

Abstract
The proinflammatory cytokine interleukin-17A (IL-17A) is known to mediate antimicrobial activity, but its role during rhinovirus (RV) infections and in asthma needs further investigation. Therefore, we addressed the role of IL-17A during allergic asthma and antiviral immune response in human and murine immunocompetent cells. In this study we found that asthmatic children with a RV infection in their upper airways have upregulated mRNA levels of the antiviral cytokine interferon type I (IFN)-β and the transcription factor T-box 21 (TBX21) and reduced levels of IL-17A protein in their peripheral blood mononuclear cells (PBMCs). We also found that IL-17A inhibited RV1b replication in infected human lung epithelial cells A549. Furthermore, by using gene array analysis we discovered that targeted deletion of Il17a in murine lung CD4+ T cells impaired Oas1g mRNA downstream of Ifnβ, independently from RV infection. Additionally, in PBMCs of children with a RV infection in their nasalpharyngeal fluid OAS1 gene expression was found downregulated. Finally RV1b inhibited IL-17A production in lung CD4+ T cells in a setting of experimental asthma. These results indicate that the RV1b inhibits IL-17A in T helper type 17 cells and IL-17A clears RV1b infection in epithelial cells. In both cases IL-17A contributes to fend off RV1b infection by inducing genes downstream of interferon type I pathway.
Supplementary information
The online version of this article (doi:10.1038/mi.2015.130) contains supplementary material, which is available to authorized users.
Subject terms: Cell death and immune response, Viral infection, Mucosal immunology, Asthma
INTRODUCTION
Asthma bronchiale is a worldwide common chronic disease of the airways that affects 300 million children and adults and its incidence was found to be associated with viral infection in the upper airways, especially in children.1, 2, 3, 4 Human rhinovirus (RV) is a member of the Picornaviridae family of viruses. RVs of the minor subgroup (e.g., RV1b) adhere to proteins of the low-density lipoprotein receptor family. The primary entrance of RV is the upper respiratory tract, where it attaches to its receptors on epithelial cells.5, 6 The infected cells then release distress signals like chemokines and cytokines.7, 8, 9, 10, 11
Interleukin-17A (IL-17A) is a cytokine with proinflammatory functions, whose role in asthma is not completely understood. It is produced by T helper type 17 (Th17) cells and it is also associated with autoimmune diseases.12 IL-17A was found upregulated in moderate to severe asthma where it contributes, e.g., to accumulation of neutrophils in the airways.13 Schnyder-Candrian et al.14 showed that exogenous rmIL-17A during allergen challenge decreased airway hyperresponsiveness (AHR). However, IL-17A released by gamma delta cells was also shown to be important for the development of asthma.15 IL-17A is also involved in mucosal and epithelial host defense against fungi and extracellular bacteria.16, 17 Furthermore, Th17 cells and IL-17A seem also to modulate immune pathophysiology of viral infections.18
The 2′-5′ oligoadenylate synthetase (OAS) pathway functions as an innate host defense in response to viral infections. Its expression is upregulated by type I interferons. Via polymerization of ATP into short 2′-5′-linked oligomers (2-5A), OAS1 activates latent ribonuclease L (RNaseL), which in turn cleaves viral and cellular single-stranded RNAs.19, 20, 21
Given that there is an ongoing clinical trial to block IL-17A in asthma, the role of this cytokine in the context of RV infection needs to be clarified.22, 23
RESULTS
Clinical outcome of the cohorts of children analyzed in this study
In this study, we analyzed 17 control children and 20 children with asthma. The clinical data of these cohorts of children are reported in Supplementary Tables S1 and S2 online. The average age of controls and cases was 4.8 years (±0.2 years). In total, 70.6% of the control children and 60% of the asthmatic children were males and thus 29.4% of the controls and 40% of children with asthma were females. By rating the severity of the disease according to Global initiative for Asthma guidelines (2005), 60% of the children have intermittent, 25% mild persistent, and 10% moderate persistent asthma. In 65% of the cases a viral infection was a triggering factor for the development of the disease. A combined treatment with steroid and non-steroid drugs was used by 50% of children with asthma. In 45% of the cases the asthma was controlled and partially controlled, respectively (see Supplementary Table S1). Analysis of the lung function of cases and controls showed that 76.5% of controls and only 40% of the cases had a FEV1 above 100%. Both, control and asthmatic children, suffered from several upper respiratory infections (e.g., common cold) during the last 12 months before they were included in the PreDicta (post-infectious immune reprogramming and its association with persistence and chronicity of respiratory allergic diseases) study. By contrast, children with asthma were more susceptible to infections of the lower respiratory tract such as pneumonia (50% compared with 5%) (see Supplementary Table S2). This can also be seen in Figure 1a, where the average number of lower respiratory infections during the last 12 months was significantly increased in cases compared with control children. Furthermore, control children as well as children with asthma suffered more often from upper respiratory infections than lower respiratory infections during the last 12 months before the time of recruitment (Figure 1a).
Figure 1.
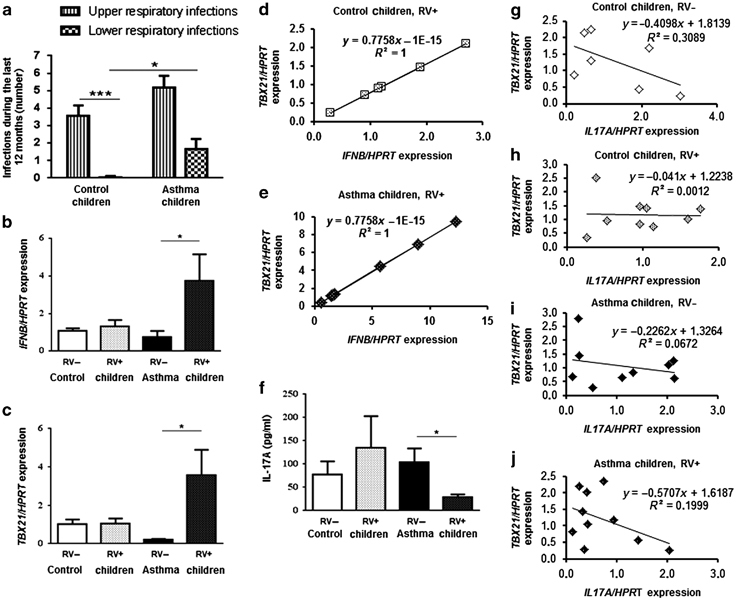
Rhinovirus infection in the upper airways is associated with lower release of interleukin-17A (IL-17A) but increased expression of IFNB and TBX21 in peripheral blood mononuclear cells (PBMCs) of preschool children with allergic asthma. (a) Number of upper (stripped bars) and lower respiratory infections (chequered bars) during the last 12 months before PreDicta started (control children n=17 and asthma children n=19–20). (b, c, f) Control (C, white bars) and asthmatic children (A, black bars) were further subdivided with regard to rhinovirus detection (RV+, dotted bars) or (RV−) in their nasopharyngeal fluid (NPF). (b) IFNB mRNA expression in PBMCs after 48 h cell culture (C RV−: n=8; C RV+: n=9, A RV−: n=11, A RV+: n=9). (c) TBX21 mRNA expression in PBMCs after 48 h cell culture (C RV−: n=2; C RV+: n=6; A RV−: n=6, RV+: n=7). (d, e) Correlation of IFNB and TBX21 mRNA expression in PBMCs after cell culture for 48 h of control children (d; n=6) and asthmatic children (e; n=7) who were RV+. (f) IL-17A ELISA in supernatants of PBMCs after 24 h cell culture (C RV−: n= 4, C RV+: n=2, A RV−: n=8, A RV+: n=4). (g–j) Correlation of TBX21 and IL17A mRNA expression in whole-blood samples of control children and children with asthma: C, RV− (g; n=7), C, RV+ (h; n=9), A, RV− (i; n=9) and A, RV+ (j; n=10). Data are provided as mean±s.e.m. * P≤0.05; *** P≤0.001 (Student’s t-test).
RV infection in the upper airways is associated with increased type I INFB and TBX21 mRNA expression but decreased IL-17A protein in PBMCs of preschool asthmatic children
Within the PreDicta study we collected nasal pharyngeal fluid (NPF) for the analysis of RV infection (see Supplementary Figure S1a and b) in the cohort of children described above, at the time of the recruitment into the study. Furthermore, at the same time point, whole blood was drawn and PBMCs were isolated for further analysis (see Supplementary Figure S1a and c).
We next further classified our two cohorts of children (healthy and asthmatics) into four groups: control and asthmatic children, whose NPF was found to be positive or negative for local RV colonization. Here, we detected an increased expression of the antiviral cytokine type I interferon-β (IFNB) in the PBMCs of children with asthma and a positive RV test in their NPF at the time of recruitment (RV+; Figure 1b). Furthermore, we found that TBX21, the main transcription factor for IFNG24 and an inhibitor of IL-17A,25 was upregulated in the PBMCs of these children (Figure 1c). Finally, we found a perfect positive correlation between TBX21 and IFNB mRNA production in the PBMCs of these children with a RV infection in their NPF (Figure 1d and e). Consistent with the latter findings, the PBMCs of these children also released significantly less IL-17A compared with those isolated from asthmatic children without RV infection in their nasopharyngeal fluid (Figure 1f), at the same time point. Furthermore, by correlating the gene expression levels of TBX21 and IL17A in whole-blood samples (Figure 1g–j), we could observe an inverse correlation between these two factors in control children without a viral infection in the upper airways (Figure 1g). In control children, the presence of RV in their NPF abolished this negative correlation (Figure 1h). By contrast, children with asthma and no RV infection in the upper airways showed only a slight inverse correlation between TBX21 and IL17A (Figure 1i), whereas the negative correlation was found in children with asthma and an additional viral infection in their NPF (Figure 1j).
The involvement of the RV in the regulation of these genes became even clearer by analyzing these markers without taking into account a viral infection in the upper airways. In this case, we could not observe any significant difference between control and asthmatic children (see Supplementary Figure 1d–g). Taken together these data demonstrate that a RV infection in the upper airways corresponds with an increase of IFNB and TBX21 mRNA and with a suppression of IL-17A in asthmatic children.
IL-17A inhibited RV1b replication in infected lung epithelial cells
As mentioned above, OAS1 is a key factor in the antiviral immunity and its transcription is induced by type I IFN signaling. Therefore, we also analyzed OAS1 mRNA expression in the PBMCs of the PreDicta children at our department (MP-UK-ER). Here, we observed a significant downregulation of OAS1 gene expression in the PBMCs of children with asthma and an additional RV infection in the upper airways compared with asthmatic children without a viral infection (Figure 2a). As both factors, OAS1 and IL-17A, were decreased in asthmatic children with a RV infection in the upper airways, we thought about a direct impact of IL-17A on OAS1 expression. Therefore, we started to analyze the epithelial cell line A549, as airway epithelial cells are the first cell type encountered by infectious agents entering the airways. We then asked about the role of IL-17A on RV1b infection in lung epithelial cells. With this aim we infected the human lung epithelial cell line A549 with RV1b and then cultured them for 24 h with increasing concentrations of rhIL-17A. Here we found a significant upregulation of OAS1 gene expression after treating the A549 cells with 25 ng/ml of rhIL-17A (Figure 2b). To find out, if we could gain an even better induction of the antiviral immunity with higher concentrations of rhIL-17A, we treated the cells, after infection with RV1b, with up to 100 ng/ml rhIL-17A and concluded that 25 ng/ml of rhIL-17A were sufficient to reduce RV1b mRNA (Figure 2c). To determine whether IL-17A decreased RV1b expression via OAS1, we first treated A549 cells with OAS1 siRNA or non-targeting (NT) siRNA, infected them with RV1b, and afterwards cultured these cells with increasing concentrations of rhIL-17A. In these experiments, we also analyzed the impact of IL-17A concentrations inferior to 25 ng/ml on OAS1 gene expression. We could only observe a significant increase of OAS1 mRNA expression in A549 cells infected with RV1b and treated with NT siRNA and 25 ng/ml rhIL-17A compared with A549 cells infected with RV1b and treated with NT siRNA and lower amounts of rhIL-17A (Figure 2d and see Supplementary Figure S2). We could also observe that this effect was IL-17A dependent and RV1b independent, as we saw a similar induction after infection with UV-RV1b (see Supplementary Figure S2). Next, we measured RV1b mRNA expression in A549 cells infected with RV1b and treated with OAS1 siRNA and 25 ng/ml rIL17A. We found out that IL-17A had also in the absence of OAS1 an impact on viral replication, as we observed a significantly downregulation of RV1b mRNA expression in A549 cells transfected with OAS1 siRNA and 25 ng/ml rhIL-17A, indicating that IL-17A influences viral replication also by affecting other antiviral yet unidentified factors (Figure 2e). In conclusion, these experiments show that antiviral properties of IL-17A are linked at least in part to OAS1 in lung epithelial cells.
Figure 2.
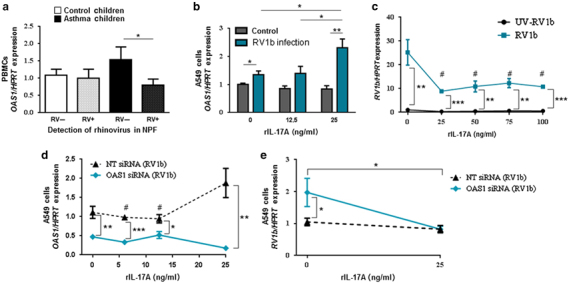
Interleukin-17A (IL-17A) inhibited RV1b replication in epithelial cells and induced OAS1. (a) Control (C, white bars) and children with asthma (A, black bars) were further subdivided with regard to rhinovirus detection (RV+, dotted bars) or (RV−) in their nasopharyngeal fluid OAS1 mRNA expression in peripheral blood mononuclear cells (PBMCs) after cell culture for 48 h (C RV−: n= 8, C RV+: n=9, A RV−: n=11, A RV+: n=9). (b) OAS1 mRNA expression in A549 cells after infection with RV1b or control medium and responding to different doses of rIL-17A (n=3 per group). (c) Infection of A549 cells with RV1b or UV-RV1b and subsequent cell culture for 24 h with different concentrations (0/25/50/75/100 ng/ml) of rhIL17A (n=3 per group). (d–e) Transfection of A549 cells with either OAS1 siRNA or non-targeting (NT) siRNA. Subsequent infection with RV1b or UV-RV1b and cell culture responding to different doses of rhIL-17A (d: 0/6/12.5/25 ng/ml rhIL-17A; e: 0 /25 ng/ml rhIL-17A). (d) OAS1 mRNA expression in A549 cells after infection with RV1b and transfection with either NT or OAS1 siRNA (n=3 per group). (e) RV1b mRNA expression in A549 cells after infection with RV1b and transfection with OAS1 or NT siRNA (n=7 per group). Data are provided as mean±s.e.m. *P≤0.05; **P≤0.01, ***P≤0.001. c: #P<0.05 compared with cells infected with RV1b not stimulated with rhIL17A (Student’s t-test). d: #P<0.05 compared with NT siRNA stimulated with 25 ng/ml rhIL17A (Student’s t-test).
Analysis of Il17a (−/−) mice in a murine model of allergic asthma
To investigate the role of Il17a during antiviral immune responses in asthma, we first analyzed wild-type (wt) and Il17a (−/−) mice in a murine model of allergic asthma (see Supplementary Figure S3). In this model, we found that lung CD4+ T cells secreted significantly increased amounts of IL-17A when purified from the lung of OVA-sensitized mice (see Supplementary Figure S3a). Moreover, by using an invasive plethysmography method, we found a significant increase in AHR in Il17a (−/−) mice at lower doses of methacholine (MCh), whereas at higher doses of MCh the wild-type littermates showed an increased AHR as compared with the Il17a-deficient mice (see Supplementary Figure S3b). Consistent with a role of the Th2-type cytokines in allergic asthma, the Th2 cytokine IL-4 was found to be significantly induced in the airways of OVA-sensitized Il17a (−/−) mice as compared with wild-type littermates (see Supplementary Figure S3c). Consistent with this increase in IL-4 OVA-treated Il17a (−/−) mice showed a significant decrease of IFN-γ in BALF (see Supplementary Figure 3d). IL-5 and IL-13, two cytokines produced by Th2 cells and by the innate ILC2 cells,26, 27 were found to be similarly induced in the airways of OVA-sensitized wild-type and Il17a-deficient mice (see Supplementary Figure S 3e and f). Consistent with the levels of IL-5 in the airways, the amount of eosinophils was equivalent in OVA-sensitized Il17a (−/−) and wild-type mice (see Supplementary Figure S4a). By contrast, Il17a (−/−) OVA-sensitized mice have reduced neutrophils in their airways (see Supplementary Figure S4b) and a reduced number of mucus producing cells in the airways as compared with their wild-type littermates (see Supplementary Figure S4c). Both these latter components are known to be involved in the clearance of infectious microorganisms.28, 29 IL-4 is known to induce IgE immunoglobulin class switching in B cells.30 However, in our model system, the serum levels of IgE in OVA-sensitized Il17a (−/−) mice were comparable to those measured in the wild-type littermates (see Supplementary Figure S4d). We further investigated Batf mRNA expression, a transcription factor31 that is expressed in lymphocytes and is also involved in immunoglobulin rearrangements and thus in IgE induction in allergic asthma.32 Furthermore, it could be shown that BATF is increased in asthma.32 Consistently in this murine model of allergic asthma Batf mRNA was found to be increased in allergic asthma in an Il17a-independent manner (see Supplementary Figure S4e).
Antiviral Oas1 genes are inhibited in lung CD4+ T cells obtained from OVA-sensitized Il17a (−/−) mice
Aeroallergen sensitization is one of the strongest risk factors for asthma.33 However, viral infection has also been emerged as a crucial factor in the pathogenesis of this disease.34 To understand the role of IL-17A in the regulation of different genes that are involved in the response to viral infections, we used a gene array approach in our study. Thus, we sorted out lung CD4+ T cells from OVA-sensitized wild-type and Il17a (−/−) mice and isolated total RNA and performed gene array analysis. By using this method, we found in Il17a- deficient lung CD4+ T cells a significant downregulation of antiviral Oas family members such as Oas1a, Oas1c, and Oas1g as well as Cxcr3 and Rnasel (see Supplementary Figure S5a and b) and an upregulation of proinflammatory genes such as Il6, Il13, Nfil3, and PDC-Trem. By intracellular flow cytometry analysis we also found an upregulation of IL-13 in CD4+ T cells of naïve Il17a (−/−) mice (see Supplementary Figure S5c). The downregulation of Oas1a and Oas1g was verified by real-time PCR in lung CD4+ T cells isolated from naïve and OVA-sensitized wild-type and Il17a (−/−) mice (see Supplementary Figure S5d and e).
We also observed a significant decrease of Oas1g mRNA in whole-lung tissue and draining lymph nodes of naïve and OVA-sensitized Il17a (−/−) mice (see Supplementary Figure S5g). Furthermore, in lung CD4+ T cells Ifnb mRNA, which is the inducing factor of Oas1g, was found significantly downregulated in naïve Il17a (−/−) mice. Treatment with OVA resulted indeed in an inhibition of the Ifnb mRNA expression in wild type as well in Il17a (−/−) mice, but we could not detect significant differences within this group any longer (see Supplementary Figure S5h).
Taken together, we observed defective expression of Oas1g and Ifnb, two genes involved in the antiviral immune response, in the lung of Il17a-deficient mice in a setting of allergic asthma. These data indicate a genetic control of Il17a on the antiviral gene Oas1.
RV1b infection downregulated IL-17A in lung CD4+ T cells isolated from OVA-sensitized wild-type mice
It is already known that lymphocytes express the low-density lipoprotein receptor, which is the portal of entry for minor-group RVs such as RV1b. Furthermore, it could also be demonstrated that RV, in contrast to RSV, is able to directly infect and activate CD4+ T cells.35, 36, 37 To address the role of IL-17A during RV infection in asthma, we infected lung CD4+ T cells obtained from OVA-sensitized wild type and Il17a (−/−) mice with RV1b and cultured them for 24 h (Figure 3a). By analyzing these cells we could observe that Il17a-deficient lung CD4+ T cells from naïve mice express significantly more Ldlr than the corresponding wt mice. Furthermore, confrontation with the allergen OVA resulted in a significant upregulation of the expression of this receptor in CD4+ T cells of wt mice, whereas Il17a-deficient CD4+ T cells show a downregulation of the Ldlr mRNA expression (Figure 3b). By using PCR analysis (see Supplementary Figure S6) we could confirm the infection with RV1b and after quantification of the PCR bands we found out that lung CD4+ T cells of naïve Il17a (−/−) mice are more susceptible to viral infection than lung CD4+ T cells of naïve wt mice (Figure 3c). Moreover, we could observe a significant upregulation of the viral load in lung CD4+ T cells obtained from OVA-sensitized wt mice compared with the cells of naïve wt mice, whereas there was no change in the viral load in lung CD4+ T cells of OVA-sensitized Il17a (−/−) mice, neither to OVA-sensitized wt cells nor to cells of naïve Il17a-deficient mice (Figure 3c). The increase of viral load observed in CD4+ T cells of OVA-sensitized wt mice could be due to the reduced release of IL-17A by these cells after infection with RV1b (Figure 3d).
Figure 3.
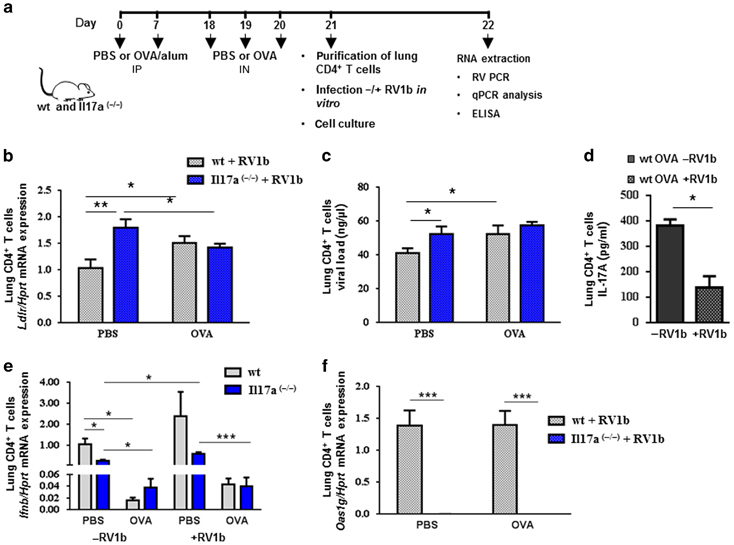
Infection of lung CD4+ T cells with RV1b. (a) Experimental design: Induction of allergic asthma and subsequent purification of lung CD4+ T cells with magnetic anit-CD4 beads from naïve and asthmatic wt and Il17a-deficient mice and successively infection with RV1b in vitro and cell culture for 24 h. (b) Ldlr mRNA expression in lung CD4+ T cells of wt and Il17a-deficient mice after infection with RV1b (n=4) (c) PCR to detect and quantify the rhinovirus infection in lung CD4+ T cells (n=4). (d) IL-17A protein in supernatants of lung CD4+ T cells from OVA-sensitized wild-type mice with and without RV1b infection (n=2–4). (e) Ifnb mRNA expression in lung CD4+ T cells of wt and Il17a (−/−) mice with or without infection with RV1b (n=2–4). (f) Oas1g mRNA expression in lung CD4+ T cells of wt and Il17a (−/−) mice after infection with RV1b (n=2–4). *P≤0.05, **P≤0.01, ***P≤0.001 (c–f: ANOVA, b: Student’s t-test).
Furthermore, the deficit of Ifnb mRNA expression, which was seen after confrontation with the allergen OVA in wild-type and Il17a (−/−) lung CD4+ T cells, could not be restored after infection with RV1b (Figure 3e). Tallying with this, we still observed the defect in Oas1g gene expression in Il17a-deficient lung CD4+ T cells after viral infection (Figure 3f). Taken together, these data indicate that RV1b targets Th17 cells in the lung of OVA-sensitized mice.
The described data lead to the suggestion that in the case of a RV infection in the upper airways, IL-17A inhibits the expression of LDL-R and thus the cellular entry of the RV in airway epithelial cells (Figure 4). Furthermore, by inducing the expression of IFN-β and OAS1, IL-17A also contributes to the suppression of viral replication. Subramaniam and colleagues3839 observed that IL-17A induced—amongst others—in a time-dependent manner the tyrosine phosphorylation of JAK1 and TYK2 as well as of STAT1 and STAT2. This might be the underlying signaling mechanism by which IL-17A induces OAS1 gene expression. Furthermore, it is already known that IL-17A activates nuclear factor-κB40 and this transcription factor is involved in the transcription of IFN-β,41 which in turn leads to the activation of antiviral factors such as OAS1 via JAK1/TYK2, STAT1/2 and IRF9. But our data also suggest that RV is able to inhibit the release of IL-17A and therefore escaping the immune system (Figure 4).To elucidate these molecular mechanisms in more detail, further investigations are needed.
Figure 4.
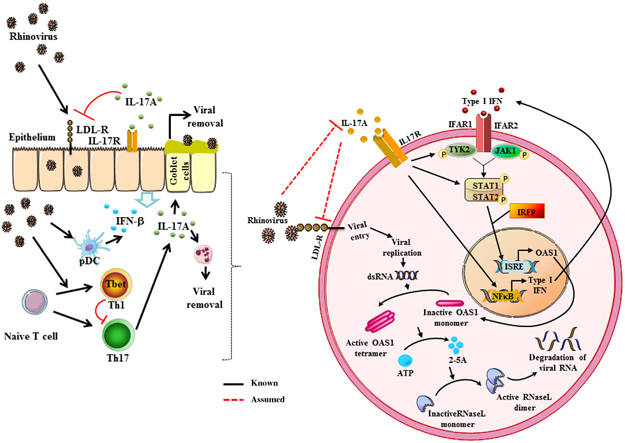
Interleukin-17A (IL-17A) mediated regulation of rhinovirus -induced inflammation. The IFN-induced OAS1-RNaseL antiviral pathway: Type I IFNs bind to the heterodimer of IFNα receptor 1 (IFNAR1) and IFNAR2 which initiates the signal transduction via the tyrosine kinases JAK1 (Janus kinase 1) and TYK2 (Tyrosine kinase 2). In the nucleus, OAS1 (2′5′-oligoadenylate synthetase 1) is transcribed upon induction of IFN-stimulated response elements (ISREs). In the cell cytoplasm OAS1 protein accumulates as an inactive monomer. Upon binding of double-stranded RNA (dsRNA), the enzyme oligomerizes to form a catalytic active tetramer. This active form synthesizes 2′5′ oligoadenylates (2-5A) using ATP as substrate. 2-5A binds to inactive RNaseL and triggers the dimerization of monomers. RNaseL then degrades RNA of viral and also cellular origin. This leads to the inhibition of viral propagation.20, 61, 62, 63 We assume that IL-17A could induce IFN-β and OAS1 expression and inhibits low-density lipoprotein receptor expression, whereas in turn, rhinovirus could suppress IL-17A.
DISCUSSION
In the present study we discovered a defect of IL-17A as well as interferon-induced OAS1, an antiviral enzyme, situated downstream of IFN-β (Figure 4), in the PBMCs of pre-school asthmatic children with RV infection in the upper airways. OAS1 activates RNaseL, which mediates RNA virus degradation.19, 20 The reduced release of IL-17A by the PBMCs of these children might be due to the induction of TBX21 in the PBMCs of these children, a transcription factor that is known to negatively regulate IL-17A.25 Furthermore, we found that IL-17A inhibited RV replication in human lung epithelial cell line infected with RV1b. We could also show that IL-17A upregulated OAS1 in this setting. This IL-17A/OAS1 pathway seems to be independent of IFNB, as we found increased expression of IFNB in children with asthma and an additional RV infection in their upper airways, whereas IL-17A and OAS1 were reduced. This observation is in line with previous findings on increased levels of INFB in PBMCs after infection with RV in vitro.42, 43 In contrast to these results in PBMCs, in bronchial epithelial cells and in the lung epithelial cell line A549 it has been previously reported that RV inhibits type I interferons.43, 44, 45, 46 Therefore, it is possible that epithelial cells, which are the first target of the RV, exhibit different immune response as compared with PBMCs. Finally, it is also possible that the usage of different RV strains influences the overall results.
We also observed increased TBX21 mRNA expression in PBMCs of children with asthma and a RV infection in their upper airways. We previously demonstrated that this Th1 specific transcription factor inhibits IL-17A.25 In addition, we could also show that TBX21 and IFNB expression perfectly correlated in healthy and asthmatic children with a viral infection in the upper airways.
By using gene array, we found that Il17a-deficient lung CD4+ T cells isolated from OVA-sensitized mice do not express Oas1g and consequently the expression of RNasel which is downstream of Oas1 and responsible for viral RNA degradation was also found downregulated. This might be due to either a direct or indirect effect secondary to the inhibited Ifnb expression seen in these mice. This might also lead to the assumption that IL-17A is involved in the induction of type I interferons. This is in line with findings of Wang and colleagues in BXD2 mice, where they could show that IL-17A favors the expression of Ifnb.47, 48 Further experiments need to be performed to demonstrate this.
The influence of IL-17A on AHR is controversially discussed. These observations seem to depend on the time point of IL-17A release.49 By using the invasive measurement of AHR, we found increased responses to low doses of MCh in OVA-sensitized Il17a-deficient mice as compared with wild-type littermates. At higher doses of MCh OVA-sensitized wild-type mice showed an increased hyperreactivity than the Il17a-deficient mice. The reason of this dual response is at the moment not clear. However, we found increased IL-4 levels and decreased IFN-γ in the lung of Il17a-deficient mice which could explain the different responses to MCh. It is possible that IL-4 is acting on the AHR at low doses of MCh and other not yet analyzed factors such as IL-33 and IL-25.50, 51, 52, 53 In addition, it is already known that IL-17A has an effect on IL-13-induced AHR.54 Therefore, the induced airway reactivity at low doses seen in the Il17a-deficient mice might be due to the increased combined amounts of IL-13 and IL-4.26
Additionally, we found less PAS+ cells and airway neutrophils in the absence of Il17a. These two components are known to be involved in the clearance of pathogens.28, 29 Therefore, this mechanism of host defence is also impaired in the absence of IL-17A.
It is already known that RV could bypass antigen presentation and could directly infect T cells.37 We thus next ask whether the RV targets Th17 cells in asthma. We found that infection of lung CD4+ T cells with RV1b resulted in a significant reduction of IL-17A. These results indicate that Th17 cells could be the target of RV in asthma. Consistent with this finding, lung CD4+ T cells from OVA-sensitized wild-type mice infected with RV1b have the same viral load as the infected lung CD4+ T cells from Il17a-deficient mice. These observations could be due to particular expression of Ldlr, which was found upregulated in lung CD4+ T cells of naïve Il17a-deficient mice, whereas confrontation with the allergen promoted the Ldlr mRNA expression in wt cells but inhibited it in Il17a-deficient cells. Furthermore, we could show by analysing CD4+ T cells of naïve and OVA-sensitized mice that treatment with OVA negatively regulated Ifnb mRNA expression independently of infection with RV1b.
Epithelial cells are the first cells that the RV encounters after inhalation and the RV has a positive tropism for this cell type.6, 55 These results clearly demonstrate a crucial mechanism on how the RV escapes the immune system by inhibiting IL-17A production by Th17 cells and thus depriving epithelial cells from IL-17A resulting in reduction of OAS1 and thus creating a good cellular microenvironment where the virus can replicate. Taken together, these results demonstrate that the RV infection targets Th17 cells in the airways of OVA-sensitized mice. Similarly to what we observed in the PBMCs of asthmatic children, RV1b reduces IL-17A production in infected CD4+ T cells from OVA-sensitized mice. This results in IL-17A deprivation which cannot anymore signal in cells of the innate immune response like epithelial cells. We further demonstrated that IL-17A via OAS1 activation shreds the RV RNA which replicates preferentially in epithelial cells. These findings have important implication for the treatment of RV-induced asthma in children.
METHODS
Human studies: detection of OAS1, TBX21 and IFNB mRNA and IL-17A protein in PBMCs. Within the PreDicta study (Post-infectious immune reprogramming and its association with persistence and chronicity of respiratory allergic diseases) we analyzed two cohorts of children with and without asthma at the age of 4–6 years. The study was approved by the ethics committee of the Friedrich-Alexander University Erlangen-Nürnberg, Germany (Re-No 4435). Informed consent was obtained from the parents of all participants of this study (Study PreDicta, Registration number “Deutsches Register Klinischer Studien” (German Clinical Trials Register) DRKS00004914). We isolated PBMCs from the whole blood using Ficoll and density gradient centrifugation. After cell culture we analyzed them by ELISA and quantitative PCR. The clinical outcomes of the recruited children in this study are reported in Supplementary Table S1 and S2.
Nasopharyngeal fluid collection with swab and RV detection. For the detection of the RV in the upper airways, a nasopharyngeal specimen from control children and asthmatic children was collected using a pernasal applicator swab, which has a tip with flocked soft nylon fiber (E-Swab 482CE; Copan, Brescia, Italy).
RV detection was performed at the Department of Virology, University of Turku (Finland). Therefore, nucleic acids were extracted using easyMag extractor (BioMeriex, Marcy l’Etoile, France) from 200 μl of the medium according to the manufacturer’s instruction. An in house PCR method was used to detect enteroviruses, RV s and respiratory syncytial viruses as described earlier.56 A commercial test kit (Anyplex II RV16 Detection; Seegene, Seoul, Korea) was used to detect 16 respiratory viruses (adenovirus; influenza A and B viruses; parainfluenza virus 1, 2, 3, and 4; RV A, B, and C; respiratory syncytial virus A and B; bocavirus 1,2,3,4; coronavirus 229E, NL63, and OC43; metapneumovirus; and enteroviruses) (see Supplementary Figure S1c).
Culture of human A549 lung epithelial cells and murine lung CD4+ T cells and infection with RV1b. To infect the cells with RV1b, cells were shook for 1 h at room temperature with RV1b suspension. RV1b was grown as previously described.57 Comparable treatment with UV-irradiated RV1b or medium served as a control. After RV infection, the cells were washed with medium. A549 cells were cultured for 24 h with medium alone or with an increasing concentration of rhIL-17A as indicated. Lung CD4+ T cells were cultured without additional stimuli for 24 h.
Mice. Mixed gender of Il17a (−/−) mice (generously provided to us by Yoichiro Iwakura) and wild-type mice on a Balb/c genetic background were used at the age of 6–8 weeks. The mice were maintained under specific pathogen-free conditions and all experiments were undertaken with approved license (54-2532.1-2/10 from the government of Mittelfranken, Bavaria, Germany).
OVA sensitization and challenge. Mice received intraperitoneal (IP) injections either of phosphate-buffered saline or of 500 μg/ml ovalbumin (OVA) complexed with 10% alum on days 0 and 7 as described previously.26, 58 On days 18, 19, and 20 the animals were treated intranasally (IN) with phosphate-buffered saline alone or OVA in phosphate-buffered saline (2 mg OVA/ml phosphate-buffered saline in solution).
Gene Array in purified lung CD4+ T cells from wild type and Il17a deficient OVA-sensitized mice. The gene array was performed at the Institute of Human Genetics in Erlangen. Briefly, RNA from lung CD4+ T cells from OVA-sensitized Il17a (−/−) and wild-type mice was isolated, the quality control was performed and the GeneChip Gene 1.0 ST Array was used for further analysis as previously described.59
Silencing of OAS1 in A549 cells infected with RV1b. Lung epithelial cells A549 were transfected with OAS1 siRNA (Dharmacon ON-TARGETplus SMART pool, Lafayette, CO) and afterwards they were cultured and infected with RV1b in the presence of increasing concentrations of rhIL-17A for 24 h. For supervisory purpose and to visualize effective transfection we also transfected the cells with a non-targeting (NT) control (Dharmacon ON-TARGETplus Control Pool, Non-Targeting pool).
RV PCR and real-time PCR. To verify that the infection with RV1b was successful a PCR was performed as described previously with some modifications.57, 60 Therefore, cDNA from infected cells and primers, which are complementary to the antisense RNA at positions 542–557 and 169–185 in the 5′-noncoding region of RV1b, called OL26 and OL27, was used. A 380-bp amplicon was generated and the samples were analysed and quantified using the QIAxcel Advanced System (Quiagen, Hilden, Germany). For detection of RV1b via real-time PCR, RNA was reverse-transcribed into cDNA. The resulting template cDNA was amplified by quantitative real-time PCR using SsoFast EvaGreen Supermix (Bio-Rad Laboratories, München, Germany) and 200 nM primers.
Statistical analysis. Differences were evaluated for significance (*P≤0.05; **P≤0.01, ***P≤0.001) by using one-way ANOVA or the Student’s t-test for independent events (Excel, Microsoft, version 2003; Microsoft, Redmond, Washington), as indicated in the figure legends. Data are given as mean values±s.e.m.
Supplementary information
Acknowledgements
The authors are grateful to C. Holzinger, B. Kroß, S. Mittler, S. Trump, A. Geiger, D. Andreev, S. Mousset, A. Maier, and K. Rauh at the Division of Molecular Pneumology and to E. Muschiol, I. Jawa, and L. Schramm at the Paediatric Pneumology-Allergology, Department of Paediatrics and Adolescent Medicine, Universitätsklinikum Erlangen, Erlangen Children Hospital in Erlangen for their technical support. Moreover, the authors thank M. Wölfel, A. Neubert, and W. Rascher from the Department of Paediatrics and Adolescent Medicine, Universitätsklinikum Erlangen, Erlangen for their assistance in the PreDicta study. Furthermore, we thank R. Stergiou from the Allergy and Clinical Immunology Unit of the National and Kapodistrian University of Athens for the RV1b. We are also grateful to R. Rieker and the Institute of Pathology of the University Hospital in Erlangen for his support on histology. In addition, We thank Y. Iwakura from the Center for Experimental Medicine and Systems Biology, The Institute of Medical Science at The University of Tokyo in Japan for donating the Il17a (−/−) mice. This work was supported by the European Study PreDicta and by the SFB-GK 643 at the University of Erlangen.
Author Contributions
A.G. and S.F. designed the experiments and A.G. performed the experiments. S.N. did the invasive measurement and analysis of the AHR. A.E and F.F. performed the gene array analysis. T.Z. and V.O.M were responsible for the clinical part WP1-UK-ER-of the PreDicta study in Erlangen. T.V. did the RV analysis of the NPF. N.P. provided the protocol for the in vitro cell infection with rhinovirus and S.T. expanded the RV and provided us a large amount to perform our complete study. S.F. and A.G. wrote the manuscript. S.F. directed the research.
PowerPoint slides
Competing interests
The authors declare no conflict of interest.
Footnotes
SUPPLEMENTARY MATERIAL is linked to the online version of the paper
References
- 1.Edwards MR, Bartlett NW, Hussell T, Openshaw P, Johnston SL. The microbiology of asthma. Nat. Rev. Microbiol. 2012;10:459–471. doi: 10.1038/nrmicro2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fuchs R, Blaas D. Productive entry pathways of human rhinoviruses. Adv. Virol. 2012;2012:826301. doi: 10.1155/2012/826301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Johnston SL. Community study of role of viral infections in exacerbations of asthma in 9-11 year old children. BMJ. 1995;310:1225–1229. doi: 10.1136/bmj.310.6989.1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nicholson KG, Kent J, Ireland DC. Respiratory viruses and exacerbations of asthma in adults. BMJ. 1993;307:982–986. doi: 10.1136/bmj.307.6910.982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Winther B. Rhinovirus infections in the upper airway. Proc. Am. Thorac. Soc. 2011;8:79–89. doi: 10.1513/pats.201006-039RN. [DOI] [PubMed] [Google Scholar]
- 6.Subauste MC, Jacoby DB, Richards SM, Proud D. Infection of a human respiratory epithelial cell line with rhinovirus. Induction of cytokine release and modulation of susceptibility to infection by cytokine exposure. J. Clin. Invest. 1995;96:549–557. doi: 10.1172/JCI118067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Deszcz L, Gaudernak E, Kuechler E, Seipelt J. Apoptotic events induced by human rhinovirus infection. J. Gen. Virol. 2005;86:1379–1389. doi: 10.1099/vir.0.80754-0. [DOI] [PubMed] [Google Scholar]
- 8.Hadfield AT. The refined structure of human rhinovirus 16 at 2.15A resolution: implications for the viral life cycle. Structure. 1997;5:427–441. doi: 10.1016/S0969-2126(97)00199-8. [DOI] [PubMed] [Google Scholar]
- 9.Kapikian AZ, Almeida JD, Stott EJ. Immune electron microscopy of rhinoviruses. J. Virol. 1972;10:142–146. doi: 10.1128/jvi.10.1.142-146.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Newcomb DC. Human rhinovirus 1B exposure induces phosphatidylinositol 3-kinase-dependent airway inflammation in mice. Am. J. Respir. Crit. Care Med. 2008;177:1111–1121. doi: 10.1164/rccm.200708-1243OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Savolainen C, Mulders MN, Hovi T. Phylogenetic analysis of rhinovirus isolates collected during successive epidemic seasons. Virus Res. 2002;85:41–46. doi: 10.1016/S0168-1702(02)00016-3. [DOI] [PubMed] [Google Scholar]
- 12.Reppert S, Zinser E, Holzinger C, Sandrock L, Koch S, Finotto S. NFATc1 deficiency in T cells protects mice from experimental autoimmune encephalomyelitis. Eur. J. Immunol. 2015;45:1426–1440. doi: 10.1002/eji.201445150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aujla SJ, Alcorn JF. T(H)17 cells in asthma and inflammation. Biochim. Biophys. Acta. 2011;1810:1066–1079. doi: 10.1016/j.bbagen.2011.02.002. [DOI] [PubMed] [Google Scholar]
- 14.Schnyder-Candrian S. Interleukin-17 is a negative regulator of established allergic asthma. J. Exp. Med. 2006;203:2715–2725. doi: 10.1084/jem.20061401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Murdoch JR, Lloyd CM. Resolution of allergic airway inflammation and airway hyperreactivity is mediated by IL-17-producing {gamma}{delta}T cells. Am. J. Respir. Crit. Care Med. 2010;182:464–476. doi: 10.1164/rccm.200911-1775OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hirota K, Ahlfors H, Duarte JH, Stockinger B. Regulation and function of innate and adaptive interleukin-17-producing cells. EMBO Rep. 2012;13:113–120. doi: 10.1038/embor.2011.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hirota K. Fate mapping of IL-17-producing T cells in inflammatory responses. Nat. Immunol. 2011;12:255–263. doi: 10.1038/ni.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van de Veerdonk FL, Gresnigt MS, Kullberg BJ, van der Meer JW, Joosten LA, Netea MG. Th17 responses and host defense against microorganisms: an overview. BMB Rep. 2009;42:776–787. doi: 10.5483/BMBRep.2009.42.12.776. [DOI] [PubMed] [Google Scholar]
- 19.Kumar S, Mitnik C, Valente G, Floyd-Smith G. Expansion and molecular evolution of the interferon-induced 2'-5' oligoadenylate synthetase gene family. Mol. Biol. Evol. 2000;17:738–750. doi: 10.1093/oxfordjournals.molbev.a026352. [DOI] [PubMed] [Google Scholar]
- 20.Sadler AJ, Williams BR. Interferon-inducible antiviral effectors. Nat. Rev. Immunol. 2008;8:559–568. doi: 10.1038/nri2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Courtney SC, Di H, Stockman BM, Liu H, Scherbik SV, Brinton MA. Identification of novel host cell binding partners of Oas1b, the protein conferring resistance to flavivirus-induced disease in mice. J. Virol. 2012;86:7953–7963. doi: 10.1128/JVI.00333-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miossec P, Kolls JK. Targeting IL-17 and TH17 cells in chronic inflammation. Nat. Rev. Drug Discov. 2012;11:763–776. doi: 10.1038/nrd3794. [DOI] [PubMed] [Google Scholar]
- 23.Busse WW. Randomized, double-blind, placebo-controlled study of brodalumab, a human anti-IL-17 receptor monoclonal antibody, in moderate to severe asthma. Am. J. Respir. Crit. Care Med. 2013;188:1294–1302. doi: 10.1164/rccm.201212-2318OC. [DOI] [PubMed] [Google Scholar]
- 24.Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, Glimcher LH. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell. 2000;100:655–669. doi: 10.1016/S0092-8674(00)80702-3. [DOI] [PubMed] [Google Scholar]
- 25.Reppert S. A role for T-bet-mediated tumour immune surveillance in anti-IL-17A treatment of lung cancer. Nat. Commun. 2011;2:600. doi: 10.1038/ncomms1609. [DOI] [PubMed] [Google Scholar]
- 26.Finotto S. Asthmatic changes in mice lacking T-bet are mediated by IL-13. Int. Immunol. 2005;17:993–1007. doi: 10.1093/intimm/dxh281. [DOI] [PubMed] [Google Scholar]
- 27.Klein Wolterink RG. Pulmonary innate lymphoid cells are major producers of IL-5 and IL-13 in murine models of allergic asthma. Eur. J. Immunol. 2012;42:1106–1116. doi: 10.1002/eji.201142018. [DOI] [PubMed] [Google Scholar]
- 28.Abraham SN, St John AL. Mast cell-orchestrated immunity to pathogens. Nat. Rev. Immunol. 2010;10:440–452. doi: 10.1038/nri2782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Segal AW. How neutrophils kill microbes. Annu. Rev. Immunol. 2005;23:197–223. doi: 10.1146/annurev.immunol.23.021704.115653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dottorini T. Serum IgE reactivity profiling in an asthma affected cohort. PloS One. 2011;6:e22319. doi: 10.1371/journal.pone.0022319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dorsey MJ, Tae HJ, Sollenberger KG, Mascarenhas NT, Johansen LM, Taparowsky EJ. B-ATF: a novel human bZIP protein that associates with members of the AP-1 transcription factor family. Oncogene. 1995;11:2255–2265. [PubMed] [Google Scholar]
- 32.Ubel C. The activating protein 1 transcription factor basic leucine zipper transcription factor, ATF-like (BATF), regulates lymphocyte- and mast cell-driven immune responses in the setting of allergic asthma. J. Allergy Clin. Immunol. 2014;133:198–206. doi: 10.1016/j.jaci.2013.09.049. [DOI] [PubMed] [Google Scholar]
- 33.Andreev K, Graser A, Maier A, Mousset S, Finotto S. Therapeutical measures to control airway tolerance in asthma and lung cancer. Front. Immunol. 2012;3:216. doi: 10.3389/fimmu.2012.00216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Heymann PW, Kennedy JL. Rhinovirus-induced asthma exacerbations during childhood: the importance of understanding the atopic status of the host. J. Allergy Clin. Immunol. 2012;130:1315–1316. doi: 10.1016/j.jaci.2012.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fazio S, Hasty AH, Carter KJ, Murray AB, Price JO, Linton MF. Leukocyte low density lipoprotein receptor (LDL-R) does not contribute to LDL clearance in vivo: bone marrow transplantation studies in the mouse. J. Lipid Res. 1997;38:391–400. [PubMed] [Google Scholar]
- 36.Ho YK, Brown S, Bilheimer DW, Goldstein JL. Regulation of low density lipoprotein receptor activity in freshly isolated human lymphocytes. J. Clin. Invest. 1976;58:1465–1474. doi: 10.1172/JCI108603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ilarraza R, Wu Y, Skappak CD, Ajamian F, Proud D, Adamko DJ. Rhinovirus has the unique ability to directly activate human T cells in vitro. J. Allergy Clin. Immunol. 2013;131:395–404. doi: 10.1016/j.jaci.2012.11.041. [DOI] [PubMed] [Google Scholar]
- 38.Subramaniam SV, Pearson LL, Adunyah SE. Interleukin-17 induces rapid tyrosine phosphorylation and activation of raf-1 kinase in human monocytic progenitor cell line U937. Biochem. Biophys. Res. Commun. 1999;259:172–177. doi: 10.1006/bbrc.1999.0746. [DOI] [PubMed] [Google Scholar]
- 39.Subramaniam SV, Cooper RS, Adunyah SE. Evidence for the involvement of JAK/STAT pathway in the signaling mechanism of interleukin-17. Biochem. Biophys. Res. Commun. 1999;262:14–19. doi: 10.1006/bbrc.1999.1156. [DOI] [PubMed] [Google Scholar]
- 40.Yao Z. Herpesvirus Saimiri encodes a new cytokine, IL-17, which binds to a novel cytokine receptor. Immunity. 1995;3:811–821. doi: 10.1016/1074-7613(95)90070-5. [DOI] [PubMed] [Google Scholar]
- 41.Jiang Z, Mak TW, Sen G, Li X. Toll-like receptor 3-mediated activation of NF-kappaB and IRF3 diverges at Toll-IL-1 receptor domain-containing adapter inducing IFN-beta. Proc. Natl Acad. Sci. USA. 2004;101:3533–3538. doi: 10.1073/pnas.0308496101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Khaitov MR. Respiratory virus induction of alpha-, beta- and lambda-interferons in bronchial epithelial cells and peripheral blood mononuclear cells. Allergy. 2009;64:375–386. doi: 10.1111/j.1398-9995.2008.01826.x. [DOI] [PubMed] [Google Scholar]
- 43.Sykes A. Rhinovirus 16-induced IFN-alpha and IFN-beta are deficient in bronchoalveolar lavage cells in asthmatic patients. J. Allergy Clin. Immunol. 2012;129:1506–1514. doi: 10.1016/j.jaci.2012.03.044. [DOI] [PubMed] [Google Scholar]
- 44.Kotla S, Peng T, Bumgarner RE, Gustin KE. Attenuation of the type I interferon response in cells infected with human rhinovirus. Virology. 2008;374:399–410. doi: 10.1016/j.virol.2008.01.022. [DOI] [PubMed] [Google Scholar]
- 45.Edwards MR. Impaired innate interferon induction in severe therapy resistant atopic asthmatic children. Mucosal Immunol. 2013;6:797–806. doi: 10.1038/mi.2012.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Baraldo S. Deficient antiviral immune responses in childhood: distinct roles of atopy and asthma. J. Allergy Clin. Immunol. 2012;130:1307–1314. doi: 10.1016/j.jaci.2012.08.005. [DOI] [PubMed] [Google Scholar]
- 47.Wang J, Mountz J. IL-17 augment type IIFN-mediated antibody production in BXD2 autoimmune mouse. J. Invest. Med. 2008;56:430–430. [Google Scholar]
- 48.Wang JH. IL-17 promotes type I interferon signaling in autoimmune BXD2 mice. Arthritis Rheum. 2008;58:S181–S181. [Google Scholar]
- 49.Iwakura Y, Nakae S, Saijo S, Ishigame H. The roles of IL-17A in inflammatory immune responses and host defense against pathogens. Immunol. Rev. 2008;226:57–79. doi: 10.1111/j.1600-065X.2008.00699.x. [DOI] [PubMed] [Google Scholar]
- 50.Glaab T, Taube C, Braun A, Mitzner W. Invasive and noninvasive methods for studying pulmonary function in mice. Respir. Res. 2007;8:63. doi: 10.1186/1465-9921-8-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kaur D. IL-33 drives airway hyper-responsiveness through IL-13-mediated mast cell: airway smooth muscle crosstalk. Allergy. 2015;70:556–567. doi: 10.1111/all.12593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kondo Y. Administration of IL-33 induces airway hyperresponsiveness and goblet cell hyperplasia in the lungs in the absence of adaptive immune system. Int. Immunol. 2008;20:791–800. doi: 10.1093/intimm/dxn037. [DOI] [PubMed] [Google Scholar]
- 53.Stock P, Lombardi V, Kohlrautz V, Akbari O. Induction of airway hyperreactivity by IL-25 is dependent on a subset of invariant NKT cells expressing IL-17RB. J. Immunol. 2009;182:5116–5122. doi: 10.4049/jimmunol.0804213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kinyanjui MW, Shan J, Nakada EM, Qureshi ST, Fixman ED. Dose-dependent effects of IL-17 on IL-13-induced airway inflammatory responses and airway hyperresponsiveness. J. Immunol. 2013;190:3859–3868. doi: 10.4049/jimmunol.1200506. [DOI] [PubMed] [Google Scholar]
- 55.Vareille M, Kieninger E, Edwards MR, Regamey N. The airway epithelium: soldier in the fight against respiratory viruses. Clin. Microbiol. Rev. 2011;24:210–229. doi: 10.1128/CMR.00014-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Peltola V, Waris M, Osterback R, Susi P, Ruuskanen O, Hyypia T. Rhinovirus transmission within families with children: incidence of symptomatic and asymptomatic infections. J. Infect. Dis. 2008;197:382–389. doi: 10.1086/525542. [DOI] [PubMed] [Google Scholar]
- 57.Tuthill TJ. Mouse respiratory epithelial cells support efficient replication of human rhinovirus. J. Gen. Virol. 2003;84:2829–2836. doi: 10.1099/vir.0.19109-0. [DOI] [PubMed] [Google Scholar]
- 58.Finotto S. Treatment of allergic airway inflammation and hyperresponsiveness by antisense-induced local blockade of GATA-3 expression. J. Exp. Med. 2001;193:1247–1260. doi: 10.1084/jem.193.11.1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kraft M. Disruption of the histone acetyltransferase MYST4 leads to a Noonan syndrome-like phenotype and hyperactivated MAPK signaling in humans and mice. J. Clin. Invest. 2011;121:3479–3491. doi: 10.1172/JCI43428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Papadopoulos NG. Rhinoviruses infect the lower airways. J. Infect. Dis. 2000;181:1875–1884. doi: 10.1086/315513. [DOI] [PubMed] [Google Scholar]
- 61.Hornung V, Hartmann R, Ablasser A, Hopfner KP. OAS proteins and cGAS: unifying concepts in sensing and responding to cytosolic nucleic acids. Nat. Rev. Immunol. 2014;14:521–528. doi: 10.1038/nri3719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Silverman RH. Viral encounters with 2',5'-oligoadenylate synthetase and RNase L during the interferon antiviral response. J. Virol. 2007;81:12720–12729. doi: 10.1128/JVI.01471-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhao L. Antagonism of the interferon-induced OAS-RNase L pathway by murine coronavirus ns2 protein is required for virus replication and liver pathology. Cell Host Microbe. 2012;11:607–616. doi: 10.1016/j.chom.2012.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.