

Abstract
As the infection of 2019-nCoV coronavirus is quickly developing into a global pneumonia epidemic, careful analysis of its transmission and cellular mechanisms is sorely needed. In this report, we first analyzed two recent studies which concluded that snakes are the intermediate hosts of 2019-nCoV and that the 2019-nCoV spike protein insertions shared a unique similarity to HIV-1. The re-implementation of the analyses, built on larger-scale datasets using state-of-the-art bioinformatics methods and databases, present however clear evidences rebutting these conclusions. Next, using metagenomic samples from Manis javanica we assembled a draft genome of the 2019-nCoV-like coronavirus, which shows 73% coverage and 91% sequence identity to the 2019-nCoV genome. In particular, the alignments of the spike surface glycoprotein receptor binding domain revealed 4-fold more variations in the bat coronavirus RaTG13 than those in the Manis coronavirus compared to 2019-nCoV, suggesting the pangolin as a missing link in the transmission of 2019-nCoV from bats to human.
Keywords: 2019-nCoV, metagenome assembly, Malayan pangolins, spike protein
Graphical Abstract
INTRODUCTION
The 2019 novel coronavirus (2019-nCoV), also known as SARS-CoV-21 and HCoV-192, is the pathogen behind COVID-19, a new type of pneumonia initially causing an outbreak in Wuhan, China, which has since spread to most countries in the world. The rapid transmission across country borders and the large number of confirmed cases prompted the World Health Organization (WHO) to declare COVID-19 as a global pandemic in March 11, 2020. As of March 20, there are at least 266,073 and 11,184 patients diagnosed with and died of COVID-19 worldwide, respectively. Among affected countries, China has the largest population of confirmed cases (81,416) and the second highest death toll (3,261). Meanwhile, Europe and North America were also hit hard: 47,021 and 15,219 cases were confirmed in Italy and the US, which are the nations with the highest number of 2019-nCoV infected patients in their respective continents, with the number of deaths in Italy (4,032) surpassing that of China. Understanding the viral infection mechanisms and animal hosts are of high urgency for the control and treatment of the 2019-nCoV virus. While it is now commonly recognized that bats such as Rhinolophus affinis may serve as the natural reservoir of 2019-nCoV,3 it is still unclear which animal serves as an intermediate host that brought the bat coronavirus to human hosts. While multiple studies suggest the Malayan pangolin (Manis javanica) as another host,4–6 some studies propose that the pangolin may be a natural host rather than an intermediate host.7–8
During the 2019-nCoV’s infection of host cells, a critical virion component is the spike surface glycoprotein, also known as the S protein. Spike proteins constitute the outermost component in a coronavirus virion particle and are responsible for viral recognition of Angiotensin Converting Enzyme 2 (ACE2), a transmembrane receptor on mammalian hosts that is utilized by the coronavirus to enter the host cells.3, 9 Therefore, the spike protein largely determines host specificity and infectivity of a coronavirus.
In this report, we first analyzed the results of two recent studies,10–11 which have spurred numerous interests and discussions in the community and society, regarding the sequence and structure of the spike protein in 2019-nCoV and the identification of its intermediate hosts. In particular, the study by Pradhan et al. reported the identification of four unique insertions that were shared only with HIV-1 and “unlikely to be fortuitous in nature”.10 Although the work has been questioned by the scientific community, rumors and conspiracy theories based on these studies still widely circulate among the general public.12 We therefore believe that there is an urgent need to systematically examine the bases and conclusions of these studies in serious scientific reports. To further examine the animal hosts of the 2019-nCoV spread, we next assembled the draft genome of a highly related coronavirus using metagenomic samples from Manis javanica. The alignment results of the assembled genome sequences, in particular on the spike proteins, suggest the importance of pangolins in the evolution of 2019-nCoV and its transmission from bats to humans.
MATERIALS AND METHODS
Protein Sequence Alignment
Global protein sequence alignment of the full-length coronavirus spike proteins was performed by MUSCLE13 and visualized by SeaView14.
Structure Prediction of Spike-ACE2 complex
We used C-I-TASSER15 to create structural models of the full-length spike protein. Here, C-I-TASSER is an extended pipeline of I-TASSER16 and utilizes the deep convolutional neural-network based contact-maps17 to guide the Monte Carlo fragment assembly simulations. Since the RBD domain of spike exhibits different conformations relative to the remaining portion of the protein, the DEMO pipeline18 was then used to re-assemble the domains and to construct a complex structure consisting of spike trimer and the extracellular domain of human ACE2, using the ACE2-bound conformation 2 of the SARS-CoV spike glycoprotein (PDB ID: 6ACJ) as a template. Our complex modeling did not use the template originally used in the Pradhan et al. study (PDB ID: 6ACD) because it did not include the ACE2 receptor.
Relative Synonymous Codon Usage (RSCU) Analysis
As per the previous study11, the RSCU for codon in a species is calculated as
| (1) |
where is the number of codons synonymous to codon (including itself), is the probability of the respective amino acid being encoded by codon among all synonymous codons in the CDSs of the whole genome. The difference of codon usage in two different species (a virus versus a vertebrate in our case) is defined by squared Euclidean distance of RSCU, i.e.,
| (2) |
Here, is the number of codons that encodes amino acids, thereby excluding the 3 stop codons. and are the RSCU for codon in the virus and in the vertebrate, respectively. In our report, the codon usages of all vertebrates are taken from the CoCoPUTS19 database, which was last updated in January 2020. This database was therefore much more recent than the Codon Usage Database20 last updated in 2007 that was used in the previous research11. To obtain the codon usage of coronaviruses, we imported the GenBank annotations of the three coronavirus genomes to SnapGene (GSL Biotech LLC) to export the codon usage table based on GenBank annotations. CodonW21 was not used for codon usage calculation as in the previous study because it cannot account for the −1 frameshift translation of the first open reading frame (ORF) in the coronavirus genome.
RESULTS AND DISCUSSION
2019-nCoV Spike Protein does not Include Insertions Unique to HIV-1
In a recent manuscript entitled “Uncanny similarity of unique inserts in the 2019-nCoV spike protein to HIV-1 gp120 and Gag”10, Pradhan et al. presented a discovery of four novel insertions unique to 2019-nCoV spike protein (Figure 1). They further concluded that these four insertions are part of the receptor binding site of 2019-nCoV, and that these insertions shared “uncanny similarity” to Human Immunodeficiency Virus 1 (HIV-1) proteins but not to other coronaviruses. These claims have resulted in considerable public panic and controversy in the community,12 even after the manuscript was withdrawn. To investigate whether the conclusions by Pradhan et al. are scientifically precise, we re-analyzed the structural location and sequence homology of the four spike protein insertions discussed therein.
Figure 1.

Sequence alignment of spike proteins from 2019-nCoV (NCBI accession: QHD43416) and SARS-CoV (UniProt ID: P59594). The four “novel” insertions “GTNGTKR” (IS1), “YYHKNNKS” (IS2), “GDSSSG” (IS3) and “QTNSPRRA” (IS4) by Pradhan et al. are highlighted in dashed rectangles. We noted that these fragments are not bona fide “insertions”; in fact, at least three out of all four fragments are also shared with Bat Coronavirus RaTG13 spike glycoprotein (NCBI accession: QHR63300.1), as shown in Table 1. Nevertheless, we still refer these fragments as “insertions” in this manuscript for consistency with the original report. The receptor binding domain of spike is marked within the solid box, which corresponds to residue positions 323 to 545 in the above alignment. A pair of arrows immediately following IS4 indicates the protease cleavage site by which spike proteins are cut into S1 and S2 isoforms.
Since the full length structure of the spike protein in 2019-nCoV was not available at the beginning of this study, we used C-I-TASSER15 to model its tertiary structure, as part of our effort for full genome structure and function analyses of 2019-nCoV, which is available at https://zhanglab.ccmb.med.umich.edu/C-I-TASSER/2019-nCoV/. The 2019-nCoV spike model was then assembled with the human ACE2 structure (PDB ID: 6ACJ)22 by DEMO18 to form a spike-ACE2 complex. In Figure 2A, we present a cartoon superposition of the C-I-TASSER model with a recently solved spike structure,23 where the C-I-TASSER model shares a high structure similarity with TM-score=0.95 24–25 to the cryo-EM structure. Since the experimental structure only covers 75% of residues in the full length sequence with several important residues on the Receptor Binding Domain (RBD) of the spike protein missing, our following analysis will be mainly built on the C-I-TASSER reconstructed full-length model. We note that C-I-TASSER, also known as “Zhang-Server”, is the top ranked automated server for protein structure prediction in the Critical Assessment of protein Structure Prediction round 13 (CASP13) challenge (http://www.predictioncenter.org/casp13/zscores_final.cgi?model_type=best&gr_type=server_only) among all 39 servers from the community. C-I-TASSER improves our previously-developed I-TASSER structure prediction protocol26 by incorporating deep learning-based contact map prediction17, 27. On all 121 CASP13 targets, the average TM-score of C-I-TASSER first model (0.674) is 8.0% higher than that of I-TASSER (0.624), and 0.15% higher than that of C-QUARK (0.673), which is our only other automated CASP13 server and was ranked in the second place in CASP13.
Figure 2.
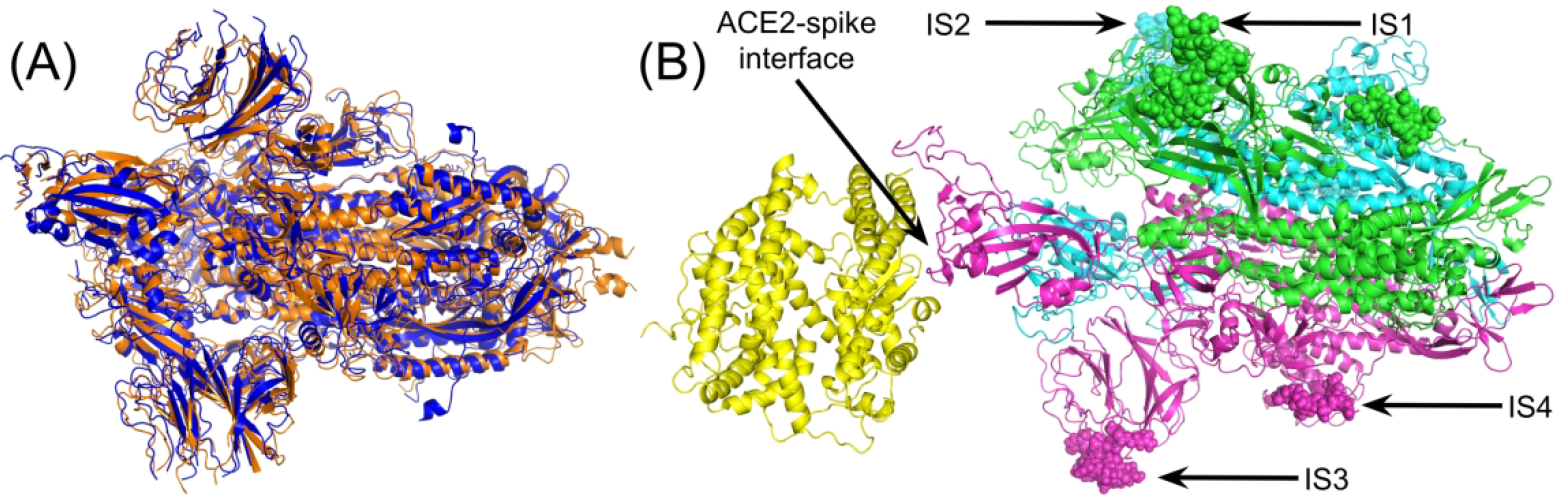
Structure of the 2019-nCoV spike protein trimer. (A) Superposition between C-I-TASSER constructed model (blue) and experimental structure (orange, PDB ID 6vsb), which was determined after our model was predicted. Only residues common to both structures are shown. (B) Complex structure model between human ACE2 (left yellow) and the spike protein trimer (right, with three chains colored in magenta, cyan, and blue respectively) constructed by C-I-TASSER. The four insertions are shown as spheres. During different stages of coronavirus infection, the spike proteins may be post-processed (i.e. cleaved) to produce different isoforms. Therefore, the eventual spike complex might not include all residues of a full-length spike protein. Nevertheless, we construct the complex model using full-length spike sequence to illustrate the locations of the four insertions.
As shown in Figure 2B, all four insertions in the C-I-TASSER/DEMO structural models are located outside the RBD of the spike protein, in contrast to the original conclusion made by Pradhan et al. which stated that the insertions are located on the interface with ACE2. Here it is important to note that, following ACE2 receptor binding, the spike protein can be cleaved by host proteases such as Cathepsin L (CTSL) to produce the S1 and S2 isoforms to facilitate viral entry into host cells 28–29. Since this cutting site follows immediately insertion 4 (IS4) (Figure 1 arrow) along the 2019-nCoV spike protein sequence, there is a possibility that IS4 could affect the cleavage of the spike protein. Regardless, all the insertions are not directly related to receptor binding.
To investigate viral homologs of the four insertions, we further performed a BLAST sequence search of these four insertions against the non-redundant (NR) sequence database, restricting the search results to viruses (taxid: 10239), but leaving other search parameters at default values. The top 5 sequence homologs (including the query itself) identified for each insertion are listed in Table 1. In contrast to the previous claim that the four insertions are unique to 2019-nCoV and HIV-1, all four insertion fragments can be found in other viruses. In fact, an HIV-1 protein is among the top BLAST hits for only one of the four insertion fragments, while three of the four insertion fragments are found in bat coronavirus RaTG13. Moreover, partly due to the very short length of these insertions, which range from 6 to 8 amino acids, the E-value of the BLAST hits, which is a parameter used by BLAST for assessing the statistical significance of the alignments and usually needs to be below 0.01 to be considered as significant,30 are all greater than 4, except for a bat coronavirus hit for IS2. These high E-values suggest that the majority of these similarities are likely to be coincidental.
Table 1.
BLAST search result for IS1. If there are multiple redundant hits for the same gene from different strains of the same species removed, only one hit is shown. Sequence identity is calculated as the number of identical residues divided by query length. Only the sequence portion aligned to the query is shown. In this table, we also list the closest BLAST hit from bat coronavirus RaTG13, which is known to be closely related to 2019-nCoV.3
| IS | NCBI accession |
Sequence | E- value |
Sequence Identity |
Species |
|---|---|---|---|---|---|
|
| |||||
| Query | GTNGTKR | 27 | 1.00 | 2019-nCoV | |
| APC94153 | GTNGTKR | 28 | 1.00 | uncultured marine virus | |
| AFU28737 | -TNGTKR | 224 | 0.86 | Human immunodeficiency virus 1 | |
| IS1 | AVE17137 | GTDGTKR | 224 | 0.86 | Rat astrovirus Rn/S510/Guangzhou |
| QBX18329 | -TNGTKR | 224 | 0.86 | Streptococcus phage Javan411 | |
| QHR63300 | GTNGIKR | 643 | 0.86 | Bat coronavirus RaTG13 | |
|
| |||||
| Query | YYHKNNKS | 0.13 | 1.00 | 2019-nCoV | |
| QHR63300 | YYHKNNKS | 0.13 | 1.00 | Bat coronavirus RaTG13 | |
| IS2 | AUL79732 | -YHKNNKS | 4.2 | 0.88 | Tupanvirus deep ocean |
| YP_007007173 | YYHKDNK- | 8.7 | 0.75 | Klebsiella phage vB_KleM_RaK2 | |
| ALS03575 | YYHKNN-- | 12 | 0.75 | Gokushovirus WZ-2015a | |
|
| |||||
| Query | GDSSSG | 1004 | 1.00 | 2019-nCoV | |
| QAU19544 | GDSSSG | 1003 | 1.00 | Orthohepevirus C | |
| IS3 | AYV78550 | GDSSSG | 1004 | 1.00 | Edafosvirus sp. |
| QHR63300 | GDSSSG | 1004 | 1.00 | Bat coronavirus RaTG13 | |
| QDP55596 | GDSSSG | 1004 | 1.00 | Prokaryotic dsDNA virus sp. | |
|
| |||||
| Query | QTNSPRRA | 1.0 | 1.00 | 2019-nCoV | |
| IS4 | |||||
| YP_009226728 | QTNSPRR- | 8.5 | 0.88 | Staphylococcus phage SPbeta-like | |
| BAF95810 | QTNSPRRA | 35 | 1.00 | Bovine papillomavirus type 9 | |
| ARV85991 | ETNSPRR- | 106 | 0.75 | Peach associated luteovirus | |
| QDH92312 | QTNAPRKA | 142 | 0.75 | Gordonia phage Spooky | |
Given that 3 out of the 4 insertion fragments are found in the bat coronavirus RaTG13, it is tempting to assume that these “insertions” may be directly inherited from bat coronaviruses. Currently, there are at least 7 known human coronaviruses (2019-nCoV, SARS-CoV, MERS-CoV, HCoV-229E, HCoV-OC43, HCoV-NL63 and HCoV-HKU1), where many of them, including Severe Acute Respiratory Syndrome-related Coronavirus (SARS-CoV) and Middle East Respiratory Syndrome-related Coronavirus (MERS-CoV), were shown to be transmitted from bats.3, 31–34 To further examine the evolutionary relationship between the 2019-nCoV and the bat coronavirus, in comparison with other human coronaviruses, we used MUSCLE to create a multiple sequence alignment (MSA) presented in Figure 3 for all the 7 human coronaviruses and two bat coronaviruses, RaTG13 and RsSHC014, which have been considered to be the ancestors of 2019-nCoV and SARS-CoV, respectively.3, 31, 34 Among the 4 “insertions” (ISs) of the 2019-nCoV, IS1 has only 1 residue different from the bat coronavirus, and 3 out of 7 residues are identical with MERS-CoV. IS2 and IS3 are all identical to the bat coronavirus. For IS4, although the local sequence alignment by BLAST did not hit the bat coronavirus in Table 1, it has a close evolutionary relation with the bat coronavirus in the MSA. In particular, the first 6 residues in the IS4 fragment “QTQTNSPRRA” from 2019-nCoV are identical to RaTG13, while the last 4 residues, which were absent in the bat coronavirus or SARS-CoV, have at least 50% identity to MERS-CoV and HCoV-HKU1.
Figure 3.
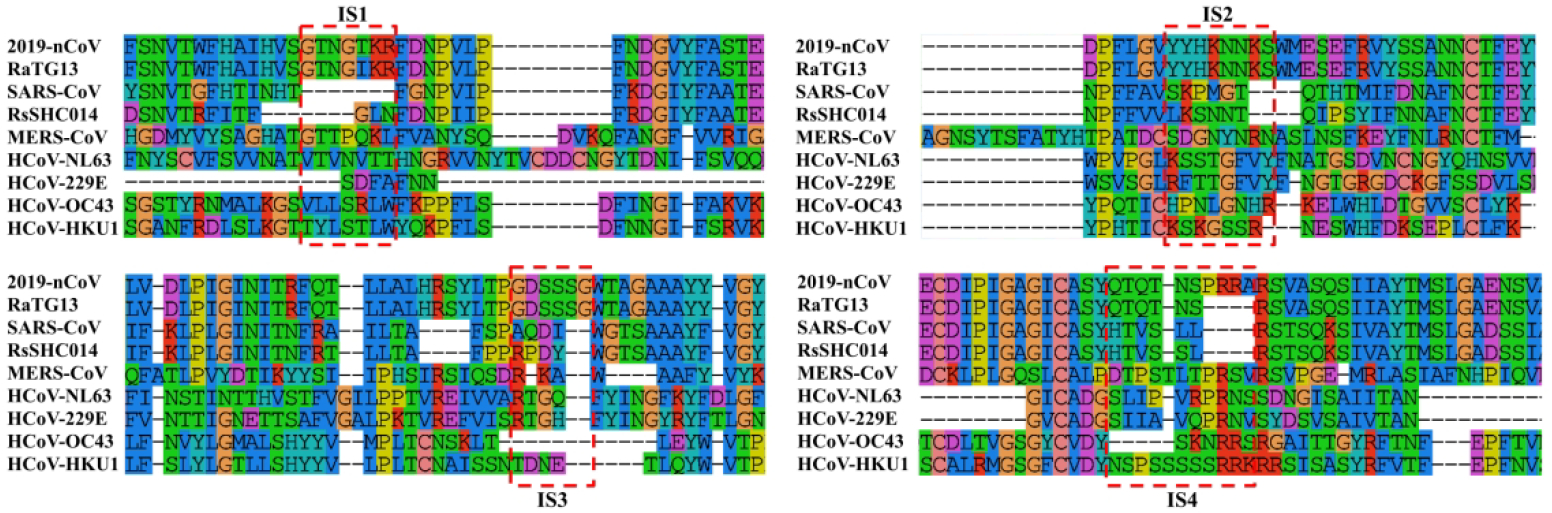
Multiple sequence alignment for the spike proteins of 7 known human coronaviruses. 2019-nCoV (QHD43416.1), SARS-CoV (P59594), MERS-CoV (YP_009047204.1), HCoV-NL63 (YP_003767.1), HCoV-229E (NP_073551.1), HCoV-OC43 (YP_009555241.1) and HCoV-HKU1 (YP_173238.1) sequences are downloaded from the NCBI and UniProt databases. RaTG13 (QHR63300.1) and RsSHC014 (AGZ48806.1), the two bat coronaviruses thought to be the ancestors of 2019-nCoV and SARS-CoV, are also included. For brevity, only the regions near the four “insertions” are displayed in the figure.
Putting these together, we believe that there is a close evolutionary relation between 2019-nCoV and bat coronavirus RaTG13. The four insertions highlighted by Pradhan et al. in the spike protein are not unique to 2019-nCoV and HIV-1. In fact, the similarities in the sequence-based alignments built on these very short fragments are statistically insignificant, as assessed by the BLAST E-values, and such similarities are shared in many other viruses including the bat coronavirus. Structurally, these “insertions” are far away from the binding interface of the spike protein with the ACE2 receptor, as shown in Figure 2, which is also contradictory to the conclusion made by Pradhan et al.
Relative Synonymous Codon Usage Cannot Identify Intermediate Hosts of Coronaviruses
Another early study attempting to understand the infection of 2019-nCoV was performed by Ji et al.11. In this study, the authors analyzed the Relative Synonymous Codon Usage (RSCU) of 2019-nCoV and 8 vertebrates, including two species of snakes (Bungarus multicinctus and Naja atra), hedgehog (Erinaceus europaeus), bat (Rhinolophus sinicus), marmot (Marmota), pangolin (Manis javanica), chicken (Gallus gallus), and human (Homo sapiens). Among these vertebrates, snakes have the smallest codon usage difference (squared Euclidean distance of RSCU) from 2019-nCoV and were therefore proposed by Ji et al. as the intermediate hosts of 2019-nCoV.
This conclusion is however controversial among virologists, due to the lack of prior biological evidence that zoonotic coronavirus can infect animals other than mammals and birds 35. Moreover, recent studies showed preliminary evidence that pangolins are the likely hosts of 2019-nCoV-like coronaviruses,4–6 further invalidating Ji et al.’s conclusion. While the conclusion of snakes being intermediate hosts seems commonly questioned by the scientific community, it is still important to carefully examine the base and reliability of the RSCU approach, which should help prevent such biased analyses from misleading the community and general public. In this report, we scrutinize the bioinformatics approach and the underlying biological assumptions through a large-scale replication of the RSCU analysis.
The bioinformatics analysis performed in the Ji et al. study has several limitations. First, there are only approximately 300 protein coding sequences (CDSs) in the NCBI GenBank for the snake species (Bungarus multicinctus and Naja atra) which the authors chose for their analysis. These CDSs represent <2% of all protein coding genes in a typical snake genome; the genome of King cobra (Naja hannah), for example, encodes 18,387 proteins according to UniProt (https://www.uniprot.org/proteomes/UP000018936). The limited numbers of known CDSs in Bungarus multicinctus and Naja atra mean that the RSCU statistics may not reflect the actual RSCU distribution in the whole genome. Second, the Codon Usage Database20 used in the analysis of Ji et al. has not been updated since 2007; a re-analysis using more recent codon usage database such as CoCoPUTs19 is therefore needed. Third, only 8 vertebrates were analyzed in their study, while there are >100,000 vertebrates with at least one CDS in the NCBI GenBank database. Finally, there is no established evidence that viruses evolve their codon usage to resemble that of their animal hosts36; this calls for a careful benchmark of RSCU analysis in terms of its ability to re-discover known hosts of characterized viruses.
To address these issues, we re-implemented the RSCU comparison algorithm proposed by Ji et al. to analyze the codon usage in the 2019-nCoV genome (NCBI accession MN908947.3) and those of all 102,367 vertebrate species in the CoCoPUTS database. To test whether this kind of analysis can recover known hosts of well-studied coronavirus, SARS-CoV (NCBI accession NC_004718) and MERS-CoV (NCBI accession NC_019843) are also included. Codon usage frequency are converted to squared Euclidean distance of RSCU in two separate analyses: one based on all vertebrates (Supplementary Figure S1A–C) and the other on the subset of vertebrates with enough statistics, i.e. >2000 known CDSs (Suppelemtnary Figure S1D–F), roughly corresponding to 10% of all protein coding genes in a typical vertebrate genome.
As shown in Figure 4A, snakes are not the vertebrates with the lowest RSCU distances to 2019-nCoV, suggesting that the implementation of RSCU analysis by Ji et al. was incomplete. More importantly, the data in Figure 4 shows that animals unrelated to coronavirus transmission, such as frogs and snakes, consistently have smaller RSCU distances to known hosts of all three coronaviruses. For example, the top-ranking vertebrates with the lowest RSCU distances to the three different coronaviruses are two kinds of frogs (Megophrys feae and Liophryne schlaginhaufeni), while another frog (Xenopus laevis) has the smallest RSCU distances among all vertebrates with sufficient sequences. Part of the reason for the failure of RSCU in intermediate host identification, as shown in Supplementary Table S1, is that different coronaviruses, such as SARS-CoV and MERS-CoV, that are known to utilize different intermediate hosts (Paguma larvata and Camelus dromedarius, respectively), have almost no difference in RSCU (squared RSCU distance=0.12). These data suggest that the RSCU analysis on its own is not specific enough to discriminate coronaviruses from different vertebrate hosts. In this regard, the failure is not merely due to the use of outdated databases or the small number of species included in the original analysis, but in fact caused by the incorrect biological assumption that coronaviruses will evolve their RSCU to resemble that of their hosts.
Figure 4.
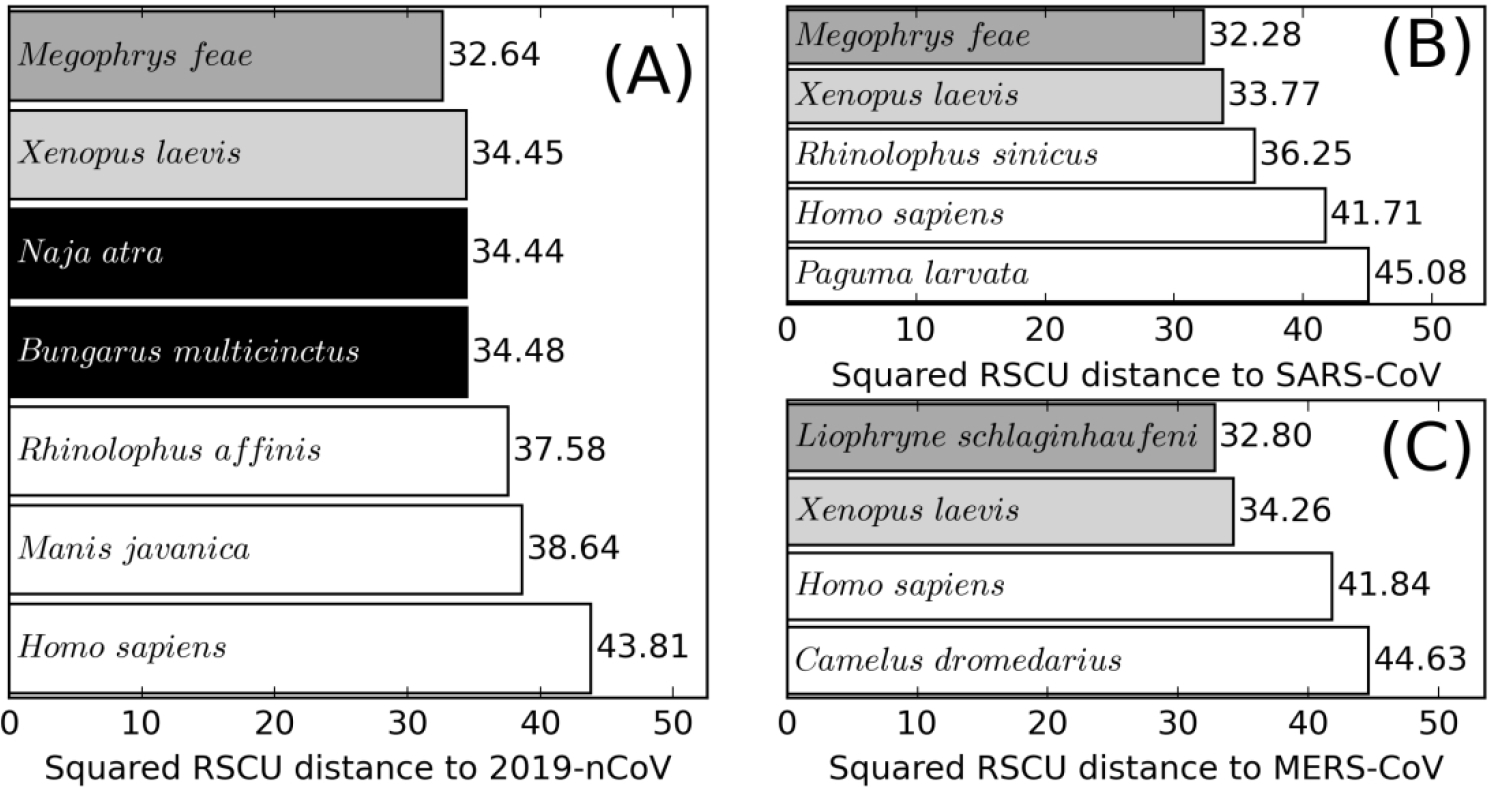
Inability of RSCU analysis for coronavirus host identification for 2019-nCoV (A), SARS-CoV (B) and MERS-CoV (C). The vertebrate species (frogs) with the lowest squared Euclidean distances of RSCU (x-axis) to the coronavirus is colored in dark grey, while the vertebrate (frog) with the lowest RSCU distance and have sufficient statistics is colored in light grey. The snakes proposed by Ji et al. as intermediate hosts (Naja atra and Bungarus multicinctus snakes) are colored in black. Confirmed hosts (Rhinolopus affinis and Manis javanica for 2019-nCoV, Rhinolopus sinicus and Paguma larvata for SARS-CoV, and Camelus dromedarius for MERS-CoV, as well as Homo sapiens for all three coronaviruses) are colored in white. These data shows not only that snakes are not the vertebrates with the lowest RSCU distances to 2019-nCoV, but also that unrelated species such as frogs and snakes have smaller RSCU distances to known hosts of all three coronaviruses. These data suggest that the closeness of RSCU is not indicative of potential pathogen-host relation.
Metagenome Assembly Suggests Pangolins as Potential Hosts of 2019-nCoV
In a recent study,6 Xiao et al. first identified coronavirus sequences in pangolins that are highly similar to the 2019-nCoV. In addition, three independent groups also reported the identification of 2019-nCoV-like coronaviruses sequences from metagenomics samples taken from the Malayan pangolin (Manis javanica),4–5, 7 making the pangolin a likely intermediate host of the 2019-nCoV.
To further examine the possibility, we tried to re-assemble a draft genome sequence of the coronavirus using the metagenomic samples of Manis javanica. To this end, we first collected a set of all publicly available metagenome samples for pangolin, including 11 samples from lung, 8 samples from spleen, 2 samples from lymph (NCBI accession PRJNA573298)37 and 4 samples for feces (NCBI accession PRJNA476660)38, from the NCBI Sequence Read Archive (SRA) database39 using the prefetch command of SRA Toolkit version 2.10.3. These samples are converted into paired end sequencing read conversion in fastq format by faster-dump. A quality check by FastQC version 0.11.9 showed that, while the 4 samples from PRJNA476660 do not contain adaptor sequences, all 21 samples from PRJNA573298 contain Illumina universal adaptors. Therefore, for these 21 samples, Trimmomatic version 0.39 40 was used to remove adaptor sequences using the flag “ILLUMINACLIP:adapters.fa:2:30:10:2: keepBothReads LEADING:3 TRAILING:3 MINLEN:36”. To remove contaminations from host and from human researchers, only read pairs that cannot be mapped to Manis javanica or Homo sapiens genomes by bowtie41 version 2.3.5.1 are retained for further analysis. These sequences are converted from sam format of bowtie2 back to fastq format by samtools42 version 1.10 and bedtools43 version 2.29.2. Following these quality control processes, we next determined which of the 25 above-mentioned samples include 2019-nCoV-like sequence by two searches at the protein- and nucleotide-levels. In the protein-level search, the 2019-nCoV spike protein sequence was searched by blastp30 through protein sequences directly assembled from sequencing reads of a metagenome sample by Plass, a protein-level metagenome sequence assembler,44 to identify if there are any close hits with E-value <0.01. Meanwhile, the nucleotide-level search selected samples where more than one pair of sequencing reads can be mapped to the 2019-nCoV genome (NCBI accession: MN908947.3) by bowtie. Both searches consistently reported that only the lung samples (SRA accessions: SRR10168376, SRR10168377, and SRR10168378) contain 2019-nCoV-like sequences. Therefore, the sequences were assembled into nucleotide and protein contigs by MEGAHITS and Plass, respectively. The assembled nucleotide and protein sequences were then aligned by blastn and blastp to the whole genome and the spike protein of 2019-nCoV, respectively, at an E-value cutoff <0.01. Finally, we separately merged all nucleotide and protein alignments into a single pairwise alignment between 2019-nCoV and the Manis coronavirus (Manis-CoV); when multiple Manis-CoV hits cover the same 2019-nCoV region, the hit with the highest sequence identity of 2019-nCoV is used in the merged alignment.
Figure 5A presents a sketch of the draft genome for the Manis-CoV, as compared to the released 2019-nCoV genome45. Overall, the assembled sequences cover 73% of the 2019-nCoV genome with 91% sequence identity. More importantly, the protein sequences assembled from these Manis lung samples includes a partial pangolin coronavirus spike protein that is 92% identical to the 2019-nCoV spike protein (Figure 5B). This sequence identity is relatively high, considering that spike proteins are critical for the coronaviruses to invade into host cells and have the largest diversity in coronavirus genomes due to evolutionary pressure for adapting to receptors on different hosts. Notably, there are only 5 residue positions in the Manis coronavirus that that are different from 2019-nCoV on the Spike receptor binding domain, compared to the 19 different residue positions between 2019-nCoV and bat coronavirus RaTG13 for the same domain (Figure 5B black box). These data imply that pangolins such as Manis javanica can either be the intermediate hosts of 2019-nCoV between the transmission of bat coronaviruses to human, or serve as alternative natural hosts, together with bats, to provide the genetic material for the origin of 2019-nCoV. Nevertheless, considering that Manis javanica individuals with coronavirus infections are usually in poor or even critical health condition,37 and previously known natural coronaviruses’ hosts (such as bats) are usually asymptotic after infection, thus allowing long term virus-host coexistence and coevolution, we believe it is more likely that Manis javanica is an intermediate host rather than natural host.
Figure 5.

Alignment between 2019-nCoV and the coronavirus infecting Manis javanica lung (Manis-CoV). (A) Schematic of alignment between 2019-nCoV full genome (thin black line) and draft genome of Manis-CoV, where thick black lines are aligned regions. Protein coding genes are indicated by thick arrows. (B) MSA of spike proteins (marked by “s” in panel (A)) from 2019-nCoV, bat coronavirus RaTG13, and Manis-CoV. Since only 78% of the spike Manis-CoV sequence can be assembled, it contains several gaps in this MSA. Nevertheless, the sequence of the spike RBD domain (solid box) can be fully assembled, where 20 residue positions (marked by arrow pairs) are different between 2019-nCoV and the other two related coronaviruses.
Approximately one quarter of nucleotides are missing in our assembled Manis coronavirus draft genome, partly because compared to whole-genome sequencing, metagenome sequencing usually has lower read depth and more assembly errors caused by the mixture of diverse species in the samples. A higher quality genome with better coverage should in theory be attainable if the Manis coronavirus can be isolated and cultured in vitro using a mammalian cell line and subjected to whole-genome sequencing.
CONCLUSIONS
Due to the scarcity of experimental and clinical data, as well as the urgency to understand the infectivity of the deadly coronaviruses, we have been increasingly relying on computational analyses to study the 2019-nCoV virus in terms of protein structures, functions, phylogeny, and interactions at both molecular and organismal levels. Indeed, within less than a month of the publication of the 2019-nCoV genome in January 2020, multiple bioinformatics analyses regarding 2019-nCoV have been either published or posted as preprint. While such expeditious analyses provide much needed insights into the biology of the 2019-nCoV virus, there is a caution to avoid over-interpretation of the data at the absence of comprehensive benchmarks or follow-up experimental validations.
In this report, we have investigated two recently published computational analyses regarding intermediate host identification and the analysis of spike protein insertions. In both cases, we found that the conclusions proposed by the original studies do not hold in the face of more comprehensive replications of these analyses. In particular, we found that the unique sequence “inserts” found by Pradhan et al. are in fact shared by multiple viruses, especially with the segments from the Bat coronavirus RaTG13, revealing the close evolutionary relation to the latter species. In addition, our benchmark results showed that the data based on RSCU are not specific enough to discriminate the relation between coronaviruses and vertebrates, which contradicts with the conclusion by Ji et al. regarding snakes as immediate host of the 2019-nCoV.
Finally, we assembled a draft genome of the 2019-nCoV-like coronavirus using the metagenomic samples from the lung of Manis javanica, which shows an overall coverage of 73% of 2019-nCoV with 91% sequence identity. In particular, the spike protein in the assembled genome, which is critical for the virus to recognize host receptors and therefore bears a high speed of variation, share a high sequence identity to the 2019-nCoV with only 5 residue position difference, compared to 19 differences between the 2019-nCoV and the bat coronavirus RaTG13. These data provide evidences on the possible evolutionary relations between RaTG13, the Manis Coronavirus and 2019-nCoV.
While current evidence mainly points to the pangolin as the most likely intermediate host, it is possible for other animals to also serve as intermediate hosts for the following two reasons. Firstly, coronaviruses are known to have multiple intermediate hosts. For example, SARS-CoV, of which the palm civet (Paguma larvata) is the most well-known intermediate host, is also reported to use a raccoon dog (Nyctereutes procyonoides) and a ferret badger (Melogale moschata) as intermediate hosts.46 Secondly, the 91% sequence identity between the Manis coronavirus and 2019-nCoV is high enough for confirmation of evolutionary relation between the two viruses, but not high enough to consider them as the same viral species. To put this into perspective, the viral sequence from intermediate hosts of SARS-CoV and MERS-CoV are 99.8% and 99.9% identical to their human versions, respectively.46–47 Therefore, even with the discovery of the Manis Coronavirus, further searching for other potential intermediate hosts should be continued.
Supplementary Material
Figure S1. Top 20 vertebrate species ranked in ascending order of squared Euclidean distance of RSCU to 2019-nCoV, SARS-CoV, and MERS-CoV.
Table S1. Squared Euclidean distances of RSCU among coronaviruses and representative vertebrate species.
ACKNOWLEDGMENTS
We thank Drs. Gibert S. Omenn and Xiaoqiong Wei for critical review of this manuscript. This work used the Extreme Science and Engineering Discovery Environment (XSEDE),48 which is supported by National Science Foundation (ACI1548562).
Funding Sources
This work is supported in part by the National Institute of General Medical Sciences (GM083107, GM116960), the National Institute of Allergy and Infectious Diseases (AI134678), and the National Science Foundation (DBI1564756, IIS1901191).
ABBREVIATIONS
- 2019-nCoV
2019 novel coronavirus
- ACE2
angiotensin converting enzyme 2
- HIV-1
human immunodeficiency virus 1
- IS
insertion
- SARS-CoV
severe acute respiratory syndrome-related coronavirus
- RBD
receptor binding domain
- CDS
protein coding sequences
- MERS-CoV
Middle East respiratory syndrome-related coronavirus
- RSCU
relative synonymous codon usage
- Manis-CoV
the coronavirus infecting Manis javanica lung
Footnotes
The authors declare no competing financial interest.
REFERENCES
- 1.Gorbalenya AE; Baker SC; Baric RS; de Groot RJ; Drosten C; Gulyaeva AA; Haagmans BL; Lauber C; Leontovich AM; Neuman BW; Penzar D; Perlman S; Poon LLM; Samborskiy D; Sidorov IA; Sola I; Ziebuhr J, Severe acute respiratory syndrome-related coronavirus: The species and its viruses – a statement of the Coronavirus Study Group. bioRxiv 2020, 2020.02.07.937862. [Google Scholar]
- 2.Jiang S; Shi Z; Shu Y; Song J; Gao GF; Tan W; Guo D, A distinct name is needed for the new coronavirus. The Lancet 2020, 395 (10228), 949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhou P; Yang X-L; Wang X-G; Hu B; Zhang L; Zhang W; Si H-R; Zhu Y; Li B; Huang C-L; Chen H-D; Chen J; Luo Y; Guo H; Jiang R-D; Liu M-Q; Chen Y; Shen X-R; Wang X; Zheng X-S; Zhao K; Chen Q-J; Deng F; Liu L-L; Yan B; Zhan F-X; Wang Y-Y; Xiao G-F; Shi Z-L, A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wahba L; Jain N; Fire AZ; Shoura MJ; Artiles KL; McCoy MJ; Jeong DE, Identification of a pangolin niche for a 2019-nCoV-like coronavirus through an extensive meta-metagenomic search. bioRxiv 2020, 2020.02.08.939660. [Google Scholar]
- 5.Lam TT-Y; Shum MH-H; Zhu H-C; Tong Y-G; Ni X-B; Liao Y-S; Wei W; Cheung WY-M; Li W-J; Li L-F; Leung GM; Holmes EC; Hu Y-L; Guan Y, Identification of 2019-nCoV related coronaviruses in Malayan pangolins in southern China. bioRxiv 2020, 2020.02.13.945485. [Google Scholar]
- 6.Xiao K; Zhai J; Feng Y; Zhou N; Zhang X; Zou J-J; Li N; Guo Y; Li X; Shen X; Zhang Z; Shu F; Huang W; Li Y; Zhang Z; Chen R-A; Wu Y-J; Peng S-M; Huang M; Xie W-J; Cai Q-H; Hou F-H; Liu Y; Chen W; Xiao L; Shen Y, Isolation and Characterization of 2019-nCoV-like Coronavirus from Malayan Pangolins. bioRxiv 2020, 2020.02.17.951335. [Google Scholar]
- 7.Wong MC; Cregeen SJJ; Ajami NJ; Petrosino JF, Evidence of recombination in coronaviruses implicating pangolin origins of nCoV-2019. bioRxiv 2020, 2020.02.07.939207. [Google Scholar]
- 8.Liu P; Jiang J-Z; Hua Y; Wang X; Hou F; Wan X-F; Chen J; Zou J; Chen J, Are pangolins the intermediate host of the 2019 novel coronavirus (2019-nCoV) ? bioRxiv 2020, 2020.02.18.954628. [Google Scholar]
- 9.Letko M; Munster V, Functional assessment of cell entry and receptor usage for lineage B β-coronaviruses, including 2019-nCoV. bioRxiv 2020, 2020.01.22.915660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pradhan P; Pandey AK; Mishra A; Gupta P; Tripathi PK; Menon MB; Gomes J; Vivekanandan P; Kundu B, Uncanny similarity of unique inserts in the 2019-nCoV spike protein to HIV-1 gp120 and Gag. bioRxiv 2020, 2020.01.30.927871. [Google Scholar]
- 11.Ji W; Wang W; Zhao X; Zai J; Li X, Cross-species transmission of the newly identified coronavirus 2019-nCoV. Journal of Medical Virology 2020, 92 (4), 433–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Calisher C; Carroll D; Colwell R; Corley RB; Daszak P; Drosten C; Enjuanes L; Farrar J; Field H; Golding J; Gorbalenya A; Haagmans B; Hughes JM; Karesh WB; Keusch GT; Lam SK; Lubroth J; Mackenzie JS; Madoff L; Mazet J; Palese P; Perlman S; Poon L; Roizman B; Saif L; Subbarao K; Turner M, Statement in support of the scientists, public health professionals, and medical professionals of China combatting COVID-19. The Lancet 2020, 395 (10226), e42–e43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Edgar RC, MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research 2004, 32 (5), 1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gouy M; Guindon S; Gascuel O, SeaView Version 4: A Multiplatform Graphical User Interface for Sequence Alignment and Phylogenetic Tree Building. Molecular Biology and Evolution 2009, 27 (2), 221–224. [DOI] [PubMed] [Google Scholar]
- 15.Zheng W; Li Y; Zhang CX; Pearce R; Mortuza SM; Zhang Y, Deep-learning contact-map guided protein structure prediction in CASP13. Proteins 2019, 87 (12), 1149–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang J; Yan R; Roy A; Xu D; Poisson J; Zhang Y, The I-TASSER Suite: protein structure and function prediction. Nature Methods 2015, 12 (1), 7–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li Y; Zhang C; Bell EW; Yu DJ; Zhang Y, Ensembling multiple raw coevolutionary features with deep residual neural networks for contact-map prediction in CASP13. Proteins 2019, 87 (12), 1082–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhou XG; Hu J; Zhang CX; Zhang GJ; Zhang Y, Assembling multidomain protein structures through analogous global structural alignments. Proceedings of the National Academy of Sciences of the United States of America 2019, 116 (32), 15930–15938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alexaki A; Kames J; Holcomb DD; Athey J; Santana-Quintero LV; Lam PVN; Hamasaki-Katagiri N; Osipova E; Simonyan V; Bar H, Codon and Codon-Pair Usage Tables (CoCoPUTs): facilitating genetic variation analyses and recombinant gene design. Journal of molecular biology 2019, 431 (13), 2434–2441. [DOI] [PubMed] [Google Scholar]
- 20.Nakamura Y; Gojobori T; Ikemura T, Codon usage tabulated from international DNA sequence databases: status for the year 2000. Nucleic Acids Research 2000, 28 (1), 292–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Peden JF Analysis of codon usage. University of Nottingham, Nottingham, England, 1999. [Google Scholar]
- 22.Song W; Gui M; Wang X; Xiang Y, Cryo-EM structure of the SARS coronavirus spike glycoprotein in complex with its host cell receptor ACE2. PLoS Pathog 2018, 14 (8), e1007236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wrapp D; Wang N; Corbett KS; Goldsmith JA; Hsieh C-L; Abiona O; Graham BS; McLellan JS, Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, eabb2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang Y; Skolnick J, Scoring function for automated assessment of protein structure template quality. Proteins 2004, 57 (4), 702–710. [DOI] [PubMed] [Google Scholar]
- 25.Xu J; Zhang Y, How significant is a protein structure similarity with TM-score = 0.5? Bioinformatics 2010, 26 (7), 889–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang J; Yan R; Roy A; Xu D; Poisson J; Zhang Y, The I-TASSER Suite: protein structure and function prediction. Nat Methods 2015, 12 (1), 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li Y; Hu J; Zhang C; Yu D-J; Zhang Y, ResPRE: high-accuracy protein contact prediction by coupling precision matrix with deep residual neural networks. Bioinformatics 2019, btz291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Simmons G; Gosalia DN; Rennekamp AJ; Reeves JD; Diamond SL; Bates P, Inhibitors of cathepsin L prevent severe acute respiratory syndrome coronavirus entry. Proceedings of the National Academy of Sciences of the United States of America 2005, 102 (33), 11876–11881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang I-C; Bosch BJ; Li F; Li W; Lee KH; Ghiran S; Vasilieva N; Dermody TS; Harrison SC; Dormitzer PR, SARS coronavirus, but not human coronavirus NL63, utilizes cathepsin L to infect ACE2-expressing cells. Journal of Biological Chemistry 2006, 281 (6), 3198–3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Altschul SF; Madden TL; Schäffer AA; Zhang J; Zhang Z; Miller W; Lipman DJ, Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Research 1997, 25 (17), 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li W; Shi Z; Yu M; Ren W; Smith C; Epstein JH; Wang H; Crameri G; Hu Z; Zhang H; Zhang J; McEachern J; Field H; Daszak P; Eaton BT; Zhang S; Wang L-F, Bats Are Natural Reservoirs of SARS-Like Coronaviruses. Science 2005, 310 (5748), 676. [DOI] [PubMed] [Google Scholar]
- 32.Wang Q; Qi J; Yuan Y; Xuan Y; Han P; Wan Y; Ji W; Li Y; Wu Y; Wang J; Iwamoto A; Woo Patrick C. Y.; Yuen K-Y; Yan J; Lu G; Gao George F., Bat Origins of MERS-CoV Supported by Bat Coronavirus HKU4 Usage of Human Receptor CD26. Cell Host & Microbe 2014, 16 (3), 328–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Corman VM; Baldwin HJ; Tateno AF; Zerbinati RM; Annan A; Owusu M; Nkrumah EE; Maganga GD; Oppong S; Adu-Sarkodie Y; Vallo P; da Silva Filho LVRF; Leroy EM; Thiel V; van der Hoek L; Poon LLM; Tschapka M; Drosten C; Drexler JF, Evidence for an Ancestral Association of Human Coronavirus 229E with Bats. Journal of Virology 2015, 89 (23), 11858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hu B; Zeng L-P; Yang X-L; Ge X-Y; Zhang W; Li B; Xie J-Z; Shen X-R; Zhang Y-Z; Wang N; Luo D-S; Zheng X-S; Wang M-N; Daszak P; Wang L-F; Cui J; Shi Z-L, Discovery of a rich gene pool of bat SARS-related coronaviruses provides new insights into the origin of SARS coronavirus. PLOS Pathogens 2017, 13 (11), e1006698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Callaway E; Cyranoski D, Why snakes probably aren’t spreading the new China virus. Nature 2020, 577 (7792), 1. [DOI] [PubMed] [Google Scholar]
- 36.Meintjes PL; Rodrigo AG, Evolution of relative synonymous codon usage in Human Immunodeficiency Virus type-1. Journal of bioinformatics and computational biology 2005, 3 (01), 157–168. [DOI] [PubMed] [Google Scholar]
- 37.Liu P; Chen W; Chen JP, Viral Metagenomics Revealed Sendai Virus and Coronavirus Infection of Malayan Pangolins (Manis javanica). Viruses-Basel 2019, 11 (11), 979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ma JE; Jiang HY; Li LM; Zhang XJ; Li GY; Li HM; Jin XJ; Chen JP, The Fecal Metagenomics of Malayan Pangolins Identifies an Extensive Adaptation to Myrmecophagy. Front Microbiol 2018, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kodama Y; Shumway M; Leinonen R; C INSD, The sequence read archive: explosive growth of sequencing data. Nucleic Acids Research 2012, 40 (D1), D54–D56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bolger AM; Lohse M; Usadel B, Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30 (15), 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Langmead B; Salzberg SL, Fast gapped-read alignment with Bowtie 2. Nat Methods 2012, 9 (4), 357–U54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li H; Handsaker B; Wysoker A; Fennell T; Ruan J; Homer N; Marth G; Abecasis G; Durbin R; Proc GPD, The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25 (16), 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Quinlan AR; Hall IM, BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26 (6), 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Steinegger M; Mirdita M; Soding J, Protein-level assembly increases protein sequence recovery from metagenomic samples manyfold. Nat Methods 2019, 16 (7), 603–+. [DOI] [PubMed] [Google Scholar]
- 45.Wu F; Zhao S; Yu B; Chen Y-M; Wang W; Song Z-G; Hu Y; Tao Z-W; Tian J-H; Pei Y-Y, A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Guan Y; Zheng BJ; He YQ; Liu XL; Zhuang ZX; Cheung CL; Luo SW; Li PH; Zhang LJ; Guan YJ; Butt KM; Wong KL; Chan KW; Lim W; Shortridge KF; Yuen KY; Peiris JSM; Poon LLM, Isolation and characterization of viruses related to the SARS coronavirus from animals in Southern China. Science 2003, 302 (5643), 276–278. [DOI] [PubMed] [Google Scholar]
- 47.Hemida MG; Chu DKW; Poon LLM; Perera RAPM; Alhammadi MA; Ng HY; Siu LY; Guan Y; Alnaeem A; Peiris M, MERS Coronavirus in Dromedary Camel Herd, Saudi Arabia. Emerg Infect Dis 2014, 20 (7), 1231–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Towns J; Cockerill T; Dahan M; Foster I; Gaither K; Grimshaw A; Hazlewood V; Lathrop S; Lifka D; Peterson GD; Roskies R; Scott JR; Wilkins-Diehr N, XSEDE: Accelerating Scientific Discovery. Comput Sci Eng 2014, 16 (5), 62–74. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Top 20 vertebrate species ranked in ascending order of squared Euclidean distance of RSCU to 2019-nCoV, SARS-CoV, and MERS-CoV.
Table S1. Squared Euclidean distances of RSCU among coronaviruses and representative vertebrate species.