

Abstract
Aim:
The recurring resistance of the malaria parasite to many drugs compels the design of innovative chemical entities in antimalarial research. Pan-histone deacetylase inhibitors (pan-HDACis) have recently been presented in the literature as powerful novel antimalarials, although their application is hampered due to toxic side effects. This drawback might be neutralized by the deployment of isoform-selective HDACis.
Results:
In this study, 42 thiaheterocyclic benzohydroxamic acids, 17 of them being potent and selective hHDAC6 inhibitors, were tested to investigate a possible correlation between hHDAC6 inhibition and antiplasmodial activity.
Conclusion:
Four hHDAC6 inhibitors showed submicromolar potency against both a chloroquine-sensitive and a chloroquine-resistant strain of Plasmodium falciparum with high selectivity indices, pointing to the relevance of exploring hHDAC6 inhibitors as potential new antiplasmodial agents.
Keywords: : benzohydroxamic acids, HDAC6, malaria, Plasmodium falciparum, thiaheterocycles
Graphical abstract
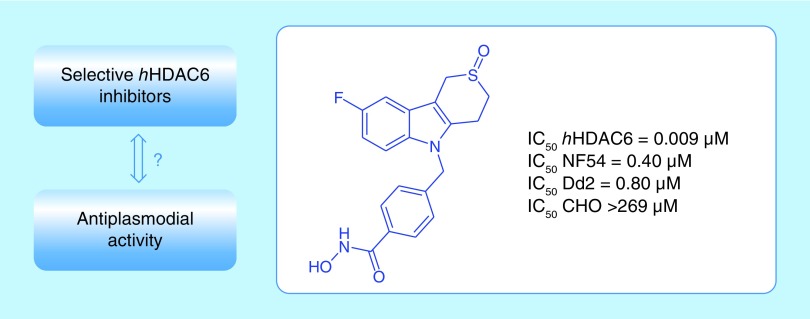
First draft submitted: 16 November2016; Accepted for publication: 12 January 2017; Published online: 6 March 2017
Malaria is a devastating parasitic disease, exemplified by the fact that roughly 3.2 billion people are at risk of contracting malaria and that this disease caused roughly 438,000 deaths in 2015, with an estimated 306,000 casualties in the group of children under the age of five (WHO) [1]. The main culprit causing this infection is the protozoan species Plasmodium falciparum, transmitted by mosquitoes of the genus Anopheles [2]. In the past 15 years (2000–2015), considerable progress has been made toward revoking this infection, as illustrated by a declining number of malaria cases and deaths (18 and 48%, respectively) [1]. However, there still is a pressing need to reduce the number of victims even further and to find solutions to address all challenges associated with this disease. A pertinent challenge relates to the expanding resistance of the Plasmodium parasite toward several treatment regimes. Indeed, resistance has emerged with respect to the standard antimalarials chloroquine (CQ), sulfadoxine, pyrimethamine and, more recently, artemisinin [3]. The acquired artemisinin resistance is particularly alarming, since artemisinin combination therapies represent the first-in-line treatment option for malaria nowadays.
A consequence of this recurring resistance is the urgent need to develop new medicines with alternative mechanisms of action, in order to impede or deter the parasite from developing resistance by applying combination therapies. In that regard, histone deacetylase inhibitors (HDACis) might offer new treatment opportunities, as several known HDAC inhibitors have recently been shown to demonstrate a promising activity against P. falciparum and other malarial strains [4–7]. HDACs and HATs function as regulators of lysine acetylation, an important post-translational modification responsible for the neutralization of the positive charges on lysine residues, and as such adjusting the exact mode of action of the targeted protein [8]. HDACs were first been discovered as histone lysine modifying enzymes but are now generally accepted to be lysine deacetylases, also deacetylating several nonhistone proteins [9]. In humans, this group of enzymes comprises four classes, with class I (HDAC1, 2, 3 and 8), IIa (HDAC4, 5, 7 and 9), IIb (HDAC6 and 10) and IV (HDAC11) employing zinc as an essential cofactor, while class III (SIRT1–7) uses NAD+ for its deacetylase activity [10]. On the other hand, five HDAC isoforms are known for P. falciparum: PfHDAC1, with homology to human class I, PfHDAC2 and 3, with homology to human class II, and PfSir2A and PfSir2B, with homology to human class III [11]. So far, mainly pan-HDACi's have been tested for their activity against P. falciparum, revealing high toxicities in the (low) nanomolar range toward the malaria parasite [4–7]. A major drawback associated with these broad-spectrum HDAC inhibitors involves the interaction with all human Zn2+-dependent HDACs, culminating in a higher risk to elicit toxic side effects upon administration. Therefore, the selective inhibition of pfHDACs over hHDAC isoforms represents a relevant challenge in antimalarial drug discovery and has led to the assessment of many hHDAC inhibitors as potential antiplasmodial agents. In that respect, a library screen of 2000 compounds has revealed (E)-7-[2-(2-bromobenzylidene)hydrazinyl]-N-hydroxy-7-oxoheptanamide to be such a selective compound and, in another report, a specific class of methylamides has been shown to be pfHDAC selective [12,13].
An alternative strategy could imply the examination of selective hHDAC inhibitors (instead of pan-hHDAC inhibitors) as novel antimalarial compounds. This approach lowers the risk of host toxicity without potentially compromising a pronounced antiplasmodial activity [14]. Selective human HDAC6 inhibitors could possibly serve this goal as it is known that mice lacking HDAC6 develop rather normally [15], so minor to no side effects are expected upon deployment of these agents. Bearing this rationale in mind, we decided to test a series of benzohydroxamic acids (previously developed by us) for their antiplasmodial activity, with several representatives being highly potent and selective hHDAC6 inhibitors. Indeed, a systematic exploration of the possible correlation between hHDAC6 inhibitors and antiplasmodial activity has not been performed so far and could reveal new opportunities in antimalarial drug development.
Materials & methods
Antiplasmodial assay
Continuous in vitro cultures of asexual erythrocyte stages of P. falciparum were maintained using a modified method of Trager and Jensen [16]. Quantitative assessment of antiplasmodial activity in vitro was determined via the parasite lactate dehydrogenase assay using a modified method described by Makler [17]. The test samples were prepared to a 20 mg/ml stock solution in 100% DMSO. Stock solutions were stored at -20°C. Further dilutions were prepared in complete medium on the day of the experiment. CQ and artesunate were used as the reference drugs. A full dose–response was performed to determine the concentration inhibiting 50% of parasite growth (IC50 value). Test samples were tested at a starting concentration of 100 μg/ml, which was then serially diluted twofold in complete medium to give 10 concentrations; with the lowest concentration being 0.2 μg/ml. The same dilution technique was used for all samples. References were tested at a starting concentration of 1 μg/ml. The highest concentration of solvent to which the parasites were exposed to had no measurable effect on the parasite viability (data not shown).
MTT assay
Test samples were screened for in vitro cytotoxicity against a mammalian cell-line, Chinese Hamster Ovary (CHO), using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide (MTT)-assay. The MTT-assay is used as a colorimetric assay for cellular growth and survival, and compares well with other available assays [18,19]. The tetrazolium salt MTT was used to measure all growth and chemosensitivity. The test samples were tested in triplicate on one occasion. The same stock solutions prepared for antiplasmodial evaluation were used for cytotoxicity testing. Test compounds were stored at -20°C until use. Dilutions were prepared on the day of the experiment. Emetine was used as the reference drug in all experiments. The starting concentration was 100 μg/ml, which was serially diluted in complete medium with tenfold dilutions to give six concentrations, the lowest being 0.001 μg/ml. The highest concentration of solvent to which the cells were exposed to had no measurable effect on the cell viability (data not shown). The IC50 values were obtained from full dose–response curves, using a nonlinear dose–response curve fitting analysis via GraphPad Prism v.4 software.
Results & discussion
This brief article focuses on the antiplasmodial evaluation of three innovative classes of benzohydroxamic acids 1–3, all featuring a different thiahetero(bi- or tri-)cyclic ‘cap group’ (Figure 1). Class 1 consists of molecules containing a saturated thiaheterocyclic ring annulated onto an indole core (designated as Tubathians), class 2 comprises benzothiophenes embodying a nitrogen atom in the linker region and class 3 includes cycloalkane-annulated 1,5-benzothiazepine scaffolds. Because of small structural modifications with respect to the ‘mother structure’ within each class (Figure 1 & Table 1), a broad set of 42 compounds with divergent decoration patterns is synthetically available. The preparation of these compounds 1–3 has previously been described, together with a detailed account on their HDAC6 selectivity, cellular activity (α-tubulin acetylation, a known substrate of HDAC6) and mutagenicity [20–23]. For Tubathian structures 1, additional information concerning the ADME/Tox properties has been disclosed as well [22]. These different classes include a number of highly potent and selective hHDAC6 inhibitors (Table 2), which have in common a para-substituted benzohydroxamic acid fragment, no substituents in the meta-position with respect to the hydroxamic acid group, and superior HDAC6 inhibitory activity for sulfoxides and sulfones over the corresponding sulfides.
Figure 1. . Available thiaheterocyclic benzohydroxamic acids 1–3.

Table 1. . Substitution pattern of thiaheterocyclic benzohydroxamic acids 1–3.
| Compound | R1 | R2 | R3 | x | n | Config.† |
|---|---|---|---|---|---|---|
| 1a | H | H | – | 0 | 1 | Para |
| 1b | H | OMe | – | 0 | 1 | Para |
| 1c | F | H | – | 0 | 1 | Para |
| 1d | F | OMe | – | 0 | 1 | Para |
| 1e | H | H | – | 2 | 1 | Para |
| 1f | H | OMe | – | 2 | 1 | Para |
| 1g | F | H | – | 2 | 1 | Para |
| 1h | F | OMe | – | 2 | 1 | Para |
| 1i | Br | H | – | 2 | 1 | Para |
| 1j | H | H | – | 1 | 1 | Para |
| 1k | F | H | – | 1 | 1 | Para |
| 1l | H | H | – | 2 | 0 | Para |
| 1m | F | H | – | 2 | 0 | Para |
| 1n | H | H | – | 0 | 1 | Meta |
| 1o | F | H | – | 0 | 1 | Meta |
| 1p | H | H | – | 2 | 1 | Meta |
| 1q | F | H | – | 2 | 1 | Meta |
| 1r | Ph | H | – | 2 | 1 | Meta |
| 1s | H | H | – | 2 | 0 | Meta |
| 1t | F | H | – | 2 | 0 | Meta |
| 1u | Br | H | – | 1 | 0 | Meta |
| 2a | H | H | H | – | – | – |
| 2b | H | Bn | H | – | – | – |
| 2c | 5-Br | H | H | – | – | – |
| 2d | 5-Br | Bn | H | – | – | – |
| 2e | 5-Ph | H | H | – | – | – |
| 2f | 5-Ph | Bn | H | – | – | – |
| 2g | 6-Br | H | H | – | – | – |
| 2h | 6-Br | Bn | H | – | – | – |
| 2i | 6-Ph | H | H | – | – | – |
| 2j | 6-Ph | Bn | H | – | – | – |
| 2k | H | H | Me | – | – | – |
| 3a | H | – | – | 0 | 1 | Cis |
| 3b | Cl | – | – | 0 | 1 | Cis |
| 3c | CF3 | – | – | 0 | 1 | Cis |
| 3d | H | – | – | 2 | 1 | Cis |
| 3e | Cl | – | – | 2 | 1 | Cis |
| 3f | CF3 | – | – | 2 | 1 | Cis |
| 3g | H | – | – | 1 | 1 | Cis |
| 3h | H | – | – | 0 | 2 | Cis |
| 3i | Cl | – | – | 0 | 1 | Trans |
| 3j | H | – | – | 0 | 2 | Trans |
†The para- and meta-configuration for compound 1 refers to the position of the hydroxamic acid group on the aromatic ring with respect to the aminomethyl substituent.
Table 2. . IC50 values (μM) of compounds 1–3 determined for a normal (NF54) and chloroquine-resistant (Dd2) Plasmodium falciparum strain, Chinese hamster ovary cells and HDAC6.
| Cmpd | NF54 | Dd2 | CHO | SI‡ | RI§ | HDAC6 |
|---|---|---|---|---|---|---|
| 1a | 37.5 | – | – | – | – | 0.015 |
| 1b | 14.0 | – | – | – | – | – |
| 1c | 2.2 | 3.1 | 105.2 | 48 | 1.4 | 0.022 |
| 1d | 23.2 | – | – | – | – | – |
| 1e | 10.8 | – | – | – | – | 0.002 |
| 1f | 21.0 | – | – | – | – | 2.0 |
| 1g | 15.8 | – | – | – | – | 0.004 |
| 1h | 32.7 | – | – | – | – | 1.3 |
| 1i | 0.11 | 0.43 | 109.0 | 991 | 3.9 | 0.003 |
| 1j | 1.28 | 1.3 | >282 | >217 | 1.0 | 0.014 |
| 1k | 0.40 | 0.80 | >269 | >673 | 2.0 | 0.009 |
| 1l | 0.92 | 0.66 | >281 | >305 | 0.7 | 0.008 |
| 1m | 1.07 | 1.55 | >267 | >250 | 1.7 | 0.016 |
| 1n | 1.48 | 2.18 | >295 | >199 | 1.5 | – |
| 1o | 1.32 | 1.44 | 48.1 | 36 | 1.1 | – |
| 1p | 5.45 | – | – | – | – | – |
| 1q | 7.84 | – | – | – | – | – |
| 1r | 8.13 | – | – | – | – | – |
| 1s | 12.2 | – | – | – | – | – |
| 1t | 9.80 | – | – | – | – | – |
| 1u | 11.9 | – | – | – | – | – |
| 2a | 1.60 | 2.13 | 12.7 | 8 | 1.3 | 0.014 |
| 2b | 32.4 | – | – | – | – | – |
| 2c | 1.02 | 2.4 | 31.0 | 30 | 2.4 | 0.037 |
| 2d | 17.8 | – | – | – | – | – |
| 2e | 5.07 | – | – | – | – | – |
| 2f | 5.75 | – | – | – | – | – |
| 2g | 1.30 | 1.58 | 46.1 | 35 | 1.2 | 0.064 |
| 2h | 8.45 | – | – | – | – | – |
| 2i | 3.34 | 1.14 | 31.2 | 9 | 0.3 | – |
| 2j | 5.02 | – | – | – | – | – |
| 2k | 36.8 | – | – | – | – | – |
| 3a | 1.59 | >2.7 | 103.9 | 65 | – | 0.036 |
| 3b | >2.48 | >2.48 | 41.4 | – | – | 0.650 |
| 3c | 1.53 | >2.29 | 61.5 | 40 | – | 0.200 |
| 3d | 0.36 | 0.94 | 107.6 | 303 | 2.6 | 0.008 |
| 3e | 0.47 | 0.44 | 35.4 | 75 | 0.9 | 0.068 |
| 3f | 0.87 | 0.70 | 56.8 | 65 | 0.8 | 0.011 |
| 3g | 1.25 | >2.60 | 172.9 | 138 | – | 0.006 |
| 3h | >2.61 | >2.61 | 87.0 | – | – | 0.033 |
| 3i | >2.48 | 1.57 | 50.9 | – | – | 0.160 |
| 3j | >2.61 | >2.61 | 61.7 | – | – | 0.092 |
Bold: IC50-value of the hydroxamic acid lower than 1 μM against both P. falciparum strains. ‘–’ is not determined.
‡SI (selectivity index) = IC50 (CHO)/IC50 (NF54).
§RI (resistance index) = IC50 (Dd2)/IC50 (NF54).
Chloroquine IC50-NF54 = 0.01 μM, IC50-Dd2 = 0.175 μM; Artesunate IC50-NF54 < 0.01 μM, IC50-Dd2 = 0.016 μM; Emetine IC50-CHO = 0.112 μM.
CHO: Chinese hamster ovary; RI: Resistance index; SI: Selectivity index.
The antiplasmodial activity of this set of structures was first determined through a modified parasite lactate dehydrogenase assay against a CQ-sensitive (CQS) strain of P. falciparum (NF54) [16,17]. When the molecules proved to be reasonably active against this strain (IC50 <5 μM), a second assay was performed against a CQ-resistant (CQR) strain of P. falciparum (Dd2), as well as an MTT-assay (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) on CHO cells to assess their mammalian cytotoxicity (Table 2) [18]. Finally, a selectivity index (SI = IC50 CHO/IC50 NF54) and a resistance index (RI = IC50 Dd2/IC50 NF54) was calculated to be able to easily compare the therapeutic window (active concentration vs toxic concentration) and sensitivity toward resistance developing. Table 2 shows that all 42 benzohydroxamic acids 1–3 display interesting antiplasmodial activities (IC50 values against the CQS strain between 0.11 and 37.5 μM). The potent HDAC6 inhibitors 1i, 1k, 1l, 3d, 3e and 3f were also found to be highly active against both CQS and CQR parasitic strains (IC50 CQS and CQR < 1 μM, IC50 HDAC6 < 0.07 μM). However, other active HDAC6 inhibitors did not demonstrate a distinct submicromolar parasitic toxicity (e.g., 1a, 1e and 1g). Thus, no consistent correlation can be drawn between hHDAC6 inhibition and antiplasmodial activity, which could be expected considering the inevitable differences between human and parasite HDAC isoforms [11]. On the other hand, it is remarkable to note that the most effective antiplasmodial compounds all are powerful hHDAC6 inhibitors, and none of the less active hHDAC6 inhibitors showed submicromolar antiplasmodial potency. Based on these observations, it can be suggested that strong hHDAC6 inhibitory activity is a necessary but not a sufficient condition for thiaheterocyclic benzohydroxamic acids to exert submicromolar antiplasmodial activity as well. From the six compounds showing the most promising antiplasmodial activity, four molecules have excellent selectivity indices higher than 300 (1i, 1k, 1l and 3d), which means that the concentration at which they kill the parasite is at least 300-times lower than their toxic concentration for CHO cells. Comparison of the resistance indices (RI) suggests that the tested molecules have comparable activity (RI = 0.3–3.9) against both strains (CQS and CQR). This is in marked contrast to the control drug CQ, which is 17-times less active against the CQR strain (RI = 17.5).
Conclusion
42 thiaheterocyclic benzohydroxamic acids 1–3, 17 of them previously being identified as highly potent and selective hHDAC6 inhibitors, were assessed in terms of their antiplasmodial profile. This study revealed six selective HDAC6 inhibitors to demonstrate submicromolar antiplasmodial potency against both a CQS and a CQR strain, and four of these structures (1i, 1k, 1l and 3d) also proved to have an excellent therapeutic window (SI > 300). On the other hand, hydroxamic acids which do not strongly inhibit hHDAC6, appear to possess only moderate antiplasmodial effects. Thus, potent and selective hHDAC6 inhibitory activity of thiaheterocyclic benzohydroxamic acids seems to be a necessary but not a sufficient condition to elicit pronounced antiplasmodial activity as well. Moreover, selective hHDAC6 inhibitors can induce powerful P. falciparum toxicity without being toxic for CHO cells (as a model for mammalian cytotoxicity). In conclusion, hHDAC6 inhibitory activity and antiplasmodial activity are somehow interconnected, and these HDAC6i new chemical entities can certainly be considered a valuable starting point for further medicinal chemistry investigation en route to novel types of antiplasmodial drugs.
Future perspective
The evaluation of isoform-selective hHDAC inhibitors, in particular hHDAC6is, as antiplasmodial agents represents a new and promising approach in antimalarial research, and further study is desirable to unravel the specific interplay between these compounds and their antiplasmodial activity (structure–activity relationship). In addition to this phenotypic approach, more information on the structure and function of pfHDACs should be obtained, which will enable rational design of pfHDAC inhibitors.
Executive summary.
Antimalarial drug resistance encourages the need for innovative chemical entities with alternative modes of action.
hHDAC6 inhibitors are proposed as a novel strategy against Plasmodium falciparum.
Results
Three classes of thiaheterocyclic benzohydroxamic acids were tested against P. falciparum.
Four potent and selective hHDAC6 inhibitors were shown to be highly active as antiplasmodial agents, displaying a good therapeutic window and resistance index as well.
Conclusion
Potent and selective hHDAC6 inhibitory activity of thiaheterocyclic benzohydroxamic acids is a necessary but not a sufficient condition to elicit pronounced antiplasmodial activity.
hHDAC6 inhibitors can induce powerful P. falciparum toxicity without being toxic for CHO cells.
Footnotes
Financial & competing interests disclosure
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
No writing assistance was utilized in the production of this manuscript.
References
Papers of special note have been highlighted as: • of interest; •• of considerable interest
- 1.World malaria report 2015. WHO. 2015:1–243. [Google Scholar]
- 2.White NJ, Pukrittayakamee S, Hien TT, Faiz MA, Mokuolu OA, Dondorp AM. Malaria. Lancet. 2014;383(9918):723–735. doi: 10.1016/S0140-6736(13)60024-0. [DOI] [PubMed] [Google Scholar]; •• A comprehensive overview of new developments and insights in malaria research.
- 3.Miller LH, Ackerman HC, Su XZ, Wellems TE. Malaria biology and disease pathogenesis: insights for new treatments. Nat. Med. 2013;19(2):156–167. doi: 10.1038/nm.3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Andrews KT, Haque A, Jones MK. HDAC inhibitors in parasitic diseases. Immunol. Cell Biol. 2012;90(1):66–77. doi: 10.1038/icb.2011.97. [DOI] [PubMed] [Google Scholar]
- 5.Aneja B, Kumar B, Jairajpuri MA, Abid M. A structure guided drug-discovery approach towards identification of Plasmodium Inhibitors. RSC Adv. 2016;6:18364–18406. [Google Scholar]; • Development of new antimalarial agents from a medicinal chemistry perspective.
- 6.Andrews KT, Tran TN, Wheatley NC, Fairlie DP. Targeting histone deacetylase inhibitors for anti-malarial therapy. Curr. Top. Med. Chem. 2009;9(3):292–308. doi: 10.2174/156802609788085313. [DOI] [PubMed] [Google Scholar]; • Importance of histone deacetylase inhibitors as new tools for malaria treatment.
- 7.Wang Q, Rosa BA, Nare B, et al. Targeting Lysine Deacetylases (KDACs) in Parasites. PLoS Negl. Trop. Dis. 2015;9(9):e0004026. doi: 10.1371/journal.pntd.0004026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat. Rev. Genet. 2009;10(1):32–42. doi: 10.1038/nrg2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Choudhary C, Kumar C, Gnad F, et al. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325(5942):834–840. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- 10.De Ruijter AJM, Van Gennip AH, Caron HN, Kemp S, Van Kuilenburg ABP. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem. J. 2003;370(3):737–749. doi: 10.1042/BJ20021321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Engel JA, Jones AJ, Avery VM, et al. Profiling the anti-protozoal activity of anti-cancer HDAC inhibitors against Plasmodium and Trypanosoma parasites. Int. J. Parasitol. Drugs Drug Resist. 2015;5(3):117–126. doi: 10.1016/j.ijpddr.2015.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Patel V, Mazitschek R, Coleman B, et al. Identification and characterization of small molecule inhibitors of a class I histone deacetylase from Plasmodium falciparum . J. Med. Chem. 2009;52(8):2185–2187. doi: 10.1021/jm801654y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ontoria JM, Paonessa G, Ponzi S, et al. Discovery of a selective series of inhibitors of Plasmodium falciparum HDACs. ACS Med. Chem. Lett. 2016;7(5):454–459. doi: 10.1021/acsmedchemlett.5b00468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hansen FK, Sumanadasa SD, Stenzel K, et al. Discovery of HDAC inhibitors with potent activity against multiple malaria parasite life cycle stages. Eur. J. Med. Chem. 2014;82:204–213. doi: 10.1016/j.ejmech.2014.05.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang Y, Kwon S, Yamaguchi T, et al. Mice lacking histone deacetylase 6 have hyperacetylated tubulin but are viable and develop normally. Mol. Cell. Biol. 2008;28(5):1688–1701. doi: 10.1128/MCB.01154-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Trager W, Jensen JB. Human malaria parasites in continuous culture. Science. 1976;193(4254):673–675. doi: 10.1126/science.781840. [DOI] [PubMed] [Google Scholar]
- 17.Makler MT, Ries JM, Williams JA, et al. Parasite lactate dehydrogenase as an assay for Plasmodium falciparum drug sensitivity. Am. J. Trop. Med. Hyg. 1993;48(6):739–741. doi: 10.4269/ajtmh.1993.48.739. [DOI] [PubMed] [Google Scholar]
- 18.Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Methods. 1983;65(1–2):55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 19.Rubinstein LV, Shoemaker RH, Paull KD, et al. Comparison of in vitro anticancer-drug-screening data generated with a tetrazolium assay versus a protein assay against a diverse panel of human tumor cell lines. J. Natl Cancer Inst. 1990;82(13):1113–1117. doi: 10.1093/jnci/82.13.1113. [DOI] [PubMed] [Google Scholar]
- 20.De Vreese R, Verhaeghe T, Desmet T, D'hooghe M. Potent and selective HDAC6 inhibitory activity of N-(4-hydroxycarbamoylbenzyl)-1,2,4,9-tetrahydro-3-thia-9-azafluorenes as novel sulfur analogues of Tubastatin A. Chem. Commun. 2013;49(36):3775–3777. doi: 10.1039/c3cc41422a. [DOI] [PubMed] [Google Scholar]; • Synthesis and biological assessment of some of the chemical compounds used in this work.
- 21.De Vreese R, Van Steen N, Verhaeghe T, et al. Synthesis of benzothiophene-based hydroxamic acids as potent and selective HDAC6 inhibitors. Chem. Commun. 2015;51(48):9868–9871. doi: 10.1039/c5cc03295d. [DOI] [PubMed] [Google Scholar]; • Synthesis and biological assessment of some of the chemical compounds used in this work.
- 22.De Vreese R, Depetter Y, Verhaeghe T, et al. Synthesis and SAR assessment of novel Tubathian analogs in the pursuit of potent and selective HDAC6 inhibitors. Org. Biomol. Chem. 2016;14(8):2537–2549. doi: 10.1039/c5ob02625c. [DOI] [PubMed] [Google Scholar]; • Synthesis and biological assessment of some of the chemical compounds used in this work.
- 23.De Vreese R, Galle L, Depetter Y, et al. Synthesis of potent and selective HDAC6 inhibitors bearing a cyclohexane- or cycloheptane-annulated 1,5-benzothiazepine scaffold. Chem. Eur. J. 2017;23(1):128–136. doi: 10.1002/chem.201604167. [DOI] [PubMed] [Google Scholar]; • Synthesis and biological assessment of some of the chemical compounds used in this work.