

Abstract
Dysfunction of excitatory amino acid transporters (EAATs) has been implicated in the pathogenesis of various neurological disorders, such as stroke, brain trauma, epilepsy, and several neurodegenerative disorders. EAAT2 is the main transporter subtype responsible for glutamate clearance in the brain, and plays a key role in regulating neurotransmission and preventing excitotoxicity. Therefore, compounds that increase the activity of EAAT2 have therapeutic potential for neuroprotection. In previous studies, we used virtual screening approaches to identify novel positive allosteric modulators (PAMs) of EAAT2. These compounds were shown to selectively increase the activity of EAAT2 and increase Vmax of transport, without changing substrate affinity. In this work, our major effort was to investigate whether increasing the activity of EAAT2 by allosteric modulation would translate to neuroprotection in in vitro primary culture models of excitotoxicity. To investigate potential neuroprotective effects of one EAAT2 PAM, GT949, we subjected cultures to acute and prolonged excitotoxic insults by exogenous application of glutamate, or oxidative stress by application of hydrogen peroxide. GT949 administration did not result in neuroprotection in the oxidative stress model, likely due to damage of the glutamate transporters. However, GT949 displayed neuroprotective properties after acute and prolonged glutamate-mediated excitotoxicity. We propose that this compound prevents excess glutamate signaling by increasing the rate of glutamate clearance by EAAT2, thereby preventing excitotoxic damage and cell death. This novel class of compounds is therefore an innovative approach for neuroprotection with potential for translation in in vivo animal models of excitotoxicity.
Keywords: EAAT2, glutamate transporter, glutamate uptake, transport enhancement, EAAT2 activators, glutamate excitotoxicity
INTRODUCTION
Glutamate is the predominant excitatory amino acid neurotransmitter in the mammalian central nervous system (CNS), and is essential for normal brain functions including cognition, memory, learning, developmental plasticity, and long-term potentiation1–3. Glutamate neurotransmission is terminated by presynaptic and astrocytic excitatory amino acid transporters (EAATs)4–6, which remove glutamate from the synaptic cleft. This process involves co-transport of glutamate and three sodium ions, followed by counter transport of one potassium7–9. There are five structurally distinct subtypes of Na+-dependent EAATs, including (rat/human homolog): GLAST/EAAT1/, GLT-1/EAAT2, EAAC1/EAAT3, EAAT4 and EAAT5, which allows for precise spatial and temporal regulation of glutamate neurotransmission4. The subtype GLT-1/EAAT2 plays a central role in maintenance of extracellular glutamate homeostasis and prevention of excitotoxicity10–14. EAAT2 accounts for approximately 95% of the total glutamate transport activity and 1% of total brain protein in the CNS15–19. This transporter is primarily expressed in astrocytes and select neurons and oligodendrocytes of the brain and spinal cord14, 20.
Glutamate excitotoxicity is caused by sustained elevation of extracellular glutamate levels and activation of postsynaptic glutamate receptors, resulting in excessive Ca2+ influx21 and activation of a cascade of phospholipases, endonucleases, and proteases that can lead to apoptotic and necrotic cell death22–24. Sustained elevation of glutamate levels may occur due to failure of astrocytes or other cells to take up excess synaptic or ambient glutamate through EAATs. Glutamate excitotoxicity plays a key role in secondary damage following acute pathologies, including traumatic brain injury (TBI)25–31, hyper-excitability and seizures32, 33, stroke11, 34–37, epilepsy33, 38, 39, and cerebral and retinal ischemia16, 26, 27, 40–44. Excitotoxicity also produces secondary damage in chronic pathologies, including Amyotrophic lateral sclerosis (ALS)45, 46, Alzheimer’s disease47, Huntington’s disease48, 49, neuropathic pain50 and HIV-associated neurocognitive disorders (HAND)51–53.
Approaches that aim to mitigate excitotoxicity, such as glutamate receptor antagonists28, 54–61 and inhibition of calcium influx62, 63 alleviate cellular damage and neuronal deficits after brain trauma, but their use in the clinic has been severely limited due to serious side effects58, 64. On the other hand, approaches that focus on removing excess glutamate through the stimulation of glutamate transporters may offer a safe and effective treatment for excitotoxicity. In this regard, approaches that increase EAAT2 protein expression13, 65 are currently being pursued. Several transcriptional or translational upregulators of EAAT2, such as GPI-104666, ceftriaxone37, 67–70, harmine71, 72 and pyridazine derivatives73, 74 were shown to be neuroprotective through augmentation of glutamate transport. Although EAAT2 upregulation is a promising approach to treat excitotoxicity, it is most relevant for chronic conditions or prophylactic use75.
Other approaches that relieve excitotoxicity by directly or indirectly increasing EAAT2 activity are also being pursued. Several compounds acutely stimulate glutamate uptake by indirect modulation of transporter activity, such as MS-15376, riluzole77, 78, guanosine79, 80, and nicergoline81 resulting in neuroprotective outcomes. However, clinical use of these compounds is limited due to poor target specificity and side effects, highlighting the need for novel target-specific treatments. Previous studies from our group have identified a natural compound isolated from Parawixia bistriata spider venom that enhances glutamate uptake through EAAT2, and has neuroprotective properties82, 83. Using this compound, we identified the molecular determinants of EAAT2 transport enhancement in a mutagenesis guided study84. This unique structural information was then used in a virtual screening approach to identify molecules that interact with EAAT2 allosteric sites, using a structural model based on the 3D crystal structure of the bacterial EAAT2 homolog GltPh85–88. These compounds were then characterized in glutamate uptake assays in transfected cells and in glial cultures, as allosteric modulators (AM) of EAAT289.
In this study, we evaluated potential neuroprotective properties of one of these EAAT2 positive allosteric modulators (PAM), GT94989, in primary neuronal culture models of excitotoxicity. Our hypothesis is that positive allosteric modulation of EAAT2 activity will rapidly remove excessive glutamate from the synapse and significantly improve neuronal survival and disease outcome in excitotoxic conditions. This novel approach may result in a new generation of safe and effective therapies for CNS disorders with comorbid excitotoxicity.
RESULTS AND DISCUSSION
In this study, we evaluated GT949, a PAM of glutamate transporter EAAT2, for neuroprotective effects after acute or prolonged insults with application of L-glutamate or H2O2. Two primary culture approaches were used: a bilaminar neuron-glia co-culture and a mixed neuron-glia culture. As positive control for neuroprotection, we used AP-V (DL-2-Amino-5-phosphonopentanoic acid), an NMDA receptor blocker. As negative control, we used GT996, an analog of GT949 that does not increase glutamate uptake89. We exposed these cultures to glutamate or H2O2, and examined the neuroprotective potential of GT949 by immunocytochemical analysis, comparing MAP-2 expression90 among different experimental groups.
Effects of GT949 in primary cultures subjected to acute glutamate-mediated excitotoxicity and oxidative stress insults
We began our analysis examining the effects of GT949 in a glutamate-mediated excitotoxicity model using a bilaminar co-culture system, where primary cortical neurons plated on coverslips were cultured in the presence of a glial feeder layer. This allowed us to separate the neurons and glia for treatments and downstream analysis as needed.
In the neuronal coverslips, ~97% of counted cells were MAP-2 positive, indicating that the vast majority of cells are neurons, and ~2–3% of cells were GFAP positive, indicating minimal astroglia contamination (< 3%, supplemental figure 1), which is in agreement with other studies using the same system91. The glial feeder is comprised of 100% GFAP positive cells, also in accordance to previous literature92, 93.
To rule out any toxicity by compound GT949 in astrocytes, we incubated pure glia cultures with increasing concentrations of GT949 (up to 1 μM). Supplemental figure 2A shows images of pure glia cultures incubated with vehicle (control) and 1 μM GT949 for 24 hours, indicating preservation of glial morphology. The quantification of GFAP + cells in figure 2B indicates no toxicity of the compound, as there no significant changes between the number of cells between control (vehicle), 100 nM and 1 μM GT949.
Figure 2. Neuroprotective properties of GT949 in bilaminar cultures after acute exposure to glutamate (in the absence of glia).
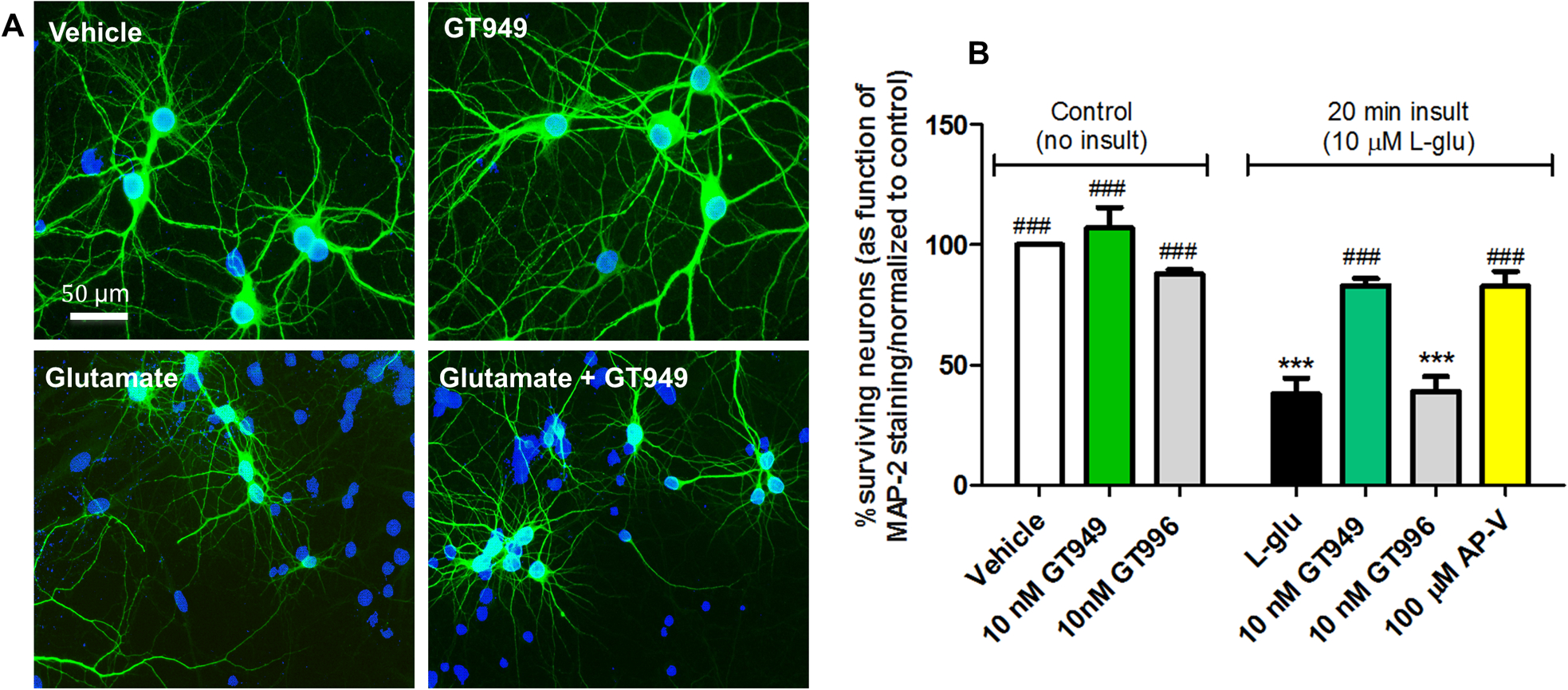
A. Representative images of cultures after different treatments in the absence of glia. The neuronal layer of bilaminar cultures was immunostained against MAP-2 (green), and counterstained with the nuclear marker DAPI (blue). Cultures exposed to vehicle, or 100 nM GT949 alone do not display obvious degeneration or cell death. Cultures acutely exposed to L-glutamate (20 min, 10 μM) show increased DAPI positive, MAP-2 negative cells, indicative of toxicity and neuronal death. However, co-exposure of 10 nM GT949 reversed a portion of the cellular damage caused by L-glutamate. All images are shown at 40x magnification. Scale bar: 50 μm.
B. Quantification of neuronal survival in the absence of glia. After vehicle or L-glutamate (20 min, 10μM) exposure in glia-free conditions, coverslips were transferred back to their original glia dishes in the presence or absence of GT949 or GT996 (10nM) or the NMDA antagonist AP-V (100μM) for 24 hours. Then, cells were fixed and immunostained as in A, and analyzed for cell death. GT949 and GT996 alone did not alter neuronal survival levels. Acute L-glutamate significantly decreased neuronal survival, while GT949, and AP-V mitigated L-glutamate neurotoxicity. GT996, the inactive analog, had no effect on neuronal survival after L-glutamate exposure. Data was normalized to control, 3–4 coverslips were assessed per treatment condition and 100–150 cells were manually counted per treatment. Neuronal survival data is representative of 3 independent experiments, and control levels were not statistically different for normalization purposes. ***p<0.001, vs. vehicle (no insult), ###p<0.001, vs. insult.
Neuronal survival rates after acute glutamate insult and GT949 treatment in the presence of the glia
Neurons and glia were co-cultured for 14 DIV before treatments. At 14 DIV, there is substantial literature showing that glutamate receptors (NMDA and AMPA) are well expressed91, 94–96. Supplemental figure 1 also demonstrates the differential expression of glutamate transporters at 14 DIV, with EAAT2 being highly expressed in glia, but not in neurons, and EAAT3 being mostly expressed in neurons. These results are in accordance with previous studies that have shown that EAAT1 and EAAT2 are predominantly expressed in astroglial cells14, 97, while EAAT3 and EAAT4 are expressed in neurons98, 99.
Acute 10 μM and 50 μM L-glutamate insults in bilaminar cultures containing the glial feeder layer did not significantly alter neuronal survival compared to vehicle-treated cultures (not shown). This suggests that the glia play a crucial role in maintaining glutamate at physiological concentrations, most likely through glutamate transporter-mediated clearance. Therefore, we used an acute 100 μM L-glutamate insult for the following experiments, as this glutamate concentration produced significant neurotoxicity and cell death in the presence of glia. Neuronal survival in cultures treated with 10 nM GT949 or GT996 alone was not significantly altered, suggesting that these compounds are not neurotoxic in the presence of glia (figure 1). On the other hand, acute 100 μM glutamate insult significantly decreased neuronal survival to 55± 12% of baseline vehicle levels (figure 1, black bar). Application of 10 nM GT949 after acute glutamate insult resulted in a significant neuroprotective effect, increasing survival levels to 87± 15% of baseline, while the inactive analog GT996 did not increase cell survival levels. The positive control AP-V resulted in significant protection from acute glutamate insult, with survival at 93± 6% of baseline. These results suggest a neuroprotective effect of GT949, possibly due to its mechanism of increasing EAAT2-mediated glutamate uptake.
Figure 1. Neuroprotective properties of GT949 in bilaminar cultures after acute exposure to glutamate (in the presence of glia).
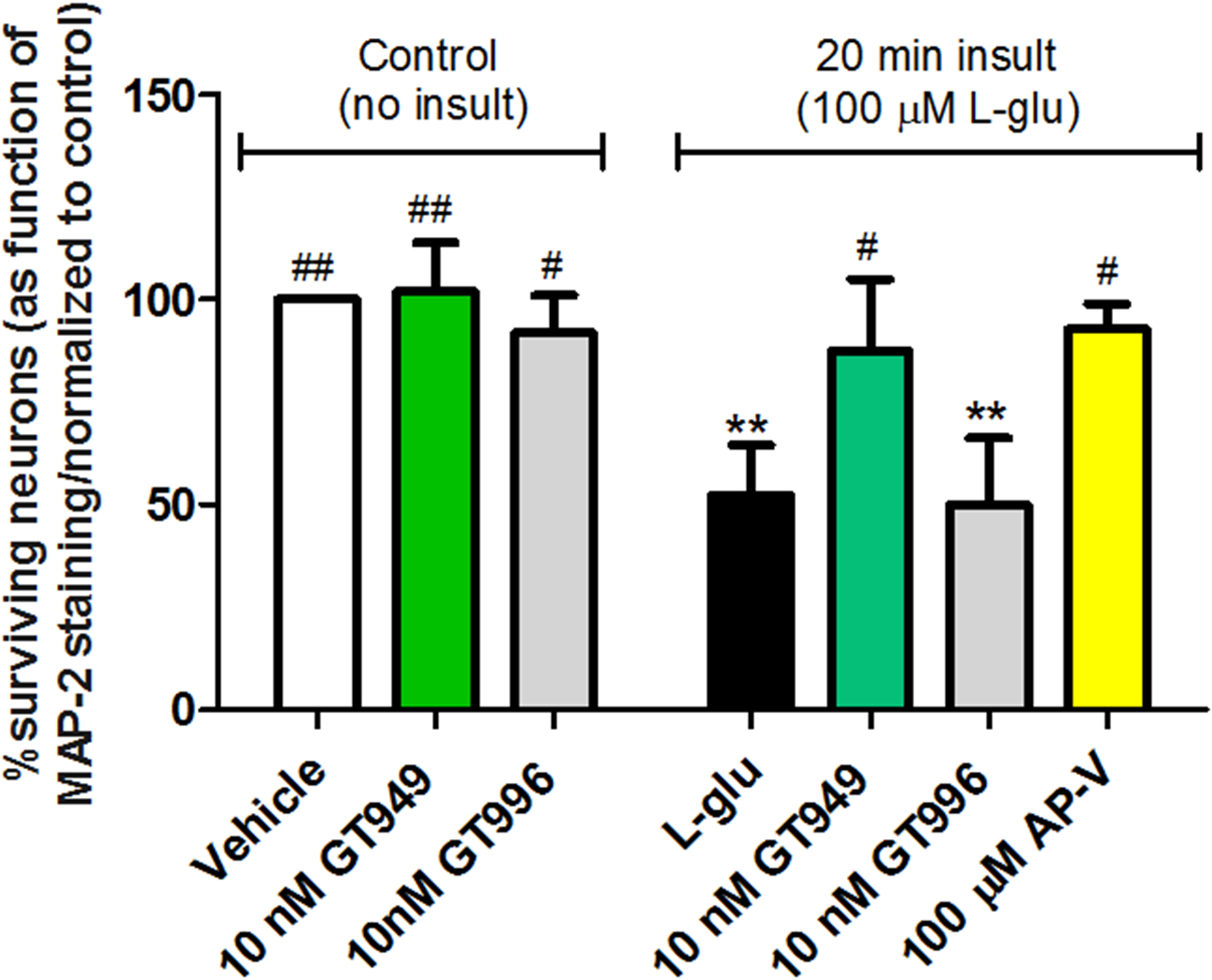
Bilaminar cultures at 14 DIV were treated with glia-conditioned medium containing L-glutamate for 20 minutes, followed by replacement of the glutamate-containing medium and compound treatment for 24 hours. Then, cells were fixed and immunostained as previously described for neuronal survival analysis. Application of 10 nM GT949 and GT996 alone had no noticeable effects on neuronal survival. Acute 100 μM L-glutamate significantly reduced neuronal survival, and GT949 and AP-V mitigated L-glutamate toxicity. GT996 also had no effect on L-glutamate toxicity. Data was normalized to control. 3–4 coverslips were assessed per treatment condition and 100–150 cells were manually counted per treatment. Neuronal survival data is representative of 3 independent experiments and control levels were not statistically different for normalization purposes. **p<0.01, vs. control (no insult), ##p<0.01, #p<0.05, vs. insult.
Neuronal survival after acute glutamate insult in the absence of glia and GT949 treatment
The bi-laminar culture approach allows for separation of the neurons and glia for treatments. We performed an acute L-glutamate insult (10 μM; 20 minutes) in the absence of glia by transferring neuronal coverslips into glia-free culture dishes containing glia-conditioned medium with L-glutamate. After the acute glutamate insult, neuronal coverslips were returned to their original bilaminar culture dishes, and treated with either GT949 or other control compounds for 24 hours. Then, neuronal coverslips were fixed and immunostained against MAP-2 and imaged by fluorescence microscopy (figure 2A) for downstream survival analysis. At baseline conditions (no glutamate insult or compound treatment - vehicle), neurons did not display pyknotic nuclei or neurite damage, which are indicators of neuronal death94, 100. Neurons treated with GT949 alone (10 nM; 24 hours) also resemble the baseline vehicle conditions, suggesting that GT949 is not neurotoxic. After acute glutamate insult, we observed reduced MAP-2 staining and DAPI positive, MAP-2 negative cells, indicators of cell death. On the other hand, neurons subjected to acute glutamate insult followed by GT949 (10 nM; 24 hours) displayed morphological characteristics similar to baseline vehicle conditions, suggesting that this compound is neuroprotective in an excitotoxic environment.
Total neuronal survival levels from different experimental conditions were then quantified (figure 2B). Similarly to figure 1, in the absence of glutamate insult, neuronal survival was not significantly altered by 10 nM GT949 or GT996, an inactive analog of GT949, suggesting that these compounds are not neurotoxic at the examined doses. On the other hand, acute, 20 minute insult with 50 and 100 μM glutamate in the absence of glia resulted in profound neuronal death (not shown) so these concentrations were not used in this experiment. Acute 10 μM glutamate insult significantly decreased neuronal survival to 38±7% of baseline, vehicle-treated cultures (figure 3B, black bar), and this concentration was used to assess the neuroprotective potential of GT949. Neuronal cultures treated with GT949 for 24h after acute glutamate insult displayed survival of 83±3% of control, a significant increase that suggests a neuroprotective effect of GT949. Cultures treated with GT996, the inactive analog of GT949, after acute glutamate insult displayed survival of 36±6% of baseline, which is not significantly different than glutamate alone. Cultures treated with the NMDA receptor antagonist AP-V (100 μM; 24 hours) provides a survival benefit similar to that of GT949 suggesting that excessive glutamate transmission through the NMDA receptor plays a crucial role in glutamate excitotoxicity and neuronal death, thus providing a positive control to measure the effect of GT949. This suggests that NMDA receptor plays a crucial role in glutamate excitotoxicity and neuronal death, likely through activation of neuronal death pathways such as calpain101, 102.
Figure 3. Lack of neuroprotective properties of GT949 in bilaminar cultures after acute exposure to H2O2-mediated oxidative stress.
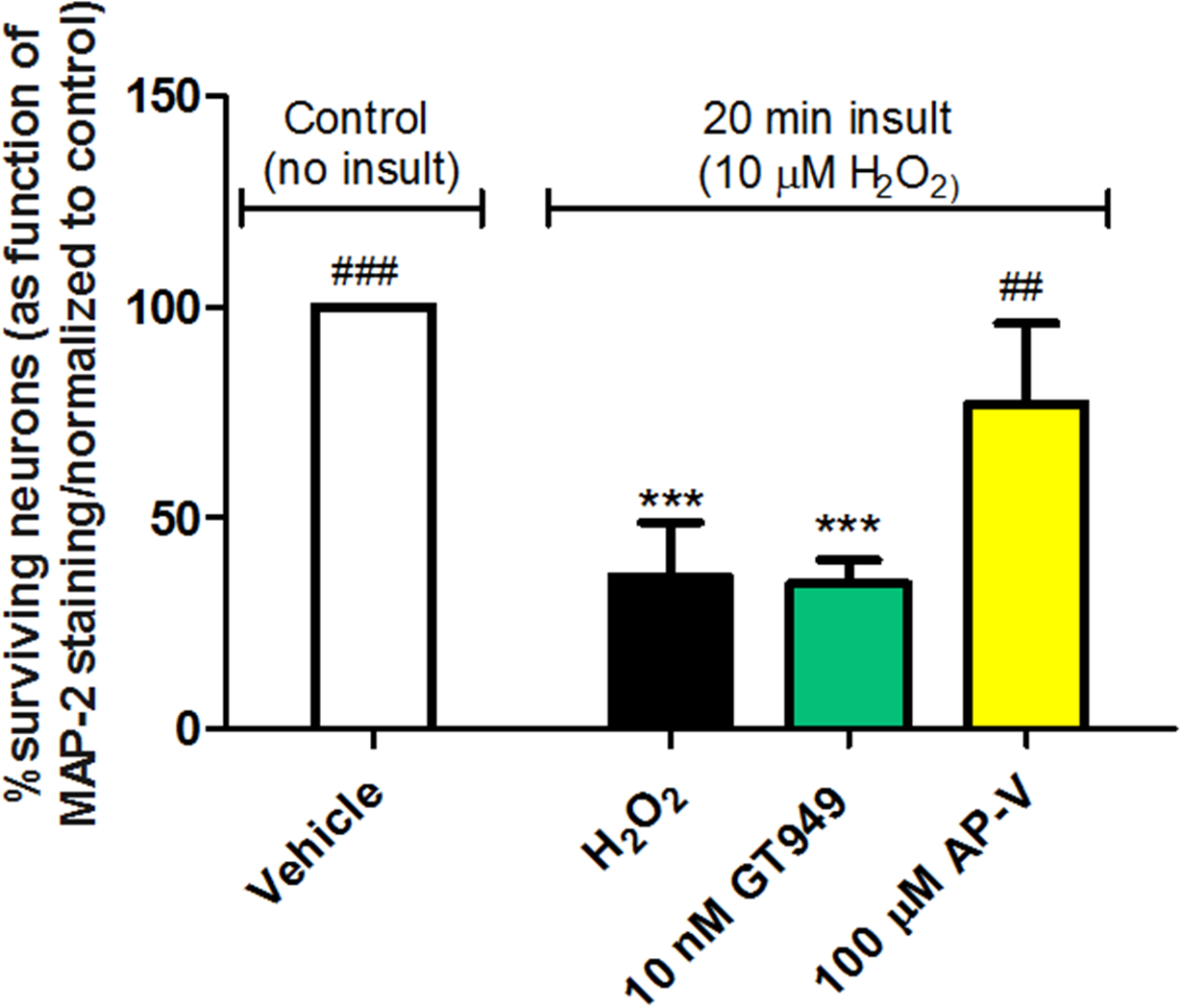
Bilaminar cultures at 14 DIV were treated with conditioned medium containing 10 μM H2O2 for 20 minutes, followed by replacement of the H2O2 medium and compound treatment for 24 hours. Neurons were then fixed and immunostained against MAP-2 for downstream survival analysis. Exposure significantly reduced neuronal survival, and co-treatment with 10 nM GT949 was not neuroprotective. Co-treatment with 10 nM GT996 also had no effect on neuronal survival after H2O2 exposure. However, Co-treatment with 100 μM AP-V did significantly reverse H2O2-induced neuronal death, suggesting that NMDA receptor activation is involved in oxidative stress-mediated neuronal toxicity. Survival quantification was normalized to control cultures. 3–4 coverslips were assessed per treatment condition and 100–150 cells were manually counted per treatment. Neuronal survival data is representative of 3 independent experiments and control levels were not statistically different for normalization purposes. ****p<0.0001, vs. control (no insult), ###p<0.001, ##p<0.01, vs. insult.
Neuronal survival rates after acute glutamate insult in mixed neuron-glia cultures
We also investigated the potential neuroprotective properties of GT949 in mixed neuron-glia cultures, a system that more closely resembles physiological conditions in the CNS. We first optimized growth conditions to result in ~15% of glia growth in the cultures after 14 DIV (supplemental figure 3). At 14 DIV, several reports show that mixed cortical neuron-glia cultures express glutamate transporters GLAST/EAAT1, GLT1/EAAT2 and EAAC1/EAAT3103–105.
Then, acute glutamate insults were performed by application of 10, 50, 100 and 1000 μM L-glutamate for 20 min. However, these insults did not result in significant levels of cell death (not shown), likely because the presence of glia conferred neuroprotection against these insults. Therefore, the neuroprotective efficacy of GT949 was evaluated only in prolonged, 24 hour glutamate insult experiments in the mixed neuron-glia cultures.
Lack of neuroprotective effects of GT949 in primary cultures subjected to H2O2-mediated oxidative stress
We expanded our analysis to test the neuroprotective potential of GT949 in an oxidative stress model in the bilaminar co-culture system. Oxidative stress promotes neuronal death by impairing mitochondrial bioenergetics function and may open the mitochondrial permeability transition pore106, 107. This results in reactive oxygen species production, energy failure, and release of pro-apoptotic factors such as cytochrome C into the cytoplasm, and damaged to nucleic acids, proteins and lipids108.
In these studies, bilaminar cultures were exposed to 10 μM H2O2 for 20 minutes in the presence of glia, followed by treatment with GT949, or other compounds for 24 hours. As before, neurons were fixed and immunostained against MAP-2 for downstream survival analysis (figure 2).
An acute H2O2 insult significantly decreased neuronal survival levels to 36 ± 13% of baseline (figure 3, black bar). Following the acute H2O2 insult with 10 nM GT949 treatment did not significantly improve neuronal survival, as the survival percentage was 35 ± 6% of baseline. However, blocking NMDA receptors after H2O2 insult with 100 μM AP-V significantly increased neuronal survival to 77 ± 19% of baseline, suggesting that glutamate does play a role in oxidative stress-induced neuronal damage.
Lack of neuroprotective effects of GT949 in mixed neuron-glia cultures subjected to H2O2-mediated oxidative stress
To further investigate whether compound GT949 had neuroprotective properties in an oxidative stress model, we next subjected 14 DIV mixed neuron-glia cultures to 100 mM H2O2 insults for 20 min, and then applied several concentrations of GT949 and 100 mM AP-V for 24 hours. Cultures were then cultures were fixed and analyzed as described before for downstream survival analysis. At baseline conditions (no insult or compound treatment - vehicle), neurons did not display pyknotic nuclei or neurite damage, similar to the observations in bilaminar cultures under control conditions (figure 2A). After acute H2O2 insult, we observed reduced MAP-2 staining and DAPI positive, MAP-2 negative cells, indicators of cell death. After insult and application of 1 μM GT949 for 24 hours, we observe cell death characteristics, like the insult alone, indicating lack of neuroprotective effect of the compound. On the other hand, neurons subjected to acute H2O2 insult followed by AP-V (100 μM; 24 hours) displayed morphological characteristics similar to baseline vehicle conditions.
Total neuronal survival levels from different experimental conditions were then quantified (figure 2B). Acute 20 minute insult with 100 μM H2O2 resulted in significant neuronal death (35±14% of baseline, red bar), and treatments with 100 nM and 1 μM GT949 for 24 hours resulted in survival of 41±10% and 35±22 % of baseline, which were not significantly different than insult alone. On the other hand, cultures treated AP-V (100 μM; 24 hours) provides a survival benefit of 102±19.9% of control, suggesting that glutamate does play a role in oxidative stress-induced neuronal damage.
We also analyzed the effect of the oxidative stress insult on EAAT2 in these cultures via western blot. 14 DIV mixed neuron-glia cultures were subjected to H2O2 insults for 20 min, media was changed to fresh media and incubated for 24 hours, then samples were collected for western blotting. Previous studies showed that under conditions of oxidative stress, the EAAT2 transporter would be cleaved and the appearance of a small band around 30 kDa appeared109. Figure 4C shows a representative western blots of samples ran in triplicate (controls, 10, 100 and 500 mM H2O2) that suggests that the transporter is cleaved at higher concentrations of H2O2 (100μM and 500μM), as we observed a band at around 30 kDa. This suggests that the lack of neuroprotective effects of GT949 could be due to the H2O2 insults resulting in damage of EAAT2.
Figure 4. Lack of neuroprotective effects of GT949 in mixed neuron-glia cultures subjected to H2O2-mediated oxidative stress.
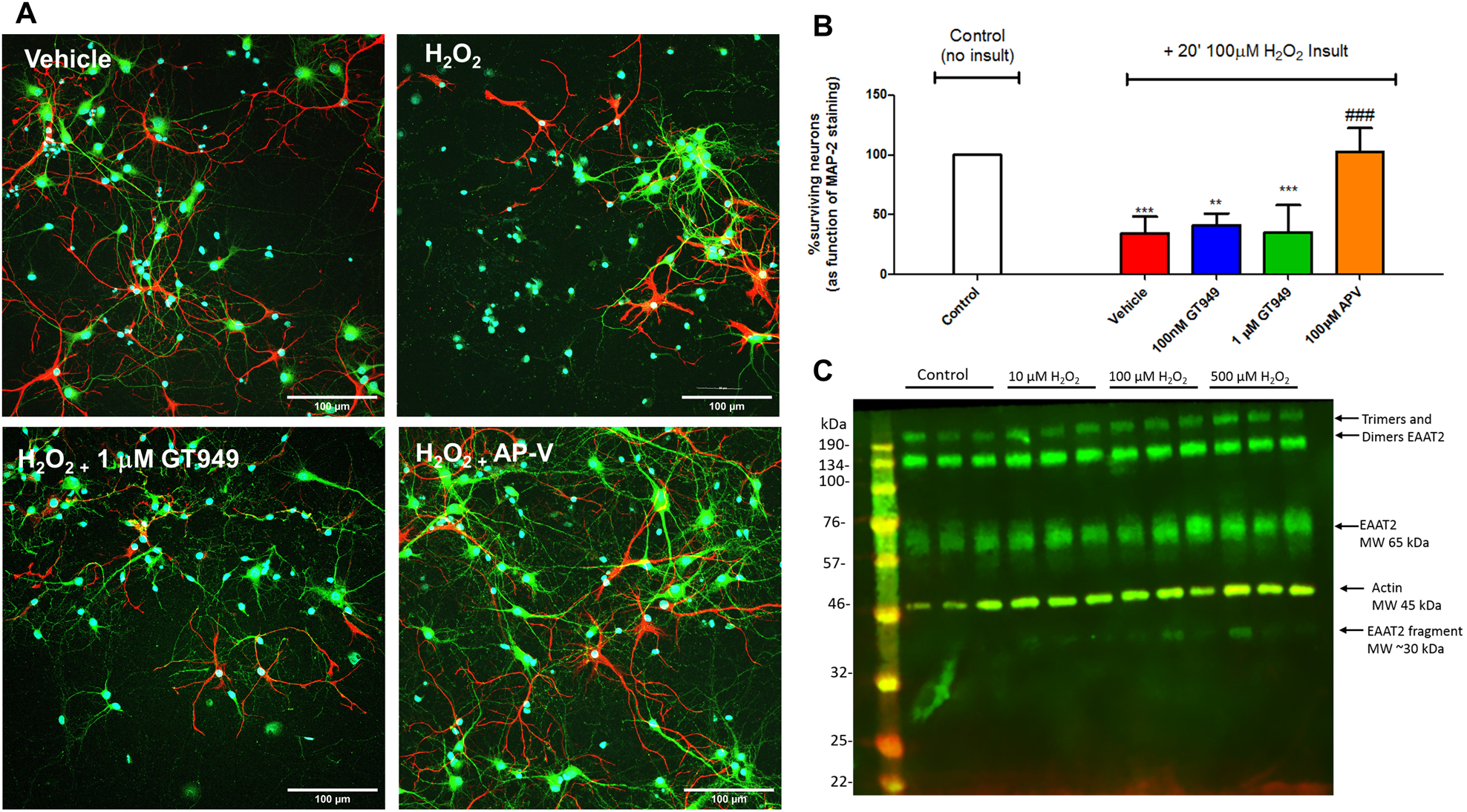
A. Representative images of cultures after different treatments, with neuronal marker MAP-2 in green, glial marker GFAP in red, and counterstaining with the nuclear marker DAPI (blue). Cultures exposed to vehicle do not display obvious degeneration or cell death. Cultures acutely exposed to H2O2 (20 min, 100 μM) show increased DAPI positive, MAP-2 negative cells, indicative of toxicity and neuronal death. Co-exposure of 1 mM GT949 did not change the level of neuronal death, whereas co-incubation with 100 mM AP-V reversed a portion of the cellular damage caused by H2O2. All images are shown at 30x magnification. Scale bar: 100 μm.
B. Quantification of neuronal survival. Cultures were treated with 100 μM H2O2 for 20 minutes, followed by media replacement and compound treatment for 24 hours. Cultures were then fixed and immunostained for neuronal death analysis as in A. Survival quantification was normalized to control cultures, with 3 coverslips assessed per treatment condition per experiment. Neuronal survival data is representative of 2 independent experiments (n=6) and control levels were not statistically different for normalization purposes. ****p<0.0001, vs. control (no insult), ###p<0.001, ##p<0.01, vs. H2O2 insult.
C. Representative immunoblot showing cleavage of the EAAT2 transporter after treatment with higher concentrations of H2O2. Each treatment group represents 3 lanes; from left to right: control (no insult), 10 μM, 100μM and 500 μM H2O2. EAAT2 expression is shown in green and beta-actin in red (used as a loading control). MW= Molecular weight.
Our results are in accordance with previous reports showing that oxidative stress increases extracellular glutamate levels, resulting in Ca2+ over-load in cultured neurons.110, 111 This is followed by activation of MAPK cascades (ERK and/or p38) that induce neuronal cell death112. In the presence of AP-V, neuronal death was mitigated, suggesting that oxidative stress-induced excitotoxicity contributes to neuronal cell death113. We hypothesize that the lack of efficacy of GT949 under oxidative stress-induced excitotoxicity could be explained by evidence that caspase-3, a key proteolytic enzyme executioner of apoptosis that is triggered by oxidative stress, cleaves EAAT2 at a single site (D505) and decreases glutamate uptake activity109, 114.
However, one study115 shows no neuroprotection in the presence of AP-V, arguing that the H2O2-induced neuronal toxicity was not a function of calcium influx into neurons. Several differences in their methodology, as they performed their assays in hippocampal slices and used a higher concentrated of H2O2, as compared to ours, could at least partially explain the different results. Further investigation is required to understand the cross talk between oxidative stress-mediated neuronal death and NMDA-mediated neuronal death in the context of excitotoxicity.
Effects of acute H2O2-mediated oxidative stress in pure glia cultures
Furthermore, lack of GT949 efficacy may also be explained by previous work suggesting that oxidative stress causes an imbalance in the mitochondrial permeability transition pore (MPTP) of astrocytes, leading to massive glial cell death115. In agreement with this, we have subjected pure glia cultures to insults with increasing concentrations of H2O2, where we demonstrate a dose dependent decrease in GFAP+ cells (Figure 5), suggesting that the lack of effect of GT949 in this model is due to damage to the glia, and fragmentation of EAAT2 (seen in figure 4).
Figure 5. H2O2 -mediated oxidative stress results in significant glial cell death.
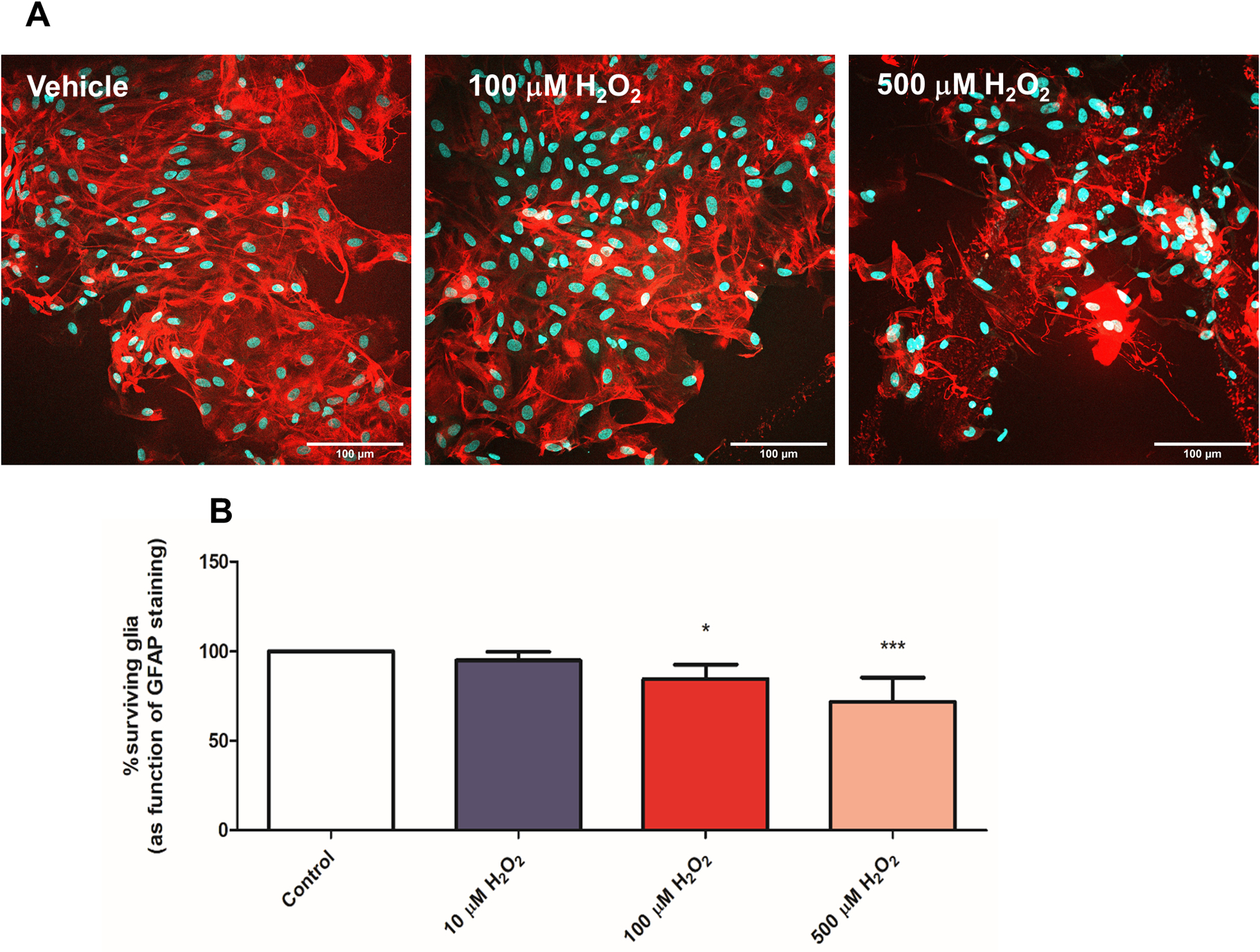
A. Representative images of pure glia cultures incubated with vehicle (control), 100 and 500μM H2O2 for 20 min, indicating loss of glia due to the insults.
B. Quantification of GFAP+ cells indicates 100 μM and 500μM H2O2 insults result in significant glial cell death in pure glial cultures. Survival quantification was determined by comparing GFAP staining to total DAPI staining and cultures were normalized to control, vehicle-treated cultures. 3 coverslips were assessed per treatment condition per experiment. Neuronal survival data is represented as the mean ± SEM of 2 independent experiments (n=6) and control levels were not statistically different for normalization purposes. ***p<0.001, *p<0.05, vs. control (no insult).
Neuroprotective effects of GT949 in primary cultures subjected to prolonged glutamate-mediated excitotoxicity insults
Neuronal survival after prolonged glutamate insult and GT949 treatment in bilaminar cultures
Since we have shown a neuroprotective effect of GT949 after an acute glutamate insult, we next determined if GT949 is also protective during a prolonged, 24 hour glutamate insult. These experiments were all performed in the presence of glia, as neurons in bilaminar cultures do not survive for an extended duration without the glial feeder layer. We began by co-treating 14 DIV bilaminar cultures with 10 μM L-glutamate and a compound of interest for 24 hours, followed by fixation and MAP-2 immunostaining as in previous experiments. As before, neuronal survival was quantified based on MAP-2 staining (figure 6). In the absence of a glutamate insult, 100 nM GT949 or GT996 treatment for 24h did not alter neuronal survival compared to the vehicle-treated baseline, suggesting that the increased dose of these compounds does not induce neurotoxicity. Cultures subjected to prolonged glutamate insult displayed significantly reduced survival, at 49±7% of baseline (figure 6, black bar). Neuronal survival was significantly increased to 80± 13% and to 98± 6% of baseline in cultures co-treated with glutamate and 10 nM or 100 nM GT949. Additionally, 100 nM GT949 produced a more significant effect on survival (##p<0.01, compared to insult), suggesting a dose response effect of the compound. In bilaminar cultures treated with glutamate and the inactive analog GT996, survival was 40 ± 16% of baseline, which was not significantly different from glutamate alone. These results suggest that GT949 is also capable of protecting neurons in a sustained excitotoxic environment, likely by increasing EAAT2-mediated uptake of glutamate.
Figure 6. Neuroprotective properties of GT949 in bilaminar cultures after prolonged exposure to glutamate (in the presence of glia).
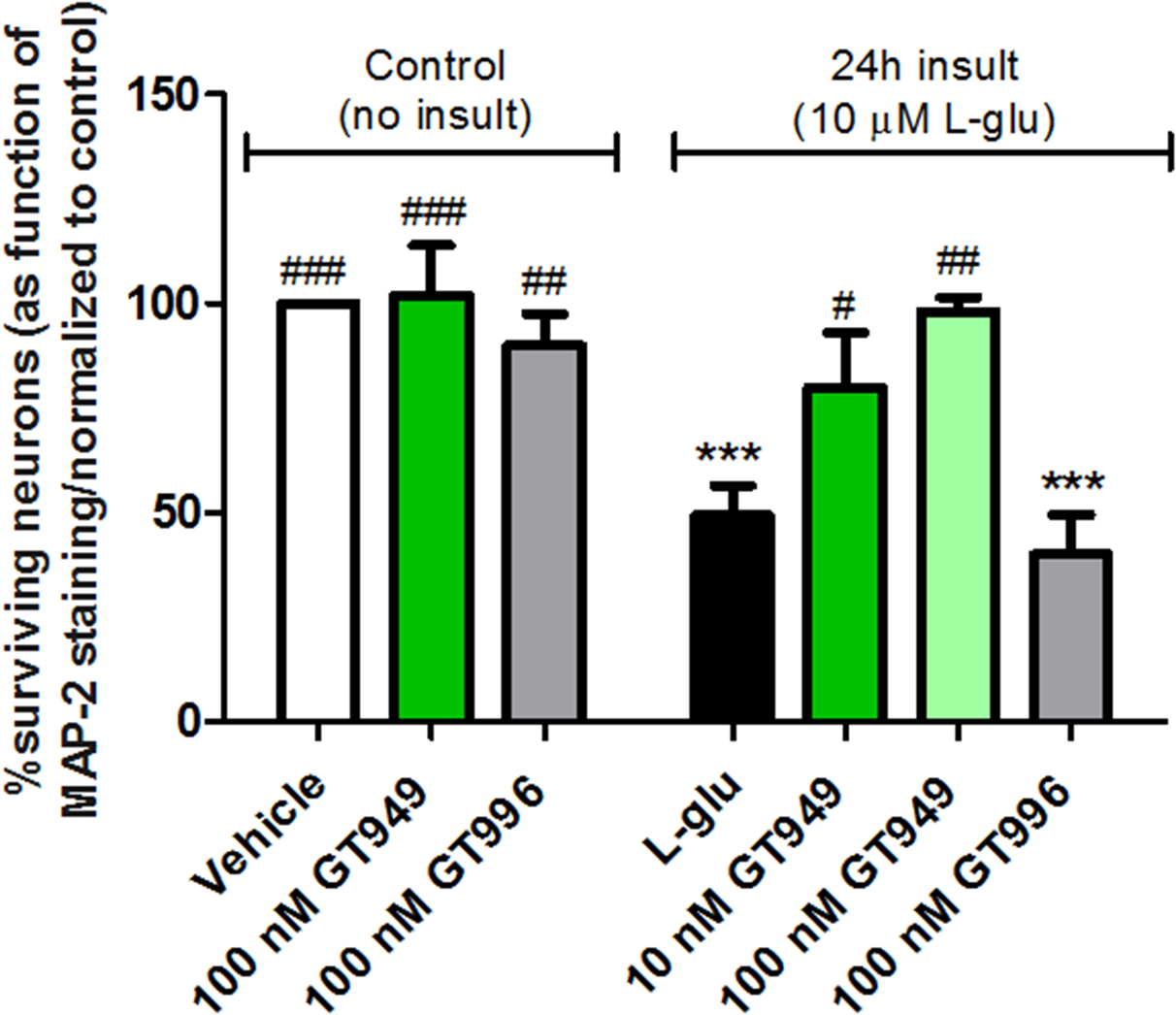
14 DIV bilaminar cultures were treated with glia-conditioned medium containing 10 μM L-glutamate and a compound of interest for 24 hours. After the treatment duration, neurons were fixed and immunostained for survival analysis. Again, treatment with 100 nM GT949 or GT996 alone did not alter neuronal survival. As expected, L-glutamate significantly decreased neuronal survival, and GT949 mitigated L-glutamate toxicity. The 100 nM dose of GT949 produced a more significant reversal of cell death than the 10 nM dose, suggesting a dose-response effect of this compound. Furthermore, the inactive GT996 has no effect on L-glutamate toxicity. Data was normalized to control cultures. 3–4 coverslips were assessed per treatment condition and 100–150 cells were manually counted per treatment. Neuronal survival data is representative of 3 independent experiments and control levels were not statistically different for normalization purposes. ***p<0.001, **p<0.01, vs. control (no insult), ##p<0.01, #p<0.05, vs. insult.
Neuronal survival and dendritic arborization after prolonged glutamate insult and GT949 treatment in mixed neuron-glia cultures
Since neurons and glia are grown on different layers in bilaminar cultures, we also wanted to examine GT949 neuroprotection in mixed neuron-glia cultures, where both cell types are in physical contact on the same layer. In these experiments, we evaluated neuronal survival with immunocytochemistry and MAP-2 ELISAs.
Mixed cultures in each treatment group were stained against the neuronal marker MAP-2 (green), the astrocyte marker GFAP (red), and the nuclear marker Hoechst (blue) (figure 7A). As in bilaminar cultures, mixed cultures with no glutamate insult or compound treatment (vehicle), as well as cultures treated with GT949 alone do not show damaged neurites or nuclei. Mixed cultures subjected to a prolonged 10 μM glutamate insult show neuronal loss, which is rescued by co-application of 10 nM GT949, suggesting a neuroprotective effect of the compound in these cultures.
Figure 7. Neuroprotective properties of GT949 in mixed neuron-glia cultures after prolonged exposure to glutamate.

A. Representative images of mixed neuron-glia cultures after treatments with 10 μM L-glutamate, and a compound of interest for 24 hours. Cells were fixed and immunostained for the neuronal marker MAP-2 (green) and the glial marker GFAP (red), and counterstaining with the nuclear marker DAPI (blue). Vehicle and GT949 treated cultures in the absence of L-glutamate do not display obvious degeneration of cell death. Cell death was evident in L-glutamate treated cultures, as assessed by the increased number of DAPI positive, MAP-2 and GFAP negative cells. Co-treatment with 100 nM GT949 reversed a portion of the L-glutamate toxicity by increasing MAP-2 and GFAP expression and reducing the number of DAPI-only cells. All images are shown at 40x magnification. Scale bar: 50 μm.
B: Quantification of neuronal survival in mixed cultures (normalized to control, vehicle-treated cultures). Treatment with 100 nM GT949, GT996, or 100 μM TBOA (glutamate transporter inhibitor) alone did not affect neuronal survival levels. L-glutamate significantly reduced neuronal survival, which was mitigated by co-treatment with GT949 and the NMDA antagonist AP-V. The 100 nM concentration of GT949 produced a more significant neuroprotective effect than the 10 nM concentration. Co-treatment with GT949 and the glutamate transporter inhibitor TBOA and the selective EAAT2 inhibitor WAY 23 213613 did not rescue neuronal damage in L-glutamate exposed cultures, suggesting that GT949 neuroprotection requires active glutamate transporters, specifically EAAT2. Co-treatment with inactive analog GT996 did not alter glutamate toxicity. 3–4 coverslips were assessed per treatment condition, and 100–150 cells were manually counted per treatment. Neuronal survival data is representative of 3 independent experiments and control levels were not statistically different for normalization purposes. ***p<0.001, *p<0.05, vs. control (no insult), ###p<0.001, ##p<0.01, vs. insult.
C. Representative images of neurons from 14 DIV mixed neuron-glia cultures under control conditions, incubated with 100 nM GT949 for 24 hours without insult, under 100 μM L-glutamate insult conditions for 24 hours, and with 100 μM L-glutamate co-applied with 100 nM GT949. Scale bar = 50μM. Sholl analysis on the right shows that glutamate insult resulted in decreased dendritic number and average dendritic length. Co-application with GT949 results in a trend in increasing the total number of dendrites, although it did not have effects on average length of dendrites. *p<0.05, vs. control (no insult), ##p<0.01, #p<0.05, vs. glutamate insult.
Next, neuronal survival was quantified in each treatment group using the immunostaining method (figure 7B). In the absence of glutamate, neuronal survival was not significantly altered after 24h incubation of 100 nM GT949 or GT996, or 100 μM DL-threo-β-benzyloxyaspartic acid (TBOA), a glutamate transport inhibitor, suggesting that these compounds are not neurotoxic at their respective doses. Cultures subjected to a 10 μM glutamate insult for 24h displayed significantly reduced neuronal survival, to 50±11% of baseline (figure 7B, black bar). However, co-treatment with 10 or 100 nM GT949 significantly increased survival levels to 78±11 % and 97±31% of baseline respectively, suggesting a neuroprotective dose response effect of GT949. To determine if the neuroprotective effect of GT949 requires an active glutamate transporter, we subjected mixed cultures to prolonged glutamate insult, followed by co-treatment with 100 nM GT949 and the glutamate transport inhibitor TBOA (100 μM), that inhibits all transporter subtypes at this concentration, and with the selective EAAT2 inhibitor WAY 213613 (1 μM). Neuronal survival in these cultures was 54±9 % of baseline in presence of TBOA, and 15±9% in presence of WAY 213613, which were not significantly different from glutamate alone, and suggests that the neuroprotective effect of GT949 requires active glutamate transporters, and specifically, EAAT2 activity. As expected, co-treatment with glutamate and the inactive analog GT996 (10 μM) did not affect survival (31±5% of baseline), while co-treatment with glutamate and the NMDA receptor antagonist AP-V (100 μM) significantly increased neuronal survival to 94±6% of baseline.
As a second measure for neuroprotection, quantification of dendritic arborization using Sholl analysis116 was performed, which includes counting the number of dendritic intersections that occur at fixed distances from the soma in concentric circles. This analysis reveals the number of branches, branch geometry, and overall branching patterns of neurons117. In this way, we evaluated changes to the dendritic arbor as a whole, after prolonged glutamate-mediated insults and GT949 treatment in mixed neuron-glia cultures.
The average dendrite length and dendrite number was decreased in response to glutamate insult (figure 7C). These results are in accordance to previous literature showing that glutamate-mediated excitotoxicity results in significant reduction in total dendrite output, which was accompanied by significant decrease in primary dendrite number and individual primary dendrite length118. Subsequent treatment with GT949 has no effect on restoring dendritic length but does restores dendritic number, as there are no significant differences between insult alone and insult with co-application of GT949. Additionally, treatment with 100 nM GT949 alone increases the number of dendrites.
We also performed MAP-2 based ELISAs as a surrogate approach to measure neuronal survival in mixed cultures (figure 8). A prolonged 100 μM glutamate insult significantly decreased MAP-2 expression to 43 ± 11% of baseline levels (figure 8, black bar), while co-treatment with 10 nM GT949 significantly increased MAP-2 expression to 86 ± 27% of baseline. Co-treatment with 10 nM GT996 resulted in survival at 52 ± 19% of baseline, which was not significantly different from glutamate alone. These results demonstrate that GT949 preserves neuronal survival and rescues expression of MAP-2 in cultures exposed to high levels of glutamate for an extended duration.
Figure 8. MAP-2 ELISA study of neuroprotective properties of GT949 in mixed neuron-glia cultures after prolonged exposure to glutamate.
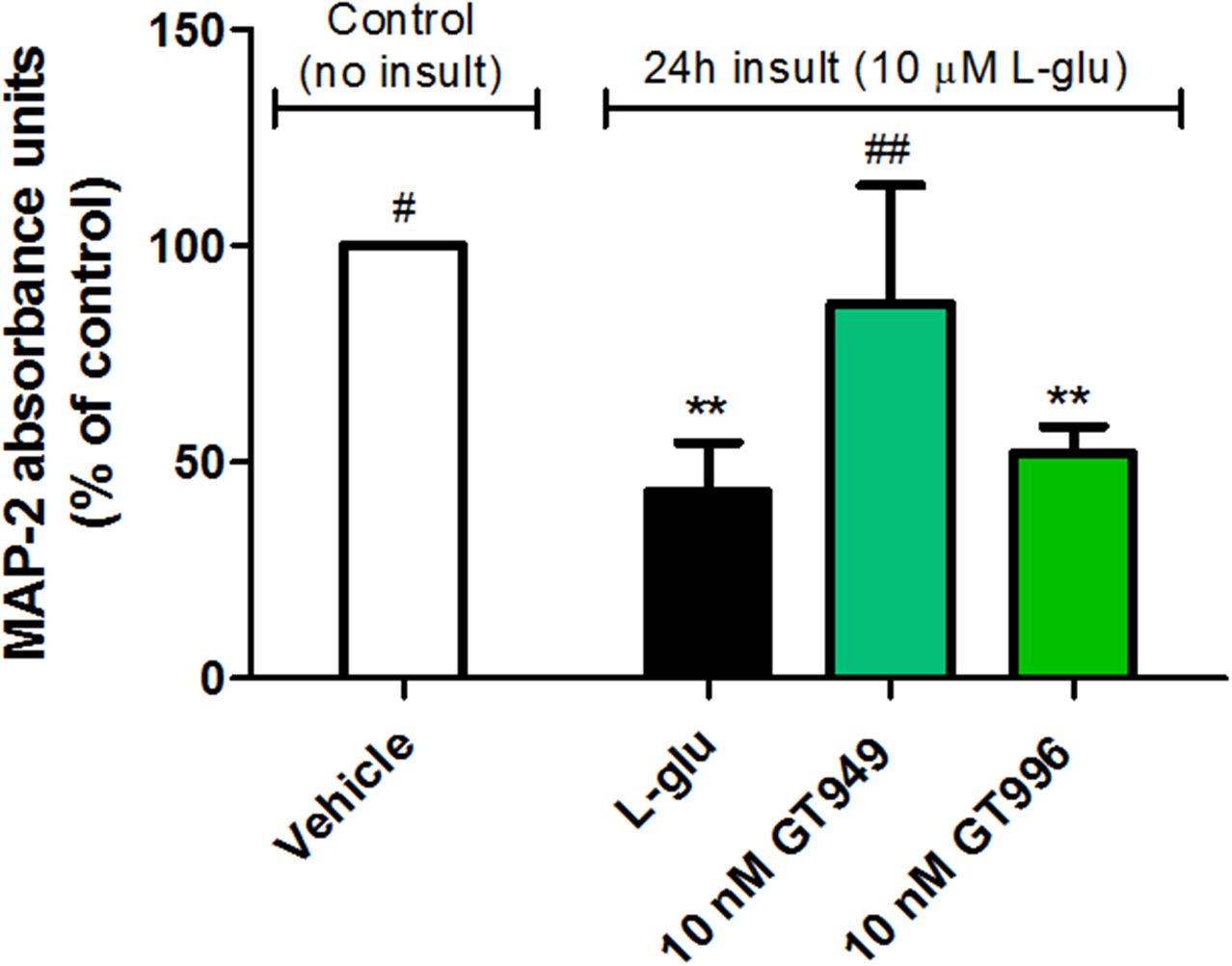
Mixed cultures were treated with 10 μM L-glutamate, and a compound of interest for 24 hours, followed by MAP-2 ELISA. L-glutamate exposure significantly reduced expression of MAP-2, as expected from previous experiments analyzing neuronal survival by MAP-2 immunostaining. Additionally, GT949 reversed the MAP-2 deficit in L-Glutamate exposed cultures, while the inactive analog GT996 had no effect. Data from at least 12 wells per treatment was plotted as a function of absorbance, which correlates to the amount of MAP-2 binding. Data was obtained from 3 independent experiments, and normalized to control values. **p<0.01, vs. control (no insult), ##p<0.01, #p<0.05, vs. insult.
In summary, we established that GT949 was neuroprotective after acute and prolonged glutamate insults in two different types of cortical neuron-glia cultures. This neuroprotective effect disappears when co-incubated with selective EAAT2 inhibitor WAY 213613 (figure 7B), indicating that the mechanism of GT949 clearly involves an active EAAT2 transporter. This suggests that increased glutamate clearance via EAAT2 activation can prevent excitotoxic damage and cell death.
The level of insult (concentration and time) results in varied levels of neuronal death, depending on the type of culture. In the bi-laminar system, the acute glutamate insult performed in the presence of glia (figure 1) required 10x higher concentrations of glutamate than in the absence of glia (figure 2) (i.e., 100 μM versus 10 μM) to result in the same level of neuronal death (~50%). This suggests a critical role of the glia in maintaining glutamate at physiological concentrations, most likely through glutamate transporter-mediated clearance. The same pattern was observed in the oxidative stress insults, when comparing the bilaminar system (figure 3), and in the mixed neuron-glia cultures (figure 4). The level of H2O2 insult had to be 10x higher in the mixed culture to reach the same level of insult as in the bilaminar culture. In the mixed neuron-glia cultures, intrinsic connections of the two different cell populations growing in the same layer makes this type of culture more physiological than the bi-laminar approach, where the two cell populations grow in different layers. However, we are very encouraged by the fact that we observe neuroprotective properties of GT949 in both approaches.
Strikingly, in the mixed neuron-glia culture system with ~15% of glia (supplemental figure 3) we were able to show neuroprotective properties of GT949 (figures 4, 7–8), suggesting that the neuroprotective effects of this class of compounds in vivo, where the proportion of glia to neurons is at least 1:1 and has been speculated to be as high as 10:1119, would be amplified.
Changes in dendritic arborization is as an important caveat as neuronal function is reliant upon synaptic connection. In this work, we observed that a glutamate insult decreased the dendrite length and number, in accordance to previous reports. there is good evidence that changes in dendritic and axonal morphology and properties take place in neurodegenerative disease in advance of neuronal loss118, 120 Subsequent treatment with GT949 showed a trend on increasing dendritic number. Additionally, treatment with 100 nM GT949 alone seems to have a beneficial effect on dendritic number, suggesting that GT949, in addition to rescue neuronal death, has effects on dendritic arborization and could modulate synaptic connection. Future studies are needed to further understand whether GT949 could function as a neuroprotective approach against progressive dendritic loss.
In this study, we subjected cultures to acute (20 min) and prolonged (24 hours) excitotoxic insults. Glutamate excitotoxicity plays a key role in secondary damage following acute pathologies, including TBI, epileptic seizures and stroke. Therefore, we propose that a compound like GT949, that has neuroprotective effects after acute insults in vitro, could be developed for the treatment of these pathologies, as long as the compound is administered within the window of opportunity, as the peak of glutamate excitotoxicity is known to be of short duration following the insults57, 121. Development of therapies for these pathologies is highly sought after, as, to date, no compelling efficacy data have been published regarding any pharmacological therapies for TBI and stroke122.
On the other hand, some CNS disorders are characterized by chronic excitotoxic events, such as ALS and Alzheimer’s disease46, 123. Because we observed a neuroprotective effect of GT949 after prolonged (24 hours) excitotoxic insults in cultures, we can also propose the development of a therapy for such disorders based on this compound. However, it is also known that the level of glutamate transporters can be downregulated in these chronic pathologies13, 124, therefore it is likely to be challenging that allosteric modulation of the glutamate transporter to be effective. In this sense, the use of transport expression enhancers (such as ceftriaxone37, 67–70 and clavulanic acid125–127) might be beneficial to restore transporter levels, and GT949 could be then used as adjuvant therapy to enhance transport activity levels and increase glutamate clearance.
The glutamate and oxidative stress insult investigated in this study are implicated in a wide range of CNS disorders, such as acute disorders like TBI128, epilepsy129 and chronic neurodegenerative disorders, like Alzheimer’s, Parkinson’s and Huntington’s, that involve neurotoxic aggregation of specific proteins in the brain130. Although the relationship between oxidative stress and development of protein aggregates is not completely established, it is speculated that simultaneously occurring glutamate toxicity, calcium imbalance, and mitochondrial impairment collectively intensify oxidative stress130. This cycle of biochemical distress in the brain might suggest that targeting the antioxidant defense may offer a better potential to limit these pathologies, rather than focusing on targeting glutamate excitotoxicity. On the other hand, some studies suggest a vicious cycle in neurodegeneration that includes glutamate excitotoxicity, oxidative stress, and mitochondrial dynamics. Therefore, possible neuroprotective strategies might include blockade of NMDA receptor activation, enhancement of SOD2 expression to clear mitochondrial ROS (reactive oxygen species), and inhibition of the downstream apoptotic cascade and positive modulation of glutamate transport46, 131, 132. Likewise, one might propose that intervention by preventing excitotoxicity through increased glutamate transport function could also break this cycle, as averting excitotoxicity would result in limiting the production of ROS if the treatment is offered at an appropriate time window. Determining the best time window for compound administration is out of the scope of these studies, as additional preclinical studies, drug development efforts and clinical trials are necessary to accurately determine this parameter. However, we suggest that the use of multiple therapies targeting these different components of the diseases’ pathology might offer potential beneficial pharmacological therapies in the future.
In this work, we focused on describing the neuroprotective effect of GT949 as associated to its actions on glutamate transporter EAAT2, but we also recognize the possibility that other mechanisms of neuroprotection could be involved. For instance, metabotropic glutamate receptors (mGluRs) can exert different actions (toxic or compensatory) in response to excitotoxic stimuli133.
Activation of group I mGluRs, which are located mostly on postsynaptic neurons, increase NMDA receptor activity and risk of excitotoxicity. Activation with agonist DHPG results in exacerbation of ischemic brain damage134, and agonist 1S,3R-ACPD induces cell death in rat striatum135. On the other hand, antagonist LY 367385 modulates presynaptic release, resulting in neuroprotection136. Activation of group II and III mGluRs, localized mostly in the presynapsis, decrease NMDA receptor activity and risk of excitotoxicity. For example, activation with selective agonists DCG-IV or L-AP4 inhibited apoptosis of primary cultured mesencephalic neurons137, and exerted neuroprotection on astrocytes via promoting glutamate uptake137, 138, and antagonists APICA or MSOP completely abolished their neuroprotective effects137. Acute or chronic exposure of astrocytes with selected mGluR II agonists attenuate glutamate release139. Moreover, glutamate transporter protein expression was shown to be regulated after stimulation with selected mGluR agonists in cultured human glial cells140, and conversely, there is evidence that astrocyte glutamate transporters regulate mGluR1-mediated excitation of hippocampal interneurons141. Collectively, these studies suggest that mGluRs are modulated by excitotoxic insults and can act as compensatory mechanism responding to elevated extracellular glutamate levels. Future studies will determine whether mGluRs modulation is involved in the action of GT949 on transporters.
Future studies will also investigate cell death mechanism in these models, if apoptotic in nature, ie., mediated by caspases or calpains142; or necrotic143. Additionaly, will also investigate intracellular pathways that play key roles after excitotoxic insults and treatments with glutamate transporter allosteric modulators. Possible pathways include ERK/12, that plays a crucial role in promoting cell death in neurodegenerative diseases144; p38, a potent regulator of stress-induced apoptosis112; Akt, one of the central kinases associated with mediating survival signaling145; and NF-kB, which ameliorates disease if inhibited in glia, and may enhance memory in neurons146.
CONCLUSIONS
In summary, our results suggest that this class of EAAT2 PAM compounds may be clinically useful for acute and chronic disorders in which glutamate excitotoxicity occurs, such as stroke, TBI and epilepsy, among others. However, we recognize that a combination of drugs targeting various aspects of neuroprotection, neuroinflammation and regeneration may be needed, as these are complex pathologies22, 147–149 Our results also suggest that we could avoid side effects that are typically associated with glutamate receptor antagonism, as our EAAT2 PAMs do not modulate the function of NMDA receptors89.
We aim to build from these initial studies by measuring glutamate concentration in culture media after insult and compound treatments, and determining the mechanism of cell death in our model of acute and prolonged glutamate insults. Additionally, we aim to determine the neuroprotective efficacy of GT949 and related compounds in other models of in vitro neurotoxicity, including NMDA-mediated insults, stretch injury (in vitro TBI150, 151), Mg2+ deprivation (in vitro epilepsy152) and oxygen-glucose deprivation (in vitro stroke36, 153). Further efforts will focus on making improved GT949 analogs with suitable drug-like properties and adequate pharmacokinetic properties for progression to in vivo studies, such as brain injury models. This will include determining the route of administration, therapeutic window, and safety in combination with other long-term treatment strategies.
Safe and effective pharmacological approaches to treat patients after excitotoxic events such as brain trauma are urgently needed121, 142, 154. Importantly, our EAAT2 PAM compounds provide a starting point for identification of a new class of neuroprotective compounds that function by enhancing the removal of excessive glutamate in the synaptic cleft and preventing glutamate-mediated excitotoxicity.36, 83, 155–157
MATERIALS AND METHODS
Compounds
The identification of allosteric modulators (AMs) of glutamate transporters has been described previously89. Compounds GT949 (positive allosteric modulator of EAAT2) and GT996 (inactive compound, used as negative control) were synthetized by the Ugi multi-component reaction158–161 using commercially available starting materials. Compounds were dissolved to 10 mM stocks in Dimethyl sulfoxide (DMSO), the maximum concentration of DMSO in the vehicle experiments was 0.01%.
L-glutamate, (2R)-amino-5-phosphonovaleric acid (AP-V), DL-threo-β-Benzyloxyaspartic acid (TBOA) and WAY 213613 were purchased from Tocris (Bristol, United Kingdom). Hydrogen peroxide solution (H2O2) was purchased from Sigma-Aldrich (St. Louis, MO).
Cell culture reagents and media
Cell culture media including high-glucose Dulbecco’s Modified Eagle Medium (DMEM) and Neurobasal medium with pyruvate, glutamine, gentamicin, HEPES (1 M), B27 and N2 supplements were purchased from Gibco (Waltham, MA, USA).
Poly-L-lysine, ovalbumin, bovine serum albumin, DNase and Cytosine-β-D-arabinofuranoside were purchased from Sigma-Aldrich (St. Louis, MO).
Fetal bovine serum and horse serum were purchase from Hyclone (South Logan, UT, USA).
Poly-lysine coated 96-well plates and phosphate-buffered saline (D-PBS) were purchased from Corning (Corning, NY, USA).
Tools for animal dissection was purchased from Biomedical Research Instruments (Silver Spring, MD, USA).
Antibodies
Primary antibodies for immunocytochemistry included polyclonal anti-microtubule associated protein-2 (MAP-2, cat: AB5622) and monoclonal anti-glial fibrillary acidic protein (GFAP, cat: MAB360), both purchased from Millipore (Temecula, CA, USA).
Secondary antibodies used for immunocytochemistry included Alexa Fluor® 488 conjugated AffiniPure Donkey Anti-Rabbit IgG and Cy™3-conjugated AffiniPure Donkey Anti-Mouse IgG. These antibodies, along with normal donkey serum and normal goat serum, were purchased from Jackson Immuno Research (West Grove, PA, USA).
Antibodies used for ELISA were MAP-2 (monoclonal, cat: 13–1500) and Goat anti-Mouse IgG (H+L) Secondary Antibody (HRP conjugate) from Thermo Fisher Scientific (Waltham, MA, USA).
Primary antibodies for western blotting EAAT1 Rabbit mAb (1:100, cat: 5684), EAAT3 Rabbit mAb (1: 100, cat: 14501) and β-actin mouse mAb (1:1,000), were purchased from Cell Signaling (Danvers, MA, USA). EAAT2 rabbit polyclonal (1: 1,000, cat: AGC-022) was purchased from Alomone, Jerusalem, Israel. Infrared-conjugated secondary antibodies [(Anti-rabbit DyLight 800 (Green) and Anti-mouse DyLight 680 (Red), both at 1:1,000)] were used for imaging using LI-COR Image Studio, version 4.0.21 (LI-COR Biosciences; Lincoln, NE, USA).
Animals
HsdHot: Holtzmann rats were purchased from Charles River (Malvern, PA, USA), and housed in Association for Assessment and Accreditation of Laboratory Animal Care-accredited facilities. Animal housing and experimental procedures were also approved by the Drexel University College of Medicine Institutional Animal Care and Use Committees (IACUC).
Primary cultures
These studies utilized two different in vitro primary neuronal cultures: a bilaminar co-culture of neurons and glia, in which neurons and glia grow in different layers; and mixed neuron-glia cultures, in which these cells grow together in the same layer.
Bilaminar co-culture of primary rat cortical neurons and glia
Bilaminar cultures were prepared as previously described91, 92, 162. Briefly, cortical neurons were obtained from late embryonic stage (E17) Holtzman rats. For immunocytochemistry, neurons were plated on poly-L-lysine-coated glass coverslips (15-mm diameter) at a density of 35,000 cells/coverslip. After 3 hours in plating medium, 3 to 5 neuronal coverslips were transferred to a single 60 mm culture dish containing an astroglia-enriched secondary culture of mixed glia. Glial cultures were prepared using cortices of newborn Holtzman rat pups (P2–4) prior to the neuronal dissection, as previously reported163. For protein isolation and western blots, neurons were plated in 60mm dishes at a density of 1×106 cells/dish. Primary glia plated on Thermanox sheets were added to the neuronal culture dish 4 hours after plating, facing the neuronal layer92.
Cytosine arabinofuranoside (AraC) was added to the cultures 24–48 h after plating to halt non-neuronal cell proliferation (2 μM for cultures with neuronal coverslips, and 10 μM for cultures with glial Thermanox coverslips). The bilaminar culture was grown for 14 DIV in a 37°C incubator (5 to 10% CO2). On day 7, 25% of the media was replaced by fresh media. The osmolarity of the culture media was assessed every 5 days, and adjusted to 300–310 mOsm if necessary. In parallel, 60 mm dishes containing only the astrocyte-enriched layer were cultured as above to generate conditioned media for further use.
In this preparation, ~97% of viable cells in these cultures are neurons (Figure 1 of supplemental material).
Mixed neuron-glia cultures
As a second model for validation of the effect of the compounds, we used mixed neuron-glia cultures, in which neurons and glia cells are grown in the same layer. Cortical cells were obtained from late embryonic stage (E17) Holtzman rats. These cells were plated at a density of 35,000 cells/coverslip in 12-well plates for immunocytochemistry assays, and 10,000 cells/well in poly-D-lysine pre-coated 96-well plates for ELISA assays, as previously described164–166, with modifications.
We did not add AraC to mixed neuron-glia cultures. Culture conditions were optimized to contain enough glia to observe suitable expression of EAAT2 and treatment effects with GT949, while maintaining a population of neurons that is large enough to accurately measure neuronal survival. This was achieved by plating cells in Neurobasal media (NB) with 2% B27 supplement and 10% Horse Serum. The media was changed 2 h after plating to NB media containing 2% B27, 2% Horse serum, 1% glutamine, 25 μM glutamate and 50μg/mL gentamicin. 4 days later, media was changed to NB media with 2% B27, 1% glutamine and 50μg/mL gentamicin. Finally, cells received fresh NB media (with 2% B27, 1% glutamine and 50μg/mL gentamicin) at 8 DIV, and cultures were exposed to excitotoxic insults and compound treatment at 14 DIV. These growth conditions produced approximately 15% glia in each culture (supplemental figure 3). This level of glia allowed for sufficient EAAT2 expression to observe an effect of GT949, while maintaining a large enough population of neurons for the quantification of MAP-2 in culture. Only cultures with consistent percentages of astrocytic populations across individual experiments and coverslips were considered. Any culture with a proportion of glia that was significantly different from a range of 7–18% (measured by One-Way ANOVA followed by Newman-Keuls posthoc test for multiple comparisons performed on the percentage of GFAP+ in each coverslip) was excluded from the analysis.
Western Blotting
Expression of glutamate transporters EAAT1–3 were analyzed in both glia and neurons from bilaminar cultures. Cell lysates were prepared from secondary glia or primary neurons grown in 60mm dishes for 14 DIV. After lysis, collection and centrifugation, protein quantification was performed by using a Pierce BCA Protein Assay Kit. Samples (40 μg protein/10μL/lane) were subjected to SDS-PAGE using NuPAGE 4–12% Bis-Tris Mini Gels. Primary antibodies for EAAT1–3 are listed above, which were incubated overnight on a rocker at 4°C. The following day, primary antibodies were removed and membranes were washed 3 times at 10 min each with PBS containing 0.05% Tween (PBS-T). After the final wash, infrared-conjugated secondary antibodies (listed above) were added and blots were placed on the rocker for 60 min at room temperature. After 3 additional washes with PBS-T, imaging and band density measurements were performed using the Odyssey imaging system (LI-COR Image Studio, version 4.0.21).”
Additionally, mixed neuron-glia cultures were plated on 6 well plates, and at 14 DIV exposed to oxidative stress insults by incubation with 10, 100 and 500 μM H2O2 for 20 min, and 24 hours later subjected to SDS- PAGE and probed with EAAT2 and actin antibodies, as described above.
In vitro excitotoxic insults (glutamate and oxidative stress) and compound administration
All insults were performed in well-differentiated cultures (14 DIV), to ensure that protein content and mitochondrial respiration were stabilized104, 167. A report shows that 14 DIV cultures also express markers of neuronal maturity, such as NeuN; NF-L; NF-M; GAD; TH; PSD-95 and synaptophysin168. All cultures were examined before each experiment for correct osmolarity and cell viability under a TCM-BR inverted brightfield microscope. Cultures that appeared unhealthy were discarded from further experiments. Insult solutions for bilaminar cultures were prepared in glia-conditioned media (collected from a glia feeder), to assure that neurons were supplied with all required factors throughout the experiment. Glutamate and compound solutions were made in fresh growth media specific for each culture.
Cultures were subjected to three types of insults, as described below.
Acute glutamate insults
Cell cultures were exposed to glutamate (10 or 100 μM), or vehicle for 20 minutes. Minor modifications were performed according to type of culture, as follows:
Bilaminar cultures in the absence of glia: neuronal coverslips were separated from the glial feeder layer and transferred to dishes containing insult solutions (10, 50 and 100 μM L-glutamate or vehicle) for 20 min. Subsequently, coverslips were returned to their original culture dishes containing the glial feeder layer and either 10 nM or 100 nM GT949 (PAM of EAAT2), GT996 (inactive analog, used as negative control), 100 μM AP-V (NMDA receptor antagonist, used as positive control) or vehicle, which were subsequently incubated for 24h until neuronal survival analysis. Previous results indicated that GT949 had an EC50 of ~1 nM89 for the effect on glutamate transport augmentation, so we tested the compound at 10 and 100X higher concentrations to ensure that the transporters were fully stimulated.
Bilaminar cultures in the presence of glia: conditioned media containing the insult solutions (L-glutamate at 10, 50 or 100 μM) or vehicle were added to dishes with neuronal coverslips and glial feeding layer. After incubation for 20 min, solutions were replaced with conditioned media containing compounds GT949, GT996, AP-V (at the same concentrations as above) or vehicle. Cultures were incubated for 24 hours, followed by downstream analyses.
Mixed neuron-glia cultures: L-glutamate (100 μM and 1mM) were directly added to the mixed cultures for 20 min. Since these insults did not result in significant levels of neuronal death (not shown), these assays were excluded from this study.
Prolonged glutamate insults
Cultures were exposed to L-glutamate (10 or 100μM, or vehicle), for 24h. Minor modifications were performed according to type of culture, as follows:
Bilaminar cultures: cultures containing neuronal coverslips and the glial feeder layer were randomly exposed to L-glutamate (10 μM or 100 μM), or vehicle in the presence of compounds (10 or 100 nM of GT949 or 100 nM GT996) or vehicle, prepared in conditioned media, for 24h.
Mixed neuron-glia cultures: L-glutamate (10 or 100μM) or vehicle was directly added to mixed cultures. Compounds GT949 or GT996 (10 nM or 100 nM), or AP-V (100 μM), were co-applied and allowed to incubate for 24h. As we also aimed to validate that the GT949 neuroprotective effect was mediated by modulation of glutamate transport, we also co-applied glutamate transport inhibitor TBOA (100 μM) and selective EAAT2 inhibitor WAY 213613 (1 μM) for 24 hours.
Additionally, to test for any possible toxicity of the experimental compounds, cultures were treated with only GT949 or GT996 (100 nM), for 24 hours.
Oxidative stress insults model of excitotoxicity
To mimic an oxidative stress model of excitotoxicity111, H2O2 was applied to bilaminar cultures in presence of glia feeder or mixed neuron-glia cultures for 20 min, as described above. Briefly, H2O2 (10 μM, 50 μM or 100 μM, for bilaminar and 100 μM for mixed neuron-glia cultures) prepared in conditioned media was added for 20 min, then removed, and compounds and positive controls were added and cells incubated for 24 hours.
Acute insult with H2O2 performed in the absence of the glia-feeding layer and prolonged insults in presence of the glia-feeding layer resulted in high levels of neuronal death (data not shown); therefore, these assays were excluded from this study.
Immunocytochemistry assays for evaluation of neuronal survival
To evaluate neuronal health status (as a function of MAP-2) and the effect of treatment with the compounds, cultures on 15 mm glass coverslips (from both bilaminar and mixed approaches) were fixed and fluorescently labeled as described90, 169, 170. As a control for initial levels of neuronal survival, neurons not subjected to treatments (time zero) were also examined. Briefly, cultures on coverslips were fixed with 2% paraformaldehyde, permeabilized with 0.1% Triton X-100, blocked with 5% Normal Donkey serum, washed and incubated overnight with primary antibodies in 5% blocking solution: anti-MAP-2 (1:1,000) and anti-GFAP (1: 400). The following secondary antibodies were used: Alexa Fluor® 488 conjugated AffiniPure Donkey Anti-Rabbit IgG for MAP-2 detection and Cy™3-conjugated AffiniPure Donkey Anti-Mouse IgG for GFAP, both at 1:500, prepared in blocking solution (5% normal donkey serum in PBS). After 30 min incubation in the dark, coverslips were washed and mounted on a slide with ProLong® Diamond Antifade Mountant with DAPI for nuclear staining. Coverslips were then sealed with clear nail polish.
Image acquisition
Cells were imaged with a fluorescent microscope to examine morphology and a confocal microscope to quantify cell survival. For the fluorescent microscope, we used the following settings: quality: 1280×1024 16-bit, 2.4x analog gain, and dynamic contrast. The exposure was adjusted between 400ms and 2s based on which time provided optimal fluorescence. For the confocal microscope, we used the following settings: quality: 1280×1024, 2.4x analog gain, and dynamic contrast. The exposure was set at 8 seconds and images were taken using the 30x zoom setting. For all experiments, 5–10 random fields/coverslip and 3–4 coverslips were evaluated per treatment condition. Images were taken in all 3 filters to visualize DAPI, Alexa-488 (for MAP-2), and CY-3 (for GFAP). For assessment of cell population, cells in the cultures were classified into neurons (MAP-2 positive cells) and glia (GFAP positive cells). The abundance of each cell type was calculated and expressed as percent of total, DAPI positive cells on a coverslip. In bilaminar cultures, ~97% of cells in neuronal layers were neurons, but a small percentage of glia and other cells were identified (DAPI positive, MAP-2 negative and GFAP negative) (supplemental figure 1). For mixed neuron-glia cultures, roughly 70% of cells were neurons, and 15% of the cells were glia (supplemental figure 3).
Neuronal survival counting and experimental blinding
Neuronal survival was evaluated by individuals blinded to the treatment conditions. Survival was determined by evaluating individual neurons for features that are indicative of neurodegeneration and apoptosis, such as pyknotic nuclei, membrane blebbing, and neuritis171, 172. Nuclei that stained with DAPI but lacked significant MAP-2 staining were also classified as dead neurons. The percentage of neuronal survival was calculated by dividing the number of MAP-2 positive neurons with no morphological signs of toxicity, by the total number of MAP-2 positive neurons on each coverslip.
For each experimental condition, 5 random fields from 3–4 coverslips were analyzed, totaling at least 100–150 neurons. Mean survival in each treatment condition was normalized to mean survival in the control group for each experiment. The normalization was performed considering the control group of each individual experiment as 100% of survival and obtaining the relative percentage of neuronal survival of each condition/treatment. Additionally, neuronal survival was evaluated before all insults (time zero) in at least 4 control coverslips, before beginning each experiment. Only sets of experiments with reasonable survival rates at time zero (>75%) were included in the final analysis of neuronal survival.
Images were processed using ImageJ® from the National Institute of Health (Bethesda, MD, USA).
Dendritic arborization analysis
As a second measure for neuroprotection, we quantified arborization, as alterations in dendritic shapes have been shown to be aberrant during disease or injury117, 173. Neurite arborization analysis was performed in mixed neuron-glia cultures subjected to control (no insult) and to acute and prolonged (??) L-glutamate insult (in presence and absence of 100nM GT949) conditions. Images were analyzed using the Neurite Tracer and Scholl Analysis plug-ins for Image-J (NIH, Bethesda, MD). 6–10 single neurons from separate images for each condition were isolated and then traced using the Neurite Tracer plug-in and were then processed by Scholl analysis116 to analyze neurite length and branching.
Immunocytochemistry assays for evaluation of toxicity of compounds and insults in pure glia cultures
To evaluate potential toxicity of compound GT949 and oxidative stress insult in pure astrocyte cultures, glia cultures were plated in coverslips, as described above. After 14 DIV, cultures were incubated with increasing concentrations of GT949 (1μM - 100μM) or H2O2 for 24h. Then coverslips were fixed, fluorescently labeled as described above, and subsequent morphological and quantification analysis of GFAP-positive cells were performed.
We also analyzed toxicity of the oxidative stress insult per se, as previous literature suggested that this type could result in glial cell death109, 114, 115. Pure glial cultures were subjected to the H2O2 insults as described above. Following the 20 min insult, media was changed to fresh glia media and incubated for 24h. Cultures were then fixed and immunocytochemistry was performed to analyze glial morphology and survival as described above.
ELISA assays
Mixed neuron-glia cultures plated in 96-well plates were subjected to cell-based MAP-2 ELISAs to evaluate neuronal survival. Briefly, after insult and compound treatments, plates were washed with phosphate buffered saline (PBS) using a plate washer, and fixed with 4% paraformaldehyde. The cell membranes were then permeabilized with 0.1% Triton-X, and non-specific antibody binding was blocked with 10% normal goat serum. Cells were then incubated with MAP-2 primary antibody (1:1,000) overnight. Primary antibody was omitted from the top two rows on each plate to allow an evaluation of the background (non-specific antibody binding). After washing, the HRP-conjugated secondary antibody (1:500 in 10% Goat Serum) was incubated for 30 min in at 37°C, and TMB substrate was added to the plates to detect horseradish peroxidase activity, yielding a blue color (Amax = 370nm and 652nm) that changes to yellow (Amax = 450nm) upon addition of 2M Sulfuric Acid solution. Absorbance was measured with a spectrophotometer at wavelengths of 450nm and 550nm. Data from at least 12 wells per treatment was plotted as a function of absorbance, which correlates to the amount of MAP-2 binding. Background (obtained in absence of the addition of primary antibody) was subtracted from the total.
Data analysis
All data were analyzed using GraphPad Prism®, version 5.0 for PC (GraphPad Software, La Jolla, CA, USA). Graphs represent average ± SEM of at least three independent experiments performed in triplicate.
Statistical significance was assessed using One-way analysis of variance (ANOVA) followed by Dunnett’s multiple-comparisons posthoc test with vehicle as control (* p <0.05), or Newman-Keuls posthoc test for multiple comparisons (# p <0.05).
Supplementary Material
Acknowledgments
The authors wish to thank Bradley Nash and Lindsay Festa in the Meucci Lab for assistance with primary culture techniques, Bradley Nash for the revision of the manuscript, and Ole. V. Mortensen for helpful discussions (all from Department of Pharmacology and Physiology, Drexel University).
Funding sources
This work was supported by Drexel University’s startup funds to ACKF, and NIH grants to OM (DA15014 and DA32444). This project is also funded, in part, under a grant with the Pennsylvania Department of Health to J.M.S. The Department specifically disclaims responsibility for any analyses, interpretations or conclusions. We would also like to acknowledge the Brazilian Scientific mobility program sponsored by CAPES & CNPQ for providing financial support to R.M.F. (Grant ID 15149399).
Abbreviations used:
- AD
Alzheimer’s disease
- ALS
Amyotrophic Lateral Sclerosis
- AMPA
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- AP-V
(2R)-amino-5-phosphonovaleric acid
- AraC
Cytosine-β−D-arabinofuranoside
- CNS
Central nervous System
- DAPI
4’,6-Diamidino-2-Phenylindole
- DIV
Days in vitro
- DL-TBOA
DL- threo-β-Benzyloxyaspartic acid
- EAAT1–5
Excitatory Amino Acid Transporters 1–5
- ELISA
Enzyme-Linked Immunosorbent Assay
- GFAP
Glial Fibrillary Acidic Protein
- GltPh
Pyrococcus horikoshii glutamate transporter
- HRP
horseradish peroxidase
- HS
horse serum
- MAP-2
Microtubule-Associated Protein 2
- NB media
Neurobasal media
- NMDA
N-methyl-D-aspartate
- OGD
Oxygen-glucose deprivation
- PAM
Positive allosteric Modulator
- PBS
Phosphate Buffer Saline
- SDS-PAGE
sodium dodecyl sulfate–polyacrylamide gel electrophoresis
- TBI
Traumatic Brain Injury
- TBOA
DL-threo-β-Benzyloxyaspartic acid
- TMB
−3,3’,5,5’-Tetramethylbenzidine, chromogenic substrate for HRP detection in ELISA
- WAY 213613
N-[4-(2-Bromo-4,5-difluorophenoxy)phenyl]-L-asparagine
Footnotes
Conflict of interests
Compounds GT949 and GT996 are protected under a patent application submitted by Drexel University to USPTO (WO application WO2018132829A1) and includes named authors J.M.S. and A.C.K.F.
References
- 1.McEntee WJ, and Crook TH (1993) Glutamate: its role in learning, memory, and the aging brain, Psychopharmacology 111, 391–401. [DOI] [PubMed] [Google Scholar]
- 2.Weiler IJ, Hawrylak N, and Greenough WT (1995) Morphogenesis in memory formation: synaptic and cellular mechanisms, Behavioural brain research 66, 1–6. [DOI] [PubMed] [Google Scholar]
- 3.Peng S, Zhang Y, Zhang J, Wang H, and Ren B (2011) Glutamate receptors and signal transduction in learning and memory, Molecular biology reports 38, 453–460. [DOI] [PubMed] [Google Scholar]
- 4.Danbolt NC (2001) Glutamate uptake, Progress in neurobiology 65, 1–105. [DOI] [PubMed] [Google Scholar]
- 5.Beart PM, and O’Shea RD (2007) Transporters for L-glutamate: an update on their molecular pharmacology and pathological involvement, British journal of pharmacology 150, 5–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vandenberg RJ, and Ryan RM (2013) Mechanisms of glutamate transport, Physiological reviews 93, 1621–1657. [DOI] [PubMed] [Google Scholar]
- 7.Amato A, Barbour B, Szatkowski M, and Attwell D (1994) Counter-transport of potassium by the glutamate uptake carrier in glial cells isolated from the tiger salamander retina, The Journal of physiology 479 (Pt 3), 371–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zerangue N, and Kavanaugh MP (1996) Flux coupling in a neuronal glutamate transporter, Nature 383, 634–637. [DOI] [PubMed] [Google Scholar]
- 9.Amara SG, and Fontana AC (2002) Excitatory amino acid transporters: keeping up with glutamate, Neurochemistry international 41, 313–318. [DOI] [PubMed] [Google Scholar]
- 10.Maragakis NJ, Dykes-Hoberg M, and Rothstein JD (2004) Altered expression of the glutamate transporter EAAT2b in neurological disease, Annals of neurology 55, 469–477. [DOI] [PubMed] [Google Scholar]
- 11.Sheldon AL, and Robinson MB (2007) The role of glutamate transporters in neurodegenerative diseases and potential opportunities for intervention, Neurochemistry international 51, 333–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lauriat TL, and McInnes LA (2007) EAAT2 regulation and splicing: relevance to psychiatric and neurological disorders, Molecular psychiatry 12, 1065–1078. [DOI] [PubMed] [Google Scholar]
- 13.Kim K, Lee SG, Kegelman TP, Su ZZ, Das SK, Dash R, Dasgupta S, Barral PM, Hedvat M, Diaz P, Reed JC, Stebbins JL, Pellecchia M, Sarkar D, and Fisher PB (2011) Role of excitatory amino acid transporter-2 (EAAT2) and glutamate in neurodegeneration: opportunities for developing novel therapeutics, Journal of cellular physiology 226, 2484–2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pines G, Danbolt NC, Bjoras M, Zhang Y, Bendahan A, Eide L, Koepsell H, Storm-Mathisen J, Seeberg E, and Kanner BI (1992) Cloning and expression of a rat brain L-glutamate transporter, Nature 360, 464–467. [DOI] [PubMed] [Google Scholar]
- 15.Haugeto O, Ullensvang K, Levy LM, Chaudhry FA, Honore T, Nielsen M, Lehre KP, and Danbolt NC (1996) Brain glutamate transporter proteins form homomultimers, The Journal of biological chemistry 271, 27715–27722. [DOI] [PubMed] [Google Scholar]
- 16.Tanaka K, Watase K, Manabe T, Yamada K, Watanabe M, Takahashi K, Iwama H, Nishikawa T, Ichihara N, Kikuchi T, Okuyama S, Kawashima N, Hori S, Takimoto M, and Wada K (1997) Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1, Science 276, 1699–1702. [DOI] [PubMed] [Google Scholar]
- 17.Lehre KP, and Danbolt NC (1998) The number of glutamate transporter subtype molecules at glutamatergic synapses: chemical and stereological quantification in young adult rat brain, The Journal of neuroscience : the official journal of the Society for Neuroscience 18, 8751–8757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Suchak SK, Baloyianni NV, Perkinton MS, Williams RJ, Meldrum BS, and Rattray M (2003) The ‘glial’ glutamate transporter, EAAT2 (Glt-1) accounts for high affinity glutamate uptake into adult rodent nerve endings, Journal of neurochemistry 84, 522–532. [DOI] [PubMed] [Google Scholar]
- 19.Scofield MD, and Kalivas PW (2014) Astrocytic dysfunction and addiction: consequences of impaired glutamate homeostasis, The Neuroscientist : a review journal bringing neurobiology, neurology and psychiatry 20, 610–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou Y, and Danbolt NC (2013) GABA and Glutamate Transporters in Brain, Frontiers in endocrinology 4, 165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nilsson E, Alafuzoff I, Blennow K, Blomgren K, Hall CM, Janson I, Karlsson I, Wallin A, Gottfries CG, and Karlsson JO (1990) Calpain and calpastatin in normal and Alzheimer-degenerated human brain tissue, Neurobiology of aging 11, 425–431. [DOI] [PubMed] [Google Scholar]
- 22.Raghupathi R (2004) Cell death mechanisms following traumatic brain injury, Brain Pathol 14, 215–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sattler R, and Tymianski M (2001) Molecular mechanisms of glutamate receptor-mediated excitotoxic neuronal cell death, Molecular neurobiology 24, 107–129. [DOI] [PubMed] [Google Scholar]
- 24.Choi DW, Maulucci-Gedde M, and Kriegstein AR (1987) Glutamate neurotoxicity in cortical cell culture, The Journal of neuroscience : the official journal of the Society for Neuroscience 7, 357–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Palmer AM, Marion DW, Botscheller ML, Swedlow PE, Styren SD, and DeKosky ST (1993) Traumatic brain injury-induced excitotoxicity assessed in a controlled cortical impact model, Journal of neurochemistry 61, 2015–2024. [DOI] [PubMed] [Google Scholar]
- 26.Brown JI, Baker AJ, Konasiewicz SJ, and Moulton RJ (1998) Clinical significance of CSF glutamate concentrations following severe traumatic brain injury in humans, Journal of neurotrauma 15, 253–263. [DOI] [PubMed] [Google Scholar]
- 27.Vespa P, Prins M, Ronne-Engstrom E, Caron M, Shalmon E, Hovda DA, Martin NA, and Becker DP (1998) Increase in extracellular glutamate caused by reduced cerebral perfusion pressure and seizures after human traumatic brain injury: a microdialysis study, Journal of neurosurgery 89, 971–982. [DOI] [PubMed] [Google Scholar]
- 28.Baker AJ, Moulton RJ, MacMillan VH, and Shedden PM (1993) Excitatory amino acids in cerebrospinal fluid following traumatic brain injury in humans, Journal of neurosurgery 79, 369–372. [DOI] [PubMed] [Google Scholar]
- 29.Nilsson P, Hillered L, Ponten U, and Ungerstedt U (1990) Changes in cortical extracellular levels of energy-related metabolites and amino acids following concussive brain injury in rats, Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism 10, 631–637. [DOI] [PubMed] [Google Scholar]
- 30.Yamamoto T, Rossi S, Stiefel M, Doppenberg E, Zauner A, Bullock R, and Marmarou A (1999) CSF and ECF glutamate concentrations in head injured patients, Acta neurochirurgica. Supplement 75, 17–19. [DOI] [PubMed] [Google Scholar]
- 31.Matute C, Domercq M, and Sanchez-Gomez MV (2006) Glutamate-mediated glial injury: mechanisms and clinical importance, Glia 53, 212–224. [DOI] [PubMed] [Google Scholar]
- 32.Seal RP, and Amara SG (1999) Excitatory amino acid transporters: a family in flux, Annual review of pharmacology and toxicology 39, 431–456. [DOI] [PubMed] [Google Scholar]
- 33.Jabs R, Seifert G, and Steinhauser C (2008) Astrocytic function and its alteration in the epileptic brain, Epilepsia 49 Suppl 2, 3–12. [DOI] [PubMed] [Google Scholar]
- 34.Yatomi Y, Tanaka R, Shimura H, Miyamoto N, Yamashiro K, Takanashi M, Urabe T, and Hattori N (2013) Chronic brain ischemia induces the expression of glial glutamate transporter EAAT2 in subcortical white matter, Neuroscience. [DOI] [PubMed] [Google Scholar]
- 35.Hazell AS (2007) Excitotoxic mechanisms in stroke: an update of concepts and treatment strategies, Neurochemistry international 50, 941–953. [DOI] [PubMed] [Google Scholar]
- 36.Lipski J, Wan CK, Bai JZ, Pi R, Li D, and Donnelly D (2007) Neuroprotective potential of ceftriaxone in in vitro models of stroke, Neuroscience 146, 617–629. [DOI] [PubMed] [Google Scholar]
- 37.Chu K, Lee ST, Sinn DI, Ko SY, Kim EH, Kim JM, Kim SJ, Park DK, Jung KH, Song EC, Lee SK, Kim M, and Roh JK (2007) Pharmacological Induction of Ischemic Tolerance by Glutamate Transporter-1 (EAAT2) Upregulation, Stroke; a journal of cerebral circulation 38, 177–182. [DOI] [PubMed] [Google Scholar]
- 38.Wang Y, and Qin ZH (2010) Molecular and cellular mechanisms of excitotoxic neuronal death, Apoptosis : an international journal on programmed cell death 15, 1382–1402. [DOI] [PubMed] [Google Scholar]
- 39.Coutinho-Netto J, Abdul-Ghani AS, Collins JF, and Bradford HF (1981) Is glutamate a trigger factor in epileptic hyperactivity?, Epilepsia 22, 289–296. [DOI] [PubMed] [Google Scholar]
- 40.Louzada-Junior P, Dias JJ, Santos WF, Lachat JJ, Bradford HF, and Coutinho-Netto J (1992) Glutamate release in experimental ischaemia of the retina: an approach using microdialysis, Journal of neurochemistry 59, 358–363. [DOI] [PubMed] [Google Scholar]
- 41.Sisk DR, and Kuwabara T (1985) Histologic changes in the inner retina of albino rats following intravitreal injection of monosodium L-glutamate, Graefe’s archive for clinical and experimental ophthalmology = Albrecht von Graefes Archiv fur klinische und experimentelle Ophthalmologie 223, 250–258. [DOI] [PubMed] [Google Scholar]
- 42.Faden AI, Demediuk P, Panter SS, and Vink R (1989) The role of excitatory amino acids and NMDA receptors in traumatic brain injury, Science 244, 798–800. [DOI] [PubMed] [Google Scholar]
- 43.Rothstein JD, Van Kammen M, Levey AI, Martin LJ, and Kuncl RW (1995) Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis, Annals of neurology 38, 73–84. [DOI] [PubMed] [Google Scholar]
- 44.Dunlop J, Beal McIlvain H, She Y, and Howland DS (2003) Impaired spinal cord glutamate transport capacity and reduced sensitivity to riluzole in a transgenic superoxide dismutase mutant rat model of amyotrophic lateral sclerosis, The Journal of neuroscience : the official journal of the Society for Neuroscience 23, 1688–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rothstein JD, Martin LJ, and Kuncl RW (1992) Decreased glutamate transport by the brain and spinal cord in amyotrophic lateral sclerosis, The New England journal of medicine 326, 1464–1468. [DOI] [PubMed] [Google Scholar]
- 46.Mehta A, Prabhakar M, Kumar P, Deshmukh R, and Sharma PL (2013) Excitotoxicity: bridge to various triggers in neurodegenerative disorders, European journal of pharmacology 698, 6–18. [DOI] [PubMed] [Google Scholar]
- 47.Hardy J, Cowburn R, Barton A, Reynolds G, Lofdahl E, O’Carroll AM, Wester P, and Winblad B (1987) Region-specific loss of glutamate innervation in Alzheimer’s disease, Neuroscience letters 73, 77–80. [DOI] [PubMed] [Google Scholar]
- 48.Massieu L, and Garcia O (1998) The role of excitotoxicity and metabolic failure in the pathogenesis of neurological disorders, Neurobiology (Bp) 6, 99–108. [PubMed] [Google Scholar]
- 49.Estrada-Sanchez AM, and Rebec GV (2012) Corticostriatal dysfunction and glutamate transporter 1 (GLT1) in Huntington’s disease: interactions between neurons and astrocytes, Basal ganglia 2, 57–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Maeda S, Kawamoto A, Yatani Y, Shirakawa H, Nakagawa T, and Kaneko S (2008) Gene transfer of GLT-1, a glial glutamate transporter, into the spinal cord by recombinant adenovirus attenuates inflammatory and neuropathic pain in rats, Molecular pain 4, 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sabri F, Titanji K, De Milito A, and Chiodi F (2003) Astrocyte activation and apoptosis: their roles in the neuropathology of HIV infection, Brain Pathol 13, 84–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Potter MC, Figuera-Losada M, Rojas C, and Slusher BS (2013) Targeting the glutamatergic system for the treatment of HIV-associated neurocognitive disorders, Journal of neuroimmune pharmacology : the official journal of the Society on NeuroImmune Pharmacology 8, 594–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kovalevich J, and Langford D (2012) Neuronal toxicity in HIV CNS disease, Future virology 7, 687–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.McIntosh TK, Vink R, Soares H, Hayes R, and Simon R (1990) Effect of noncompetitive blockade of N-methyl-D-aspartate receptors on the neurochemical sequelae of experimental brain injury, Journal of neurochemistry 55, 1170–1179. [DOI] [PubMed] [Google Scholar]
- 55.Okiyama K, Smith DH, White WF, Richter K, and McIntosh TK (1997) Effects of the novel NMDA antagonists CP-98,113, CP-101,581 and CP-101,606 on cognitive function and regional cerebral edema following experimental brain injury in the rat, Journal of neurotrauma 14, 211–222. [DOI] [PubMed] [Google Scholar]
- 56.Okiyama K, Smith DH, White WF, and McIntosh TK (1998) Effects of the NMDA antagonist CP-98,113 on regional cerebral edema and cardiovascular, cognitive, and neurobehavioral function following experimental brain injury in the rat, Brain research 792, 291–298. [DOI] [PubMed] [Google Scholar]
- 57.Ginsberg MD (2008) Neuroprotection for ischemic stroke: past, present and future, Neuropharmacology 55, 363–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ikonomidou C, and Turski L (2002) Why did NMDA receptor antagonists fail clinical trials for stroke and traumatic brain injury?, Lancet neurology 1, 383–386. [DOI] [PubMed] [Google Scholar]
- 59.Vetiska S, and Tymianski M (2014) Neuroprotectants Targeting NMDA Receptor Signaling In Handbook of Neurotoxicity (Kostrzewa RM, Ed.) pp 1381–1402, Springer New York. [Google Scholar]
- 60.Hicks RR, Smith DH, and McIntosh TK (1995) Temporal response and effects of excitatory amino acid antagonism on microtubule-associated protein 2 immunoreactivity following experimental brain injury in rats, Brain research 678, 151–160. [DOI] [PubMed] [Google Scholar]
- 61.Minger SL, Geddes JW, Holtz ML, Craddock SD, Whiteheart SW, Siman RG, and Pettigrew LC (1998) Glutamate receptor antagonists inhibit calpain-mediated cytoskeletal proteolysis in focal cerebral ischemia, Brain Res 810, 181–199. [DOI] [PubMed] [Google Scholar]
- 62.Okiyama K, Smith DH, Thomas MJ, and McIntosh TK (1992) Evaluation of a novel calcium channel blocker, (S)-emopamil, on regional cerebral edema and neurobehavioral function after experimental brain injury, Journal of neurosurgery 77, 607–615. [DOI] [PubMed] [Google Scholar]
- 63.Cheney JA, Brown AL, Bareyre FM, Russ AB, Weisser JD, Ensinger HA, Leusch A, Raghupathi R, and Saatman KE (2000) The novel compound LOE 908 attenuates acute neuromotor dysfunction but not cognitive impairment or cortical tissue loss following traumatic brain injury in rats, Journal of neurotrauma 17, 83–91. [DOI] [PubMed] [Google Scholar]
- 64.Maas AIR (2000) Assessment of Agents for the Treatment of Head Injury: Problems and Pitfalls in Trial Design, CNS drugs 13, 139–154. [Google Scholar]
- 65.Dunlop J (2006) Glutamate-based therapeutic approaches: targeting the glutamate transport system, Current opinion in pharmacology 6, 103–107. [DOI] [PubMed] [Google Scholar]
- 66.Ganel R, Ho T, Maragakis NJ, Jackson M, Steiner JP, and Rothstein JD (2006) Selective up-regulation of the glial Na+-dependent glutamate transporter GLT1 by a neuroimmunophilin ligand results in neuroprotection, Neurobiology of disease 21, 556–567. [DOI] [PubMed] [Google Scholar]
- 67.Lee SG, Su ZZ, Emdad L, Gupta P, Sarkar D, Borjabad A, Volsky DJ, and Fisher PB (2008) Mechanism of ceftriaxone induction of excitatory amino acid transporter-2 expression and glutamate uptake in primary human astrocytes, The Journal of biological chemistry 283, 13116–13123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rothstein JD, Patel S, Regan MR, Haenggeli C, Huang YH, Bergles DE, Jin L, Dykes Hoberg M, Vidensky S, Chung DS, Toan SV, Bruijn LI, Su ZZ, Gupta P, and Fisher PB (2005) Beta-lactam antibiotics offer neuroprotection by increasing glutamate transporter expression, Nature 433, 73–77. [DOI] [PubMed] [Google Scholar]
- 69.Wei J, Pan X, Pei Z, Wang W, Qiu W, Shi Z, and Xiao G (2012) The beta-lactam antibiotic, ceftriaxone, provides neuroprotective potential via anti-excitotoxicity and anti-inflammation response in a rat model of traumatic brain injury, The journal of trauma and acute care surgery. [DOI] [PubMed] [Google Scholar]
- 70.Pan XD, Wei J, and Xiao GM (2011) [Effects of beta-lactam antibiotics ceftriaxone on expression of glutamate in hippocampus after traumatic brain injury in rats], Zhejiang da xue xue bao. Yi xue ban = Journal of Zhejiang University. Medical sciences 40, 522–526. [DOI] [PubMed] [Google Scholar]
- 71.Li Y, Sattler R, Yang EJ, Nunes A, Ayukawa Y, Akhtar S, Ji G, Zhang PW, and Rothstein JD (2011) Harmine, a natural beta-carboline alkaloid, upregulates astroglial glutamate transporter expression, Neuropharmacology 60, 1168–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhong Z, Tao Y, and Yang HUI (2015) Treatment with harmine ameliorates functional impairment and neuronal death following traumatic brain injury, Molecular medicine reports 12, 7985–7991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Xing X, Chang LC, Kong Q, Colton CK, Lai L, Glicksman MA, Lin CL, and Cuny GD (2011) Structure-activity relationship study of pyridazine derivatives as glutamate transporter EAAT2 activators, Bioorganic & medicinal chemistry letters 21, 5774–5777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kong Q, Chang LC, Takahashi K, Liu Q, Schulte DA, Lai L, Ibabao B, Lin Y, Stouffer N, Mukhopadhyay CD, Xing X, Seyb KI, Cuny GD, Glicksman MA, and Lin CL (2014) Small-molecule activator of glutamate transporter EAAT2 translation provides neuroprotection, The Journal of clinical investigation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nederkoorn PJ, Westendorp WF, Hooijenga IJ, de Haan RJ, Dippel DW, Vermeij FH, Dijkgraaf MG, Prins JM, Spanjaard L, and van de Beek D (2011) Preventive antibiotics in stroke study: rationale and protocol for a randomised trial, International journal of stroke : official journal of the International Stroke Society 6, 159–163. [DOI] [PubMed] [Google Scholar]
- 76.Shimada F, Shiga Y, Morikawa M, Kawazura H, Morikawa O, Matsuoka T, Nishizaki T, and Saito N (1999) The neuroprotective agent MS-153 stimulates glutamate uptake, European journal of pharmacology 386, 263–270. [DOI] [PubMed] [Google Scholar]
- 77.Azbill RD, Mu X, and Springer JE (2000) Riluzole increases high-affinity glutamate uptake in rat spinal cord synaptosomes, Brain research 871, 175–180. [DOI] [PubMed] [Google Scholar]
- 78.Mu X, Azbill RD, and Springer JE (2000) Riluzole and methylprednisolone combined treatment improves functional recovery in traumatic spinal cord injury, Journal of neurotrauma 17, 773–780. [DOI] [PubMed] [Google Scholar]
- 79.Frizzo ME, Schwalm FD, Frizzo JK, Soares FA, and Souza DO (2005) Guanosine enhances glutamate transport capacity in brain cortical slices, Cellular and molecular neurobiology 25, 913–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Frizzo ME, Lara DR, Dahm KC, Prokopiuk AS, Swanson RA, and Souza DO (2001) Activation of glutamate uptake by guanosine in primary astrocyte cultures, Neuroreport 12, 879–881. [DOI] [PubMed] [Google Scholar]
- 81.Nishida A, Iwata H, Kudo Y, Kobayashi T, Matsuoka Y, Kanai Y, and Endou H (2004) Nicergoline enhances glutamate uptake via glutamate transporters in rat cortical synaptosomes, Biological & pharmaceutical bulletin 27, 817–820. [DOI] [PubMed] [Google Scholar]
- 82.Fontana AC, Guizzo R, de Oliveira Beleboni R, Meirelles ESAR, Coimbra NC, Amara SG, dos Santos WF, and Coutinho-Netto J (2003) Purification of a neuroprotective component of Parawixia bistriata spider venom that enhances glutamate uptake, British journal of pharmacology 139, 1297–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Fontana AC, de Oliveira Beleboni R, Wojewodzic MW, Ferreira Dos Santos W, Coutinho-Netto J, Grutle NJ, Watts SD, Danbolt NC, and Amara SG (2007) Enhancing glutamate transport: mechanism of action of Parawixin1, a neuroprotective compound from Parawixia bistriata spider venom, Molecular pharmacology 72, 1228–1237. [DOI] [PubMed] [Google Scholar]
- 84.Mortensen OV, Liberato JL, Coutinho-Netto J, Dos Santos WF, and Fontana AC (2015) Molecular determinants of transport stimulation of EAAT2 are located at interface between the trimerization and substrate transport domains, Journal of neurochemistry 133, 199–210. [DOI] [PubMed] [Google Scholar]
- 85.Yernool D, Boudker O, Jin Y, and Gouaux E (2004) Structure of a glutamate transporter homologue from Pyrococcus horikoshii, Nature 431, 811–818. [DOI] [PubMed] [Google Scholar]
- 86.Boudker O, Ryan RM, Yernool D, Shimamoto K, and Gouaux E (2007) Coupling substrate and ion binding to extracellular gate of a sodium-dependent aspartate transporter, Nature 445, 387–393. [DOI] [PubMed] [Google Scholar]
- 87.Reyes N, Ginter C, and Boudker O (2009) Transport mechanism of a bacterial homologue of glutamate transporters, Nature 462, 880–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Akyuz N, Altman RB, Blanchard SC, and Boudker O (2013) Transport dynamics in a glutamate transporter homologue, Nature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kortagere S, Mortensen OV, Xia J, Lester W, Fang Y, Srikanth Y, Salvino JM, and Fontana ACK (2018) Identification of Novel Allosteric Modulators of Glutamate Transporter EAAT2, ACS chemical neuroscience 9, 522–534. [DOI] [PubMed] [Google Scholar]
- 90.Cook DR, Gleichman AJ, Cross SA, Doshi S, Ho W, Jordan-Sciutto KL, Lynch DR, and Kolson DL (2011) NMDA receptor modulation by the neuropeptide apelin: implications for excitotoxic injury, Journal of neurochemistry 118, 1113–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Cook A, Hippensteel R, Shimizu S, Nicolai J, Fatatis A, and Meucci O (2010) Interactions between chemokines: regulation of fractalkine/CX3CL1 homeostasis by SDF/CXCL12 in cortical neurons, The Journal of biological chemistry 285, 10563–10571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Shimizu S, Abt A, and Meucci O (2011) Bilaminar co-culture of primary rat cortical neurons and glia, Journal of visualized experiments : JoVE, e3257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Burbassi S, Sengupta R, and Meucci O (2010) Alterations of CXCR4 function in mu-opioid receptor-deficient glia, The European journal of neuroscience 32, 1278–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Nicolai J, Burbassi S, Rubin J, and Meucci O (2010) CXCL12 inhibits expression of the NMDA receptor’s NR2B subunit through a histone deacetylase-dependent pathway contributing to neuronal survival, Cell death & disease 1, e33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Meucci O, Fatatis A, Simen AA, Bushell TJ, Gray PW, and Miller RJ (1998) Chemokines regulate hippocampal neuronal signaling and gp120 neurotoxicity, Proceedings of the National Academy of Sciences of the United States of America 95, 14500–14505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.O’Donnell LA, Agrawal A, Jordan-Sciutto KL, Dichter MA, Lynch DR, and Kolson DL (2006) Human immunodeficiency virus (HIV)-induced neurotoxicity: roles for the NMDA receptor subtypes, The Journal of neuroscience : the official journal of the Society for Neuroscience 26, 981–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lehre KP, Levy LM, Ottersen OP, Storm-Mathisen J, and Danbolt NC (1995) Differential expression of two glial glutamate transporters in the rat brain: quantitative and immunocytochemical observations, The Journal of neuroscience : the official journal of the Society for Neuroscience 15, 1835–1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kanai Y, and Hediger MA (1992) Primary structure and functional characterization of a high-affinity glutamate transporter, Nature 360, 467–471. [DOI] [PubMed] [Google Scholar]
- 99.Rothstein JD, Martin L, Levey AI, Dykes-Hoberg M, Jin L, Wu D, Nash N, and Kuncl RW (1994) Localization of neuronal and glial glutamate transporters, Neuron 13, 713–725. [DOI] [PubMed] [Google Scholar]
- 100.Chernova T, Steinert JR, Guerin CJ, Nicotera P, Forsythe ID, and Smith AG (2007) Neurite Degeneration Induced by Heme Deficiency Mediated via Inhibition of NMDA Receptor-Dependent Extracellular Signal-Regulated Kinase 1/2 Activation, The Journal of Neuroscience 27, 8475–8485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hardingham GE, and Bading H (2010) Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders, Nature reviews. Neuroscience 11, 682–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Arundine M, and Tymianski M (2004) Molecular mechanisms of glutamate-dependent neurodegeneration in ischemia and traumatic brain injury, Cellular and molecular life sciences : CMLS 61, 657–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Gegelashvili G, Danbolt NC, and Schousboe A (1997) Neuronal soluble factors differentially regulate the expression of the GLT1 and GLAST glutamate transporters in cultured astroglia, Journal of neurochemistry 69, 2612–2615. [DOI] [PubMed] [Google Scholar]
- 104.Guillet B, Lortet S, Masmejean F, Samuel D, Nieoullon A, and Pisano P (2002) Developmental expression and activity of high affinity glutamate transporters in rat cortical primary cultures, Neurochemistry International 40, 661–671. [DOI] [PubMed] [Google Scholar]
- 105.Velly Lionel J. M.D., Guillet Benjamin A. Ph.D., Masmejean Frederique M., Nieoullon André L. Ph.D., Bruder Nicolas J. M.D., Gouin François M. M.D., and Pisano Pascale M. Ph.D. (2003) Neuroprotective Effects of Propofol in a Model of Ischemic Cortical Cell Cultures: Role of Glutamate and Its Transporters, Anesthesiology: The Journal of the American Society of Anesthesiologists 99, 368–375. [DOI] [PubMed] [Google Scholar]
- 106.Kis B, Nagy K, Snipes JA, Rajapakse NC, Horiguchi T, Grover GJ, and Busija DW (2004) The mitochondrial K(ATP) channel opener BMS-191095 induces neuronal preconditioning, Neuroreport 15, 345–349. [DOI] [PubMed] [Google Scholar]
- 107.Polster BM, Basanez G, Etxebarria A, Hardwick JM, and Nicholls DG (2005) Calpain I induces cleavage and release of apoptosis-inducing factor from isolated mitochondria, The Journal of biological chemistry 280, 6447–6454. [DOI] [PubMed] [Google Scholar]
- 108.Kowaltowski AJ, Castilho RF, and Vercesi AE (2001) Mitochondrial permeability transition and oxidative stress, FEBS letters 495, 12–15. [DOI] [PubMed] [Google Scholar]
- 109.Boston-Howes W, Gibb SL, Williams EO, Pasinelli P, Brown RH Jr., and Trotti D (2006) Caspase-3 cleaves and inactivates the glutamate transporter EAAT2, The Journal of biological chemistry 281, 14076–14084. [DOI] [PubMed] [Google Scholar]
- 110.Miralles VJ, Martinez-Lopez I, Zaragoza R, Borras E, Garcia C, Pallardo FV, and Vina JR (2001) Na+ dependent glutamate transporters (EAAT1, EAAT2, and EAAT3) in primary astrocyte cultures: effect of oxidative stress, Brain research 922, 21–29. [DOI] [PubMed] [Google Scholar]
- 111.Lee Y, Aono M, Laskowitz D, Warner DS, and Pearlstein RD (2004) Apolipoprotein E protects against oxidative stress in mixed neuronal-glial cell cultures by reducing glutamate toxicity, Neurochemistry international 44, 107–118. [DOI] [PubMed] [Google Scholar]
- 112.Takeda K, and Ichijo H (2002) Neuronal p38 MAPK signalling: an emerging regulator of cell fate and function in the nervous system, Genes to cells : devoted to molecular & cellular mechanisms 7, 1099–1111. [DOI] [PubMed] [Google Scholar]
- 113.Odaka H, Numakawa T, Adachi N, Ooshima Y, Nakajima S, Katanuma Y, Inoue T, and Kunugi H (2014) Cabergoline, Dopamine D2 Receptor Agonist, Prevents Neuronal Cell Death under Oxidative Stress via Reducing Excitotoxicity, PLoS One 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Trotti D, Aoki M, Pasinelli P, Berger UV, Danbolt NC, Brown RH Jr., and Hediger MA (2001) Amyotrophic lateral sclerosis-linked glutamate transporter mutant has impaired glutamate clearance capacity, The Journal of biological chemistry 276, 576–582. [DOI] [PubMed] [Google Scholar]
- 115.Feeney CJ, Frantseva MV, Carlen PL, Pennefather PS, Shulyakova N, Shniffer C, and Mills LR (2008) Vulnerability of glial cells to hydrogen peroxide in cultured hippocampal slices, Brain research 1198, 1–15. [DOI] [PubMed] [Google Scholar]
- 116.Sholl DA (1953) Dendritic organization in the neurons of the visual and motor cortices of the cat, Journal of anatomy 87, 387–406. [PMC free article] [PubMed] [Google Scholar]
- 117.O’Neill KM, Akum BF, Dhawan ST, Kwon M, Langhammer CG, and Firestein BL (2015) Assessing effects on dendritic arborization using novel Sholl analyses, Frontiers in cellular neuroscience 9, 285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Monnerie H, Shashidhara S, and Le Roux PD (2003) Effect of excess extracellular glutamate on dendrite growth from cerebral cortical neurons at 3 days in vitro: Involvement of NMDA receptors, Journal of neuroscience research 74, 688–700. [DOI] [PubMed] [Google Scholar]
- 119.von Bartheld CS, Bahney J, and Herculano-Houzel S (2016) The search for true numbers of neurons and glial cells in the human brain: A review of 150 years of cell counting, J Comp Neurol 524, 3865–3895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Luebke JI, Weaver CM, Rocher AB, Rodriguez A, Crimins JL, Dickstein DL, Wearne SL, and Hof PR (2010) Dendritic vulnerability in neurodegenerative disease: insights from analyses of cortical pyramidal neurons in transgenic mouse models, Brain Struct Funct 214, 181–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Marklund N, and Hillered L (2011) Animal modelling of traumatic brain injury in preclinical drug development: where do we go from here?, British journal of pharmacology 164, 1207–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Patel RAG, and McMullen PW (2017) Neuroprotection in the Treatment of Acute Ischemic Stroke, Progress in cardiovascular diseases 59, 542–548. [DOI] [PubMed] [Google Scholar]
- 123.Lewerenz J, and Maher P (2015) Chronic Glutamate Toxicity in Neurodegenerative Diseases-What is the Evidence?, Frontiers in neuroscience 9, 469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Berry JD, Shefner JM, Conwit R, Schoenfeld D, Keroack M, Felsenstein D, Krivickas L, David WS, Vriesendorp F, Pestronk A, Caress JB, Katz J, Simpson E, Rosenfeld J, Pascuzzi R, Glass J, Rezania K, Rothstein JD, Greenblatt DJ, and Cudkowicz ME (2013) Design and initial results of a multi-phase randomized trial of ceftriaxone in amyotrophic lateral sclerosis, PloS one 8, e61177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kim J, John J, Langford D, Walker E, Ward S, and Rawls SM (2016) Clavulanic acid enhances glutamate transporter subtype I (GLT-1) expression and decreases reinforcing efficacy of cocaine in mice, Amino acids 48, 689–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Huh Y, Ju MS, Park H, Han S, Bang Y-M, Ferris CF, Koppell GA, King JA, Kim ML, Kim DJ, Ahn CH, and Oh M-S (2010) Clavulanic acid protects neurons in pharmacological models of neurodegenerative diseases, Drug Development Research 71, 351–357. [Google Scholar]
- 127.Hajhashemi V, and Dehdashti K (2014) Antinociceptive effect of clavulanic acid and its preventive activity against development of morphine tolerance and dependence in animal models, Research in pharmaceutical sciences 9, 315–321. [PMC free article] [PubMed] [Google Scholar]
- 128.Sullivan PG, Keller JN, Mattson MP, and Scheff SW (1998) Traumatic brain injury alters synaptic homeostasis: implications for impaired mitochondrial and transport function, Journal of neurotrauma 15, 789–798. [DOI] [PubMed] [Google Scholar]
- 129.Vishnoi S, Raisuddin S, and Parvez S (2016) Glutamate Excitotoxicity and Oxidative Stress in Epilepsy: Modulatory Role of Melatonin, Journal of environmental pathology, toxicology and oncology : official organ of the International Society for Environmental Toxicology and Cancer 35, 365–374. [DOI] [PubMed] [Google Scholar]
- 130.Salim S (2017) Oxidative Stress and the Central Nervous System, The Journal of pharmacology and experimental therapeutics 360, 201–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Nguyen D, Alavi MV, Kim KY, Kang T, Scott RT, Noh YH, Lindsey JD, Wissinger B, Ellisman MH, Weinreb RN, Perkins GA, and Ju WK (2011) A new vicious cycle involving glutamate excitotoxicity, oxidative stress and mitochondrial dynamics, Cell death & disease 2, e240–e240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Kumari S, Mehta SL, and Li PA (2012) Glutamate Induces Mitochondrial Dynamic Imbalance and Autophagy Activation: Preventive Effects of Selenium, PloS one 7, e39382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Niswender CM, and Conn PJ (2010) Metabotropic Glutamate Receptors: Physiology, Pharmacology, and Disease, Annual review of pharmacology and toxicology 50, 295–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Nik Ramli NN, and Siran R (2015) The neuroprotective effects of (S)-3,5-dihydroxyphenylglycine preconditioning in middle cerebral artery occluded rats: a perspective as a contrivance for stroke, Neural regeneration research 10, 1221–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Wang Y, Qin ZH, Nakai M, and Chase TN (1997) Glutamate metabotropic receptor agonist 1S,3R-ACPD induces internucleosomal DNA fragmentation and cell death in rat striatum, Brain research 772, 45–56. [DOI] [PubMed] [Google Scholar]
- 136.Vergassola M, Olivero G, Cisani F, Usai C, Bossi S, Puliti A, and Pittaluga A (2018) Presynaptic mGlu1 Receptors Control GABA(B) Receptors in an Antagonist-Like Manner in Mouse Cortical GABAergic and Glutamatergic Nerve Endings, Frontiers in molecular neuroscience 11, 324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Zhou F, Yao HH, Wu JY, Yang YJ, Ding JH, Zhang J, and Hu G (2006) Activation of Group II/III metabotropic glutamate receptors attenuates LPS-induced astroglial neurotoxicity via promoting glutamate uptake, Journal of neuroscience research 84, 268–277. [DOI] [PubMed] [Google Scholar]
- 138.Yao HH, Ding JH, Zhou F, Wang F, Hu LF, Sun T, and Hu G (2005) Enhancement of glutamate uptake mediates the neuroprotection exerted by activating group II or III metabotropic glutamate receptors on astrocytes, Journal of neurochemistry 92, 948–961. [DOI] [PubMed] [Google Scholar]
- 139.Ye ZC, and Sontheimer H (1999) Metabotropic glutamate receptor agonists reduce glutamate release from cultured astrocytes, Glia 25, 270–281. [PubMed] [Google Scholar]
- 140.Aronica E, Gorter JA, Ijlst-Keizers H, Rozemuller AJ, Yankaya B, Leenstra S, and Troost D (2003) Expression and functional role of mGluR3 and mGluR5 in human astrocytes and glioma cells: opposite regulation of glutamate transporter proteins, The European journal of neuroscience 17, 2106–2118. [DOI] [PubMed] [Google Scholar]
- 141.Huang YH, Sinha SR, Tanaka K, Rothstein JD, and Bergles DE (2004) Astrocyte glutamate transporters regulate metabotropic glutamate receptor-mediated excitation of hippocampal interneurons, J Neurosci 24, 4551–4559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Lau A, and Tymianski M (2010) Glutamate receptors, neurotoxicity and neurodegeneration, Pflugers Archiv : European journal of physiology 460, 525–542. [DOI] [PubMed] [Google Scholar]
- 143.Niquet J, Baldwin RA, Allen SG, Fujikawa DG, and Wasterlain CG (2003) Hypoxic neuronal necrosis: Protein synthesis-independent activation of a cell death program, Proceedings of the National Academy of Sciences 100, 2825–2830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Subramaniam S, and Unsicker K (2010) ERK and cell death: ERK1/2 in neuronal death, The FEBS journal 277, 22–29. [DOI] [PubMed] [Google Scholar]
- 145.Ahn J-Y (2014) Neuroprotection Signaling of Nuclear Akt in Neuronal Cells, Experimental Neurobiology 23, 200–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Kaltschmidt B, and Kaltschmidt C (2009) NF-κB in the Nervous System, Cold Spring Harbor Perspectives in Biology 1, a001271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Barkhoudarian G, Hovda DA, and Giza CC (2011) The molecular pathophysiology of concussive brain injury, Clinics in sports medicine 30, 33–48, vii–iii. [DOI] [PubMed] [Google Scholar]
- 148.Jain KK (2008) Neuroprotection in traumatic brain injury, Drug discovery today 13, 1082–1089. [DOI] [PubMed] [Google Scholar]
- 149.Barker-Haliski M, and White HS (2015) Glutamatergic Mechanisms Associated with Seizures and Epilepsy, Cold Spring Harbor perspectives in medicine 5, a022863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Magou GC, Pfister BJ, and Berlin JR (2015) Effect of acute stretch injury on action potential and network activity of rat neocortical neurons in culture, Brain research. [DOI] [PubMed] [Google Scholar]
- 151.Shahlaie K, Lyeth BG, Gurkoff GG, Muizelaar JP, and Berman RF (2010) Neuroprotective Effects of Selective N-Type VGCC Blockade on Stretch-Injury-Induced Calcium Dynamics in Cortical Neurons, Journal of neurotrauma 27, 175–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Xie N, Wang C, Wu C, Cheng X, Gao Y, Zhang H, Zhang Y, and Lian Y (2016) Mdivi-1 Protects Epileptic Hippocampal Neurons from Apoptosis via Inhibiting Oxidative Stress and Endoplasmic Reticulum Stress in Vitro, Neurochemical research 41, 1335–1342. [DOI] [PubMed] [Google Scholar]
- 153.Goldberg MP, and Choi DW (1993) Combined oxygen and glucose deprivation in cortical cell culture: calcium-dependent and calcium-independent mechanisms of neuronal injury, The Journal of neuroscience : the official journal of the Society for Neuroscience 13, 3510–3524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Bunch L, Erichsen MN, and Jensen AA (2009) Excitatory amino acid transporters as potential drug targets, Expert opinion on therapeutic targets 13, 719–731. [DOI] [PubMed] [Google Scholar]
- 155.Lin CL, Kong Q, Cuny GD, and Glicksman MA (2012) Glutamate transporter EAAT2: a new target for the treatment of neurodegenerative diseases, Future medicinal chemistry 4, 1689–1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Verma R, Mishra V, Sasmal D, and Raghubir R (2010) Pharmacological evaluation of glutamate transporter 1 (GLT-1) mediated neuroprotection following cerebral ischemia/reperfusion injury, European journal of pharmacology 638, 65–71. [DOI] [PubMed] [Google Scholar]
- 157.Chao XD, Fei F, and Fei Z (2010) The role of excitatory amino acid transporters in cerebral ischemia, Neurochemical research 35, 1224–1230. [DOI] [PubMed] [Google Scholar]
- 158.Gunawan S, Ayaz M, De Moliner F, Frett B, Kaiser C, Patrick N, Xu Z, and Hulme C (2012) Synthesis of Tetrazolo-Fused Benzodiazepines and Benzodiazepinones by a Two-Step Protocol Using an Ugi-Azide Reaction for Initial Diversity Generation, Tetrahedron 68, 5606–5611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Maleki A, and Sarvary A (2015) Synthesis of tetrazoles via isocyanide-based reactions, RSC Advances 5, 60938–60955. [Google Scholar]
- 160.Patil P, Khoury K, Herdtweck E, and Domling A (2015) MCR synthesis of a tetracyclic tetrazole scaffold, Bioorganic & medicinal chemistry 23, 2699–2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Zhao T, Boltjes A, Herdtweck E, and Domling A (2013) Tritylamine as an ammonia surrogate in the Ugi tetrazole synthesis, Organic letters 15, 639–641. [DOI] [PubMed] [Google Scholar]
- 162.Khan MZ, Brandimarti R, Shimizu S, Nicolai J, Crowe E, and Meucci O (2008) The chemokine CXCL12 promotes survival of postmitotic neurons by regulating Rb protein, Cell death and differentiation 15, 1663–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Sengupta R, Burbassi S, Shimizu S, Cappello S, Vallee RB, Rubin JB, and Meucci O (2009) Morphine increases brain levels of ferritin heavy chain leading to inhibition of CXCR4-mediated survival signaling in neurons, The Journal of neuroscience : the official journal of the Society for Neuroscience 29, 2534–2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Carrier RL, Ma TC, Obrietan K, and Hoyt KR (2006) A sensitive and selective assay of neuronal degeneration in cell culture, Journal of neuroscience methods 154, 239–244. [DOI] [PubMed] [Google Scholar]
- 165.Chen S-H, Oyarzabal EA, and Hong J-S (2013) Preparation of Rodent Primary Cultures for Neuron–Glia, Mixed Glia, Enriched Microglia, and Reconstituted Cultures with Microglia, Methods in molecular biology (Clifton, N.J.) 1041, 231–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Banker GA, and Cowan WM (1977) Rat hippocampal neurons in dispersed cell culture, Brain research 126, 397–342. [DOI] [PubMed] [Google Scholar]
- 167.Plachez C, Danbolt NC, and Recasens M (2000) Transient expression of the glial glutamate transporters GLAST and GLT in hippocampal neurons in primary culture, Journal of neuroscience research 59, 587–593. [DOI] [PubMed] [Google Scholar]
- 168.Yoshikawa K (2000) Cell cycle regulators in neural stem cells and postmitotic neurons, Neuroscience research 37, 1–14. [DOI] [PubMed] [Google Scholar]
- 169.Wang Y, White MG, Akay C, Chodroff RA, Robinson J, Lindl KA, Dichter MA, Qian Y, Mao Z, Kolson DL, and Jordan-Sciutto KL (2007) Activation of cyclin-dependent kinase 5 by calpains contributes to human immunodeficiency virus-induced neurotoxicity, Journal of neurochemistry 103, 439–455. [DOI] [PubMed] [Google Scholar]
- 170.White MG, Wang Y, Akay C, Lindl KA, Kolson DL, and Jordan-Sciutto KL (2011) Parallel high throughput neuronal toxicity assays demonstrate uncoupling between loss of mitochondrial membrane potential and neuronal damage in a model of HIV-induced neurodegeneration, Neuroscience research 70, 220–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Takahashi K, Kong Q, Lin Y, Stouffer N, Schulte DA, Lai L, Liu Q, Chang L-C, Dominguez S, Xing X, Cuny GD, Hodgetts KJ, Glicksman MA, and Lin C-LG (2015) Restored glial glutamate transporter EAAT2 function as a potential therapeutic approach for Alzheimer’s disease, The Journal of Experimental Medicine 212, 319–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Nakajima Y, Iguchi H, Kamisuki S, Sugawara F, Furuichi T, and Shinoda Y (2016) Low doses of the mycotoxin citrinin protect cortical neurons against glutamate-induced excitotoxicity, The Journal of toxicological sciences 41, 311–319. [DOI] [PubMed] [Google Scholar]
- 173.Jones KA, Sumyia M, Woolfrey KM, Srivastava DP, and Penzes P (2019) Loss of EPAC2 alters dendritic spine morphology and inhibitory synapse density, Molecular and cellular neurosciences. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.