

Abstract
The heritability of disordered eating increases during puberty; however, prior studies have largely examined a composite score of disordered eating, rather than specific symptoms. Body weight and shape concerns cut across all eating disorder diagnoses and are some of the strongest prospective risk factors for the development of eating disorders. Yet, little is known about potential developmental increases or decreases in genetic and environmental influences for these key symptoms. This study examined differences in genetic and environmental effects on a range of body weight and shape concerns during puberty and compared results to findings for overall levels of disordered eating symptoms. Participants were 926 same-sex female twins (ages 8–16) from the Michigan State University Twin Registry. Well-validated questionnaires were used to examine pubertal maturation, overall levels of disordered eating, and a range of cognitive body weight/shape constructs: body dissatisfaction, weight/shape concerns, and weight preoccupation. Findings for overall levels of disordered eating were very similar to those obtained in previous work, with significantly increased genetic effects in girls at more advanced pubertal development. Importantly, these same pubertal increases in genetic influences were observed for body dissatisfaction and weight/shape concerns. However, no pubertal moderation of genetic effects was observed for weight preoccupation; instead, pubertal moderation of nonshared and shared environmental effects was observed. Our findings point to differences in the extent to which genetic and environmental factors contribute to various cognitive body weight and shape symptoms during puberty.
Keywords: body dissatisfaction, weight preoccupation, eating disorders, genetic, adolescence, puberty
Developmental twin studies have shown increases in the heritability of eating disorder symptoms during puberty, with nominal genetic influences (~0%) during pre-puberty and substantial genetic influences (~50%) in mid-late puberty (Culbert, et al., 2009; Klump et al., 2007a, 2007b, 2012, 2017a). Despite consistent findings that genetic influences increase across puberty, prior studies have largely focused on a composite score of disordered eating (i.e., an omnibus measure that includes binge eating, body dissatisfaction, weight preoccupation, compensatory behaviors; Culbert et al., 2009; Klump et al., 2007a, 2007b). Only one study has explored a specific disordered eating symptom: binge eating. Using the same sample as the present study, Klump et al (2017a) found pubertal moderation of genetic effects on binge eating that was similar to those observed previously for composite disordered eating scores (i.e., genetic influences increasing during mid-puberty).
Notably, weight and shape concerns cut across all eating disorder diagnoses and are some of the strongest prospective risk factors for the development of clinical eating pathology (Jacobi, 2005; Killen et al., 1996; Stice & Shaw, 2002). In order to develop more comprehensive etiologic models of eating pathology, it is important to understand if genetic influences operate solely at the level of behavior (e.g., binge eating) or if weight/shape concerns also demonstrate etiologic shifts in heritability across puberty.
No study has explored etiologic differences underlying body weight and shape concerns across pubertal development. Three developmental twin studies explored changes in the heritability across adolescent age, but results were mixed. Two studies found negligible genetic influences (<1%) in pre-adolescence (i.e., ages 10–13) and significant genetic influences (22–50%) in early-to-late adolescence (~ages 13–16; Klump et al, 2010a; Wade et al., 2012) for weight and shape concerns. A third study observed more modest developmental differences for body dissatisfaction (e.g., 49% heritability at age 11; 60% at age 17) and no significant age differences in weight preoccupation (i.e., 47% heritability at age 11; 47% at age 17) (Klump et al., 2000), yet lower power may have affected detection of significant age differences that appeared to be present (i.e., twin correlations suggested lower heritability at age 11 compared to age 17) for weight preoccupation (Klump et al., 2000). Nonetheless, the variability in results suggests that additional studies are needed to clarify developmental differences in genetic effects. Pubertal development tends to be a better predictor of changes in eating pathology both phenotypically (Killen et al., 1992) and etiologically (Klump, 2013) than age; thus, examining differences across pubertal development may more accurately depict developmental shifts in etiologic effects.
Given the above, the present study examined differences in genetic and environmental influences on body weight/shape concerns and overall levels of disordered eating across pubertal development in a large sample of adolescent same-sex female twins (N = 926). A range of weight and shape concerns subscales were assessed using two different measures to examine replicability and convergence of findings, as well as possible unique effects captured across the constructs. In order to compare these effects to those observed previously, we also investigated pubertal differences for overall levels of disordered eating symptoms. Inclusion of a composite disordered eating score allowed for replication of prior work in a new sample of twins, a reproducibility effort that fits with recent calls for replication in clinical science (Pashler & Wagenmaker, 2012; Spellman, 2013). Finally, we capitalized on the availability of a sample that spanned the full range of pubertal development (i.e., from pre-puberty to post-puberty) to ensure that we captured both very early and late developmental differences.
Methods
Participants
The sample consisted of 926 same-sex female twins (MZ n=466, 50.3%; DZ n=460, 49.7%) ranging from age 8 to 16 (M=11.71, SD=2.01; for additional details see Supplemental Table 1) from the Michigan State University Twin Registry (MSUTR; Burt & Klump, 2013, 2019; Klump & Burt, 2006). The MSUTR is a population-based twin registry that recruits twins (ages 3–50) using birth records in collaboration with the Michigan Department of Health and Human Services (see Burt & Klump, 2013 for recruitment details). The current study included archival data from the Twin Study of Mood, Behavior, and Hormones during Puberty (see O’Connor et al., 2016) within the MSUTR. The Michigan State University Institutional Review Board approved this study and all procedures complied with the Helsinki Declaration of 1975, as revised in 2008. Parents of twins provided informed consent and twins provided assent.
Notably, participants had to meet several inclusion/exclusion criteria (e.g., no recent psychotropic, steroid, or other medication use that is known to influence hormone functioning), as a primary aim of this MSUTR study was to examine ovarian hormone influences on disordered eating. However, past studies have shown that MSUTR twins who met inclusion/exclusion criteria for these and other studies (Klump et al., 2013a, 2013b, 2015) are not significantly different from non-participating MSUTR families in terms of body weight/shape concerns (e.g., body dissatisfaction, over-evaluation of weight/shape, etc.; d’s = .02-.14, all p>.05). Twins from the MSUTR are demographically representative of the recruitment region (see Burt & Klump, 2019), as are the twins in the current study (i.e., twins identified as White (82.7%), African American (8.0%), Asian (0.6%), American Indian/Alaskan Native (0.2%), multiracial (8.2%), and Latinx (3.9%).
Measures
Zygosity Determination.
Zygosity was determined using a well-validated physical similarity questionnaire (Lykken et al., 1990) shown to be over 95% accurate (Peeters et al.,1998). This questionnaire was completed independently by trained research assistants and the twins’ parent (the mother in over 95% of cases). Reports were compared amongst raters, and discrepancies were resolved using questionnaire responses, review of twin photographs by principal investigators (KLK), and/or DNA analysis (i.e., twin concordance across several single nucleotide polymorphisms (Burt & Klump, 2013; Klump & Burt, 2006)).
Disordered Eating.
The Minnesota Eating Behavior Survey (MEBS; von Ranson et al., 2005)1 and the Youth Eating Disorder Examination Questionnaire (YEDE-Q; Goldfein et al., 2005; Goldschmidt et al., 2007) were used to assess disordered eating. The MEBS is a 30-item self-report questionnaire consisting of true/false questions. This measure was developed for use with adults and children as young as 10-years-old and has been shown to be appropriate for use in children as young as 8-years-old (e.g., Luo et al., 2016; O’Connor et al., 2016). The present study focused on weight and shape concerns and thus, examined the body dissatisfaction (i.e., assessing discontent with body size and shape) and weight preoccupation (i.e., assessing preoccupation with weight, eating, and dieting) subscales. The MEBS Total Score (i.e., sum of all 30 items including items assessing body dissatisfaction, weight preoccupation, binge eating, and compensatory behaviors) was also examined to allow for comparisons with past studies. Good three-year stabilities and criterion-related validity has been demonstrated in past studies (von Ranson et al., 2005). Internal consistencies for these MEBS scales have ranged from .78-.89 in prior research (von Ranson et al., 2005) and were .75-.87 in the current study.
The YEDE-Q (Goldfein et al., 2005; Goldschmidt et al., 2007) is a self-report questionnaire that was adapted from the Eating Disorder Examination Questionnaire (EDE-Q; Fairburn & Beglin, 1994) for use in children. The YEDE-Q assesses disordered eating attitudes and behaviors over the past 28 days. The present study focused on the shape concerns (e.g., desire for a flat stomach, influence of body shape on self-evaluation) and weight concerns (e.g., dissatisfaction with weight, influence of weight on self-evaluation) items. Notably, factor analyses within clinical and community samples have suggested weight and shape concern items load on one-factor (Allen et al., 2011), and the shape concerns and weight concerns subscales were highly correlated (r=.90) in our sample. Thus, similar to past studies (e.g., Mond et al., 2007; van Zutven et al., 2014), we created a composite weight/shape concerns subscale by averaging the items from the two subscales. Excellent internal consistency has been demonstrated for this combined weight/shape subscale in past studies (alphas=.92-.95; Klump et al., 2010a) and in the current sample (alpha=.94).
Based on item content, body dissatisfaction appears to tap how acceptable the participant finds her body size/shape, whereas weight preoccupation appears to tap the amount an individual thinks about or fears weight change. The weight and shape concerns subscale appears to tap both the amount an individual thinks about her weight/shape and the extent weight/shape concerns impact the participant’s view of themselves (i.e., overvaluation of weight/shape). Correlations between the different body weight and shape subscales seemed to confirm that there are discernible differences between the scales. These correlations ranged from r=.57-.70, indicating that only 33–49% of the variance in these scales is shared (see Supplemental Table 2). That leaves over 50% (range = 51–62%) of unique variance in each scale that could translate into etiologic differences across puberty for the different weight and shape scales. This degree of shared/unique variance allows for examining replication of effects across different weight/shape concern constructs, as well as identifying any unique etiologic factors that may be present.
Pubertal Development.
Pubertal development was assessed using the self-report Pubertal Development Scale (PDS; Petersen et al., 1988), which asks participants to rate their development across several physical markers of puberty (i.e., height spurts, body hair growth, skin changes, breast development, and onset of menarche) on a 4-point scale: (1) development has not yet begun; (2) development has barely started; (3) development is definitely underway; and (4) development seems completed. Menses was coded dichotomously as either absent (1) or present (4). The ratings of each physical marker are summed and averaged to obtain an overall continuous PDS score, with higher scores representing more advanced pubertal development. The continuous PDS score allowed us to evaluate effects across different degrees of maturation. PDS scores correlate with physician ratings of pubertal development (r=.61-.67; Petersen et al., 1988) and exhibit good internal consistency in past research (alphas= .76-.83; O’Connor et al., 2016; Petersen et al., 1988) and the present sample (alpha = .84).
Body Mass Index (BMI).
BMI was calculated (weight in kg/ height in m2) using height and weight measured with a wall-mounted ruler and digital scale, respectively.
Statistical Analyses
Prior to analyses, body dissatisfaction, weight preoccupation, weight/shape concerns, and PDS scores were log transformed to account for positive skew. Past research has demonstrated significant associations between BMI and disordered eating (Jones et al., 2001; Keel et al., 1997) and between age and puberty (r=.79 within the current sample). Thus, consistent with past studies (e.g., Culbert et al., 2009; Klump et al, 2010b, 2017a), BMI and age were regressed out of each twins’ disordered eating scores to ensure that differences across puberty were not unduly influenced by these potentially important variables.
We used extended, univariate twin moderation models (see Figure 1; van der Sluis et al., 2012) to directly examine differences in additive genetic (A; effects that add or sum across genes), shared environmental (C; environmental factors that are common to siblings growing up in the same family and contribute to their behavioral similarity), and nonshared environmental (E; factors that are unique to siblings growing up in the same family and contribute to behavioral differences, including measurement error) influences on each weight/shape concern symptom, as well as overall disordered eating, across pubertal development. A strength of twin moderation models is that all twins, irrespective of twin concordance/discordance on pubertal development, can be included in analyses. These models estimate: 1) path coefficients (i.e., a, c, and e) assessing the degree of genetic/environmental influences at the lowest level of the moderator (i.e., pubertal development); 2) linear moderators (i.e., βX, βY, and βZ,) assessing whether genetic/environmental influences increase or decrease linearly across pubertal development; and 3) quadratic moderators (i.e., βX2, βY2, and βZ2) assessing non-linear changes in genetic/environmental influences.
Figure 1. Path Diagram for the Full Moderation Model and Extended Moderation Model for One Twin Only.
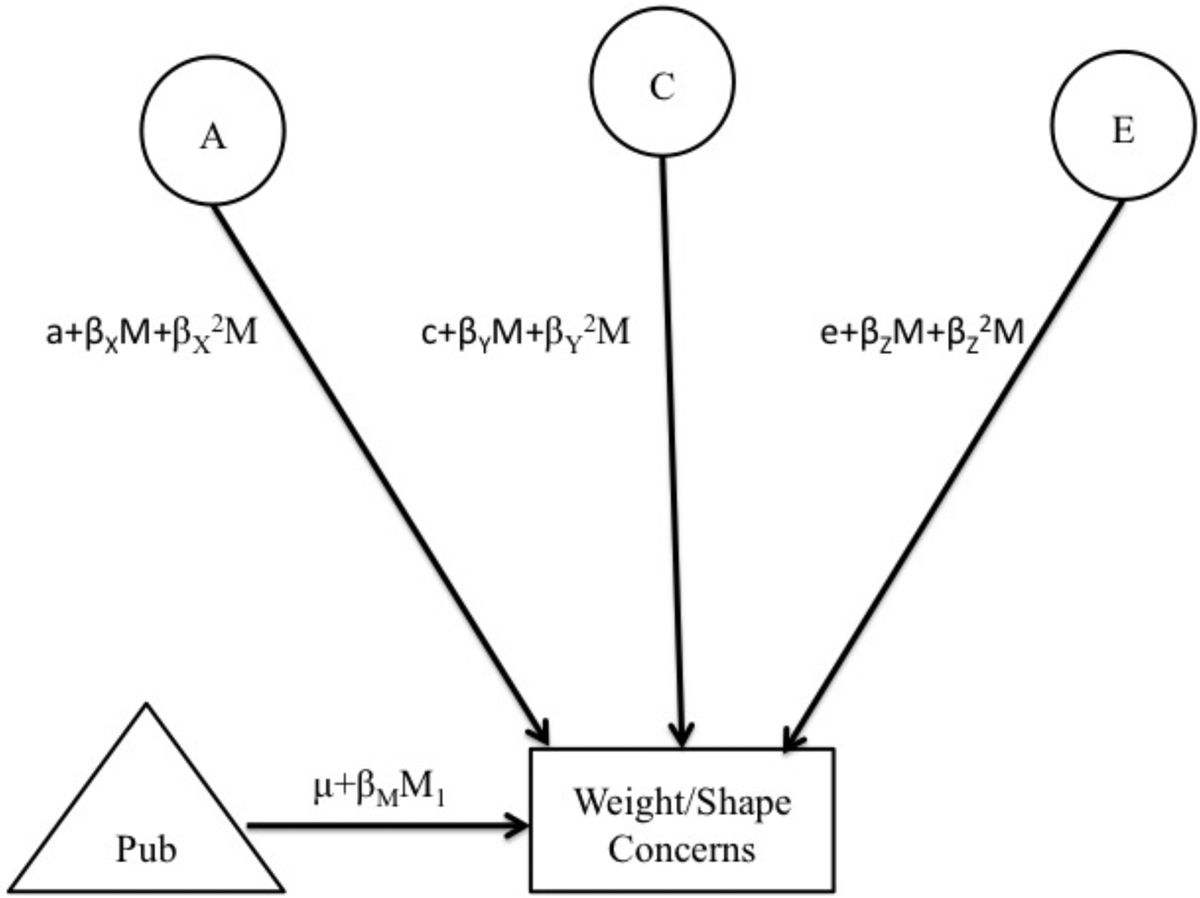
Abbreviations include: Pub = puberty; A = additive genetic effects; C = shared environmental effects; E = non-shared environmental effects; M1 and M2 =moderator for twin 1 and twin 2, respectively; βM = phenotypic regression coefficient; a, c, and e = paths or intercepts; βX, βY, βZ = linear moderators; βX2, βY2, βZ2 = quadratic moderators. The model is depicted for only one twin, but parameters are fit separately for MZ and DZ twins.
We first fit the “full” moderation model that included all three sets of parameters (see Mx script in Supplemental Materials). We then directly tested our study hypotheses by comparing the fit of this model to one that constrained all linear and quadratic moderators to 0 (i.e., no moderation: genetic and environmental influences do not significantly change across puberty). Since the no moderation model provided poor fit to the data for all of the scales examined (see below), we fit nested models that differentially constrained linear and non-linear moderators. Because of the large number of submodels that could be fit, we used parameter estimates from the full model to identify appropriate submodels in order to test theoretically relevant submodels without unduly increasing the number of tests. Given our use of the full model, both full and best-fitting models are presented in our results (see Tables 1 and 2).
Table 1.
Model Fit Comparisons (N=926 twins)
| Model | −2lnL (df) | χ2 Δ (df) | p | AIC | BIC | SABIC | DIC |
|---|---|---|---|---|---|---|---|
| MEBS Total Score | |||||||
| Full Model | 2409.82(909) | -- | -- | 591.82 | −1584.69 | −142.22 | −749.37 |
| Nested Submodels: | |||||||
| No moderation | 2466.60(915) | 56.78(6) | <.01 | 636.60 | −1574.71 | −122.72 | −733.88 |
| Constrain all nonlinear mods to 0 | 2416.46(912) | 6.64(3) | .08 | 592.46 | −1590.57 | −143.35 | −752.50 |
| Constrain all nonlinear and linear A mods to 0 | 2424.12(913) | 14.30(4) | .01 | 598.12 | −1589.82 | −141.00 | −750.82 |
| Constrain all nonlinear and linear C mods to 0 | 2417.94(913) | 8.12(4) | .09 | 591.94 | −1592.90 | −144.09 | −753.91 |
| Constrain all nonlinear and linear E mods to 0 | 2416.82(913) | 7.00(4) | .14 | 590.82 | −1593.46 | −144.65 | −754.47 |
| Constrain all nonlinear & CE linear mods to 0 | 2418.33(914) | 8.51(5) | .13 | 590.33 | −1595.78 | −145.38 | −755.87 |
| Body Dissatisfaction | |||||||
| Full Model | 2328.20(905) | -- | -- | 518.20 | −1613.22 | −177.10 | −781.58 |
| Nested Submodels: | |||||||
| No moderation | 2400.59(911) | 72.39(6) | <.01 | 578.59 | −1595.44 | −149.80 | −758.29 |
| Constrain all nonlinear mods to 0 | 2331.32(908) | 3.12(3) | .37 | 515.32 | −1620.87 | −179.99 | −786.48 |
| Constrain all nonlinear and linear A mods to 0 | 2335.82(909) | 7.62(4) | .11 | 517.82 | −1621.69 | −179.22 | −786.37 |
| Constrain all nonlinear and linear C mods to 0 | 2335.04(909) | 6.84(4) | .14 | 517.04 | −1622.08 | −179.61 | −786.76 |
| Constrain all nonlinear and linear E mods to 0 | 2340.81(909) | 12.61(4) | .01 | 522.81 | −1619.19 | −176.73 | −783.88 |
| Constrain AE nonlinear mods to 0 | 2330.46(907) | 2.26(2) | .32 | 516.46 | −1618.23 | −178.94 | −784.75 |
| Weight/ Shape Concerns | |||||||
| Full Model | 2444.40 (910) | 624.40 | −1570.47 | −126.42 | −734.23 | ||
| Nested Submodels: | |||||||
| No moderation | 2490.23(916) | 45.83(6) | <.01 | 658.23 | −1565.97 | −112.39 | −724.22 |
| Constrain all nonlinear mods to 0 | 2448.21(913) | 3.81(3) | .28 | 622.21 | −1577.77 | −128.95 | −738.78 |
| Constrain all nonlinear and linear A mods to 0 | 2454.42(914) | 10.02(4) | .04 | 626.42 | −1577.73 | −127.33 | −737.82 |
| Constrain all nonlinear and linear C mods to 0 | 2448.65(914) | 4.25(4) | .37 | 620.65 | −1580.62 | −130.22 | −740.71 |
| Constrain all nonlinear and linear E mods to 0 | 2449.93(914) | 5.53(4) | .24 | 621.93 | −1579.98 | −129.58 | −740.07 |
| Constrain all nonlinear & CE linear mods to 0 | 2450.51 (915) | 6.11(5) | .30 | 620.51 | −1582.76 | −130.77 | −741.93 |
| Weight Preoccupation | |||||||
| Full Model | 2396.48(904) | -- | -- | 588.48 | −1576.01 | −141.48 | −745.29 |
| Nested Submodels: | |||||||
| No moderation | 2427.09(910) | 30.61(6) | <.01 | 607.09 | −1579.12 | −135.07 | −742.89 |
| Constrain all non-linear mods to 0 | 2410.59(907) | 14.11(3) | <.01 | 596.59 | −1578.17 | −138.87 | −744.69 |
| Constrain all A mods to 0 | 2396.84(906) | 0.36(2) | .84 | 584.84 | −1581.97 | −144.26 | −749.41 |
| Constrain all C mods to 0 | 2406.90(906) | 10.42(2) | <.01 | 594.90 | −1576.94 | −139.24 | −744.38 |
| Constrain all E mods to 0 | 2401.08(906) | 4.60(2) | .10 | 589.08 | −1579.85 | −142.15 | −747.29 |
| Constrain A nonlinear mod to 0 | 2396.82(905) | 0.34(1) | .56 | 586.82 | −1578.91 | −142.80 | −747.27 |
| Constrain E nonlinear mod to 0 | 2400.50(905) | 4.02(1) | .04 | 590.50 | −1577.07 | −140.96 | −745.44 |
| Constrain A nonlinear and all E mods to 0 | 2401.39(907) | 4.91(3) | .18 | 587.39 | −1582.77 | −143.47 | −749.29 |
| Constrain E nonlinear and all A mods to 0 | 2402.16(907) | 5.68(3) | .13 | 588.16 | −1582.38 | −143.09 | −748.90 |
Note. −2lnL = minus twice the log-likelihood; χ2 Δ= chi-square change; AIC = Akaike Information Criterion; Full Model = model with paths, linear, and quadratic moderators; MEBS= Minnesota Eating Behavior Survey; A = additive genetic effects; C = shared environmental effects; E = nonshared environmental effects; mod(s) = moderator(s). Each nested submodel is compared to the full model when calculating the χ2 Δ and degrees of freedom. The best-fitting models, as determined by a non-significant chi-square change test and the lowest AIC, BIC, SABIC, and DIC values, are noted by the outlined and bolded text. If fit statistics identify more than one model as best fitting, the model with the most model fit statistics indicating best fit was selected.
Table 2.
Unstandardized Path and Moderator Estimates for the Full and Best-Fitting Pubertal Moderation Models (N=926 twins)
| Path Estimates | Linear Moderator Estimates | Nonlinear Moderator Estimates | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Model | a | c | e | βX | βY | βZ | βX2 | βY2 | βZ2 |
| MEBS Total Score | |||||||||
| Full Model | −.01 | .22 | .68 | 2.14 | 2.13 | −0.86 | −1.15 | −3.19 | 1.73 |
| (−.38, .37) | (−.15, .45) | (.56, .80) | (−4.76, 4.76) | (−.72, 4.42) | (−1.95, .26) | (−8.78, 8.78) | (−7.48, 2.46) | (−.26, 3.50) | |
| *No nonlinear | −.08 | .40 | .63 | −1.61 | - | - | - | - | - |
| or CE linear | (−.33, .17) | (.09, .53) | (.58, .68) | (−2.14,−1.11) | - | - | - | - | - |
| Body Dissatisfaction | |||||||||
| Full Model | .03 | .47 | −.46 | −2.41 | −.60 | −1.10 | 1.99 | −2.43 | 1.06 |
| (−.46, .43) | (.06, .63) | (−.59, −.35) | (−5.09, .92) | (−3.06, 1.65) | (−2.23, .09) | (−3.59, 7.46) | (−6.41, 5.84) | (−1.11, 3.17) | |
| *No nonlinear | .07 | −.34 | .55 | −1.58 | - | .44 | - | - | - |
| or C linear | (−.40, .35) | (−.44, .44) | (.47, .65) | (−2.21, −.67) | - | (.11, .79) | - | - | - |
| Weight/Shape Concerns | |||||||||
| Full Model | −.03 | .36 | .57 | 2.84 | 1.37 | .14 | −1.97 | −3.23 | 0.05 |
| (−.43, .55) | (−.21, .57) | (.43, .71) | (−2.57, 5.00) | (−4.95, 4.95) | (−1.04, 1.45) | (−6.68, 6.17) | (−8.44, 8.85) | (−2.14, 2.10) | |
| *No nonlinear | .25 | −.34 | −.62 | 1.34 | - | - | - | - | - |
| or CE linear | (−.02, .49) | (−.52, .52) | (−.68, −.58) | (.88, 1.87) | - | - | - | - | - |
| Weight Preoccupation | |||||||||
| Full Model | .01 | .13 | .74 | 1.20 | 3.63 | −.91 | −3.19 | −4.96 | 1.78 |
| (−.46, .47) | (−.14, .36) | (.60, .86) | (−5.00, 5.00) | (1.27, 5.40) | (−1.50, .20) | (−9.00, 9.00) | (−8.24, −.97) | (−.05, 3.00) | |
| *No A mods | .00 | .14 | .75 | - | 3.48 | −.94 | - | −4.61 | 1.88 |
| (−.36, .36) | (−.09, .34) | (.64, .86) | - | (1.96, 5.00) | (−1.50, .00) | - | (−7.07, −2.16) | (.28, 3.00) | |
Note. MEBS= Minnesota Eating Behavior Survey; a = genetic path estimate; c = shared environmental path estimate; e = non-shared environmental path estimate; βX = linear moderator of genetic path estimate; βY = linear moderator of shared environmental path estimate; βZ =linear moderator of non-shared environmental path estimates; βX2= nonlinear moderator of genetic path estimate; βY2= nonlinear moderator of shared environmental path estimate; βZ2= nonlinear moderator of non-shared environmental path estimate. Estimates are followed by 95% confidence intervals in parentheses. Confidence intervals that do not overlap with zero indicate statistical significance (bolded) at p <.05. In the “Full” model, genetic, shared environmental, and non-shared environmental estimates are allowed to vary both linearly and nonlinearly across levels of the moderator (i.e., pubertal development). The best fitting model is listed below the full model and denoted by an asterisk.
All models were fit to the raw data using the full-information maximum-likelihood (FIML) option in Mx (Neale et al., 1999), which allowed for retention of any twin pairs that had missing data for one co-twin (n=1 to 7; 0.2%−1.5% of the total sample). Model fit comparisons were made by taking the difference in minus twice the log-likelihood (−2lnL) between the full and nested models, which is chi-squared distributed under the null hypothesis implied by the reduced model. Large (statistically significant) differences led to a rejection of the nested model. Akaike’s information criterion (AIC; Akaike, 1987), Bayesian information criterion (BIC; Raftery, 1995), sample-size adjusted BIC (SABIC; Sclove, 1987), and deviance information criterion (DIC; Spiegelhalter et al., 2002) were also used to select the best fitting model, where models that minimized these scores were preferred. Prior to model-fitting, scores on the disordered eating subscales were standardized, and PDS scores were “floored” to zero to ease interpretation. Following previous recommendations (Purcell, 2002), unstandardized estimates are reported in figures, as they more accurately depict absolute differences in etiologic effects than standardized estimates that represent differences as proportions of the total variance. Nonetheless, we report standardized estimates in the text where appropriate, as these estimates allow for a direct comparison of our findings to previous twin studies.2
Results
Descriptive Analyses
Variability in MEBS and YEDE-Q scores was present for all scales across pre-early puberty and mid-late puberty (see Supplemental Table 1), and 3.9% of the sample scored above the clinical cut-off on the MEBS (total score ≥ 15.55; von Ranson et al., 2005). Consistent with past studies, (e.g., Culbert et al., 2009; Klump et al., 2003), higher mean scores were found in mid-late puberty (PDS score > 2.5; N= 333) than pre-early puberty (PDS score < 2.5; N= 593) for overall disordered eating (d=.30, p<.01), body dissatisfaction (d=.51, p<.01), weight/shape concerns (d=.47, p<.01), and weight preoccupation (d=.17, p=.01).
Twin Moderation Models
For all outcome variables, moderation models suggested pubertal differences in etiologic effects. The no moderation models provided a poor fit to the data when compared to the full model (i.e., significant chi-square change tests and larger AIC, BIC, SABIC and DIC values were present; see Table 2). Additional submodels were then fit to identify the specific types of moderation that were significant across development. Importantly, with one clear exception (i.e., weight preoccupation – see below), there appeared to be minimal non-linear moderation for any of the genetic, shared environmental, or nonshared environmental factors; thus, we also fit a model that constrained all non-linear moderators to 0 for each of the scales.
Similar to previous work (Culbert et al., 2009; Klump et al., 2007a, 2007b, 2012), the full model for MEBS total score showed mainly linear increases in genetic and non-shared environmental effects across pubertal development (see Figure 2). However, unlike previous work, there were minimal differences in shared environmental influences. Based on this full model, we fit a series of submodels that tested whether constraining the non-linear moderators and individually constraining the linear genetic, shared environmental, and non-shared environmental influences improved model fit. As shown in Table 2, the model that constrained the linear shared environmental and non-shared environmental moderators (and all non-linear moderators) provided the best fit to the data. Unstandardized (see Figure 2) and standardized (see parentheses) estimates from the best fitting model showed significant linear increases in genetic effects (from 1% to 66%) across pubertal development and no differences in shared environmental (10–28%) and non-shared environmental (24–70%) influences3.
Figure 2. Unstandardized Estimates of Additive Genetic (a), Shared Environmental (c), and Nonshared Environmental (e) Effects from the Full and Best-Fitting Models for Total Score, Body Dissatisfaction, Weight/Shape Concerns, and Weight Preoccupation.
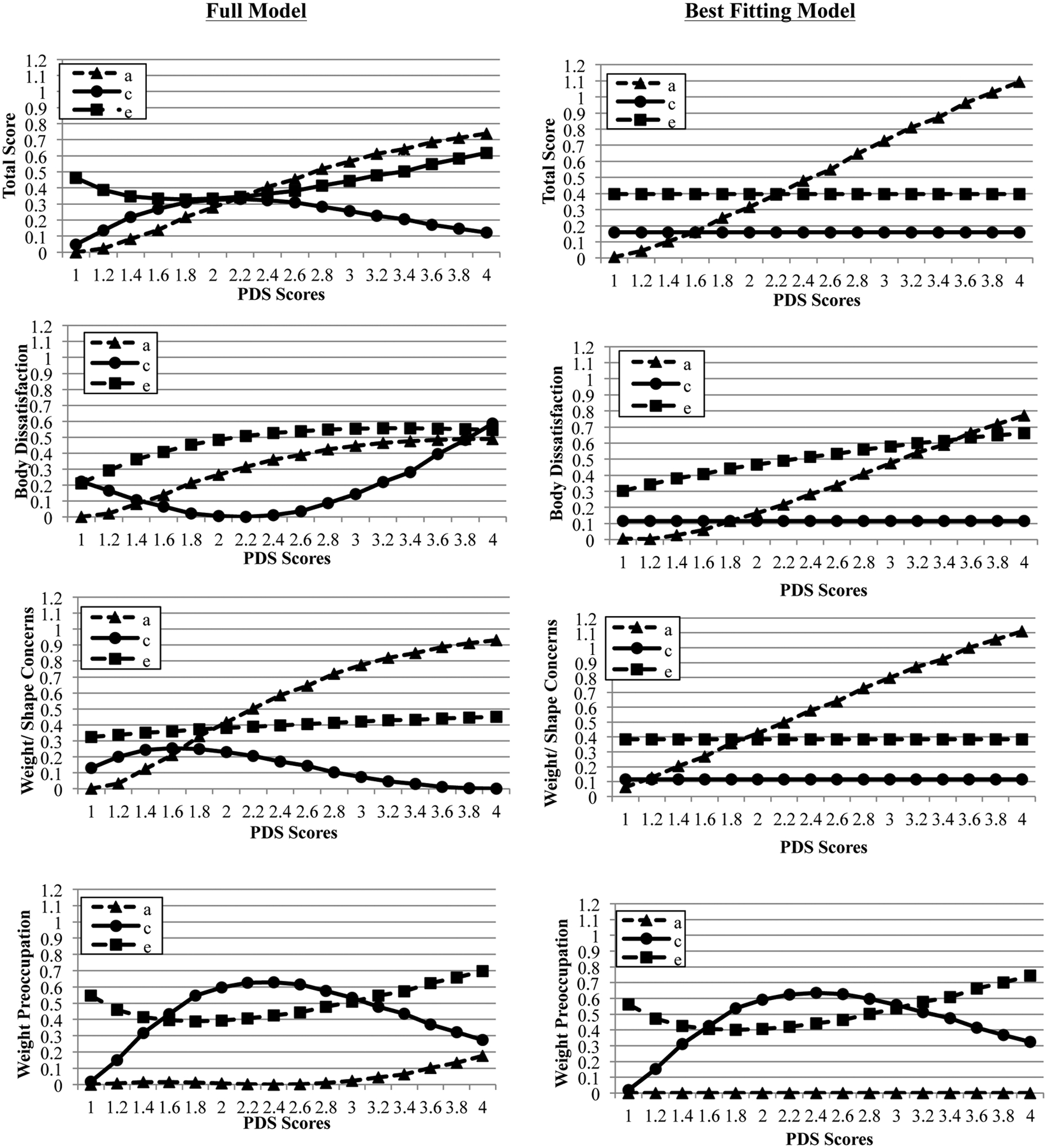
Although log-transformed PDS scores that were floored to 0 were used in all models (see Methods), raw PDS scores are depicted here for ease of interpretation.
Findings for body dissatisfaction and weight/shape concerns showed a similar pattern of results with graphs of the full model indicating increases in genetic effects across pubertal development (see Figure 2). For both body dissatisfaction and weight/shape concerns, the best-fitting model constrained the linear shared environmental (and all non-linear moderators; see Table 2)4. Additionally, the best fitting model for weight/shape concerns constrained the nonlinear environmental moderation to zero. Unstandardized estimates (see Figure 2) and standardized estimates (in parentheses) from the best-fitting models showed significant linear increases in genetic effects from pre-puberty to post-puberty (body dissatisfaction: 1% to 50%; weight/shape concerns: 11% to 69%), and no difference in shared environmental influences (body dissatisfaction: 7–27%; weight/shape concerns 7–21%). Significant linear increases in non-shared environmental influences are shown across puberty for body dissatisfaction (despite the proportion of attributable variance decreasing for the standardized estimates: 72–74% to 43%), whereas no differences in non-shared environmental influences are shown for weight/shape concerns (24–68%).
A different pattern of effects was observed for weight preoccupation. The graph of the full model suggested small increases in genetic influences after mid-puberty, non-linear changes (increasing then decreasing) in shared environmental influences, and increases in non-shared environmental influences (see Figure 2). The best-fitting submodel constrained genetic moderators to zero (see Table 2 and Figure 2). Unstandardized estimates from the best fitting model (see Figure 2) and standardized estimates (in parentheses) showed significant shared environmental effects increasing (from 3% to 60%) until mid-puberty then decreasing (to 30%) in late puberty. Non-shared environmental effects significantly decreased from early to mid-puberty (from 97% to 40%) then increased (to 70%) in late puberty.
Post-Hoc Analyses of Binge Eating
Prior studies have found significant pubertal effects on the heritability of binge eating, with dramatic increases across pubertal development that are similar to the differences in genetic effects described above (e.g., Klump et al., 2017a). Importantly, binge eating (measured with the MEBS binge eating score that assesses the tendency to think about or engage in binge eating symptoms) is moderately correlated with body dissatisfaction (r=.38, p<.001), weight/shape concerns, (r=.51, p<.001), and weight preoccupation (r=.50, p<.001) in our sample. Consequently, we conducted post hoc analyses in which we re-fit the moderation models to the weight/shape concerns subscales after regressing out MEBS binge eating scores5 in order to confirm that pubertal differences in genetic effects on weight/shape concerns were not completely due to genetic effects on binge eating.
Graphical depictions of genetic effects from the full models (see Supplemental Figure 1) were largely similar to those obtained in initial analyses (see Figure 2). Genetic effects on body dissatisfaction and weight/shape concerns appeared to linearly increase from pre-puberty to post-puberty (Supplemental Figure 1), although the magnitude of genetic effects was somewhat attenuated. Indeed, comparison of the model fit between submodels indicated that the no moderation model was best-fitting (see Supplemental Table 3). It is likely that the more attenuated differences in genetic (and other) effects across puberty (see Supplemental Figure 1) decreased our power to detect significant moderating effects. Full model results for the modified weight preoccupation scale showed some increases in genetic effects later in puberty (see Supplemental Figure 1) that were similar to those of the original weight preoccupation scale (see Figure 2). However, results also revealed a more complex and non-linear pattern of genetic effects that is difficult to detect without larger sample sizes (i.e., the no moderation model was best-fitting for this scale as well- see Supplemental Table 3). Overall, these results suggest that the general pattern of pubertal moderation of genetic effects was similar across the scales with binge eating regressed out, although pubertal differences in genetic effects were somewhat attenuated.
As a final check on our results, we also re-fit the twin moderation models for binge eating (as conducted in Klump et al. (2017a)) after regressing out the weight/shape concern scores to ensure that pubertal differences in genetic effects on binge eating were independent of weight/shape concerns. We fit the same models as those included in the original paper (see Supplemental Table 3 and Klump et al. (2017a)) and found that in all cases, the same model that was best-fitting in the original paper was best-fitting with the weight/shape scales regressed out (see Supplemental Table 3). These best-fitting models indicated significant linear increases in genetic effects on binge eating from pre-puberty to post-puberty (see Supplemental Table 3; Supplemental Figure 1). Taken together, there appear to be unique (albeit, attenuated) pubertal moderation of genetic effects on weight/shape concerns that are independent of binge eating, and pubertal moderation of genetic effects on binge eating that are independent of weight/shape concerns.
Discussion
Results confirm that weight and shape concerns show etiologic differences across pubertal development. These data are the first to demonstrate that genetic effects on body dissatisfaction and weight/shape concerns increase during puberty, and since age was regressed out of weight/shape scores, our results highlight that puberty likely accounts for the age-related effects found in prior studies (Klump et al., 2000, 2010a; Wade et al., 2012). This pubertal emergence of genetic effects mimics effects found for overall disordered eating (in this study and past work: Culbert et al., 2009; Klump et al., 2007a, 2007b) and binge eating (e.g., Klump et al., 2017a). Post-hoc analyses revealed that genetic moderation underlying weight/shape concerns exist independently of binge eating, although effects were somewhat attenuated when controlling for binge eating.
Animal and human studies have highlighted the likely role of ovarian hormones during puberty for the genetic diathesis of binge eating (Klump et al., 2018; for reviews, see Culbert et al., 2018; Klump, 2013, Klump et al., 2017b). Specifically, genetic and hormonal changes during puberty have been linked to changes in basic appetitive or reward processes that may increase intake for palatable food. While these reward and appetitive processes are unlikely to be important for weight/shape concerns, hormones have wide-reaching effects, including effects on social/emotional processing that could drive pubertal effects for weight/shape concerns. Changes in social and affective processing begin around the onset of puberty (Crone & Dahl, 2012). Theoretically, it is possible that these pubertal changes in affective processing and heightened attention to social stimuli may make one more vulnerable to perceived pressure to be thin. In this case, genetic/hormonal changes driving changes in cognitive processing and attention could underlie the increased genetic influence across puberty observed in this study. Notably, the influence of ovarian hormones on social and affective processing may be more complicated than processes affecting reward and appetitive processes (see Crone & Dahl, 2012), which may explain the more attenuated and, in some cases, complicated nature (i.e., for weight preoccupation) of genetic influences across puberty after controlling for binge eating. Larger samples are needed to increase power to detect these more complex effects. Moreover, longitudinal studies are needed that carefully parse out temporal ordering of binge eating and weight/shape concerns to examine the emergence of these concerns across time and better understand their phenotypic and genetic links across pubertal development.
Interestingly, unlike past studies that have demonstrated pubertal moderation of shared environment influences for overall disordered eating and binge eating (e.g., Klump et al., 2017a), the present study generally did not detect significant differences in shared environmental influences across puberty. One exception were for weight preoccupation, for which significant pubertal moderation of both shared and non-shared environmental effects and negligible genetic influences were suggested. Larger samples are needed to clarify whether the general lack of pubertal moderation of shared environmental influences is due to low power, as graphs of the full model depict some shifts in shared environmental effects for nearly all measures.
The absence of genetic contributions to weight preoccupation during puberty was somewhat surprising. Prior studies have generally demonstrated significant genetic influences on weight preoccupation in females (e.g., heritability= 47%−66%; Klump et al., 2000, 2009; Spanos et al., 2010). However, while the best-fitting model constrained genetic influences across puberty to be zero, it is notable that the visual depiction of the raw data (see Full Model in Figure 2) was suggestive of possible increases in genetic influences in late/post-puberty. Thus, genetic influences on weight preoccupation may become activated late in pubertal development. Large-scale developmental twin studies are needed to test this hypothesis.
Indeed, future research is needed to understand the differences in etiologic patterns for weight preoccupation compared to weight/shape concerns and body dissatisfaction. Evaluation of item content on the various subscales suggests that weight/shape concerns and body dissatisfaction assesses the extent to which an individual is distressed or impacted by their weight/shape concerns or view their bodies as unacceptable, whereas the weight preoccupation subscale assesses the extent to which an individual thinks about weight or weight loss. Further exploration is needed to understand how differences in these subscales may map on to the observed differences in etiologic effects.
Before ending, we should note a few limitations. We used a community sample of twins and thus, it is unclear whether results generalize to clinical populations. However, weight and shape concerns are both symptoms of, and precursors to, clinical eating disorders (Killen et al., 1996; Stice & Shaw, 2002), and recent data suggest that disordered eating exists on a continuum with eating disorders (Luo et al., 2016). Although additional studies are needed, our results may generalize across the spectrum of eating pathology. We used a cross-sectional design and thus, longitudinal studies are needed to confirm that differences in genetic influence observed across pubertal development occur within the same sample assessed repeatedly across pubertal development. Finally, we only examined female twins, and past studies suggest sex differences in etiologic shifts for overall levels of eating pathology across pubertal development (see Culbert et al., 2018). These past studies suggest that earlier developmental periods may be important for males (i.e., adrenarche) and consequently, it will be critical for future work to examine the full range of pubertal development (i.e., adrenarche and gonadarche) to comprehensively examine sex differences in effects.
Despite these limitations, our study had many strengths (e.g., large sample size, examination of a range of body weight/shape concerns) that allowed us to comprehensively examine differences in etiologic factors across pubertal development in girls. Our findings confirm that puberty is a critical developmental period for etiologic shifts in risk for the weight/shape concerns that are central to eating disorders. These etiologic shifts appear to occur even when controlling for binge eating (albeit, attenuated, as demonstrated by smaller increases in genetic effects across puberty). Studies focused on identifying the factors contributing to these shifts (e.g., changes in social and affective processing) are needed to build comprehensive models of pubertal risk. Although more work is needed before these findings can directly inform prevention or intervention efforts, our findings add to the growing recognition (e.g., Ciao et al., 2014) that these efforts should be aimed at girls across the developmental spectrum (i.e., pre-puberty, puberty, young adulthood) in order to directly address the range and shifting nature of etiologic risk factors for eating disorders for females.
Supplementary Material
Footnotes
Declarations of Interest: None
The Minnesota Eating Behavior Survey (MEBS; previously known as the Minnesota Eating Disorder Inventory [M-EDI]) was adapted and reproduced by special permission of Psychological Assessment Resources, Inc., 16204 North Florida Avenue, Lutz, FL 33549, from the Eating Disorder Inventory (collectively, EDI and EDI-2) by Garner, Olmstead, and Polivy (1983) by the Psychological Assessment Resources, Inc. Further reproduction of the MEBS is prohibited without prior permission from Psychological Assessment Resources, Inc.
Notably, twin moderation models rest on one important assumption – that the moderator (i.e., pubertal development) is genetically independent of the dependent variable (i.e., body dissatisfaction, weight preoccupation, weight/shape concerns, overall disordered eating). If there is not independence, then genetic mediation (i.e., termed gene-environment correlations or rGE) may be present and could “masquerade” as puberty effects. Consequently, we fit a series of “GxE in the presence of rGE” models that tested for genetic mediation and found that the parameter that tests for these effects (i.e., the moderation of the covariance path) could be constrained to 0 (comparison with full model: for MEBS total score: χ2Δ(1) = .18, p = .67; body dissatisfaction: χ2Δ(1) = .02, p = .89; for weight/shape concerns: χ2Δ(1) = .02, p = .90; and for weight preoccupation: χ2Δ(3) = 1.14, p =.29.
Percentages are not identical because they are standardized estimates that are proportional to the total variance.
Notably, Figure 2 indicated that there might be some non-linear shared environmental moderation for body dissatisfaction, however the model constraining these non-linear effects (“Constrain AE nonlinear mods to 0”) produced larger BIC, SABIC, and DIC values as compared to the full model (see Table 1). Additionally, two fit indices (i.e., BIC and DIC) supported constraining linear shared environmental moderation for body dissatisfaction, while two other fit indices (i.e., AIC and SABIC) supported retaining linear shared environmental moderation (see “Constrain all nonlinear mods to 0” and “Constrain all nonlinear and linear C mods to 0” in Table 1). However, the linear shared environmental moderation estimate was non-significant when included in the full model. Thus, we selected the most parsimonious model for body dissatisfaction that constrained all non-linear and linear shared environmental moderators to zero as best-fitting.
Notably, we re-ran twin moderation models for the total score recalculated without binge eating items; however, given the lack of endorsement of compensatory behaviors, this recalculated total score largely reflects body dissatisfaction and weight preoccupation, which we provide models for separately herein.
References
- Akaike H, 1987. Factor analysis and AIC. Psychometrika. 52, 317–332. [Google Scholar]
- Allen KL, Byrne SM, Lampard A, Watson H, Fursland A, 2011. Confirmatory factor analysis of the Eating Disorder Examination Questionnaire (EDE-Q). Eat. Behav 12, 143–151. [DOI] [PubMed] [Google Scholar]
- Burt SA, Klump KL, 2013. The Michigan State University Twin Registry (MSUTR): An update. Twin Res Hum Genet. 16, 344–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burt SA, Klump KL, 2019. The Michigan State University Twin Registry (MSUTR): 15 Years of Twin and Family Research. Twin Res Hum Genet. 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciao AC, Loth K, Neumark-Sztainer D, 2014. Preventing eating disorder pathology: Common and unique features of successful eating disorders prevention programs. Curr Psychiatry Rep. 16, 453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culbert KM, Burt SA, McGue M, Iacono WG, Klump KL, 2009. Puberty and the genetic diathesis of disordered eating attitudes and behaviors. J Abnorm Psychol. 118, 788–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culbert KM, Sisk CL, Klump KL, 2018. Sex steroid hormones and differential risk for eating pathology: A review of genetic and phenotypic effects across development. Curr. Opin. Behav. Sci 23, 124–130. [Google Scholar]
- Crone EA, Dahl RE 2012. Understanding adolescence as a period of social-affective engagement and goal flexibility. Nat. Rev. Neurosci 13, 636–650. [DOI] [PubMed] [Google Scholar]
- Fairburn CG, Beglin SJ, 1994. Assessment of eating disorders: Interview or self-report questionnaire? Int J Eat Disord. 16, 363–370. [PubMed] [Google Scholar]
- Goldfein JA, Devlin MJ Kamenetz C, 2005. Eating Disorder Examination- Questionnaire with and without instructions to assess binge eating in patients with binge eating disorder. Int J Eat Disord. 37, 107–111. [DOI] [PubMed] [Google Scholar]
- Goldschmidt AB, Doyle AC, Wilfely DE, 2007. Assessment of binge eating in overweight youth using a questionnaire version of the Child Eating Disorder Examination with Instructions. Int J Eat Disord. 40, 460–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobi C, 2005. Psychosocial risk factors for eating disorders, in: Wonderlich SA, Mitchell JE, deZwaan M, Steiger H (Eds.), Eating Disorders Review, Radcliffe Publishing Ltd: Oxford, UK, pp. 59–85. [Google Scholar]
- Jones JM, Bennett S, Olmsted MP, Lawson ML, Rodin G, 2001. Disordered eating attitudes and behaviours in teenaged girls: A school-based study. Can Med Assoc J. 165, 547–552. [PMC free article] [PubMed] [Google Scholar]
- Keel PK, Fulkerson JA, Leon GR, 1997. Disordered eating precursors in pre- and early adolescent girls and boys. J Youth Adolesc. 26, 203–216. [Google Scholar]
- Killen JD, Hayward C, Litt I, Hammer L, Wilson DM, Miner B, Taylor CB, Varady A, Shisslak C, 1992. Is puberty a risk factor for eating disorders? Am J of Dis Child. 146, 323–325. [DOI] [PubMed] [Google Scholar]
- Killen JD, Taylor CB, Hayward C, Haydel KF, Wilson DM, Hammer L, Kraemer H, Blair-Greiner A, Strachowski D, 1996. Weight concerns influence the development of eating disorders: A 4-year prospective study. J Consult Clin Psychol. 64, 936–940. [DOI] [PubMed] [Google Scholar]
- Klump KL, 2013. Puberty as a critical risk period for eating disorders: A review of human and animal studies. Horm Behav. 64, 399–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klump KL, Burt SA, 2006. The Michigan State University Twin Registry (MSUTR): Genetic, environmental and neurobiological influences on behavior across development. Twin Res Hum Genet. 9, 971–977. [DOI] [PubMed] [Google Scholar]
- Klump KL, Burt SA, McGue M, Iacono WG, 2007a. Changes in genetic and environmental influences on disordered eating across adolescence: A longitudinal twin study. Arch Gen Psychiatry. 64, 1409–1415. [DOI] [PubMed] [Google Scholar]
- Klump KL, Burt SA, Spanos A, McGue M, Iacono WG, Wade TM, 2010a. Age differences in genetic and environmental influences on weight and shape concerns. Int J Eat Disord. 43, 679–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klump KL, Culbert KM, O’Connor SM, Fowler N, Burt SA, 2017a. The significant effects of puberty on the genetic diathesis of binge eating in girls. Int J Eat Disord. 50, 984–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klump KL, Culbert KM, Sisk CL, 2017b. Sex differences in binge eating: Gonadal hormone effects across development. Annu Rev Clin Psychol. 13, 183–207. [DOI] [PubMed] [Google Scholar]
- Klump KL, Culbert KM, Slane JD, Burt SA, Sisk CL, Nigg JT, 2012. The effects of puberty on genetic risk for disordered eating: Evidence for a sex difference. Psychol Med. 42, 627–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klump KL, Fowler N, Mayhall L, Sisk CL, Culbert KM, Burt SA, 2018. Estrogen moderates genetic influences on binge eating during puberty: Disruption of normative processes? J Abnorm Psychol. 127, 458–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klump KL, Hildebrant BA, O’Connor SM, Keel PK, Neale M, Sisk CL, Boker S, Burt SA, 2015. Changes in genetic risk for emotional eating across the menstrual cycle: A longitudinal Study. Psychol Med. 45, 3227–3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klump KL, Keel PK, Burt SA, Racine SE, Neale MC, Sisk CL, Boker S 2013a. Ovarian hormones and emotional eating associations across the menstrual cycle: An examination of the potential moderating effects of body mass index and dietary restraint. Int J Eat Disord. 46, 256–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klump KL, Keel PK, Racine SE, Burt SA, Neale M, Sisk CL, Boker S, Hu JY, 2013b. The interactive effects of estrogen and progesterone on changes in emotional eating across the menstrual cycle. J Abnorm Psychol. 122, 131–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klump KL, Keel PK, Sisk CL, Burt SA, 2010b. Preliminary evidence that estradiol moderates genetic influences on disordered eating attitudes and behaviors during puberty. Psychol Med. 40, 1745–1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klump KL, McGue M, Iacono W, 2000. Age differences in genetic and environmental influences on eating attitudes and behaviors in preadolescent and adolescent female twins. J Abnorm Psychol. 109, 239–251. [PubMed] [Google Scholar]
- Klump KL, McGue M, Iacono WG, 2003. Differential heritability of eating attitudes and behaviors in prepubertal versus pubertal twins. Int J Eat Disord. 33, 287–292. [DOI] [PubMed] [Google Scholar]
- Klump KL, Perkins P, Burt SA, McGue M, Iacono WG, 2007b. Puberty moderates genetic influences on disordered eating. Psychol Med. 37, 627–634. [DOI] [PubMed] [Google Scholar]
- Klump KL, Suisman JL, Burt SA, McGue M, Iacono WG, 2009. Genetic and environmental influences on disordered eating: An adoption study. J Abnorm Psychol. 118, 797–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo X, Donnellan MB, Burt SA, Klump KL, 2016. The dimensional nature of eating pathology: Evidence from a direct comparison of categorical, dimensional, and hybrid models. J Abnorm Psychol. 125, 715–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lykken DT, Bouchard TJ, McGue M, Tellegen A, 1990. The Minnesota Twin Family Registry: Some initial findings. Acta Genet Med Gemellol. 39, 35–70. [DOI] [PubMed] [Google Scholar]
- Mond JM, Rodger B, Hay PJ, Darby A, Owen C, Baune BT, Kennedy RL, 2007. Obesity and impairment in psychosocial functioning in women: The mediating role of eating disorder features. Obesity. 15, 2769–2779. [DOI] [PubMed] [Google Scholar]
- Neale MC, Boker SM, Xie G, Maes HM, 1999. Statistical modeling. Richmond, VA: Department of Psychiatry, Virginia Commonwealth University. [Google Scholar]
- O’Connor SM, Burt SA, VanHuysse JL, Klump KL, 2016. What drives the association between weight-conscious peer groups and disordered eating? Disentangling genetic and environmental selection from pure socialization effects. J Abnorm Psychol. 125, 356–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pashler H, Wagenmakers E, 2012. Editors’ introduction to the special ection on replicability in psychological science: A crisis of confidence? Perspect. Psychol. Sci 7, 528–530. [DOI] [PubMed] [Google Scholar]
- Peeters H, Van Gestel S, Vlietinck R, Derom C, Derom R, 1998. Validation of telephone zygosity questionnaire in twins of known zygosity. Behav Genet. 28, 159–161. [DOI] [PubMed] [Google Scholar]
- Petersen AC, Crockett L, Richards M, Boxer A, 1988. A self-report measure of pubertal status: Reliability, validity, and initial norms. J Youth Adolesc. 17, 117–133. [DOI] [PubMed] [Google Scholar]
- Purcell S, 2002. Variance components models for gene-environment interaction in twin analysis. Twin Res. 5, 554–571. [DOI] [PubMed] [Google Scholar]
- Raftery AE, 1995. Bayesian model selection in social research. Sociol Methodol. 25, 111–163. [Google Scholar]
- Sclove LS, 1987. Application of model-selection criteria to some problems in multivariate analysis. Psychometrika. 53, 333–343. [Google Scholar]
- Spanos A, Klump KL, Burt SA, McGue M, Iacono WG, 2010. A longitudinal investigation of the relationship between disordered eating attitudes and behaviors and parent-child conflict: A monozygotic twin differences design. J Abnorm Psychol. 119, 293–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spellman BA, 2013. Introduction to the specical section on advancing science. Perspect. Psychol. Sci 8, 412–413. [DOI] [PubMed] [Google Scholar]
- Spiegelhalter DJ, Best NG, Carlin BP, Van Der Linde A, 2002. Bayesian measures of model complexity and fit. J Royal Stat Soc. 64, 583–639. [Google Scholar]
- Stice E, Shaw HE, 2002. Role of body dissatisfaction in the onset and maintenance of eating pathology: A synthesis of research findings. J Psychosom Res. 53, 985–993. [DOI] [PubMed] [Google Scholar]
- van der Sluis S, Posthuma D, Dolan CV, 2012. A note on false positives and power in G x E modelling of twin data. Behav Genet. 42, 170–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Zutven K, Mond J, Latner J, Rodgers B, 2014. Obesity and psychosocial impairment: Mediating roles of health status, weight/shape concerns and binge eating in a community of sample of women and men. Int J Obes. 39, 346–352. [DOI] [PubMed] [Google Scholar]
- von Ranson KM, Klump KL, Iacono WG, McGue M, 2005. The Minnesota Eating Behaviors Survey: A brief measure of disordered eating attitudes and behaviors. Eat Behav. 6, 373–392. [DOI] [PubMed] [Google Scholar]
- Wade TD, Hansell NK, Crosby RD, Bryant-Waugh R, Treasure J, Nixon R, Byrne S, Martin NG, 2012. A study of changes in genetic and environmental influences on weight and shape concern across adolescence. J Abnorm Psychol. 122, 119–130. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.