

Abstract
Inflammatory lung disease to innocuous antigens or infectious pathogens is a common occurrence and in some cases, life threatening. Often, the inflammatory infiltrate that accompanies these events contributes to pathology by deleterious effects on otherwise healthy tissue and by compromising lung function by consolidating (blocking) the airspaces. A fine balance, therefore, exists between a lung immune response and immune-mediated damage, and in some the “threshold of ignorance” may be set too low. In most cases, the contributing, potentially offending, cell population or immune pathway is known, as are factors that regulate them. Why then are targeted therapeutic strategies to manipulate them not more commonplace in clinical medicine? This review highlights immune homeostasis in the lung, how and why this is lost during acute lung infection, and strategies showing promise as future immune therapeutics.
Supplementary information
The online version of this article (doi:10.1038/mi.2008.16) contains supplementary material, which is available to authorized users.
Introduction
The mucosal immune system must maintain composure in the presence of an onslaught of antigenic and potentially pathogenic material. Exposed to the outside world with, in most cases, only a single epithelial cell barrier protecting them, our mucosal surfaces have developed a sophisticated system of immune exclusion, ignorance and tolerance. The best characterized of these are described in the gastrointestinal tract. An understanding of immunity in the respiratory tract has lagged behind that of the gut, and although numerous key components have emerged, the sequence of events from initial inhalation to immune pathology in the lower respiratory tract is still unclear. Despite best efforts to maintain immune homeostasis, respiratory inflammatory disease is common and significantly life threatening. This review will highlight mechanisms that maintain lung immune homeostasis and current therapeutic efforts to contain infection-induced exaggerated acute inflammation once it occurs. The respiratory tract includes the nasopharyngeal cavity, trachea and larynx, bronchi, bronchioles, and finally the alveoli. Organized lymphoid tissue is embedded in some, but importantly not all, of these stages in the respiratory tree. Similarly, draining lymph nodes are associated with only a few of these sites. The cellular composition, requirements for activation, and expansion dynamics of respiratory tract associated lymph nodes are virtually similar to any other lymph node and will therefore not be discussed in detail here. We will focus on the regulation (or de-regulation) of immune cells embedded in the respiratory tract itself.
Respiratory Immune Compartments
Considering the total surface area of the respiratory tract constitutive, embedded organized lymphoid tissue is actually quite rare (Figure 1). Organized structured lymphoid tissue exists in the nasal cavity of rodents (nasal associated lymphoid tissue, NALT) as paired lymphoid structures at the entrance to the pharyngeal duct, but identical structures in man remain elusive (for a review, see reference Bienenstock and McDermott1). Organized lymphoid follicles are observed in post-mortem specimens extracted from 150 children that contain occasional germinal centers, which are associated with lymphocytes in the overlying nasal epithelium and the presence of high endothelial venules. However, in adults such lymphoid tissue is disseminated across the whole nasal mucosa,2 and is analogous to the less well-organized diffuse lymphoid tissue (termed D-NALT) lining the nasal passages of mice.3 In man, diffuse NALT develops after birth, likely in response to antigen, and B- and T-cell responses parallel those that occur in lymph nodes. The Waldeyer's ring comprising the nasopharyngeal (upper midline in naso-pharynx, adenoids), paired tubal (around openings of auditory tube), paired palatine (either side of the oropharynx), and lingual (under the mucosa of the posterior third of the tongue) tonsil(s) are thought of as analogous structures to NALT, but are located outside of the respiratory tract and probably also contribute to gastrointestinal immunity. Experiments with mice show that, unlike peripheral lymphoid organs, NALT develops independently of lymphotoxin-α. However, its structure and function are perturbed in lymphotoxin-α-knockout mice, possibly due to impaired expression of CXCL13, C–C chemokine ligand19 (CCL19), and CCL21, which are crucial for the recruitment and placement of lymphocytes and dendritic cells (DCs).4
Figure 1.
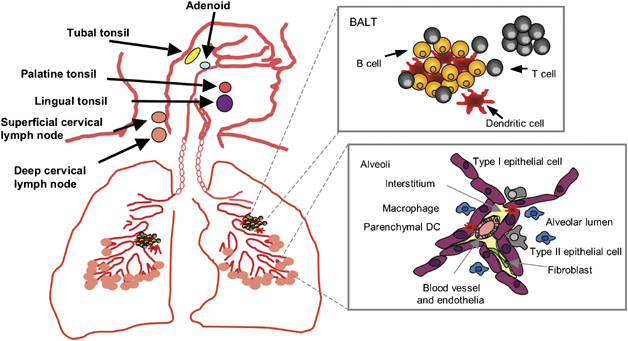
Schematic representation of organized and scattered lymphoid tissue associated with the respiratory tract. Expanded diagrams show the composition of BALT and a typical alveoli lumen containing alveolar macrophages and the dendrites of sub-mucosal DCs.
The only other organized lymphoid structure described to date located within the respiratory tract is bronchus-associated lymphoid tissue (BALT) (reviewed by Bienenstock and McDermott1). Whether it routinely contributes to primary immune responses or maintenance of T- and B-cell memory in the respiratory tract is not known.5, 6 However, a recent study in mice lacking peripheral lymphoid organs suggests that BALT can initiate anti-influenza immunity and provide sufficient T cells to mediate protection against a second infection.7 Humoral immune responses elicited by BALT are primarily mediated by immunoglobulin A (IgA) and IgG produced both locally and by BALT-derived B cells that traffic to distant mucosal sites.8, 9 Similarly located T-cell responses have been noted. On the basis of these findings, BALT can be thought of as functionally analogous to mucosal lymphoid aggregates in the intestine. Present in up to 40% of children and adolescents (to age 20), BALT is rare in the lungs of healthy adults.10, 11 Although originally described at the bifurcations of the bronchi, immediately beneath the epithelium,12, 13 in the absence of antigen BALT is rare14 and may be controlled or limited by regulatory T cells.15 Inflammation in the lung is associated with BALT neogenesis and is described in a variety of pulmonary (Moyron-Quiroz et al7 and references therein) and non-pulmonary16 inflammatory conditions. Homeostatic chemokines, including CCL19 and CCL21, in mice are required for development of such inducible BALT (iBALT).4 The extent of iBALT appears to depend on the level of inflammation in the local microenvironment, and suggests that it is developed when required. Mice lacking oxidoreductases, that protect from oxidative stress, display heightened cellularity and inflammatory cytokines and iBALT is more prevalent.17 Whether it remains associated with larger airways and persists long after resolution of inflammation is still uncertain.
Intriguingly, mice lacking peripheral lymph nodes and spleen, but retaining iBALT, clear influenza infection (albeit slower) and survive higher doses of virus than do immune-competent mice. Such lymphotoxin-knockout mice show slower generation of influenza-specific T cells that eventually reach wild-type levels, similar to antibody isotype switching to IgG and T-cell cytokine production and effector function. This indicates that immune responses generated in iBALT although slower are protective and potentially less pathologenic.7 This may represent a qualitative difference between local and peripherally derived immune cells or simply reflect the reduced magnitude of immunity when iBALT is the only inducing immune compartment. Regardless, in the case of lung immunity, secondary lymphoid tissues are not essential for the maintenance of immunological memory, since a pulmonary infection with influenza virus is handled equally as efficiently in their absence.18
In Which Compartment is Lung Inflammation Detrimental?
To date no studies have shown that organized embedded lymphoid tissue such as iBALT and NALT contribute directly to inflammatory pathology in the lung. They may initiate production of immune T and B cells that then track to less organized lung immune compartments,7, 19, 20, 21 and 22 but their neogenesis (iBALT) or continued presence (NALT), per se, is not associated with pathology. Instead, pathological lung inflammation is attributed to those compartments that lack organized lymphoid structures, the airways and lung parenchyma. This could also be said of inflammation in the gut; Peyer's Patches and mesenteric lymph nodes may not be directly associated with pathology, whereas the lamina propria is. What the lamina propria and lung parenchyma have in common is a loose scattering of non-organized immune cells and a vast surface area of potentially non-professional antigen-presenting cells (APCs); the epithelium. The epithelium expresses constitutive MHC class I and, when inflamed, MHC class II and B7 molecules.23, 24 It can, therefore, process and present antigen and activate T cells, but can it turn them off? We believe this non-professionalism and lack of immune-cell organization leads to immune dysregulation. During inflammation, the mediastinal lymph nodes and iBALT expand in an organized and precisely compartmentalized manner. Although low frequencies of antigen-specific cells can be observed in these sites,25 the ratio of immune-cell subsets does not significantly change. Contraction of lung-associated lymph nodes is also well controlled; again cell proportions are retained. In lung compartments devoid of organized lymphoid tissue however, immune cells are recruited in droves by a chemotactic gradient26, 27 and 28 from infected epithelium and/or tissue resident and alveolar macrophages. Once in the lung parenchyma or the airways, they do not form structures analogous to iBALT or lymph nodes, and it is here that pathological damage occurs, Toll-like receptors ligands dominate, and inflammatory cytokines are produced by the infiltrate in abundance. Unlike the lung-associated lymph nodes, the airways and lung parenchyma, therefore, experience dramatic shifts in their cellular composition.
This is illustrated graphically in Figure 2. The approximate cellular composition in NALT, airways, and lung is shown in homeostasis and at the peak of respiratory infections by three very different pathogens, influenza virus, the bacterium Streptococcus pneumoniae, and the fungus Cryptococcus neoformans. Obviously at other stages of the infection, slightly different cells will dominate, but only the peak of inflammation is presented for clarity. For example, natural killer cells dominate in the airways at days 3–4 of a viral infection.29 At its peak of activity, the influenza-infected airway and lung is dominated by CD8+ and CD4+ T cells,25, 30, 31 and 32 whereas during S. pneumoniae infection, macrophages, neutrophils, and γδT cells are more abundant.33, 34C. neoformans in C57BL/6 mice induces an eosinophil-dominated response in the lung and airways.35, 36 and 37 This infiltrate in the air spaces and lung for all pathogens is dramatically different to the same sites in homeostasis that contain few lymphoid cells but a prominent macrophage population.38, 39 and 40 Note that the NALT, despite being infected with all three pathogens, does not substantially alter the proportion of immune-cell subsets present, and the same is also true for the relatively non-infected lung draining lymph nodes.
This lack of control and excessive response is only observed in a minority, but when it occurs it is life threatening. For most of us, respiratory pathogens are cleared by non-inflammatory means, including IgA that does not fix complement well, but in dimeric form agglutinates and physically excludes antigen by a process known as immune exclusion.41, 42 What, therefore, goes wrong in a minority? To address some of these questions, we need to understand how immune homeostasis is maintained in health in these non-organized lung compartments (reviewed by Holt43) and what pathways contribute to immune pathology.
Epithelial cells contribute a multitude of strategies to maintain lung immune homeostasis (for a review, see reference Holt43). In addition to barrier function, they secrete a variety of antimicrobial substances (surfactant protein C, mucins, and antimicrobial peptides), affect airway smooth-muscle, DC, and memory T-cell activation via nitric oxide production;44, 45 assist in cell recruitment via production of cytokines and chemokines;46, 47 and prolong cell survival by secreting stimulating factors such as granulocyte–macrophage colony-stimulating factor.48, 49 Raz and co-workers50, 51 highlight an interesting pathway critical for maintaining alveolar macrophage homeostasis, involving integrin αvβ6 that localizes these cells next to epithelial expressed transforming growth factor-β (TGF-β). This may explain why these cells are refractory to migration to the draining lymph nodes.52 For inflammation to proceed, this inhibitory pathway must be overcome, which is mediated by a Toll-like receptor-induced conformational change of macrophages, disruption of TGF-β signaling, and reduced integrin expression. We often assume that innate immunity is inactive in the absence of antigen. However, the work of Raz et al. clearly shows that active suppression is required for homeostasis. This is also observed in mice lacking components of NADPH oxidase53 that have heightened basal levels of airway macrophage activation due to loss of feedback inhibition.
Active suppressive mechanisms, therefore, set a “threshold of ignorance”. Those that succumb continually to inflammatory lung disease may, therefore, have dysregulated homeostatic pathways or the threshold, which antigen must exceed to induce inflammation, set too low. In the cases of TGF-β-mediated suppression of alveolar macrophages, homeostasis is overcome by cleavage of the integrin tethering it to the respiratory epithelium. Homeostasis is restored when macrophage-released matrix metalloproteinases transform latent TGF-β into its active form.51 It may, therefore, be possible to harness these pathways artificially to dampen inflammatory lung disease with the caveat that pathogen clearance may be affected by such a global anti-inflammatory strategy.54
Macrophages, particularly in the airways, have long been known to have an immune-suppressive phenotype. Renewal is achieved primarily via local cell proliferation; recruitment via CCL2:CCR2 does occur, although such cells may take days to mature into the classical immune-suppressive phenotype.55 In addition to shielding the immune system from inhaled antigens,56 they display poor phagocytic activity57 and tend not to migrate well to draining lymph nodes. Macrophages held in homeostasis also affect other cell types that may otherwise be proinflammatory within the respiratory tract (for a review, see reference Holt43, 58). DC migration to the draining lymph nodes is enhanced upon macrophage depletion,52 and T-cell-mediated inflammatory disease ensues to antigens that would otherwise have been ignored,59 most likely due to the usual direct suppressive influence that alveolar macrophages have on DC function.39
Both myeloid and plasmacytoid DCs (pDCs) are present within the lung, both increase and are recruited rapidly during inflammation, and are attracted by chemokines and cytokines produced by epithelial cells and alveolar macrophages.43, 60, 61 and 62 Myeloid DC responses are similar to counterparts found elsewhere in the body. pDCs, however, also appear to play a tolerogenic role in the respiratory tract. They have poor APC activity63 (but once activated enhance CD8+ T-cell responses in vivo64 and possibly CD4+ T cells at distant sites65), promote inhalation tolerance,66 and can protect from development of allergic airway disease (reviewed by de Heer et al,66 Hammad and Lambrecht,67 and Lambrecht68). During acute respiratory viral infections, pDCs perform dual functions of promoting viral clearance by secretion of type I interferon (IFN), and limit inflammation by induction of interleukin-10 (IL-10) (reviewed by Grayson and Holtzman69). Their role in limiting lung inflammation can be clearly seen in respiratory syncytial virus (RSV)-infected mice where pDC depletion leads to increased viral replication and enhanced immunopathology in the lungs70, 71. DCs encounter antigen predominantly in the lung parenchyma, although microbial sampling, via dendrite projections through the epithelial cells into the airway lumen, may induce activation, maturation, and migration, arming them to support potent T-cell responses.72 Quite what returns these cells to homeo-stasis is unknown but may involve the level of Toll-like receptor signals and/or the influence of surface expressed inhibitory receptors such as CD200R.73 Airway epithelial cells may also control DC activity via TGF-β analogous to alveolar macrophages,51 although this has not been proven. Whether DCs actually transmigrate into the airspaces is still controversial.74, 75 and 76 CCR2 assists their transit across the endothelium and CCR6 their subsequent migration into the airway.74 The recently described CD103+ (αEβ7), CD207+, CD11blo DCs express all of the requisite chemokine receptors to draw them into the airways,77 but their presence in this compartment has not been proven.
Therapeutic Strategies to Limit Acute Lung Inflammation
Clearly, harnessing the homeostatic pathways described above may help to resolve ongoing inflammation. However, it is equally likely that immune cell types and pathways brought into the lung with inflammatory cells during inflammation also provide a therapeutic opportunity. It would not be possible to cover all of the pathways attempted to limit lung inflammation. We will, therefore, restrict our analysis to acute infectious events (i.e., not asthma) and to targeted therapeutic strategies (i.e., not global anti-inflammatories such as corticosteroids or experiments in gene-deleted animals). Immune-mediated pathology can be manipulated at any stage of its generation. From a clinical perspective, however, after onset of identifiable symptoms would be the most beneficial. For this reason it is often resolution of inflammation that is targeted, which can include modulation of cell survival, successive waves of recruitment, and ongoing innate immunity.
Modulation of Cell Survival
It is still unclear whether immune excess in the respiratory tract stems from over-exuberant recruitment, proliferation within the airspaces, and/or accumulation in the absence of clearance of innate and adaptive immune cells. Evidence suggests that the airspaces may not support efficient T-cell proliferation78, 79 and 80 despite memory T cells acquiring bromodeoxyuridine staining at the same rate as secondary lymphoid organs.25, 80
It is likely that the inflammatory cytokine and chemokine cascade that ensues upon infection81, 82, 83, 84, 85 and 86 will prolong immune-cell survival, but at the same time enhance their recruitment, either directly or by altering vascular or epithelial permeability.87, 88 An inflammatory environment is also associated with the highest expression of “late co-stimulatory molecules” on T cells that prevent activation-induced cell death and rely on cognate ligands expressed on APCs for signalling. OX40 (CD134) is one such late co-stimulator that is restricted to recently activated T cells, whereas the ligand (OX40L) is expressed on a number of cell types, predominantly APCs.89 Stimulation through OX40 promotes CD4 and CD8 T-cell survival, clonal expansion,90 inhibition of regulatory T cells,91 and enhanced immunity to a variety of pathogens.36, 92, 93 and 94 However, during acute influenza virus infection of mice, transient blockade of OX40 is more beneficial; alleviating illness and pathology to influenza, without compromising pathogen clearance or immunological memory.95 A permanent absence of multiple late co-stimulators, however, can compromise immunological memory.96, 97 and 98 Manipulation of one late co-stimulatory pathway, therefore, leaves others intact to seed the memory T-cell pool.97 This suggests that a full compliment of late co-stimulators may actually be prolonging T-cell survival and contributing to pathology. Inflammatory cytokines, such as those abundantly expressed in the airways during infection, are known to increase OX40L on APCs.99 Due to lack of immune organization in the airways, late co-stimulatory molecules and their ligands may be downregulated too late to avoid bystander tissue damage. The observation of OX40L on inflamed endothelium highlights the difficulty in separating the effects of immune modulators on cell survival vs. cell recruitment,100, 101 especially when ligation of OX40L on endothelial cells induces secretion of chemokines.101, 102 4-1BB, like OX40, is restricted to late-effector T cells and its absence or blockade impairs CD8+ T-cell responses to influenza,103 although the effect on associated respiratory pathology is not yet known. Care must be taken, however, since not all inducible co-stimulators result in a beneficial outcome when blocked. During lung influenza infection, ICOS blockade impairs respiratory T cells to such an extent that the virus escapes clearance.32 This is similar to treatment of influenza-infected mice with a CTLA4-Ig fusion protein (that blocks CD28 binding to B7 molecules)104 and CD40L-knockout mice infected with C. neoformans105 (where macrophage antimicrobial strategies are impaired). It is likely that targeting CD27 may also produce untoward side effects, since it is expressed on resting naïve and memory T cells and is crucial for the formation CD8+ T-cell responses.96, 106 Perhaps the defining line for therapeutic potential should be placed between those late co-stimulators that absolutely depend on T-cell receptor and constitutive CD28 signaling for their expression and those that can appear in a bystander fashion in the presence of inflammatory cytokines. The likely effect of co-stimulatory blockade and the site where manipulation may have the most effect is shown in Figure 3.
Figure 3.

The location of the dominant site of action of co-stimulatory molecule blockade during acute lung infection. This model assumes that antigen specific T cells are primed in the lung-associated lymph nodes (e.g., mediastinal). As such blockade of CD28 on T cells using the B7 competitor CTLA4:Ig will affect T-cell priming in organized lymphoid tissue (a). The same is also likely to be true for ICOS that is induced rapidly after T-cell receptor and CD28 signalling. By contrast, OX40 and 4-1BB are expressed at very low levels in the lung-associated lymph nodes but upregulated in the lung parenchyma and airways, most likely due to re-recognition of antigen in situ and/or the inflammatory cytokine environment. Blockade will, therefore, impact these sites rather than the lymph nodes (b). Note that OX40 and 4-1BB blockade may also affect primed T-cell migration, as their respective ligands are present on inflamed endothelium (c). In the airways, the dominant effect will be to allow activation-induced cell death to progress; in the parenchyma, however, it may be a combination of activation-induced cell death (d) and migration.
In addition to late co-stimulatory molecules, a number of other pathways affect the longevity of lung inflammation during acute infection. Whether apoptosis is beneficial or harmful depends on the specific infection and the dominant cell type that mediates its clearance.107 As a general rule, apoptosis favors the host in chronic and acute intracellular bacterial and viral infections (as the process clears the pathogen), but is detrimental for extracellular bacteria.107 For example, during infection of rats with Pneumocystis, alveolar macrophage apoptosis delays clearance of the organism that can be improved by administering caspase-9 inhibitors.108 Similarly, apoptosis of airway epithelial cells via FAS/FAS ligand is essential to prevent dissemination of Pseudomonas aeruginosa.109 However, during influenza infection it may be more advantageous to reduce cell survival, especially in the case of TNF producing CD8+ T cells. TNF receptor-II and very late antigen--1 synergize to protect CD8 T cells in the influenza virus infected airways from apoptosis,110 whereas engagement of Qa-1b by CD94/NKG2A transmits a negative signal that limits immune pathology.111 It may, therefore, be possible to resolve T-cell inflammation before bystander tissue damage occurs by blocking or enhancing these surface receptors once the viral load has reduced. This would only be useful, however, if the strategy specifically targeted a defined cell population, since apoptosis of airway epithelial cells and leukocytes may be linked to the pathology observed in those infected with highly pathogenic avian influenza.112, 113 Furthermore, apoptosis leading to systemic lymphopenia,114 observed during influenza infection, may assist virus propagation and survival. Other respiratory pathogens, such as cytomegalovirus and parainfluenza virus, use antiapoptotic strategies to prolong survival of the cells they infect.115 Chlamydia blocks apoptosis by affecting the release of cytochrome c from mito-chondria.116 The extracellular pathogens Pseudomonas cepacia and S. pneumoniae cause apoptosis of neutrophils and airway epithelial cells (Table 1),117 respectively, to aid survival. Modulation of apoptosis is, therefore, complicated; what may benefit the host for one infection would compromise it to another.
Table 1. Summary of the impact of therapeutic intervention for intracellular and extracellular acute respiratory pathogensa.
| Intracellular pathogens | Extracellular pathogens | |||
|---|---|---|---|---|
| Intervention/treatment | Pathogen clearance | Immunopathology | Pathogen clearance | Immunopathology |
| Apoptosis | ||||
| OX40 blockade | ↔ | ↓ | ↔ | ↓ |
| 4-1BB blockade | ↓ | ?a | ? | ? |
| ICOS blockade | ↓ | ↓ | ? | ? |
| CTLA-4-Ig | ↓ | ↓ | ? | ? |
| Caspase inhibitors | ? | ? | ↑ | ? |
| Migration | ||||
| Anti-MIP-2 | ↔ | ↓ | ? | ↓ |
| Met-RANTES | ↔ | ↓ | ? | ? |
| MCP-1 | ↔ | ↓ | ? | ? |
| CCR1 antagonism | ? | ↓ | ? | ? |
| Innate modulation | ||||
| Prior infection | ↔ | ↓ | ↓ | ↓ |
| Nitric oxide | ? | ? | ↑ | ? |
| Antioxidants | ? | ↓ | ? | ? |
| Free-radical scavengers | ↔ | ↓ | ? | ? |
| NF-κB inhibition | ↔ | ↓ | ↓ | ? |
| Cytokine modulation | ||||
| IFN-α | ↑ | ↓ | ? | ? |
| IL-1Ra | ? | ? | ↓ | ↔ |
| Anti-IL-12 | ? | ↓ | ↓ | ? |
| Anti-TNF | ↔ | ↓ | ↓ | ? |
| Anti-IFN-γ | ↓ | ↓ | ↓ | ? |
| Anti-IL-10 | ? | ↓ | ↑ | ↓ |
| TGF-β | ↓ | ↓ | ↓ | ↓ |
CTLA, cytotoxic T lymphocyte antigen; IFN, interferon; Ig, immunoglobulin; IL, interleukin; IL-1Ra, IL-1-receptor antagonist; MIP, macrophage inflammatory protein; NF-κB, nuclear factor-κB; TGF, transforming growth factor; TNF, tumor-necrosis factor.
aThe effect of immune manipulation conforms to general observations for the majority of intracellular or extracellular pathogens. Specific organisms may vary. ? The outcome of this manipulation is currently unknown.
Modulation of Cell Recruitment
The recruitment and trafficking of leukocytes in response to inflammation is a tightly regulated process that can tip the balance between protection from infection and immune mediated damage in the lung. In brief, it involves slow rolling (conco-mitant to the activation of leukocytes), an increase in integrin expression and avidity to regulate the rolling arrest, adhesion strengthening as well as spreading, and intravascular crawling culminating in paracellular or trans-cellular migration.118, 119
Since excessive cell recruitment is a feature of many acute lung infections, targeting specific molecules involved in any of the above-mentioned events may be beneficial. Blocking monoclonal antibodies against the integrin very late antigen-4 (natalizumab)120 and lymphocyte function-associated antigen-1 (efalizumab)121 are currently used to treat inflammatory autoimmune disorders and Crohn's disease, but are also immunosuppressive and have not been studied in the context of acute respiratory infection.122
The potential benefit of blocking chemokines or their receptors using competitive blockers or antagonistic compounds is well described (reviewed by Glass et al123 and Charo and Ransohoff124). Secreted chemokines bind glucosaminoglycans on endothelial cells, forming chemoattractant gradients that direct cells to inflammatory sites,125 and are classified into constitutive homeostatic and inflammatory (requiring a pro-inflammatory stimulus such as IFN-α, TNF, or microbial products) chemokines.126 Chemokine receptors are transmembrane G-protein-coupled molecules that trigger a signal transduction event resulting in activation and firm adhesion of the migrating cell.127 The interactions between chemokines and their receptors are functionally redundant; many chemokines bind the same receptor and one chemokine can bind several receptors. As such it can be difficult to design reliable therapeutics that disrupt the interaction of one particular chemokine with its receptor.128 Therapeutic administration of antibodies that block macrophage inflammatory protein-2 during influenza infection reduces neutrophil recruitment by 49% and improves lung pathology without altering viral clearance.129 However, this strategy requires testing in co-infection models, since neutrophils are critical for clearance of most respiratory bacteria that commonly cause secondary pneumonia in the presence of influenza virus.130, 131 RANTES (CCL5) is another potential target produced by respiratory epithelial cells during a variety of viral infections. RANTES induces CCR1-, CCR3-, and CCR5-expressing T cell, eosinophil, and monocyte recruitment to the lung.132 During RSV infection, CCL5 is expressed by infected epithelia and resident macrophages and secreted into the airway during the first 48 h of infection. Production is then taken over by newly recruited T cells. Blocking this chemokine with a competitive inhibitor (met-RANTES)133 during a primary RSV infection reduces lung immunopathology. However, heightened cell recruitment occurs during homologous RSV re-challenge, suggesting that manipulation during the first infection severely compromises immunological memory.134 The same strategy has been tested in a mouse model of pneumonia virus infection. This highly lethal mouse pathogen induces a disease closely resembling severe human RSV infection in man, that is abrogated by co-administration of the antiviral agent Rivabirin and met-RANTES.135
In contrast to inhibition of chemokines during virus-induced lung inflammation, some infections may require their administration. P. aeruginosa lung infection causes airway neutrophil infiltration that rapidly apoptose and become toxic. Administration of recombinant monocyte chemoattractant protein-1/CCL2 recruits and activates lung macrophages that clear apoptotic neutrophils before they cause pathology.136 IFN-inducible protein 10 (CXCL10) is critical not only for clearance of Klebsiella pneumoniae,137 but is also produced during lung viral infection where neutralization may be beneficial.138
Many chemokine receptors are also increased during respiratory viral infection; CXCR3, for example, is upregulated during murine gammaherpes virus b8 infection, and its absence delays viral clearance.139 Antagonism of CCR1 reduces mortality of pneumovirus infection of mice,140 and absence of CCR1 also prevents the RSV-induced exacerbation of asthma.141 Relatively little has been accomplished in this area, especially therapeutically, however, due to the paucity of reagents available for chemokine-receptor blockade.
Another method for altering lymphocyte migration is to manipulate sphingosine 1-phosphate receptors that are required for egress of lymphocytes from the thymus and peripheral lymph nodes, and impact on vascular permeability. FTY720 is a novel synthetic immunosuppressive drug that inhibits lymphocyte emigration from lymphoid organs by binding and activating sphingosine 1-phosphate receptors.142 Sphingosine 1-phosphate is known to play a role in endotoxin-induced lung injury by affecting endothelial barrier function,143 and is currently in trial for a variety of inflammatory disorders, including transplantation,144 but is yet to be tested in acute lung infection.
Modulation of Innate Immunity
Modulation of lung innate immunity represents another potential therapeutic target. However, once again, few studies have tested manipulating it after the onset of symptoms during acute respiratory infection; most examine the impact of an innate immunity-associated molecule using gene-depleted animals or prior to infection. Manipulation of innate immunity may seem to be akin to “shutting the stable door after the horse has bolted,” since innate immunity is activated first and drives subsequent adaptive immune responses. However, each epithelial cell or macrophage infected sends off the next wave of an immune response. At the time of clinical presentation, therefore, both innate and adaptive immunity will be in full swing. Manipulating innate immunity at this stage will assist the resolution process, but whether inhibition or activation is required will depend on the pathogen.
Infective influences on acute respiratory infection
One way to temper innate immunity is to mature the lung microenvironment or instill probiotic microbes that would compete with the survival of pathogenic microorganisms. Oral administration of Lactobacillus casei during lung S. pneumoniae infection is protective, resulting in more rapid clearance, a shorter period of septicemia, and decreased S. pneumoniae load in the lungs. This benefit is attributed to increased neutrophils, myeloperoxidase, and IL-10 that limits lung tissue damage and is likely mediated by migration of mature APCs from the gut to the lung that are better equipped for bacterial clearance.145 Prior infection in the lung also has a beneficial effect on some subsequent acute respiratory infections through modification or maturation of the microenvironment.146, 147, 148, 149, 150 and 151 Prior influenza infection, for example, reduces subsequent Toll-like receptor responsiveness of alveolar macrophages for prolonged periods of time.152 Administration of microbial products, such as CpG DNA or a modified bacterial labile toxin (LTK63), also protects against an array of subsequent respiratory pathogens,153, 154 as do chronic or acute infections in distant sites.155, 156 and 157 The ability of pathogen-derived proteins to modify acute respiratory infections, however, is yet to be tested therapeutically.
Resveratrol, a polyphenolic compound found in red wine, inhibits nuclear factor-κB activation, decreases mortality and pro-inflammatory cytokines (TNF, IL-1β, and IL-6) to Serratia marcescens pneumonia in rats. Though this strategy increases neutrophil numbers, they resolve more rapidly.158 Similarly, an acidic polysaccharide compound from Cordyceps militaris, an insect-borne fungus, has anti-viral properties in a murine influenza infection model.159 Intranasal administration of the fungal polysaccharide decreases mortality and influenza viral titers, while increasing lung pro-inflammatory cytokines. In vitro, influenza infected macrophage cell lines treated with Resveratrol display enhanced inducible nitric oxide synthase and nitric oxide (NO), suggesting that this compound may function through non-specific stimulation of alveolar macrophages in vivo.159 Enhanced innate and adaptive immunity and reduced microbial load is also observed upon therapeutic administration of retinoic acid in mice infected with Mycobacterium tuberculosis.160 The increased numbers of macrophages, natural killer and T cells, and increased expression of IFN-γ, TNF, inducible nitric oxide synthase, IL-10, and CXCL10 may also benefit other acute respiratory bacteria or fungi, though during viral infection it would be predicted to be detrimental. Acute respiratory infections are yet to be examined.
Manipulation of oxidative stress
A critical pathway for clearance of pathogens and infected lung epithelial cells is via NO and reactive oxygen and nitrogen species. NO contributes to host defence and mediates both pro- and anti-inflammatory effects (Reviewed in161, 162). As with many of the therapeutic treatments discussed, the timing and extent of modulation of NO and associated free radicals is critical. While there is no clear consensus in the literature, there are a few examples of successful intervention in this pathway. In rats with P. aeruginosa pneumonia, treatment with inhaled NO post-infection improves bacterial clearance through direct bactericidal effects, increased recruitment of neutrophils to the airways163 or enhanced endothelial permeability.164 During K. pneumoniae infection of rats, inhaled NO also suppresses bacterial replication and decreases lung intercellular adhesion molecule-1 expression, myeloperoxidase activity, TNF levels, and nuclear factorκB activity. Thus, NO has many paracrine effects on inflammatory cells, but also promotes bacterial clearance.165 During viral infection, the impact of NO manipulation is less clear. Inhibition of NO during RSV infection reduces pulmonary inflammation and bystander tissue damage, but viral replication increases.166
Similar therapeutic strategies have been employed to reduce local concentration of reactive oxygen species and reactive nitrogen species, which are produced by infected lung epithelium and macrophages. During RSV infection of mice, administration of the antioxidant, butylated hydroxyanisole, decreases illness scores, weight loss, and lung neutrophil recruitment. Again a multi-factorial attenuation of inflammation occurs.167 The same is observed in influenza-infected mice treated with a free-radical scavenger, manganese superoxide dismutase, within 48–96 h of infection. This enzyme has potent anti-inflammatory properties, resulting in less lung consolidation and improved arterial oxygen saturation, presumably as a result of decreased tissue damage.168 Reduced tissue damage also occurs in mice lacking superoxide dismutase or treated therapeutically with a manganic porphyrin that scavenges reactive oxygen species.53 Therefore, inhibition of reactive oxygen species/reactive nitrogen species and NO is beneficial for acute respiratory viral infection, but likely to be detrimental for concurrent respiratory bacteria.
Manipulation of innate immune receptor signalling
Modulation of immune-receptor signaling is in its infancy with regards to acute respiratory infection and may be limited by (a) the toxicity/safety profile of available drugs, (b) formulation challenges for in vivo delivery, (c) a lack of specificity due to shared receptor associations, and (d) problematic pharmacokinetics (sustained blockage will be detrimental for protection against infection and drugs may have to be delivered locally to prevent systemic effects). The only truly therapeutic manipulation, to date, is abrogation of airway fluid clearance during RSV infection, caused by interaction of uridine triphosphate with purinergic receptors, by post-infection administration of an active metabolite of leflunomide, A77-1726. Leflunomide restores airway fluid clearance, providing symptomatic relief and reduced lung inflammation and hypoxemia, without impairing viral replication or clearance.169
Although signaling molecules associated with pattern recognition and cytokine receptors are well described, little is known about the complexity of innate pathways induced by a whole pathogen, especially in the lungs.170 Controlling the signaling pathways leading to exuberant inflammation is of major interest, but a balance needs to be struck to maintain host defence against infection. A feasible approach might be by treating patients at the peak of inflammation where immune mediators are in excess and using drugs with a short half-life. This is a promising strategy, but, to our knowledge, no drugs targeting signaling components are reported efficacious in human lung infection, although they are under development.171 A comprehensive review of relevant inhibitors of inflammatory signaling pathways can be found elsewhere.172
Receptors recognizing pathogens such as Toll-like receptors or nod-like receptors, but also TNF and IL-1β, which are often involved in amplification of the inflammatory response, are potent activators of NF-κB.173 NF-κB induction is a key factor triggering inflammation in the influenza-infected epithelium.174 Mice treated with a cell-permeable peptide that reduces NF-κB levels, via an effect on I-kappa-B kinase beta, reduces pulmonary RSV-induced inflammation.175 Again, this strategy may negatively affect respiratory bacteria, since intra-tracheal adenovirus delivery of a dominant NF-κB inhibitor impairs clearance of respiratory P. aeruginosa,176 despite the reduced inflammation observed with purified bacterial products.177, 178 Peroxisome proliferator-activated receptor-γ is a nuclear receptor involved in the stress response during lung injury and attenuates inflammatory responses by inhibiting NF-κB.179, 180 Many steroids target peroxisome proliferator-activated receptor-γ and show encouraging results in models of lung inflammation.172 A series of natural and synthetic ligands for these receptors have been developed and RSV-specific responses in human lung epithelial cell lines are reduced by some of these agonists.181
Mitogen-activated protein kinase p38 and c-Jun-N-terminal kinase pathways may also provide future suitable targets. Activation of the p38 pathway is often associated with induction of NF-κB (in Toll-like receptor responses, for example) and is thought to maintain inflammatory responses by stabilizing cytokine mRNA.182 Inhibiting the mitogen-activated protein kinase pathway may, therefore, favor termination of inflammation. Inhibitors of p38 reduce the epithelial disruption caused by RSV and Bordetella pertussis.183, 184 Again, inhibition of this pathway, to our knowledge, although tested in asthma,185 has not been tested in lung infection models.
Another approach to reduce pathogen-induced immunopathology during acute infection is to target signaling molecules involved in cell migration. Downstream of chemokine receptors is the phosphatidylinositol 3-kinase, which activates protein kinase C and Rho GTPases.186 Phosphatidylinositol 3-kinase inhibitors reduce the recruitment of neutrophils and T cells in vivo.187, 188 and 189 Although this treatment is efficient in alleviating chronic inflammation such as asthma,190 it is not validated for acute microbial infection. In addition, use of phosphatidylino-sitol 3-kinase inhibitors may interfere with development of the innate response to bacteria.191
Innate (and adaptive) cytokine manipulation
With the identification of each new cytokine, a series of papers describing their manipulation in models of acute infection has followed. All of them cannot be detailed here due to space constraint, but our discussion can be limited to those that have been tested therapeutically in acute lung infection models. The first, type-I IFN, plays such an important role in limiting viral replication that they have developed strategies to avoid it. During RNA virus lung infection IFN-α is produced predominantly by alveolar macrophages (or, to a lesser extent, pDCs) whose depletion impairs viral clearance.192 Dosing with IFN-α and a double-stranded RNA IFN-α inducer, 4 h after SARS coronaviruas infection, reduces lung viral titers.193 Similarly, RSV or human metapneumovirus-infected BALB/c mice, treated intranasally with recombinant IFN-α, have reduced lung viral titers and inflammatory disease as compared with untreated controls.194 Prophylactic treatment of SARS coronavirus-infected macaques with pegylated IFN-α significantly reduces viral load and pulmonary damage; post-exposure treatment is effective, although producing intermediate results.195 Recombinant IFN-α, therefore, appears beneficial for reducing viral replication and associated pathology when administered early after infection. The influence of concomitant bacterial infection requires examination.
Other early innate cytokines important in respiratory viral and bacterial infections include IL-1, IL-12, and TNF. Their blockade or promotion, however, is complicated by the fact that respiratory bacteria tend to require them for clearance. For example, Rhinovirus induces IL-1-receptor antagonist, IL-1Ra, from airway epithelial cells, which facilitates resolution of inflammation.196 However, IL-1Ra enhances bacterial outgrowth in the lungs of mice with pneumococcal pneumonia, without the benefit of reducing the host response.197 Therefore, this pathway is predicted to be good for one lung infection but bad for another. Blockade of IL-12 is another example where neutralization benefits the severity of lung viral infection,198, 199 but impairs clearance of lung Histoplasma capsulatum200 and Legionella pneumophila201 infections. The list continues with neutralization of TNF, which benefits immune pathology induced by influenza and RSV infection,81, 202 but not respiratory bacteria or fungi,203, 204 especially if treatment is prolonged (although this will depend on the precise strategy used205). Webster and co-workers reported recently that absence of TNF and IL-6 is of no benefit during murine influenza H5N1 infection.206 However, up to 50% survival was observed in some experiments and the gene-depleted animals used may harbor other developmental abnormalities. Viruses induce local production of IFN-γ by T and non-T cells in the respiratory tract, and its neutralization not only reduces local lung cellularity and systemic humoral responses to influenza virus infection in mice,207 but may also delay viral clearance.208 IFN-γ is also required for clearance of S. pneumoniae.209 It would appear that the new cytokine on the block, IL-17, may also present opposing effects in viral and bacterial lung infection. Suitable reagents are yet to be developed for IL-17 neutralization, but in vivo blockade of IL-23p19 alone, or in combination with IL-23/IL-12p40 (required for Th17 development), significantly reduces Mycoplasma pneumoniae-induced IL-17 and subsequent bacterial clearance, possibly via reduced neutrophil activity.210K. pneumoniae clearance also depends on IL-17,211 but the influence of IL-17 or IL-23 neutralization on respiratory viral infection is unknown.
Administration of immune suppressive cytokines has been considered for infection induced lung inflammatory disease and encountered similar problems. IL-10 neutralization increases survival of mice infected with K. pneumoniae.212 In contrast, influenza induces indoleamine 2,3-dioxygenase and IL-10 production, which may limit lung inflammation. However, treatment with an indoleamine 2,3-dioxygenase inhibitor (that would reduce IL-10) induces a 20-fold reduction in lung S. pneumoniae load.213 Intranasal IL-10 treatment of RSV-infected mice reduces lung nuclear factorκB DNA-binding activity, chemokine gene expression, and airway inflammation.175 Similarly, administration of TGF-β-encoding plasmid reduces inflammation to viral and fungal lung pathogens, but, without exception, prevents their clearance.54
Concluding Remarks
Although a plethora of strategies have been used to modulate lung inflammation during acute infection, few are tested therapeutically after the onset of clinical symptoms, and even less are tested in models of common co-existing lung pathogens. Hundreds of immune modulators are, therefore, beneficial during influenza infection, but what of the bacteria that sometimes accompany them? Would immune therapeutics work best in combination with antibiotics? Equally, selection of immune modulators requires precision in determining exactly what the patient is infected with. In the absence of this knowledge, we may apply a beneficial strategy to one supposed infection, but create an altogether different type of problem. Several gaps remain in our knowledge of how immune homeostasis is maintained in the respiratory tract, inflammatory pathways that overcome them, and the precise effector/memory phenotype of immune cells within the airways and lung parenchyma. Controversy also still surrounds the potential of “acute” respiratory infections to persist, since detection of pathogen genome is common, but few studies have been able to demonstrate classical reactivation long after the primary infection. Should persistence exist then, depending on the nature of the persisting organism, immune modulators may cause their reactivation. The development of sensitive tools for pathogen detection, and elucidation of specific gene expression patterns in patients with acute infection,214 mean that future use of targeted immune modulators is not impossible as long as we are able to strike a balance between immune pathology and immune defence.
Disclosure
The authors declared no conflict of interest.
Figure 2.
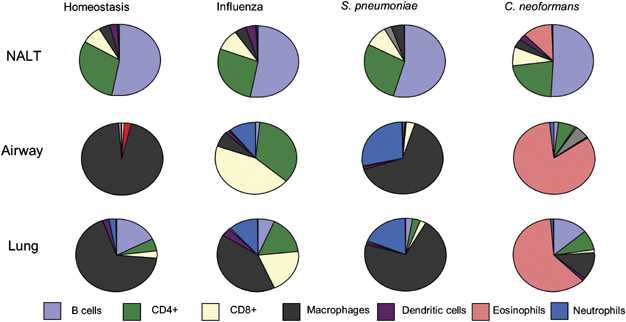
Examples of cell subtypes in the NALT,1, 144, 215 and 216 airway (by bronchoalveolar lavage) and lung parenchyma in homeostasis,38 or during influenza virus,25, 30, 31 and 32S. pneumonia,33, 34 and 217 or C. neoformans35, 36 and 37 infection. Each sector represents an approximate percentage. Alterations in dominant cell subtypes from homeostasis is shown for the peak inflammatory infiltrate of each infection (influenza=day 7, S. pneumoniae=day 4, and C. neoformans=day 12).
Acknowledgements
This work was supported by the Medical Research Council (P171/03/C1/048), the National Institute of Health (NGA: 1 U01 AI070232-01 PI: SMITH, KENDALL A), and the European Union (contract number 032296).
PowerPoint slides
References
- 1.Bienenstock J, McDermott MR. Bronchus- and nasal-associated lymphoid tissues. Immunol. Rev. 2005;206,:22–31. doi: 10.1111/j.0105-2896.2005.00299.x. [DOI] [PubMed] [Google Scholar]
- 2.Debertin AS. Nasal-associated lymphoid tissue (NALT): frequency and localization in young children. Clin. Exp. Immunol. 2003;134,:503–507. doi: 10.1111/j.1365-2249.2003.02311.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kuper CF. The role of nasopharyngeal lymphoid tissue. Immunol. Today. 1992;13,:219–224. doi: 10.1016/0167-5699(92)90158-4. [DOI] [PubMed] [Google Scholar]
- 4.Rangel-Moreno J, Moyron-Quiroz JE, Hartson L, Kusser K, Randall TD. Pulmonary expression of CXC chemokine ligand 13, CC chemokine ligand 19, and CC chemokine ligand 21 is essential for local immunity to influenza. Proc. Natl. Acad. Sci. USA. 2007;104,:10577–10582. doi: 10.1073/pnas.0700591104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pabst R. Is BALT a major component of the human lung immune system? Immunol. Today. 1992;13,:119–122. doi: 10.1016/0167-5699(92)90106-H. [DOI] [PubMed] [Google Scholar]
- 6.Xu B. Lymphocyte homing to bronchus-associated lymphoid tissue (BALT) is mediated by L-selectin/PNAd, alpha4beta1 integrin/VCAM-1, and LFA-1 adhesion pathways. J. Exp. Med. 2003;197,:1255–1267. doi: 10.1084/jem.20010685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moyron-Quiroz JE. Role of inducible bronchus associated lymphoid tissue (iBALT) in respiratory immunity. Nat. Med. 2004;10,:927–934. doi: 10.1038/nm1091. [DOI] [PubMed] [Google Scholar]
- 8.Zuercher AW, Cebra JJ. Structural and functional differences between putative mucosal inductive sites of the rat. Eur. J. Immunol. 2002;32,:3191–3196. doi: 10.1002/1521-4141(200211)32:11<3191::AID-IMMU3191>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 9.Marquez MG, Sosa GA, Roux ME. Developmental study of immunocompetent cells in the bronchus-associated lymphoid tissue (BALT) from Wistar rats. Dev. Comp. Immunol. 2000;24,:683–689. doi: 10.1016/s0145-305x(00)00003-3. [DOI] [PubMed] [Google Scholar]
- 10.Tschernig T, Kleemann WJ, Pabst R. Bronchus-associated lymphoid tissue (BALT) in the lungs of children who had died from sudden infant death syndrome and other causes. Thorax. 1995;50,:658–660. doi: 10.1136/thx.50.6.658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hiller AS, Tschernig T, Kleemann WJ, Pabst R. Bronchus-associated lymphoid tissue (BALT) and larynx-associated lymphoid tissue (LALT) are found at different frequencies in children, adolescents and adults. Scand. J. Immunol. 1998;47,:159–162. doi: 10.1046/j.1365-3083.1998.00276.x. [DOI] [PubMed] [Google Scholar]
- 12.Sminia T, van der Brugge Gamelkoorn GJ, Jeurissen SH. Structure and function of bronchus-associated lymphoid tissue (BALT) Crit. Rev. Immunol. 1989;9,:119–150. [PubMed] [Google Scholar]
- 13.Pabst R, Gehrke I. Is the bronchus-associated lymphoid tissue (BALT) an integral structure of the lung in normal mammals, including humans? Am. J. Respir. Cell Mol. Biol. 1990;3,:131–135. doi: 10.1165/ajrcmb/3.2.131. [DOI] [PubMed] [Google Scholar]
- 14.Tschernig T, Pabst R. Bronchus-associated lymphoid tissue (BALT) is not present in the normal adult lung but in different diseases. Pathobiology. 2000;68,:1–8. doi: 10.1159/000028109. [DOI] [PubMed] [Google Scholar]
- 15.Kocks JR, Davalos-Misslitz AC, Hintzen G, Ohl L, Forster R. Regulatory T cells interfere with the development of bronchus-associated lymphoid tissue. J. Exp. Med. 2007;204,:723–734. doi: 10.1084/jem.20061424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rangel-Moreno J. Inducible bronchus-associated lymphoid tissue (iBALT) in patients with pulmonary complications of rheumatoid arthritis. J. Clin. Invest. 2006;116,:3183–3194. doi: 10.1172/JCI28756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Das A. BALT development and augmentation of hyperoxic lung injury in mice deficient in NQO1 and NQO2. Free Radic. Biol. Med. 2006;40,:1843–1856. doi: 10.1016/j.freeradbiomed.2006.01.025. [DOI] [PubMed] [Google Scholar]
- 18.Moyron-Quiroz JE. Persistence and responsiveness of immunologic memory in the absence of secondary lymphoid organs. Immunity. 2006;25,:643–654. doi: 10.1016/j.immuni.2006.08.022. [DOI] [PubMed] [Google Scholar]
- 19.Rangel-Moreno J. Role of CXC chemokine ligand 13, CC chemokine ligand (CCL) 19, and CCL21 in the organization and function of nasal-associated lymphoid tissue. J. Immunol. 2005;175,:4904–4913. doi: 10.4049/jimmunol.175.8.4904. [DOI] [PubMed] [Google Scholar]
- 20.Wiley JA, Hogan RJ, Woodland DL, Harmsen AG. Antigen-specific CD8(+) T cells persist in the upper respiratory tract following influenza virus infection. J. Immunol. 2001;167,:3293–3299. doi: 10.4049/jimmunol.167.6.3293. [DOI] [PubMed] [Google Scholar]
- 21.Tamura S. Antibody-forming cells in the nasal-associated lymphoid tissue during primary influenza virus infection. J. Gen. Virol. 1998;79(Pt 2):291–299. doi: 10.1099/0022-1317-79-2-291. [DOI] [PubMed] [Google Scholar]
- 22.van der Brugge-Gamelkoorn GJ, Claassen E, Sminia T. Anti-TNP-forming cells in bronchus-associated lymphoid tissue (BALT) and paratracheal lymph node (PTLN) of the rat after intratracheal priming and boosting with TNP-KLH. Immunology. 1986;57,:405–409. [PMC free article] [PubMed] [Google Scholar]
- 23.Suda T, Sato A, Sugiura W, Chida K. Induction of MHC class II antigens on rat bronchial epithelial cells by interferon-gamma and its effect on antigen presentation. Lung. 1995;173,:127–137. doi: 10.1007/BF02981472. [DOI] [PubMed] [Google Scholar]
- 24.Hundorfean G. Luminal antigens access late endosomes of intestinal epithelial cells enriched in MHC I and MHC II molecules: in vivo study in Crohn's ileitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2007;293,:G798–G808. doi: 10.1152/ajpgi.00135.2007. [DOI] [PubMed] [Google Scholar]
- 25.Lawrence CW, Ream RM, Braciale TJ. Frequency, specificity, and sites of expansion of CD8+ T cells during primary pulmonary influenza virus infection. J. Immunol. 2005;174,:5332–5340. doi: 10.4049/jimmunol.174.9.5332. [DOI] [PubMed] [Google Scholar]
- 26.Kohlmeier JE, Woodland DL. Memory T cell recruitment to the lung airways. Curr. Opin. Immunol. 2006;18,:357–362. doi: 10.1016/j.coi.2006.03.012. [DOI] [PubMed] [Google Scholar]
- 27.Puneet P, Moochhala S, Bhatia M. Chemokines in acute respiratory distress syndrome. Am. J. Physiol Lung Cell Mol. Physiol. 2005;288,:L3–L15. doi: 10.1152/ajplung.00405.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.D’Ambrosio D, Mariani M, Panina-Bordignon P, Sinigaglia F. Chemokines and their receptors guiding T lymphocyte recruitment in lung inflammation. Am. J. Respir. Crit. Care Med. 2001;164,:1266–1275. doi: 10.1164/ajrccm.164.7.2103011. [DOI] [PubMed] [Google Scholar]
- 29.Hussell T, Openshaw PJM. Intracellular interferon-gamma expression in natural killer cells precedes lung CD8+ T cell recruitment during respiratory syncytial virus infection. J. Gen. Virol. 1998;79,:2593–2601. doi: 10.1099/0022-1317-79-11-2593. [DOI] [PubMed] [Google Scholar]
- 30.Swain SL, Dutton RW, Woodland DL. T cell responses to influenza virus infection: effector and memory cells. Viral Immunol. 2004;17,:197–209. doi: 10.1089/0882824041310577. [DOI] [PubMed] [Google Scholar]
- 31.Doherty PC. Effector CD4+ and CD8+ T-cell mechanisms in the control of respiratory virus infections. Immunol. Rev. 1997;159,:105–117. doi: 10.1111/j.1600-065x.1997.tb01010.x. [DOI] [PubMed] [Google Scholar]
- 32.Humphreys IR. A critical role for ICOS co-stimulation in immune containment of pulmonary influenza virus infection. Eur. J. Immunol. 2006;36,:2928–2938. doi: 10.1002/eji.200636155. [DOI] [PubMed] [Google Scholar]
- 33.Nakasone C. Accumulation of gamma/delta T cells in the lungs and their roles in neutrophil-mediated host defense against pneumococcal infection. Microbes. Infect. 2007;9,:251–258. doi: 10.1016/j.micinf.2006.11.015. [DOI] [PubMed] [Google Scholar]
- 34.Kirby AC, Newton DJ, Carding SR, Kaye PM. Evidence for the involvement of lung-specific gammadelta T cell subsets in local responses to Streptococcus pneumoniae infection. Eur. J. Immunol. 2007;37,:3404–3413. doi: 10.1002/eji.200737216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huffnagle GB, Yates JL, Lipscomb MF. Immunity to a pulmonary Cryptococcus neoformans infection requires both CD4+ and CD8+ T cells. J. Exp. Med. 1991;173,:793–800. doi: 10.1084/jem.173.4.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Humphreys IR. OX40 ligation on activated T cells enhances the control of Cryptococcus neoformans and reduces pulmonary eosinophilia. J. Immunol. 2003;170,:6125–6132. doi: 10.4049/jimmunol.170.12.6125. [DOI] [PubMed] [Google Scholar]
- 37.Huffnagle GB, Boyd MB, Street NE, Lipscomb MF. IL-5 is required for eosinophil recruitment, crystal deposition, and mononuclear cell recruitment during a pulmonary Cryptococcus neoformans infection in genetically susceptible mice (C57BL/6) J. Immunol. 1998;160,:2393–2400. [PubMed] [Google Scholar]
- 38.Murphy J, Summer R, Wilson AA, Kotton DN, Fine A. The prolonged life-span of alveolar macrophages. Am. J. Respir. Cell Mol. Biol. 2008;38,:380–385. doi: 10.1165/rcmb.2007-0224RC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Holt PG. Downregulation of the antigen presenting cell function(s) of pulmonary dendritic cells in vivo by resident alveolar macrophages. J. Exp. Med. 1993;177,:397–407. doi: 10.1084/jem.177.2.397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hussell T, Spender LC, Georgiou A, O’Garra A, Openshaw PJM. Th1 and Th2 cytokine induction in pulmonary T-cells during infection with respiratory syncytial virus. J. Gen. Virol. 1996;77,:2447–2455. doi: 10.1099/0022-1317-77-10-2447. [DOI] [PubMed] [Google Scholar]
- 41.Brandtzaeg P. Induction of secretory immunity and memory at mucosal surfaces. Vaccine. 2007;25,:5467–5484. doi: 10.1016/j.vaccine.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 42.Wines BD, Hogarth PM. IgA receptors in health and disease. Tissue Antigens. 2006;68,:103–114. doi: 10.1111/j.1399-0039.2006.00613.x. [DOI] [PubMed] [Google Scholar]
- 43.Holt PG, Strickland DH, Wikstrom ME, Jahnsen FL. Regulation of immunological homeostasis in the respiratory tract. Nat. Rev. Immunol. 2008;8,:142–152. doi: 10.1038/nri2236. [DOI] [PubMed] [Google Scholar]
- 44.Bilyk N, Holt PG. Inhibition of the immunosuppressive activity of resident pulmonary alveolar macrophages by granulocyte/macrophage colony-stimulating factor. J. Exp. Med. 1993;177,:1773–1777. doi: 10.1084/jem.177.6.1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Eriksson U. Human bronchial epithelium controls TH2 responses by TH1-induced, nitric oxide-mediated STAT5 dephosphorylation: implications for the pathogenesis of asthma. J. Immunol. 2005;175,:2715–2720. doi: 10.4049/jimmunol.175.4.2715. [DOI] [PubMed] [Google Scholar]
- 46.Mayer AK. Differential recognition of TLR-dependent microbial ligands in human bronchial epithelial cells. J. Immunol. 2007;178,:3134–3142. doi: 10.4049/jimmunol.178.5.3134. [DOI] [PubMed] [Google Scholar]
- 47.Pichavant M. Impact of bronchial epithelium on dendritic cell migration and function: modulation by the bacterial motif KpOmpA. J. Immunol. 2006;177,:5912–5919. doi: 10.4049/jimmunol.177.9.5912. [DOI] [PubMed] [Google Scholar]
- 48.Cox G, Gauldie J, Jordana M. Bronchial epithelial cell-derived cytokines (G-CSF and GM-CSF) promote the survival of peripheral blood neutrophils in vitro. Am. J. Respir. Cell Mol. Biol. 1992;7,:507–513. doi: 10.1165/ajrcmb/7.5.507. [DOI] [PubMed] [Google Scholar]
- 49.Bleck B, Tse DB, Jaspers I, Curotto de Lafaille MA, Reibman J. Diesel exhaust particle-exposed human bronchial epithelial cells induce dendritic cell maturation. J. Immunol. 2006;176,:7431–7437. doi: 10.4049/jimmunol.176.12.7431. [DOI] [PubMed] [Google Scholar]
- 50.Raz E. Organ-specific regulation of innate immunity. Nat. Immunol. 2007;8,:3–4. doi: 10.1038/ni0107-3. [DOI] [PubMed] [Google Scholar]
- 51.Takabayshi K. Induction of a homeostatic circuit in lung tissue by microbial compounds. Immunity. 2006;24,:475–487. doi: 10.1016/j.immuni.2006.02.008. [DOI] [PubMed] [Google Scholar]
- 52.Jakubzick C, Tacke F, Llodra J, van Rooijen N, Randolph GJ. Modulation of dendritic cell trafficking to and from the airways. J. Immunol. 2006;176,:3578–3584. doi: 10.4049/jimmunol.176.6.3578. [DOI] [PubMed] [Google Scholar]
- 53.Snelgrove RJ, Edwards L, Rae AJ, Hussell T. An absence of reactive oxygen species improves the resolution of lung influenza infection. Eur. J. Immunol. 2006;36,:1364–1373. doi: 10.1002/eji.200635977. [DOI] [PubMed] [Google Scholar]
- 54.Williams AE. TGF-beta prevents eosinophilic lung disease but impairs pathogen clearance. Microbes. Infect. 2005;7,:365–374. doi: 10.1016/j.micinf.2004.11.012. [DOI] [PubMed] [Google Scholar]
- 55.Bilyk N, Holt PG. Cytokine modulation of the immunosuppressive phenotype of pulmonary alveolar macrophage populations. Immunology. 1995;86,:231–237. [PMC free article] [PubMed] [Google Scholar]
- 56.MacLean JA. Sequestration of inhaled particulate antigens by lung phagocytes. A mechanism for the effective inhibition of pulmonary cell-mediated immunity. Am. J. Pathol. 1996;148,:657–666. [PMC free article] [PubMed] [Google Scholar]
- 57.Holt PG. Inhibitory activity of unstimulated alveolar macrophages on T-lymphocyte blastogenic response. Am. Rev. Respir. Dis. 1978;118,:791–793. doi: 10.1164/arrd.1978.118.4.791. [DOI] [PubMed] [Google Scholar]
- 58.Lambrecht BN. Alveolar macrophage in the driver's seat. Immunity. 2006;24,:366–368. doi: 10.1016/j.immuni.2006.03.008. [DOI] [PubMed] [Google Scholar]
- 59.Thepen T, van Rooijen N, Kraal G. Alveolar macrophage elimination in vivo is associated with an increase in pulmonary immune response in mice. J. Exp. Med. 1989;170,:499–509. doi: 10.1084/jem.170.2.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Demedts IK, Brusselle GG, Vermaelen KY, Pauwels RA. Identification and characterization of human pulmonary dendritic cells. Am. J. Respir. Cell Mol. Biol. 2005;32,:177–184. doi: 10.1165/rcmb.2004-0279OC. [DOI] [PubMed] [Google Scholar]
- 61.Wikstrom ME, Stumbles PA. Mouse respiratory tract dendritic cell subsets and the immunological fate of inhaled antigens. Immunol. Cell Biol. 2007;85,:182–188. doi: 10.1038/sj.icb.7100039. [DOI] [PubMed] [Google Scholar]
- 62.Holt PG, Haining S, Nelson DJ, Sedgwick JD. Origin and steady-state turnover of class II MHC-bearing dendritic cells in the epithelium of the conducting airways. J. Immunol. 1994;153,:256–261. [PubMed] [Google Scholar]
- 63.Demedts IK, Bracke KR, Maes T, Joos GF, Brusselle GG. Different roles for human lung dendritic cell subsets in pulmonary immune defense mechanisms. Am. J. Respir. Cell Mol. Biol. 2006;35,:387–393. doi: 10.1165/rcmb.2005-0382OC. [DOI] [PubMed] [Google Scholar]
- 64.Schlecht G. Murine plasmacytoid dendritic cells induce effector/memory CD8+ T-cell responses in vivo after viral stimulation. Blood. 2004;104,:1808–1815. doi: 10.1182/blood-2004-02-0426. [DOI] [PubMed] [Google Scholar]
- 65.Sapoznikov A. Organ-dependent in vivo priming of naive CD4+, but not CD8+, T cells by plasmacytoid dendritic cells. J. Exp. Med. 2007;204,:1923–1933. doi: 10.1084/jem.20062373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.de Heer HJ, Hammad H, Kool M, Lambrecht BN. Dendritic cell subsets and immune regulation in the lung. Semin. Immunol. 2005;17,:295–303. doi: 10.1016/j.smim.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 67.Hammad H, Lambrecht BN. Recent progress in the biology of airway dendritic cells and implications for understanding the regulation of asthmatic inflammation. J. Allergy Clin. Immunol. 2006;118,:331–336. doi: 10.1016/j.jaci.2006.03.041. [DOI] [PubMed] [Google Scholar]
- 68.Lambrecht BN. Dendritic cells and the regulation of the allergic immune response. Allergy. 2005;60,:271–282. doi: 10.1111/j.1398-9995.2005.00708.x. [DOI] [PubMed] [Google Scholar]
- 69.Grayson MH, Holtzman MJ. Emerging role of dendritic cells in respiratory viral infection. J. Mol. Med. 2007;85,:1057–1068. doi: 10.1007/s00109-007-0212-3. [DOI] [PubMed] [Google Scholar]
- 70.Smit JJ, Rudd BD, Lukacs NW. Plasmacytoid dendritic cells inhibit pulmonary immunopathology and promote clearance of respiratory syncytial virus. J. Exp. Med. 2006;203,:1153–1159. doi: 10.1084/jem.20052359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wang H, Peters N, Schwarze J. Plasmacytoid dendritic cells limit viral replication, pulmonary inflammation, and airway hyperresponsiveness in respiratory syncytial virus infection. J. Immunol. 2006;177,:6263–6270. doi: 10.4049/jimmunol.177.9.6263. [DOI] [PubMed] [Google Scholar]
- 72.Hammad H, Lambrecht BN. Lung dendritic cell migration. Adv. Immunol. 2007;93,:265–278. doi: 10.1016/S0065-2776(06)93007-7. [DOI] [PubMed] [Google Scholar]
- 73.Barclay AN, Wright GJ, Brooke G, Brown MH. CD200 and membrane protein interactions in the control of myeloid cells. Trends Immunol. 2002;23,:285–290. doi: 10.1016/s1471-4906(02)02223-8. [DOI] [PubMed] [Google Scholar]
- 74.Osterholzer JJ. CCR2 and CCR6, but not endothelial selectins, mediate the accumulation of immature dendritic cells within the lungs of mice in response to particulate antigen. J. Immunol. 2005;175,:874–883. doi: 10.4049/jimmunol.175.2.874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Robays LJ. Chemokine receptor CCR2 but not CCR5 or CCR6 mediates the increase in pulmonary dendritic cells during allergic airway inflammation. J. Immunol. 2007;178,:5305–5311. doi: 10.4049/jimmunol.178.8.5305. [DOI] [PubMed] [Google Scholar]
- 76.Vermaelen KY, Carro-Muino I, Lambrecht BN, Pauwels RA. Specific migratory dendritic cells rapidly transport antigen from the airways to the thoracic lymph nodes. J. Exp. Med. 2001;193,:51–60. doi: 10.1084/jem.193.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sung SS. A major lung CD103 (alphaE)-beta7 integrin-positive epithelial dendritic cell population expressing Langerin and tight junction proteins. J. Immunol. 2006;176,:2161–2172. doi: 10.4049/jimmunol.176.4.2161. [DOI] [PubMed] [Google Scholar]
- 78.Hogan RJ. Long-term maintenance of virus-specific effector memory CD8+ T cells in the lung airways depends on proliferation. J. Immunol. 2002;169,:4976–4981. doi: 10.4049/jimmunol.169.9.4976. [DOI] [PubMed] [Google Scholar]
- 79.Harris NL, Watt V, Ronchese F, Le Gros G. Differential T cell function and fate in lymph node and nonlymphoid tissues. J. Exp. Med. 2002;195,:317–326. doi: 10.1084/jem.20011558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ely KH, Cookenham T, Roberts AD, Woodland DL. Memory T cell populations in the lung airways are maintained by continual recruitment. J. Immunol. 2006;176,:537–543. doi: 10.4049/jimmunol.176.1.537. [DOI] [PubMed] [Google Scholar]
- 81.Peper RL, Van Campen H. Tumor necrosis factor as a mediator of inflammation in influenza A viral pneumonia. Microb. Pathog. 1995;19,:175–183. doi: 10.1006/mpat.1995.0056. [DOI] [PubMed] [Google Scholar]
- 82.Kurokawa M, Imakita M, Kumeda CA, Shiraki K. Cascade of fever production in mice infected with influenza virus. J. Med. Virol. 1996;50,:152–158. doi: 10.1002/(SICI)1096-9071(199610)50:2<152::AID-JMV8>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 83.Kozak W, Conn CA, Klir JJ, Wong GH, Kluger MJ. TNF soluble receptor and antiserum against TNF enhance lipopolysaccharide fever in mice. Am. J. Physiol. 1995;269,:R23–R29. doi: 10.1152/ajpregu.1995.269.1.R23. [DOI] [PubMed] [Google Scholar]
- 84.Kozak W. Sickness behavior in mice deficient in interleukin-6 during turpentine abscess and influenza pneumonitis. Am. J. Physiol. 1997;272,:R621–R630. doi: 10.1152/ajpregu.1997.272.2.R621. [DOI] [PubMed] [Google Scholar]
- 85.Cook DN. The role of MIP-1 alpha in inflammation and hematopoiesis. J. Leukoc. Biol. 1996;59,:61–66. doi: 10.1002/jlb.59.1.61. [DOI] [PubMed] [Google Scholar]
- 86.Rutigliano JA, Graham BS. Prolonged production of TNF-alpha exacerbates illness during respiratory syncytial virus infection. J. Immunol. 2004;173,:3408–3417. doi: 10.4049/jimmunol.173.5.3408. [DOI] [PubMed] [Google Scholar]
- 87.Sedgwick JB, Menon I, Gern JE, Busse WW. Effects of inflammatory cytokines on the permeability of human lung microvascular endothelial cell monolayers and differential eosinophil transmigration. J. Allergy Clin. Immunol. 2002;110,:752–756. doi: 10.1067/mai.2002.128581. [DOI] [PubMed] [Google Scholar]
- 88.Mazzon E, Cuzzocrea S. Role of TNF-alpha in lung tight junction alteration in mouse model of acute lung inflammation. Respir. Res. 2007;8,:75. doi: 10.1186/1465-9921-8-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Watts TH. TNF/TNFR family members in costimulation of T cell responses. Annu. Rev. Immunol. 2005;23,:23–68. doi: 10.1146/annurev.immunol.23.021704.115839. [DOI] [PubMed] [Google Scholar]
- 90.Bansal-Pakala P, Halteman BS, Cheng MH, Croft M. Costimulation of CD8 T cell responses by OX40. J. Immunol. 2004;172,:4821–4825. doi: 10.4049/jimmunol.172.8.4821. [DOI] [PubMed] [Google Scholar]
- 91.Ito T. OX40 ligand shuts down IL-10-producing regulatory T cells. Proc. Natl. Acad. Sci. USA. 2006;103,:13138–13143. doi: 10.1073/pnas.0603107103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Serghides L. Evaluation of OX40 ligand as a costimulator of human antiviral memory CD8 T cell responses: comparison with B7.1 and 4-1BBL. J. Immunol. 2005;175,:6368–6377. doi: 10.4049/jimmunol.175.10.6368. [DOI] [PubMed] [Google Scholar]
- 93.Zubairi S, Sanos SL, Hill S, Kaye PM. Immunotherapy with OX40L-Fc or anti-CTLA-4 enhances local tissue responses and killing of Leishmania donovani. Eur. J. Immunol. 2004;34,:1433–1440. doi: 10.1002/eji.200324021. [DOI] [PubMed] [Google Scholar]
- 94.Munks MW, Mourich DV, Mittler RS, Weinberg AD, Hill AB. 4-1BB and OX40 stimulation enhance CD8 and CD4 T-cell responses to a DNA prime, poxvirus boost vaccine. Immunology. 2004;112,:559–566. doi: 10.1111/j.1365-2567.2004.01917.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Humphreys IR. A critical role for OX40 in T cell-mediated immunopathology during lung viral infection. J. Exp. Med. 2003;198,:1237–1242. doi: 10.1084/jem.20030351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hendriks J. During viral infection of the respiratory tract, CD27, 4-1BB, and OX40 collectively determine formation of CD8+ memory T cells and their capacity for secondary expansion. J. Immunol. 2005;175,:1665–1676. doi: 10.4049/jimmunol.175.3.1665. [DOI] [PubMed] [Google Scholar]
- 97.Dawicki W, Bertram EM, Sharpe AH, Watts TH. 4-1BB and OX40 act independently to facilitate robust CD8 and CD4 recall responses. J. Immunol. 2004;173,:5944–5951. doi: 10.4049/jimmunol.173.10.5944. [DOI] [PubMed] [Google Scholar]
- 98.Vidric M, Suh WK, Dianzani U, Mak TW, Watts TH. Cooperation between 4-1BB and ICOS in the immune response to influenza virus revealed by studies of CD28/ICOS-deficient mice. J. Immunol. 2005;175,:7288–7296. doi: 10.4049/jimmunol.175.11.7288. [DOI] [PubMed] [Google Scholar]
- 99.Zaini J. OX40 ligand expressed by DCs costimulates NKT and CD4+ Th cell antitumor immunity in mice. J. Clin. Invest. 2007;117,:3330–3338. doi: 10.1172/JCI32693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lane P. Role of OX40 signals in coordinating CD4 T cell selection, migration, and cytokine differentiation in T helper (Th)1 and Th2 cells. J. Exp. Med. 2000;191,:201–206. doi: 10.1084/jem.191.2.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kotani A, Hori T, Matsumura Y, Uchiyama T. Signaling of gp34 (OX40 ligand) induces vascular endothelial cells to produce a CC chemokine RANTES/CCL5. Immunol. Lett. 2002;84,:1–7. doi: 10.1016/s0165-2478(02)00082-2. [DOI] [PubMed] [Google Scholar]
- 102.Sugamura K, Ishii N, Weinberg AD. Therapeutic targeting of the effector T-cell co-stimulatory molecule OX40. Nat. Rev. Immunol. 2004;4,:420–431. doi: 10.1038/nri1371. [DOI] [PubMed] [Google Scholar]
- 103.Bertram EM, Lau P, Watts TH. Temporal segregation of 4-1BB versus CD28-mediated costimulation: 4-1BB ligand influences T cell numbers late in the primary response and regulates the size of the T cell memory response following influenza infection. J. Immunol. 2002;168,:3777–3785. doi: 10.4049/jimmunol.168.8.3777. [DOI] [PubMed] [Google Scholar]
- 104.Lumsden JM, Roberts JM, Harris NL, Peach RJ, Ronchese F. Differential requirement for CD80 and CD80/CD86-dependent costimulation in the lung immune response to an influenza virus infection. J. Immunol. 2000;164,:79–85. doi: 10.4049/jimmunol.164.1.79. [DOI] [PubMed] [Google Scholar]
- 105.Pietrella D. Disruption of CD40/CD40L interaction influences the course of Cryptococcus neoformans infection. FEMS Immunol. Med. Microbiol. 2004;40,:63–70. doi: 10.1016/S0928-8244(03)00297-9. [DOI] [PubMed] [Google Scholar]
- 106.Hendriks J, Xiao Y, Borst J. CD27 promotes survival of activated T cells and complements CD28 in generation and establishment of the effector T cell pool. J. Exp. Med. 2003;198,:1369–1380. doi: 10.1084/jem.20030916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Behnia M, Robertson KA, Martin WJ. Lung infections: role of apoptosis in host defense and pathogenesis of disease. Chest. 2000;117,:1771–1777. doi: 10.1378/chest.117.6.1771. [DOI] [PubMed] [Google Scholar]
- 108.Lasbury ME. Suppression of alveolar macrophage apoptosis prolongs survival of rats and mice with Pneumocystis pneumonia. J. Immunol. 2006;176,:6443–6453. doi: 10.4049/jimmunol.176.11.6443. [DOI] [PubMed] [Google Scholar]
- 109.Grassme H. CD95/CD95 ligand interactions on epithelial cells in host defense to Pseudomonas aeruginosa. Science. 2000;290,:527–530. doi: 10.1126/science.290.5491.527. [DOI] [PubMed] [Google Scholar]
- 110.Richter MV, Topham DJ. The alpha1beta1 integrin and TNF receptor II protect airway CD8+ effector T cells from apoptosis during influenza infection. J. Immunol. 2007;179,:5054–5063. doi: 10.4049/jimmunol.179.8.5054. [DOI] [PubMed] [Google Scholar]
- 111.Zhou J, Matsuoka M, Cantor H, Homer R, Enelow RI. Cutting edge: engagement of NKG2A on CD8+ effector T cells limits immunopathology in influenza pneumonia. J. Immunol. 2008;180,:25–29. doi: 10.4049/jimmunol.180.1.25. [DOI] [PubMed] [Google Scholar]
- 112.Uiprasertkul M. Apoptosis and pathogenesis of avian influenza A (H5N1) virus in humans. Emerg. Infect. Dis. 2007;13,:708–712. doi: 10.3201/eid1305.060572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Lam WY. Avian influenza A/HK/483/97(H5N1) NS1 protein induces apoptosis in human airway epithelial cells. J. Virol. 2008;82,:2741–2751. doi: 10.1128/JVI.01712-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Tumpey TM, Lu X, Morken T, Zaki SR, Katz JM. Depletion of lymphocytes and diminished cytokine production in mice infected with a highly virulent influenza A (H5N1) virus isolated from humans. J. Virol. 2000;74,:6105–6116. doi: 10.1128/jvi.74.13.6105-6116.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Sieg S, King C, Huang Y, Kaplan D. The role of interleukin-10 in the inhibition of T-cell proliferation and apoptosis mediated by parainfluenza virus type 3. J. Virol. 1996;70,:4845–4848. doi: 10.1128/jvi.70.7.4845-4848.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Fan T. Inhibition of apoptosis in chlamydia-infected cells: blockade of mitochondrial cytochrome c release and caspase activation. J. Exp. Med. 1998;187,:487–496. doi: 10.1084/jem.187.4.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Hakansson A. Aspects on the interaction of Streptococcus pneumoniae and Haemophilus influenzae with human respiratory tract mucosa. Am. J. Respir. Crit. Care Med. 1996;154,:S187–S191. doi: 10.1164/ajrccm/154.4_Pt_2.S187. [DOI] [PubMed] [Google Scholar]
- 118.Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat. Rev. Immunol. 2007;7,:678–689. doi: 10.1038/nri2156. [DOI] [PubMed] [Google Scholar]
- 119.Butcher EC, Picker LJ. Lymphocyte homing and homeostasis. Science. 1996;272,:60–66. doi: 10.1126/science.272.5258.60. [DOI] [PubMed] [Google Scholar]
- 120.Stuve O, Bennett JL. Pharmacological properties, toxicology and scientific rationale for the use of natalizumab (Tysabri) in inflammatory diseases. CNS Drug Rev. 2007;13,:79–95. doi: 10.1111/j.1527-3458.2007.00003.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Dedrick RL, Walicke P, Garovoy M. Anti-adhesion antibodies efalizumab, a humanized anti-CD11a monoclonal antibody. Transpl. Immunol. 2002;9,:181–186. doi: 10.1016/s0966-3274(02)00029-1. [DOI] [PubMed] [Google Scholar]
- 122.Li YY, Perez HD, Zollner TM. Fatalities in natalizumab treatment—a ‘no go’ for leukocyte recirculation approaches? Expert Opin. Ther. Targets. 2006;10,:489–499. doi: 10.1517/14728222.10.4.489. [DOI] [PubMed] [Google Scholar]
- 123.Glass WG, Rosenberg HF, Murphy PM. Chemokine regulation of inflammation during acute viral infection. Curr. Opin. Allergy Clin. Immunol. 2003;3,:467–473. doi: 10.1097/00130832-200312000-00008. [DOI] [PubMed] [Google Scholar]
- 124.Charo IF, Ransohoff RM. The many roles of chemokines and chemokine receptors in inflammation. N. Engl. J. Med. 2006;354,:610–621. doi: 10.1056/NEJMra052723. [DOI] [PubMed] [Google Scholar]
- 125.Proudfoot AE, Power CA, Rommel C, Wells TN. Strategies for chemokine antagonists as therapeutics. Semin. Immunol. 2003;15,:57–65. doi: 10.1016/s1044-5323(02)00128-8. [DOI] [PubMed] [Google Scholar]
- 126.Moser B. Chemokines: role in immune cell traffic. Eur. Cytokine Netw. 2003;14,:204–210. [PubMed] [Google Scholar]
- 127.Kinashi T. Intracellular signalling controlling integrin activation in lymphocytes. Nat. Rev. Immunol. 2005;5,:546–559. doi: 10.1038/nri1646. [DOI] [PubMed] [Google Scholar]
- 128.Devalaraja MN, Richmond A. Multiple chemotactic factors: fine control or redundancy? Trends Pharmacol. Sci. 1999;20,:151–156. doi: 10.1016/s0165-6147(99)01342-5. [DOI] [PubMed] [Google Scholar]
- 129.Sakai S. Therapeutic effect of anti-macrophage inflammatory protein 2 antibody on influenza virus-induced pneumonia in mice. J. Virol. 2000;74,:2472–2476. doi: 10.1128/jvi.74.5.2472-2476.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Brundage JF. Interactions between influenza and bacterial respiratory pathogens: implications for pandemic preparedness. Lancet Infect. Dis. 2006;6,:303–312. doi: 10.1016/S1473-3099(06)70466-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Beadling C, Slifka MK. How do viral infections predispose patients to bacterial infections? Curr. Opin. Infect. Dis. 2004;17,:185–191. doi: 10.1097/00001432-200406000-00003. [DOI] [PubMed] [Google Scholar]
- 132.Schall TJ, Bacon K, Toy KJ, Goeddel DV. Selective attraction of monocytes and T lymphocytes of the memory phenotype by cytokine RANTES. Nature (London) 1990;347,:669–671. doi: 10.1038/347669a0. [DOI] [PubMed] [Google Scholar]
- 133.Proudfoot AE. Extension of recombinant human RANTES by the retention of the initiating methionine produces a potent antagonist. J. Biol. Chem. 1996;271,:2599–2603. doi: 10.1074/jbc.271.5.2599. [DOI] [PubMed] [Google Scholar]
- 134.Culley FJ. Role of CCL5 (RANTES) in viral lung disease. J. Virol. 2006;80,:8151–8157. doi: 10.1128/JVI.00496-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Rosenberg HF, Bonville CA, Easton AJ, Domachowske JB. The pneumonia virus of mice infection model for severe respiratory syncytial virus infection: identifying novel targets for therapeutic intervention. Pharmacol. Ther. 2005;105,:1–6. doi: 10.1016/j.pharmthera.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 136.Amano H. Essential contribution of monocyte chemoattractant protein-1/C–C chemokine ligand-2 to resolution and repair processes in acute bacterial pneumonia. J. Immunol. 2004;172,:398–409. doi: 10.4049/jimmunol.172.1.398. [DOI] [PubMed] [Google Scholar]
- 137.Zeng X. Interferon-inducible protein 10, but not monokine induced by gamma interferon, promotes protective type 1 immunity in murine Klebsiella pneumoniae pneumonia. Infect. Immun. 2005;73,:8226–8236. doi: 10.1128/IAI.73.12.8226-8236.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Culley FJ, Pennycook AM, Tregoning JS, Hussell T, Openshaw PJ. Differential chemokine expression following respiratory virus infection reflects Th1- or Th2-biased immunopathology. J. Virol. 2006;80,:4521–4527. doi: 10.1128/JVI.80.9.4521-4527.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Lee BJ. Role of CXCR3 in the immune response to murine gammaherpesvirus 68. J. Virol. 2005;79,:9351–9355. doi: 10.1128/JVI.79.14.9351-9355.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Bonville CA. Functional antagonism of chemokine receptor CCR1 reduces mortality in acute pneumovirus infection in vivo. J. Virol. 2004;78,:7984–7989. doi: 10.1128/JVI.78.15.7984-7989.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.John AE. Respiratory syncytial virus-induced exaggeration of allergic airway disease is dependent upon CCR1-associated immune responses. Eur. J. Immunol. 2005;35,:108–116. doi: 10.1002/eji.200425439. [DOI] [PubMed] [Google Scholar]
- 142.Matloubian M. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature (London) 2004;427,:355–360. doi: 10.1038/nature02284. [DOI] [PubMed] [Google Scholar]
- 143.Jacobson JR, Garcia JG. Novel therapies for microvascular permeability in sepsis. Curr. Drug Targets. 2007;8,:509–514. doi: 10.2174/138945007780362719. [DOI] [PubMed] [Google Scholar]
- 144.Rodriguez-Monroy MA, Rojas-Hernandez S, Moreno-Fierros L. Phenotypic and functional differences between lymphocytes from NALT and nasal passages of mice. Scand. J. Immunol. 2007;65,:276–288. doi: 10.1111/j.1365-3083.2006.01898.x. [DOI] [PubMed] [Google Scholar]
- 145.Racedo S. Lactobacillus casei administration reduces lung injuries in a Streptococcus pneumoniae infection in mice. Microbes. Infect. 2006;8,:2359–2366. doi: 10.1016/j.micinf.2006.04.022. [DOI] [PubMed] [Google Scholar]
- 146.Selin LK. Memory of mice and men: CD8+ T-cell cross-reactivity and heterologous immunity. Immunol. Rev. 2006;211,:164–181. doi: 10.1111/j.0105-2896.2006.00394.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Kamradt T, Goggel R, Erb KJ. Induction, exacerbation and inhibition of allergic and autoimmune diseases by infection. Trends Immunol. 2005;26,:260–267. doi: 10.1016/j.it.2005.03.009. [DOI] [PubMed] [Google Scholar]
- 148.Walzl G, Tafuro S, Moss P, Openshaw PJ, Hussell T. Influenza virus lung infection protects from respiratory syncytial virus-induced immunopathology. J. Exp. Med. 2000;192,:1317–1326. doi: 10.1084/jem.192.9.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Chen HD. Memory CD8+ T cells in heterologous antiviral immunity and immunopathology in the lung. Nat. Immunol. 2001;2,:1067–1076. doi: 10.1038/ni727. [DOI] [PubMed] [Google Scholar]
- 150.Chen HD, Fraire AE, Joris I, Welsh RM, Selin LK. Specific history of heterologous virus infections determines antiviral immunity and immunopathology in the lung. Am. J. Pathol. 2003;163,:1341–1355. doi: 10.1016/S0002-9440(10)63493-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Walzl G. Prior exposure to live Mycobacterium bovis BCG decreases Cryptococcus neoformans-induced lung eosinophilia in a gamma interferon-dependent manner. Infect. Immun. 2003;71,:3384–3391. doi: 10.1128/IAI.71.6.3384-3391.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Didierlaurent A. Sustained desensitization to bacterial Toll-like receptor ligands after resolution of respiratory influenza infection. J. Exp. Med. 2008;205,:323–329. doi: 10.1084/jem.20070891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Edwards L. Stimulation via Toll-like receptor 9 reduces Cryptococcus neoformans-induced pulmonary inflammation in an IL-12-dependent manner. Eur. J. Immunol. 2005;35,:273–281. doi: 10.1002/eji.200425640. [DOI] [PubMed] [Google Scholar]
- 154.Williams AE. Innate imprinting by the modified heat-labile toxin of Escherichia coli (LTK63) provides generic protection against lung infectious disease. J. Immunol. 2004;173,:7435–7443. doi: 10.4049/jimmunol.173.12.7435. [DOI] [PubMed] [Google Scholar]
- 155.Maizels RM. Infections and allergy—helminths, hygiene and host immune regulation. Curr. Opin. Immunol. 2005;17,:656–661. doi: 10.1016/j.coi.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 156.Furze RC, Hussell T, Selkirk ME. Amelioration of influenza-induced pathology in mice by coinfection with Trichinella spiralis. Infect. Immun. 2006;74,:1924–1932. doi: 10.1128/IAI.74.3.1924-1932.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Williams AE, Edwards L, Hussell T. Colonic bacterial infection abrogates eosinophilic pulmonary disease. J. Infect. Dis. 2006;193,:223–230. doi: 10.1086/498915. [DOI] [PubMed] [Google Scholar]
- 158.Lu CC, Lai HC, Hsieh SC, Chen JK. Resveratrol ameliorates Serratia marcescens-induced acute pneumonia in rats. J. Leukoc. Biol. 2008;83,:1028–1037. doi: 10.1189/jlb.0907647. [DOI] [PubMed] [Google Scholar]
- 159.Ohta Y. In vivo anti-influenza virus activity of an immunomodulatory acidic polysaccharide isolated from Cordyceps militaris grown on germinated soybeans. J. Agric. Food Chem. 2007;55,:10194–10199. doi: 10.1021/jf0721287. [DOI] [PubMed] [Google Scholar]
- 160.Yamada H, Mizuno S, Ross AC, Sugawara I. Retinoic acid therapy attenuates the severity of tuberculosis while altering lymphocyte and macrophage numbers and cytokine expression in rats infected with Mycobacterium tuberculosis. J. Nutr. 2007;137,:2696–2700. doi: 10.1093/jn/137.12.2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Liew FY, Cox FE. Nonspecific defence mechanism: the role of nitric oxide. Immunol. Today. 1991;12,:A17–A21. doi: 10.1016/S0167-5699(05)80006-4. [DOI] [PubMed] [Google Scholar]
- 162.Nagata M. Inflammatory cells and oxygen radicals. Curr. Drug Targets Inflamm. Allergy. 2005;4,:503–504. doi: 10.2174/1568010054526322. [DOI] [PubMed] [Google Scholar]
- 163.Jean D. Beneficial effects of nitric oxide inhalation on pulmonary bacterial clearance. Crit. Care Med. 2002;30,:442–447. doi: 10.1097/00003246-200202000-00029. [DOI] [PubMed] [Google Scholar]
- 164.Ader F. Inhaled nitric oxide increases endothelial permeability in Pseudomonas aeruginosa pneumonia. Intensive Care Med. 2007;33,:503–510. doi: 10.1007/s00134-006-0497-7. [DOI] [PubMed] [Google Scholar]
- 165.Sun Z, Sun B, Wang X, Wang W, Zhu L. Anti-inflammatory effects of inhaled nitric oxide are optimized at lower oxygen concentration in experimental Klebsiella pneumoniae pneumonia. Inflamm. Res. 2006;55,:430–440. doi: 10.1007/s00011-006-6029-7. [DOI] [PubMed] [Google Scholar]
- 166.Stark JM. Immune and functional role of nitric oxide in a mouse model of respiratory syncytial virus infection. J. Infect. Dis. 2005;191,:387–395. doi: 10.1086/427241. [DOI] [PubMed] [Google Scholar]
- 167.Castro SM. Antioxidant treatment ameliorates respiratory syncytial virus-induced disease and lung inflammation. Am. J. Respir. Crit. Care Med. 2006;174,:1361–1369. doi: 10.1164/rccm.200603-319OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Sidwell RW. Inhibitory effects of recombinant manganese superoxide dismutase on influenza virus infections in mice. Antimicrob. Agents Chemother. 1996;40,:2626–2631. doi: 10.1128/aac.40.11.2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Davis IC. Post-infection A77-1726 blocks pathophysiologic sequelae of respiratory syncytial virus infection. Am. J. Respir. Cell Mol. Biol. 2007;37,:379–386. doi: 10.1165/rcmb.2007-0142OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Brown KL, Cosseau C, Gardy JL, Hancock RE. Complexities of targeting innate immunity to treat infection. Trends Immunol. 2007;28,:260–266. doi: 10.1016/j.it.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 171.Park GY, Christman JW. Nuclear factor kappa B is a promising therapeutic target in inflammatory lung disease. Curr. Drug Targets. 2006;7,:661–668. doi: 10.2174/138945006777435317. [DOI] [PubMed] [Google Scholar]
- 172.O’Neill LA. Targeting signal transduction as a strategy to treat inflammatory diseases. Nat. Rev. Drug Discov. 2006;5,:549–563. doi: 10.1038/nrd2070. [DOI] [PubMed] [Google Scholar]
- 173.Lee MS, Kim YJ. Signaling pathways downstream of pattern-recognition receptors and their cross talk. Annu. Rev. Biochem. 2007;76,:447–480. doi: 10.1146/annurev.biochem.76.060605.122847. [DOI] [PubMed] [Google Scholar]
- 174.Bernasconi D, Amici C, La Frazia S, Ianaro A, Santoro MG. The IkappaB kinase is a key factor in triggering influenza A virus-induced inflammatory cytokine production in airway epithelial cells. J. Biol. Chem. 2005;280,:24127–24134. doi: 10.1074/jbc.M413726200. [DOI] [PubMed] [Google Scholar]
- 175.Haeberle HA. IkappaB kinase is a critical regulator of chemokine expression and lung inflammation in respiratory syncytial virus infection. J. Virol. 2004;78,:2232–2241. doi: 10.1128/JVI.78.5.2232-2241.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Sadikot RT. Targeted immunomodulation of the NF-kappaB pathway in airway epithelium impacts host defense against Pseudomonas aeruginosa. J. Immunol. 2006;176,:4923–4930. doi: 10.4049/jimmunol.176.8.4923. [DOI] [PubMed] [Google Scholar]
- 177.Cheng DS. Airway epithelium controls lung inflammation and injury through the NF-kappa B pathway. J. Immunol. 2007;178,:6504–6513. doi: 10.4049/jimmunol.178.10.6504. [DOI] [PubMed] [Google Scholar]
- 178.Everhart MB. Duration and intensity of NF-kappaB activity determine the severity of endotoxin-induced acute lung injury. J. Immunol. 2006;176,:4995–5005. doi: 10.4049/jimmunol.176.8.4995. [DOI] [PubMed] [Google Scholar]
- 179.Zingarelli B, Cook JA. Peroxisome proliferator-activated receptor-gamma is a new therapeutic target in sepsis and inflammation. Shock. 2005;23,:393–399. doi: 10.1097/01.shk.0000160521.91363.88. [DOI] [PubMed] [Google Scholar]
- 180.Michalik L, Wahli W. Involvement of PPAR nuclear receptors in tissue injury and wound repair. J. Clin. Invest. 2006;116,:598–606. doi: 10.1172/JCI27958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Arnold R, Neumann M, Konig W. Peroxisome proliferator-activated receptor-gamma agonists inhibit respiratory syncytial virus-induced expression of intercellular adhesion molecule-1 in human lung epithelial cells. Immunology. 2007;121,:71–81. doi: 10.1111/j.1365-2567.2006.02539.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Ashwell JD. The many paths to p38 mitogen-activated protein kinase activation in the immune system. Nat. Rev. Immunol. 2006;6,:532–540. doi: 10.1038/nri1865. [DOI] [PubMed] [Google Scholar]
- 183.Singh D, McCann KL, Imani F. MAPK and heat shock protein 27 activation are associated with respiratory syncytial virus induction of human bronchial epithelial monolayer disruption. Am. J. Physiol. Lung Cell Mol. Physiol. 2007;293,:L436–L445. doi: 10.1152/ajplung.00097.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Garcia JG. Critical involvement of p38 MAP kinase in pertussis toxin-induced cytoskeletal reorganization and lung permeability. FASEB J. 2002;16,:1064–1076. doi: 10.1096/fj.01-0895com. [DOI] [PubMed] [Google Scholar]
- 185.Duan W. Inhaled p38alpha mitogen-activated protein kinase antisense oligonucleotide attenuates asthma in mice. Am. J. Respir. Crit. Care Med. 2005;171,:571–578. doi: 10.1164/rccm.200408-1006OC. [DOI] [PubMed] [Google Scholar]
- 186.Kehrl JH. Chemoattractant receptor signaling and the control of lymphocyte migration. Immunol. Res. 2006;34,:211–227. doi: 10.1385/IR:34:3:211. [DOI] [PubMed] [Google Scholar]
- 187.Thomas MJ. Airway inflammation: chemokine-induced neutrophilia and the class I phosphoinositide 3-kinases. Eur. J. Immunol. 2005;35,:1283–1291. doi: 10.1002/eji.200425634. [DOI] [PubMed] [Google Scholar]
- 188.Pinho V. Tissue- and stimulus-dependent role of phosphatidylinositol 3-kinase isoforms for neutrophil recruitment induced by chemoattractants in vivo. J. Immunol. 2007;179,:7891–7898. doi: 10.4049/jimmunol.179.11.7891. [DOI] [PubMed] [Google Scholar]
- 189.Bonnans C, Fukunaga K, Keledjian R, Petasis NA, Levy BD. Regulation of phosphatidylinositol 3-kinase by polyisoprenyl phosphates in neutrophil-mediated tissue injury. J. Exp. Med. 2006;203,:857–863. doi: 10.1084/jem.20052143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Myou S. Blockade of inflammation and airway hyperresponsiveness in immune-sensitized mice by dominant-negative phosphoinositide 3-kinase-TAT. J. Exp. Med. 2003;198,:1573–1582. doi: 10.1084/jem.20030298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Maus UA. Importance of phosphoinositide 3-kinase gamma in the host defense against pneumococcal infection. Am. J. Respir. Crit. Care Med. 2007;175,:958–966. doi: 10.1164/rccm.200610-1533OC. [DOI] [PubMed] [Google Scholar]
- 192.Kumagai Y. Alveolar macrophages are the primary interferon-alpha producer in pulmonary infection with RNA viruses. Immunity. 2007;27,:240–252. doi: 10.1016/j.immuni.2007.07.013. [DOI] [PubMed] [Google Scholar]
- 193.Barnard DL. Evaluation of immunomodulators, interferons and known in vitro SARS-coV inhibitors for inhibition of SARS-coV replication in BALB/c mice. Antivir. Chem. Chemother. 2006;17,:275–284. doi: 10.1177/095632020601700505. [DOI] [PubMed] [Google Scholar]
- 194.Guerrero-Plata A. Activity and regulation of alpha interferon in respiratory syncytial virus and human metapneumovirus experimental infections. J. Virol. 2005;79,:10190–10199. doi: 10.1128/JVI.79.16.10190-10199.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Haagmans BL. Pegylated interferon-alpha protects type 1 pneumocytes against SARS coronavirus infection in macaques. Nat. Med. 2004;10,:290–293. doi: 10.1038/nm1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Yoon HJ, Zhu Z, Gwaltney JM, Jr, Elias JA. Rhinovirus regulation of IL-1 receptor antagonist in vivo and in vitro: a potential mechanism of symptom resolution. J. Immunol. 1999;162,:7461–7469. [PubMed] [Google Scholar]
- 197.Rijneveld AW. Interleukin-1 receptor antagonist transiently impairs antibacterial defense but not survival in murine pneumococcal pneumonia. Eur. Cytokine Netw. 2003;14,:242–245. [PubMed] [Google Scholar]
- 198.Kostense S. Interleukin 12 administration enhances Th1 activity but delays recovery from influenza A virus infection in mice. Antiviral Res. 1998;38,:117–130. doi: 10.1016/s0166-3542(98)00019-9. [DOI] [PubMed] [Google Scholar]
- 199.Hussell T, Khan U, Openshaw PJM. IL-12 treatment attenuates Th2 and B cell responses but does not improve vaccine-enhanced lung illness. J. Immunol. 1997;159,:328–334. [PubMed] [Google Scholar]
- 200.Cain JA, Deepe GS., Jr Interleukin-12 neutralization alters lung inflammation and leukocyte expression of CD80, CD86, and major histocompatibility complex class II in mice infected with Histoplasma capsulatum. Infect. Immun. 2000;68,:2069–2076. doi: 10.1128/iai.68.4.2069-2076.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Brieland JK, Remick DG, LeGendre ML, Engleberg NC, Fantone JC. In vivo regulation of replicative Legionella pneumophila lung infection by endogenous interleukin-12. Infect. Immun. 1998;66,:65–69. doi: 10.1128/iai.66.1.65-69.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Hussell T, Pennycook A, Openshaw PJ. Inhibition of tumor necrosis factor reduces the severity of virus-specific lung immunopathology. Eur. J. Immunol. 2001;31,:2566–2573. doi: 10.1002/1521-4141(200109)31:9<2566::aid-immu2566>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 203.Kirby AC, Raynes JG, Kaye PM. The role played by tumor necrosis factor during localized and systemic infection with Streptococcus pneumoniae. J. Infect. Dis. 2005;191,:1538–1547. doi: 10.1086/429296. [DOI] [PubMed] [Google Scholar]
- 204.Herring AC. Transient neutralization of tumor necrosis factor alpha can produce a chronic fungal infection in an immunocompetent host: potential role of immature dendritic cells. Infect. Immun. 2005;73,:39–49. doi: 10.1128/IAI.73.1.39-49.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Plessner HL. Neutralization of tumor necrosis factor (TNF) by antibody but not TNF receptor fusion molecule exacerbates chronic murine tuberculosis. J. Infect. Dis. 2007;195,:1643–1650. doi: 10.1086/517519. [DOI] [PubMed] [Google Scholar]
- 206.Salomon R, Hoffmann E, Webster RG. Inhibition of the cytokine response does not protect against lethal H5N1 influenza infection. Proc. Natl. Acad. Sci. USA. 2007;104,:12479–12481. doi: 10.1073/pnas.0705289104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Baumgarth N, Kelso A. In vivo blockade of gamma interferon affects the influenza virus-induced humoral and the local cellular immune response in lung tissue. J. Virol. 1996;70,:4411–4418. doi: 10.1128/jvi.70.7.4411-4418.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Sarawar SR, Sangster M, Coffman RL, Doherty PC. Administration of anti-IFN-gamma antibody to b2-microglobulin-deficient mice delays influenza virus clearance but does not switch the response to a T helper cell 2 phenotype. J. Immunol. 1994;153,:1246–1253. [PubMed] [Google Scholar]
- 209.Nakamatsu M. Role of interferon-gamma in Valpha14+ natural killer T cell-mediated host defense against Streptococcus pneumoniae infection in murine lungs. Microbes. Infect. 2007;9,:364–374. doi: 10.1016/j.micinf.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 210.Wu Q. IL-23-dependent IL-17 production is essential in neutrophil recruitment and activity in mouse lung defense against respiratory Mycoplasma pneumoniae infection. Microbes. Infect. 2007;9,:78–86. doi: 10.1016/j.micinf.2006.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Happel KI. Cutting edge: roles of Toll-like receptor 4 and IL-23 in IL-17 expression in response to Klebsiella pneumoniae infection. J. Immunol. 2003;170,:4432–4436. doi: 10.4049/jimmunol.170.9.4432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Greenberger MJ. Neutralization of IL-10 increases survival in a murine model of Klebsiella pneumonia. J. Immunol. 1995;155,:722–729. [PubMed] [Google Scholar]
- 213.van der Sluijs KF. Influenza-induced expression of indoleamine 2,3-dioxygenase enhances interleukin-10 production and bacterial outgrowth during secondary pneumococcal pneumonia. J. Infect. Dis. 2006;193,:214–222. doi: 10.1086/498911. [DOI] [PubMed] [Google Scholar]
- 214.Ramilo O. Gene expression patterns in blood leukocytes discriminate patients with acute infections. Blood. 2007;109,:2066–2077. doi: 10.1182/blood-2006-02-002477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Heritage PL, Underdown BJ, Arsenault AL, Snider DP, McDermott MR. Comparison of murine nasal-associated lymphoid tissue and Peyer's patches. Am. J. Respir. Crit. Care Med. 1997;156,:1256–1262. doi: 10.1164/ajrccm.156.4.97-03017. [DOI] [PubMed] [Google Scholar]
- 216.Asanuma H. Isolation and characterization of mouse nasal-associated lymphoid tissue. J. Immunol. Methods. 1997;202,:123–131. doi: 10.1016/s0022-1759(96)00243-8. [DOI] [PubMed] [Google Scholar]
- 217.Blair C, Naclerio RM, Yu X, Thompson K, Sperling A. Role of type 1 T helper cells in the resolution of acute Streptococcus pneumoniae sinusitis: a mouse model. J. Infect. Dis. 2005;192,:1237–1244. doi: 10.1086/444544. [DOI] [PubMed] [Google Scholar]