

Abstract
Genome-wide association studies have identified dozens of loci that alter the risk to develop Alzheimer’s disease. However, with the exception of the APOE-ε4 allele, most variants bear only little individual effect and have, therefore, limited diagnostic and prognostic value. Polygenic risk scores aim to collate the disease risk distributed across the genome in a single score. Recent works have demonstrated that polygenic risk scores designed for Alzheimer’s disease are predictive of clinical diagnosis, pathology confirmed diagnosis and changes in imaging biomarkers. Methodological innovations in polygenic risk modelling include the polygenic hazard score, which derives effect estimates for individual single nucleotide polymorphisms from survival analysis, and methods that account for linkage disequilibrium between genomic loci. In this work, using data from the Alzheimer’s disease neuroimaging initiative, we compared different approaches to quantify polygenic disease burden for Alzheimer’s disease and their association (beyond the APOE locus) with a broad range of Alzheimer’s disease-related traits: cross-sectional CSF biomarker levels, cross-sectional cortical amyloid burden, clinical diagnosis, clinical progression, longitudinal loss of grey matter and longitudinal decline in cognitive function. We found that polygenic scores were associated beyond APOE with clinical diagnosis, CSF-tau levels and, to a minor degree, with progressive atrophy. However, for many other tested traits such as clinical disease progression, CSF amyloid, cognitive decline and cortical amyloid load, the additional effects of polygenic burden beyond APOE were of minor nature. Overall, polygenic risk scores and the polygenic hazard score performed equally and given the ease with which polygenic risk scores can be derived; they constitute the more practical choice in comparison with polygenic hazard scores. Furthermore, our results demonstrate that incomplete adjustment for the APOE locus, i.e. only adjusting for APOE-ε4 carrier status, can lead to overestimated effects of polygenic scores due to APOE-ε4 homozygous participants. Lastly, on many of the tested traits, the major driving factor remained the APOE locus, with the exception of quantitative CSF-tau and p-tau measures.
Keywords: Alzheimer’s disease, polygenic risk score, polygenic hazard score, biomarker, imaging genetics
Altmann et al. compared different approaches to quantify polygenic disease burden for Alzheimer’s disease and their association with a broad range of Alzheimer’s disease-related traits. Beyond the APOE locus, polygenic scores were associated with clinical diagnosis, CSF-tau and p-tau and progressive atrophy, but not with amyloid beta pathology.
Graphical Abstract
Introduction
The capability to predict disease risk from a person’s genome has invaluable applications, ranging from stratifying people for interventions, enrolling them in screening programmes to capture the first signs of disease onset and enriching clinical trials. Accurate prediction of both developing disease and approximate timing can be made in familial diseases where a single genetic variant is causative of the disorder, e.g. APP, PSEN1 and PSEN2 mutations in familial Alzheimer’s disease (Bateman et al., 2012) and the HTT trinuclear repeat in Huntington’s disease (MacDonald et al., 1993). The ability to predict disease risk or the age of symptom onset from genetic information is more complex in the sporadic manifestations of various neurodegenerative disorders, despite, in the case of Alzheimer’s disease, it being highly heritable (h2 range 0.58–0.79; Gatz et al., 2006) and highly polygenic (Kunkle et al., 2019). In the context of late-onset Alzheimer’s disease, with a minor allele frequency of 0.14, the ε4 allele of the APOE gene is the strongest common genetic risk factor in people with central European ancestry (odds ratio about 3.5). Consequently, the effect of APOE-ε4 on various Alzheimer’s disease biomarkers has been studied extensively over the last decades [reviewed by Belloy et al. (2019)] and modifying demographic factors such as ethnicity and sex have been identified (Farrer et al., 1997; Ungar et al., 2013; Altmann et al., 2014).
Despite genetic studies with ever-increasing size (Lambert et al., 2013; Jansen et al., 2019; Kunkle et al., 2019) other variants with equivalent risk to APOE-ε4 have failed to materialize. Most variants discovered through genome-wide association studies (GWAS) exhibit only minor effects on Alzheimer’s disease risk (odds ratios between 0.8 and 1.2; Kunkle et al., 2019). One notable exception is the R47H substitution in the TREM2 gene which confers a similar level of risk to develop Alzheimer’s disease as APOE-ε4 (odds ratio about 2.0) but occurs very rarely in the population (global minor allele frequency of 0.0025; Guerreiro et al., 2013). However, if a single variant cannot deliver predictive accuracy equivalent to APOE-ε4, then a combination of known risk variants could be of use. This is the rationale behind genome-wide polygenic risk scores (PRSs), which combine the effects of multiple independent risk variants in a person’s genome into a single score capturing an individual’s overall genetic disease risk (International Schizophrenia Consortium et al., 2009). Owing to the ease of access to results from GWAS and the availability of dedicated software (Euesden et al., 2015; Vilhjálmsson et al., 2015), such scores can be built easily and have been adopted rapidly in the genetics field. In recent reports, their capacity to predict disease in common disorders was found to be equivalent to that of (rare) monogenic mutations (Khera et al., 2018). In the context of Alzheimer’s disease PRS have been shown to be associated with clinical diagnosis (Escott-Price et al., 2015), cortical thickness (Sabuncu et al., 2012), memory, hippocampal volume and cognitive decline (Mormino et al., 2016), disease progression (Scelsi et al., 2018) and lastly pathological confirmed cases of Alzheimer’s disease (Escott-Price et al., 2017). Furthermore, Alzheimer’s disease PRSs, which are conceptually based on GWAS of late-onset Alzheimer’s disease, modulate disease risk and age of disease onset in familial late-onset Alzheimer’s disease as well as sporadic early-onset Alzheimer’s disease (Tosto et al., 2017; Cruchaga et al., 2018).
Methods for developing and refining PRS have gained increased attention over the recent years and ever-increasing sample sizes in GWAS result in better-calibrated risk scores for out of sample predictions (Dudbridge, 2013). Beyond these improved effect estimates for single variants, methodological improvements have included the explicit modelling of linkage disequilibrium dependencies between variants (Vilhjálmsson et al., 2015; Ge et al., 2019). A recent conceptual novelty in this domain is the polygenic hazard score (PHS), which seeks to gain predictive power by deriving effect sizes for genetic variants from the time to disease onset in a survival analysis, rather than from wide-spread case–control analyses that are traditionally used (Desikan et al., 2017). A series of recent studies have demonstrated very strong associations between a PHS for Alzheimer’s disease and a range of quantitative measurements related to Alzheimer’s disease pathology including clinical dementia rating (CDR), clinical conversion rates, cognitive tests, cortical in vivo amyloid uptake, change in cortical volumes and post-mortem neuropathology (Tan et al., 2017, 2018, 2019). The PHS introduced by Desikan et al. (2017) comprises 33 genetic markers (two of which are APOE-ε4 and APOE-ε2) and is highly correlated with APOE-ε4 burden [Supplementary Fig. 1 in Desikan et al. (2017)]. Previous works claimed that the PHS is superior to PRS (Tan and Desikan, 2018) and exhibits predictive power beyond APOE; however, the published analyses controlled only for APOE-ε4 status rather than APOE-ε4 allele burden (Desikan et al., 2017; Tan et al., 2017, 2018, 2019).
We hypothesized that by failing to adjust for APOE genotype—i.e. adjusting for ε2 and ε4 allele count rather than only for ε4 carrier status—previous reports on the predictive power of PHS over APOE were overestimating the impact of PHS. In this work, we investigated whether PRS or PHS are associated with Alzheimer’s disease-related biomarkers and clinical outcomes beyond the APOE locus.
Methods
Data
Data used in the preparation of this article were obtained from the ADNI database (http://adni.loni.usc.edu). The ADNI was launched in 2003 as a public–private partnership, led by Principal Investigator Michael W. Weiner, MD. The primary goal of the ADNI has been to test whether serial MRI, PET, other biological markers, and clinical and neuropsychological assessment can be combined to measure the progression of mild cognitive impairment (MCI) and early Alzheimer’s disease. For up-to-date information, see www.adni-info.org. ADNI study data were accessed through the R-package ADNIMERGE (accessed: 7 March 2019).
Preparation of genetic data
At the time of this study, single nucleotide polymorphism (SNP) genotyping data were available for n = 1674 subjects across all ADNI phases. Genotyping was conducted using three different platforms: Human610-Quad, HumanOmniExpress and Omni 2.5 M (Illumina; Saykin et al., 2010). Prior to imputation subject-level quality control (QC) steps based on call rate (10% cutoff) and concordance between chip-inferred sex and self-reported sex were performed separately for each genotyping chip. On SNP-level standard QC steps ensuring compatibility with the reference panel used for imputation (strand consistency, allele names, position, Ref/Alt assignments and minor allele frequency discrepancy (MAF; 0.2 cutoff) were conducted. Imputation was carried out using the Sanger Imputation Server (https://imputation.sanger.ac.uk/) with SHAPEIT for phasing (Delaneau et al., 2012), Positional Burrows-Wheeler Transform (Durbin, 2014) for imputation and the entire Haplotype Reference Consortium (release 1.1) reference panel (McCarthy et al., 2016). Data from the three different genotyping platforms were imputed separately. As part of post-imputation QC, multi-allelic variants and SNPs with imputation INFO score <0.3 were removed and genotype calls with posterior probability <0.9 were set to missing (i.e. hard called). Following the initial QC, genotypes from the three platforms were merged. Further information on the imputation and QC process is detailed in Scelsi et al. (2018) and https://rpubs.com/maffleur/452627. Using the merged data, we retained SNPs with MAF ≥1% and genotyping rate >0.9.
SNPweights (Chen et al., 2013) was used to infer genetic ancestry from genotyped SNPs using the reference panel comprising Central European, Yoruba Africans and East Asian samples from HapMap 3 (The International HapMap 3 Consortium et al., 2010) and native Americans from Reich et al. (2012). Subjects with predicted central European ancestry of 80% or more were retained. Next, using the imputed and merged data genetic relatedness between central European subjects was computed. First, the SNP content was restricted to SNPs with MAF ≥5% and LD-pruning was carried out in PLINK v1.9 (–indep-pairwise 1000 50 0.1). The genetic relatedness matrix was computed using the remaining autosomal SNPs and the dataset was trimmed to remove subjects with relatedness >0.1 (–rel-cutoff 0.1). For the remaining subjects, the first five PCA components were computed in PLINK v1.9 and were used to account for population structure in the data.
Polygenic risk scores
PRSs were computed using the software PRSice v2.1.9 (Euesden et al., 2015). As base GWAS the stage 1 results of the most recent Alzheimer’s disease GWAS featuring a pure Alzheimer’s disease phenotype was used [Kunkle et al., 2019; this is opposed to a larger GWAS including subjects with parental Alzheimer’s disease diagnosis resulting in diagnosis-by-proxy (Jansen et al., 2019)]. For PRS computation, SNPs with MAF ≥5% were considered and SNPs were selected using LD-clumping (1000 KB, R2 of 0.1 and P-value threshold of 1.0), missing SNPs were simply ignored on subject level (using the setting–missing SET_ZERO) and the APOE region was excluded (hg19 coordinates chr19 from 44 400 000 to 46 500 000). Effects for APOE-ε2 and APOE-ε4 were manually added using effect sizes from (Kunkle et al., 2019): 1.2017 (ε4) and -0.4673 (ε2). For this study, two P-value inclusion cut-offs were used: SNPs passing genome-wide suggestive (P = 1.0e-05) and P = 0.5, which was found to be an optimal choice in an earlier study (Escott-Price et al., 2015). The resulting scores are referred to as PRS1 and PRS2, respectively. In addition to this conventional approach, PRS were generated using posterior effect size estimates generated with PRS-CS (Ge et al., 2019). The effect estimates were generated using the auto setting and the PRS are referred to as PRS-cs. PRSs were computed for all ADNI participants with genome-wide genotyping data.
Polygenic hazard score
The derivation of the PHS was described in detail in Desikan et al. (2017). The conceptual difference between PRS and PHS is that PHS uses SNP-level effect size estimates from survival analysis (Cox proportional hazards model) as opposed to odds ratios from case–control analyses. The second difference is that effect sizes in PRS were derived univariately while the hazard ratios used for PHS were derived using a multivariate approach, i.e. effects for each SNP are estimated in the presence of the remaining ones. For this work, we extracted the PHS values provided by the ADNI database (desikanlab table).
Statistical analyses
A series of association analyses were conducted to measure the effect of PHS on imaging, CSF and cognitive biomarkers in Alzheimer’s disease. First, we aimed to replicate the results on ADNI data presented in Tan et al. (2019) by only correcting for APOE-ε4 carrier status (i.e. including a variable that takes the values 0 or 1), referred to as ‘PHS (status)’. Next, the association strength of PHS beyond the APOE locus was tested by accounting for APOE-ε4 and APOE-ε2 dosage (i.e. including two variables that take values 0, 1 or 2), referred to as ‘PHS (burden)’. In addition, in absence of a real polygenic score, the effect of APOE-ε4 dosage beyond the effect of APOE-ε4 carrier status was quantified, referred to as ‘APOE-ε44’. That is, in this setting APOE-ε44 acts as a ‘polygenic’ score that only counts the number of APOE-ε4 alleles. Lastly, PHS was compared with standard PRS at two P-value cut-offs (1e-05 and 0.5), referred to as ‘PRS1’ and ‘PRS2’, respectively, and PRS-cs. All these methods were adjusted for APOE locus. However, previously ADNI contributed 441 samples to the Alzheimer’s Disease Genetics Consortium (ADGC), which in turn contributed to the IGAP study. Base GWAS and target data must be independent from each other to avoid overly optimistic results due to overfitting (Dudbridge, 2013). Thus, n = 441 subjects that contributed to the ADGC GWAS were removed for the comparison between PHS and PRS. Furthermore, analyses were restricted to subjects with predicted central European ancestry and self-reported white non-Hispanic ethnicity. Using large P-value thresholds (e.g. P < 0.05) results in the PRS comprising many SNPs and potentially makes the score more susceptible to geographical differences in genetic structure (Kerminen et al.,). To account for this bias in PRS2 and PRS-cs, we additionally tested associations while also adjusting for population structure by including the first five principal components of the genetic relatedness matrix in the corresponding models.
Disease status
Association between polygenic scores and latest recorded clinical diagnosis were conducted using logistic regression. In particular, four contrasts were explored: CN versus Alzheimer’s disease, CN versus MCI, MCI versus Alzheimer’s disease and CN versus MCI or Alzheimer’s disease. The model was adjusted for sex, years of education and age in addition to APOE (either for ‘status’ or for ‘burden’). For subjects with the CN diagnosis the latest recorded age was used; for Alzheimer’s disease subjects the earliest age with recorded Alzheimer’s disease diagnosis was used; for MCI subjects the earliest date of MCI diagnosis was used in the comparison to CN subjects (CN versus MCI and CN versus MCI or Alzheimer’s disease contrasts) and the latest age was used in the comparison with Alzheimer’s disease. In addition to these statistical association tests, we estimated the predictive performance of the logistic regression using 10-fold cross-validation. We used receiver operating characteristics (ROC) curves as well as the area under the ROC curve (AUC) as means to quantify and compare the model performance. Improvement by including polygenic scores over the ‘status’ or the ‘burden’ model was tested using a signed paired Wilcoxon rank-sum test on the 10 AUC estimates obtained from the cross-validation.
Baseline CSF biomarkers
The ADNI database provides data on Alzheimer’s disease-related CSF biomarkers amyloid β protein fragment 1–42 (Aβ), total tau (tau) and tau phosphorylated at threonine 181 (p-tau). The association between polygenic scores and baseline CSF biomarkers was tested using linear regression models. Prior to the analysis, the biomarker values were log-transformed to render their distribution more normal. Analyses were adjusted for age at baseline visit, sex, years of education and APOE (either for ‘status’ or for ‘burden’). Moreover, genetic influence may vary by disease stage, thus linear regression models were estimated for each clinical disease group (CN, MCI and Alzheimer’s disease); effect sizes were combined using the inverse variance method for meta-analyses to obtain an overall effect size and P-value. In addition to these statistical association tests, we estimated the predictive performance of the linear regression using 10-fold cross-validation. We used Pearson’s correlation coefficient as means to quantify the model performance. Improvement by including polygenic scores over the ‘status’ or the ‘burden’ model was tested using a signed paired Wilcoxon rank-sum test on the 10 correlation coefficient estimates obtained from the cross-validation.
Cross-sectional amyloid PET
We followed the analytical setup by Tan et al. (2019): associations of the polygenic scores with regional load of amyloid were computed. Amyloid load was derived from PET using the florbetapir tracer (AV45). Florbetapir synthesis and image acquisition details are described in detail elsewhere (http://adni-info.org, Landau et al., 2013). Processed regional AV45 levels were obtained from ‘ucberkeleyav45’ table providing values for cortical regions of the Desikan-Killiany Atlas (Desikan et al., 2006). Regional AV45 levels were divided by AV45 level in the cerebellum region to obtain regional SUVRs. Values for the right and left hemisphere were averaged. For each of the 34 cortical regions, a linear regression was estimated to quantify the effect of polygenic score on regional amyloid load. The models were adjusted for age at imaging, sex, years of education as well as APOE (either for ‘status’ or for ‘burden’). In addition to the setup by Tan et al. (2019) and as in the case for the CSF biomarkers, an analysis stratified by disease status was carried out. Multiple-testing correction for the tests across the 34 regions was conducted using false discovery rate (FDR).
Disease progression
Longitudinal data from ADNI were used to quantify the association of polygenic scores with clinical disease progression, i.e. clinical conversion. The effect of polygenic scores on clinical conversion was tested in a Cox proportional hazards model using subjects without a dementia diagnosis at the baseline visit. We defined clinical conversion as a dementia diagnosis in previously not demented participants. A left-truncated (age at entry) right-censored (age at event or last visit) design was chosen (Altmann et al., 2014). The model was adjusted for age at baseline, sex and years of education, APOE (either for ‘status’ or for ‘burden’) and stratified for baseline diagnosis (CN or MCI). In addition to these statistical association tests, we estimated the predictive performance of the Cox regression using 10-fold cross-validation. We used the concordance index, which is conceptually related to AUC, as means to quantify the model performance. Improvement by including polygenic scores over the ‘status’ or the ‘burden’ model was tested using a signed paired Wilcoxon rank-sum test on the 10 concordance indices obtained from the cross-validation.
Cognitive decline
We followed the analytical setup by Tan et al. (2019): The effect of polygenic scores on longitudinal measures of cognitive performance was assessed using linear mixed-effects models in non-demented people. In particular, the longitudinal development of clinical dementia rating sum of boxes (CDR-SB) obtained from ADNIMERGE and composite scores for memory (MEM) and executive function (EF) derived using item response theory methods (Crane et al., 2012) and obtained through ADNIMERGE (table uwnpsychsum). The first model was exactly specified as in Tan et al. (2019), where the target variable was the change from the baseline cognitive measurement (Δc) and the fixed effects were interactions between time since baseline (t) and sex, years of education, age, volume of the entorhinal cortex, amyloid burden in the frontal lobe, APOE (either for ‘status’ or for ‘burden’) and PHS and a random intercept for each subject. The variable of interest was the interaction of time and PHS. This model is referred to as ‘intercept only’. In addition, a linear mixed-effects model that included random effects for subject-level decline (random slopes) was used. Model likelihoods were compared with determine which model provides the better fit for the data. Random slope models have been previously used to explore longitudinal changes in ADNI (Holland et al., 2013). Moreover, we added fixed effects that did not interact with time as well as time itself as a fixed effect. These full models were compared with models lacking the polygenic score-by-time interaction using a likelihood ratio test to assess the effect of polygenic scores on longitudinal cognitive decline.
Longitudinal decline in grey matter
The longitudinal change in grey matter loss was quantified using the boundary shift integral (BSI; Leung et al., 2010). We accessed precomputed BSI values through ADNIMERGE (foxlabbsi table). BSI measures from volumetric T1-weighted MRIs were used that did not use the accelerated acquisition. Moreover, if for the same baseline/repeat pair a set of 1.5T as well as a set of 3.0T scans were available, the 3.0T scan was used. The association between BSI and polygenic scores was tested using a linear mixed-effects model as described above (with random intercepts and slopes). In particular, the BSI represents directly the Δc value, the model was adjusted for age, sex, years of education, APOE (either for ‘status’ or for ‘burden’) as well as their interactions with time since baseline. In addition to the whole brain BSI (BBSI), which was derived using the KN-BSI method (Leung et al., 2010) (KMNDBCB in the foxlabbsi table), BSI for the ventricles (VBSI), left (HLBSI) and right (HRBSI) hippocampi were tested as well.
Data availability
Data used in the preparation of this article were obtained from the ADNI database (http://adni.loni.usc.edu) and are freely available after registration. Derived polygenic scores (PRS1, PRS2 and PRS-cs) and R-scripts to analyse the data and to produce the results presented here are available at https://github.com/andrealtmann/polygenic_AD/
Results
Whole genotyping data were available for n = 1674 Alzheimer’s Disease Neuroimaging Initiative (ADNI) participants spread over three different genotyping platforms. QC and imputation were carried out per platform and were merged into a single file. PRSs were computed for all ADNI participants with genotyping data using the stage 1 results from a recent Alzheimer’s disease GWAS (Kunkle et al., 2019) at P-value cut-offs of 1e-05 (PRS1) and 0.5 (PRS2), resulting in PRS based on 55 and 101 450 SNPs, respectively. We also computed a PRS using the novel Bayesian regression and shrinkage priors (PRS-cs) method (Ge et al., 2019). PHS were obtained from the corresponding table in ADNIMERGE provided by Desikan et al. (2017). A total of 157 subjects were removed on the basis of non-European ancestry as inferred by SNPweights (Chen et al., 2013), and, n = 15 subjects were removed due to relatedness. Next, only those participants who self-reported as white non-Hispanic/Latino ethnicity were retained (n = 49 removed). Finally, n = 48 subjects lacking a PHS were excluded from the analyses, leaving a total of 1405 eligible ADNI participants for this study. The baseline demographics for the entire cohort are listed in Table 1.
Table 1.
Baseline demographics of the full cohort
| CN | MCI | Alzheimer’s disease | Total | P-value (F-value) | |
|---|---|---|---|---|---|
| N | 417 | 712 | 275 | 1404 | |
| Female (%) | 205 (49) | 279 (39) | 118 (43) | 603 (43) | 0.004 (5.37) |
| Age (SD) | 74.5 (5.6) | 73.1 (7.5) | 75.1 (7.7) | 73.9 (7.1) | 4.28e-05 (10.13) |
| APOE-ε4 (0/1/2) | 297/110/10 | 347/286/79 | 90/134/51 | 735/530/140 | <2e-16 (64.67) |
| MMSE (IQR) | 29 (29–30) | 28 (26–29) | 23 (22–25) | 28 (26–29) | <2e-16 (1061) |
| Education | 16.48 (2.64) | 16.02 (2.84) | 15.27 (2.92) | 16.01 (2.83) | 2.37e-07 (15.42) |
| Amyloid PET (−/+/NA) | 146/76/195 | 171/216/325 | 14/107/154 | 331/399/674 | <2e-16 (53.24) |
Comprising n = 1404 individuals after QC of the genetic data. Age given as mean, MMSE given as median, years of education as mean, amyloid PET as negative (−) or positive (+) and unavailable (NA). CN = cognitively normal; MCI = mild cognitive impairment.
The 441 ADNI subjects who contributed to the Alzheimer’s Disease Genetics Consortium (ADGC), which fed into stage 1 of the recent GWAS (Kunkle et al., 2019) were excluded from analyses involving the PRS (at either cut-off) and PRS-cs; affecting n = 410 participants in the dataset after the detailed QC. Baseline demographics for the reduced cohort are listed in Supplementary Table 1. We could confirm a very high degree of correlation between PHS and PRS1 (r2 = 0.75, Supplementary Fig. 1) as previously reported by Leonenko et al. (2019b), whereas there was no correlation between either PRS2 and either PHS or PRS1 (r2 ≤ 0.018). PRS-cs was highly correlated with PHS and PRS1 (r2 ≥ 0.57) and moderately correlated with PRS2 (r2 = 0.17). Moreover, PHS, PRS1 and PRS-cs exhibited a strong grouping effect by APOE-ε4 burden, which was not observed for PRS2 (Supplementary Fig. 1). This grouping effect is reflected in the correlation between APOE-ε4 burden and PHS (r2 = 0.84), PRS1 (r2 = 0.72) and PRS-cs (r2 = 0.58). PRS2 was not correlated with APOE-ε4 burden (r2 = 0.017), but instead showed correlation with the first four principal components of population structure (r2 > 0.05); max r2 for any of the other polygenic scores with any principal component was 0.0073.
Polygenic scores are associated with clinical diagnosis beyond APOE
The association between polygenic scores and most recent diagnosis was tested for four contrasts using logistic regression. After adjusting for age, sex, years of education and APOE-ε4 carrier status, PHS (status) showed a strong association in all three contrasts involving CN subjects (P ≤ 4.32e-06), while there was only a nominally significant effect between MCI and Alzheimer’s disease (P = 0.044; Table 2). APOE-ε4 burden (i.e. number of ε4 alleles), in addition to APOE-ε4 status, showed the same pattern, although at a reduced magnitude: P ≤ 7.74e-03 for contrasts involving CN subjects and P = 0.306 for the association between MCI and Alzheimer’s disease, confirming that APOE-ε4 burden was predictive of diagnosis beyond APOE-ε4 status. Owing to this pronounced effect of APOE-ε4 homozygous carriers, the association of PHS with diagnosis was tested after comprehensive adjustment for the APOE locus, i.e. accounting for APOE-ε4 burden as well as APOE-ε2 burden. The association of PHS (burden) was markedly reduced but remained significant for contrasts involving CN subjects P ≤ 1.54e-03; and at trend level for the contrast between MCI and Alzheimer’s disease (P = 0.092; Table 2). Inclusion of PHS in the model using APOE status improved predictive performance in all four settings (AUC improvement ranging from 0.015 to 0.025; Supplementary Table 2 and Fig. 2). Adding PHS to the model that uses APOE burden led only to statistically significant improvement for the CN versus Alzheimer’s disease classification (AUC increased from 0.765 to 0.777; P < 0.006; Supplementary Table 2 and Fig. 2).
Table 2.
Association of polygenic scores with most recent diagnosis
| Score | CN|AD | CN|MCI | MCI|AD | CN|MCI+AD | |
|---|---|---|---|---|---|
| Entire cohort (n = 1404) | PHS (status) | 9.46E-10 | 4.32E-06 | 0.044 | 1.48E-09 |
| PHS (burden) | 8.45E-09 | 1.54E-03 | 0.092 | 2.21E-05 | |
| APOE-ε44 | 6.50E-04 | 7.74E-03 | 0.30 | 5.99E-04 | |
| Reduced cohort (n = 994) | PHS (burden) | 1.28E-03 | 6.75E-05 | 0.32 | 8.49E-05 |
| PRS1 | 0.028 | 7.78E-03 | 0.68 | 5.99E-03 | |
| PRS2 | 0.031 | 2.91E-04 | 0.14 | 4.98E-04 | |
| PRS2* | 4.12E-07 | 8.26E-03 | 0.010 | 3.44E-05 | |
| PRS-cs | 7.02E-03 | 2.80E-05 | 0.30 | 3.06E-05 | |
| PRS-cs* | 2.69E-04 | 7.37E-04 | 0.43 | 2.80E-05 |
Results show the uncorrected P-values obtained from logistic regression while adjusting for age, sex, education and APOE (either for ‘status’ or for ‘burden’). Scores with * are additionally adjusted for five principal components reflecting population structure. Reduced cohort does not include subjects who contributed to ADGC. CN = cognitively normal; MCI = mild cognitive impairment; AD = Alzheimer’s disease.
After removal of ADGC participants, PHS (burden) was compared with PRS1, PRS2 and PRS-cs on the same dataset including comprehensive modelling of the APOE locus for all scores. PHS on the reduced dataset showed comparable results to the full dataset, i.e. significant P ≤ 1.28e-03 effects for contrasts involving CN subjects. PRS1 and PRS2 both replicated the pattern of associations seen with PHS: associations on contrasts including CN (P ≤ 0.031) but not detectable between MCI and Alzheimer’s disease (P ≥ 0.14). PRS-cs showed P-values of the same magnitude as PHS (burden). After adjusting the model for PRS2 in addition for population structure the association with diagnosis was more pronounced (P = 4.12e-07 with population structure compared with P = 0.031 without) for the other contrasts no such pronounced effect was observed. The PRS-cs model with additional adjustment for population structure showed slightly improved P-values over the model without adjustment. However, owing due to only a marginal change in results, the adjustment for population structure in the PRS-cs model was not investigated further.
Predictive performance over the model using APOE burden was improved by adding PHS when classifying CN versus MCI (AUC increase of 0.018) or by including PHS or PRS-cs when classifying CN versus MCI or Alzheimer’s disease (AUC increase of 0.011 or 0.02; Supplementary Table 2 and Fig. 2).
Polygenic scores are associated with CSF tau but not CSF Aβ beyond APOE
The association of polygenic scores with log-transformed baseline CSF biomarkers (Aβ, tau and p-tau) was tested using linear models stratified by disease group. When adjusting only for APOE ε4 status (i.e. ε4+/−) PHS (status) showed a strong association with baseline CSF Aβ levels (Table 3; P = 9.99e-07). However, the effect was driven by homozygous APOE-ε4 carriers (APOE-ε44, P = 2.54e-10). After comprehensive adjustment for the APOE locus, the association between PHS (burden) and Aβ disappeared (P = 0.87). This held true also on the reduced dataset for PHS (burden) as well as PRS1, PRS2 and PRS-cs (Table 3; P > 0.31). In contrast, the effect of polygenic scores (PHS and PRS) on either tau and p-tau was not driven by APOE-ε4 homozygotes (P ≥ 0.067). PHS (status) showed significant associations with both CSF tau (P = 1.06e-03) and p-tau levels (P = 9.25e-04), which remained significant after comprehensive adjustment for the APOE locus in PHS (burden) (P < 3.15e-03). This held true also for the PHS (burden) and PRS1 on the reduced dataset (Table 3). These associations were corroborated by the performance of the predictive models where adding PHS and PRS1 to the APOE burden baseline model improved the model for tau and p-tau but not Aβ (Supplementary Table 3).
Table 3.
Associations between polygenic scores and CSF biomarkers Aβ, total tau and phosphorylated tau
| Sample size (N) | Score | Aβ | Tau | p-tau |
|---|---|---|---|---|
| 1008 | PHS (status) | 9.99E-07 | 1.06E-03 | 9.25E-04 |
| PHS (burden) | 0.87 | 2.33E-03 | 3.15E-03 | |
| APOE-ε44 | 2.54E-10 | 0.067 | 0.073 | |
| 807 | PHS (burden) | 0.37 | 1.04E-03 | 6.85E-04 |
| PRS1 | 0.31 | 1.33E-04 | 5.96E-05 | |
| PRS2 | 0.49 | 0.053 | 0.087 | |
| PRS2* | 0.42 | 0.95 | 0.95 | |
| PRS-cs | 0.82 | 0.15 | 0.10 |
Biomarker values were log-transformed. The table depicts uncorrected P-values from linear models adjusted for age, sex, education and APOE (either for ‘status’ or for ‘burden’). Results in the top part are for the entire cohort (n = 1008) and in the bottom part after excluding ADGC subjects (n = 807). The model indicated with * was additionally adjusted for genetic population structure.
Polygenic scores show only moderate association with cortical amyloid beyond APOE
In addition to CSF Aβ1–42, cortical amyloid load was quantified using Florbetapir (18F) PET (AV45) and tested for association with polygenic scores for Alzheimer’s disease. There were n = 917 subjects with AV45 scans (730 obtained at baseline visit). When adjusting for APOE-ε4 status there were significant associations between PHS (status) and regional amyloid in all 34 cortical ROIs (PFDR range from 1.41e-03 to 6.22e-07; Supplementary Fig. 3 and Table 4) confirming earlier results by Tan et al. (2019). Similar to CSF amyloid, cortical amyloid was significantly associated with APOE-ε4 burden over APOE-ε4 status (28 of 34 regions with PFDR from 0.035 to 6.65e-04; Supplementary Fig. 3 and Table 4). Consequently, comprehensive adjustment for the APOE locus reduced the association strength of PHS (burden) with regional amyloid, but all of the 34 ROIs remained significantly associated (PFDR < 0.0478; Supplementary Fig. 3 and Table 4). Additional stratification by disease status at amyloid imaging, left only one significant ROI with an PFDR = 0.0444: transverse temporal gyrus; another 26 ROIs were associated at trend level (PFDR<0.1; Supplementary Fig. 3 and Table 4). This pattern remained unchanged on the reduced dataset without the ADGC participants (Fig. 1; Supplementary Table 5) comprising n = 822 participants with AV45 scans (730 obtained at baseline visit). On the same dataset, PRS1 showed consistent association with regional amyloid despite comprehensive adjustment for APOE burden and stratification by disease group (26 of 34 ROIs; PFDR from 0.0446 to 4.00e-04; Fig. 1; Supplementary Table 5). Neither, PRS-cs nor PRS2 (with or without correction for population structure) showed any association with regional amyloid load (Supplementary Table 5).
Table 4.
Effect of polygenic scores on clinical conversion obtained from Cox proportional hazards models
| Sample size (N) | Score | Beta | SE | P-value |
|---|---|---|---|---|
| 1092 | PHS (status) | 0.290 | 0.09 | 0.002 |
| PHS (burden) | 0.372 | 0.15 | 0.013 | |
| APOE-ε44 | 0.295 | 0.17 | 0.088 | |
| 813 | PHS (burden) | −0.005 | 0.20 | 0.99 |
| PRS1 | −0.029 | 0.15 | 0.84 | |
| PRS2 | −0.016 | 0.08 | 0.84 | |
| PRS2* | 0.290 | 0.11 | 0.012 | |
| PRS-cs | −0.181 | 0.12 | 0.11 |
Clinical conversion was defined as a dementia diagnosis in previously not demented participants. Rows correspond to different polygenic scores and estimates for the full dataset (top) and after removal of ADGC subjects (bottom). Beta = log hazards ratio; SE = standard error. *was additionally adjusted for genetic population structure.
Figure 1.
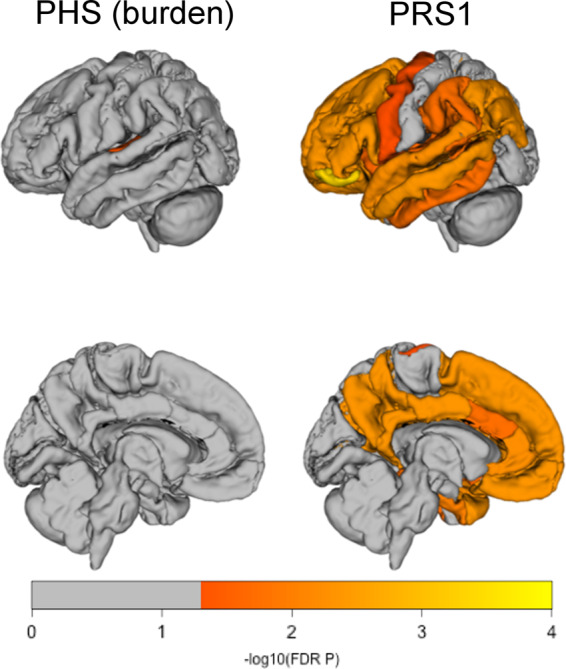
Regional associations between polygenic risk scores and regional uptake of amyloid. The colour code represents the -log10 FDR-corrected P-values obtained from linear models adjusted for age, sex, education, APOE-ε4 count, APOE-ε2 count and stratified by disease group at the time of imaging. There were no associations with PFDR < 0.05 for PRS2 and PRS-cs.
Clinical conversion
The longitudinal design of the ADNI study was leveraged to test the association of polygenic scores with clinical progression to Alzheimer’s disease in non-demented participants using Cox proportional hazards models. There were 1092 non-demented subjects (406 CN, 686 MCI) of whom 318 converted to Alzheimer’s disease (29 CN, 289 MCI). When adjusting for APOE-ε4 status in the Cox regression, PHS (status) showed a significant effect on clinical progression (P = 0.002), with higher polygenic hazard increasing the risk of conversion, while APOE-ε4 burden showed a trend over APOE-ε4 status (P = 0.09). Comprehensive adjustment for the APOE locus led to a decline in association of PHS (burden) with clinical conversion but remained significant (P = 0.013). On the reduced dataset, there were 813 subjects (258 CN, 555 MCI) of whom 179 converted to Alzheimer’s disease (11 CN, 168 MCI). None of the scores showed a significant association with clinical conversion (P ≥ 0.11, Table 4). However, PRS2 adjusted for population structure showed an association with clinical conversion (P = 0.012; Table 4). None of the polygenic scores improved the predictive performance of the baseline models in the Cox regression (Supplementary Table 6).
Cognitive decline
The effect of polygenic burden on cognitive decline was assessed on 4465 observations obtained from 601 non-demented subjects (220 CN, 381 MCI) using linear mixed effect models with an initial setup as described in Tan et al. (2019) followed by a more refined model (see Methods section). Initially, in a model adjusted for APOE-ε4 status and using only random intercepts PHS (status) showed a very strong association with decline in CDR-SB (P = 7.8e-09), memory (P = 8.15e-03) and EF (P = 1.32e-03) replicating earlier results (Tan et al., 2019). Using the same model with only random intercepts, APOE-ε4 burden exhibited effects of similar strength (Table 5). Consequently, after comprehensive adjustment for APOE, PHS (burden) only showed moderate association with CDR-SB (P = 3.95e-03) and none for memory or EF (P > 0.10). Using a linear mixed-effects model with random slopes and random intercepts (while adjusting only for APOE-ε4 status), substantially reduced the association between PHS (status) and CDR-SB (P = 5.92e-03), memory (P = 0.012) and marginally for EF (P = 3.7e-03) compared with random intercepts model (Table 5). Models using random slopes and random intercepts showed significantly higher likelihood (P < 2.2e-16) than models using only random intercepts (Supplementary Table 7). Combining comprehensive adjustment for the APOE locus and the linear mixed effects model with random slopes and random intercept left no significant association (P ≥ 0.08) between the PHS (burden) and decline in cognitive scores. On the same dataset (i.e. there were no ADGC participants), PRS1 showed a moderate significant association with decline in CDR-SB (P = 3.57e-03) but not with decline in memory or EF (P ≥ 0.42). There was neither a significant association for PRS2 (P ≥ 0.21) nor for PRS-cs (P ≥ 0.34). After additional adjustment for population structure, PRS2 showed a trending association with decline in CDR-SB (P = 0.059) but neither with memory nor with EF decline (P ≥ 0.40).
Table 5.
Associations between polygenic scores and cognitive decline
| Model | Score | CDR-SB | MEM | EF |
|---|---|---|---|---|
| Random intercept | PHS (status) | 7.80E-09 | 8.15E-03 | 1.32E-03 |
| PHS (burden) | 3.95E-03 | 0.20 | 0.10 | |
| APOE-ε44 | 2.09E-08 | 2.02E-03 | 9.19E-05 | |
| Random intercept + slope | PHS (status) | 5.92E-03 | 0.012 | 3.70E-03 |
| PHS (burden) | 0.080 | 0.47 | 0.55 | |
| PRS1 | 3.57E-03 | 0.42 | 0.65 | |
| PRS2 | 0.45 | 0.21 | 0.71 | |
| PRS2* | 0.059 | 0.40 | 0.43 | |
| PRS-cs | 0.34 | 0.66 | 0.65 |
P-values for the score-by-time interaction on change in CDR sum of boxes) and composite scores for memory and executive function. Rows correspond to different polygenic scores. Linear mixed-effects models were estimated with either random intercept or random intercept and random slope (for time). CDR-SB = clinical dementia rating sum of boxes; MEM = memory; EF = executive function. *was additionally adjusted for genetic population structure.
Grey matter decline
The BSI (Leung et al., 2010) was used to quantify longitudinal decline in grey matter for the whole brain, the ventricles as well as left and right hippocampus. On the entire dataset, there were n = 1250 subjects with 3909 scans for whole brain and ventricular BSI. A reduced number of subjects (n = 618) and scans (n = 1714) was available for hippocampal BSI. Using a linear mixed-effects model with random intercepts and random slopes, there was a strong association between PHS (status) and whole brain and ventricular BSI (P ≤ 2.38e-05) and trending for hippocampal BSI (P ≤ 0.091). Some of these effects were driven by APOE-ε4 homozygotes (Table 6) and consequently, the association between PHS (burden) and BSI measures was reduced (P ≤ 4.12e-03 for whole brain and ventricular; P ≥ 0.3 for hippocampi). On the dataset lacking the ADGC participants, there was no significant association between PHS (burden) or PRS2 and any BSI measures (P ≥ 0.19). PRS1 was associated with whole brain and ventricular BSI (P ≤ 1.7e-03) and marginally for right hippocampus and trending for left hippocampus (Table 6). Similarly, PRS-cs showed at least trend-level association (P ≤ 0.061) for any BSI.
Table 6.
Associations between polygenic scores and atrophy
| Subjects (scans) | Score | BBSI | VBSI | HRBSI* | HLBSI* |
|---|---|---|---|---|---|
| n = 1250 (n = 3909) | PHS (status) | 1.54e-5 | 2.38e-5 | 9.14e-2 | 5.45e-2 |
| PHS (burden) | 2.56e-3 | 4.12e-3 | 0.82 | 0.30 | |
| APOE-ε44 | 4.93e-3 | 1.50e-2 | 1.25e-2 | 2.96e-2 | |
| n = 878 (n = 2754) | PHS (burden) | 0.19 | 0.42 | 0.82 | 0.30 |
| PRS1 | 6.7e-4 | 1.7e-3 | 0.02 | 0.065 | |
| PRS2 | 0.65 | 0.91 | 0.88 | 0.29 | |
| PRS2** | 0.02 | 7.7e-3 | 0.10 | 0.15 | |
| PRS-cs | 0.0041 | 0.031 | 0.061 | 0.004 |
P-values for a time-by-polygenic score interaction effect on longitudinal changes in grey matter quantified by the BSI. Columns correspond to different target regions and/or measures of BSI. BBSI = whole brain BSI; VBSI = Ventricular BSI; HRBSI = Right side hippocampal BSI; HLBSI = Left side hippocampal BSI. * indicates a reduced dataset with n = 618 subjects and n = 1714 scans. ** was additionally adjusted for genetic population structure.
Discussion
In a series of statistical tests, we investigated whether polygenic scores are associated with a range of Alzheimer’s disease-related traits beyond the APOE locus. We found that polygenic scores were associated beyond APOE with clinical diagnosis, CSF-tau levels and, to a minor degree, with progressive atrophy. However, for many other tested traits such as clinical disease progression, CSF amyloid, cognitive decline and cortical amyloid load, the additional effects of polygenic scores beyond APOE were of minor nature. Our results demonstrate that incomplete adjustment for the APOE locus can lead to severely overestimated effects of polygenic scores due to APOE-ε4 homozygous participants. For instance, while our analysis confirmed the PHS’ association with a range of Alzheimer’s disease-related measures beyond APOE-ε4 carrier status first reported by Tan et al. (2019), these effects mostly disappeared when using a comprehensive correction for the APOE locus comprising the number of APOE-ε2 and APOE-ε4 alleles, mainly owing to the high correlation between PHS and APOE-ε4 burden. Conversely, on the same cohort of subjects, we tested a simple ‘polygenic’ score comprising only the APOE-ε4 allele count and could demonstrate its statistical effects beyond APOE-ε4 carrier status. Thus, taken together these results suggest that the previously observed associations of PHS with Alzheimer’s disease-related measures (Tan et al., 2017, 2018, 2019) were partially driven by APOE-ε4 homozygous participants. Our analysis also uncovered a second source of statistical modelling that led to overestimated effects of PHS on longitudinal changes in cognition and atrophy. The linear mixed-effects models used in PHS analyses (Desikan et al., 2017; Tan et al., 2017, 2018, 2019) only allowed for subject-specific intercepts, i.e. requiring that the rate of decline is shared between all subjects regardless of disease group, baseline characteristics and genetics, and only modulated by the few variables included as fixed effects. More flexibility is provided by allowing individual rates of decline that are subject-specific (random slopes) in addition to random intercepts. Such random slope linear mixed-effects models have been used recently to analyse longitudinal changes in ADNI data (Holland et al., 2013; Mormino et al., 2016) and are likely to more accurately model ‘real-life’ scenarios where individuals decline at very heterogenous rates. Indeed, the addition of random slopes resulted in a significantly better model fit (P < 2.2e-16) on our data than models using only random intercepts (Supplementary Table 7). Here, using random slopes in addition to random intercepts led to dramatically reduced associations between PHS and longitudinal changes in cognitive decline. In combination with a corrected adjustment for APOE-ε4 burden, this led to a complete lack of association between the PHS-by-time interaction and CDR-SB (P = 0.08), which showed initially a very strong association (P = 7.80E-09) with the same setup as in Tan et al. (2019) (who reported P = 2.44e-10).
Overall, we found that PHS and PRS1, which was based on SNPs exceeding the genome-wide suggestive threshold (P = 1e-05), were highly correlated (r2 = 0.75; Supplementary Fig. 1). This confirms earlier results comparing PHS and PRS, which reported that despite the different nature of the statistical analysis, the estimated effects per SNP were very similar (Leonenko et al., 2019b). Consequently, the association profile of the two scores was quite similar across the tested Alzheimer’s disease-related measures. In addition to PRS1, we tested two other PRS scores: PRS2, which was based on SNPs exceeding the P = 0.5 threshold, and PRS-cs using a novel method that adjust GWAS-derived effect size estimates for local LD structure. PRS-cs was based on all available SNP data, making it in theory more similar to PRS2, but the correction effect, which emphasized variants with strong effects, rendered the final score to be more correlated to PRS1 rather than to PRS2.
PRS2 showed poorer association with traits compared with PRS1. However, PRS2 was quite sensitive to the correction of population structure (as reflected by substantial correlations between PRS2 and principal components reflecting population structure). The effect was most pronounced when testing for an association with clinical diagnosis where the P-value improved from P = 0.031 (without correction) to P = 4.12e-07 (with correction) in the CN versus Alzheimer’s disease contrast (Table 2). A recent study in the Finnish population demonstrated that polygenic scores are sensitive to geographic patterns even in relatively homogenous populations (Kerminen et al., 2019). Furthermore, the observed bias increased with the inclusion of more variants in the polygenic scores. Thus, while PRS1 (and PHS) may be relatively robust to population bias, PRS2 is more sensitive but requires additional correction for population structure. Overall PRS-cs was less biased by this effect, probably owing to the emphasis on high effect size variants, which is expressed by the high correlation with PHS and PRS1.
After comprehensive adjustment for the APOE locus and stratification for disease group, there was no association between polygenic scores and amyloid (measured in CSF or through amyloid PET). In initial reports, PHS showed a strong association with cortical amyloid (Tan et al., 2019, with strongest association seen in Rostral Middle Frontal gyrus; PFDR = 5.82E-08; replicated in this study with PFDR=6.21e-07). Implementing adjustments for APOE locus and disease status, only one ROI remained significant (Transverse Temporal gyrus, PFDR = 0.038). Likewise, CSF Aβ did not show any association with polygenic scores beyond APOE (P > 0.34). Only CSF tau and p-tau were consistently associated with polygenic risk beyond APOE and clinical diagnosis, with the strongest effects for PHS and PRS1. We found that the genes contributing to PRS1 were significantly enriched (P = 4.1e-04) among the protein-interaction partners of tau (Supplementary results). The association with tau rather than amyloid pathology confirms recently published results by Leonenko et al. (2019a) showing that amyloid pathology is mainly driven by APOE genotype and that polygenic risk contributes to risk further conversion to Alzheimer’s disease. Moreover, recent works suggest that brain amyloid deposition is driven by the APOE locus (Yan et al., 2018) and that a genome-wide significant variant in the Alzheimer’s disease risk gene BIN1 (rs744373) is associated with tau pathology instead of amyloid pathology (Franzmeier et al., 2019).
For some analyses, we conducted predictive modelling to assess the effect of polygenic scores in addition to the statistical association tests. The results supported our findings: using APOE burden as opposed to APOE-ε4 status provided better diagnostic prediction, however, adding PRSs to models already using APOE burden yielded only marginal gains (Supplementary Table 2). CSF tau and p-tau were better predicted by adding PHS or PRS1 to the models, but it was not the case for CSF Aβ (Supplementary Table 3). Lastly, there was no predictive gain by using polygenic scores on clinical progression in the survival analysis (Supplementary Table 6).
One limitation of this study was that, owing to sample overlap, the entire ADNI cohort could not be used for the PHS and PRS comparison. However, in order to demonstrate the effect of being APOE-ε4 homozygous, we tested APOE-ε4 burden in addition to APOE-ε4 status and compared the results to PHS on the entire cohort, finding broadly consistent results.
In summary, this work presents a comprehensive comparison of PHS, PRS and PRS-cs and their relation to a range of Alzheimer’s disease-related traits within the ADNI cohort. The results showed that there are not many effective differences between PHS and PRS1 in terms of associations with disease stage or biomarkers. In fact, many associations of PHS reported in earlier works were inflated due to lack of adjustment for the APOE locus, clinical diagnosis or underspecified statistical models. Given the ease with which PRS can be derived (i.e. from publicly available summary statistics) compared with PHS (i.e. requires reanalysis of the GWAS data), we believe the PRS constitutes the more practical choice. Moreover, on many of the tested traits, the major driving factor remained the APOE locus, with the sole exception of quantitative CSF-tau and p-tau measures.
Funding
A.A. holds a Medical Research Council eMedLab Medical Bioinformatics Career Development Fellowship. This work was supported by the Medical Research Council [grant number MR/L016311/1]. M.A.S. acknowledges financial support the Engineering and Physical Sciences Research Council (EPSRC)-funded UCL Centre for Doctoral Training in Medical Imaging (EP/L016478/1). L.M.A. has received funding European Union’s Horizon 2020 research and innovation programme under grant agreement No. 666992. E.d.S. acknowledges supported from the National Institute for Health University College London Hospitals Biomedical Centre. D.M.C. is supported by grants from the Alzheimer Society (AS-PG-15-025), Alzheimer’s Research UK (ARUK-PG2014-1946) and Medical Research Council UK (MR/M023664/1). J.H. was supported in part by the Dolby Family Fund, the UK Dementia Research Institute and the Health University College London Hospitals Biomedical Centre as well as by an anonymous foundation. J.M.S. acknowledges the support of the National Institute for Health Research University College London Hospitals Biomedical Research Centre, Wolfson Foundation, Engineering and Physical Sciences Research Council (EPSRC) (EP/J020990/1), Medical Research Council (MRC) Dementias Platform UK (MR/L023784/1), Alzheimer’s Research UK (ARUK-PG2017-1946), Brain Research UK (UCC14191), Weston Brain Institute (UB170045) and European Union’s Horizon 2020 research and innovation programme (Grant 666992).
Data collection and sharing for this project were funded by the Alzheimer’s Disease Neuroimaging Initiative (ADNI; National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH-12-2-0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol-Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann-La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California.
Competing interests
J.M.S. has received research funding and PET tracer from AVID Radiopharmaceuticals (a wholly owned subsidiary of Eli Lilly); has consulted for Roche, Eli Lilly, Biogen and Merck, GE; received royalties from Oxford University Press and Henry Stewart Talks; given education lectures sponsored by Eli Lilly, Biogen and GE; and serves on a Data Safety Monitoring Committee for Axon Neuroscience SE.
Supplementary Material
Glossary
- Aβ
amyloid beta
- ADGC
Alzheimer’s disease Genetics Consortium
- ADNI
Alzheimer’s disease neuroimaging initiative
- APOE
Apolipoprotein E gene
- AUC
area under the ROC curve
- BSI
boundary shift integral
- CDR
clinical dementia rating
- CN
cognitively normal
- CSF
cerebrospinal fluid
- EF
executive function
- GWAS
genome-wide association study
- LD
linkage disequilibrium
- MAF
minor allele frequency
- MCI
mild cognitive impairment
- MEM
memory
- PHS
polygenic hazard score
- PRS
polygenic risk score
- QC
quality control
- ROC
Receiver Operating Characteristics
- ROI
region of interest
- SNP
single nucleotide polymorphism
- SUVR
standardized uptake value ratio
Appendix: ADNI collaborators
Part A: Leadership and Infrastructure
Principal Investigator
Michael W. Weiner, MD UC San Francisco
ATRI PI and Director of Coordinating Center Clinical Core
Paul Aisen, MD University of Southern California
Executive Committee
Michael Weiner, MD UC San Francisco
Paul Aisen, MD University of Southern California
Ronald Petersen, MD, PhD Mayo Clinic, Rochester
Clifford R. Jack, Jr., MD Mayo Clinic, Rochester
William Jagust, MD UC Berkeley
John Q. Trojanowki, MD, PhD U Pennsylvania
Arthur W. Toga, PhD USC
Laurel Beckett, PhD UC
Davis Robert C. Green, MD, MPH Brigham and Women’s Hospital/Harvard Medical School
Andrew J. Saykin, PsyD Indiana University
John Morris, MD Washington University St. Louis
Leslie M. Shaw University of Pennsylvania
ADNI External Advisory Board (ESAB)
Zaven Khachaturian, PhD Prevent Alzheimer’s Disease 2020 (Chair)
Greg Sorensen, MD Siemens
Maria Carrillo, PhD Alzheimer’s Association
Lew Kuller, MD University of Pittsburgh
Marc Raichle, MD Washington University St. Louis
Steven Paul, MD Cornell University
Peter Davies, MD Albert Einstein College of Medicine of Yeshiva University
Howard Fillit, MD AD Drug Discovery Foundation
Franz Hefti, PhD Acumen Pharmaceuticals
David Holtzman, MD Washington University St. Louis
M. Marcel Mesulam, MD Northwestern University
William Potter, MD National Institute of Mental Health
Peter Snyder, PhD Brown University
ADNI 3 Private Partner Scientific Board (PPSB)
Veronika Logovinsky, MD, PhD Eli Lilly (Chair)
Data and Publications Committee
Robert C. Green, MD, MPH BWH/HMS (Chair)
Resource Allocation Review Committee
Tom Montine, MD, PhD University of Washington (Chair)
Clinical Core Leaders
Ronald Petersen, MD, PhD Mayo Clinic, Rochester (Core PI)
Paul Aisen, MD University of Southern California
Clinical Informatics and Operations
Gustavo Jimenez, MBS USC
Michael Donohue, PhD USC
Devon Gessert, BS USC
Kelly Harless, BA USC
Jennifer Salazar, MBS USC
Yuliana Cabrera, BS USC
Sarah Walter, MSc USC
Lindsey Hergesheimer, BS USC
Biostatistics Core Leaders and Key Personnel
Laurel Beckett, PhD UC Davis (Core PI)
Danielle Harvey, PhD UC Davis
Michael Donohue, PhD UC San Diego
MRI Core Leaders and Key Personnel
Clifford R. Jack, Jr., MD Mayo Clinic, Rochester (Core PI)
Matthew Bernstein, PhD Mayo Clinic, Rochester
Nick Fox, MD University of London
Paul Thompson, PhD UCLA School of Medicine
Norbert Schuff, PhD UCSF MRI
Charles DeCArli, MD UC Davis
Bret Borowski, RT Mayo Clinic
Jeff Gunter, PhD Mayo Clinic
Matt Senjem, MS Mayo Clinic
Prashanthi Vemuri, PhD Mayo Clinic
David Jones, MD Mayo Clinic
Kejal Kantarci Mayo Clinic
Chad Ward Mayo Clinic
PET Core Leaders and Key Personnel
William Jagust, MD UC Berkeley (Core PI)
Robert A. Koeppe, PhD University of Michigan
Norm Foster, MD University of Utah
Eric M. Reiman, MD Banner Alzheimer’s Institute
Kewei Chen, PhD Banner Alzheimer’s Institute
Chet Mathis, MD University of Pittsburgh
Susan Landau, PhD UC Berkeley
Neuropathology Core Leaders
John C. Morris, MD Washington University St. Louis
Nigel J. Cairns, PhD, FRCPath Washington University St. Louis
Erin Franklin, MS, CCRP Washington University St. Louis
Lisa Taylor-Reinwald, BA, HTL Washington University St. Louis (ASCP)—Past Investigator
Biomarkers Core Leaders and Key Personnel
Leslie M. Shaw, PhD UPenn School of Medicine
John Q. Trojanowki, MD, PhD UPenn School of Medicine
Virginia Lee, PhD, MBA UPenn School of Medicine
Magdalena Korecka, PhD UPenn School of Medicine
Michal Figurski, PhD UPenn School of Medicine
Informatics Core Leaders and Key Personnel
Arthur W. Toga, PhD USC (Core PI)
Karen Crawford USC
Scott Neu, PhD USC
Genetics Core Leaders and Key Personnel
Andrew J. Saykin, PsyD Indiana University
Tatiana M. Foroud, PhD Indiana University
Steven Potkin, MD UC UC Irvine
Li Shen, PhD Indiana University
Kelley Faber, MS, CCRC Indiana University
Sungeun Kim, PhD Indiana University
Kwangsik Nho, PhD Indiana University
Initial Concept Planning & Development
Michael W. Weiner, MD UC San Francisco
Lean Thal, MD UC San Diego
Zaven Khachaturian, PhD Prevent Alzheimer’s Disease 2020
Early Project Proposal Development
Leon Thal, MD UC San Diego
Neil Buckholtz National Institute on Aging
Michael W. Weiner, MD UC San Francisco
Peter J. Snyder, PhD Brown University
William Potter, MD National Institute of Mental Health
Steven Paul, MD Cornell University
Marilyn Albert, PhD Johns Hopkins University
Richard Frank, MD, PhD Richard Frank Consulting
Zaven Khachaturian, PhD Prevent Alzheimer’s Disease 2020
NIA
John Hsiao, MD National Institute on Aging
Part B: Investigators by Site
Oregon Health & Science University:
Joseph Quinn, MD
Lisa C. Silbert, MD
Betty Lind, BS
Jeffrey A. Kaye, MD, A.—Past Investigator
Raina Carter, BA—Past Investigator
Sara Dolen, BS—Past Investigator
University of Southern California:
Lon S. Schneider, MD
Sonia Pawluczyk, MD
Mauricio Becerra, BS
Liberty Teodoro, RN
Bryan M. Spann, DO, PhD—Past Investigator
University of California—San Diego:
James Brewer, MD, PhD
Helen Vanderswag, RN
Adam Fleisher, MD—Past Investigator
University of Michigan:
Jaimie Ziolkowski, MA, BS, TLLP
Judith L. Heidebrink, MD, MS
Joanne L. Lord, LPN, BA, CCRC—Past Investigator
Mayo Clinic, Rochester:
Ronald Petersen, MD, PhD
Sara S. Mason, RN
Colleen S. Albers, RN
David Knopman, MD
Kris Johnson, RN—Past Investigator
Baylor College of Medicine:
Javier Villanueva-Meyer, MD
Valory Pavlik, PhD
Nathaniel Pacini, MA
Ashley Lamb, MA
Joseph S. Kass, MD, LD, FAAN
Rachelle S. Doody, MD, PhD—Past Investigator
Victoria Shibley, MS—Past Investigator
Munir Chowdhury, MBBS, MS—Past Investigator
Susan Rountree, MD—Past Investigator
Mimi Dang, MD—Past Investigator
Columbia University Medical Center:
Yaakov Stern, PhD
Lawrence S. Honig, MD, PhD
Karen L. Bell, MD
Randy Yeh, MD
Washington University, St. Louis:
Beau Ances, MD, PhD, MSc
John C. Morris, MD
David Winkfield, BS
Maria Carroll, RN, MSN, GCNS-BC
Angela Oliver, RN, BSN, MSG
Mary L. Creech, RN, MSW—Past Investigator
Mark A. Mintun, MD—Past Investigator
Stacy Schneider, APRN, BC, GNP—Past Investigator
University of Alabama—Birmingham:
Daniel Marson, JD, PhD
David Geldmacher, MD
Marissa Natelson Love, MD
Randall Griffith, PhD, ABPP—Past Investigator
David Clark, MD—Past Investigator
John Brockington, MD—Past Investigator
Mount Sinai School of Medicine:
Hillel Grossman, MD
Effie Mitsis, PhD—Past Investigator
Rush University Medical Center:
Raj C. Shah, MD
Melissa Lamar, PhD
Patricia Samuels
Wien Center:
Ranjan Duara, MD
Maria T. Greig-Custo, MD
Rosemarie Rodriguez, PhD
Johns Hopkins University:
Marilyn Albert, PhD
Chiadi Onyike, MD
Daniel D’Agostino II, BS
StepMohammed O. Sheikh, MD
Jamika Singleton-Garvin, CCRP
Anaztasia Ulysse
Mrunalini Gaikwad
Duke University Medical Center:
P. Murali Doraiswamy, MBBS, FRCP
Jeffrey R. Petrella, MD
Olga James, MD
Salvador Borges-Neto, MD
Terence Z. Wong, MD—Past Investigator
Edward Coleman—Past Investigator
University of Pennsylvania:
Jason H. Karlawish, MD
David A. Wolk, MD
Sanjeev Vaishnavi, MD
Christopher M. Clark, MD—Past Investigator
Steven E. Arnold, MD—Past Investigator
University of Kentucky:
Charles D. Smith, MD
Greg Jicha, MD
Peter Hardy, PhD
Riham El Khouli, MD
Elizabeth Oates, MD
Gary Conrad, MD
University of Pittsburgh:
Oscar L. Lopez, MD
MaryAnn Oakley, MA
Donna M. Simpson, CRNP, MPH
University of Rochester Medical Center:
Anton P. Porsteinsson, MD
Kim Martin, RN
Nancy Kowalksi, MS, RNC
Melanie Keltz, RN
Bonnie S. Goldstein, MS, NP—Past Investigator
Kelly M. Makino, BS—Past Investigator
M. Saleem Ismail, MD—Past Investigator
Connie Brand, RN—Past Investigator
University of California Irvine IMIND:
Gaby Thai, MD
Aimee Pierce, MD
Beatriz Yanez, RN
Elizabeth Sosa, PhD
Megan Witbracht, PhD
University of Texas Southwestern Medical School:
Kyle Womack, MD
Dana Mathews, MD, PhD
Mary Quiceno, MD
Emory University:
Allan I. Levey, MD, PhD
James J. Lah, MD, PhD
Janet S. Cellar, DNP, PMHCNS-BC
University of Kansas, Medical Center:
Jeffrey M. Burns, MD
Russell H. Swerdlow, MD
William M. Brooks, PhD
University of California, Los Angeles:
Ellen Woo, PhD
Daniel H.S. Silverman, MD, PhD
Edmond Teng, MD, PhD
Sarah Kremen, MD
Liana Apostolova, MD—Past Investigator
Kathleen Tingus, PhD—Past Investigator
Po H. Lu, PsyD—Past Investigator
George Bartzokis, MD—Past Investigator
Mayo Clinic, Jacksonville:
Neill R Graff-Radford, MBBCH, FRCP (London)
Francine Parfitt, MSH, CCRC
Kim Poki-Walker, BA
Indiana University:
Martin R. Farlow, MD
Ann Marie Hake, MD
Brandy R. Matthews, MD—Past Investigator
Jared R. Brosch, MD
Scott Herring, RN, CCRC
Yale University School of Medicine:
Christopher H. van Dyck, MD
Richard E. Carson, PhD
Pradeep Varma, MD
McGill Univ., Montreal-Jewish General Hospital:
Howard Chertkow, MD
Howard Bergman, MD
Chris Hosein, MEdhanie Kielb, BS—Past Investigator
Sunnybrook Health Sciences, Ontario:
Sandra Black, MD, FRCPC
Bojana Stefanovic, PhD
Chris (Chinthaka) Heyn, BSC, PhD, MD, FRCPC
U.B.C. Clinic for AD & Related Disorders:
Ging-Yuek Robin Hsiung, MD, MHSc, FRCPC
Benita Mudge, BS
Vesna Sossi, PhD
Howard Feldman, MD, FRCPC—Past Investigator
Michele Assaly, MA—Past Investigator
Cognitive Neurology—St. Joseph's, Ontario:
Elizabeth Finger, MD
Stephen Pasternack, MD, PhD
William Pavlosky, MD
Irina Rachinsky, MD—Past Investigator
Dick Drost, PhD—Past Investigator
Andrew Kertesz, MD—Past Investigator
Cleveland Clinic Lou Ruvo Center for Brain Health:
Charles Bernick, MD, MPH
Donna Munic, PhD
Northwestern University:
Marek-Marsel Mesulam, MD
Emily Rogalski, PhD
Kristine Lipowski, MA
Sandra Weintraub, PhD
Borna Bonakdarpour, MD
Diana Kerwin, MD—Past Investigator
Chuang-Kuo Wu, MD, PhD—Past Investigator
Nancy Johnson, PhD—Past Investigator
Premiere Research Inst (Palm Beach Neurology):
Carl Sadowsky, MD
Teresa Villena, MD
Georgetown University Medical Center:
Raymond Scott Turner, MD, PhD
Kathleen Johnson, NP
Brigid Reynolds, NP
Brigham and Women's Hospital:
Reisa A. Sperling, MD
Keith A. Johnson, MD
Gad A. Marshall, MD
Stanford University:
Jerome Yesavage, MD
Joy L. Taylor, PhD
Steven Chao, MD, PhD
Barton Lane, MD—Past Investigator
Allyson Rosen, PhD—Past Investigator
Jared Tinklenberg, MD—Past Investigator
Banner Sun Health Research Institute:
Edward Zamrini, MD
Christine M. Belden, PsyD
Sherye A. Sirrel, CCRC
Boston University:
Neil Kowall, MD
Ronald Killiany, PhD
Andrew E. Budson, MD
Alexander Norbash, MD—Past Investigator
Patricia Lynn Johnson, BA—Past Investigator
Howard University:
Thomas O. Obisesan, MD, MPH
Ntekim E. Oyonumo, MD, PhD
Joanne Allard, PhD
Olu Ogunlana, BPharm
Case Western Reserve University:
Alan Lerner, MD
Paula Ogrocki, PhD
Curtis Tatsuoka, PhD
Parianne Fatica, BA, CCRC
University of California, Davis—Sacramento:
Evan Fletcher, PhD
Pauline Maillard, PhD
John Olichney, MD
Charles DeCarli, MD
Owen Carmichael, PhD—Past Investigator
Neurological Care of CNY:
Smita Kittur, MD—Past Investigator
Parkwood Institute:
Michael Borrie, MB ChB
T-Y Lee, PhD
Dr Rob Bartha, PhD
University of Wisconsin:
Sterling Johnson, PhD
Sanjay Asthana, MD
Cynthia M. Carlsson, MD, MS
Banner Alzheimer's Institute:
Pierre Tariot, MD
Anna Burke, MD
Joel Hetelle, BS
Kathryn DeMarco, BS
Nadira Trncic, MD, PhD, CCRC—Past Investigator
Adam Fleisher, MD—Past Investigator
Stephanie Reeder, BA—Past Investigator
Dent Neurologic Institute:
Vernice Bates, MD
Horacio Capote, MD
Michelle Rainka, PharmD, CCRP
Ohio State University:
Douglas W. Scharre, MD
Maria Kataki, MD, PhD
Rawan Tarawneh, MD
Albany Medical College:
Earl A. Zimmerman, MD
Dzintra Celmins, MD
David Hart, MD
Hartford Hospital, Olin Neuropsychiatry Research Center:
Godfrey D. Pearlson, MD
Karen Blank, MD
Karen Anderson, RN
Dartmouth-Hitchcock Medical Center:
Laura A. Flashman, PhD
Marc Seltzer, MD
Mary L. Hynes, RN, MPH
Robert B. Santulli, MD—Past Investigator
Wake Forest University Health Sciences:
Kaycee M. Sink, MD, MAS
Mia Yang, MD
Akiva Mintz, MD, PhD
Rhode Island Hospital:
Brian R. Ott, MD
Geoffrey Tremont, PhD
Lori A. Daiello, Pharm.D, ScM
Butler Hospital:
Stephen Salloway, MD, MS
Paul Malloy, PhD
Stephen Correia, PhD
Athena Lee, PhD
UC San Francisco:
Howard J. Rosen, MD
Bruce L. Miller, MD
David Perry, MD
Medical University South Carolina:
Jacobo Mintzer, MD, MBA
Kenneth Spicer, MD, PhD
David Bachman, MD
St. Joseph’s Health Care:
Elizabeth Finger, MD
Stephen Pasternak, MD
Irina Rachinsky, MD
John Rogers, MD
Andrew Kertesz, MD—Past Investigator
Dick Drost, MD—Past Investigator
Nathan Kline Institute:
Nunzio Pomara, MD
Raymundo Hernando, MD
Antero Sarrael, MD
University of Iowa College of Medicine:
Delwyn D. Miller, PharmD, MD
Karen Ekstam Smith, RN
Hristina Koleva, MD
Ki Won Nam, MD
Hyungsub Shim, MD
Susan K. Schultz, MD—Past Investigator
Cornell University:
Norman Relkin, MD, PhD
Gloria Chiang, MD
Michael Lin, MD
Lisa Ravdin, PhD
University of South Florida: USF Health Byrd Alzheimer’s Institute:
Amanda Smith, MD
Christi Leach, MD
Balebail Ashok Raj, MD—Past Investigator
Kristin Fargher, MD—Past Investigator
Courtney Bodge, PhD
Footnotes
*Data used in preparation of this article were obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in analysis or writing of this report. A complete listing of ADNI investigators can be found at http://adni.loni.usc.edu/wp-content/uploads/how_to_apply/ADNI_Acknowledgement_List.pdf. The members of Alzheimer’s Disease Neuroimaging Initiative are listed in the Appendix.
References
- Altmann A, Tian L, Henderson VW, Greicius MD; Alzheimer’s Disease Neuroimaging Initiative Investigators. Sex modifies the APOE-related risk of developing Alzheimer disease. Ann Neurol 2014; 75: 563–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman RJ, Xiong C, Benzinger TLS, Fagan AM, Goate A, Fox NC, et al. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N Engl J Med 2012; 367: 795–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belloy ME, Napolioni V, Greicius MD.. A quarter century of APOE and Alzheimer’s disease: progress to date and the path forward. Neuron 2019; 101: 820–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C-Y, Pollack S, Hunter DJ, Hirschhorn JN, Kraft P, Price AL.. Improved ancestry inference using weights from external reference panels. Bioinformatics 2013; 29: 1399–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crane PK, Carle A, Gibbons LE, Insel P, Mackin RS, Gross A, et al. ; for the Alzheimer’s Disease Neuroimaging Initiative. Development and assessment of a composite score for memory in the Alzheimer’s Disease Neuroimaging Initiative (ADNI). Brain Imaging Behav 2012; 6: 502–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruchaga C, Del-Aguila JL, Saef B, Black K, Fernandez MV, Budde J, et al. Polygenic risk score of sporadic late-onset Alzheimer’s disease reveals a shared architecture with the familial and early-onset forms. Alzheimer’s Dement 2018; 14: 205–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaneau O, Marchini J, Zagury JF.. A linear complexity phasing method for thousands of genomes. Nat Methods 2012; 9: 179–81. [DOI] [PubMed] [Google Scholar]
- Desikan RS, Fan CC, Wang Y, Schork AJ, Cabral HJ, Cupples LA, et al. Genetic assessment of age-associated Alzheimer disease risk: development and validation of a polygenic hazard score. PLoS Med 2017; 14: e1002258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desikan RS, Ségonne F, Fischl B, Quinn BT, Dickerson BC, Blacker D, et al. An automated labeling system for subdividing the human cerebral cortex on MRI scans into gyral based regions of interest. Neuroimage 2006; 31: 968–80. [DOI] [PubMed] [Google Scholar]
- Dudbridge F. Power and predictive accuracy of polygenic risk scores. PLoS Genet 2013; 9: e1003348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durbin R. Efficient haplotype matching and storage using the positional Burrows-Wheeler transform (PBWT). Bioinformatics 2014; 30: 1266–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escott-Price V, Myers AJ, Huentelman M, Hardy J.. Polygenic risk score analysis of pathologically confirmed Alzheimer disease. Ann Neurol 2017; 82: 311–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escott-Price V, Sims R, Bannister C, Harold D, Vronskaya M, Majounie E, et al. Common polygenic variation enhances risk prediction for Alzheimer’s disease. Brain 2015; 138: 3673–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Euesden J, Lewis CM, O’Reilly PF.. PRSice: polygenic risk score software. Bioinformatics 2015; 31: 1466–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrer LA, Cupples LA, Haines JL, Hyman B, Kukull WA, Mayeux R, et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA 1997; 278: 1349–56. [PubMed] [Google Scholar]
- Franzmeier N, Rubinski A, Neitzel J, Ewers M; The Alzheimer’s Disease Neuroimaging Initiative (ADNI). The BIN1 rs744373 SNP is associated with increased tau-PET levels and impaired memory. Nat Commun 2019; 10: 1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatz M, Reynolds CA, Fratiglioni L, Johansson B, Mortimer JA, Berg S, et al. Role of genes and environments for explaining Alzheimer disease. Arch Gen Psychiatry 2006; 63: 168–74. [DOI] [PubMed] [Google Scholar]
- Ge T, Chen CY, Ni Y, Feng YCA, Smoller JW.. Polygenic prediction via Bayesian regression and continuous shrinkage priors. Nat Commun 2019; 10: 1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, et al. TREM2 variants in Alzheimer’s disease. N Engl J Med 2013; 368: 117–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland D, Desikan RS, Dale AM, McEvoy LK.. Higher rates of decline for women and apolipoprotein e ε4 carriers. AJNR Am J Neuroradiol 2013; 34: 2287–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Schizophrenia Consortium, Purcell SM, Wray NR, Stone JL, Visscher PM, O’Donovan MC, et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature 2009; 460: 748–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen IE, Savage JE, Watanabe K, Bryois J, Williams DM, Steinberg S, et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat Genet 2019; 51: 404–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerminen S, Martin AR, Koskela J, Ruotsalainen SE, Havulinna AS, Surakka I, et al. Geographic variation and bias in polygenic scores of complex diseases and traits in Finland. Am J Hum Genet 2019; 104: 1169–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khera AV, Chaffin M, Aragam KG, Haas ME, Roselli C, Choi SH, et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat Genet 2018; 50: 1219–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkle BW, Grenier-Boley B, Sims R, Bis JC, Damotte V, Naj AC, et al. ; Alzheimer Disease Genetics Consortium (ADGC). Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat Genet 2019; 51: 414–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert J-C, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet 2013; 45: 1452–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landau SM, Breault C, Joshi AD, Pontecorvo M, Mathis CA, Jagust WJ, et al. Amyloid-β imaging with Pittsburgh compound B and florbetapir: comparing radiotracers and quantification methods. J Nucl Med 2013; 54: 70–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonenko G, Shoai M, Bellou E, Sims R, Williams J, Hardy J, et al. ; the Alzheimer’s Disease Neuroimaging Initiative. Genetic risk for Alzheimer’s disease is distinct from genetic risk for amyloid deposition. Ann Neurol 2019. a; 86: 427–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonenko G, Sims R, Shoai M, Frizzati A, Bossù P, Spalletta G, et al. Polygenic risk and hazard scores for Alzheimer’s disease prediction. Ann Clin Transl Neurol 2019b; 6: 456–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung KK, Clarkson MJ, Bartlett JW, Clegg S, Jack CR, Weiner MW, et al. Robust atrophy rate measurement in Alzheimer’s disease using multi-site serial MRI: tissue-specific intensity normalization and parameter selection. Neuroimage 2010; 50: 516–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald ME, Ambrose CM, Duyao MP, Myers RH, Lin C, Srinidhi L, et al. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 1993; 72: 971–83. [DOI] [PubMed] [Google Scholar]
- McCarthy S, Das S, Kretzschmar W, Delaneau O, Wood AR, Teumer A, et al. ; Haplotype Reference Consortium. A reference panel of 64, 976 haplotypes for genotype imputation. Nat Genet 2016; 48: 1279–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mormino EC, Sperling RA, Holmes AJ, Buckner RL, De Jager PL, Smoller JW, et al. ; For the Alzheimer’s Disease Neuroimaging Initiative. Polygenic risk of Alzheimer disease is associated with early- and late-life processes. Neurology 2016; 87: 481–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich D, Patterson N, Campbell D, Tandon A, Mazieres S, Ray N, et al. Reconstructing Native American population history. Nature 2012; 488: 370–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabuncu MR, Buckner RL, Smoller JW, Lee PH, Fischl B, Sperling RA; Alzheimer’s Disease Neuroimaging Initiative. The association between a polygenic Alzheimer score and cortical thickness in clinically normal subjects. Cereb Cortex 2012; 22: 2653–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saykin AJ, Shen L, Foroud TM, Potkin SG, Swaminathan S, Kim S, et al. Alzheimer’s Disease Neuroimaging Initiative biomarkers as quantitative phenotypes: genetics core aims, progress, and plans. Alzheimer’s Dement 2010; 6: 265–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scelsi MA, Khan RR, Lorenzi M, Christopher L, Greicius MD, Schott JM, et al. Genetic study of multimodal imaging Alzheimer’s disease progression score implicates novel loci. Brain 2018; 141: 2167–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan CH, Bonham LW, Fan CC, Mormino EC, Sugrue LP, Broce IJ, et al. ; for the Alzheimer’s Disease Neuroimaging Initiative. Polygenic hazard score, amyloid deposition and Alzheimer’s neurodegeneration. Brain 2019; 142: 460–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan CH, Desikan RS.. Interpreting Alzheimer disease polygenic scores. Ann Neurol 2018; 83: 443–5. [DOI] [PubMed] [Google Scholar]
- Tan CH, Fan CC, Mormino EC, Sugrue LP, Broce IJ, Hess CP, et al. ; For the Alzheimer’s Disease Neuroimaging Initiative. Polygenic hazard score: an enrichment marker for Alzheimer’s associated amyloid and tau deposition. Acta Neuropathol 2018; 135: 85–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan CH, Hyman BT, Tan JJX, Hess CP, Dillon WP, Schellenberg GD, et al. Polygenic hazard scores in preclinical Alzheimer disease. Ann Neurol 2017; 82: 484–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The International HapMap 3 Consortium; Altshuler DM, Gibbs RA, Peltonen L, Altshuler DM, Gibbs RA, Peltonen L, et al. Integrating common and rare genetic variation in diverse human populations. Nature 2010; 467: 52–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tosto G, Bird TD, Tsuang D, Bennett DA, Boeve BF, Cruchaga C, et al. Polygenic risk scores in familial Alzheimer disease. Neurology 2017; 88: 1180–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungar L, Altmann A, Greicius M.. Apolipoprotein E, gender, and Alzheimer’s disease: an overlooked, but potent and promising interaction. Brain Imaging and Behavior 2013; 8: 262–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilhjálmsson BJ, Yang J, Finucane HK, Gusev A, Lindstrom S, Ripke S, et al. Modeling linkage disequilibrium increases accuracy of polygenic risk scores. Am J Hum Genet 2015; 97: 576–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Q, Nho K, Del-Aguila JL, Wang X, Risacher SL, Fan K-H, et al. Genome-wide association study of brain amyloid deposition as measured by Pittsburgh Compound-B (PiB)-PET imaging. Mol Psychiatry 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data used in the preparation of this article were obtained from the ADNI database (http://adni.loni.usc.edu) and are freely available after registration. Derived polygenic scores (PRS1, PRS2 and PRS-cs) and R-scripts to analyse the data and to produce the results presented here are available at https://github.com/andrealtmann/polygenic_AD/