

ABSTRACT
The global spread of the 2019-nCoV is continuing and is fast moving, as indicated by the WHO raising the risk assessment to high. In this article, we provide a preliminary phylodynamic and phylogeographic analysis of this new virus. A Maximum Clade Credibility tree has been built using the 29 available whole genome sequences of 2019-nCoV and two whole genome sequences that are highly similar sequences from Bat SARS-like Coronavirus available in GeneBank. We are able to clarify the mechanism of transmission among the countries which have provided the 2019-nCoV sequence isolates from their patients. The Bayesian phylogeographic reconstruction shows that the 2019–2020 nCoV most probably originated from the Bat SARS-like Coronavirus circulating in the Rhinolophus bat family. In agreement with epidemiological observations, the most likely geographic origin of the new outbreak was the city of Wuhan, China, where 2019-nCoV time of the most recent common ancestor emerged, according to molecular clock analysis, around November 25th, 2019. These results, together with previously recorded epidemics, suggest a recurring pattern of periodical epizootic outbreaks due to Betacoronavirus. Moreover, our study describes the same population genetic dynamic underlying the SARS 2003 epidemic, and suggests the urgent need for the development of effective molecular surveillance strategies of Betacoronavirus among animals and Rhinolophus of the bat family.
KEYWORDS: 2019-nCoV, molecular Epidemiology, phylogeny, SARS
Introduction
Emerging viruses and pathogens represent a global public health threat. The recent Coronavirus epidemic outbreak, reported for the first time in late 2019 in Wuhan, Hubei province, China, is rapidly becoming of worldwide concern. Coronaviruses are single, plus-stranded RNA viruses belonging to the family Coronaviridae including MERS (MERS-CoV) and SARS (SARS-CoV). Coronavirus cause different disease with respiratory, enteric, hepatic and neurological clinical symptoms [1,2]. In December 2019, several clusters of patients with pneumonia of unknown origin, epidemiologically associated with a seafood and animal market in Wuhan, were described, calling the attention of the Chinese Center for Disease Control (Report of clustering pneumonia of unknown etiology in Wuhan City (Wuhan Municipal Health Commission, 2019; http://wjw .wuhan .gov.cn/front/web/showDetail/2019123108989), leading to the isolation of a new coronavirus, named 2019-nCoV, distinct from both MERS-CoV and SARS [3].
Since December 2019, the number of ascertained cases of infection has been daily increasing, with 4,593 confirmed cases and 106 deaths up to the time of writing (ECDC, Jan 26, 2020) (https://www.ecdc.europa.eu/en/publications-data/risk-assessment-outbreak-acute-respiratory-syndrome-associated-novel-0)
Based on epidemiological analysis, animal-to-human transmission seems to be the likely origin of the epidemic, as the first cases were detected in patients with recent history of visits to Wuhan fish and wild markets. Evidences for animal-to-human and subsequent human-to-human transmission of the virus were also reported, even if the transmission dynamics are not completely understood and significant knowledge gaps still need to be filled in [4–6]. In this short report, initial phylodynamic and phylogeography analyses of the 2019-nCoV were performed on the full genome sequences currently available, in order to clarify virus transmission dynamics and trace its initial epidemic spread.
Materials and methods
The dataset comprised all currently available (n = 29) full genome sequences from the current (2019–2020) nCoV epidemic, as well as closely related (n = 2) bat strains (SARS-like CoV) retrieved from NCBI (http://www.ncbi.nlm.nih.gov/genbank/) and GISAID (https://www.gisaid.org/) databases. Alignment was performed using MAFFT online program [7]. The complete dataset was assessed for presence of phylogenetic signal by applying the likelihood mapping analysis implemented in the IQ-TREE 1.6.8 software (http://www.iqtree.org) [8]. A maximum likelihood (ML) phylogeny was reconstructed using IQ-TREE 1.6.8 software under the HKY nucleotide substitution model with four gamma categories (HKY+G4), which was inferred in jModelTest (https://github.com/ddarriba/jmodeltest2) as the best fitting model [9].
In order to investigate the temporal signal, we regressed root-to-tip genetic distances from this ML tree against sample collection dates using TempEst v 1.5.1 (http://tree.bio.ed.ac.uk) [10]. The ML phylogeny was used as a starting tree for Bayesian time-scaled phylogenetic analysis using BEAST 1.10.4 (http://beast.community/index.html) [11]. We employed a stringent model selection analysis using both path-sampling (PS) and stepping stone (SS) procedures to estimate the most appropriate molecular clock model for the Bayesian phylogenetic analysis [12]. We tested a) the strict molecular clock model, which assumes a single rate across all phylogeny branches, and b) the more flexible uncorrelated relaxed molecular clock model with a lognormal rate distribution (UCLN) [13]. Both SS and PS estimators indicated the uncorrelated relaxed molecular clock (Bayes Factor = 4.3) as the best fitted model to the dataset under analysis. Besides, we have used the he HKY+G4 codon partitioned (CP)1 + 2,3 substitution model and the Bayesian Skyline coalescent model of population size and growth [14]. We computed MCMC (Markov chain Monte Carlo) duplicate runs of 100 million states each, sampling every 10,000 steps. Convergence of MCMC chains was checked using Tracer v.1.7.1 [14]. Proper mixing of the MCMC was checked for ESS values >200 for each estimated parameter using Tracer 1.7. Systematic Biology. 2018;67(5):901–4). A Maximum Clade Credibility (MCC) trees was obtained from the tree posterior distribution using TreeAnnotator (http://beast.community/index.html) after 10% burn-in.
Results
Despite the short time since the beginning of the epidemic, the isolates analyzed have already exhibited a substantial degree of heterogeneity with differences in 15% of the sites, 11% of which were parsimony informative, thus indicating the presence of sufficient phylogenetic signal for further analysis, in agreement with the low level of phylogenetic noise shown by likelihood mapping (<7%). The root-to-tip vs. divergence plot of the full dataset showed high correlation between sampling time and genetic distance to the root of the ML tree of the available sequences (R-squared 0.85), indicating substantial temporal signal and the possibility to calibrate a reliable molecular clock, despite the limited number of sequences and short sampling interval available.
Bayesian model selection chose the Bayesian Skyline demographic model with an uncorrelated relaxed clock as the one that best fit the data. Molecular clock calibration estimated the evolutionary rate of the 2019-nCoV whole genome sequences at 6.58 × 10−3 substitutions site per year (95% HPD 5.2 × 10−3 – 8.1 × 10−3).
Figure 1 A,B shows the MCC tree with Bayesian phylogeographic reconstruction of 2019-nCoV isolates. The probable origin of 2019-nCoV is, as expected, Wuhan with a state posterior probability (spp) of 0.93 dating back the time of the most recent common ancestor (MRCA) of the human outbreak to November 25, 2019 (95%HPD: September 28, 2019; December 21, 2019), while the MRCA of Bat SARS-like Coronavirus and related 2019-nCoV lineages dates back to February 22, 2011 (95%HPD: September 20, 2008; August 15, 2014) (Figure S1), which may suggest a relatively extended period of sub-epidemic circulation before the most recent events. The first evidence of 2019-nCoV dissemination appears to be, according to our phylogeographic reconstruction, from Wuhan, China, to Nonthamburi, Thailand, with an spp. of 0.96, followed by the emergence of two distinct lineages, one with further spreading in Nonthamburi, and the second one following a more complex pattern: from Nonthamburi to Zhejiang, Huangzhou (spp = 0.47), as well as from Zhejiang to Kanagawa, Kanto (spp = 0.62) and from Nonthamburi to Guandong, Zhuhai (spp = 0.45). The first reported US cases, in Chicago, Illinois and Seattle, Washington, appeared to be linked to Guandong, Zhuhai isolates, in agreement with reports of patients traveling back from that region of China before being diagnosed. Finally, our analysis identified the Bat SARS-like Coronavirus as the most probable origin of the 2019-nCoV (spp = 0.99).
Figure 1.
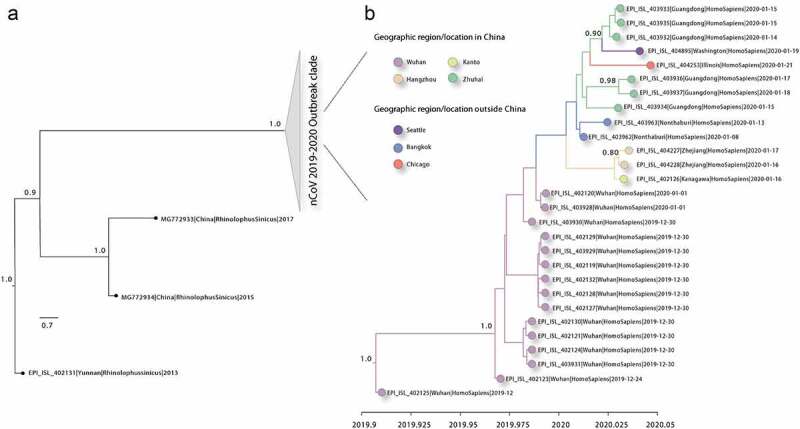
(a) Maximum clade credibility (MCC) tree estimated from complete or near-complete nCoV virus genomes available by enforcing a relaxed molecular clock. Triangular clades represent the nCoV 2019–2020 outbreak clade. (b) Expansion of the clade containing the novel genomes sequences from the nCoV 2019–2020 epidemic. Clade posterior probabilities are shown at well supported nodes. Internal branches were colored by ancestral state reconstruction with support shown when greater than 0.8.
Discussion
Very little is known about 2019-nCoV virus, including basic biology, animal source or any specific treatment. The substantial degree of genetic heterogeneity (15%) accumulated among human isolates during the past few months of the ongoing epidemic outbreak is not necessarily surprising for an RNA virus that has been shown to be a measurably evolving population over short time spans [15]. However, our findings underscore the urgent need for further molecular surveillance and the development of appropriate and an in-depth monitoring system capable of investigating viral mutation and transmission capabilities as 2019-nCoV unfortunately keeps spreading at a regional and potential global level. In other words, given the virus’s fast evolutionary rate and population dynamic, tracking the emergence of novel transmission routes and/or patterns should be considered a significant priority.
The results of our Bayesian phylogeographic reconstruction seem to be in agreement with a recent report of Benvenuto et al. (https://www.biorxiv.org/) suggesting that a Bat SARS-like coronavirus sequence is homologous and genetically more similar to the 2019-nCoV than other Bat SARS-like coronavirus sequences, but very distant from sequences isolated in SARS 202/2003 epidemic and MERS coronavirus. The finding may imply a most recent common ancestor between 2019-nCoV and the Bat SARS-like Coronavirus circulating in the Rhinolophus bat family. We also identify, in agreement with epidemiological reports, the city of Wuhan as the most likely origin of the human epidemic, dating back to the end of November 2019. In 2010, a previous article has suggested that the emergence of diverse virus strains within a few decades, in the different Rhinolophus species, may be the result of rapid evolution generating variants with the ability of easily crossing species barriers. The same study has shown that the epizootic transmission of the SARS from bat to human during the 2003 epidemic may have actually occurred up to 8 years earlier than the actual human outbreak [16]. Moreover, the migration map of the Rhinolophus bats in China involves almost the same geographic areas of the 2019-nCoV epidemic (http://www.bio.bris.ac.uk/research/bats/China%20bats/rhinolophussinicus.htm). Taken together, these results indicate a recurring pattern among the sub-genre of the Betacoronavirus leading to periodical epizootic epidemics.
In the present 2019-nCoV epidemic, WHO estimates R0, the basic reproduction number, as 1.4 to 2.5 less than SARS (2 to 5); but, this number can grow if the epidemic is not controlled by applying quarantine and isolation strategies. Purely epidemiological data such as incidence reports and contact tracing can provide background information on individual cases and population level transmission. However, molecular epidemiological data analyses, when sampling strategies are appropriate and representative of the full genetic heterogeneity of the pathogen population, can circumvent human error and present quantitative information about an infectious agent.
Combining epidemiology with molecular evolutionary data in a holistic approach is valuable for understanding the virus epidemic history and transmission in order to implement effective public health measures and prevent future epidemics like SARS-CoV and 2019-nCoV. Using phylodynamic analysis to investigate 2019-nCoV evolutionary history will add indispensable details to curb the current outbreak by identifying most closely related cases and providing crucial information of transmission and evolutionary patterns.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Weiss SR, Leibowitz JL.. Coronavirus pathogenesis. Adv Virus Res. 2011;81:85–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Drosten C, Günther S, Preiser W, et al. Identification of a novel coronavirus associated with severe acute respiratory syndrome. N Engl J Med. 2003;348(20):1967–1976. [DOI] [PubMed] [Google Scholar]
- [3].Zhu N, Zhang D, Wang W, et al. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med. 2020. January 24. DOI: 10.1056/NEJMoa2001017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Lu H, Stratton CW, Tang YW.. Outbreak of pneumonia of unknown etiology in Wuhan China: the mystery and the miracle. J Med Virol. 2020. DOI: 10.1002/jmv.25678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Ji W, Wang W, Zhao X, et al. Homologous recombination within the spike glycoprotein of the newly identified coronavirus may boost cross‐species transmission from snake to human. J Med Virol. 2020. January 22. DOI: 10.1002/jmv.25682 [DOI] [Google Scholar]
- [6].Heymann DL. Emerging understandings of 2019-nCoV. Lancet. 2020. January 24. DOI: 10.1016/S0140-6736(20)30186-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Katoh K, Rozewicki J, Yamada KD. MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief Bioinform. 2017; 20(4): 1160–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Nguyen LT, Schmidt HA, von Haeseler A, et al. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 2015;32(1):268–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Darriba D, Taboada GL, Doallo R, et al. jModelTest 2: more models, new heuristics and parallel computing. Nat Methods. 2012;9(8):772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Rambaut A, Lam TT, Max Carvalho L, et al. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2016;2(1):vew007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Suchard MA, Lemey P, Baele G, et al. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018;4(1):vey016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Baele G, Li WL, Drummond AJ, et al. Accurate model selection of relaxed molecular clocks in Bayesian phylogenetics. Mol Biol Evol. 2013;30(2):239–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Drummond AJ, Ho SY, Phillips MJ, et al. Relaxed phylogenetics and dating with confidence. PLoS Biol. 2006;4(5):e88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Rambaut A, Drummond AJ, Xie D, et al. Posterior summarization in Bayesian phylogenetics using tracer 1.7. Syst Biol. 2018;67(5):901–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Salemi M, Fitch WM, Ciccozzi M, et al. SARS-CoV sequence characteristics and evolutionary rate estimate from maximum likelihood analysis. J Virol. 2004;78:1602–1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Lau SK, Li KS, Huang Y, et al. Ecoepidemiology and complete genome comparison of different strains of severe acute respiratory syndrome-related Rhinolophus bat coronavirus in China reveal bats as a reservoir for acute, self-limiting infection that allows recombination events. J Virol. 2010;84(6):2808–2819. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.