

Abstract
Background
Nucleic acid sequence-based amplification (NASBA) offers huge potential for low-cost, point-of-care (POC) diagnostic devices, but has been limited by high false-positive rates and the challenges of primer design.
Objective
We offer a systematic analysis of NASBA design with a view toward expanding its applicability.
Methods
We examine the parameters that effect dimer formations, and we provide a framework for designing NASBA primers that will reduce false-positive results and make NASBA suitable for more POC diagnostic applications. Then we compare three different oligonucleotide sets to examine (1) the inhibitory effect of dimer formations, (2) false positives with poorly designed primers, and (3) the effect of beacon target location during real-time NASBA. The required T7 promoter sequence adversely affects the reaction kinetics, although the common abridged sequence can improve kinetics without sacrificing accuracy.
Results
We demonstrate that poorly designed primers undergo real-time exponential amplification in the absence of target RNA, resulting in false positives with a time to half of the peak value (t 1/2) of 50 min compared to 45 min for true positives. Redesigning the oligonucleotides to avoid inhibitory dimers eliminated false positives and reduced the true positive t 1/2 by 10 min. Finally, we confirm the efficacy of two molecular beacon design schemes and discuss their multiplexing utility in two clinical scenarios.
Conclusion
This study provides a pathway for using NASBA in developing POC diagnostic assays.
Electronic supplementary material
The online version of this article (doi:10.1007/s40291-013-0029-4) contains supplementary material, which is available to authorized users.
Keywords: West Nile Virus, Severe Acute Respiratory Syndrome, Molecular Beacon, Severe Acute Respiratory Syndrome, Hybridization Energy
Introduction
Since the early 1990s [1], nucleic acid sequence-based amplification (NASBA) has provided an attractive alternative to PCR for the amplification of RNA (Fig. 1). It is routinely used in the clinical diagnosis of many single-stranded RNA viruses, including influenza A [2], foot-and-mouth virus [3, 4], severe acute respiratory syndrome (SARS), coronavirus [5, 6], West Nile virus [7], human bocavirus (HBoV) [8], and most importantly HIV [9, 10]. NASBA offers the potential for low-cost, portable diagnostic devices, because unlike PCR, NASBA is isothermal. Additionally, NASBA often runs faster than PCR, it compares favorably with regard to sensitivity and convenience [11], and it is preferred over PCR for clinical HIV samples [10, 12]. Because of advances in microfluidics, NASBA is poised to offer major advantages in sequence-based diagnosis.
Fig. 1.
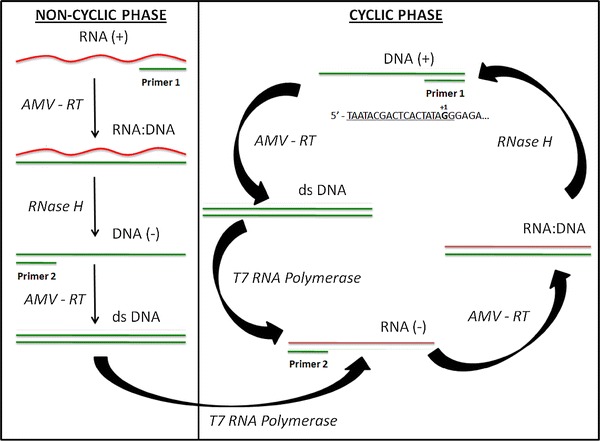
Traditional NASBA begins with a non-cyclic phase, which creates the RNA product (RNA(−)). T7 polymerase uses the dsDNA from the non-cyclic phase as a template to transcribe RNA(−), which initiates the cyclic phase. RNA(−) is the product and template of the cyclic amplification phase and can be detected by end-point gel electrophoresis or in real-time NASBA using molecular beacons. The T7 promoter sequence must be incorporated at the 5′ end of primer 1 (as shown in the cyclic phase). The underlined sequence represents the abridged sequence, and the +1 base is the first base incorporated into the RNA during transcription
Despite these advantages, NASBA has been limited by the challenges of primer design. Hybridization sites on long, viral RNA strands require rigid oligonucleotide design, and only a small fraction of oligonucleotides hybridize efficiently to RNA [13], making many conserved regions unsuitable as NASBA priming sites. Even heating NASBA reagents to 65 °C before adding enzymes [14] and selecting primer sites 100–200 nucleotides apart fail to prevent inhibitory secondary structures [15]. Most importantly, primer dimerization can significantly increase the rate of false positives, which is extremely dangerous in certain clinical tests.
Primer-beacon and other non-specific hybridizations are bound to occur in a dynamic system like NASBA. These non-specific, confounding hybridizations are considered non-inhibitory when their melting temperatures are well below the amplification reaction temperature (41 °C). In contrast, inhibitory dimer formations involve non-specific hybridizations characterized by T m values close, equal to, or exceeding the amplification reaction temperature. Of all of the possible dimer formations, the most likely to disturb normal reaction kinetics involves the primer containing the T7 promoter sequence, because this sequence is directly involved in the amplification process. The T7 promoter sequence, which must be incorporated into one of the primers, is inherently prone to forming dimers.
NASBA can yield endpoint results or real-time results, making it analogous to PCR and real-time PCR, respectively. Endpoint NASBA is usually accompanied by a gel electrophoresis, which determines the size of nucleotides and detects size-specific sequences. Real-time NASBA is often advantageous because it does not require gel electrophoresis: it uses molecular beacons or other forms of fluorophores for sequence detection. The use of fluorophores is conducive to inexpensive diagnostic devices utilizing optics, such as LEDs and photodiodes. However, due to the lack of nucleotide size detection, real-time NASBA is more susceptible to false positives.
New RNA detection techniques [16, 17] amplify a short ssDNA probe molecule using NASBA. The short probe serves as an indicator and amplicon for the much longer target RNA. These techniques overcome some of the traditional limitations of NASBA: (1) primers for the short amplicon are easy to design and use with excellent hybridization kinetics, (2) pre-heating is not required because there is no inherent secondary structure of the short probe, and (3) the relatively long non-cyclic phase of NASBA is bypassed because the short probe (DNA(+) from Fig. 1) is the amplicon used during the cyclic phase rather than the longer RNA, resulting in faster amplification and detection. These new detection techniques still have the potential to undergo non-specific amplification if the primers are not designed deliberately as outlined below.
Although NASBA has been widely explored and developed for a variety of microfluidic diagnostic applications, the formation of inhibitory dimers and their effect on the reaction kinetics in traditional or short amplicon NASBA have not been adequately addressed. Most literature denotes the importance of avoiding dimer formations during PCR and NASBA, but a systematic description of dimer formations that occur in NASBA and design techniques to avoid such dimers is not yet available.
In this article, we detail the cause and effect of dimer formations, and we give examples of dimers that interfere with real-time NASBA and non-specific hybridizations of molecular beacons resulting in false positives. We suggest ways to avoid dimerizations and false positives caused by non-specific molecular beacon binding. Additionally, we discuss molecular beacon design parameters for effective multiplex NASBA assays.
Materials and Methods
We engineered an HIV-1 oligonucleotide set without systematic consideration of primer design (Old), a systematically designed HIV-1 set (New) and an H3 Influenza set (Table 1). Both HIV-1 sets were used for real-time NASBA detection using beacon design (i), while the H3 influenza set used beacon design (ii) as shown in Fig. 2. As the starting template, 1 nM of amplicon probe was used for each set in our experiments. These synthetic oligonucleotides were purchased from Integrated DNA Technologies (Coralville, IA, USA), and their nucleotide sequences are available in the supplementary section. Each oligonucleotide set was analyzed to predict hybridization energies and potential dimer formations and illustrated using DI-nucleic acid hybridization and melting prediction (DINAMelt Server, RNA Institute, SUNY Albany, Albany, NY; available at http://mfold.rna.albany.edu) [18]. The reagents used for real-time NASBA and their concentrations at the final reaction volume (80 μL) were 40 mM Tris (pH 8.0), 12.5 mM MgCl2, 73.5 mM CH3COOK (KOAc), 5.25 mM dithiothreitol, 1.05 mM dNTP, 2.1 mM rNTP, 0.21 μM each primer, 52.5 nM molecular beacon, 15.75 % dimethyl sulfoxide, 1.68 U/μL T7 RNA polymerase, 0.34 U/μL AMV-RT, 0.005 μ/μL RNase-H, and 0.11 mg/mL bovine serum albumin (BSA). Samples were amplified in real-time for 90 min in BRAND disposable UV micro cuvettes (Wertheim, Germany) using a Photon Technology International QuantamasterTM spectrofluorometer (Birmingham, NJ, USA) maintained at 41 °C. Enzymes and reagents used for real-time NASBA were purchased from Promega (Madison, WI, USA).
Table 1.
Theoretical hybridization ΔG and T m values for all potential primer dimers and/or non-specific hybridizations for HIV-1 K103N oligonucleotide set (OLD), redesigned HIV-1 K103N oligonucleotide set (NEW), and H3 influenza oligonucleotide set
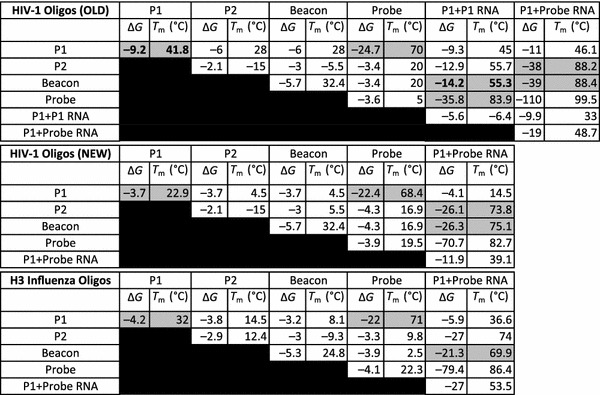
Energy rules for DNA/RNA and RNA/RNA: RNA @ 41 °C, C T = 0.001 μM, [Na+] = 1 M (fixed), [Mg++] = 0 M (fixed). Energy rules for DNA/DNA: DNA @ 41 °C, C T = 10 μM, [Na+] = 73.5 mM, [Mg++] = 12.5 mM
Fig. 2.
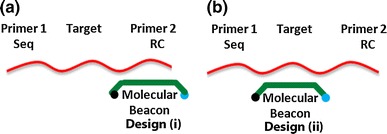
Molecular beacon designs. a Beacon design (i) is designed for multiplexing. A variety of probes can be used to target a group of individual sequences, all of which are indicated with one molecular beacon. b Beacon design (ii) is designed for targeting a specific sequence rather than targeting a group of sequences
Results
Hybridization energies and melting temperatures for the T7 full sequence and abridged sequence were calculated to be ΔG = −9.7 J/mol, T m = 50.6 °C and ΔG = −3.7 J/mol, T m = 22.9 °C, respectively. Hybridization energies for the Old primers were ΔG = −4.0 J/mol, T m = 7.3 °C without any promoter sequence; ΔG = −7.6 J/mol, T m = 41.8 °C with the abridged T7 promoter sequence; and ΔG = −11.3 J/mol, T m = 48.0 °C with the full T7 promoter sequence (Fig. 3). Table 1 outlines the hybridization energies and melting points for the all oligonucleotide sets. In the poorly designed HIV-1 oligonucleotide set (Old) primer 1 notably forms a dimer with ΔG = −9.2 J/mol, T m = 41.8 °C. Furthermore, this P1-P1 dimer was calculated to form a secondary hybridization to the beacon at ΔG = −14.0 J/mol, T m = 55.3 °C. In contrast, analysis of the redesigned HIV-1 oligos (New) only predicted a strong hybridization between primer 1 and the probe—ΔG = −22.4 J/mol, T m = 68.4 °C. The H3 influenza analysis also predicted only one significant hybridization between primer 1 and the probe—ΔG = −22 J/mol, T m = 71.0 °C. Real-time NASBA amplification of six oligonucleotide sets resulted in an exponential increase in signal from the Old, New, and H3 influenza sets when probe was included; a similar exponential increase was observed in the Old set when probe was not included (Fig. 4). The New and H3 sets failed to show a significant increase in signal when probe was not included. Time to half of the peak value (t 1/2) was 45 min for Old, 35 min for New, 18 min for H3 influenza, and 50 min for Old without probe.
Fig. 3.
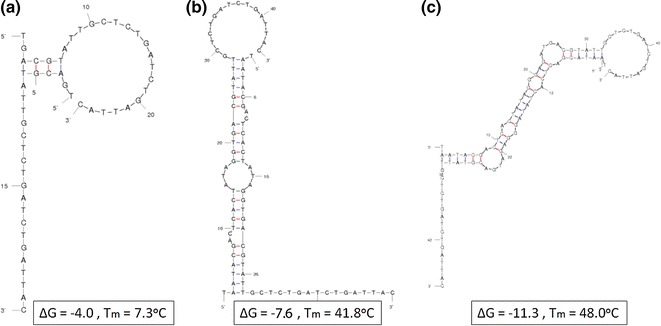
Primer dimer theoretical hybridization energies simulated with DINAMelt. Energy rules: DNA @ 41 °C, C T = 10 μM, [Na+] = 73.5 mM, [Mg++] = 12.5 mM. a Primer alone b primer with abridged T7 promoter c primer with full T7 promoter
Fig. 4.
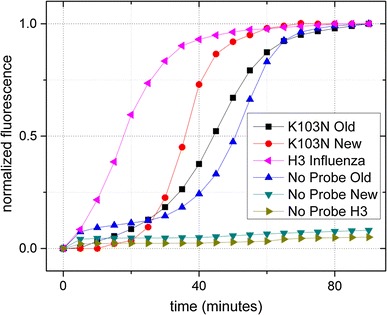
Real-time NASBA of poorly designed HIV-1 K103N oligonucleotide set (Old) vs. redesigned HIV-1 K103N set (New) vs. H3 influenza oligonucleotide set. 1nM probes used and no probe controls included. Standard deviation was within 5 % of results (error bars not shown)
Discussion
Thermodynamic Evaluation of the T7 Promoter Sequence Using Hybridization and Melting Predictions
Potential hybridizations/dimer formations were theoretically evaluated. They are quantitatively represented by hybridization energy (ΔG) and melting temperature (T m). A negative ΔG value indicates that the hybridization is energetically favorable, and the T m represents the system in equilibrium at which half of the two oligonucleotides hybridize and the other half remains single stranded.
The T7 promoter sequence alone forms a very strong theoretical dimer with itself, even under optimal NASBA conditions. Although the T7 promoter sequence must be used to catalyze T7 RNA polymerase, an abridged version may be used for efficient transcription [19]. The abridged version significantly reduces the hybridization energy (increasing ΔG) and reduces T m to the point where it would be considered non-inhibitory. The example primer shown in Fig. 3 is from the poorly designed oligonucleotide set (Old) for the NASBA amplification of an HIV-1 K103N sequence. This primer initially is non-inhibitory; however, the addition of the abridged T7 sequence causes an inhibitory dimer formation. This is further exacerbated by the full T7 promoter sequence.
Thermodynamic Evaluation of the Old Primer Set
Screening for unwanted formations involves cross checking the ΔG and T m values for all oligonucleotides used in the NASBA reaction at the appropriate reaction conditions. Table 1 shows these values for all oligonucleotide sets. The poorly designed oligonucleotide set (Old) undergoes inhibitory dimerization during NASBA. The primer 1 (P1) and probe ΔG and T m values (highlighted grey) show that P1 has a very strong affinity to its intended target probe. P1 also forms a strong self-dimer (highlighted grey and bold), albeit nowhere near as strong as the P1-Probe hybridization. In the presence of large quantities of probe, the P1 dimer will remain non-inhibitory. However, as the concentration of probe decreases, the P1 dimer has a greater inhibitory effect. For negative controls (i.e., no probe), the P1 dimer will become completely dominant. The P1 dimer is capable of producing its own RNA product through NASBA amplification, because it contains a T7 promoter region (Table 2).
Table 2.
Poorly designed HIV-1 K103 N oligonucleotide set (Old), redesigned HIV-1 K103N oligonucleotide set (New), and H3 influenza oligonucleotide set
| Oligonucleotide | HIV-1 K103N sequences (old) |
|---|---|
| Probe | 5′-TCAAGAGTAGACACCTGTTACCGAT TTGTTCTTCTTTAACCCTG GTAATCAGATCAGAGCAATACGTCA-3′ |
| Primer 1 (PI) | 5′-taatacgactcactatagg TGACGTATTGCTCTGATCTGATTAC-3′ |
| P1-Probe RNA product | 5′-gg UGACGUAUUGCUCUGAUCUGAUUAC CAGGGUUAAAGAAGAACAA AUCGGUAACAGGUGUCUACUCUUGA-3′ |
| Primer 2 (P2) | 5′-TCAAGAGTAGACACCTGTTACCGAT-3′ |
| Molecular beacon | 5′-/6-FAM/CGCG TCAAGAGTAGACACCTGTTACCGAT CGCG/3IAB1k_FQ/-3′ |
| Oligonucleotide | HIV-1 K103N sequences (new) |
|---|---|
| Probe | 5′-TCAAGAGTAGACACCTGTTACCGAT TTGTTCTTCTTTAACCCTG TATATTTTGGTGGTTGCAGAATAT -3′ |
| Primer 1 (PI) | 5′-taatacgactcactatagg ATATTCTGCAACCACCAAAATATA-3′ |
| P1-Probe RNA product | 5′-gg AUAUUCUGCAACCACCAAAAUAUA CAGGGUUAAAGAAGAACAA AUCGGUAACAGGUGUCUACUCUUGA- 3′ |
| Primer 2 (P2) | 5′-TCAAGAGTAGACACCTGTTACCGAT-3′ |
| Molecular beacon | 5′-/6-FAM/CGCG TCAAGAGTAGACACCTGTTACCGAT CGCG/3IABIk_FQ/-3′ |
| Oligonucleotide | H3 influenza sequences |
|---|---|
| Probe | 5′-TCAACTCATCACACAGGATCAGCAT ACTCCA AATGGAAGCATTCCCAATG ATTTTGGTGGTTGCAGAATCT-3′ |
| Primer 1 (PI) | 5′-taatacgactcactatagg AGATTCTGCAACCACCAAAAT-3′ |
| P1 probe RNA product | 5′-gg AGAUUCUGCAACCACCAAAAU CAUUGGGAAUGCUUCCAUUUGGAGU AUGCUGAUCCUGUGUGAUGAGUUGA-3′ |
| Primer 2 (P2) | 5′-TCAACTCATCACACAGGATCAGCAT-3′ |
| Molecular beacon | 5′-/6-FAM/CGCG AAATGGAAGCATTCCCAATG CGCG/3IAB1k FO/-3′ |
The probe sequences contain an underlined sequence, which hybridizes with P1. The P1 sequences contain the T7 promoter sequence (lower case and bold). The transcribed P1-Probe RNA product hybridizes with P2 (italics). In the HIV-1 oligonucleotide sets, the molecular beacon (italics) competes with P2 to hybridize with the P1-Probe RNA product. In the influenza oligonucleotide set, the molecular beacon (underlined and bold) is designed to be sequence specific and hybridizes with the target region (underlined and bold) of the transcribed P1-Probe RNA product. Molecular beacons contain the fluorophore, 6-FAM, at their 5′ end and the fluorescence quencher, 3IABlk_FQ, at their 3′ end
Specifically, the P1 dimer is reverse transcribed to form a dsDNA template containing the T7 promoter region in two locations (Fig. 5a). Meanwhile, the P1-Probe hybrid is reverse transcribed, and then T7 polymerase transcribes RNA as desired (Fig. 5b). The transcribed RNA from the P1 dimer dsDNA template will interfere with the reaction kinetics because it contains two T7 promoter regions and misappropriates rNTPs. It becomes impossible to distinguish between a false-positive control (no probe) and a positive sample because real-time NASBA results only indicate fluorescence values, not oligonucleotide lengths.
Fig. 5.
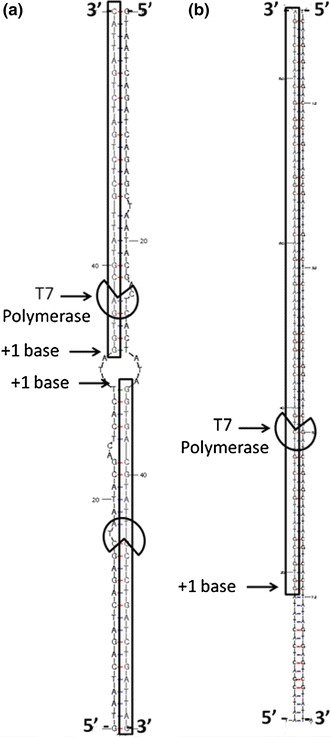
a T7 polymerization of unexpected P1-P1 RNA. There are two locations for the T7 polymerase to polymerize RNA since the T7 promoter exists on both strands 5′ → 3′. b T7 polymerization of expected P1-Probe RNA
Secondary Hybridizations
The P1+probe RNA product is the desired RNA product of the NASBA reaction, and it forms a strong bond with the molecular beacon (highlighted gray in Table 1), indicating effective fluorescence detection. The P1+probe RNA should also form a hybrid of similar strength with primer 2 (P2), which is desirable. However, DINAMelt analysis shows that the P1+Probe RNA actually forms its strongest bond with additional free-floating probe. This hybridization is understandable because P1 and the probe are perfect reverse compliments. However, in practice the much shorter and abundant P2 and beacon molecules are more likely hybridize with the P1+Probe RNA because of their relatively small size and much greater quantity than the probe.
The P1+P1 RNA product forms a relatively weak bond (highlighted gray and bold) with the molecular beacon. However, when there is a low concentration of probe, this relatively weak bond becomes dominant, and the molecular beacon will non-specifically hybridize with the P1-P1 RNA product, resulting in false-positive fluorescence detection. Additionally, the P1-P1 RNA product forms a relatively strong bond (gray) with the probe, which likely stifles the reaction kinetics. Both primary and secondary dimers in the Old set have the potential to result in false positives.
Redesigning the Primer Avoids Inhibitory Dimer Formation
The oligonucleotides can be redesigned to undergo fewer potential dimers and non-specific hybridizations. This is achieved by manually manipulating individual bases in the sequence of P1 so that there are fewer potential adenine-thymine and guanine–cytosine hydrogen bonds in the DINAmelt predicted hybridization. The required T7 promoter sequence gives primer 1 inherent minimum ΔG and T m values that make it susceptible to dimerization. After manipulating the original P1 sequence used, we achieve ΔG and T m values (Table 1) for the P1 dimer matching those of the abridged T7 promoter sequence dimer alone (ΔG = −3.7, T m = 22.9 °C); thus, we have designed P1 such that dimerization is as unfavorable as theoretically possible. Although there is a slight decrease in ΔG and T m for the P1-Probe hybridization, the potential for P1 inhibitory dimerization has been eliminated because the dimers dissociate far below the reaction temperature. Finally, in the H3 influenza set (Table 1), the P1-P1 dimer dissociates well below the reaction temperature, and P1-Probe hybridization is relatively strong and occurs well above the reaction temperature.
Molecular Beacon Design for NASBA
In NASBA, the molecular beacon is designed to either hybridize with (i) the primer 2 reverse complement (RC) region or (ii) the middle target region of the target RNA, as shown in Fig. 2. In design (i), primer 2 is competitive with the molecular beacon to hybridize with the primer 2 RC region, sacrificing reaction kinetics. However, design (i) enables effective multiplexing—an unlimited number of potential target sequences may be used with only one molecular beacon. In design (ii), there is no competition for binding sites. However, because the beacons are target-specific, one beacon design may only be used for one sequence-specific target. Both beacon system designs have their ideal applications. For example, design (i) is ideal for HIV-1 mutation detection: a large number of HIV-1 mutations impart drug resistance, so it is very important to screen for many mutations in parallel (multiplexing). Although design (i) can target all known mutations, a positive result does not indicate which mutation is present. Design (ii) is ideal for influenza virus sub-typing. This design yields sequence-specific results, indicating which types of influenza are present within a given sample, (H1N1, H3, H5, etc.), and it avoids premature transcription abortion by the T7 RNA polymerase, which has been observed in previous work [16].
Real-time NASBA Using Molecular Beacon Detection
Three real-time NASBA runs of each oligonucleotide set were performed (nine runs in total), and the average of the real-time fluorescence results for each of the samples is shown in Fig. 4. As predicted by DINAmelt analysis, the Old set has an invalid negative control, but the New set had a valid negative control. While the influenza oligonucleotide set did not offer any significant improvements in predicted hybridization energies compared to the New oligonucleotide set, it did show dramatic improvements in NASBA reaction speed. The only significant difference in the H3 oligonucleotide design was the beacon hybridization location. The improved reaction kinetics of the H3 influenza set suggest that the beacon location has a significant effect on the reaction speed.
Conclusion
Careful considerations of ΔG and T m should be made when designing real-time NASBA systems. Poorly considered primers can not only decrease reaction kinetics, but can also increase the rate of false positives, which is dangerous in the context of clinical HIV testing, for example. Two approaches to primer design significantly improve reaction kinetics and test accuracy—reducing the length of the T7 promoter sequence to the abridged version and reducing the potential number of adenine-thymine and guanine–cytosine hydrogen bonds in the primer. Furthermore, effective primer design requires DINAMelt analysis of the bond strength not only between primers and probes, but also between potential dimers and the beacon. A safe approach to improve reaction kinetics is to redesign the probes to avoid dimers at 41 °C, because that is the optimal temperature of the reaction enzymes. Salt ion concentrations may also be altered but have significant implications for all hybridization energies and should not be altered if possible [20]. The application of the NASBA assay is an important design parameter to consider since we have shown that a real-time NASBA system capable of detecting a group of sequences is less efficient than a real-time NASBA system only detecting one target. Successful primer design is not simply a matter of improving the efficiency of NASBA-based clinical assays; it is crucial to their accuracy as well.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
The authors acknowledge the financial support of the National Institute of Health (Grant No. 1R21 A1073808-01 A1) and The RNA Institute, College of Arts and Sciences, State University of New York at Albany, for use of their DINAMelt web server. The authors have no conflicts of interest that are directly relevant to the content of this article.
References
- 1.Compton J. Nucleic-acid sequence-based amplification. Nature. 1991;350(6313):91–92. doi: 10.1038/350091a0. [DOI] [PubMed] [Google Scholar]
- 2.Collins RA, Ko LS, So KL, Ellis T, Lau LT, Yu AC. Detection of highly pathogenic and low pathogenic avian influenza subtype H5 (Eurasian lineage) using NASBA. J Virol Methods. 2002;103(2):213–225. doi: 10.1016/S0166-0934(02)00034-4. [DOI] [PubMed] [Google Scholar]
- 3.Collins RA, Ko LS, Fung KY, Lau LT, Xing J, Yu AC. A method to detect major serotypes of foot-and-mouth disease virus. Biochem Biophys Res Commun. 2002;297(2):267–274. doi: 10.1016/S0006-291X(02)02178-2. [DOI] [PubMed] [Google Scholar]
- 4.Lau LT, Reid SM, King DP, Lau AM, Shaw AE, Ferris NP, et al. Detection of foot-and-mouth disease virus by nucleic acid sequence-based amplification (NASBA) Vet Microbiol. 2008;126(1–3):101–110. doi: 10.1016/j.vetmic.2007.07.008. [DOI] [PubMed] [Google Scholar]
- 5.Chantratita W, Pongtanapisit W, Piroj W, Srichunrasmi C, Seesuai S. Development and comparison of the real-time amplification based methods—NASBA-Beacon, RT-PCR taqman and RT-PCR hybridization probe assays–for the qualitative detection of sars coronavirus. Southeast Asian J Trop Med Public Health. 2004;35(3):623–629. [PubMed] [Google Scholar]
- 6.Keightley MC, Sillekens P, Schippers W, Rinaldo C, George KS. Real-time NASBA detection of SARS-associated coronavirus and comparison with real-time reverse transcription-PCR. J Med Virol. 2005;77(4):602–608. doi: 10.1002/jmv.20498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lanciotti RS, Kerst AJ. Nucleic acid sequence-based amplification assays for rapid detection of West Nile and St. Louis encephalitis viruses. J Clin Microbiol. 2001;39(12):4506–4513. doi: 10.1128/JCM.39.12.4506-4513.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bohmer A, Schildgen V, Lusebrink J, Ziegler S, Tillmann RL, Kleines M, et al. Novel application for isothermal nucleic acid sequence-based amplification (NASBA) J Virol Methods. 2009;158(1–2):199–201. doi: 10.1016/j.jviromet.2009.02.010. [DOI] [PubMed] [Google Scholar]
- 9.Vandamme AM, Schmit JC, Van Dooren S, Van Laethem K, Gobbers E, Kok W, et al. Quantification of HIV-1 RNA in plasma: comparable results with the NASBA HIV-1 RNA QT and the AMPLICOR HIV monitor test. J Acquir Immune Defic Syndr Hum Retrovirol. 1996;13(2):127–139. doi: 10.1097/00042560-199610010-00003. [DOI] [PubMed] [Google Scholar]
- 10.Vandamme AM, Van Dooren S, Kok W, Goubau P, Fransen K, Kievits T, et al. Detection of HIV-1 RNA in plasma and serum samples using the NASBA amplification system compared to RNA-PCR. J Virol Methods. 1995;52(1–2):121–132. doi: 10.1016/0166-0934(94)00151-6. [DOI] [PubMed] [Google Scholar]
- 11.Schneider P, Wolters L, Schoone G, Schallig H, Sillekens P, Hermsen R, et al. Real-time nucleic acid sequence-based amplification is more convenient than real-time PCR for quantification of Plasmodium falciparum. J Clin Microbiol. 2005;43(1):402–405. doi: 10.1128/JCM.43.1.402-405.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dyer JR, Gilliam BL, Eron JJ, Grosso L, Cohen MS, Fiscus SA. Quantitation of human immunodeficiency virus type 1 RNA in cell free seminal plasma: comparison of NASBA™ with Amplicor™ reverse transcription-PCR amplification and correlation with quantitative culture. J Virol Methods. 1996;60(2):161–170. doi: 10.1016/0166-0934(96)02063-0. [DOI] [PubMed] [Google Scholar]
- 13.Luebke KJ, Balog RP, Garner HR. Prioritized selection of oligodeoxyribonucleotide probes for efficient hybridization to RNA transcripts. Nucl Acids Res. 2003;31(2):750–758. doi: 10.1093/nar/gkg133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guatelli JC, Whitfield KM, Kwoh DY, Barringer KJ, Richman DD, Gingeras TR. Isothermal, in vitro amplification of nucleic acids by a multienzyme reaction modeled after retroviral replication. Proc Natl Acad Sci. 1990;87(5):1874–1878. doi: 10.1073/pnas.87.5.1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Deiman B, van Aarle P, Sillekens P. Characteristics and applications of nucleic acid sequence-based amplification (NASBA) Mol Biotechnol. 2002;20(2):163–179. doi: 10.1385/MB:20:2:163. [DOI] [PubMed] [Google Scholar]
- 16.McCalla SE, Ong C, Sarma A, Opal SM, Artenstein AW, Tripathi A. A simple method for amplifying RNA targets (SMART) J Mol Diagn. 2012;14(4):328–335. doi: 10.1016/j.jmoldx.2012.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ong C, Tai W, Sarma A, Opal SM, Artenstein AW, Tripathi A. Ligation with Nucleic Acid Sequence-Based Amplification. J Mol Diagn. 2012;14(3):206–213. doi: 10.1016/j.jmoldx.2012.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Markham NR. DI-Nucleic Acid hybridization and melting prediction. The UNAFold Web Server. University at Albany: The RNA Institute College of Arts and Sciences. 2011. http://mfold.rna.albany.edu/?q=dinamelt. Accessed June 2011.
- 19.Technologies L. The Basics: in vitro transcription (cited 2011). http://www.invitrogen.com/site/us/en/home/References/Ambion-Tech-Support/probe-labeling-systems/general-articles/the-basics-in-vitro-transcription.html.
- 20.Owczarzy R, Moreira BG, You Y, Behlke MA, Walder JA. Predicting stability of DNA duplexes in solutions containing magnesium and monovalent cations. Biochemistry-Us. 2008;47(19):5336–5353. doi: 10.1021/bi702363u. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.