

Abstract
Human plasma-derived proteins, such as immunoglobulins, coagulation factors, α1-antitrypsin, fibrin sealants, and albumin, are widely used as therapeutics for many serious and life-threatening medical conditions. The human origin of these proteins ensures excellent efficacy and compatibility but may also introduce the risk of unintentional disease transmission. Historically, only viruses, particularly hepatitis and HIV, have posed serious threats to the safety of these therapeutics. Fortunately, between 1970 and 1990, the molecular biology of each of the major viruses was elucidated. These advances led to the development and implementation of effective donor screening tests, mainly based on immunoassays and nucleic acid testing, which resulted in a significant reduction of disease transmission risk. In addition, viral inactivation and removal steps were implemented and validated by manufacturers, further reducing the risk associated with known, as well as unidentified, viruses. Since the late 1990s, a different class of transmissible agent, referred to as prions, has been identified as a new risk for disease transmission. However, prion diseases are very rare, and prion transmission through plasma-derived proteins has not been reported to date. The prion-related risk is minimized by deferring donors with certain key risk factors, and by the manufacturing processes that are capable of removing prions. Advances in science and pathogen safety-related technology, compliance with good manufacturing practices by manufacturers, and increasingly stringent regulatory oversight, has meant that plasma-derived proteins have been developed into today’s highly effective therapeutics with very low risk of disease transmission.
Keywords: Bovine Spongiform Encephalopathy, Scrapie, Bovine Viral Diarrhea Virus, Caprylate, Virus Reduction
In the mid-1940s, the Cohn-Oncley fractionation process was developed to isolate therapeutic proteins from human plasma, based on the solubility differences between individual proteins under a given solvent condition.[1] Subsequently, techniques such as filtration, ultrafiltration, and chromatography have been incorporated in manufacturing processes to increase the purity and yield of products. These advances have led to a continuous supply of human plasma-derived proteins that are essential to the treatment of many serious medical conditions and diseases, including albumin for shock and burn treatment, immunoglobulins for immunodeficiencies, coagulation factors for hemophilia, and α1-antitrypsin for hereditary emphysema. Although human plasma-derived proteins are administered to save millions of lives every year, all biologic products carry the risk of pathogen contamination. Table I lists some of the pathogens that have been shown to be transmissible or have the potential for transmission by blood. Historically, since large pathogens such as bacteria and parasites are removed by sterile filtration or destroyed by a freeze-thaw cycle, only viruses have posed serious threats to the safety of plasma-derived proteins.
Table I.

Pathogens transmissible or potentially transmissible by blood
The first of many blood-borne viruses to be identified was the hepatitis B virus (HBV),[6] whose surface antigen was discovered in the mid-1960s.[7] After the implementation of HBV testing in the 1970s, another blood-borne hepatitis virus, which was referred to as ‘non-A, non-B’ hepatitis and later identified as mainly caused by the hepatitis C virus (HCV), became more prominent.[8,9] In 1982, the first of many HIV infections transmitted by blood products among hemophilic patients was reported.[10] The awareness of potential virus transmission by blood and blood products prompted active research and development efforts in the field to reduce, remove, or inactivate these viruses as well as any emerging viruses.
One of the risk-reduction measures is the initial questioning to identify and defer disease-carrying or high-risk donors. This is particularly important when a disease is associated with a particular behavior(s) or geographic region(s), or when a disease-specific diagnostic test is not available. The regulations for donor qualification are well established in major industrial countries and are continuously revised to accommodate emerging diseases. The source-plasma and therapeutics industry, as represented by the Plasma Protein Therapeutics Association (PPTA), has also implemented initiatives to improve the donor selection process. One example is the compilation of the National Donor Deferral Registry (NDDR), a database of permanently deferred donors in North America, available since December 1993.
Donor selection is only the first layer of defence against the transmission of diseases. Subsequently, the collected donations are tested for major clinically relevant viruses (table I) and positive or elevated plasma units are removed. In addition, removal and inactivation steps are incorporated into manufacturing processes to target pathogens that might have escaped detection or to prevent transmission of emerging viruses of unknown nature. Testing, removal, and inactivation of viruses, and the measures that are currently being used to reduce the theoretical risk of prion transmission, are discussed in detail in the following sections.
1. Detection of Blood-Borne Viral Pathogens
Traditionally, a common perspective has been used to address pathogen detection issues associated with whole blood and plasma. Although whole blood and plasma products are both regulated by many of the same agencies worldwide and often by the same policies, there are profound differences in the way whole blood and plasma are collected, stored, and processed. Unlike blood, the process of collecting plasma removes cellular components, thereby reducing the risk for contamination by cell-associated pathogens. While whole blood cannot be frozen, plasma for fractionation is stored at sub-freezing temperatures. The long-term storage of frozen plasma allows the implementation of a holding period during which donor qualification is reassessed (commonly referred to as ‘donor look-back’) and disqualified plasma units are removed. This procedure is very effective in lowering residual risk that may be associated with early infections during the pre-seroconversion ‘window period’, when the markers of infection are undetectable by a specific test.[11]
The duration of a window period is defined by the performance specifications of the technology used for detection. For serology-based tests (i.e. tests that detect viral-specific antigens and antibodies), a window period could be the result of limited test sensitivity for viral antigens or the result of delayed antibody response in patients. Improvements in existing technologies and the development of new ones, such as tests based on nucleic acid technology (NAT), increase the opportunities to shorten the window period. Depending on the targeted viral infection, acute viremia may be assessed by the detection of viral antigens or nucleic acids in whole blood or plasma samples, whereas resolved infections or chronic viremia are more readily indicated by the presence of antibody.
1.1 Detection of Virus-Specific Antibodies
The detection of antibodies in response to viral infection remains one of the most conventional and widely used approaches to determine the prevalence of relevant human pathogens within specific populations. The detection of antibodies as a diagnostic indicator remains an important tool, particularly when faced with newly emerging pathogens whose characteristics are poorly defined or unknown. Moreover, the detection of different classes of virus-specific antibodies offers predictive value in addition to diagnostic value when applied to donor or patient history. Virus-specific antibody testing remains an important component of current donor screening programs targeting severe human pathogens, such as HIV-1/2 and HCV, which typically establish debilitating chronic infections. Because accuracy in diagnosis of these types of infections is critical, antibody testing protocols typically involve a combination of donor screening and confirmatory assays.
1.1.1 Detection of HIV-Specific Antibodies
Since 1985, several enzyme immunoassay-based methods have been approved as primary donor screening tests for the detection of HIV-specific antibodies.[12] Three additional test systems, including Western blot analysis and indirect immunofluorescence, have been approved for confirmatory testing. Infection with HIV-1 is characterized by an acute primary phase during which patients exhibit a wide range of flu-like symptoms over a 2- to 3-week period.[13] Seroconversion typically follows this period, marking the end of the pre-seroconversion window. However, incubation times may vary widely, from a few days up to 3 months, and seroconversion may occur between 1 and 10 weeks.[14] A long-term asymptomatic phase that is characteristic of latent or low-level persistent infection typically follows seroconversion.[15] Estimates of residual risk among US blood donors (e.g. the probability that a donor is donating during the pre-seroconversion window) suggest a dramatic decrease since the introduction of donor HIV protocols in 1985,[16] from approximately 1 per 50 000 donors to <1 per 500 000 donors.
1.1.2 Detection of Hepatitis C Virus-Specific Antibodies
The screening of blood and plasma donors for HCV was implemented in 1990, following the identification of the virus responsible for the majority of the cases of non-A, non-B viral hepatitis and the development of analyte-specific reagents based on viral antigens.[8,17] The first-generation test was based on a single peptide, but subsequent improvements were introduced and two tests that utilize multiple peptides for antibody capture have been approved.[18] Analyses in which different versions of the test were compared indicate that the incremental modifications have improved assay performance.[19] The pre-seroconversion window for HCV is relatively long, averaging approximately 80 days.[20] Infected donors are typically unaware of their infections because few symptoms are evident during this period despite fluctuating viral loads that sometimes exceed 108 copies of the viral genome/mL.[21] These conditions contribute to an elevated residual risk (1 per 103 000 US volunteer blood donors) associated with this pre-seroconversion window.[20]
1.2 Detection of Viral Antigens
Viral antigens are detected early in infection, before the appearance of circulating antibodies. This period frequently overlaps the prodrome, and many donors are likely to appear asymptomatic. Viral replication can be particularly robust during this period, with viral loads typically increasing exponentially during a relatively short period of time. Understandably, this phase of infection may pose the greatest level of risk to both public health and product safety. The direct detection of specific viral antigens provides the opportunity to notify infected donors and to remove potentially infectious plasma prior to entry into the manufacturing inventory.
1.2.1 Detection of Hepatitis B Virus Surface Antigen
The conventional paradigm for the routine detection of active viremia in human blood and plasma was established approximately 30 years ago with the implementation of radioimmune assays (RIA) specific for the HBV surface antigen (HBsAg),[22] which was initially referred to as the Australia (Au) antigen or the hepatitis-associated antigen.[23,24] The association of this viral marker with the transmission of infectious hepatitis through plasma-based therapies generated interest in how infectious viral particles partitioned during the plasma fractionation process. The ability to analyze the partitioning of pathogens through the tracking of defined markers created new opportunities to design fractionation and purification processes with pathogen safety as a primary consideration.[25,26] Moreover, the systematic detection of HBsAg provided the foundation for the development of screening and quality control strategies designed to improve the quality of plasma prior to the production of derivatives.[27]
The detection of HBsAg using specific monoclonal or polyclonal antibodies has a firmly established history in transfusion medicine and plasma industry practice. High levels of empty virus particles, in addition to high titers of intact, infectious virus particles, means that the detection of surface antigen is generally an effective measure for the interdiction of donations from donors in the acute phase of infection.[28] Initial efforts to develop and implement screening tests involved multiple technology platforms. However, only a few could satisfy the operational requirements associated with the screening of whole blood and plasma units. Assays that rely on solid-phase immunochemistry for the detection of HBsAg, such as the RIA and ELISA, gained early favor over more traditional assays that included complement fixation and counterimmunodiffusion methods.[29] Currently, several test systems from different manufacturers are licensed by the US FDA for use in donor screening and confirmatory testing.[12,30] The approval of more sensitive test systems is pending.[31] Although the incidence of HBV infections varies significantly worldwide, representative studies of US volunteer blood donors indicate an infection rate of 1 per 63 000 donors.[20]
1.2.2 Detection of HIV P24 Antigen
Drawing from the screening paradigm already established for HBV, early emphasis was placed on the development of assays for the direct detection of HIV-1. Public and political pressures heightened the sense of urgency to close the infectious window beyond what had been achieved through the implementation of anti-HIV antibody screening.[32] However, ambiguities surrounding the outcome of several large clinical studies instilled a sense of uncertainty about the potential benefits associated with p24-antigen screening.[33,34] Despite the doubts about the utility of p24-antigen testing in the field, the FDA adopted p24-antigen screening as an interim measure to facilitate an expanded donor HIV screening protocol.[32,35] However, other regional regulatory agencies, most notably those associated with Europe and Japan, declined to adopt this protocol. Because the safety benefit associated with the implementation of this expanded protocol has been generally perceived as marginal, the replacement of the p24-antigen test with more effective screening technology has been an industry and a regulatory goal. The recent licensure of a number of NAT testing systems specific for the detection of HIV-1 RNA has provided the opportunity to eliminate p24-antigen testing from the donor HIV screening protocol.[36,37]
1.3 Detection of Viral Nucleic Acids
NAT offers a sensitive and flexible platform for the detection of viremia in both blood and plasma donors and creates additional opportunities to remove plasma units contaminated with clinically relevant pathogens, such as HBV, HCV, and HIV, and to defer infected donors. With experience and further demonstration of efficacy, NAT testing has the potential to supplant viral antigen testing as a direct indicator of viremia, allowing for the elimination of less effective, redundant tests. NAT testing is also emerging as an important technology for the detection of less severe viral pathogens, such as parvovirus B19 and hepatitis A virus (HAV). Infections with these pathogens are generally self-limiting because of an effective immune response. The use of NAT testing to identify only viremic donations for removal ensures that convalescent donors whose plasma may contain protective, virus-specific antibodies are retained. NAT is also playing an increasingly important role in resolving issues particular to emerging pathogens for which the timely development and implementation of testing are critical.[3,4,38]
1.3.1 Trends in the Development of Nucleic Acid Technology (NAT)
Nucleic acids have several properties that make them effective targets for detection. The ability of DNA and RNA to hybridize with complementary molecules means that a great deal of control can be exerted over assay specificity when target nucleic acid sequences are known. NAT methods typically combine nucleic acid hybridization with amplification technologies, driven either enzymatically or chemically, to achieve highly sensitive detection of specific nucleic acids. The PCR is the earliest and the most widely recognized method that has been applied to the detection of limited quantities of viral nucleic acids.[39,40] However, other methods offer alternative technology platforms with similar performance specifications for the amplification of nucleic acid targets. These systems include transcription mediated amplification (TMA),[41] nucleic acid sequence based amplification (NASBA),[42] thermophilic strand-displacement amplification (tSDA),[43] ligase chain reaction (LCR),[44] and branched DNA assay (bDNA).[45] When coupled with efficient nucleic acid isolation methods, any of these technologies could provide a foundation for a highly efficient high-throughput testing platform.
NAT methods also provide opportunities to introduce economy into a high-throughput testing environment. The increased sensitivity associated with NAT methods allows for the testing of pooled samples; individual samples need to be tested only when suspected positive for a targeted pathogen. Because the vast majority of samples are negative, pooling strategies greatly enhance operational efficiency by reducing the number of test samples, thus, effectively increasing throughput. The utilization of fluorescent or chemiluminescent detection chemistries with NAT methods facilitates process consolidation and reduces assay time. Additionally, probe-specific detection and target differentiation facilitate the multiplexing of assays thereby increasing the array of targets while reducing the number of tests performed. Finally, the progressive introduction of automation will gradually reduce the number of labor-intensive steps and further decrease the time required to perform an assay.
Since 1995, an active dialog has been underway among scientists from the blood and plasma industries, molecular diagnostic developers, and regulatory agencies worldwide concerning the introduction of NAT methods into protocols established for testing blood-borne viral pathogens. Semi-annual meetings and international collaborations have helped to establish standardized, uniform policies.[46] The collaborative development of WHO international standards for NAT methods (table II) has provided the basis for various regional regulatory policies and industry initiatives. The application of the international unit, a description of empirically derived, consensus-based potency, rather than a putative physical entity, has enabled the creation of performance standards across multiple technology platforms. Legislation and guidance documents frequently specify performance standards for various regions of the world.
Table II.

WHO international standards for nucleic acid testing methods
1.3.2 Applications of NAT
The EU’s codification of specifications for the detection of pathogenic viral nucleic acids in pooled human plasma[54] opened the door to a new era in the detection of viral pathogens in plasma used in the manufacture of protein therapeutics. The plasma therapeutics industry has applied NAT testing for HCV in pooled plasma samples to comply with European and US initiatives.[55] NAT testing has also been implemented to improve the interdiction and removal of HIV-, HBV-, parvovirus-B19-, and HAV-contaminated units from plasma inventories and manufacturing streams. These NAT methods enhance the margin of safety of blood and plasma products by allowing the detection of viral pathogens earlier in the window period than can be achieved using current antigen and antibody tests. For example, the window period for HIV-1 associated with antibody assays is approximately 22 days.[14] The HIV-1 p24-antigen assay reduces the window period to 16 days.[56] The application of NAT assays for HIV-1 RNA further reduces the window period to between 7 and 11 days,[57,58] depending on the pooling strategy employed. In the US, the implementation of licensed HIV-1 NAT assays allows for a reduction in testing redundancies through the discontinuation of p24-antigen testing. However, in many developing countries, the implementation of NAT techniques is hindered by its complexity and high cost.
The implementation of new testing platforms and the upgrade of older ones are consistent with the strong emphasis on continuous improvement to which the biologic products industry has committed itself during the past decade. These testing procedures complement the viral removal and inactivation measures designed into product-specific manufacturing streams to provide better safety features for plasma-derived therapeutic proteins.
2. Removal and Inactivation of Viruses
2.1 Viral Validation Studies
By the early 1980s, the plasma industry had initiated viral inactivation studies,[10] and in 1991, the EU Committee for Proprietary Medicinal Products (CPMP) required all manufacturers of blood and plasma products to implement viral inactivation/removal steps in their processes. In accordance with regulatory requirements, viral validation studies are performed to estimate the overall level of virus reduction across manufacturing processes. During these studies, known amounts of virus are deliberately added (spiked) into production intermediates and the materials are treated according to manufacturing specifications, using bench-scale models of the production process. Virus reduction is calculated by comparing the level of virus infectivity remaining at the end of the process with the level in the starting material. Since most manufacturing processes consist of multiple steps, challenging the system with virus at the beginning of a process and sampling the final product is not practical. Such a strategy would require massive amounts of virus, something that is difficult to obtain. As a result, individual steps of a process are usually evaluated. Breaking down the process into individual steps is not only technically more reasonable, but it also allows for more accurate characterization of the conditions critical for viral reduction and a better understanding of the mechanisms for viral clearance.
Virus reduction is expressed on a logarithmic scale. The overall virus reduction capacity of a process is defined as the sum of the log reduction factors from individual steps. However, a simple summation of the individual reduction factors should not be used as the only measure of viral safety. Individual clearance steps yielding <1 log10 are considered negligible and should not be included in the overall clearance for a process. Adding numerous steps with low individual reduction factors or adding steps that share the same mechanism of virus clearance may overestimate the virus reduction capacity of a process. An effective step is one that clears at least 4 log10 units of virus (i.e. a 10 000-fold reduction in titer) and a robust step is one that clears virus regardless of variability in production parameters. In general, a single effective and robust step provides more assurance of viral safety than several steps with the same overall log reduction. Most regulatory authorities recommend incorporating at least two effective steps with unrelated clearance mechanisms in each manufacturing process and at least one of the steps should be effective against non-enveloped viruses.[59]
Ideally, a pathogenic virus should be directly tested to determine the viral clearance values. However, not all clinically relevant viruses can be propagated easily in vitro. As a result, specific model viruses with similar physicochemical characteristics are used to represent the relevant but uncultivable viruses. In addition, non-specific model viruses with different physicochemical characteristics are included to evaluate the theoretical capability of the manufacturing process to clear unknown or undetected viruses. Thus, no single indicator virus is used to assure the viral clearance capacity of a manufacturing process for known and unknown viruses. Instead, a test panel of relevant and model viruses is used in validation studies to determine the capacity of viral reduction in manufacturing processes. Table III is a short list of common model (test) viruses that are used in viral validation studies to mimic blood-borne viruses with potential for causing clinical infections.
Table III.

Models used in viral validation studies
2.2 Virus Removal Steps
Reduction in virus infectivity can be achieved through routine processing and purification operations or by dedicated virus reduction steps. Depending on the method, clearance strategies can be classified as removal (physical separation) or inactivation (irreversible loss of viral infectivity). Virus removal is often achieved by partitioning of viruses from the proteins of interest by precipitation, chromatography, or filtration. It should be emphasized that, to assure effective virus reduction by removal steps, processing conditions need to be carefully defined, controlled, and carried out in compliance with good manufacturing practices (GMP) regulations.
2.2.1 Cold Alcohol (Ethanol) Precipitation
Cohn-Oncley fractionation is a well established and efficient method that allows for multiple stepwise extractions of therapeutic proteins (figure 1). Frozen plasma is thawed slowly at 35.6–39.2°F (2–4°C) and centrifuged to remove cryoprecipitate, the cold insoluble plasma fraction that contains fibrinogen, Factor VIII (FVIII) and von Willebrand (vWB) factor. The solubility of different protein fractions in the remaining supernatant (effluent) is then manipulated by adding different amounts of alcohol (ethanol) and changing the temperature, pH, and/or ionic strength conditions. After each alcohol precipitation, the fractions of different proteins are separated (partitioned) by centrifugation or filtration.
Fig. 1.
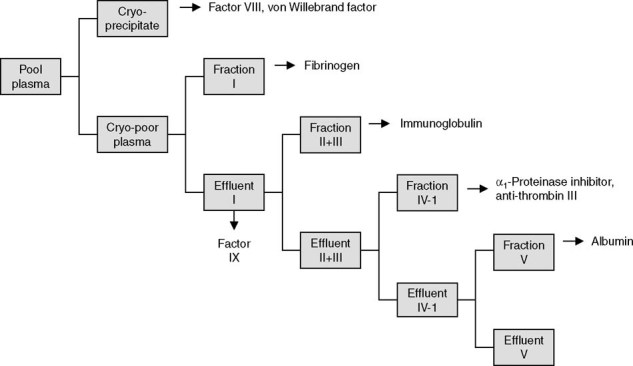
A Cohn-Oncley plasma fractionation scheme.
Although not designed specifically for virus removal, some fractionation processes possess varying capacities for clearing viruses. Viral validation studies help to explain how FVIII concentrates, produced before the implementation of viral inactivation measures, became contaminated with multiple viruses and why immunoglobulins produced during the same period never transmitted HIV.[60] The early FVIII concentrates were derived from cryoprecipitate, and viral validation studies showed no viral clearance across the initial precipitation step to generate cryoprecipitate. In contrast, immunoglobulins, which are derived from Fraction II+III, undergo an additional precipitation step (Fraction III) that not only purifies product but also removes >4 log10 of some model non-enveloped and >3 log10 of some model enveloped viruses.[61] It must be emphasized that fractionation steps are not effective against all viruses and that HCV was transmitted by immunoglobulins that were manufactured by a process that relied entirely on fractionation to remove virus. Consequently, additional viral inactivation/removal steps, with different mechanisms of action, are required in manufacturing processes to assure viral safety.[62]
2.2.2 Caprylate (Octanoic Acid) Precipitation
Caprylate (octanoic acid) has been used to precipitate and remove lipids/lipoproteins during the purification processes of intravenous immunoglobulin (IVIG).[63–66] Under acidic conditions, this treatment also inactivates/removes at least 4 log10 of enveloped viruses.[67–70] When coupled with depth filtration, 2–4 log10 of non-enveloped viruses can also be removed.[67–70]
2.2.3 Polyethylene Glycol Precipitation
Proteins are usually soluble in water solutions because their hydrophilic amino acids interact with water molecules. Polyethylene glycol (PEG) is an inert, nondenaturing, water-soluble polymer that precipitates proteins (and viruses) from aqueous solutions by excluding protein interactions with water. As an example, the PEG precipitation step is capable of removing 3–4 log10 viruses during the α1-antitrypsin purification process.[61]
2.2.4 Chromatography
Although traditional purification schemes relied exclusively on alcohol precipitations, many newly developed processes for FVIII and immune globulins use chromatography steps to remove contaminants, including viruses. However, because of the complexity of biologic process streams, chromatography is not typically used for primary extraction, but is more often employed as a downstream polishing step of partially purified fractions. Chromatographic methods separate proteins based on differences in their physicochemical characteristics, and since viruses consist of many different proteins, they may be separated by many different kinds of chromatographic methods.
Affinity chromatography uses biologic ligands, such as antibodies and co-factors, to recognize and bind their complementary proteins.[71] For example, during one purification process of FVIII, immobilized monoclonal antibodies specifically captured FVIII, allowing contaminating proteins and 2.8 log10 porcine parvovirus (PPV) and 3.7 log10 poliovirus (both small non-enveloped viruses) to pass through.[72]
Ion-exchange chromatography uses differences in charge to separate proteins. Although the binding characteristics of proteins during chromatographic processes are well understood, virus binding is not. Consequently many of the ligands used for viral clearance have relatively general binding properties and involve combinations of ionic, hydrophobic, hydrogen bonding, or van der Waals interactions. During one new IVIG purification process, the chromatography step not only separates impurities from IgG, but also clears >4 log10 bovine viral diarrhea virus (BVDV), a surrogate for HCV and PPV, a model for human parvovirus B19 and removes HIV, pseudorabies virus (PRV, model for human herpes viruses), Reovirus, and HAV to below detection.[70]
Depth filters were originally developed for the plasma fractionation industry to replace asbestos prefilters as clarifying filters and basically consist of diatomaceous earth, cellulose, and polymeric binding resins. Some binding resins also confer positive charges to the matrix, enabling the filters to function as an ion exchange chromatographic medium. Depth filtration effectively removes enveloped and non-enveloped viruses from products such as IVIG, albumin, and α1-antitrypsin.[61]
2.2.5 Nanofiltration
Unlike chromatography steps that were developed for protein purification and where virus removal was coincidental, virus retentive filters, or nanofilters, were developed specifically to remove viruses. Viruses larger than the pore size of the nanofilter are retained by a combination of size exclusion and adsorption mechanisms while the smaller less rigid plasma proteins pass through. Nanofiltration can be performed under relatively mild physiologic conditions (pressure, pH, osmolarity, and temperature) and does not require stabilizers or other chemical agents. As a result, the biologic integrity of a product is not compromised and adverse biologic and immunologic reactions are not induced.[73] Nanofiltration, in theory, is limited only by pore size, so the smaller the pore, the smaller the viruses that are retained. However, size-based exclusion applies to plasma proteins as well as to viruses, and currently only the smallest plasma products pass easily through the filters. As nanofiltration technology improves, it is reasonable to expect the availability of membranes capable of separating viruses as small as parvovirus from proteins as large as IgG or FVIII.
2.3 Viral Inactivation Methods
Inactivation methods render viruses non-infectious by physical or chemical means that, by themselves, may adversely affect the protein product. Examples of adverse effects include protein denaturation and loss of biologic activity, alteration of antigenicity, and production of neoantigens and induction of antibodies or inhibitors in the recipient.[74] Stabilizers are usually added to protect biologic activity or to reduce protein aggregation. If the added stabilizers or inactivating chemicals must be removed from the product, the subsequent manufacturing steps to remove or reduce their levels can result in recovery losses and extended processing times. The most commonly used chemical inactivation steps are treatment with solvent/detergent and low pH. Acetone is used in some albumin processes and caprylate has been used to inactivate enveloped viruses in immune globulin solutions. Licensed methods to physically inactivate viruses also include pasteurization (heating in solution),vapor heating, and terminal dry heat.
2.3.1 Solvent/Detergent Treatment
Solvent/detergent (SD) treatment, which disrupts the lipid coat of enveloped viruses, consists of gently stirring a plasma protein solution in a non-volatile organic solvent, tri-n-butyl-phosphate (TNBP) and detergent (Tween® 1 80, Triton® X-100, or sodium cholate). SD treatment was first licensed by the FDA in 1985 and is considered a breakthrough in viral safety because of its simplicity and effectiveness to inactivate enveloped viruses while maintaining high protein recovery.[75] HIV, BVDV, and PRV were inactivated to below detection after SD treatment of FVIII and IVIG process intermediates. The downstream processes required to remove the SD agents are often existing steps of removing protein contaminants. The main limitation is its failure to inactivate non-enveloped viruses. While there have been no HIV, HCV, or HBV transmissions by SD-treated plasma products since its introduction, there have been several cases of HAV and parvovirus B19 transmission from SD-treated clotting factor concentrates.[76]
2.3.2 Caprylate Inactivation
In addition to the capacity of inducing precipitation (section 2.2.2) caprylate can also inactivate viruses. The proposed mechanism for inactivation is that lipophilic non-ionized caprylate disrupts viral lipid membranes and renders the virus non-infectious.[68] Caprylate inactivation studies for one new IVIG process showed that HIV, BVDV, and PRV were completely inactivated after exposure for only 3 minutes.[70] Variations in incubation time, temperature, pH, protein concentration, and caprylate concentration within manufacturing specifications did not affect virus inactivation, demonstrating the potential of caprylate treatment for use as a robust alternative to SD treatment.[77]
2.3.3 Acetone
Although not very commonly used in manufacturing processes, the washing of albumin paste with acetone has been shown to inactivate ≥5.1 log10 HIV, 4.5 log10 BVDV, and ≥4.2 log10 PRV.[61]
2.3.4 Low pH Incubation
Similar to the fortuitous utility of alcohol precipitation as a virus removal step, low pH incubation serves two functions. Incubation of IVIG at low pH (pH 4.0–4.3) was initially developed to reduce anti-complementary activity, avoid aggregates, and permit intravenous infusion. However, incubation of IVIG at low pH also inactivates substantial amounts of enveloped viruses. During the low pH final incubation step of one IVIG purification process, HIV, BVDV, and PRV were inactivated to below detection after 14, 21, and 4 days, respectively.[61]
2.3.5 Pasteurization (Heating in Solution)
Pasteurization, the heating of aqueous, stabilized protein solutions, was developed during World War II when albumin was shipped all over the world to treat cases of shock.[78] No cases of HIV, HBV, or HCV have ever been traced to a transfusion of pasteurized albumin. Pasteurization in the final container is the approved pharmacopeial method for viral inactivation in albumin preparations.[79]
The results of viral validation studies support the virucidal capacity of albumin pasteurization and extend its effectiveness to some non-enveloped viruses. Pasteurization of albumin final container inactivated enveloped viruses, HIV, BVDV, PRV, and non-enveloped viruses, Reovirus and HAV, to near or below detection.[61] In contrast, only 1.6 log10 PPV reduction was achieved after heating at 140°F (60°C) for 10 hours. Blumel et al.[80] confirmed the resistance of PPV during albumin pasteurization but also reported that parvovirus B19, unlike the model animal virus, was inactivated after heating for only 10 minutes. These results indicate that the model virus, PPV, is more resistant to heat treatment than parvovirus B19 and suggest the effectiveness of pasteurization to inactivate both enveloped and non-enveloped viruses in albumin. However, pasteurization in the presence of sucrose was less effective against non-enveloped viruses, as Reovirus reduction was only 1.0 log10. Similarly, a recent report of parvovirus B19 transmission by a pasteurized immunoglobulin, in the presence of stabilizers, indicates that heating in solution may not be effective in eliminating high concentrations of non-enveloped viruses such as parvovirus B19.[81]
2.3.6 Vapor Heating
Pressurized steam, or vapor heat, has been used to treat coagulation concentrates. Two-stage vapor heating, involving 10 hour, 140°F (60°C), at 1190 mbar, followed by 1 hour, 176°F (80°C), at 1375 mbar, inactivated 10.9 log10 HIV, >12.4 log10 tickborne encephalitis virus, >11.5 log10 PRV, and >11.0 log10 equine rhinopneumonitis virus (model for HAV).[82] While vapor heating inactivates enveloped viruses in general,[83] and some non-enveloped viruses including HAV,[84] the treatment may not inactivate parvovirus B19 completely.[85]
2.3.7 Terminal Dry Heat Treatment
During terminal dry heat treatment, final containers of freeze dried product are heated at temperatures ranging from 140°F (60°C) to 212°F (100°C) for varying lengths of time.[76] Terminal dry heat treatments were developed to inactivate HIV in coagulation factor concentrates and involved heating at 140–154.4°F (60–68°C).[10] However, later studies showed that inactivation of other enveloped viruses, such as HBV and HCV, required more stringent conditions. As a result, some FVIII products are treated by dry heat at even higher temperatures (e.g. 176°F [80°C]) for a longer period of time (e.g. 72 hours). Such treatment inactivates significant levels of both enveloped and non-enveloped viruses, but the effectiveness of the step is dependent on residual cake moisture[86] and formulation.[72]
2.4 Viral Reduction Across Manufacturing Processes
Experience with hepatitis viruses and HIV has led to the implementation of many viral removal and inactivation technologies in the plasma fractionation industry. Currently, there is no ‘magic bullet’ to inactivate all viruses in all processes and such a technique is not anticipated in the near future. Thus, new production processes are designed to contain orthogonal reduction steps to assure broad and effective virus reduction within specific product streams. Table IV summarizes viral reduction across several representative manufacturing processes. The overall global reduction across each manufacturing process was calculated by adding all reduction factors from individual steps that were ≥1.0 log10. These values are the collective results of the methods described in the previous sections.
Table 4.

Reduction of enveloped and non-enveloped viruses across manufacturing processes[1,9]
For enveloped viruses, EU regulatory agencies now recommend that manufacturing processes contain at least two effective and mechanistically independent steps that have 4 log10 or more clearance capacity. These steps include chemical and physical treatment, precipitation, and filtration integrated to inactivate and remove enveloped viruses. As shown in table IV, the capacity of manufacturing processes to inactivate/remove enveloped viruses, such as HIV, HCV, and HBV, is very high.
Clearance of non-enveloped viruses, such as parvovirus B19 and HAV, is more difficult across manufacturing processes. Still, significant reduction can be observed (table IV). Non-enveloped virus reduction is achieved by a combination of removal steps, such as precipitation and filtration, nanofiltration, and inactivation steps, such as pasteurization and dry heat treatment. For this group of viruses, which often leads to less severe clinical outcomes, EU regulatory agencies require at least one effective (4 log10 or more) reduction step.
The presence of multiple steps that are capable of removing or inactivating a wide variety of viruses by completely different mechanisms provides the greatest assurance that any virus surviving one step will be cleared by another step. However, viral inactivation or removal steps are useful only if they are implemented properly in the production and carried out in adherence with current GMPs (cGMPs).[87] Manufacturers of plasma products have made major investments to improve elements critical for product integrity and viral safety, such as facility and equipment design, qualification and validation, process control and monitoring, quality assurance, standard operating procedures, and staff training.[88] Increasingly rigorous regulatory inspections during the past decade also played a key role in promoting and enforcing cGMP compliance of the industry.[89]
3. Theoretical Risk of Prion Transmission
Prion diseases, or transmissible spongiform encephalopathies (TSEs), are a group of fatal neurodegenerative disorders linked to misfolded prion proteins.[90–93] The most notable TSEs include scrapie in sheep, bovine spongiform encephalopathy (BSE) or ‘mad cow disease’ in cattle, and Creutzfeldt-Jakob Disease (CJD) in humans. All these diseases feature a long latency period ranging from years to decades and the accumulation of the pathogenic prion protein (scrapie form) [PrPSc] mainly in the CNS.
Following an epidemic of BSE in the UK in the 1980s, a new form of CJD appeared mainly in the UK with distinctive clinical and pathologic features, including the appearance of PrPSc in lymphoid tissues that had not been described in classical CJD cases. This new illness was named variant CJD, or vCJD, to distinguish it from other CJDs. Epidemiologic and experimental evidence strongly suggested a causal link between BSE and vCJD, possibly due to the consumption of BSE-contaminated meat products.[94–96]
The emergence of vCJD, along with its possible oral route of transmission and peripheral lymphoid tissue involvement,[97,98] prompted concerns that vCJD may pose a higher risk level than classical CJD in terms of transmission through blood or blood products. Therefore it is important to examine such a risk based on epidemiologic and experimental evidence.
3.1 Blood Infectivity, Testing, and Donor Deferral
3.1.1 Blood Infectivity
From an epidemiologic point of view, if classical CJD can be transmitted through blood or blood products, a few cases over a long period of time would be expected among frequent blood or blood product recipients, such as patients with hemophilia, thalassemia, or sickle cell disease. However, the rate of classical CJD occurrence is not elevated in these populations.[99–102] Retrospective studies have also been conducted on transfusion recipients from donors who later developed classical CJD. Again, no classical CJD transmission was detected.[103,104] For vCJD, epidemiology data remain limited because of the disease’s short history, long latency period, and small number of cases.[105] However, during 2004, two cases of presumptive vCJD transmission through the use of packed red blood cells were reported in the UK.[106,107] Therefore, vCJD transmission by blood is possible, although very rare.
There are no conclusive positive results from many early attempts to transfer CJD to animals through blood.[108–110] However, a relatively recent sheep model study has convincingly demonstrated the transmission of TSE by whole blood transfusion. Some of the blood samples were collected at pre-clinical stages, indicating that asymptomatic animals can carry the infectious agent.[99,111,112] By using intracerebral inoculations, infectivity has also been detected in blood samples from rodent TSE models.[113–116]
3.1.2 Testing for Creutzfeldt-Jakob Disease (CJD)
An obvious solution for the management of CJD-related risk is to implement a screening assay capable of detecting a marker for the illness at the pre-clinical stage. In the US alone, it is estimated that ~5% of blood donors are deferred because of the theoretical risk of vCJD transmission.[117] Since vCJD is virtually non-existent in the US, the donor loss could be avoided if an accurate screening test were available.
However, the pathogenic prion protein has so far evaded detection in blood samples and no practical ante-mortem screening test for this pathogen is currently available.[118–124] On the other hand, the growing practice of using post-mortem tests to exclude infected beef from human consumption reduces the risk of transmitting BSE to human populations, thus indirectly improving the safety of blood or blood products.
3.1.3 CJD-Related Donor Deferral Policies
In the absence of a reliable ante-mortem, in vitro diagnostic test for CJD, a deferral policy is the only way to exclude high-risk donors. Donors with a family history of CJD and patients who have undergone certain medical procedures that are associated with the transmission of CJD (dura mater transplant or human pituitary-derived growth hormone administration) are excluded. A decision has also been made to stop processing UK plasma into derivatives.[125]
A major challenge is how to defer donors who have an elevated risk of having been exposed to a BSE agent. After balancing the theoretical risk of CJD transmission with the need for a stable blood supply, regulatory agencies around the world have recommended geographical deferral policies. These policies mainly exclude donors who spent a certain amount of time in the UK during the BSE epidemic period between the beginning of 1980 and the end of 1996. The US has the most stringent criteria, which exclude donors who stayed in the UK for a cumulative 3 months, or in France for a cumulative 5 years, or on military bases in Europe for a cumulative 6 months during the risk period. The US policy also excludes donors who had blood transfusions or received bovine insulin prepared in the UK from 1980 to present.[117] Australia and Japan both defer donors who spent more than 6 months in the UK during 1980–1996. These policies are likely to be revised as the mechanism of CJD transmission becomes better understood.
3.2 Removal of Prions
3.2.1 Prion Removal Versus Inactivation
The prion agent is very robust, and treatments such as heat, solvent detergent, and ionizing radiation that inactivate viruses cannot be relied upon to eliminate the TSE infectivity. TSE inactivation can be achieved using extreme methods, such as treatment with caustics and autoclaving, methods that will also destroy the biologic product of interest.[126] Therefore, the removal of the prion agent is the only viable means to reduce the theoretical risk associated with the transmission of TSEs through plasma-derived proteins.
3.2.2 Bench-Scale Models for Prion Clearance Studies
To determine the capacity of prion removal for a manufacturing process, prion clearance studies are performed. These studies are analogous to viral clearance studies where bench-scale models of manufacturing processes are used (section 2.1). The TSE spikes are typically derived from infected brain homogenate because of the high titers of the prion protein and infectivity (also see section 3.2.5).
The partitioning of TSE infectivity during a purification process can be monitored by rodent bioassay. Although sensitive, such an assay typically requires a number of months to complete. Alternatively, a sensitive Western Blot assay has been developed and used to monitor the partitioning of the proteinase resistant form of prion, or PrPRes, as a surrogate marker for TSE infectivity.[127,128] The correlation between the clearance of PrPRes and infectivity in plasma purification processes has been demonstrated.[129] A prion confirmation-dependent immunoassay has also been developed and used for bench-scale clearance studies.[130]
3.2.3 Methods of Prion Removal
Separation of prions from other protein entities may be accomplished by exploiting the prions’ structural and solution-state properties. Prions are hydrophobic aggregates of the β-sheet form of the prion protein and are often associated with cellular components such as membranes. These aggregates vary in size from nanometers to microns depending on the source and preparation of the tissue. If the desired therapeutic protein is small compared with the prion particles, sedimentation or filtration can be used to remove the prion impurity. Use of well defined solution conditions (pH, organic agents, and temperature) can promote the aggregation/precipitation of the prion, which can be removed using sedimentation or filtration (table V). At this time, the degree of prion clearance cannot be reliably predicted on the basis of the conditions used to purify proteins. Thus, it is a prudent practice to evaluate the removal of the prion agent on a case-by-case basis, and not rely on analogy.
Table V.

Removal of prion protein scrapie form and purification process parameters
3.2.4 Examples of Prion Removal
One of the best studied processes for prion clearance is the Cohn-Oncley fractionation. The general methodology of this process (see section 2.2.1) is to lower the temperature of plasma, increase the alcohol concentration, and make pH adjustments to selectively precipitate classes of proteins. A number of laboratories have investigated the inherent ability of the Cohn-Oncley process to remove spiked prion protein and infectivity.[114,115,127,131–134] Cryoseparation and Fraction I separation yield Factor VIII/vWB and Fibrinogen/Factor IX, respectively (figure 1). These two plasma intermediates have the highest protein concentrations, and the clearance of spiked prions is ≤1 log10. Following the Fraction I separation is the Fraction II+III separation, which is the first significant prion removal step. The effect of 20% (volume/volume) alcohol combined with low temperature and centrifugation results in ≥4.7 log10 of removal of PrPSc with respect to the effluent.[61] This suggests that prions can be precipitated or aggregated using specific solution-state conditions. The Fraction II+III clearance is clearly beneficial for the downstream products (α1-antitrypsin, plasma protein fraction, and albumin) in the Cohn-Oncley process. However, this removal is not in favor of the immunoglobulin product, as the prion spike copurifies with the Fraction II+III paste, which is the starting material for the IVIG process. Fortunately, there are steps during immunoglobulin purification that significantly clear spiked prions from the immunoglobulin product. For example, the Fraction III separation relies on solution conditions that maintain the solubility of IgGs in 17% alcohol at low temperatures while precipitating the prions, which are removed (≈4 log10) using centrifugal sedimentation.
Factors that control the precipitation of PrPSc during the Cohn-Oncley fractionation have been investigated in some detail.[131] Several factors were identified that controlled the precipitation of PrPSc in solution; among these were pH, alcohol content, and salt concentration. Other parameters such as total protein concentration, specific protein constituents, and temperature were found to be less important in terms of precipitating PrPSc.
A new IVIG product has been developed using a chromatographic process as the major purification method.[65] In this process, prions are effectively precipitated at low pH in presence of caprylate and removed by a subsequent filtration step. A significant prion reduction factor of >4 log10 has been validated for this step in the process.[61] Other organic agents, such as PEG, can also be used to effectively remove spiked prions from process intermediates. In one such purification process, the addition of 11.5% PEG 3350 yielded ≥4.9 log10 of PrPSc removal.[61,129]
Utilizing precipitation coupled to sedimentation and filtration are not the only effective means to remove prions and reduce the risk of TSE transmission. Nanofiltration, a size exclusion-based method, has also demonstrated a significant (≥4 log10) potential for the removal of prions. For example, the Viresolve® NFP (Normal Flow Parvovirus) and Viresolve® 180 membranes (Millipore, Billerica, MA, USA) yield removal of various preparations of prion spikes from model and process solutions.[61] Similar removal values were measured using Novasip™ DV20 (Pall Corp., East Hills, NY, USA) and Planova® 15N (Asahi Kasei Pharma, Tokyo, Japan) nanofilters.[135] Chromatography methods may also be effective in removing prions to various degrees.[132]
3.2.5 Relevance of Spiking Material
An issue related to the investigation of prion removal is the use of spiking material derived from rodent brain tissue. Typically, hamster 263K, which is a PrPSc strain derived from sheep scrapie, is used in clearance studies. However, it is important to establish hamster scrapie as a valid model for the partitioning of human prions. A comparative study of the partitioning of human, sheep, and hamster forms of PrPSc was conducted.[125] Three plasma purification steps were chosen that demonstrated low (cryoseparation, about 1 log10), intermediate (3% PEG precipitation, 2–3 log10), and high (11.5% PEG precipitation, 3 log10) clearance of PrPSc in the hamster scrapie spike model. In brief, it was found that PrPSc derived from vCJD, classical CJD, sporadic CJD, or Gerstmann Sträussler Sheinker disease human brain tissue partitioned quantitatively similarly to PrPSc derived from sheep or hamster scrapie.[133] These data indicate that in the case of biochemical separations, hamster scrapie can be used to predict the partitioning of human and other species of prions.
The preparation of the spiking material is a related issue.[132,134] The physical properties of circulating TSE infectivity in blood have not been measured. Brain homogenate may or may not be an ideal model of endogenous circulating TSE infectivity. Microsomal and highly purified preparations from brain homogenate have been proposed and investigated as alternatives to the crude brain homogenate. As a rule, the membrane-bound forms of the TSE infectivity and prion protein (i.e. crude, clarified, or microsomal fractions of brain homogenate) result in the lowest removal measured in spiking studies. Highly purified forms, such as fibrils or full length scrapie protein, tend to result in very high levels of removal. Thus the use of less purified fractions represents the most conservative estimation of prion removal.
4. Conclusion
During the past 2 decades, the overall risk of acquiring serious viral diseases through plasma-derived proteins has been reduced significantly. In fact, no confirmed transmission of HBV, HCV, or HIV by US licensed, virally reduced plasma derivatives has been reported in recent years.[60] This favorable status is the result of a combination of measures, including the screening and deferring of high-risk donors, the testing of collected donations using immunoassays and NAT, the incorporation and validation of virus removal and inactivation steps in production processes, and the establishment of increasingly stringent pathogen safety regulations.
In spite of these advances, the risk of pathogen transmission from plasma products can never be assumed to be nonexistent. Currently unknown viruses could emerge, known viruses could redistribute geographically or mutate into new strains. The development of testing methods that could be used to screen plasma requires time. The reduction of these risks largely relies on the viral removal and inactivation steps integrated into the manufacturing processes. However, these steps are effective against some but not all virus families. Therefore, it is important to validate each of these steps for its viral clearance capacity. For enveloped viruses, robust clearance steps are available for nearly all of the products examined (table IV). Therefore, it can be predicted with confidence that emerging enveloped viruses, such as severe acute respiratory syndrome (SARS), monkeypox, and West Nile virus are likely to be reduced significantly during manufacturing processes. On the other hand, non-enveloped viruses, especially small DNA viruses, are more difficult to inactivate (table IV).
For the special case of the pathogenic prions, collective experimental data indicate that the likelihood of transmission through plasma products is extremely low. This assumption is substantiated by the fact that the transmission of CJD through plasma products in human populations has never been identified and that various manufacturing steps are capable of removing pathogenic prions.
To this end, manufacturers of plasma-derived biologic products continue to implement new measures to decrease the potential for disease transmission. Such measures may include the addition of testing procedures for new viruses as well as the addition of processing steps dedicated solely to the removal of viruses and prions (e.g. nanofiltration). Because plasma-derived therapeutic products are used to treat life-threatening diseases, it is imperative that diligence in the area of pathogen safety continues. Recent history has demonstrated that the threat of pathogen transmission by these products can be significantly mitigated with appropriate measures implemented at various stages of the product life cycle.
Acknowledgments
The authors thank Michael Fournel, Senior Vice President of Bayer Biological Products; Dr Wendy P. Osheroff; Dr Kathryn M. Remington; and Dr Jeanette L.C. Miller for their critical review of this manuscript.
The authors are researchers employed by Bayer Biological Products.
No sources of funding were used to assist in the preparation of this review. The authors have no conflicts of interest that are directly relevant to the content of this review.
Footnotes
The use of trade names is for product identification purposes only and does not imply endorsement.
References
- 1.Cohn E.J., Strong L.E., Hughes W.L., et al. Preparation and properties of serum and plasma proteins: IV. A system for the separation into fractions of the protein and lipoprotein components of biological tissues and fluids. J Am Chem Soc. 1946;68:459–75. doi: 10.1021/ja01207a034. [DOI] [PubMed] [Google Scholar]
- 2.Campbell G.L., Martin A.A., Lanciotti R.S., et al. West Nile virus. Lancet Infect Dis. 2002;2(9):519–29. doi: 10.1016/S1473-3099(02)00368-7. [DOI] [PubMed] [Google Scholar]
- 3.Drosten C., Gunther S., Preiser W., et al. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N Engl J Med. 2003;348(20):1967–76. doi: 10.1056/NEJMoa030747. [DOI] [PubMed] [Google Scholar]
- 4.Ksiazek T.G., Erdman D., Goldsmith C.S., et al. A novel coronavirus associated with severe acute respiratory syndrome. N Engl J Med. 2003;348(20):1953–66. doi: 10.1056/NEJMoa030781. [DOI] [PubMed] [Google Scholar]
- 5.Wagner S.J., Friedman L.I., Dodd R.Y. Transfusion-associated bacterial sepsis. Clin Microbiol Rev. 1994;7(3):290–302. doi: 10.1128/cmr.7.3.290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nordenfelt E., Kjellen L. Presence and persistence of Australian antigen in a Swedish hepatitis series. Acta Pathol Microbiol Scand. 1969;77(3):489–94. doi: 10.1111/j.1699-0463.1969.tb04255.x. [DOI] [PubMed] [Google Scholar]
- 7.Alter H.J., Blumberg B.S. Further studies on a ‘new’ human isoprecipitin system (Australia antigen) Blood. 1966;27(3):297–309. [PubMed] [Google Scholar]
- 8.Choo Q.L., Kuo G., Weiner A.J., et al. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science. 1989;244(4902):359–62. doi: 10.1126/science.2523562. [DOI] [PubMed] [Google Scholar]
- 9.Alter H.J., Purcell R.H., Shih J.W., et al. Detection of antibody to hepatitis C virus in prospectively followed transfusion recipients with acute and chronic non-A, non-B hepatitis. N Engl J Med. 1989;321(22):1494–500. doi: 10.1056/NEJM198911303212202. [DOI] [PubMed] [Google Scholar]
- 10.Leveton L.B., Sox Jr H.C., Stoto M.A., editors. HIV and the blood supply: an analysis of crisis decision making. Washington, DC: National Academy Press; 1995. [DOI] [PubMed] [Google Scholar]
- 11.Kleinman S., Busch M.P., Korelitz J.J., et al. The incidence/window period model and its use to assess the risk of transfusion-transmitted human immunodeficiency virus and hepatitis C virus infection. Transfus Med Rev. 1997;11(3):155–72. doi: 10.1053/tmrv.1997.0110155. [DOI] [PubMed] [Google Scholar]
- 12.US Food and Drug Administration, Center for Biologics Evaluation and Research (CBER). Licensed/approved HIV, HTLV and hepatitis tests [online]. Available from URL: http://www.fda.gov/cber/products/testkits.htm [Accessed 2005 Feb 22]
- 13.Urba W.J., Longo D.L. Clinical spectrum of human retroviral-induced diseases. Cancer Res. 1985;45(9Suppl.):4637s–43s. [PubMed] [Google Scholar]
- 14.Cooper D.A., Imrie A.A., Penny R. Antibody response to human immunodeficiency virus after primary infection. J Infect Dis. 1987;155(6):1113–8. doi: 10.1093/infdis/155.6.1113. [DOI] [PubMed] [Google Scholar]
- 15.Pantaleo G., Fauci A.S. Immunopathogenesis of HIV infection. Annu Rev Microbiol. 1996;50:825–54. doi: 10.1146/annurev.micro.50.1.825. [DOI] [PubMed] [Google Scholar]
- 16.Busch M.P., Kleinman S.H., Nemo G.J. Current and emerging infectious risks of blood transfusions. JAMA. 2003;289(8):959–62. doi: 10.1001/jama.289.8.959. [DOI] [PubMed] [Google Scholar]
- 17.Kuo G., Choo Q.L., Alter H.J., et al. An assay for circulating antibodies to a major etiologic virus of human non-A, non-B hepatitis. Science. 1989;244(4902):362–4. doi: 10.1126/science.2496467. [DOI] [PubMed] [Google Scholar]
- 18.Hosein B., Fang C.T., Popovsky M.A., et al. Improved serodiagnosis of hepatitis C virus infection with synthetic peptide antigen from capsid protein. Proc Natl Acad sci U S A. 1991;88(9):3647–51. doi: 10.1073/pnas.88.9.3647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vrielink H., Reesink H.W., van den Burg P.J., et al. Performance of three generations of anti-hepatitis C virus enzyme-linked immunosorbent assays in donors and patients. Transfusion. 1997;37(8):845–9. doi: 10.1046/j.1537-2995.1997.37897424409.x. [DOI] [PubMed] [Google Scholar]
- 20.Schreiber G.B., Busch M.P., Kleinman S.H., et al. The risk of transfusion-transmitted viral infections: the Retrovirus Epidemiology Donor Study. N Engl J Med. 1996;334(26):1685–90. doi: 10.1056/NEJM199606273342601. [DOI] [PubMed] [Google Scholar]
- 21.Nubling C.M., Scitz R., Lower J. Application of nucleic acid amplification techniques for blood donation screening. Infusionsther Transfusionsmed. 1998;25:86–90. [Google Scholar]
- 22.Walsh J.H., Yalow R., Berson S.A. Detection of Australia antigen and antibody by means of radioimmunoassay techniques. J Infect Dis. 1970;121(5):550–4. doi: 10.1093/infdis/121.5.550. [DOI] [PubMed] [Google Scholar]
- 23.Cossart Y.E., Field A.M. Virus-like particles in serum of patients with Australia-antigen-associated hepatitis [letter] Lancet. 1970;I(7651):848. doi: 10.1016/S0140-6736(70)92460-8. [DOI] [PubMed] [Google Scholar]
- 24.Dane D.S., Cameron C.H., Briggs M. Virus-like particles in serum of patients with Australia-antigen-associated hepatitis. Lancet. 1970;I(7649):695–8. doi: 10.1016/S0140-6736(70)90926-8. [DOI] [PubMed] [Google Scholar]
- 25.Schroeder D.D., Mozen M.M. Australia antigen: distribution during Cohn ethanol fractionation of human plasma. Science. 1970;168(938):1462–4. doi: 10.1126/science.168.3938.1462. [DOI] [PubMed] [Google Scholar]
- 26.Andrassy K., Ritz E., Sanwald R. Australia antigen in various plasma fractions. Vox Sang. 1970;19(3):357–8. doi: 10.1111/j.1423-0410.1970.tb01539.x. [DOI] [PubMed] [Google Scholar]
- 27.Blumberg B.S., London W.T., Sutnick A.I. Practical applications of the Australia antigen test. Postgrad Med. 1971;50(6):70–6. doi: 10.1080/00325481.1971.11697697. [DOI] [PubMed] [Google Scholar]
- 28.Hollinger F.B., Vorndam V., Dreesman G.R. Assay of Australia antigen and antibody employing double-antibody and solid-phase radioimmunoassay techniques and comparison with the passive hemagglutination methods. J Immunol. 1971;107(4):1099–111. [PubMed] [Google Scholar]
- 29.Kliman A. Australia antigen in volunteer and paid blood donors [letter] N Engl J Med. 1971;284(2):109. doi: 10.1056/NEJM197101142840220. [DOI] [PubMed] [Google Scholar]
- 30.Palmer D.R., Perry K.R., Mortimer P.P., et al. Variation in the sensitivity of HBsAg screening kits. Transfus Med. 1996;6(4):311–7. doi: 10.1111/j.1365-3148.1996.tb00089.x. [DOI] [PubMed] [Google Scholar]
- 31.Biswas R., Tabor E., Hsia C.C., et al. Comparative sensitivity of HBV NATs and HBsAg assays for detection of acute HBV infection. Transfusion. 2003;43(6):788–98. doi: 10.1046/j.1537-2995.2003.00424.x. [DOI] [PubMed] [Google Scholar]
- 32.AuBuchon J.P., Birkmeyer J.D., Busch M.P. Cost-effectiveness of expanded human immunodeficiency virus-testing protocols for donated blood. Transfusion. 1997;37(1):45–51. doi: 10.1046/j.1537-2995.1997.37197176950.x. [DOI] [PubMed] [Google Scholar]
- 33.Busch M.P., Taylor P.E., Lenes B.A., et al. Screening of selected male blood donors for p24 antigen of human immunodeficiency virus type 1: the Transfusion Safety Study Group. N Engl J Med. 1990;323(19):1308–12. doi: 10.1056/NEJM199011083231904. [DOI] [PubMed] [Google Scholar]
- 34.Alter H.J., Epstein J.S., Swenson S.G., et al. Prevalence of human immunodeficiency virus type 1 p24 antigen in US blood donors: an assessment of the efficacy of testing in donor screening. The HIV-Antigen Study Group. N Engl J Med. 1990;323(19):1312–7. doi: 10.1056/NEJM199011083231905. [DOI] [PubMed] [Google Scholar]
- 35.US Food and Drug Administration, Center for Biologics Evaluation and Research (CBER). Recommendations for donor screening with a licensed test for HIV-1 antigen [online]. Available from URL: http://www.fda.gov/cber/bldmem/hivag.pdf [Accessed 2005 Feb 22]
- 36.US Food and Drug Administration, Center for Biologics Evaluation and Research (CBER). Product approval information, Licenses #1582, #1592, #1636 [online]. Available from URL: http://www.fda.gov/cber/efoi/approve.htm [Accessed 2005 Feb 22]
- 37.US Food and Drug Administration, Center for Biologies Evaluation and Research (CBER). Criteria for discontinuation of HIV-1 p24 antigen screening of source plasma: current thinking, 1999 [online]. Available from URL: http://www.fda.gov/ohrms/dockets/ac/00/backgrd/3649blg.doc [Accessed 2005 Feb 22]
- 38.Lanciotti R.S., Kerst A.J. Nucleic acid sequence-based amplification assays for rapid detection of West Nile and St Louis encephalitis viruses. J Clin Microbiol. 2001;39(12):4506–13. doi: 10.1128/JCM.39.12.4506-4513.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Murakawa G.J., Zaia J.A., Spallone P.A., et al. Direct detection of HIV-1 RNA from AIDS and ARC patient samples. DNA. 1988;7(4):287–95. doi: 10.1089/dna.1988.7.287. [DOI] [PubMed] [Google Scholar]
- 40.Kubo Y., Takeuchi K., Boonmar S., et al. A cDNA fragment of hepatitis C virus isolated from an implicated donor of post-transfusion non-A, non-B hepatitis in Japan. Nucleic Acids Res. 1989;17(24):10367–72. doi: 10.1093/nar/17.24.10367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Giachetti C., Linnen J.M., Kolk D.P., et al. Highly sensitive multiplex assay for detection of human immunodeficiency virus type 1 and hepatitis C virus RNA. J Clin Microbiol. 2002;40(7):2408–19. doi: 10.1128/JCM.40.7.2408-2419.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kievits T., van Gemen B., van Strijp D., et al. NASBA isothermal enzymatic in vitro nucleic acid amplification optimized for the diagnosis of HIV-1 infection. J Virol Methods. 1991;35(3):273–86. doi: 10.1016/0166-0934(91)90069-C. [DOI] [PubMed] [Google Scholar]
- 43.Ichiyama S., Ito Y., Sugiura F., et al. Diagnostic value of the strand displacement amplification method compared to those of Roche Amplicor PCR and culture for detecting mycobacteria in sputum samples. J Clin Microbiol. 1997;35(12):3082–5. doi: 10.1128/jcm.35.12.3082-3085.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Marshall R.L., Laffler T.G., Cerney M.B., et al. Detection of HCV RNA by the asymmetric gap ligase chain reaction. PCR Methods Appl. 1994;4(2):80–4. doi: 10.1101/gr.4.2.80. [DOI] [PubMed] [Google Scholar]
- 45.Terrault N.A., Dailey P.J., Ferrell L., et al. Hepatitis C virus: quantitation and distribution in liver. J Med Virol. 1997;51(3):217–24. doi: 10.1002/(SICI)1096-9071(199703)51:3<217::AID-JMV13>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 46.Robertson J.S. International standardization of gene amplification technology. Biologicals. 1998;26(2):111–3. doi: 10.1006/biol.1998.0136. [DOI] [PubMed] [Google Scholar]
- 47.Holmes H., Davis C., Heath A., et al. An international collaborative study to establish the 1st international standard for HIV-1 RNA for use in nucleic acid-based techniques. J Virol Methods. 2001;92(2):141–50. doi: 10.1016/S0166-0934(00)00283-4. [DOI] [PubMed] [Google Scholar]
- 48.Davis C., Heath A., Best S. Calibration of HIV-1 working reagents for nucleic acid amplification techniques against the 1st international standard for HIV-1 RNA. J Virol Methods. 2003;107(1):37–44. doi: 10.1016/S0166-0934(02)00187-8. [DOI] [PubMed] [Google Scholar]
- 49.Saldanha J., Gerlich W., Lelie N., et al. An international collaborative study to establish a World Health Organization international standard for hepatitis B virus DNA nucleic acid amplification techniques. Vox Sang. 2001;80(1):63–71. doi: 10.1046/j.1423-0410.2001.00003.x. [DOI] [PubMed] [Google Scholar]
- 50.Saldanha J., Lelie N., Heath A. Establishment of the first international standard for nucleic acid amplification technology (NAT) assays for HCV RNA: WHO Collaborative Study Group. Vox Sang. 1999;76(3):149–58. doi: 10.1046/j.1423-0410.1999.7630149.x. [DOI] [PubMed] [Google Scholar]
- 51.Saldanha J., Heath A., Lelie N., et al. Calibration of HCV working reagents for NAT assays against the HCV international standard: the Collaborative Study Group. Vox Sang. 2000;78(4):217–24. doi: 10.1046/j.1423-0410.2000.7840217.x. [DOI] [PubMed] [Google Scholar]
- 52.Saldanha J, Heath A, Lelie N, et al. A World Health Organisation (WHO) international standard for hepatitis A virus (HAV) RNA nucleic acid amplification technology (NAT) assays. Vox Sang. In press 2005 [DOI] [PubMed]
- 53.Saldanha J., Lelie N., Yu M.W., et al. Establishment of the first World Health Organization International Standard for human parvovirus B19 DNA nucleic acid amplification techniques. Vox Sang. 2002;82(1):24–31. doi: 10.1046/j.1423-0410.2002.00132.x. [DOI] [PubMed] [Google Scholar]
- 54.European Agency for the Evaluation of Medicinal Products/Committee for Proprietory Medical Products. The introduction of nucleic acid amplification technology (NAT) for the detection of hepatitis C virus RNA in plasma pools (CPMP/BWP/390/97): addendum to Note for guidance on plasma derived medicinal products, (CPMP/BWP/269/95) [online]. Available from URL: http://www.tga.gov.au/docs/pdf/euguide/bwp/039097en.pdf [Accessed 2005 Feb 22]
- 55.US Food and Drug Administration, Center for Biologics Evaluation and Research (CBER). Guidance for industry: use of nucleic acid tests on pooled samples from source plasma donors to adequately and appropriately reduce the risk of transmission of HIV-1 and HCV, 2001 [online]. Available from URL: http://www.fda.gov/cber/gdlns/hivhcvnatbld.htm [Accessed 2005 Feb 22]
- 56.Busch M.P., Lee L.L., Satten G.A., et al. Time course of detection of viral and serologic markers preceding human immunodeficiency virus type 1 seroconversion: implications for screening of blood and tissue donors. Transfusion. 1995;35(2):91–7. doi: 10.1046/j.1537-2995.1995.35295125745.x. [DOI] [PubMed] [Google Scholar]
- 57.Piatak M., Jr, Saag M.S., Yang L.C., et al. High levels of HIV-1 in plasma during all stages of infection determined by competitive PCR. Science. 1993;259(5102):1749–54. doi: 10.1126/science.8096089. [DOI] [PubMed] [Google Scholar]
- 58.Morandi P.A., Schockmel G.A., Yerly S., et al. Detection of human immunodeficiency virus type 1 (HIV-1) RNA in pools of sera negative for antibodies to HIV-1 and HIV-2. J Clin Microbiol. 1998;36(6):1534–8. doi: 10.1128/jcm.36.6.1534-1538.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.European Agency for the Evaluation of Medicinal Products/Committee for Proprietory Medical Products. Note for guidance on virus validation studies: the design, contribution and interpretation of studies validating the inactivation and removal of viruses [online]. Available from URL: http://www.emea.eu.int/pdfs/human/bwp/026895en.pdf [Accessed 2005 Feb 22]
- 60.Tabor E. The epidemiology of virus transmission by plasma derivatives: clinical studies verifying the lack of transmission of hepatitis B and C viruses and HIV type 1. Transfusion. 1999;39(11–12):1160–8. doi: 10.1046/j.1537-2995.1999.39111160.x. [DOI] [PubMed] [Google Scholar]
- 61.Internal viral clearance reports. Research Triangle Park (NC): Bayer Biological Products, 2003. (Data on file)
- 62.Scheiblauer H., Nubling M., Willkommen H., et al. Prevalence of hepatitis C virus in plasma pools and the effectiveness of cold ethanol fractionation. Clin Ther. 1996;18(Suppl.B):59–70. doi: 10.1016/S0149-2918(96)80196-2. [DOI] [PubMed] [Google Scholar]
- 63.Steinbuch M., Audran R. Isolation of IgG immunoglobulin from human plasma using caprylic acid [in French] Rev Fr Etud Clin Biol. 1969;14(10):1054–8. [PubMed] [Google Scholar]
- 64.Habeeb A.F., Francis R.D. Preparation of human immunoglobulin by caprylic acid precipitation. Prep Biochem. 1984;14(1):1–17. doi: 10.1080/10826068408070610. [DOI] [PubMed] [Google Scholar]
- 65.Stephan W., Dichtelmuller H., Schedel I. Properties and efficacy of a human immunoglobulin M preparation for intravenous administration [in German] Arzneimittel Forschung. 1985;35(6):933–6. [PubMed] [Google Scholar]
- 66.Lebing W., Remington K.M., Schreiner C., et al. Properties of a new intravenous immunoglobulin (IGIV-C, 10%) produced by virus inactivation with caprylate and column chromatography. Vox Sang. 2003;84(3):193–201. doi: 10.1046/j.1423-0410.2003.00285.x. [DOI] [PubMed] [Google Scholar]
- 67.Horowitz B., Piet M.P., Prince A.M., et al. Inactivation of lipid-enveloped viruses in labile blood derivatives by unsaturated fatty acids. Vox Sang. 1988;54(1):14–20. doi: 10.1111/j.1423-0410.1988.tb01606.x. [DOI] [PubMed] [Google Scholar]
- 68.Lundblad J.L., Seng R.L. Inactivation of lipid-enveloped viruses in proteins by caprylate. Vox Sang. 1991;60(2):75–81. doi: 10.1111/j.1423-0410.1991.tb00878.x. [DOI] [PubMed] [Google Scholar]
- 69.Dichtelmuller H., Rudnick D., Kloft M. Inactivation of lipid enveloped viruses by octanoic acid treatment of immunoglobulin solution. Biologicals. 2002;30(2):135–42. doi: 10.1006/biol.2002.0332. [DOI] [PubMed] [Google Scholar]
- 70.Trejo S.R., Hotta J.A., Lebing W., et al. Evaluation of virus and prion reduction in a new intravenous immunoglobulin manufacturing process. Vox Sang. 2003;84(3):176–87. doi: 10.1046/j.1423-0410.2003.00279.x. [DOI] [PubMed] [Google Scholar]
- 71.Burnouf T., Radosevich M. Affinity chromatography in the industrial purification of plasma proteins for therapeutic use. J Biochem Biophys Methods. 2001;49(1-3):575–86. doi: 10.1016/S0165-022X(01)00221-4. [DOI] [PubMed] [Google Scholar]
- 72.Chandra S., Groener A., Feldman F. Effectiveness of alternative treatments for reducing potential viral contaminants from plasma-derived products. Thromb Res. 2002;105(5):391–400. doi: 10.1016/S0049-3848(02)00044-0. [DOI] [PubMed] [Google Scholar]
- 73.Burnouf T., Radosevich M. Nanofiltration of plasma-derived biopharmaceutical products. Haemophilia. 2003;9(1):24–37. doi: 10.1046/j.1365-2516.2003.00701.x. [DOI] [PubMed] [Google Scholar]
- 74.Barrowcliffe T.W. Viral inactivation vs biological activity. Dev Biol Stand. 1993;81:125–35. [PubMed] [Google Scholar]
- 75.Mannucci P.M. Viral safety of coagulation factor concentrates. In: Brown F., editor. Virological safety aspects of plasma derivative. Basel: Karger; 1993. pp. 253–9. [Google Scholar]
- 76.Burnouf T., Radosevich M. Reducing the risk of infection from plasma products: specific preventative strategies. Blood Rev. 2000;14(2):94–110. doi: 10.1054/blre.2000.0129. [DOI] [PubMed] [Google Scholar]
- 77.Korneyeva M., Hotta J., Lebing W., et al. Enveloped virus inactivation by caprylate: a robust alternative to solvent-detergent treatment in plasma derived intermediates. Biologicals. 2002;30(2):153–62. doi: 10.1006/biol.2002.0334. [DOI] [PubMed] [Google Scholar]
- 78.Edsall J.T. Stabilization of serum albumin to heat, and inactivation of the hepatitis virus. Vox Sang. 1984;46(5):338–40. doi: 10.1111/j.1423-0410.1984.tb00096.x. [DOI] [PubMed] [Google Scholar]
- 79.European Agency for the Evaluation of Medicinal Products/Committee for Proprietory Medical Products. Note for guidance on plasma-derived medicinal products [online]. Available from URL: http://www.emea.eu.int/pdfs/human/bwp/026995en.pdf [Accessed 2005 Feb 22]
- 80.Blumel J., Schmidt I., Willkommen H., et al. Inactivation of parvovirus B19 during pasteurization of human serum albumin. Transfusion. 2002;42(8):1011–8. doi: 10.1046/j.1537-2995.2002.00158.x. [DOI] [PubMed] [Google Scholar]
- 81.Hayakawa F., Imada K., Towatari M., et al. Life-threatening human parvovirus B19 infection transmitted by intravenous immune globulin. Br J Haematol. 2002;118(4):1187–9. doi: 10.1046/j.1365-2141.2002.03741.x. [DOI] [PubMed] [Google Scholar]
- 82.Horwith G., Revie D.R. Efficacy of viral clearance methods used in the manufacture of activated prothrombin complex concentrates: focus on AUTOPLEX T. Haemophilia. 1999;5(Suppl.3):19–23. doi: 10.1046/j.1365-2516.1999.00033.x. [DOI] [PubMed] [Google Scholar]
- 83.Mannucci P.M., Schimpf K., Abe T., et al. Low risk of viral infection after administration of vapor-heated factor VIII concentrate: International Investigator Group. Transfusion. 1992;32(2):134–8. doi: 10.1046/j.1537-2995.1992.32292180141.x. [DOI] [PubMed] [Google Scholar]
- 84.Barrett P.N., Meyer H., Wachtel I., et al. Inactivation of hepatitis A virus in plasma products by vapor heating. Transfusion. 1997;37(2):215–20. doi: 10.1046/j.1537-2995.1997.37297203527.x. [DOI] [PubMed] [Google Scholar]
- 85.Blumel J., Schmidt I., Effenberger W., et al. Parvovirus B19 transmission by heat-treated clotting factor concentrates. Transfusion. 2002;42(11):1473–81. doi: 10.1046/j.1537-2995.2002.00221.x. [DOI] [PubMed] [Google Scholar]
- 86.Savage M., Torres J., Franks L., et al. Determination of adequate moisture content for efficient dry-heat viral inactivation in lyophilized factor VIII by loss on drying and by near infrared spectroscopy. Biologicals. 1998;26(2):119–24. doi: 10.1006/biol.1998.0140. [DOI] [PubMed] [Google Scholar]
- 87.World Health Organization. Guidelines on viral inactivation and removal procedures intended to assure the viral safety of human blood plasma products. Geneva: Expert Committee on Biological Standardization, 2001: 40–44
- 88.Ways J.P., Preston M.S., Baker D., et al. Good manufacturing practice (GMP) compliance in the biologics sector: plasma fractionation. Biotechnol Appl Biochem. 1999;30:257–65. [PubMed] [Google Scholar]
- 89.US General Accounting Office. Blood plasma safety: plasma product risks are low if good manufacturing practices are followed. Report to the Chairman, Subcommittee on Human Resources, Committee on Government Reform and Oversight, House of Representatives, Sep 1998 [online]. Available from URL: http://www.gao.gov/archive/1998/he98205.pdf [Accessed 2005 Feb 22]
- 90.Aguzzi A., Weissmann C. Prion diseases. Haemophilia. 1998;4(4):619–27. doi: 10.1046/j.1365-2516.1998.440619.x. [DOI] [PubMed] [Google Scholar]
- 91.Prusiner S.B. Prions. Proc Natl Acad sci U S A. 1998;95(23):13363–83. doi: 10.1073/pnas.95.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Chesebro B. Prion protein and the transmissible spongiform encephalopathy diseases. Neuron. 1999;24(3):503–6. doi: 10.1016/S0896-6273(00)81105-8. [DOI] [PubMed] [Google Scholar]
- 93.Goldfarb L.G., Brown P. The transmissible spongiform encephalopathies. Annu Rev Med. 1995;46:57–65. doi: 10.1146/annurev.med.46.1.57. [DOI] [PubMed] [Google Scholar]
- 94.Hill A.F., Desbruslais M., Joiner S., et al. The same prion strain causes vCJD and BSE. Nature. 1997;389(6650):448–50. doi: 10.1038/38925. [DOI] [PubMed] [Google Scholar]
- 95.Bruce M.E., Will R.G., Ironside J.W., et al. Transmissions to mice indicate that ‘new variant’ CJD is caused by the BSE agent. Nature. 1997;389(6650):498–501. doi: 10.1038/39057. [DOI] [PubMed] [Google Scholar]
- 96.Scott M.R., Will R., Ironside J., et al. Compelling transgenetic evidence for transmission of bovine spongiform encephalopathy prions to humans. Proc Natl Acad sci U S A. 1999;96(26):15137–42. doi: 10.1073/pnas.96.26.15137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Glatzel M., Aguzzi A. Peripheral pathogenesis of prion diseases. Microbes Infect. 2000;2(6):613–9. doi: 10.1016/S1286-4579(00)00364-6. [DOI] [PubMed] [Google Scholar]
- 98.Ramasamy I., Law M., Collins S., et al. Organ distribution of prion proteins in variant Creutzfeldt-Jakob disease. Lancet Infect Dis. 2003;3(4):214–22. doi: 10.1016/S1473-3099(03)00578-4. [DOI] [PubMed] [Google Scholar]
- 99.Holman R.C., Khan A.S., Kent J., et al. Epidemiology of Creutzfeldt-Jakob disease in the United States, 1979-1990: analysis of national mortality data. Neuroepidemiology. 1995;14(4):174–81. doi: 10.1159/000109793. [DOI] [PubMed] [Google Scholar]
- 100.Gibbons R.V., Holman R.C., Belay E.D., et al. Creutzfeldt-Jakob disease in the United States: 1979-1998. JAMA. 2000;284(18):2322–3. doi: 10.1001/jama.284.18.2322. [DOI] [PubMed] [Google Scholar]
- 101.Evatt B., Austin H., Barnhart E., et al. Surveillance for Creutzfeldt-Jakob disease among persons with hemophilia. Transfusion. 1998;38(9):817–20. doi: 10.1046/j.1537-2995.1998.38998409000.x. [DOI] [PubMed] [Google Scholar]
- 102.Lee C.A., Ironside J.W., Bell J.E., et al. Retrospective neuropathological review of prion disease in UK haemophilic patients. Thromb Haemost. 1998;80(6):909–11. [PubMed] [Google Scholar]
- 103.National Blood Data Resource Center. Creutzfeldt-Jakob Disease investigational lookback study [online]. Available from URL: http://www.nbdrc.org/research/cjd.htm [Accessed 2005 Feb 22]
- 104.Heye N., Hensen S., Muller N. Creutzfeldt-Jakob disease and blood transfusion. Lancet. 1994;343(8892):298–9. doi: 10.1016/S0140-6736(94)91148-7. [DOI] [PubMed] [Google Scholar]
- 105.National CJD Surveillance Unit, Western General Hospital. Creutzfeldt-Jakob disease surveillance in the UK: twelfth annual report [online]. Available from URL: http://www.cjd.ed.ac.uk/twelfth/rep2003.htm [Accessed 2005 Feb 22]
- 106.Llewelyn C.A., Hewitt P.E., Knight R.S., et al. Possible transmission of variant Creutzfeldt-Jakob disease by blood transfusion. Lancet. 2004;363:417–21. doi: 10.1016/S0140-6736(04)15486-X. [DOI] [PubMed] [Google Scholar]
- 107.Peden A.H., Head M.W., Ritchie D.L., et al. Preclinical vCJD after blood transfusion in a PRNP codon 129 heterozygous patient. Lancet. 2004;364:527–9. doi: 10.1016/S0140-6736(04)16811-6. [DOI] [PubMed] [Google Scholar]
- 108.Brown P., Cervenakova L., Diringer H. Blood infectivity and the prospects for a diagnostic screening test in Creutzfeldt-Jakob disease. J Lab Clin Med. 2001;137(1):5–13. doi: 10.1067/mlc.2001.111951. [DOI] [PubMed] [Google Scholar]
- 109.Brown P., Gibbs Jr C.J., Rodgers-Johnson P., et al. Human spongiform encephalopathy: the National Institutes of Health series of 300 cases of experimentally transmitted disease. Ann Neurol. 1994;35(5):513–29. doi: 10.1002/ana.410350504. [DOI] [PubMed] [Google Scholar]
- 110.Bruce M.E., McConnell I., Will R.G., et al. Detection of variant Creutzfeldt-Jakob disease infectivity in extraneural tissues. Lancet. 2001;358(9277):208–9. doi: 10.1016/S0140-6736(01)05411-3. [DOI] [PubMed] [Google Scholar]
- 111.Houston F., Foster J.D., Chong A., et al. Transmission of BSE by blood transfusion in sheep. Lancet. 2000;356(9234):999–1000. doi: 10.1016/S0140-6736(00)02719-7. [DOI] [PubMed] [Google Scholar]
- 112.Hunter N., Foster J., Chong A., et al. Transmission of prion diseases by blood transfusion. J Gen Virol. 2002;83:2897–905. doi: 10.1099/0022-1317-83-11-2897. [DOI] [PubMed] [Google Scholar]
- 113.Kuroda Y., Gibbs C.J., Jr, Amyx H.L., et al. Creutzfeldt-Jakob disease in mice: persistent viremia and preferential replication of virus in low-density lymphocytes. Infect Immun. 1983;41(1):154–61. doi: 10.1128/iai.41.1.154-161.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Brown P., Rohwer R.G., Dunstan B.C., et al. The distribution of infectivity in blood components and plasma derivatives in experimental models of transmissible spongiform encephalopathy. Transfusion. 1998;38(9):810–6. doi: 10.1046/j.1537-2995.1998.38998408999.x. [DOI] [PubMed] [Google Scholar]
- 115.Brown P., Cervenakova L., McShane L.M., et al. Further studies of blood infectivity in an experimental model of transmissible spongiform encephalopathy, with an explanation of why blood components do not transmit Creutzfeldt-Jakob disease in humans. Transfusion. 1999;39(11–12):1169–78. doi: 10.1046/j.1537-2995.1999.39111169.x. [DOI] [PubMed] [Google Scholar]
- 116.Holada K., Vostal J.G., Theisen P.W., et al. Scrapie infectivity in hamster blood is not associated with platelets. J Virol. 2002;76(9):4649–50. doi: 10.1128/JVI.76.9.4649-4650.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.US Food and Drug Administration, Center for Biologies Evaluation and Research (CBER). Guidance for industry: revised preventive measures to reduce the possible risk of transmission of Creutzfeldt-Jakob Disease (CJD) and variant Creutzfeldt-Jakob disease (vCJD) by blood and blood products, 2002 [online]. Available from URL: httpV/www.fda.gov/cber/gdlns/cjdvcjd.htm [Accessed 2005 Feb 22]
- 118.Saborio G.P., Permanne B., Soto C. Sensitive detection of pathological prion protein by cyclic amplification of protein misfolding. Nature. 2001;411(6839):810–3. doi: 10.1038/35081095. [DOI] [PubMed] [Google Scholar]
- 119.Schmitt J., Beekes M., Brauer A., et al. Identification of scrapie infection from blood serum by Fourier transform infrared spectroscopy. Anal Chem. 2002;74(15):3865–8. doi: 10.1021/ac015688s. [DOI] [PubMed] [Google Scholar]
- 120.Safar J., Wille H., Itri V., et al. Eight prion strains have PrP (Sc) molecules with different conformations. Nat Med. 1998;4(10):1157–65. doi: 10.1038/2654. [DOI] [PubMed] [Google Scholar]
- 121.Bieschke J., Giese A., Schulz-Schaeffer W., et al. Ultrasensitive detection of pathological prion protein aggregates by dual-color scanning for intensely fluorescent targets. Proc Natl Acad sci U S A. 2000;97(10):5468–73. doi: 10.1073/pnas.97.10.5468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Miele G., Manson J., Clinton M. A novel erythroid-specific marker of transmissible spongiform encephalopathies. Nat Med. 2001;7(3):361–4. doi: 10.1038/85515. [DOI] [PubMed] [Google Scholar]
- 123.European Commission. The evaluation of tests for the diagnosis of transmissible spongiform encephalophathy in bovines: Jul 1999 [online]. Available from URL: http://europa.eu.int/comm/food/fs/bse/bsel2_en.pdf [Accessed 2005 Feb 22]
- 124.European Commission. The evaluation of five rapid tests for the diagnosis of transmissible spongiform encephalophathy in bovines: 2nd study [online]. Available from URL: http://europa.eu.int/comm/food/fs/bse/bse42_en.pdf [Accessed 2005 Feb 22]
- 125.European Medicines Agency. CPMP position statement on Creutzfeldt-Jakob disease and plasma-derived and urine-derived medicinal products [online]. Available from URL: http://www.emea.eu.int/pdfs/human/press/pos/287902en.pdf [Accessed 2005 Feb 22]
- 126.Taylor D.M. Inactivation of transmissible degenerative encephalopathy agents: a review. Vet J. 2000;159(1):10–7. doi: 10.1053/tvjl.1999.0406. [DOI] [PubMed] [Google Scholar]
- 127.Lee D.C., Stenland C.J., Hartwell R.C., et al. Monitoring plasma processing steps with a sensitive Western blot assay for the detection of the prion protein. J Virol Methods. 2000;84(1):77–89. doi: 10.1016/S0166-0934(99)00135-4. [DOI] [PubMed] [Google Scholar]
- 128.Rapid method of determining clearance of prion protein. US Patent 6605445. 2003
- 129.Lee D.C., Stenland C.J., Miller J.L., et al. A direct relationship between the partitioning of the pathogenic prion protein and transmissible spongiform encephalopathy infectivity during the purification of plasma proteins. Transfusion. 2001;41(4):449–55. doi: 10.1046/j.1537-2995.2001.41040449.x. [DOI] [PubMed] [Google Scholar]
- 130.Bellon A., Seyfert-Brandt W., Lang W., et al. Improved conformation-dependent immunoassay: suitability for human prion detection with enhanced sensitivity. J Gen Virol. 2003;84:1921–5. doi: 10.1099/vir.0.18996-0. [DOI] [PubMed] [Google Scholar]
- 131.Cai K., Miller J.L., Stenland C.J., et al. Solvent-dependent precipitation of prion protein. Biochim Biophys Acta. 2002;1597(1):28–35. doi: 10.1016/S0167-4838(02)00282-0. [DOI] [PubMed] [Google Scholar]
- 132.Foster P.R., Welch A.G., McLean C., et al. Studies on the removal of abnormal prion protein by processes used in the manufacture of human plasma products. Vox Sang. 2000;78(2):86–95. doi: 10.1046/j.1423-0410.2000.7820086.x. [DOI] [PubMed] [Google Scholar]
- 133.Stenland C.J., Lee D.C., Brown P., et al. Partitioning of human and sheep forms of the pathogenic prion protein during the purification of therapeutic proteins from human plasma. Transfusion. 2002;42(11):1497–500. doi: 10.1046/j.1537-2995.2002.00216.x. [DOI] [PubMed] [Google Scholar]
- 134.Vey M., Baron H., Weimer T., et al. Purity of spiking agent affects partitioning of prions in plasma protein purification. Biologicals. 2002;30(3):187–96. doi: 10.1006/biol.2002.0327. [DOI] [PubMed] [Google Scholar]
- 135.Tateishi J., Kitamoto T., Mohri S., et al. Scrapie removal using planova virus removal filters. Biologicals. 2001;29(1):17–25. doi: 10.1006/biol.2001.0269. [DOI] [PubMed] [Google Scholar]