associated with each other, beginning with the earliest stages of tauopathy pathogenesis. Axonal swellings and accumulation of vesicular organelles also suggest abnormalities in FAT. Collectively, these observations suggest that pathological forms of tau may exert toxic effects through disruption of normal processes in the axon. In fact, as discussed below, evidence from several model systems supports this hypothesis and indicates that regulation of FAT appears to be disturbed by several disease-associated pathological changes to tau.
Tau-Based Effects on Fast Axonal Transport
Tau and Regulatory Signaling Pathways for Fast Axonal Transport
Accumulating evidence clearly implicates a number of kinase and phosphatase signaling pathways in the regulation of motor complex-based transport of organelles along axons. The squid isolated axoplasm model of FAT was instrumental in several significant discoveries of the biology of FAT, including the initial discovery of conventional kinesin, the most abundant member of the kinesin superfamily [11]. In this model, the giant axons are removed and the axoplasm extruded, which allows the study of axon-autonomous molecular events including FAT [44, 85]. Video-enhanced contrast-differential interference contrast microscopy is used to visualize and measure the velocity of membrane-bound organelle cargoes undergoing FAT along microtubules [85]. Over the past several years the squid axoplasm assay also was used to elucidate a number of signaling pathways regulating aFAT and rFAT through different mechanisms. For example, activation of the p38α MAPK pathway results in inhibition of kinesin-based aFAT, while activation of CK2 or JNK3 causes inhibition of both aFAT and rFAT [61, 64, 66, 72]. The squid axoplasm preparation was particularly important in elucidating tau’s role in regulating FAT through the PP1-GSK3 signaling pathway. This pathway involves activation of PP1 and subsequent dephosphorylation of Ser9 in GSK3β, an event that results in activation of this kinase (Fig. 7.4) [62]. Active GSK3 regulates aFAT by phosphorylating kinesin light chains and causing release of conventional kinesin holoenzymes from their transported cargo [61, 97]. Importantly, several disease-relevant pathological modifications of tau were shown to disrupt FAT through alterations of this signaling cascade [40, 50].
Fig. 7.4.
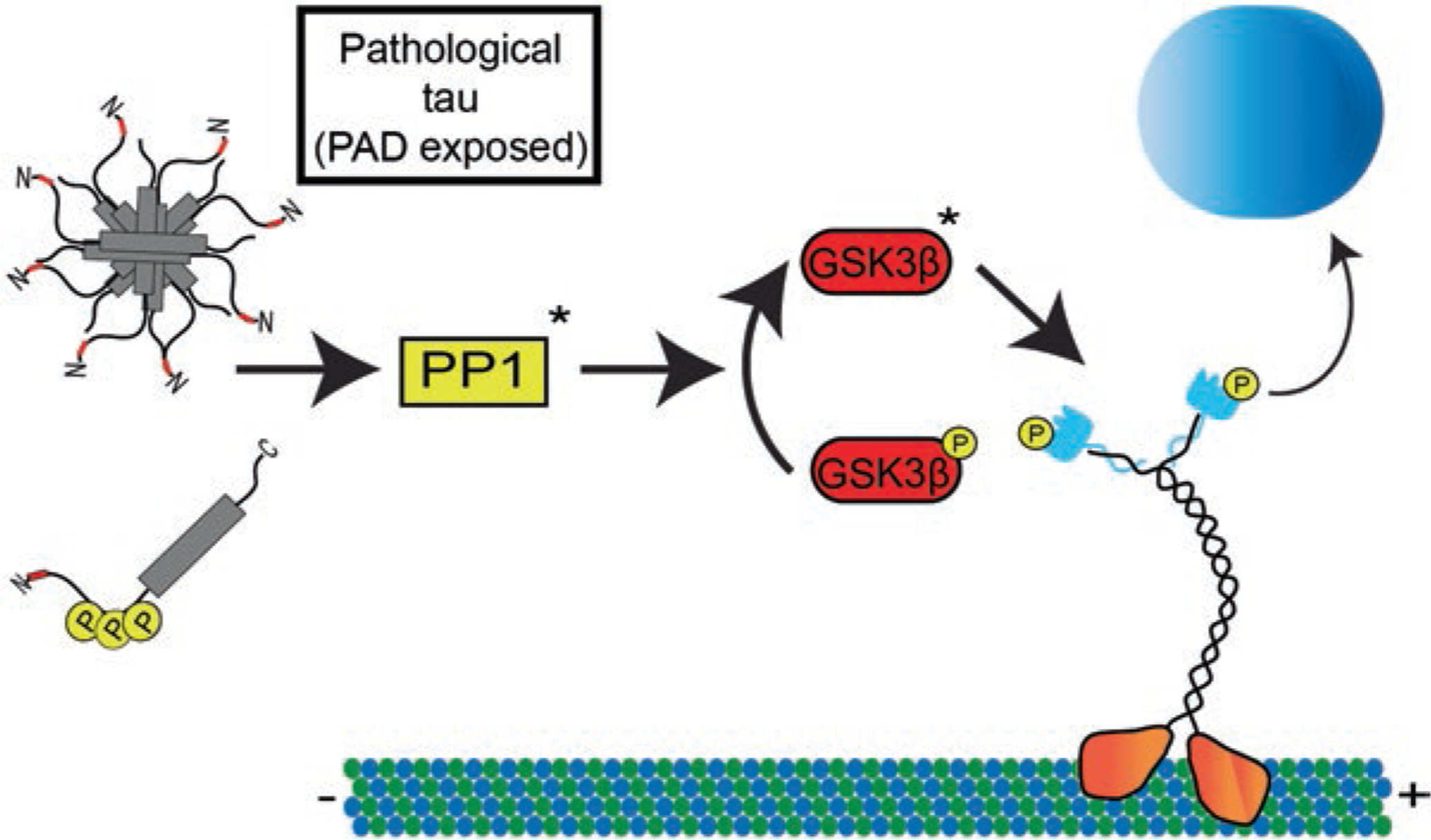
Tau alters kinesin-cargo interactions through modulation of a PP1-GSK3β signaling pathway. Tau contains a phosphatase-activating domain (PAD) within amino acids 2–18 at the extreme N-terminus of tau (shown in red). Under normal conditions this epitope is obscured allowing for normal kinesin-based transport along the microtubules. In disease conditions tau can undergo a variety of modifications including aggregation or specific phosphorylation events that can aberrantly expose the PAD epitope. In the proposed model, these forms of tau can disrupt normal kinesin-based transport by activating protein phosphatase 1 (PP1). This in turn dephosphorylates an inhibitory phosphate on GSK3β in order to activate it. GSK3β then phosphorylates kinesin light chain inducing a release of cargo and disruption of FAT
Initial studies using the squid axoplasm focused on testing effects of tau monomers on FAT, which had been proposed to compete with conventional kinesin for microtubule binding [81]. Introduction of soluble tau monomers at concentrations from 1–25 μM tau had no apparent effect on FAT in either direction, while much lower concentration of a dynein microtubule-binding domain peptide blocked both directions [63]. These studies raised questions about whether tau aggregates would affect FAT. Recombinant wild-type tau aggregates (a mixed population of oligomers and filaments) specifically inhibited aFAT but did not affect rFAT, while monomeric tau had no effect on FAT in either direction [18, 50]. The toxic effect of aggregated tau on aFAT was blocked when tau aggregates were co-perfused into isolated axoplasm with either PP1-specific or GSK3-specific inhibitors demonstrating activation of a PP1-GSK3 pathway as the underlying mechanism [40, 50]. Additional work found that tau-mediated transport toxicity was dependent upon a 17 amino acid motif in the extreme amino terminus of tau (i.e. aa 2–18), which was since termed the phosphatase-activating domain (PAD). Additional experiments showed that the PAD was sufficient to disrupt aFAT through this pathway [40, 50]. Relevant to tauopathies, the PAD epitope was normally sequestered in control tau, but exposed in pathological forms of tau. This was true for both isolated tau and tau aggregates in vitro [40] and in vivo for all tauopathies examined to date [16, 17, 43]. Collectively, this body of work provided a new framework for understanding tau-mediated toxicity. Specifically, disease-associated tau modifications (e.g. aggregation, specific phosphorylation events, etc.) alter tau structure in a way that leads to conformation-dependent exposure of PAD. Subsequently, aberrant PAD exposure is directly linked to a specific molecular pathway of toxicity (i.e. the PP1-GSK3 pathway) that results in FAT impairment. Tau contains three putative RVxF PP1-interacting domains (including one within PAD) and is able to localize the phosphatase to microtubules, which is consistent with a role for tau in regulating this pathway [24, 52]. The biological activity of the PAD suggests that physiological changes in tau could lead to tightly regulated exposure, which may allow delivery of selected organelles at specific axonal subdomains under normal conditions (reviewed in [67]).
Further studies tested the PAD-dependent mechanism of toxicity using two specific tau modifications that were predicted to facilitate PAD exposure by impairing tau paperclip folding. First, a key set of studies showed that phospho-mimicking triple phosphorylation at AT8 sites in tau (i.e. Ser199, Ser202 and Thr205) disrupted the paperclip conformation causing extension of the extreme amino-terminus [39] and subsequently PAD exposure. Consistent with enhanced PAD exposure, when applied to squid axoplasm the phosphomimetic AT8 tau monomers disrupted aFAT [40]. Second, an FTDP-17 mutation was described that causes deletion of exons 6–9 comprising the first microtubule-binding region and the proline-rich region, the domain of tau that allows flexibility for the N-terminus to fold onto the C-terminus in the paperclip conformation [77]. The prediction that the monomeric form of this protein would significantly impair aFAT was confirmed in squid axoplasm as well [40]. A role of tau-mediated FAT toxicity in AD was consistent with studies in mammalian neurons, where toxic effects of oligomeric Aβ on FAT were dependent on both tau and GSK3β [102]. Finally, PAD-dependent impairments in synaptic transmission were found in the squid giant synapse [60] and other studies demonstrate oligomeric tau (a multimeric form with increased PAD exposure) is toxic to synaptic function suggesting PAD exposure may disrupt both axonal and synaptic functions in neurons. These studies not only confirmed this novel PAD-mediated mechanism of tau toxicity but also indicated that modifications of monomeric tau can confer toxicity independently of aggregation.
Human tissue studies further confirmed this working model of tau toxicity in multiple tauopathies, demonstrating that tau species shown to impair FAT manifest early during disease progression. For example, the TNT1 and TNT2 antibodies are markers of conformation-dependent PAD exposure (Fig. 7.3) [17, 40]. These antibodies label the earliest forms of tau deposition, pre-tangles, and robustly label neuropil threads early in AD as well as the hallmark pathologies in AD and other tauopathies. Importantly, these antibodies show little to no detection of normal tau in post-mortem human brains or in non-denaturing in vitro assays [16–18, 40] (see Fig. 7.3). This pattern of staining is very similar to the robust labeling of pre-tangle inclusions and neuropil threads observed early during disease progression with the AT8 phospho-tau antibody [8, 40] and TOC1, a tau oligomer-specific antibody [18, 71, 104]. The conformation- and disease-specific labeling with these specific tau antibodies suggests that changes in the global conformation of tau leads to exposure of the PAD motif early in disease.
Activation of other pathways by mutant forms of tau have also been proposed. Transgenic mice expressing a K369I FTDP-17 mutation displayed aFAT defects that were proposed to be associated with tau interacting with JIP1 [37]. However, this suggestion was based on overexpression of mutant tau and cannot rule out a role for PAD exposure in the FAT changes. Expression of another FTDP-17 mutant tau, A152T, in C. elegans neurons led to abnormal localization of synaptic vesicles that may be due to minor disruptions in aFAT and rFAT independent of any tau aggregation or changes to microtubule binding [14, 73]. These studies do not identify a specific signaling pathway, but the conclusions may be similar to observed impairment of aFAT and rFAT by tau filaments phosphorylated at S422 [93], a disease-specific phosphorylation event that occurs early in pre-tangles and robustly labels neuropil threads in areas of the brain involved in memory and cognition [32, 94]. Such observations suggest that certain modifications in tau may expose PAD and other domain(s) that in turn activate alternative signaling pathways.
Tau May Physically Interfere with Kinesin Binding to Microtubules
Several studies reported that increased levels of tau can alter the behavior of motor proteins, while others have failed to see such effects. Overexpression of fluorescently-tagged tau in primary neurons was reported to inhibit aFAT of amyloid precursor protein (APP) and similar effects on kinesin-based transport of mitochondria were observed in other cell lines [23, 87]. In complementary in vitro studies, the presence of tau reduced the processivity of multiple motor proteins but did not affect their overall speed [96]. All of these effects were proposed to occur through tau-based hindrance of kinesin’s binding sites [54]. A similar mechanism was proposed based on tau’s interference with kinesin activity and dynein reversals on stabilized microtubules [22]. However, other studies have not supported this hypothesis. The kinesin- and dynein-binding sites on microtubules are overlapping, so addition of the dynein microtubule-binding domain effectively blocks both kinesin- and dynein-based motility [63]. In contrast, levels of tau as high as 1 tau per tubulin dimer had no effect on FAT in the squid axoplasm preparation, providing strong evidence against the notion that tau competes with kinesin for microtubule binding [63]. Also, tau binding to microtubules did not alter kinesin speed or run length and only marginally affected microtubule-binding at tau:tubulin concentration ratios much higher than physiological levels [50, 63, 81]. Studies in tau-overexpressing transgenic mice also found that increasing levels of normal tau had no effect on kinesin-based transport in vivo [110]. Efforts to explain these discordant results suggested that tau may interact differentially with microtubules under varying conditions. For example, the nucleotide-binding state of microtubules were reported to alter tau effects as tau inhibited kinesin-based transport on GDP-microtubules but not on GTP-microtubules and in an isoform-dependent manner [57]. In this case the shortest 3R tau induced a more severe phenotype than the longest 4R isoform. However, it is difficult to find a physiological role for these effects and the high levels of tau and the distinctive conditions involved are not known to exist in any disease condition. Overall, definitive evidence for direct physical interference of tau with microtubule-based motor proteins in vivo is lacking, but more complicated interactions under non-physiological conditions cannot be ruled out.
Tau Isoforms Differentially Affect Fast Axonal Transport
Other studies have also examined isoform-specific effects of tau. Expression of wild-type human 3R tau, but not 4R tau, induced an accumulation of vesicles in the axons of Drosophila larva motor neurons [80]. This effect was exacerbated by tau phosphorylation and reversed upon inhibition of GSK3β, potentially implicating it in the mechanistic pathway [68]. In human induced pluripotent stem cell (iPSC) dopaminergic cultures, shRNA-mediated knockdown of only 4R isoforms reduced the average velocity of axonal mitochondria compared to control and total tau knockdown neurons [4]. The shortest 3R tau isoform shortened kinesin run length to a greater degree than the longest 4R isoform in an in vitro assay, an effect that was dependent upon the number of motor proteins bound to the beads and was interpreted to be a result of tau reducing kinesin binding to microtubules [100]. Therefore, the local tau isoform composition was suggested to act as a regulatory factor in influencing cargo travel and final destinations within the axon, although a mechanism for creating such differential distributions of tau isoforms in cells remains to be identified.
The functional implications of having different tau isoforms remain unclear and the pathognomonic inclusions of different tauopathies are typically composed of specific isoforms. For example, AD and CTE are a mixture of 4R and 3R inclusions, while PSP and CBD are primarily 4R inclusions and PiD is primarily a 3R inclusion disease [13, 27, 83, 109]. To evaluate the effects of different tau isoforms, preparations of monomeric and aggregated tau were generated for each of the 6 isoforms found in human CNS. Although there were differences in their relative toxicity, aggregated forms of the six tau isoforms similarly impaired aFAT in the squid axoplasm preparation, suggesting the PAD-dependent PP1-GSK3 mechanism of toxicity may be a common element among tauopathies independent of the tau isoforms that comprise the pathology [18]. Further work is required to fully appreciate the normal and pathological functions of each tau isoform.
Other Mechanisms of Fast Axonal Transport Regulation by Tau
Tau may also alter FAT through direct effects on microtubule organization. Tau, in an isoform-specific manner, may act as a microtubule-spacer that simultaneously bundles microtubules and prevents them from overcrowding which could facilitate transport under normal cellular conditions [58]. Microtubule-based effects were also examined by overexpressing or deleting the fly homologue of human tau in Drosophila melanogaster neurons [91]. Increasing tau expression levels resulted in increased pause times of vesicles being transported in aFAT and rFAT. This was associated with changes to microtubule density and axon caliber as well, which may have influenced FAT. Unlike humans, flies do not express redundant MAPs so it is difficult to know how these findings translate to mammalian neurons. Other studies have failed to see a significant effect of tau overexpression on either direction transport in mouse axons [110].
Tau may also mediate effects of other AD-related molecules. In addition to tau pathology, diseases such as AD are characterized by the accumulation of pathological amyloid-β (Aβ) peptides in plaques [106]. Treatment of primary neurons with Aβ oligomers inhibited FAT of mitochondria and TrkA and the effect was dependent upon presence of tau [101]. A cross of MAPTP301L mice, an FTDP-17 tau line, with TgCRND8, a mouse line expressing mutant amyloid precursor protein, resulted in more severe decrease of mitochondrial transport while in the tau line alone aFAT was slightly increased before decreasing with age [1, 26]. Synergistic effects of oligomeric Aβ have been noted in a variety of other studies as well. In particular, GSK3 phosphorylation of kinesin light chains requires a priming phosphorylation event [61], which is efficiently provided via activation of CK2 by oligomeric Aβ [72]. The result of activating both pathways at the same time would exacerbate inhibition of FAT in AD (reviewed in [9]).
Reinforcing the notion of tau playing a role in FAT regulation, a post-translational modification within the PAD was shown to modulate tau’s effects. Specifically, phosphorylation at Tyr18 rescued PAD-mediated aFAT impairment in the isolated squid axoplasm model [41]. Further, phospho-Tyr18 co-localized with TNT1 immunoreactive tau inclusions in human disease [41]. Thus, Tyr18 phosphorylation may represent a normal regulatory mechanism that attenuates tau’s ability to cause dissociation of kinesin from cargoes for delivery, and in pathological contexts may be neuroprotective and block the aberrant activation the PP1-GSK3 pathway caused by PAD-exposed tau species. Accordingly, surviving neurons at late AD stages display strong immunoreactivity when using an anti-phospho-Tyr18 antibody [41]. Phosphorylation at this site may affect other mechanisms of tau-mediated transport regulation. For example, pseudophosphorylation at Tyr18 rescued inhibition of kinesin-1 motility induced by the shortest 3R isoform [89].
Conclusion
Based upon the evidence presented in this chapter, pathological forms of the tau protein found in AD and other tauopathies may induce toxicity by disrupting FAT. Tau modifications, such as conformational changes and increased phosphorylation, are detected within axons very early in the course of disease progression. In addition, the initial signs of neurodegeneration typically occur with synaptic dysfunction followed by a “dying back” degeneration of axons. Given that pathological tau and neurodegeneration first occur in the axon, it is natural to ask if the protein may exert toxic effects through disruption of a critical cellular process like FAT. In fact, many pathological forms of tau were found to disrupt FAT across multiple model systems ranging from in vitro biochemical systems to squid axoplasm and mouse models. Several potential mechanisms of tau toxicity, not mutually exclusive, are proposed and supported experimentally. Given the discovery of biological activity for the PAD, it is reasonable to speculate that tau may help regulate FAT under normal conditions and exert toxicity in disease through a hypermorphic gain-of-function mechanism. Tau’s normal function may take the form of modulating signaling pathways that regulate FAT by modulating kinesin-cargo interactions and/or kinesin-microtubule interactions. Further studies will provide a better understanding of tau’s normal and pathological roles, potentially providing a specific mechanistic framework for the development of effective therapeutic strategies to treat tauopathies.
Acknowledgments
Supported by NIH grants (AG044372 and NS082730), the BrightFocus Foundation, the Secchia Family Foundation and the Rainwater Foundation.
Contributor Information
Benjamin Combs, Department of Translational Neuroscience, College of Human Medicine, Michigan State University, Grand Rapids, MI, USA.
Rebecca L. Mueller, Department of Translational Neuroscience, College of Human Medicine, Michigan State University, Grand Rapids, MI, USA Neuroscience Program, Michigan State University, East Lansing, MI, USA.
Nicholas M. Kanaan, Department of Translational Neuroscience, College of Human Medicine, Michigan State University, Grand Rapids, MI, USA Neuroscience Program, Michigan State University, East Lansing, MI, USA; Hauenstein Neuroscience Center, Mercy Health Saint Mary’s, Grand Rapids, MI, USA.
References
- 1.Adalbert R, Milde S, Durrant C, Ando K, Stygelbout V, Yilmaz Z, Gould S, Brion JP, Coleman MP. Interaction between a MAPT variant causing frontotemporal dementia and mutant APP affects axonal transport. Neurobiol Aging. 2018;68:68–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ahmed Z, Josephs KA, Gonzalez J, DelleDonne A, Dickson DW. Clinical and neuropathologic features of progressive supranuclear palsy with severe pallido-nigro-luysial degeneration and axonal dystrophy. Brain. 2008;131:460–72. [DOI] [PubMed] [Google Scholar]
- 3.Amaratunga A, Morin PJ, Kosik KS, Fine RE. Inhibition of kinesin synthesis and rapid anterograde axonal transport in vivo by an antisense oligonucleotide. J Biol Chem. 1993;268:17427–30. [PubMed] [Google Scholar]
- 4.Beevers JE, Lai MC, Collins E, Booth HDE, Zambon F, Parkkinen L, Vowles J, Cowley SA, Wade-Martins R, Caffrey TM. MAPT genetic variation and neuronal maturity Alter isoform expression affecting axonal transport in iPSC-derived dopamine neurons. Stem Cell Rep. 2017;9:587–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Binder LI, Frankfurter A, Rebhun LI. The distribution of tau in the mammalian central nervous system. J Cell Biol. 1985;101:1371–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Black MM. Axonal transport: the orderly motion of axonal structures. Methods Cell Biol. 2016;131:1–19. [DOI] [PubMed] [Google Scholar]
- 7.Braak H, Braak E. Staging of Alzheimer’s disease-related neurofibrillary changes. Neurobiol Aging. 1995;16:271–8.. discussion 278–284 [DOI] [PubMed] [Google Scholar]
- 8.Braak E, Braak H, Mandelkow EM. A sequence of cytoskeleton changes related to the formation of neurofibrillary tangles and neuropil threads. Acta Neuropathol (Berl). 1994;87:554–67. [DOI] [PubMed] [Google Scholar]
- 9.Brady ST, Morfini GA. Regulation of motor proteins, axonal transport deficits and adult-onset neurodegenerative diseases. Neurobiol Dis. 2017;105:273–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brady ST, Sperry AO. Biochemical and functional diversity of microtubule motors in the nervous system. Curr Opin Neurobiol. 1995;5:551–8. [DOI] [PubMed] [Google Scholar]
- 11.Brady ST, Lasek RJ, Allen RD. Fast axonal transport in extruded axoplasm from squid giant axon. Science. 1982;218:1129–31. [DOI] [PubMed] [Google Scholar]
- 12.Bramblett GT, Goedert M, Jakes R, Merrick SE, Trojanowski JQ, Lee VM. Abnormal tau phosphorylation at Ser396 in Alzheimer’s disease recapitulates development and contributes to reduced microtubule binding. Neuron. 1993;10:1089–99. [DOI] [PubMed] [Google Scholar]
- 13.Buee L, Delacourte A. Comparative biochemistry of tau in progressive supranuclear palsy, corticobasal degeneration, FTDP-17 and Pick’s disease. Brain Pathol. 1999;9:681–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Butler VJ, Salazar DA, Soriano-Castell D, Alves-Ferreira M, Dennissen FJA, Vohra M, Oses-Prieto JA, Li KH, Wang AL, Jing B, Li B, Groisman A, Gutierrez E, Mooney S, Burlingame AL, Ashrafi K, Mandelkow EM, Encalada SE, Kao AW. Tau/MAPT disease-associated variant A152T alters tau function and toxicity via impaired retrograde axonal transport. Hum Mol Genet. 2018; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carmel G, Mager EM, Binder LI, Kuret J. The structural basis of monoclonal antibody Alz50’s selectivity for Alzheimer’s disease pathology. J Biol Chem. 1996;271:32789–95. [DOI] [PubMed] [Google Scholar]
- 16.Combs B, Kanaan NM. Exposure of the amino terminus of tau is a pathological event in multiple Tauopathies. Am J Pathol. 2017;187:1222–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Combs B, Hamel C, Kanaan NM. Pathological conformations involving the amino terminus of tau occur early in Alzheimer’s disease and are differentially detected by monoclonal antibodies. Neurobiol Dis. 2016;94:18–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cox K, Combs B, Abdelmesih B, Morfini G, Brady ST, Kanaan NM. Analysis of isoform-specific tau aggregates suggests a common toxic mechanism involving similar pathological conformations and axonal transport inhibition. Neurobiol Aging. 2016;47:113–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.DeBerg HA, Blehm BH, Sheung J, Thompson AR, Bookwalter CS, Torabi SF, Schroer TA, Berger CL, Lu Y, Trybus KM, Selvin PR. Motor domain phosphorylation modulates kinesin-1 transport. J Biol Chem. 2013;288:32612–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.DeBoer SR, You Y, Szodorai A, Kaminska A, Pigino G, Nwabuisi E, Wang B, Estrada-Hernandez T, Kins S, Brady ST, Morfini G. Conventional kinesin holoenzymes are composed of heavy and light chain homodimers. Biochemistry. 2008;47:4535–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.DeKosky ST, Scheff SW. Synapse loss in frontal cortex biopsies in Alzheimer’s disease: correlation with cognitive severity. Ann Neurol. 1990;27:457–64. [DOI] [PubMed] [Google Scholar]
- 22.Dixit R, Ross JL, Goldman YE, Holzbaur EL. Differential regulation of dynein and kinesin motor proteins by tau. Science. 2008;319:1086–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ebneth A, Godemann R, Stamer K, Illenberger S, Trinczek B, Mandelkow E. Overexpression of tau protein inhibits kinesin-dependent trafficking of vesicles, mitochondria, and endoplasmic reticulum: implications for Alzheimer’s disease. J Cell Biol. 1998;143:777–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Egloff MP, Johnson DF, Moorhead G, Cohen PT, Cohen P, Barford D. Structural basis for the recognition of regulatory subunits by the catalytic subunit of protein phosphatase 1. EMBO J. 1997;16:1876–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ghoshal N, Garcia-Sierra F, Wuu J, Leurgans S, Bennett DA, Berry RW, Binder LI. Tau conformational changes correspond to impairments of episodic memory in mild cognitive impairment and Alzheimer’s disease. Exp Neurol. 2002;177:475–93. [DOI] [PubMed] [Google Scholar]
- 26.Gilley J, Seereeram A, Ando K, Mosely S, Andrews S, Kerschensteiner M, Misgeld T, Brion JP, Anderton B, Hanger DP, Coleman MP. Age-dependent axonal transport and locomotor changes and tau hypo-phosphorylation in a “P301L” tau knockin mouse. Neurobiol Aging. 2012;33:621 e621–15. [DOI] [PubMed] [Google Scholar]
- 27.Goedert M, Spillantini MG, Cairns NJ, Crowther RA. Tau proteins of Alzheimer paired helical filaments: abnormal phosphorylation of all six brain isoforms. Neuron. 1992;8:159–68. [DOI] [PubMed] [Google Scholar]
- 28.Gong CX, Lidsky T, Wegiel J, Zuck L, Grundke-Iqbal I, Iqbal K. Phosphorylation of microtubule-associated protein tau is regulated by protein phosphatase 2A in mammalian brain. Implications for neurofibrillary degeneration in Alzheimer’s disease. J Biol Chem. 2000;275:5535–44. [DOI] [PubMed] [Google Scholar]
- 29.Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci U S A. 1986a;83:4913–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grundke-Iqbal I, Iqbal K, Quinlan M, Tung YC, Zaidi MS, Wisniewski HM. Microtubule-associated protein tau. A component of Alzheimer paired helical filaments. J Biol Chem. 1986b;261:6084–9. [PubMed] [Google Scholar]
- 31.Guillozet-Bongaarts AL, Garcia-Sierra F, Reynolds MR, Horowitz PM, Fu Y, Wang T, Cahill ME, Bigio EH, Berry RW, Binder LI. Tau truncation during neurofibrillary tangle evolution in Alzheimer’s disease. Neurobiol Aging. 2005;26:1015–22. [DOI] [PubMed] [Google Scholar]
- 32.Guillozet-Bongaarts AL, Cahill ME, Cryns VL, Reynolds MR, Berry RW, Binder LI. Pseudophosphorylation of tau at serine 422 inhibits caspase cleavage: in vitro evidence and implications for tangle formation in vivo. J Neurochem. 2006;97:1005–14. [DOI] [PubMed] [Google Scholar]
- 33.Hauw JJ, Verny M, Delaere P, Cervera P, He Y, Duyckaerts C. Constant neurofibrillary changes in the neocortex in progressive supranuclear palsy. Basic differences with Alzheimer’s disease and aging. Neurosci Lett. 1990;119:182–6. [DOI] [PubMed] [Google Scholar]
- 34.Horowitz PM, Patterson KR, Guillozet-Bongaarts AL, Reynolds MR, Carroll CA, Weintraub ST, Bennett DA, Cryns VL, Berry RW, Binder LI. Early N-terminal changes and caspase-6 cleavage of tau in Alzheimer’s disease. J Neurosci. 2004;24:7895–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hutton M, et al. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393:702–5. [DOI] [PubMed] [Google Scholar]
- 36.Ikeda K, Akiyama H, Haga C, Kondo H, Arima K, Oda T. Argyrophilic thread-like structure in corticobasal degeneration and supranuclear palsy. Neurosci Lett. 1994;174:157–9. [DOI] [PubMed] [Google Scholar]
- 37.Ittner LM, Ke YD, Gotz J. Phosphorylated tau interacts with c-Jun N-terminal kinase-interacting protein 1 (JIP1) in Alzheimer disease. J Biol Chem. 2009;284:20909–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jeganathan S, von Bergen M, Brutlach H, Steinhoff HJ, Mandelkow E. Global hairpin folding of tau in solution. Biochemistry. 2006;45:2283–93. [DOI] [PubMed] [Google Scholar]
- 39.Jeganathan S, Hascher A, Chinnathambi S, Biernat J, Mandelkow EM, Mandelkow E. Proline-directed pseudo-phosphorylation at AT8 and PHF1 epitopes induces a compaction of the paperclip folding of tau and generates a pathological (MC-1) conformation. J Biol Chem. 2008;283:32066–76. [DOI] [PubMed] [Google Scholar]
- 40.Kanaan NM, Morfini GA, LaPointe NE, Pigino GF, Patterson KR, Song Y, Andreadis A, Fu Y, Brady ST, Binder LI. Pathogenic forms of tau inhibit kinesin-dependent axonal transport through a mechanism involving activation of axonal phosphotransferases. J Neurosci. 2011;31:9858–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kanaan NM, Morfini G, Pigino G, LaPointe NE, Andreadis A, Song Y, Leitman E, Binder LI, Brady ST. Phosphorylation in the amino terminus of tau prevents inhibition of anterograde axonal transport. Neurobiol Aging. 2012;33(826):e815–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kanaan NM, Pigino GF, Brady ST, Lazarov O, Binder LI, Morfini GA. Axonal degeneration in Alzheimer’s disease: when signaling abnormalities meet the axonal transport system. Exp Neurol. 2013;246:44–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kanaan NM, Cox K, Alvarez VE, Stein TD, Poncil S, McKee AC. Characterization of early pathological tau conformations and phosphorylation in chronic traumatic encephalopathy. J Neuropathol Exp Neurol. 2016;75:19–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kang M, Baker L, Song Y, Brady ST, Morfini G. Biochemical analysis of axon-specific phosphorylation events using isolated squid axoplasms. Methods Cell Biol. 2016;131:199–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kneynsberg A, Combs B, Christensen K, Morfini G, Kanaan NM. Axonal degeneration in Tauopathies: disease relevance and underlying mechanisms. Front Neurosci. 2017;11:572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kovacs GG. Invited review: neuropathology of tauopathies: principles and practice. Neuropathol Appl Neurobiol. 2015;41:3–23. [DOI] [PubMed] [Google Scholar]
- 47.Kowall NW, Kosik KS. Axonal disruption and aberrant localization of tau protein characterize the neuropil pathology of Alzheimer’s disease. Ann Neurol. 1987;22:639–43. [DOI] [PubMed] [Google Scholar]
- 48.Kraus MF, Susmaras T, Caughlin BP, Walker CJ, Sweeney JA, Little DM. White matter integrity and cognition in chronic traumatic brain injury: a diffusion tensor imaging study. Brain. 2007;130:2508–19. [DOI] [PubMed] [Google Scholar]
- 49.Kreutzberg GW. Neuronal dynamics and axonal flow. IV. Blockage of intra-axonal enzyme transport by colchicine. Proc Natl Acad Sci U S A. 1969;62:722–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.LaPointe NE, Morfini G, Pigino G, Gaisina IN, Kozikowski AP, Binder LI, Brady ST. The amino terminus of tau inhibits kinesin-dependent axonal transport: implications for filament toxicity. J Neurosci Res. 2009;87:440–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lasek RJ, Garner JA, Brady ST. Axonal transport of the cytoplasmic matrix. J Cell Biol. 1984;99:212s–21s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liao H, Li Y, Brautigan DL, Gundersen GG. Protein phosphatase 1 is targeted to microtubules by the microtubule-associated protein tau. J Biol Chem. 1998;273:21901–8. [DOI] [PubMed] [Google Scholar]
- 53.Ling H Untangling the tauopathies: current concepts of tau pathology and neurodegeneration. Parkinsonism Relat Disord. 2018;46(Suppl 1):S34–8. [DOI] [PubMed] [Google Scholar]
- 54.Mandelkow EM, Stamer K, Vogel R, Thies E, Mandelkow E. Clogging of axons by tau, inhibition of axonal traffic and starvation of synapses. Neurobiol Aging. 2003;24:1079–85. [DOI] [PubMed] [Google Scholar]
- 55.Martin L, Latypova X, Terro F. Post-translational modifications of tau protein: implications for Alzheimer’s disease. Neurochem Int. 2011;58:458–71. [DOI] [PubMed] [Google Scholar]
- 56.McKee AC, Cantu RC, Nowinski CJ, Hedley-Whyte ET, Gavett BE, Budson AE, Santini VE, Lee HS, Kubilus CA, Stern RA. Chronic traumatic encephalopathy in athletes: progressive tauopathy after repetitive head injury. J Neuropathol Exp Neurol. 2009;68:709–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.McVicker DP, Chrin LR, Berger CL. The nucleotide-binding state of microtubules modulates kinesin processivity and the ability of tau to inhibit kinesin-mediated transport. J Biol Chem. 2011;286:42873–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mephon-Gaspard A, Boca M, Pioche-Durieu C, Desforges B, Burgo A, Hamon L, Pietrement O, Pastre D. Role of tau in the spatial organization of axonal microtubules: keeping parallel microtubules evenly distributed despite macromolecular crowding. Cell Mol Life Sci. 2016;73:3745–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Miki H, Okada Y, Hirokawa N. Analysis of the kinesin superfamily: insights into structure and function. Trends Cell Biol. 2005;15:467–76. [DOI] [PubMed] [Google Scholar]
- 60.Moreno H, Morfini G, Buitrago L, Ujlaki G, Choi S, Yu E, Moreira JE, Avila J, Brady ST, Pant H, Sugimori M, Llinas RR. Tau pathology-mediated presynaptic dysfunction. Neuroscience. 2016;325:30–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Morfini G, Szebenyi G, Elluru R, Ratner N, Brady ST. Glycogen synthase kinase 3 phosphorylates kinesin light chains and negatively regulates kinesin-based motility. EMBO J. 2002;21:281–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Morfini G, Szebenyi G, Brown H, Pant HC, Pigino G, DeBoer S, Beffert U, Brady ST. A novel CDK5-dependent pathway for regulating GSK3 activity and kinesin-driven motility in neurons. EMBO J. 2004;23:2235–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Morfini G, Pigino G, Mizuno N, Kikkawa M, Brady ST. Tau binding to microtubules does not directly affect microtubule-based vesicle motility. J Neurosci Res. 2007;85:2620–30. [DOI] [PubMed] [Google Scholar]
- 64.Morfini GA, You YM, Pollema SL, Kaminska A, Liu K, Yoshioka K, Bjorkblom B, Coffey ET, Bagnato C, Han D, Huang CF, Banker G, Pigino G, Brady ST. Pathogenic huntingtin inhibits fast axonal transport by activating JNK3 and phosphorylating kinesin. Nat Neurosci. 2009a;12:864–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Morfini GA, Burns M, Binder LI, Kanaan NM, LaPointe N, Bosco DA, Brown RH Jr, Brown H, Tiwari A, Hayward L, Edgar J, Nave KA, Garberrn J, Atagi Y, Song Y, Pigino G, Brady ST. Axonal transport defects in neurodegenerative diseases. J Neurosci. 2009b;29:12776–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Morfini GA, Bosco DA, Brown H, Gatto R, Kaminska A, Song Y, Molla L, Baker L, Marangoni MN, Berth S, Tavassoli E, Bagnato C, Tiwari A, Hayward LJ, Pigino GF, Watterson DM, Huang CF, Banker G, Brown RH Jr, Brady ST. Inhibition of fast axonal transport by pathogenic SOD1 involves activation of p38 MAP kinase. PLoS One. 2013;8:e65235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Morfini G, Schmidt N, Weissmann C, Pigino G, Kins S. Conventional kinesin: biochemical heterogeneity and functional implications in health and disease. Brain Res Bull. 2016;126:347–53. [DOI] [PubMed] [Google Scholar]
- 68.Mudher A, Shepherd D, Newman TA, Mildren P, Jukes JP, Squire A, Mears A, Drummond JA, Berg S, MacKay D, Asuni AA, Bhat R, Lovestone S. GSK3beta inhibition reverses axonal transport defects and behavioural phenotypes in Drosophila. Mol Psychiatry. 2004;9:522–30. [DOI] [PubMed] [Google Scholar]
- 69.Mukrasch MD, Bibow S, Korukottu J, Jeganathan S, Biernat J, Griesinger C, Mandelkow E, Zweckstetter M. Structural polymorphism of 441-residue tau at single residue resolution. PLoS Biol. 2009;7:e34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Padovani A, Borroni B, Brambati SM, Agosti C, Broli M, Alonso R, Scifo P, Bellelli G, Alberici A, Gasparotti R, Perani D. Diffusion tensor imaging and voxel based morphometry study in early progressive supranuclear palsy. J Neurol Neurosurg Psychiatry. 2006;77:457–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Patterson KR, Remmers C, Fu Y, Brooker S, Kanaan NM, Vana L, Ward S, Reyes JF, Philibert K, Glucksman MJ, Binder LI. Characterization of prefibrillar tau oligomers in vitro and in Alzheimer disease. J Biol Chem. 2011;286:23063–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pigino G, Morfini G, Atagi Y, Deshpande A, Yu C, Jungbauer L, LaDu M, Busciglio J, Brady S. Disruption of fast axonal transport is a pathogenic mechanism for intraneuronal amyloid beta. Proc Natl Acad Sci U S A. 2009;106:5907–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pir GJ, Choudhary B, Mandelkow E, Mandelkow EM. Tau mutant A152T, a risk factor for FTD/PSP, induces neuronal dysfunction and reduced lifespan independently of aggregation in a C. elegans Tauopathy model. Mol Neurodegener. 2016;11:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Probst A, Tolnay M, Langui D, Goedert M, Spillantini MG. Pick’s disease: hyperphosphorylated tau protein segregates to the somatoaxonal compartment. Acta Neuropathol (Berl). 1996;92:588–96. [DOI] [PubMed] [Google Scholar]
- 75.Ray K, Perez SE, Yang Z, Xu J, Ritchings BW, Steller H, Goldstein LS. Kinesin-II is required for axonal transport of choline acetyltransferase in Drosophila. J Cell Biol. 1999;147:507–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Reck-Peterson SL, Redwine WB, Vale RD, Carter AP. The cytoplasmic dynein transport machinery and its many cargoes. Nat Rev Mol Cell Biol. 2018;19:382–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rovelet-Lecrux A, Lecourtois M, Thomas-Anterion C, Le Ber I, Brice A, Frebourg T, Hannequin D, Campion D. Partial deletion of the MAPT gene: a novel mechanism of FTDP-17. Hum Mutat. 2009;30:E591–602. [DOI] [PubMed] [Google Scholar]
- 78.Roy S Seeing the unseen: the hidden world of slow axonal transport. Neuroscientist RevJ Bring Neurobiol Neurol Psychiat. 2014;20:71–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Roy S, Zhang B, Lee VM, Trojanowski JQ. Axonal transport defects: a common theme in neurodegenerative diseases. Acta Neuropathol. 2005;109:5–13. [DOI] [PubMed] [Google Scholar]
- 80.Sealey MA, Vourkou E, Cowan CM, Bossing T, Quraishe S, Grammenoudi S, Skoulakis EMC, Mudher A. Distinct phenotypes of three-repeat and four-repeat human tau in a transgenic model of tauopathy. Neurobiol Dis. 2017;105:74–83. [DOI] [PubMed] [Google Scholar]
- 81.Seitz A, Kojima H, Oiwa K, Mandelkow EM, Song YH, Mandelkow E. Single-molecule investigation of the interference between kinesin, tau and MAP 2c. EMBO J. 2002;21:4896–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sengupta A, Kabat J, Novak M, Wu Q, Grundke-Iqbal I, Iqbal K. Phosphorylation of tau at both Thr 231 and Ser 262 is required for maximal inhibition of its binding to microtubules. Arch Biochem Biophys. 1998;357:299–309. [DOI] [PubMed] [Google Scholar]
- 83.Sergeant N, Wattez A, Delacourte A. Neurofibrillary degeneration in progressive supranuclear palsy and corticobasal degeneration: tau pathologies with exclusively “exon 10” isoforms. J Neurochem. 1999;72:1243–9. [DOI] [PubMed] [Google Scholar]
- 84.Simic G, Babic Leko M, Wray S, Harrington C, Delalle I, Jovanov-Milosevic N, Bazadona D, Buee L, de Silva R, Di Giovanni G, Wischik C, Hof PR. Tau protein hyperphosphorylation and aggregation in Alzheimer’s disease and other Tauopathies, and possible neuroprotective strategies. Biomol Ther. 2016;6:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Song Y, Kang M, Morfini G, Brady ST. Fast axonal transport in isolated axoplasm from the squid giant axon. Methods Cell Biol. 2016;131:331–48. [DOI] [PubMed] [Google Scholar]
- 86.Soppina V, Verhey KJ. The family-specific K-loop influences the microtubule on-rate but not the super-processivity of kinesin-3 motors. Mol Biol Cell. 2014;25:2161–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Stamer K, Vogel R, Thies E, Mandelkow E, Mandelkow EM. Tau blocks traffic of organelles, neurofilaments, and APP vesicles in neurons and enhances oxidative stress. J Cell Biol. 2002;156:1051–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Stenoien DL, Brady ST. Immunochemical analysis of kinesin light chain function. Mol Biol Cell. 1997;8:675–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Stern JL, Lessard DV, Hoeprich GJ, Morfini GA, Berger CL. Phosphoregulation of tau modulates inhibition of kinesin-1 motility. Mol Biol Cell. 2017;28:1079–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Szodorai A, Kuan YH, Hunzelmann S, Engel U, Sakane A, Sasaki T, Takai Y, Kirsch J, Muller U, Beyreuther K, Brady S, Morfini G, Kins S. APP anterograde transport requires Rab3A GTPase activity for assembly of the transport vesicle. J Neurosci. 2009;29:14534–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Talmat-Amar Y, Arribat Y, Parmentier ML. Vesicular axonal transport is modified in vivo by tau deletion or overexpression in Drosophila. Int J Mol Sci. 2018;19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Tanaka Y, Kanai Y, Okada Y, Nonaka S, Takeda S, Harada A, Hirokawa N. Targeted disruption of mouse conventional kinesin heavy chain, kif5B, results in abnormal perinuclear clustering of mitochondria. Cell. 1998;93:1147–58. [DOI] [PubMed] [Google Scholar]
- 93.Tiernan CT, Combs B, Cox K, Morfini G, Brady ST, Counts SE, Kanaan NM. Pseudophosphorylation of tau at S422 enhances SDS-stable dimer formation and impairs both anterograde and retrograde fast axonal transport. Exp Neurol. 2016;283:318–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Tiernan CT, Ginsberg SD, He B, Ward SM, Guillozet-Bongaarts AL, Kanaan NM, Mufson EJ, Counts SE. Pretangle pathology within cholinergic nucleus basalis neurons coincides with neurotrophic and neurotransmitter receptor gene dysregulation during the progression of Alzheimer’s disease. Neurobiol Dis. 2018;117:125–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Trabzuni D, Wray S, Vandrovcova J, Ramasamy A, Walker R, Smith C, Luk C, Gibbs JR, Dillman A, Hernandez DG, Arepalli S, Singleton AB, Cookson MR, Pittman AM, de Silva R, Weale ME, Hardy J, Ryten M. MAPT expression and splicing is differentially regulated by brain region: relation to genotype and implication for tauopathies. Hum Mol Genet. 2012;21:4094–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Trinczek B, Ebneth A, Mandelkow EM, Mandelkow E. Tau regulates the attachment/detachment but not the speed of motors in microtubule-dependent transport of single vesicles and organelles. J Cell Sci. 1999;112(Pt 14):2355–67. [DOI] [PubMed] [Google Scholar]
- 97.Tsai MY, Morfini G, Szebenyi G, Brady ST. Release of kinesin from vesicles by hsc70 and regulation of fast axonal transport. Mol Biol Cell. 2000;11:2161–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Uchida A, Alami NH, Brown A. Tight functional coupling of kinesin-1A and dynein motors in the bidirectional transport of neurofilaments. Mol Biol Cell. 2009;20:4997–5006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Uryu K, Chen XH, Martinez D, Browne KD, Johnson VE, Graham DI, Lee VM, Trojanowski JQ, Smith DH. Multiple proteins implicated in neurodegenerative diseases accumulate in axons after brain trauma in humans. Exp Neurol. 2007;208:185–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Vershinin M, Carter BC, Razafsky DS, King SJ, Gross SP. Multiple-motor based transport and its regulation by tau. Proc Natl Acad Sci U S A. 2007;104:87–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Vossel KA, Zhang K, Brodbeck J, Daub AC, Sharma P, Finkbeiner S, Cui B, Mucke L. Tau reduction prevents Abeta-induced defects in axonal transport. Science. 2010;330:198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Vossel KA, Xu JC, Fomenko V, Miyamoto T, Suberbielle E, Knox JA, Ho K, Kim DH, Yu GQ, Mucke L. Tau reduction prevents Abeta-induced axonal transport deficits by blocking activation of GSK3beta. J Cell Biol. 2015;209:419–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wang Y, Mandelkow E. Tau in physiology and pathology. Nat Rev Neurosci. 2016;17:5–21. [DOI] [PubMed] [Google Scholar]
- 104.Ward SM, Himmelstein DS, Lancia JK, Fu Y, Patterson KR, Binder LI. TOC1: characterization of a selective oligomeric tau antibody. J Alzheimers Dis. 2013;37:593–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Weingarten MD, Lockwood AH, Hwo SY, Kirschner MW. A protein factor essential for microtubule assembly. Proc Natl Acad Sci U S A. 1975;72:1858–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wong CW, Quaranta V, Glenner GG. Neuritic plaques and cerebrovascular amyloid in Alzheimer disease are antigenically related. Proc Natl Acad Sci U S A. 1985;82:8729–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wood JG, Mirra SS, Pollock NJ, Binder LI. Neurofibrillary tangles of Alzheimer disease share antigenic determinants with the axonal microtubule-associated protein tau (tau) [published erratum appears in Proc Natl Acad Sci U S A 1986 Dec;83(24):9773]. Proc Natl Acad Sci U S A. 1986;83:4040–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Yamakawa K, Takanashi M, Watanabe M, Nakamura N, Kobayashi T, Hasegawa M, Mizuno Y, Tanaka S, Mori H. Pathological and biochemical studies on a case of pick disease with severe white matter atrophy. Neuropathol Off J Jpn Soc Neuropathol. 2006;26:586–91. [DOI] [PubMed] [Google Scholar]
- 109.Yoshida M Cellular tau pathology and immunohistochemical study of tau isoforms in sporadic tauopathies. Neuropathol Off J Jpn Soc Neuropathol. 2006;26:457–70. [DOI] [PubMed] [Google Scholar]
- 110.Yuan A, Kumar A, Peterhoff C, Duff K, Nixon RA. Axonal transport rates in vivo are unaffected by tau deletion or overexpression in mice. J Neurosci. 2008;28:1682–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Zhang Y, Walter R, Ng P, Luong PN, Dutt S, Heuer H, Rojas-Rodriguez JC, Tsai R, Litvan I, Dickerson BC, Tartaglia MC, Rabinovici G, Miller BL, Rosen HJ, Schuff N, Boxer AL. Progression of microstructural degeneration in progressive Supranuclear palsy and Corticobasal syndrome: a longitudinal diffusion tensor imaging study. PLoS One. 2016;11:e0157218. [DOI] [PMC free article] [PubMed] [Google Scholar]