

Abstract
Antibodies are an effective line of defense in preventing infectious diseases. Highly potent neutralizing antibodies can intercept a virus before it attaches to its target cell and, thus, inactivate it. This ability is based on the antibodies’ specific recognition of epitopes, the sites of the antigen to which antibodies bind. Thus, understanding the antibody/epitope interaction provides a basis for the rational design of preventive vaccines. It is assumed that immunization with the precise epitope, corresponding to an effective neutralizing antibody, would elicit the generation of similarly potent antibodies in the vaccinee. Such a vaccine would be a ‘B-cell epitope-based vaccine’, the implementation of which requires the ability to backtrack from a desired antibody to its corresponding epitope.
In this article we discuss a range of methods that enable epitope discovery based on a specific antibody. Such a reversed immunological approach is the first step in the rational design of an epitope-based vaccine. Undoubtedly, the gold standard for epitope definition is x-ray analyses of crystals of antigen: antibody complexes. This method provides atomic resolution of the epitope; however, it is not readily applicable to many antigens and antibodies, and requires a very high degree of sophistication and expertise. Most other methods rely on the ability to monitor the binding of the antibody to antigen fragments or mutated variations. In mutagenesis of the antigen, loss of binding due to point modification of an amino acid residue is often considered an indication of an epitope component. In addition, computational combinatorial methods for epitope mapping are also useful. These methods rely on the ability of the antibody of interest to affinity isolate specific short peptides from combinatorial phage display peptide libraries. The peptides are then regarded as leads for the definition of the epitope corresponding to the antibody used to screen the peptide library. For epitope mapping, computational algorithms have been developed, such as Mapitope, which has recently been found to be effective in mapping conformational discontinuous epitopes. The pros and cons of various approaches towards epitope mapping are also discussed.
Keywords: Severe Acute Respiratory Syndrome, Severe Acute Respiratory Syndrome, Epitope Mapping, Contact Residue, Receptor Binding Domain
Without a doubt, the greatest advancement in managing public health has been the practice of vaccination. Ever since Jenner’s classical demonstration of the protective benefit of vaccination (in Gloucestershire, England — 1796), scores of vaccines have been developed which have saved the lives of hundreds of millions of people worldwide. The success of vaccines has even gone beyond its original objective (to empower individuals with immunity against various pathogens), and today it appears that through persistent vaccination some diseases have been eradicated from the face of the earth.[1–4] The most common implementation of vaccination has been to expose the vaccinee to a killed intact pathogen or one that has been attenuated. In either case, there is the need to culture the pathogen in high quantity, effectively inactivate it and ultimately subject the vaccinee to the full spectrum of the pathogen’s antigens in all their complexity. However, over the years it has become clear that not all antigens provide the same degree of protection and, in fact, in some cases there may be disadvantages in raising an immune response to specific aspects of the pathogen (as in the case of inducing autoimmunity due to pathogen mimicry of native antigens[5–7]). Therefore, and with the advent of recombinant DNA technologies, a new concept in vaccination emerged in which isolated antigens replace intact pathogens for vaccination. This obviously affords the ability to focus the immune response on more effective targets and also benefits from the advantages in manufacturing recombinant proteins as opposed to having to culture infectious agents and processing them for safety. Indeed, vaccines based on isolated antigens, referred to as subunit vaccines, have become very effective and their use is ever more expanding.[8–10]
Taking such a reductionist approach one step further, one can propose the use of selected isolated epitopes as vaccine modalities.[10–12] Once an effective subunit vaccine is identified, one might be able to gain advantages by identifying the specific epitopes that provide the protection, produce them individually and use the isolated epitopes as immunogens. Conceptually, the simplest way for identifying a desirable epitope is to isolate a monoclonal antibody (mAb) that binds to it. One would expect that such a mAb represents the ultimate desired effect of vaccinating with its corresponding epitope. Thus, when a highly potent neutralizing mAb is identified, it is proposed that immunizing an individual with the epitope of that mAb should elicit the same potent neutralizing activity characteristic of the original mAb. If the mAb is also highly cross reactive, in this case a desirable attribute as it enables the mAb to bind a diversity of genetic variants of the pathogen it neutralizes, those individuals immunized with the mAb epitope would also acquire the ability to broadly cross neutralize divergent pathogens. Thus, the crux of the problem for such a ‘reversed immunological approach’ is to be able to backtrack from a mAb of desired activity to its corresponding epitope. This is ‘Step 1’ in the development of epitope-based vaccines and the subject of this review.
1. The Advantages of Epitope-Based Vaccines
Although no examples of commercially available epitope-based vaccines currently exist, we nonetheless believe that this type of vaccine has intrinsic advantages that will be realized in the future. Some of these are listed and discussed in this section.
1.1 Focusing the Immune Response
In the natural pathogen, there is a plethora of antigens, each comprising tens or hundreds of potential epitopes. Certainly, not all antigens or epitopes are equally useful in generating protective immunity. It is also assumed that an individual does not mount a strong, high-titer response to all potential epitopes. In fact, the complexity of antibodies in an individual’s polyclonal serum corresponds to a relatively limited collection of epitopes that may not necessarily include those that are most important for protection. Thus, by creating a cocktail of defined epitopes selected for their ability to afford proven protection, one should be able to focus the immune system to generate a preferred repertoire of antibodies, ‘hand picked to do the job’.
1.2 Enhancing Immunity
Within the diversity of antigens and epitopes of the pathogen, not all are equal in their ability to elicit antibody production. Very often we find that some epitopes seem to be dominant and the strong response towards them may be at the expense of being able to respond to other epitopes of the pathogen. One thing that is certain is that the most dominant epitopes do not necessarily correspond to the most effective neutralizing ones. In fact, natural selection pressure tends to drive occlusion of the most neutralizing epitopes of the pathogen. The pathogen gains a selective advantage when it is able to obscure its ‘weak points’ and distract the host’s immune system by devising ‘seductive’ epitopes with little protective value. This can be achieved by creating‘baits’ of sorts that are particularly surface-accessible and hydrophilic, yet amenable to constant genetic variation. For example, the five variable loops of HIV-1 gp120 are highly immunogenic but, because of their ever changing nature, allow the swarm of HIV to evade immune surveillance by constantly mutating out of the binding capacity of the antibodies produced after first encounters.[13–15]
Epitope-based vaccines would contribute to overcoming this problem. It is envisaged that epitopes would be selected for their ability to elicit potent neutralization rather than their natural surface accessibility. Such epitopes are most likely to correspond to conserved aspects of the pathogen that cannot tolerate modification and through natural selection have evolved to be less immunogenic. For example, those aspects of HIV-1 gp120 that must function in receptor recognition are required to correspond to the invariant structures of the host (e.g. CD4 and CCR5[16]). These same epitopes are buried in gp120 and, as a result, are comparatively less immunogenic, leading to the production of relatively few antibodies against them in the natural progression of disease.[15] By isolating these epitopes and presenting them out of context, i.e. immunizing with them in the absence of the rest of the virus, one should gain a desired effect of enhancing their immunogenicity. Producing epitope-based vaccines derived from the most conserved aspects of the viral antigens would not only promise a more focused, and hopefully more functional, protective immunity but also a more efficient immune response towards them.
1.3 Avoiding Undesirable Epitopes
The generation of antibodies in response to infection is not always advantageous. Whereas the intent of the immune response is to neutralize the pathogen, the ultimate consequence of the antibodies produced cannot be anticipated by the immune system. Thus, deleterious effects might also occur.
Two types of undesirable effects have been recognized. In the first situation, the generation of antibodies against non-effective epitopes can, for example, lead to infection of macrophages due to Fc-receptor-mediated endocytosis of immunocomplexed pathogens[17] Such enhancing antibodies have been proposed as a means for infection via an alternate route (via the ‘back door’). This has been proposed as a mechanism that might explain CD4 independent infection with HIV-1.[18,19] Epitope-based vaccines could be designed to reduce the amount of ineffective antibodies in the infected individual and, thus, increase the specific activity of neutralizing antibodies.
The second danger in mounting antibodies against pathogens stems from the possibility of epitope mimetics of native host proteins. The development of autoimmunity has been connected with viral infections,[6,7] the idea being that in the event of a foreign antigen having structural similarities to native host proteins, mounting antibodies towards these similar structures may lead to cross-reactive binding of the antibodies with the auto-antigens leading to pathological autoimmunity; note, that in this case cross-reactivity is detrimental as compared with the desirable effects described previously. A case in point has recently been described for the vaccines developed against Borrelia burgdorferi (the agent causing Lyme disease). The outer surface protein A (OspA) of the bacterium contains a short sequence of nine amino acids, which is homologous to the human leukocyte function associated antigen-1 (hLFA-1). As a result, the Lyme vaccine, based on recombinant OspA, has been postulated to induce an autoimmune reaction to the auto-antigen, resulting in arthritic symptoms. Targeted deletion of the nonapeptide epitope from the OspA has been proposed as a solution to this problem.[20–22] Obviously, in developing epitope-based vaccines one would be able to rationally design the epitope cocktail to exclude epitopes that could be associated with auto-antigens.
1.4 Reducing Costs
The production of vaccines can be technically complicated and biohazardous when manufacturers are required to culture large volumes of pathogens. Moreover, each pathogen may have its own idiosyncrasies that translate into the development of custom tailored protocols for production, requiring specific conditions and reagents. Assuming an effective epitope-based vaccine is available, its production is perceived as being less complicated, much safer and certainly cheaper in the long run.
2. Three Steps in Epitope-Based Vaccine Production
As discussed above, the first step in developing an epitope-based vaccine is to identify the epitope itself. Obviously, the epitope should correspond to the ultimate immune response desired, i.e. the broad cross neutralization (BCN) of the genetic diversity of pathogens for which protection is required. The discovery of such epitopes could be serendipitous through experimentation, trial and error. However, rational design of such vaccines would be preferable and the concept of backtracking from mAbs of proven BCN activity seems the most efficient means for singling out epitope candidates for vaccine production.
Once such an epitope has been defined, the second step would be to reconstitute the epitope into a functional immunogen. There are a number of considerations that must be taken into account. First, B-cell epitopes (as opposed to T-cell epitopes) are very often discontinuous and highly conformational.[23] In fact, even in the event that a significant portion of an epitope is a short linear peptide, this does not promise that such a peptide represents the entire epitope or that it does not require a distinct conformation. We recognize that even short linear peptides can greatly depend on their three dimensional conformation for bioactivity. Thus, for example, we do not presume that short peptide hormones or neurotransmitters, such as leu- or met-enkephalin (5 residues),[24] angiotensin II (8 residues)[25] or substance P (11 residues)[26] are exempt from specific conformational requirements for their physiological function.
B-cell epitopes typically comprise some 15–20 residues derived from 2–3 discontinuous segments of the antigen brought together through folding to produce a contiguous surface that is recognized by the antibody.[23] The task of reconstituting an effective epitope-based vaccine must take these facts into consideration and attempt to position the critical contact residues in a proper spatial orientation. This is by no means an easy task. The final reconstituted epitope could either be based on the actual antigen segments tacked onto and supported by some scaffold of sorts or, alternatively, may functionally be similar to the epitope but structurally an unrelated mimetic. In this instance it is presumed that functional moieties of the epitope are effectively situated in space using alternative residues or chemistries to recreate a landscape similar to that of the epitope but not using the same composition. Finally, a simpler yet less comprehensive approach would be to attempt to reconstitute only partial structural elements of the epitope. For example, if an epitope is comprised of a series of anti-parallel β strands forming a number of juxtaposed β hairpins, one might consider attempting to reconstruct each β hairpin separately instead of trying to encompass the epitope in its entirety.
Once a mimetic or reconstituted epitope is produced, the third step in the development of an epitope-based vaccine would be to produce effective immunogens. For this, the epitopes would need to be mounted onto carriers or scaffolds and one would be confronted with the task of developing the most effective immunization schemes considering routes of immunization, adjuvants, and immunization schedules.
3.‘Step 1’: Monoclonal Antibody-Guided Mapping of B-Cell Epitopes
3.1 Pepscan
Given that a target mAb exists, the task before us is to map its corresponding epitope within the antigen it binds. In the simplest case, a major element of the epitope may be a linear segment of the antigen that in its isolated form continues to bind the mAb with detectable affinity. Discovery of the segment can be accomplished by screening the mAb against a ‘Geysen pepscan’. In 1984 Mario Geysen introduced the idea of generating a series of overlapping linear peptides that cover the entirety of the antigen being studied.[27] In such a case, the array of peptides is reacted with the antibody and those segments that continue to bind represent a significant aspect of the epitope. This linear peptide can then be tested for its capacity to elicit the desired effect when used as a peptide immunogen.[23] Such pepscans can also indicate components of discontinuous epitopes in the event that two distant peptides each contain sufficient structural elements that allow them to bind the mAb separately. One would then conclude that these two peptides contribute to the intact epitope. For these types of analyses there is no requirement for a solved atomic structure of the antigen.
In recent years, classical pepscans have been incorporated into microarray applications (for examples see Cretich et al.[28] and Poetz et al.[29]) and are used in high throughput systems as well as for multiplexing.[30]
3.2 Biophysical Methods: Co-Crystallization and NMR
Most B-cell epitopes are not simple linear sequences but rather highly conformational discontinuous structures and therefore are rarely compatible with pepscan analyses.
3.2.1 Co-Crystallization
The gold standard for epitope definition has become the co-crystallization of the antigen:antibody complex followed by solution of its atomic structure using x-ray diffraction and analysis. In these systems, highly purified antigen and corresponding antibody are allowed to co-crystallize to form an antigen:antibody complex. The crystals are then interrogated with x-rays and due to the highly structured repeated nature of the crystal unit, diffraction patterns are generated. These can then be related by the Fourier transform to the 3-dimensional coordinates of the electron densities of the amino acids composing the antigen and its bound antibody. The epitope is in essence the antigen component of the antigen:an-tibody interface. Whereas epitope characterization derived by x-ray crystallography is spectacular, the method is highly sophisticated, tedious, demanding, and rather capricious.
Of the hundreds of thousands of antibodies that have been produced over the years, only some 70 unique co-crystals have been generated and solved, illustrating the exceptionally low overall efficiency of this technique.[31] Nonetheless, when a co-crystal is obtained one can assume that its atomic structure faithfully represents the genuine structures of the antigen and antibody, their relative orientation with respect to one another, and defines with precision the interface between them (i.e. the surface of the epitope). The designation of actual contact residues is then a matter of interpretation.
3.2.2 Nuclear Magnetic Resonance
A second biophysical approach towards epitope mapping uses nuclear magnetic resonance (NMR). In contrast to crystallography, NMR gives a dynamic picture of the antigen:antibody complex in solution. The structure of the complex and, as a result, the atomic definition of the antigen:antibody interface, is derived from measuring the effect of magnetic fields and pulsed electromagnetic radiation on the protons of the protein complex.
The structures are derived from the specific manner in which the protons associated with each amino acid absorb electromagnetic radiation when the sample is subjected to a high magnetic field. Modulation of the absorbance spectrum for each residue is caused by neighboring residues and the intensity of these effects correlates with the distance between residues and their neighbors. Thus, through measured distance and angle restraints, 3-dimensional models of the proteins can be generated.
The major limitation of this method, aside from a high degree of sophistication, demanding technical expertise, and expensive instrumentation, is that it is typically restricted to relatively small proteins (<30 kDa).[32] Nonetheless, when NMR is used to study the binding of epitope fragments to Fabs, one can learn much about the nature of epitope recognition.
Generally, comparing the structures obtained by crystallography versus NMR, one can conclude that the two methods complement each other well and generate very similar structures, although differences in details do exist. A case in point is the binding of the carbohydrate-mimetic peptide: M-D-W-N-M-H-A-A, to the anti-carbohydrate antibody SYA/J6, directed against the O-polysaccharide of Shigella flexneri Y.[33] NMR analyses would indicate that the γ-methylene proton at 2.3 ppm of the Met-1 residue clearly contacts the antibody. However, this is not supported in the crystal structure, where this methyl group is situated more than 5 Å away from any residue of the antibody. On the other hand, the crystal structure strongly suggests the designation of residues Asn-4 and His-6 as clear contact residues, yet these two residues show absolutely no enhancement in the NMR analyses, arguing against their forming contacts (i.e. in close proximity) with the Fab. Thus, the two biophysical methods are excellent in delineating the boundaries of the antigen: antibody interface, while the definition of which residues actually contribute to the binding and, to what degree, may still be a matter of controversy.
3.3 Computational Docking
Less demanding than co-crystallization is the production of two separate crystal structures: one of the antigen and the other of the antibody. Satisfaction of this pre-requisite opens the option of computational docking as a means of epitope discovery. In general, there is an abundance of structures for isolated antibodies and Fabs, and the production of new structures either experimentally or by computer modeling has become reasonably efficient. Thus, in the event that a structure can be obtained for the antigen in question, generating a model of the specific corresponding antibody should not pose a problem. Once the two structures exist separately, a battery of computer algorithms exist that can be used to attempt to dock in silico one structure onto the other. The ultimate goal of all docking algorithms is to determine the structure of a complex from the separately determined coordinates of its components. In 1982 Kuntz and his colleagues came out with the first widely used docking program, named DOCK.[34] Since then the docking field has flourished with many new algorithms.
Docking predictions are based on the assumption that the structure of a complex represents the lowest free energy state accessible to the system, which is often achieved through geometrical complementarities. Docking algorithms can differ in the methods used to generate the candidate solutions and in the scoring functions applied to evaluate them. The search methods can be classified into three categories: brute-force, local feature algorithms, and nondeterministic methods.[35] After a panel of possible solutions is generated, the candidate solutions are scored from the least likely to the most likely to represent the genuine complex. The scoring functions can take into account one or more features such as: shape complementarity, contact area, hydrogen bonding, electrostatic interactions, salvation energy, etc.[36,37] Once the docking prediction is completed, two questions can be formulated, given, of course, that experimental coordinates of the genuine complex are available: ‘what is the ranking number of the solution closest to the bonafide complex?’ and ‘what is the quality of this solution (how closely does it compare to the genuine interface)?’
Although considerable improvement has been achieved, the scoring functions are still the weakest component of most docking algorithms and even when a near-native complex is found, it is not necessarily ranked as one of the top scoring solutions.[38,39]
Docking procedures are most successful for small molecule docking; for example, predicting enzyme-inhibitor/ligand complexes generates relatively accurate results.[40] In this case, the small molecule is rigid and its corresponding binding site is a well structured pocket that closely follows the contours of the ligand, a ‘negative relief’ that complements its physicochemical requirements (such as electrostatic charge complementation of negative and positive charges). Antibody-hapten complexes have also been reproduced successfully. For example, phosphocholine binding to the immunoglobulin McPC603,[41] or steroid binding to the antibody DB3.[42] Here too, the target molecules are rigid, allowing one to introduce flexibility of the antibody’s binding site without generating an infinite number of solutions. Flexibility of the binding site helps to predict the conformational rearrangements occurring during antibody recognition.
Compared with small molecule docking, protein:protein interfaces are relatively large, both partners are flexible and tend to undergo conformational changes, which makes docking extremely difficult.[43,44] In fact, accurate results are best obtained when the starting point is two already co-crystallized protein structures (often called ‘bound docking’[39]). However, ‘unbound docking’applications involving the structure of separately crystallized proteins (also called “predictive docking”) yield many false-positive solutions which have theoretically good surface complementarity and, as such, score extremely high but are in actuality totally irrelevant to the native complex.[39,43] Therefore, it appears that whereas the computational docking method is extremely attractive conceptually, in the case of antibody-antigen docking the results, for the moment, are less satisfying.[43]
3.4 Binding Analyses
A number of methods for epitope characterization rely on functional binding of the mAb to the antigen or its derivatives. One approach is to limit the scope of the problem of epitope mapping by testing the ability of the antibody to continue to bind to fragments of the antigen. Thus, binding assays such as ELISA, dot blot, or western blot assays can provide insights as to the location of the epitope within the gross anatomy of the antigen. For example, the epitope of the CD4-specific mAb OKT4A can be localized to the first 86 residues of CD4, as cleavage of the antigen into fragments or expression of truncated versions of the antigen illustrate that the first, D1, domain of the antigen binds the antibody.[18] Similarly, systematic biochemical analyses and enzymatic degradation of the α subunit of the nicotinic acetylcholine receptor,[45] use of synthetic peptides,[46,47] and expression of fusion proteins,[48,49] were able to localize the ligand binding site to a segment of the α subunit of the receptor that contained <50 amino acid residues. In principle, so long as one can generate fragments that continue to bind, one can attempt to identify the smallest segment of the receptor or antigen that continues to be recognized. Thus, binding per se serves as an extremely effective monitor for the presence of functional aspects of the epitope. Exploiting this operational definition of the epitope, as a segment that continues to bind the antibody, a number of experimental strategies have been employed to map epitopes.
3.5 Mutagenesis
One of the simplest and most popular approaches towards epitope mapping with regard to its technical implementation is site directed mutagenesis.[50] The concept is straightforward in that one can replace any amino acid at a given position in the antigen with another, and then test the binding capacity of the modified protein. Loss of binding is taken to indicate that the modified residue could be associated with the epitope in question. Generally, two approaches are possible, alanine scanning mutagenesis (ASM) and saturating mutagenesis.
3.5.1 Alanine Scanning Mutagenesis
ASM (or ‘alanine walking’) is the most systematic approach for mutagenesis, in that one introduces an alanine for every residue in a given sequence, one at a time (positions containing alanine in the original sequence are usually replaced with glycine).[51,52] Thus, a suspected stretch of the antigen can be analyzed in the hope of identifying specific residues that are critical for mAb recognition. ASM provides important information on the protein-binding interface, but the method is laborious. Many mutants must be produced, purified and evaluated regarding the structural integrity and binding constant of each mutant. Furthermore, in ASM experiments, the interpretation of results can be confusing as they often do not correspond well with the physical binding site defined by crystallographic studies.
A case in point is the analysis of the severe acute respiratory syndrome (SARS) coronavirus spike glycoprotein receptor binding domain (RBD) complexed to its receptor, angiotensin converting enzyme II (ACE2). Here 32 residues of the RBD were subjected to ASM,[53] and of these, six were found to be components of the actual binding surface as ultimately determined by co-crystallization. A total of 11 of the 32 exchanges indeed had a dramatic effect on RBD binding to ACE2. Surprisingly, however, only three of these were actual contact residues (3 of the 6 mentioned above). Thus, modification of the other three residues of the binding site had no impact on binding at all. Moreover, the remaining eight mutations that had a profound inhibitory effect on binding were in fact situated outside of the boundaries of the binding surface. This goes to illustrate that mutations of residues outside the bona fide epitope can result in loss of binding, possibly implying their importance for the structural integrity of the functional epitope rather than a role as a contact residue per se.[53,54] With no structural information available, such residues would mistakenly be assigned to the binding epitope. Alternatively, as in the case of the S ARS RBD-ACE2 interface, mutation of bona fide contact residues to alanine does not necessarily interfere with binding.[54–56]
Another example is the anti-HEL (hen egg-white lysozyme) antibody D1.3 and HEL interface which turns out to be highly tolerant to mutations.[55] Only 4 alanine mutations of 12 contact residues (as determined from the co-crystal) on HEL, significantly reduced antibody binding. Alternatively, mutating the critical contact residue Asp-18 to Ala, resulting in the loss of one hydrogen bond and seven van der Waals contacts with the antibody, has no impact on binding at all. This can be explained by the fact that three water molecules fill the void created by the substitution of Asp-18 for Ala and, therefore, compensate for the lack of the functional Asp moiety, and allow binding activity to persist. This indicates that only a few of the many residues which comprise an epitope are absolutely critical for interaction, providing essential contacts required for antibody recognition. As a result, in the absence of a co-crystal, contact residues that are tolerant to mutations would not be included in the predicted epitope.
Mutations can also be directed in a more targeted fashion. Namely, based on assumptions of the potential involvement of a given residue or area of the antigen in mAb binding, one can systematically construct mutations to specifically test the effect of alteration of selected residues. For example, in order to localize the X5 Fab epitope on HIV-1 gp120,[57] 55 residues of gp120 were selected for ‘alanine exchange’. The mutagenesis was directed by structural information on the epitope recognized by a competing antibody. Of the mutants constructed, eight resulted in significant reduction of antibody binding. Subsequently, when the crystal structure of gp120-X5 Fab-CD4 was solved,[58] it became apparent that of the 55 substitutions made, only 6 residues fell within the genuine binding epitope. Nonetheless, of these six alanine mutations, four had no effect on binding and had not been predicted as part of the epitope, whereas two mutations did impair binding significantly. Moreover, six mutations outside of the epitope did have a significant effect on antibody binding. Once again we realize that the majority of the mutations that affected binding were actually not part of the genuine epitope.
3.5.2 Saturating Mutagenesis
An alternative to ASM is ‘saturating mutagenesis’ which is a less systematic approach to introducing single residue modifications. Technically, there are a number of methods that can be used to randomly insert mutations throughout the sequence of an antigen. One method for the production of peptides with a limited number of mutations per molecule is ‘biased random mutagenesis’.[59–63] In this technique, corresponding oligonucleotides are synthesized using phosphoramidite monomer precursors contaminated with regulated amounts of the other three nucleotide precursors. The degree of contaminating phosphoramidites needed to achieve the desirable mutation frequency per peptide can be calculated using an algorithm described by Ophir and Gershoni.[63] Random mutations can also be achieved using chemical reagents such as sodium bisulfite,[64] hydroxylamine,[65] and nitrous acid.[66] These reagents induce structural modifications of specific bases in the DNA sequence, resulting in an alternative base pairing and, thus, changing the DNA sequence.[67,68] Additionally, chemical reagents such as formic acid[69,70] and hydrazine[71] are capable of damaging DNA bases, thus preventing Watson-Crick pairing and ultimately causing mis-incorporation of a nucleotide.[68] Error-prone polymerase chain reaction (PCR) is widely used as a mutagenesis technique and is implemented by using a low-fidelity DNA polymerase that efficiently introduces random mutations along the amplified sequence.[72]
3.5.3 Mutations and mAb Binding
Whatever the method, the ultimate goal is to randomly cover the surface of the antigen with mutations and score mAb binding. Here too, inhibition of binding is taken to indicate a role for the original residue, which has been mutated, in antibody recognition. Thus, epitope mapping stems from the identification of a cluster of mutations that strongly affect mAb binding. Once again, the problem is that mutations in contact residues do not always result in loss of mAb binding. Moreover, loss of binding is often the result of modification of residues that lie outside of the epitope. For instance, the degree of success of random mutagenesis as a means to predict epitopes was determined in the study of the crystal structure of histidine phospho-carrier protein (HPr)-Jel42 mAb complex.[73,74] Among the 34 random mutations made to the surface of HPr, 12 were contact residues as defined by the co-crystal. Thirteen of the 34 mutations resulted in reduction of Jel42 mAb binding. Of these, nine residues lay within the genuine binding epitope. However, the remaining four inhibitory mutations were of residues totally removed from the binding interface. Furthermore, mutations of three bona fide contact residues had virtually no effect on Jel42 binding, once again indicating that substitutions of epitope-residues can often be accommodated.
In summary, targeted or random mutagenesis are easily performed methods, and can often generate dramatic effects on mAb binding to its mutated antigen. However, in view of the fact that mutated contact residues can either inhibit binding, enhance binding, or have no effect at all, the interpretation of mutagenesis experiments can be confusing. All the more so when residues outside the area of the epitope can also impact on mAb binding. Thus, epitope predictions by mutagenesis should at best be considered with caution.
3.6 Combinatorial Approaches
In contrast to the biophysical or biochemical/immuno-binding approaches in which the analyses stem from examining the precise structure/sequence of the antigen in question, a totally different concept has also proven to be effective for epitope mapping. The idea is to test the binding capacity of vast collections of random peptides that represent a huge diversity of possible combinations of amino acid sequences. Smith and Petrenko[75] illustrated that one can exploit phage display systems for this purpose, where short peptides can be expressed on the surface of filamentous bacteriophages.
3.6.1 Phage Display Peptide Libraries
Phage display libraries of random peptides are constructed using phage display vectors in which a short oligonucleotide (e.g. 18 bases corresponding to six codons creating hexa-peptides) is cloned at the 5′ end of either the protein 3 or protein 8 genes of the phage. The random nature is produced by allowing each position in the oligonucleotide to assume any of the four bases (GATC). In this manner, all possible combinations of bases, and consequently all amino acids can be expressed. Each phage thus displays the peptide that is coded for in its single stranded genome. This physical linkage between the peptide and its genetic code allows one to affinity isolate phages that bind a mAb of interest and then elucidate the sequence of the peptide by standard DNA sequencing.
Over the years, numerous phage display peptide libraries have been produced and used extensively to characterize B-cell epitopes.[76–78] Generally, the procedure is straightforward. The mAb to be analyzed is immobilized on a solid surface and reacted with a comprehensive library of random peptides (usually in the order of 108–1010 different peptides that are typically 6–15 residues long and may or may not be disulfide constrained);[79,80] this process has been coined ‘biopanning’. The result is that those random peptides that resemble the mAb epitope in some manner are bound by the immobilized mAb and the others are washed away. The affinity bound phages are then collected and their peptide sequences determined. When the sequence of the affinity purified phages corresponds to linear stretches of the antigen, epitope mapping becomes self evident. However, generally one obtains a collection of phages that do not necessarily resemble the antigen at all. In this situation, one can assume that the phages that specifically bind the mAb reflect its epitope in some fashion.
3.6.2 Analysis and Interpretation of Phage Display Data
The question becomes how to extract from the panel of mAb binding peptides information pertaining to the native epitope being sought.
There are basically two approaches:
The first is more empirical. Through repeated rounds of biopanning one selects the peptide that binds the antibody most efficiently, irrespective of the fact that it may not be homologous to any segment of the linear sequence of the antigen. This best binding peptide is considered a peptidomimetic of the epitope that, although very different in composition and structure compared with the genuine epitope, is able to mimic the latter functionally.[81–83] Thus, the mimetic is viewed as being able to place critical contact moieties in spatially defined positions that correspond to similar structures of the epitope. In this situation, the best binding mimetic is considered as a potential surrogate for the genuine epitope and can be used as an immu-nogen in the hope that it will elicit antibodies that can favorably cross react with the original antigen. Some success has been reported with this approach.[83]
The second application of random peptide libraries is to actually regard the affinity purified panel of mAb binding peptides as a dataset that can be used for epitope mapping within the atomic structure of the antigen. Here also, two approaches have been described. In the first, one seeks to align the peptide with areas in the 3-dimensional structure of the antigen.[84,85] Thus, taking the sequence of a given peptide one tries to match it residue for residue on the surface of the antigen irrespective of linear connectivity. A number of algorithms have been developed to do this automatically.[84,85] The basis for these is that a peptide or significant fragment thereof is regarded as the unit of information. In contrast to this approach, an alternative computational predictive algorithm has been developed in which the panel of peptides is analyzed collectively.[76,86]
The Mapitope Algorithm
Mapitope is a novel computer algorithm for the prediction of discontinuous B-cell epitopes based on the notion that the panel of peptides derived from a random peptide library collectively represents the epitope of the mAb that they bind.[76,86]
The underlying principle of Mapitope is that the simplest meaningful fragment of an epitope is an ‘amino acid pair’ (AAP) of residues that lie within the footprint of the epitope. These AAPs can be related to one another on the surface of the antigen such that a cluster of pairs is defined which constitutes the majority of the epitope footprint, i.e. the predicted epitope is in essence a cluster of connected AAPs. The AAPs of the epitope need not be consecutive tandem residues of the antigen, but often are the result of juxtaposition of distant residues brought together through folding of the polypeptide chain. AAPs of the epitope are simulated by tandem residues of the peptides, affinity selected from a random library.
In order to identify the most meaningful AAPs present in the panel of peptides, the peptides are deconvoluted into pairs. For example, a peptide of the sequence ABCDE… would be written as the series of pairs, AB BC CD DE, etc. All AAPs derived from a panel of peptides are then pooled and the frequency of each type is calculated and determined whether its representation in the pool is higher than its random expectation (based on the theoretical amino acid frequencies of the phage library). The most significant AAPs are considered as statistically significant pairs (SSPs). Thus, each peptide is assumed to contain one or more epitope-relevant AAPs, which are the basis for mAb recognition of that peptide.
Figure 1 demonstrates the fundamental concept of the algorithm; that is, the role of SSPs in mAb recognition. Note that the mutation of a residue that is a member of an SSP greatly impacts on antibody binding, whereas alteration of a residue that is not part of an SSP does not affect binding.[87] After compiling the list of SSPs, the algorithm seeks these pairs on the surface of the antigen and attempts to link them into clusters (see figure 2 and Tarnovit-ski et al.[88]).
Fig. 1.
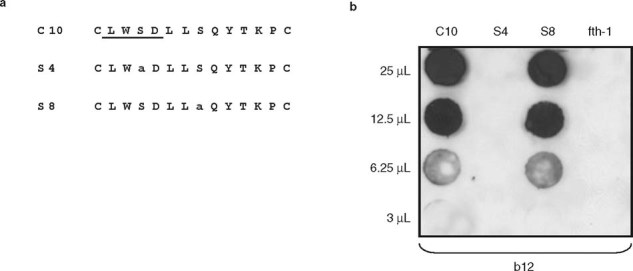
Mutagenic comparison of serine-containing statistically significant pairs (SSPs) versus amino acid pairs. (a) Amino acid sequences of b12 monoclonal antibody (mAb) affinity purified phages: C10 (wild-type phage), S4 and S8 in which Ser-4 and Ser-8 are converted to alanine. The LWSDL segment of phage C10, determined to contain SSPs which are important for b12 binding, is underlined. (b) Two-fold dilutions of equal amounts of phages were dot-blotted onto a nitrocellulose membrane filter and reacted with the b12 mAb. The fth-1 phage, containing no insert, was used as a negative control, and signals were produced using enhanced chemiluminescence (from Bublil et al.,[87] with permission).
Fig. 2.
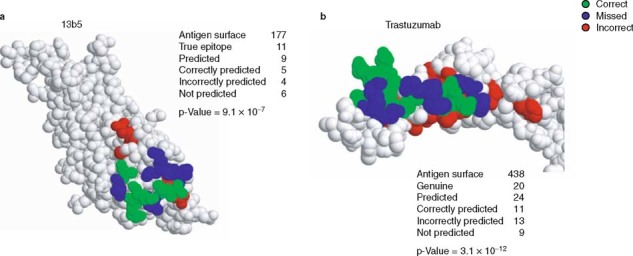
Space-filling representation of Mapitope predictions of (a) the monoclonal antibody 13b5 epitope on the surface of HIV-1 p24 antigen and (b) the trastuzumab epitope on the surface of the HER-2/neu receptor.[78] The number of amino acids comprising each antigen’s surface, genuine and predicted epitopes are given. Also given is the number of correctly predicted residues (indicated in green), over-predicted residues (indicated in red) and residues that were missed (indicated in blue). The p-values express a hyper-geometric distribution (the probability of randomly predicting the epitope) and were calculated as described by Mayrose et al.[85]
In summary, a mAb is used to screen a random peptide library to generate a panel of affinity selected peptides. These peptides are deconvoluted into AAPs and the most SSPs are identified. The SSPs are then mapped on the surface of the atomic structure of the antigen and the most elaborate and diverse clusters identified. These are regarded as the predicted epitope candidates.
4. Conclusions
Backtracking from mAbs to their corresponding epitopes provides a means for the rational design of vaccines. The use of epitope-based vaccines, although not yet implemented commercially, should prove to be effective and in some cases obviate problems that exist in the more traditional vaccine paradigms. Hence, the first real challenge in developing an epitope-based vaccine is to be able to identify the epitope of interest with reasonable precision. Undoubtedly, the most effective means for this is through the physical x-ray analysis of the antigen:antibody co-crystal. However, this is often not easily achievable. The other empirical approaches described above are certainly more readily accessible, yet each has its advantages and disadvantages.
The prospects of using the combinatorial approach, selecting for mAb-specific peptides and analyzing them via computational algorithms such as Mapitope, seems very promising. Once epitopes are identified and understood at the atomic level, the next step and challenge will be to reconstitute them so as to generate effective immunogens. Hopefully as these initial steps become routine, epitope-based vaccines will be able to contribute to the long tradition of vaccination for the betterment of human health.
Acknowledgments
Parts of this article are derived from research supported by grants from the Israel Science Foundation and the Binational Israel-USA Science Foundation awarded to Dr Gershoni. Natalie Tarnovitski Freund is the recipient of the Jakov, Mirianna and Jorge Saia Doctoral Prize. Dror Siman-Tov is supported by the Marian Gertner Institute for Medical Nanosystems.
The authors have no conflicts of interest that are directly relevant to the content of this review.
References
- 1.Henderson D.A. Smallpox eradication. Public Health Rep. 1980;95:422–6. [PMC free article] [PubMed] [Google Scholar]
- 2.Henderson D.A. Eradication: lessons from the past. Bull World Health Organ. 1998;76(Suppl.2):17–21. [PMC free article] [PubMed] [Google Scholar]
- 3.Hovi T. Inactivated poliovirus vaccine and the final stages of poliovirus eradication. Vaccine. 2001;19:2268–72. doi: 10.1016/S0264-410X(00)00515-6. [DOI] [PubMed] [Google Scholar]
- 4.John T.J. The final stages of the global eradication of polio. N Engl J Med. 2000;343:806–7. doi: 10.1056/NEJM200009143431111. [DOI] [PubMed] [Google Scholar]
- 5.Albert L.J., Inman R.D. Molecular mimicry and autoimmunity. N Engl J Med. 1999;341:2068–74. doi: 10.1056/NEJM199912303412707. [DOI] [PubMed] [Google Scholar]
- 6.Bogdanos D.P., Choudhuri K., Vergani D. Molecular mimicry and autoimmune liver disease: virtuous intentions, malign consequences. Liver. 2001;21:225–32. doi: 10.1034/j.1600-0676.2001.021004225.x. [DOI] [PubMed] [Google Scholar]
- 7.Olson J.K., Croxford J.L., Calenoff M.A., et al. A virus-induced molecular mimicry model of multiple sclerosis. J Clin Invest. 2001;108:311–8. doi: 10.1172/JCI13032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Andersen P. Effective vaccination of mice against Mycobacterium tuberculosis infection with a soluble mixture of secreted mycobacterial proteins. Infect Immun. 1994;62:2536–44. doi: 10.1128/iai.62.6.2536-2544.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Babai I., Samira S., Barenholz Y., et al. A novel influenza subunit vaccine composed of liposome-encapsulated haemagglutinin/neuraminidase and IL-2 or GM-CSF. I: vaccine characterization and efficacy studies in mice. Vaccine. 1999;17:1223–38. doi: 10.1016/S0264-410X(98)00346-6. [DOI] [PubMed] [Google Scholar]
- 10.Nemchinov L.G., Liang T.J., Rifaat M.M., et al. Development of a plant-derived subunit vaccine candidate against hepatitis C virus. Arch Virol. 2000;145:2557–73. doi: 10.1007/s007050070008. [DOI] [PubMed] [Google Scholar]
- 11.Arthur L.O., Pyle S.W., Nara P.L., et al. Serological responses in chimpanzees inoculated with human immunodeficiency virus glycoprotein (gp120) subunit vaccine. Proc Natl Acad Sci U S A. 1987;84:8583–7. doi: 10.1073/pnas.84.23.8583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sun D.X., Seyer J.M., Kovari I., et al. Localization of protective epitopes within the pilin subunit of the Vibrio cholerae toxin-coregulated pilus. Infect Immun. 1991;59:114–8. doi: 10.1128/iai.59.1.114-118.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kwong P.D., Wyatt R., Robinson J., et al. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature. 1998;393:648–59. doi: 10.1038/31405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Parren P.W., Moore J.P., Burton D.R., et al. The neutralizing antibody response to HIV-1: viral evasion and escape from humoral immunity. Aids. 1999;13(Suppl.A):S137–62. [PubMed] [Google Scholar]
- 15.Wyatt R., Kwong P.D., Desjardins E., et al. The antigenic structure of the HIV gp120 envelope glycoprotein. Nature. 1998;393:705–11. doi: 10.1038/31514. [DOI] [PubMed] [Google Scholar]
- 16.Berkower I., Smith G.E., Giri C., et al. Human immunodeficiency virus 1: predominance of a group-specific neutralizing epitope that persists despite genetic variation. J Exp Med. 1989;170:1681–95. doi: 10.1084/jem.170.5.1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fust G. Enhancing antibodies in HIV infection. Parasitology. 1997;115:S127–40. doi: 10.1017/S0031182097001819. [DOI] [PubMed] [Google Scholar]
- 18.Jacotot E., Krust B., Callebaut C., et al. HIV-1 envelope glycoproteins-mediated apoptosis is regulated by CD4 dependent and independent mechanisms. Apoptosis. 1997;2:47–60. doi: 10.1023/A:1026435625144. [DOI] [PubMed] [Google Scholar]
- 19.Montefiori D.C. Role of complement and Fc receptors in the pathogenesis of HIV-1 infection. Springer Semin Immunopathol. 1997;18:371–90. doi: 10.1007/BF00813504. [DOI] [PubMed] [Google Scholar]
- 20.Steere A.C., Gross D., Meyer A.L., et al. Autoimmune mechanisms in antibiotic treatment-resistant Lyme arthritis. J Autoimmun. 2001;16:263–8. doi: 10.1006/jaut.2000.0495. [DOI] [PubMed] [Google Scholar]
- 21.Trollmo C., Meyer A.L., Steere A.C., et al. Molecular mimicry in Lyme arthritis demonstrated at the single cell level: LFA-1 alphaL is a partial agonist for outer surface protein A-reactive T cells. J Immunol. 2001;166:5286–91. doi: 10.4049/jimmunol.166.8.5286. [DOI] [PubMed] [Google Scholar]
- 22.Willett T.A., Meyer A.L., Brown E.L., et al. An effective second-generation outer surface protein A-derived Lyme vaccine that eliminates a potentially autoreactive T cell epitope. Proc Natl Acad Sci U S A. 2004;101:1303–8. doi: 10.1073/pnas.0305680101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Van Regenmortel M.H., Pellequer J.L. Predicting antigenic determinants in proteins: looking for unidimensional solutions to a three-dimensional problem? Pept Res. 1994;7:224–8. [PubMed] [Google Scholar]
- 24.Cai X., Dass C. Structural characterization of methionine and leucine enkephalins by hydrogen/deuterium exchange and electrospray ionization tandem mass spectrometry. Rapid Commun Mass Spectrom. 2005;19:1–8. doi: 10.1002/rcm.1739. [DOI] [PubMed] [Google Scholar]
- 25.Laporte S.A., Boucard A.A., Servant G., et al. Determination of peptide contact points in the human angiotensin II type I receptor (ATI) with photosensitive analogs of angiotensin II. Mol Endocrinol. 1999;13:578–86. doi: 10.1210/me.13.4.578. [DOI] [PubMed] [Google Scholar]
- 26.Sagan S., Quancard J., Lequin O., et al. Conformational analysis of the C-terminal Gly-Leu-Met-NH2 tripeptide of substance P bound to the NK-1 receptor. Chem Biol. 2005;12:555–65. doi: 10.1016/j.chembiol.2005.03.005. [DOI] [PubMed] [Google Scholar]
- 27.Geysen H.M., Meloen R.H., Barteling S.J., et al. Use of peptide synthesis to probe viral antigens for epitopes to a resolution of a single amino acid. Proc Natl Acad Sci U S A. 1984;81:3998–4002. doi: 10.1073/pnas.81.13.3998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cretich M., Damin F., Pirri G., et al. Protein and peptide arrays: recent trends and new directions. Biomol Eng. 2006;23:77–88. doi: 10.1016/j.bioeng.2006.02.001. [DOI] [PubMed] [Google Scholar]
- 29.Poetz O., Ostendorp R., Brocks B., et al. Protein microarrays for antibody profiling: specificity and affinity determination on a chip. Proteomics. 2005;5:2402–11. doi: 10.1002/pmic.200401299. [DOI] [PubMed] [Google Scholar]
- 30.Andresen H., Zarse K., Grotzinger C., et al. Development of peptide microarrays for epitope mapping of antibodies against the human TSH receptor. J Immunol Methods. 2006;315:11–8. doi: 10.1016/j.jim.2006.06.012. [DOI] [PubMed] [Google Scholar]
- 31.Lo C. L., Chothia C., Janin J. The atomic structure of protein-protein recognition sites. J Mol Biol. 1999;285:2177–98. doi: 10.1006/jmbi.1998.2439. [DOI] [PubMed] [Google Scholar]
- 32.Wuthrich K. Protein structure determination in solution by NMR spectroscopy. J Biol Chem. 1990;265:22059–62. [PubMed] [Google Scholar]
- 33.Johnson M.A., Pinto B.M. Saturation-transfer difference NMR studies for the epitope mapping of a carbohydrate-mimetic peptide recognized by an anti-carbohydrate antibody. Bioorg Med Chem. 2004;12:295–300. doi: 10.1016/j.bmc.2003.09.041. [DOI] [PubMed] [Google Scholar]
- 34.Kuntz I., Blaney J., Oatley S., et al. A geometric approach to macromolecule-ligand interactions. J Mol Biol. 1982;161:269–88. doi: 10.1016/0022-2836(82)90153-X. [DOI] [PubMed] [Google Scholar]
- 35.Inbar Y., Benyamini H., Nussinov R., et al. Prediction of multimolecular assemblies by multiple docking. J Mol Biol. 2005;349:435–47. doi: 10.1016/j.jmb.2005.03.039. [DOI] [PubMed] [Google Scholar]
- 36.Mendez R., Leplae R., De Maria L., et al. Assessment of blind predictions of protein-protein interactions: current status of docking methods. Proteins. 2003;52:51–67. doi: 10.1002/prot.10393. [DOI] [PubMed] [Google Scholar]
- 37.Mendez R., Leplae R., Lensink M.F., et al. Assessment of CAPRI predictions in rounds 3–5 shows progress in docking procedures. Proteins. 2005;60:150–69. doi: 10.1002/prot.20551. [DOI] [PubMed] [Google Scholar]
- 38.Gray J.J., Moughon S.E., Kortemme T., et al. Protein-protein docking predictions for the CAPRI experiment. Proteins. 2003;52:118–22. doi: 10.1002/prot.10384. [DOI] [PubMed] [Google Scholar]
- 39.Halperin I., Ma B., Wolfson H., et al. Principles of docking: an overview of search algorithms and a guide to scoring functions. Proteins. 2002;47:409–43. doi: 10.1002/prot.10115. [DOI] [PubMed] [Google Scholar]
- 40.Sotriffer C.A., Flader W., Winger R.H., et al. Automated docking of ligands to antibodies: methods and applications. Methods. 2000;20:280–91. doi: 10.1006/meth.1999.0922. [DOI] [PubMed] [Google Scholar]
- 41.Di N. A., Roccatano D., Berendsen H.J. Molecular dynamics simulation of the docking of substrates to proteins. Proteins. 1994;19:174–82. doi: 10.1002/prot.340190303. [DOI] [PubMed] [Google Scholar]
- 42.Apostolakis J., Caflisch A. Computational ligand design. Comb Chem High Throughput Screen. 1999;2:91–104. [PubMed] [Google Scholar]
- 43.Vajda S., Camacho C.J. Protein-protein docking: is the glass half-full or half-empty? Trends Biotechnol. 2004;22:110–6. doi: 10.1016/j.tibtech.2004.01.006. [DOI] [PubMed] [Google Scholar]
- 44.Wodak S.J., Mendez R. Prediction of protein-protein interactions: the CAPRI experiment, its evaluation and implications. Curr Opin Struct Biol. 2004;14:242–9. doi: 10.1016/j.sbi.2004.02.003. [DOI] [PubMed] [Google Scholar]
- 45.Wilson P.T., Gershoni J.M., Hawrot E., et al. Binding of alpha-bungarotoxin to proteolytic fragments of the alpha subunit of Torpedo acetylcholine receptor analyzed by protein transfer on positively charged membrane filters. Proc Natl Acad Sci U S A. 1984;81:2553–7. doi: 10.1073/pnas.81.8.2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Neumann D., Barchan D., Safran A., et al. Mapping of the alpha-bungarotoxin binding site within the alpha subunit of the acetylcholine receptor. Proc Natl Acad Sci U S A. 1986;83:3008–11. doi: 10.1073/pnas.83.9.3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wilson P.T., Lentz T.L., Hawrot E. Determination of the primary amino acid sequence specifying the alpha-bungarotoxin binding site on the alpha subunit of the acetylcholine receptor from Torpedo californica. Proc Natl Acad Sci U S A. 1985;82:8790–4. doi: 10.1073/pnas.82.24.8790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Barkas T., Mauron A., Roth B., et al. Mapping the main immunogenic region and toxin-binding site of the nicotinic acetylcholine receptor. Science. 1987;235:77–80. doi: 10.1126/science.2432658. [DOI] [PubMed] [Google Scholar]
- 49.Gershoni J.M. Expression of the alpha-bungarotoxin binding site of the nicotinic acetylcholine receptor by Escherichia coli transformants. Proc Natl Acad Sci U S A. 1987;84:4318–21. doi: 10.1073/pnas.84.12.4318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Benjamin D.C., Perdue S.S. Site-directed mutagenesis in epitope mapping. Methods. 1996;9:508–15. doi: 10.1006/meth.1996.0058. [DOI] [PubMed] [Google Scholar]
- 51.Cunningham B.C., Wells J.A. High-resolution epitope mapping of hGH-receptor interactions by alanine-scanning mutagenesis. Science. 1989;244:1081–5. doi: 10.1126/science.2471267. [DOI] [PubMed] [Google Scholar]
- 52.Wells J.A. Systematic mutational analyses of protein-protein interfaces. Methods Enzymol. 1991;202:390–411. doi: 10.1016/0076-6879(91)02020-A. [DOI] [PubMed] [Google Scholar]
- 53.Chakraborti S., Prabakaran P., Xiao X., et al. The SARS coronavirus S glycoprotein receptor binding domain: fine mapping and functional characterization. Virol J. 2005;2:73. doi: 10.1186/1743-422X-2-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cunningham B.C., Wells J.A. Comparison of a structural and a functional epitope. J Mol Biol. 1993;234:554–63. doi: 10.1006/jmbi.1993.1611. [DOI] [PubMed] [Google Scholar]
- 55.Dall’Acqua W., Goldman E.R., Lin W., et al. A mutational analysis of binding interactions in an antigen-antibody protein-protein complex. Biochemistry. 1998;37:7981–91. doi: 10.1021/bi980148j. [DOI] [PubMed] [Google Scholar]
- 56.Nuss J.M., Whitaker P.B., Air G.M. Identification of critical contact residues in the NC41 epitope of a subtype N9 influenza virus neuraminidase. Proteins. 1993;15:121–32. doi: 10.1002/prot.340150204. [DOI] [PubMed] [Google Scholar]
- 57.Darbha R., Phogat S., Labrijn A.F., et al. Crystal structure of the broadly cross-reactive HIV-1-neutralizing Fab X5 and fine mapping of its epitope. Biochemistry. 2004;43:1410–7. doi: 10.1021/bi035323x. [DOI] [PubMed] [Google Scholar]
- 58.Huang C.C., Tang M., Zhang M.Y., et al. Structure of a V3-containing HIV-1 gp120 core. Science. 2005;310:1025–8. doi: 10.1126/science.1118398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Derbyshire K.M., Salvo J.J., Grindley N.D. A simple and efficient procedure for saturation mutagenesis using mixed oligodeoxynucleotides. Gene. 1986;46:145–52. doi: 10.1016/0378-1119(86)90398-7. [DOI] [PubMed] [Google Scholar]
- 60.Fraga D., Fillingame R.H. Essential residues in the polar loop region of subunit c of Escherichia coli F1F0 ATP synthase defined by random oligonucleotide-primed mutagenesis. J Bacteriol. 1991;173:2639–43. doi: 10.1128/jb.173.8.2639-2643.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Matteucci M.D., Heyneker H.L. Targeted random mutagenesis: the use of ambiguously synthesized oligonucleotides to mutagenize sequences immediately 5′ of an ATG initiation codon. Nucleic Acids Res. 1983;11:3113–21. doi: 10.1093/nar/11.10.3113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ner S.S., Goodin D.B., Smith M. A simple and efficient procedure for generating random point mutations and for codon replacements using mixed oligodeoxynucleotides. DNA. 1988;7:127–34. doi: 10.1089/dna.1988.7.127. [DOI] [PubMed] [Google Scholar]
- 63.Ophir R., Gershoni J.M. Biased random mutagenesis of peptides: determination of mutation frequency by computer simulation. Protein Eng. 1995;8:143–6. doi: 10.1093/protein/8.2.143. [DOI] [PubMed] [Google Scholar]
- 64.Shortle D., Nathans D. Local mutagenesis: a method for generating viral mutants with base substitutions in preselected regions of the viral genome. Proc Natl Acad Sci U S A. 1978;75:2170–4. doi: 10.1073/pnas.75.5.2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shugar D., Huber C.P., Birnbaum G.I. Mechanism of hydroxylamine mutagenesis: crystal structure and conformation of 1,5-dimethyl-N4-hydroxycytosine. Biochim Biophys Acta. 1976;447:274–84. doi: 10.1016/0005-2787(76)90050-2. [DOI] [PubMed] [Google Scholar]
- 66.Fried M. Isolation of temperature-sensitive mutants of polyoma virus. Virology. 1965;25:669–71. doi: 10.1016/0042-6822(65)90098-X. [DOI] [PubMed] [Google Scholar]
- 67.Shortle D., DiMaio D., Nathans D. Directed mutagenesis. Annu Rev Genet. 1981;15:265–94. doi: 10.1146/annurev.ge.15.120181.001405. [DOI] [PubMed] [Google Scholar]
- 68.Smith M. In vitro mutagenesis. Annu Rev Genet. 1985;19:423–62. doi: 10.1146/annurev.ge.19.120185.002231. [DOI] [PubMed] [Google Scholar]
- 69.Burns E.L., Nicholas R.A., Price E.M. Random mutagenesis of the sheep Na,K-ATPase alphal subunit generating the ouabain-resistant mutant L793P. J Biol Chem. 1996;271:15879–83. doi: 10.1074/jbc.271.46.29436. [DOI] [PubMed] [Google Scholar]
- 70.Wrenn C.K., Katzenellenbogen B.S. Structure-function analysis of the hormone binding domain of the human estrogen receptor by region-specific mutagenesis and phenotypic screening in yeast. J Biol Chem. 1993;268:24089–98. [PubMed] [Google Scholar]
- 71.Brown D.M., McNaught A.D., Schell P. The chemical basis of hydrazine mutagenesis. Biochem Biophys Res Commun. 1966;24:967–71. doi: 10.1016/0006-291X(66)90345-7. [DOI] [PubMed] [Google Scholar]
- 72.Cadwell R.C., Joyce G.F. Randomization of genes by PCR mutagenesis. PCR Methods Appl. 1992;2:28–33. doi: 10.1101/gr.2.1.28. [DOI] [PubMed] [Google Scholar]
- 73.Prasad L., Sharma S., Vandonselaar M., et al. Evaluation of mutagenesis for epitope mapping: structure of an antibody-protein antigen complex. J Biol Chem. 1993;268:10705–8. [PubMed] [Google Scholar]
- 74.Sharma S., Georges F., Delbaere L.T., et al. Epitope mapping by mutagenesis distinguishes between the two tertiary structures of the histidine-containing protein HPr. Proc Natl Acad Sci U S A. 1991;88:4877–81. doi: 10.1073/pnas.88.11.4877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Smith G.P., Petrenko V.A. Phage display. Chem Rev. 1997;97:391–410. doi: 10.1021/cr960065d. [DOI] [PubMed] [Google Scholar]
- 76.Enshell-Seijffers D., Denisov D., Groisman B., et al. The mapping and reconstitution of a conformational discontinuous B-cell epitope of HIV-1. J Mol Biol. 2003;334:87–101. doi: 10.1016/j.jmb.2003.09.002. [DOI] [PubMed] [Google Scholar]
- 77.Oleksiewicz M.B., Botner A., Toft P., et al. Epitope mapping porcine reproductive and respiratory syndrome virus by phage display: the nsp2 fragment of the replicase polyprotein contains a cluster of B-cell epitopes. J Virol. 2001;75:3277–90. doi: 10.1128/JVI.75.7.3277-3290.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Riemer A.B., Kraml G., Scheiner O., et al. Matching of trastuzumab (Herceptin) epitope mimics onto the surface of Her-2/neu: a new method of epitope definition. Mol Immunol. 2005;42:1121–4. doi: 10.1016/j.molimm.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 79.Enshell-Seijffers D., Smelyanski L., Gershoni J.M. The rational design of a ‘type 88’ genetically stable peptide display vector in the filamentous bacteriophage fd. Nucleic Acids Res. 2001;29:E50. doi: 10.1093/nar/29.10.e50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Parmley S.F., Smith G.P. Antibody-selectable filamentous fd phage vectors: affinity purification of target genes. Gene. 1988;73:305–18. doi: 10.1016/0378-1119(88)90495-7. [DOI] [PubMed] [Google Scholar]
- 81.Fleming T.J., Sachdeva M., Delic M., et al. Discovery of high-affinity peptide binders to BLyS by phage display. J Mol Recognit. 2005;18:94–102. doi: 10.1002/jmr.722. [DOI] [PubMed] [Google Scholar]
- 82.Schreiber A., Humbert M., Benz A., et al. 3D-Epitope-Explorer (3DEX): localization of conformational epitopes within three-dimensional structures of proteins. J Comput Chem. 2005;26:879–87. doi: 10.1002/jcc.20229. [DOI] [PubMed] [Google Scholar]
- 83.Zwick M.B., Bonnycastle L.L., Menendez A., et al. Identification and characterization of a peptide that specifically binds the human, broadly neutralizing anti-human immunodeficiency virus type 1 antibody b12. J Virol. 2001;75:6692–9. doi: 10.1128/JVI.75.14.6692-6699.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Halperin I., Wolfson H., Nussinov R. SiteLight: binding-site prediction using phage display libraries. Protein Sci. 2003;12:1344–59. doi: 10.1110/ps.0237103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mayrose I., Shlomi T., Rubinstein N.D., et al. Epitope mapping using combinatorial phage-display libraries: a graph-based algorithm. Nucleic Acids Res. 2007;35(1):69–78. doi: 10.1093/nar/gkl975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bublil EM, Tarnovitski Freund N, Mayrose I, et al. Stepwise prediction of conformational discontinuous B-cell epitopes using the mapitope algorithm. Proteins Structure Function Bioinformatics. In press [DOI] [PubMed]
- 87.Bublil E.M., Yeger-Azuz S., Gershoni J.M. Computational prediction of the cross-reactive neutralizing epitope corresponding to the monoclonal antibody b12 specific for HIV-1 gp120. Faseb J. 2006;20:1762–74. doi: 10.1096/fj.05-5509rev. [DOI] [PubMed] [Google Scholar]
- 88.Tarnovitski N., Matthews L.J., Sui J., et al. Mapping a neutralizing epitope on the SARS coronavirus spike protein: computational prediction based on affinity-selected peptides. J Mol Biol. 2006;359:190–201. doi: 10.1016/j.jmb.2006.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]