

Abstract
Background
Chronic pulmonary infection is a hallmark of lung disease in cystic fibrosis. Infections dominated by organisms of the Burkholderia cepacia complex, a group of at least 18 closely‐related species of gram‐negative bacteria, are particularly difficult to treat. These infections may be associated with a fulminant necrotising pneumonia. Burkholderia cepacia complex bacteria are resistant to many common antibiotics and able to acquire resistance against many more. Following patient segregation in cystic fibrosis medical care, the more virulent epidemic strains are not as frequent, and new infections are more likely to be with less virulent environmentally‐acquired strains. Although evidence‐based guidelines exist for treating respiratory exacerbations involving Pseudomonas aeruginosa, these cannot be extended to Burkholderia cepacia complex infections. This review, which is an update of a previous review, aims to assess the available trial evidence for the choice and application of treatments for these infections.
Objectives
To assess the effectiveness and safety of different antibiotic regimens in people with cystic fibrosis experiencing an exacerbation and chronically infected with organisms of the Burkholderia cepacia complex.
Search methods
We searched the Cochrane Cystic Fibrosis Trials Register, compiled from electronic database searches and handsearching of journals and conference abstract books. We also searched the reference lists of relevant articles and reviews.
Date of latest search: 28 August 2015.
Selection criteria
Randomised and quasi‐randomised controlled trials of treatments for exacerbations of pulmonary symptoms in people with cystic fibrosis chronically infected with organisms of the Burkholderia cepacia complex.
Data collection and analysis
No relevant trials were identified.
Main results
No trials were included in this review.
Authors' conclusions
Burkholderia cepacia complex infections present a significant challenge for people with cystic fibrosis and their clinicians. The incidence is likely to increase as the cystic fibrosis population ages; and managing and treating these infections will become more important. There is a lack of trial evidence to guide decision making and no conclusions can be drawn from this review about the optimal antibiotic regimens for people with cystic fibrosis who have chronic Burkholderia cepacia complex infections. Clinicians must continue to assess each person individually, taking into account in vitro antibiotic susceptibility data, previous clinical responses and their own experience. Multicentre randomised clinical trials are needed to assess the effectiveness of different antibiotic regimens in people with cystic fibrosis infected with organisms of the Burkholderia cepacia complex.
Plain language summary
Which antibiotics are best to treat worsening symptoms in people with cystic fibrosis with persistent Burkholderia cepacia complex lung infection?
Review question
We looked for evidence of which antibiotics are best to treat a flare up of symptoms in people with cystic fibrosis with persistent Burkholderia cepacia complex lung infection.
Background
Cystic fibrosis is a common inherited condition where the lungs often become blocked with mucus. This harms the lungs' defences and often results in chronic, persistent infections that cannot be cleared by antibiotics. People with cystic fibrosis often need courses of antibiotics to reduce their symptoms (for instance cough, excess mucus and breathlessness) when these flare up or worsen. Such episodes are called exacerbations, and are usually treated with intravenous antibiotics (given through a drip into a vein). One group of bacteria that can infect the lungs of people with cystic fibrosis is called the Burkholderia cepacia complex. These closely‐related bacteria are found widely in the environment and do not cause infections in healthy people who do not have cystic fibrosis. They are particularly hard to treat as they are resistant to many commonly‐used antibiotics. Currently doctors do not know which antibiotics are best at treating these infections; which combinations of antibiotics should be used; how long the antibiotics should be used for; or whether there are additional treatments that might also help. This is an update of a previously published review.
Search date
The evidence is current to: 28 August 2015.
Study characteristics
We did not find any trials which compared groups of volunteers with exacerbations who were also infected with Burkholderia cepacia complex bacteria, who were given different treatments at random.
Key results
More research is needed to find out which treatments are best for improving lung function and survival and for reducing side effects and length of time spent in hospital for people infected with Burkholderia cepacia complex bacteria experiencing an exacerbation.
Background
Description of the condition
Cystic fibrosis (CF) is a complex multisystem disease, caused by defects in a single gene (Ratjen 2003). It is the most common life‐shortening genetic disorder in those of European descent, affecting 1 in 2500 live births in the UK and Western Europe, although it is also found in all other major ethnic groups (Bobadilla 2002). The effects in the lungs are responsible for most of the morbidity and mortality, and the hallmark of the condition is progressive and irreversible bronchiectasis and respiratory failure.
The primary defect in CF is in an epithelial chloride ion channel (Riordan 1989). This is expressed throughout the body, and is involved in secretory function in a number of organs, including the lung, liver, gut, pancreas and sweat glands (Hoogeveen 1991). Defective ion transport in CF is responsible for the effects of the condition in these organs. In the lung, loss of chloride excretion and uncontrolled sodium absorption at the airway surface leads to dehydration and shrinking of the thin layer of fluid that bathes the surface of the airway (Matsui 1998). This results in defective clearance of airway mucus, retention of secretions and blockage of small airways. Bacterial infections are not cleared effectively, leading to a vicious cycle of chronic lower airway infection, inflammation and tissue destruction (Boucher 2007). The end result is progressive lung damage and decline in lung function.
Burkholderia cepacia complex (Bcc) infections in CF
Respiratory infections start early in life in CF (Rosenfeld 2001). Unlike non‐CF lung infections, these tend to be persistent, sometimes involving phenotypic change in the organisms themselves (Harrison 2007). The sequence of infecting organisms also tends to follow a characteristic course, with Staphylococcus aureus in early life followed by Pseudomonas aeruginosa in later childhood or teenage years (CF Foundation 2009); although this picture has been complicated by the emergence of many new pathogens over the last two decades (Harrison 2007).
Organisms of the Bcc are a group of gram negative bacteria that comprise at least 18 closely‐related species (Coenye 2001; Vanlaere 2008; Vanlaere 2009). These are phenotypically very similar and are differentiated on the basis of genetic or biochemical tests. Some of these organisms are ubiquitous in nature, commonly found in soil or water (Mahenthiralingam 2008), though for others the reservoirs remain unidentified (including Burkholderia multivorans, a major cause of Bcc infection in CF). Clinically, Bcc are known to be responsible for chronic pulmonary infection in CF and to be rare causes of nosocomial infections in immune‐compromised individuals (Conly 1986; Pegues 1993).
Infection with Bcc organisms is particularly problematic as the organisms produce a wide variety of potential virulence factors and exhibit innate resistance to many antibiotics and disinfectants (Leitao 2010). It has been observed that Bcc are able to adhere to epithelial cells and mucin, and are also capable of invading and surviving inside airway epithelial cells and macrophages (large white blood cells, occurring principally in connective tissue and in the bloodstream, that ingest foreign particles and infectious microorganisms) (Sajjan 1995). Furthermore, Bcc form biofilms, which may protect them from both host defences and antibiotics and they have a distinct lipopolysaccharide which has also been implicated in antibiotic and antimicrobial peptide resistance (Vinion‐Dubiel 2004). In addition, they secrete a number of factors, such as catalases, proteases and siderophores, which may help them to evade host defences and survive in hostile environments (Mahenthiralingam 2005). The role of individual putative virulence factors in human infection have yet to be clearly defined, and none alone is both necessary and sufficient for virulence.
Since the mid‐1980s, Bcc have been increasingly identified as important pathogens in CF, with resultant increases in rates of mortality and morbidity (Tablan 1985). Currently around 3% of people with CF in the USA and UK are infected with Bcc organisms, though prevalence increases with age (CF Foundation 2009; CF Trust 2008). Although transient infection occurs, the majority of infections cannot be eradicated (De Boeck 2004). Clinical effects vary from asymptomatic carriage to a rapid and uncontrolled deterioration with septicaemia and necrotizing pneumonia (the so‐called "cepacia syndrome") that usually results in early death (Isles 1984). Several studies have shown that those chronically infected with Bcc have increased mortality and morbidity (Frangolias 1999; Tablan 1987). Infection with Bcc is associated with increased requirements for intravenous antibiotics (Frangolias 1999) and outpatient attendances (Jones 2004), though this is principally in those infected with Burkholderia cenocepacia (B. cenocepacia) (Jones 2004). Some of the excess mortality and morbidity associated with B. cenocepacia may be due to the dominance of the ET12 strain in these studies, and may not be true of environmentally acquired B. cenocepacia infections. Individuals with B. cenocepacia, however, do appear to have poorer post lung transplant outcomes (Chaparro 2001; De Soyza 2010; Murray 2008), and most clinical units will not consider these people for lung transplantation.
Patient‐to‐patient spread of organisms may occur through social contact (LiPuma 1990). This is particularly well described for the epidemic (ET‐12) strain of B. cenocepacia, though other epidemic strains exist, including B. multivorans and B. dolosa (Biddick 2003; Khalish 2006). The recognition of epidemic spread, and the increased mortality rates associated with the epidemic strains, has led to the policy of strict segregation in clinical units since the 1990s. As a consequence the epidemiology has altered and the most common new Bcc infection these days is with B. multivorans (Govan 2007), probably acquired from as‐yet unidentified environmental reservoirs. Unlike B. cenocepacia, B. multivorans has not been associated with significant excess change in lung function or nutritional status compared to matched Pseudomonas‐infected controls (Jones 2004; Courtney 2004). This strain has however been associated with both epidemic spread and cepacia syndrome (Govan 2007; Zahariadis 2003), and segregation policies, designed to limit the spread of the epidemic strains, are generally applied to anyone infected with any member of the Bcc. Segregation may itself cause psychological harm by excluding people from the full range of CF unit facilities (Duff 2002).
Description of the intervention
Intravenous antibiotics are given to people with CF to treat pulmonary exacerbations (CF Trust 2009; Flume 2009). Exacerbations are typically characterised by an increase in respiratory symptoms with associated deterioration in biochemical, radiological or physiological markers of disease. The precise definition of an exacerbation is contentious, often relying on clinical judgement rather than strict objective criteria, and may vary between studies. Typically, antibiotics are given for 10 to 21 days and guidelines generally recommend a combination of at least two different antibiotics (CF Trust 2009; Flume 2009). It can never be clear however, which species is responsible for the exacerbation in CF, since many (if not all) individuals are chronically infected by more than one organism, but it is common practice to choose antibiotics that are likely to be effective against the dominant bacterial species in that person. However, Bcc are innately resistant to colomycin and will often show resistance to aminoglycosides and β‐lactams (Aaron 2000). Some Bcc species are even able to use penicillin G as a sole carbon source (Beckman 1979). A specific efflux pump can also confer resistance to quinolones, chloramphenicol and trimethoprim (Burns 1996). Some strains have inducible β‐lactamases or dihydrofolate reductases. Multiple drug resistance is common in vitro, with 50% of isolates being resistant to all of 10 commonly‐used antibiotics (Aaron 2000). In vitro treatment of Bcc cultured from individuals with CF requires multiple antibiotics, typically at least three, to offer the greatest chance of bactericidal effect (Aaron 2000).
How the intervention might work
Choice of antibiotics is dependent upon a number of factors, including drug reactions, toxicities and interactions with other medications. The best choice of antibiotics would lead to the most rapid resolution of symptoms and correction of other abnormalities (e.g. radiology, lung function and inflammatory markers) with the least toxicity. At present, there is little guidance on the most effective therapies to use in people with Bcc (CF Trust 2004).
Length of treatment is also an important factor. Shorter courses of treatment may be insufficient to treat the infection adequately, but there is an increased incidence of allergic reactions with longer antibiotic courses (Parmar 2005) and an increased risk of toxicity (e.g. ototoxicity and nephrotoxicity with aminoglycosides (Prayle 2010)), or blood dyscrasias (an imbalance in the composition of blood) with chloramphenicol.
Other factors that may determine the success of a treatment strategy include the route of administration, and whether there is additional benefit to be gained from other therapies such as recombinant dornase alfa (DNase) (Grimwood 2009) or amiloride (Middleton 2005).
It is important to recognise that when a person with CF is unwell with an exacerbation of respiratory symptoms, therapy is chosen which is both broad in spectrum and which is likely to be effective against the dominant bacterial species isolated from that individual. In all cases, however, when a person is unwell, it is unethical to withhold treatments which are widely considered to be effective. Truly placebo‐controlled trials of antibiotic therapy in CF exacerbations cannot therefore be performed, and in this review we will instead consider the effects of one antibiotic regimen against another. When additional therapies are added in to a conventional regimen, then this may reasonably be compared to placebo, and we will also review any studies of this nature.
Why it is important to do this review
Since Bcc are resistant to many antibiotics, and can prove to be difficult to treat in clinical practice, it is important to review the available evidence in order to establish the optimal antibiotic therapy for treating a pulmonary exacerbation in people with CF chronically infected with Bcc organisms. This includes consideration of any factors that may impact on treatment efficacy, including the choice of antibiotics and length of treatment. Given the toxicity profiles of some of the treatments used to treat Bcc, an important part of this review is also to consider the safety profiles of different regimens.
This is an update of a previous version of this review (Horsley 2012).
Objectives
The aim of this review is to assess the effectiveness and safety of different antibiotic regimens in people with CF experiencing an exacerbation, who are chronically infected with organisms of the Bcc. This will cover only those receiving treatment for an exacerbation of pulmonary symptoms, either diagnosed by the attending clinician or by pre‐defined objective criteria, but not those receiving elective eradication therapy (which is the subject of a separate Cochrane review (Regan 2014)).
Methods
Criteria for considering studies for this review
Types of studies
Randomised and quasi‐randomised controlled trials, published or unpublished.
Types of participants
Individuals with CF, of any age, diagnosed on the basis of clinical evidence of CF‐lung disease and either genotype analysis or sweat testing, or both. Participants were also required to have evidence of pulmonary infection with organisms of the Burkholderia cepacia complex (Bcc), defined as at least two positive sputum or broncho‐alveolar lavage microbiology specimens within the last six months, grown on specialist media and confirmed by conventional molecular and microbiological techniques (see below).
The nature of the techniques used to identify and type Bcc have changed over time, and early studies may not have had access to current technology. Early studies were considered if Bcc infection was identified by culture on Bcc‐specific selective media and confirmed by a national or regional reference laboratory. For studies published after 2010 however, the techniques used to identify Bcc were required to be consistent with published microbiological guidelines, and appropriate molecular techniques (e.g. recA sequencing, species specific polymerase chain reaction (PCR) or recA PCR and restriction fragment length polymorphism) should have been employed to confirm species identification (CF Trust 2010).
Finally, participants should have been receiving therapy for an exacerbation of CF‐lung disease, defined on the basis of a combination of changes in clinical criteria, lung function and radiology. There is no universally accepted consensus as to what constitutes an exacerbation in CF. Studies will not be excluded based upon the definition of exacerbation, which could be diagnosed by the attending clinician or by pre‐defined objective criteria, providing that the defining criteria are explicit and consistent and that the intent was treatment of new symptoms rather than eradication of existing infection.
Types of interventions
Any antibiotic treatment regimen for treating an exacerbation of CF‐lung disease compared to placebo or different antibiotic regimen, regardless of frequency of administration, treatment duration, route of delivery or use of additional therapies (e.g. DNase, amiloride etc).
Types of outcome measures
Primary outcomes
-
Lung function
change in absolute values for forced expiratory volume in one second (FEV1)
change in per cent predicted values for FEV1
Survival (measured as hazard ratios)
-
Adverse events, classified as:
mild (transient and treatment continued, e.g. rash, nausea);
moderate (necessitating discontinuation of treatment, e.g. reversible renal or hepatic impairment);
severe (life threatening or seriously debilitating, e.g. aplastic anaemia, permanent hearing loss or renal impairment)
Secondary outcomes
Number of days in hospital
Time to next exacerbation (post hoc change)
-
Nutritional markers
weight (kg)
body mass index (BMI)
Acquisition or eradication of other significant CF pathogens
-
Changes in inflammatory markers
serum samples
sputum or bronchoalveolar lavage (BAL) samples
Quality of life (QoL) (as measured by a valid disease‐specific QoL tool (Gee 2000; Quittner 2009))
Search methods for identification of studies
Electronic searches
We identified relevant trials from the Group's Cystic Fibrosis Trials Register using the following terms: 'antibiotics AND burkholderia cepacia' and also 'antibiotics AND mixed infections AND (acute treatment [pulmonary exacerbations] OR unknown'.
The Cystic Fibrosis Trials Register is compiled from electronic searches of the Cochrane Central Register of Controlled Trials (CENTRAL) (updated each new issue of The Cochrane Library), weekly searches of MEDLINE, a search of Embase to 1995 and the prospective handsearching of two journals ‐ Pediatric Pulmonology and the Journal of Cystic Fibrosis. Unpublished work was identified by searching the abstract books of three major cystic fibrosis conferences: the International Cystic Fibrosis Conference; the European Cystic Fibrosis Conference and the North American Cystic Fibrosis Conference. For full details of all searching activities for the register, please see the relevant sections of the Cystic Fibrosis and Genetic Disorders Group Module.
Date of the latest search of the Cystic Fibrosis Trials Register: 28 August 2015.
Searching other resources
For the initial review we contacted experts in the field to enquire if they are aware of unpublished trial data. We invited manufacturers of antimicrobial agents known to be of use in the treatment of Bcc infections to submit data from unpublished trials. This will not be repeated in any subsequent updates. If any trials are identified in the future, we will assess the bibliographic references of these for additional trial reports.
Data collection and analysis
Selection of studies
In both the initial review and subsequent update all authors independently selected trials (published both as abstracts or full papers) to be included in the review. We reviewed abstracts initially to exclude obviously irrelevant trials; we then reviewed the full text of the remaining trials separately. We will repeat the same process at future updates of this review. We plan to resolve any disagreement through discussion and if necessary seek arbitration from an independent third party.
Data extraction and management
We were not able to include any trials in this initial version of the review; however, if we include trials in future updates of the review, we will proceed as detailed below.
All authors will independently extract data from included trials and enter these onto data extraction sheets for analysis using the review software 'Review Manager' (RevMan 2011). All authors will independently make the decision on whether trials are sufficiently similar in design and quality to be pooled; we will resolve any disagreement through discussion. It may be necessary to perform several different meta‐analyses, depending on the types of trials and recorded outcome measures available. If outcome measures differ in the way they are reported (e.g. FEV1 versus per cent predicted FEV1, survival at different time points), we will contact the primary investigators for raw data to be used in the meta‐analysis. We will consider different modes of drug delivery separately. If there are differences between trials in the length of treatment, we will report data at two weeks, four weeks and three months, but will consider presenting data at other time‐points as appropriate.
Assessment of risk of bias in included studies
All authors will independently assess the risk of bias for each included trial using the Cochrane risk of bias tool (Higgins 2011a). We will use this to generate a risk of bias table for each trial and assess the risk of bias as high, low or unclear. In particular, we will examine the following potential sources of bias: the process of randomisation; the degree of blinding in the trial; completeness of outcome data; selective outcome reporting and any other potential sources of bias.
Measures of treatment effect
We plan to pool trials of similar design and with similar outcomes and analyse data using the RevMan software (RevMan 2011). For binary outcome measures we will calculate a pooled estimate of treatment effect for each outcome using the pooled odds ratio (OR) and 95% confidence intervals (CIs).
For continuous outcomes, we will use either mean change over the course of therapy, or mean post‐treatment values, and standard deviation (SD) to calculate a pooled estimate of the treatment effect across trials by calculating the mean difference (MD) and 95% CIs.
We plan to compare survival data using hazard ratios (HR). We will combine adverse event data regardless of length of treatment and compare using risk ratios.
Unit of analysis issues
Because of the nature of the intervention and predicted length of therapy, all participants are likely to be receiving some form of active therapy. We do not consider cross‐over trials of an additional therapy during the course of a single treatment episode to be an appropriate trial design because of both the short‐term effects of the other therapies and the anticipated length of treatment. We will consider cross‐over trials that randomised to different interventions during separate exacerbation episodes if the washout period between exacerbations was appropriate (minimum four weeks).
Dealing with missing data
We will perform an intention‐to‐treat analysis. In cases of missing data, we will contact the trial primary investigators to provide this, wherever possible. If individual participant data are not available, and SDs are not quoted, we plan to estimate these as described in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011b).
Assessment of heterogeneity
We plan to assess heterogeneity between comparable trials by inspection of forest plots and application of the I2 statistic, which describes the percentage of total variation across trials that is caused by heterogeneity rather than by chance (Higgins 2003). The importance of the observed value of I2 depends on both the magnitude and direction of effects and on the strength of evidence for heterogeneity, represented by the CIs for I2. We will base our assessment of the level of heterogeneity on the following cut offs for I2, taking into account the CIs:
0% to 40%: may not be important;
30% to 60%: may represent moderate heterogeneity;
50% to 90%: may represent substantial heterogeneity;
75% to 100%: may represent considerable heterogeneity.
We will calculate CIs for the I2 values after taking statistical advice and according to the methods described by Higgins (Higgins 2002).
Assessment of reporting biases
Wherever possible, we will obtain the original trial protocols for comparison with the published papers to ensure that all outcomes are reported. If it is not possible to obtain the trial protocols, we will scrutinise the 'Methods' section of the published paper(s) to ensure full reporting of all measured variables. If negative data are not fully reported, we will contact the primary investigators for these data. If there are a sufficient number of comparable trials, we will explore these for reporting bias using a funnel plot. We will also assess publication bias by looking for evidence of conference presentations not followed by subsequent journal publications.
Data synthesis
We plan to use a fixed‐effect model to analyse data from trials. If there is evidence of substantial heterogeneity (I2 is greater than 50%), we will use a random‐effects analysis and compare the two analyses. If the results of the random‐effects analysis are different, we will review the trials to identify sources of heterogeneity.
Subgroup analysis and investigation of heterogeneity
If we identify substantial heterogeneity (I2 greater than 50%), we plan to explore this further. In the first instance, we will check data to ensure accurate recording and entry. Where there are sufficient data to permit it (minimum of 10 trials), we then plan to use subgroup analysis to examine the causes of heterogeneity. In particular, this will include a comparison of different age groups (up to 16 years and 16 years and older), lung function severity (expressed as FEV1 per cent predicted: less than 40%; 40% to 60%; 60% to 80%; and over 80%), species of Bcc (cenocepacia or multivorans or other), and presence of significant co‐morbidities (diabetes, CF‐related liver disease, BMI below 18).
Sensitivity analysis
If there are sufficient comparable trials to permit it, we will perform the following sensitivity analyses: excluding and including trials with a high risk of bias (in any of the five key domains); and analysis using a fixed‐effect versus a random‐effects model.
Results
Description of studies
Results of the search
Searching of databases, for both the initial review and update, identified 102 records. After the exclusion of 61 references, we retrieved a total of 41 references to 28 trials (Figure 1). In six cases, we previously contacted the authors for further details, where the trials have reported the inclusion of participants with "Pseudomonas cepacia" (as Bcc organisms were originally designated). We have received replies for three of these trials, all of which have subsequently been excluded as separate data are not available for participants with Bcc infections (Bosso 1987; Bosso 1988; Gold 1987). There was no response for the other three remaining trials and these have now also been excluded (Conway 1996a; Reed 1987c; Reed 1987c). On the initial review one unpublished trial was listed as 'Awaiting classification'; this trial has now been excluded (Tullis 2014).
1.
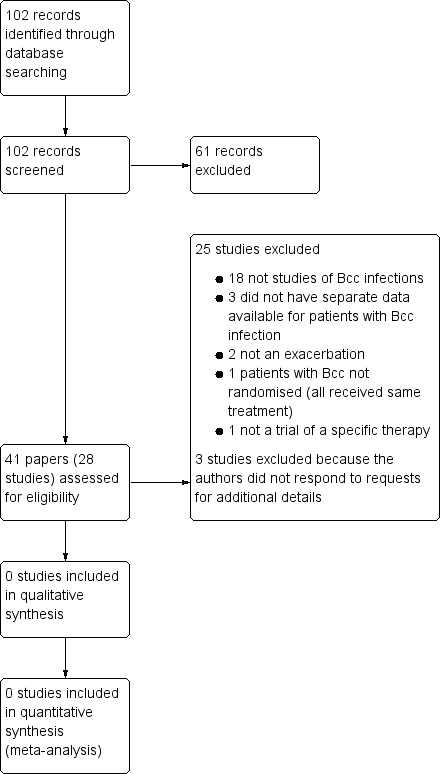
Study flow diagram.
Please note: some studies had more than one reason for exclusion. All are included here, although only the major reason is described in the text.
Included studies
We did not identify any trials for inclusion in this review.
Excluded studies
All retrieved trials were excluded. The majority of these (n = 18) reported exclusively on treatment for chronic infection with Pseudomonas aeruginosa. Three trials did not have separate data available for participants chronically infected with Bcc organisms (Bosso 1987; Bosso 1988; Gold 1987). One trial was not randomised (Blumer 2003). We were only able to identify three randomised trials of Bcc‐specific interventions, none of which met the inclusion criteria. In two trials, the treatment was only given to individuals with stable disease (Ledson 2002; Tullis 2014) and in a third the intervention was not a specific therapy regimen, but related to the use of bacterial cultures to select antibiotics (Aaron 2005). For three studies the authors were contacted but did not reply therefore they have been excluded (Conway 1996; Reed 1987a; Reed 1987b).
Risk of bias in included studies
We did not identify any trials for inclusion in this review.
Effects of interventions
We did not identify any trials for inclusion in this review.
Discussion
Despite the recognition of the clinical importance of Burkholderia cepacia complex (Bcc) infections in cystic fibrosis (CF) for over 25 years now, there have been no randomised trials to systematically evaluate the optimum treatment of exacerbations. Historically, there are several factors which may have contributed to this. Some of the antibiotics now commonly employed in these infections were not available when the importance of the Bcc infections was first recognised. In addition, the epidemiology has changed from one of a transmissible and virulent infection to one of sporadic but infrequent new acquisitions (Govan 2007). Although a small proportion of new Bcc infections may be cleared, either spontaneously or with antibiotics, the majority become chronic (Horsley 2011). With time, and with an ageing CF population, the challenges posed by managing exacerbations in individuals infected with Bcc will therefore not only persist but may even increase.
The small numbers of randomised trials in CF have understandably focused on treating individuals chronically infected with Pseudomonas aeruginosa, since this infection is both common and known to be associated with accelerated decline in lung function (Emerson 2002). The differences between the two organisms mean that outcomes from these trials cannot be applied to people with Bcc infections. We reviewed many of these trials, however, in the hope that they might also contain data on outcomes in individuals additionally infected with organisms of the Bcc. Unfortunately, where such infections were reported, the data were not analysed separately for the different infecting organisms. These studies are now at least 16 years old (and the majority much older). The antibiotic regimens were not specifically tailored to treating people with Bcc infections, and would not be likely to be considered for current management of exacerbations. We have been able to identify only three randomised trials targeted at people with Bcc infections, none of which met the the inclusion criteria for this review. The first of these was an assessment in people with stable disease (Ledson 2002) and the second was not a trial of a specific therapy, but rather of an approach to selecting the optimal treatment regimen (Aaron 2005). The third trial, not included in the previous version of this review, was a trial of inhaled aztreonam but again in people with stable disease (Tullis 2014).
Authors' conclusions
Implications for practice.
We did not identify any randomised trials of therapies for treating exacerbations in people with CF chronically infected with Bcc, which could be included. No conclusions can be drawn about the optimal antibiotic regimens for people with CF with chronic Bcc infections and clinicians must continue to assess each person individually, taking into account in vitro antibiotic susceptibility data, previous clinical responses and their own experience.
Implications for research.
Currently very little is known about how Bcc infections in CF respond to antibiotics in vivo and a number of important questions remain unanswered. There is a need for multicentre randomised clinical trials to assess the effectiveness of different antibiotic regimens in people with CF infected with organisms of the Bcc and identify: the optimum antibiotic regimen(s); the length of treatment; the incidence of adverse effects; the effects of additional therapies; and long‐term outcome data. Trials should explore these and other aspects of exacerbation therapies in people with CF chronically infected with Bcc. The most important primary outcome measures are those which reflect improvement in lung function and survival, and reduction in adverse events. Important secondary outcome measures include length of hospital stay, quality of life, nutritional status and measure of inflammation.
What's new
| Date | Event | Description |
|---|---|---|
| 6 January 2016 | New search has been performed | A search of the Cystic Fibrosis and Genetic Disorders Group's Cystic Fibrosis Register identified 21 references potentially eligible for inclusion in the review. Of these we retrieved a total of seven papers, all for the same study, which had previously been listed as 'Awaiting classification' (Tullis 2014). This was then excluded as it was not a therapy for an acute exacerbation. |
| 6 January 2016 | New citation required but conclusions have not changed | A new author (Dr R Lord) has joined the review team. No additional studies have been included in the review and our conclusions remain the same. |
Acknowledgements
The authors gratefully acknowledge the kind assistance provided by Nikki Jahnke in preparing this review.
Characteristics of studies
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Aaron 2005 | Not an assessment of a specific treatment regimen. |
| Blumer 2003 | Participants with Bcc not randomised, all given same treatment. |
| Bosso 1987 | Separate data not available for participants with Bcc infection ‐ confirmed by trial authors. |
| Bosso 1988 | Separate data not available for participants with Bcc infection ‐ confirmed by trial authors. |
| Carswell 1987 | Not a trial of participants with Bcc infections. |
| Conway 1996 | Not clear if participants had Bcc infections and no answer from trial authors after contact. |
| Cooper 1985 | Not a trial of participants with Bcc infections. |
| Gold 1987 | Separate data not available for participants with Bcc infection ‐ confirmed by trial authors. |
| Heiniger 1993 | Not a trial of participants with Bcc infections. |
| Hoogkamp‐Korstanje 1983 | Not a trial of participants with Bcc infections. |
| Huang 1979 | Not a trial of participants with Bcc infections. |
| Huang 1982 | Not a trial of participants with Bcc infections. |
| Ivanov 1997 | Not a trial of participants with Bcc infections. |
| Kapranov 1995 | Not a trial of participants with Bcc infections. |
| Knight 1979 | Not a trial of participants with Bcc infections. Assessed stable participants only, not those being treated for an exacerbation. |
| Knowles 1988 | Not a trial of participants with Bcc infections. |
| Krause 1979 | Not a trial of participants with Bcc infections. |
| Ledson 2002 | Assessed stable participants only, not those being treated for an exacerbation. |
| Loenig‐Baucke 1978 | Not a trial of participants with Bcc infections. Assessed stable participants only, not those being treated for an exacerbation. |
| Nathanson 1985 | Not a trial of participants with Bcc infections. |
| Powell1983 | Not a trial of participants with Bcc infections. |
| Reed 1987a | Not clear if participants had Bcc infections and no answer from trial authors after contact. |
| Reed 1987b | Not clear if participants had Bcc infections and no answer from trial authors after contact. |
| Rubio 1987 | Not a trial of participants with Bcc infections. |
| Salh 1992 | Not a trial of participants with Bcc infections. |
| Tullis 2014 | Not a trial of a regimen for acute exacerbations |
| Wang 1988 | Not a trial of participants with Bcc infections. |
| Wesley 1988 | Not a trial of participants with Bcc infections. |
Bcc: Burkholderia cepacia complex
Differences between protocol and review
An additional secondary outcome measure has been added to those stated in the protocol (time to next exacerbation), for analysis in future updates of this review. This is a clinically valid endpoint that was overlooked in the initial protocol.
Contributions of authors
| Roles and responsibilities | |
| TASK | WHO WILL UNDERTAKE THE TASK? |
| Protocol stage: draft the protocol | Alex Horsley (AH) |
| Review stage: select which trials to include (2 + 1 arbiter) | AH & Andrew Jones (AJ) |
| Review stage: extract data from trials (2 people) | AH & AJ |
| Review stage: enter data into RevMan | AH |
| Review stage: carry out the analysis | AH |
| Review stage: interpret the analysis | AH & AJ |
| Review stage: draft the final review | AH & AJ |
| Update stage: update the review | AH, AJ and RL |
Sources of support
Internal sources
University Hospitals South Manchester, UK.
University of Manchester, UK.
External sources
-
National Institute for Health Research Clinician Scientist Award, UK.
AH is funded by a National Institute for Health Research Clinician Scientist award (NIHR‐CS012‐13).
The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health.
-
National Institute for Health Research, UK.
This systematic review was supported by the National Institute for Health Research, via Cochrane Infrastructure funding to the Cochrane Cystic Fibrosis and Genetic Disorders Group.
Declarations of interest
AH declares that his institution has received money from Celtaxys Pharmaceuticals (consultancy), Vertex Pharamceuticals (grant to host a one day CF conference in Manchester in 2016) and Markedsmodingsfonden (grant from Danish Government to assist in improving the clinical utility of the Innocor multiple breath washout system; funds supported research nurse to use the system and feedback on how to improve usability). AH has applied to the UK CF Trust as part of two Strategic Research Consortium bids (money payable to his institution) both of which are multi‐centre collaborative efforts to address different areas of CF care, unrelated to the current review. He has personally received an honorarium from Gilead Pharmaceuticals for presenting at the Expert Viewpoints in CF meeting, 2014 and received royalties from Oxford University Press from the sale of Oxford Respiratory Medicine Library Handbook on Cystic Fibrosis, published March 2015 (2nd edition).
AJ declares the receipt of funds for advisory board work for Gilead Pharmaceuticals. His institution has received contributions from Forest Pharmaceuticals and Gilead Pharmaceuticals for AH to travel to conferences. He has participated in a multicentre project evaluating clinical outcome of use of nebulised aztreonam (sponsored by Gilead Sciences) and in January 2015 he spoke at the national Irish CF Meeting (meeting and AJ travel to there sponsored by Gilead). AJ further declares sponsorship from Forest Pharmaceuticals for the CF Centre’s annual away day team meeting and an annual meeting with regional paediatric teams (which he helps organise and run). Vertex Pharmaceuticals provided an unrestricted educational grant to his institution to fund a one‐day CF meeting in Manchester in 2016.
RL declares no known conflict of interest.
New search for studies and content updated (no change to conclusions)
References
References to studies excluded from this review
Aaron 2005 {published data only}
- Aaron S. Clinical evidence for combination antibiotic susceptibility testing (synergy testing) [abstract]. Pediatric Pulmonology 2008;43(Suppl 31):157. [CFGD Register: PI198c; MEDLINE: ] [Google Scholar]
- Aaron S, Vandemheen K, Ferris W, Tullis E, Haase D, Berthiaume Y, et al. Treatment of CF exacerbations based on multiple combination antibiotic susceptibility testing‐a randomised, double‐blind, controlled clinical trial [abstract]. Pediatric Pulmonology 2005;40(Suppl 28):304. [CFGD Register: PI198a] [Google Scholar]
- Aaron SD, Vandemheen KL, Ferris W, Fergusson D, Tullis E, Haase D, et al. Combination antibiotic susceptibility testing to treat exacerbations of cystic fibrosis associated with multiresistant bacteria: a randomised, double‐blind, controlled clinical trial. Lancet 2005;366(9484):463‐71. [CFGD Register: PI198b] [DOI] [PubMed] [Google Scholar]
Blumer 2003 {published data only}
- Blumer JL, Minkwitz M, Saiman L, San Gabriel P, Iaconis J, Melnick D. Meropenem (MEM) compared with ceftazidime (CAZ) in combination with tobramycin (TOB) for treatment of acute pulmonary exacerbations (APE) in patients with cystic fibrosis (CF) infected with Pseudomonas aeruginosa (PA) or Burkholderia cepacia (BC) [abstract]. Pediatric Pulmonology 2003;36(Suppl 25):294. [CFGD Register: PI179] [Google Scholar]
Bosso 1987 {published data only}
- Bosso JA, Black PG, Matsen JM. Ciprofloxacin versus tobramycin plus azlocillin in pulmonary exacerbations in adult patients with cystic fibrosis. American Journal of Medicine 1987;82(4A):180‐4. [CFGD Register: PI47] [PubMed] [Google Scholar]
Bosso 1988 {published data only}
- Bosso JA, Black PG. A comparative trial of aztreonam and tobramycin plus azlocillin [abstract]. Proceedings of 10th International Cystic Fibrosis Congress; 1988 March 5‐10; Sydney, Australia. Excerpta Medica, Asia Pacific Congress Series. 1988:R(c)17. [CFGD Register: PI159a]
- Bosso JA, Black PG. Controlled trial of aztreonam vs. tobramycin and azlocillin for acute pulmonary exacerbations of cystic fibrosis. Pediatric Infectious Disease Journal 1988;7(3):171‐6. [CFGD Register: PI59b] [DOI] [PubMed] [Google Scholar]
Carswell 1987 {published data only}
- Carswell F, Ward C, Cook DA, Speller DC. A controlled trial of nebulized aminoglycoside and oral flucloxacillin versus placebo in the outpatient management of children with cystic fibrosis. British Journal of Diseases of the Chest 1987;81(4):356‐60. [CFGD Register: PI54] [DOI] [PubMed] [Google Scholar]
Conway 1996 {published data only}
- Conway SP. Ceftazidime 3G BD is as effective as ceftazidime 2G TDS in the treatment of respiratory exacerbations in cystic fibrosis [abstract]. Israel Journal of Medical Sciences 1996;32(Suppl):S256. [CFGD Register: PI78] [Google Scholar]
Cooper 1985 {published data only}
- Cooper DM, Harris M, Mitchell I. Comparison of intravenous and inhalation antibiotic therapy in acute pulmonary deterioration in cystic fibrosis [abstract]. American Review of Respiratory Disease 1985;131:A242. [CFGD Register: PI129] [Google Scholar]
Gold 1987 {published data only}
- Gold R, Carpenter S, Heurter H, Corey M, Levison H. Randomized trial of ceftazidime versus placebo in the management of acute respiratory exacerbations in patients with cystic fibrosis. Journal of Pediatrics 1987;111(6 pt 1):907‐13. [CFGD Register: PI57] [DOI] [PubMed] [Google Scholar]
Heiniger 1993 {published data only}
- Heininger U, Bowing B, Stehr K, Solbach W. Aminoglycosides in patients with mucoviscidosis and pulmonary exacerbation. Comparison of once or three times daily administration [Aminoglykoside bei Patienten mit Mukoviszidose und pulmonaler Exazerbation: Vergleich von Einmal‐ und Dreimalgabe]. Klinische Padiatrie 1993;205(1):18‐22. [CFGD Register: PI74] [DOI] [PubMed] [Google Scholar]
Hoogkamp‐Korstanje 1983 {published data only}
- Hoogkamp‐Korstanje JA, Laag J. Piperacillin and tobramycin in the treatment of Pseudomonas lung infections in cystic fibrosis. Journal of Antimicrobial Chemotherapy 1983;12(2):175‐83. [CFGD Register: PI140; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Huang 1979 {published data only}
- Huang N, Palmer J, Schidlow D, Hsuan F, Hsu C, Goldberg M, et al. Evaluation of antibiotic therapy in patients with cystic fibrosis [abstract]. Chest 1979;76(3):354‐5. [CFGD Register: PI113] [Google Scholar]
Huang 1982 {published data only}
- Huang NN, Palmer J, Braverman S, Keith HH, Schidlow D. Therapeutic efficacy of ticarcillin and carbenicillin in patients with cystic fibrosis: a double blind study [abstract]. Proceedings of 23rd Annual Meeting Cystic Fibrosis Club Abstracts; 1982 May 14; Washington DC. 1982:124. [CFGD Register: PI114]
Ivanov 1997 {published data only}
- Ivanov V, Shabalova L, Holmes C, Hill C, Illangovan P, O'Hara S, et al. Ceftazidime bid vs tid in the treatment of bacterial respiratory exacerbations in Russian cystic fibrosis [abstract]. Pediatric Pulmonology 1997;24(Suppl 14):292. [CFGD Register: PI125] [Google Scholar]
Kapranov 1995 {published data only}
- Kapranov NI, Belousov YB, Kashyrskaya NY, Smirnova EY. Quinoline therapy in children with cystic fibrosis [abstract]. Proceedings of 20th European Cystic Fibrosis Conference; 1995 June 18‐21; Brussels, Belgium. 1995:P19. [CFGD Register: PI104]
Knight 1979 {published data only}
- Knight RK, Batten JC, Mearns M. A double blind trial of cephalexin in cystic fibrosis patients with pseudomonas in the sputum [abstract]. Proceedings of 9th Meeting European Working Group for Cystic Fibrosis; 1979 June 12‐13; Noordwijkerhout, The Netherlands. 1979:52. [CFGD Register: PI124]
Knowles 1988 {published data only}
- Knowles NE, Hallberg TK, Colombe JL. Efficacy of aerosolized antibiotics combined with intravenous treatment of cystic fibrosis pulmonary exacerbation [abstract]. Pediatric Pulmonology 1988;5(Suppl 2):120‐1. [CFGD Register: PI87] [Google Scholar]
Krause 1979 {published data only}
- Krause PJ, Young LS, Cherry JD, Osher AB, Spencer MJ, Bryson YJ. The treatment of exacerbations of pulmonary disease in cystic fibrosis: netilmicin compared with netilmicin and carbenicillin. Current Therapeutic Research, Clinical and Experimental 1979;25:609‐17. [CFGD Register: PI166] [Google Scholar]
Ledson 2002 {published data only}
- Ledson MJ, Gallagher MJ, Cowperthwaite C, Robinson M, Convery RP, Walshaw MJ. A randomised double blind placebo controlled crossover trial of nebulised taurolidine in adult CF patients colonised with B Cepacia [abstract]. Proceedings of 22nd European Cystic Fibrosis Conference; 1998 June 13‐19; Berlin, Germany. 1998:120. [CFGD Register: PI128a]
- Ledson MJ, Gallagher MJ, Robinson M, Cowperthwaite C, Williets T, Hart CA, et al. A randomized double‐blinded placebo‐controlled crossover trial of nebulized taurolidine in adult cystic fibrosis patients infected with Burkholderia cepacia. Journal of Aerosol Medicine 2002;15(1):51‐7. [CFGD Register: PI128b] [DOI] [PubMed] [Google Scholar]
Loenig‐Baucke 1978 {published data only}
- Loening‐Baucke V, Mischler EH, Myers MG. Cephalexin compared to placebo in the management of patients with cystic fibrosis [abstract]. 19th Cystic Fibrosis Club Abstracts; 1978. 1978:69. [CFGD Register: PI19a]
- Loening‐Baucke VA, Mischler E, Myers MG. A placebo‐controlled trial of cephalexin therapy in the ambulatory management of patients with cystic fibrosis. Journal of Pediatrics 1979;95(4):630‐7. [CFGD Register: PI19b] [DOI] [PubMed] [Google Scholar]
- Loening‐Baucke VA, Mischler EH, Myers MG. Cephalexin in cystic fibrosis: a placebo‐controlled study [abstract]. Pediatric Research 1978;12(4 Pt 2):495. [CFGD Register: PI19c; MEDLINE: ] [Google Scholar]
Nathanson 1985 {published data only}
- Nathanson I, Cropp GJA, Li P, Neter E. Effectiveness of aerosolized gentamicin in cystic fibrosis (CF) [abstract]. Cystic Fibrosis Club Abstracts; 1985. 1985; Vol. 28:145. [CFGD Register: PI130]
Powell1983 {published data only}
- Powell SH, Stern RC, Thompson WL. Safety of once daily therapy with high‐dose tobramycin [abstract]. Cystic Fibrosis Club Abstracts; 1979 May 1; Atlanta, Georgia. 1979:74. [CFGD Register: PI139b]
- Powell SH, Thompson WL, Luthe MA, Stern RC, Grossniklaus DA, Bloxham DD, et al. Once‐daily vs. continuous aminoglycoside dosing: efficacy and toxicity in animal and clinical studies of gentamicin, netilmicin, and tobramycin. Journal of Infectious Diseases 1983;147(5):918‐32. [CFGD Register: PI139a; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Reed 1987a {published data only}
- Reed MD, Stern RC, O'Brien CA, Crenshaw DA, Blumer JL. Randomized double‐blind evaluation of ceftazidime dose ranging in hospitalised patients with cystic fibrosis. Antimicrobial Agents and Chemotherapy 1987;31(5):698‐702. [CFGD Register: PI49] [DOI] [PMC free article] [PubMed] [Google Scholar]
Reed 1987b {published data only}
- Reed MD, Stern RC, Myers CM, Klinger JD, Yamashita TS, Blumer JL. Therapeutic evaluation of piperacillin for acute pulmonary exacerbations in cystic fibrosis. Pediatric Pulmonology 1987;3(2):101‐9. [CFGD Register: PI46] [DOI] [PubMed] [Google Scholar]
Rubio 1987 {published data only}
- Rubio TT. Ciprofloxacin: comparative data in cystic fibrosis. American Journal of Medicine 1987;82(Suppl 4A):185‐8. [CFGD Register: PI137; MEDLINE: ] [PubMed] [Google Scholar]
Salh 1992 {published data only}
- Salh B, Bilton D, Dodd M, Abbot J, Webb K. A comparison of aztreonam and ceftazidime in the treatment of respiratory infections in adults with cystic fibrosis. Scandinavian Journal of Infectious Diseases 1992;24(2):215‐8. [CFGD Register: PI72] [DOI] [PubMed] [Google Scholar]
Tullis 2014 {published data only}
- Burns J, LiPuma JJ, Retsch‐Bogart G, Bresnik M, Henig N, McKevitt M, et al. No antibiotic cross‐resistance after 1 year of continuous aztreonam for inhalation solution (AZLI) in cystic fibrosis (CF) patients (pts) with chronic Burkholderia (BURK) infection [abstract]. Journal of Cystic Fibrosis 2012;11 Suppl 1:S71, Abstract no: 58. [CENTRAL: 867109; CFGD Register: PI259d ; CRS: 5500100000011251] [Google Scholar]
- Burns JL, LiPuma J, McKevitt M, Lewis S, Retsch‐Bogart GZ, Bresnik M, et al. The effect of Burkholderia colony morphology in cystic fibrosis (CF) patients with chronic Burkholderia species in a randomized trial of aztreonam for inhalation solution (AZLI) [abstract]. Pediatric Pulmonology 2012;47(S35):333, Abstract no: 307. [CENTRAL: 867111; CFGD Register: PI259g ; CRS: 5500100000011253] [Google Scholar]
- Tullis DE, Burns JL, Retsch‐Bogart GZ, Bresnik M, Henig NR, Lewis SA, et al. Inhaled aztreonam for chronic burkholderia infection in cystic fibrosis: A placebo‐controlled trial. Journal of Cystic Fibrosis 2014;13(3):296‐305. [CENTRAL: 984866; CFGD Register: PI259h ; CRS: 5500050000000047; EMBASE: 2014217550] [DOI] [PubMed] [Google Scholar]
- Tullis E, Burns JL, Retsch‐Bogart G, Bresnik M, Henig NR, Lewis S, et al. Aztreonam for inhalation solution (AZLI) in cystic fibrosis (CF) patients with chronic Burkholderia species (BURK) infection: final results from a randomized, placebo‐controlled trial [abstract]. Journal of Cystic Fibrosis 2012;11(Suppl 1):S11, Abstract No. WS5.4. [CFGD Register: PI259e] [Google Scholar]
- Tullis E, Burns JL, Retsch‐Bogart G, Bresnik M, Henig NR, Lewis S, et al. Aztreonam for inhalation solution (AZLI) in cystic fibrosis (CF) patients with chronic burkholderia species (Burk) infection: initial results from a randomized, placebo‐controlled trial [abstract]. Pediatric Pulmonology 2011;46 Suppl 34:296, Abstract no: 234. [CENTRAL: 867118; CFGD Register: PI259b ; CRS: 5500100000011263] [Google Scholar]
- Tullis E, Burns JL, Retsch‐Bogart G, Bresnik M, Lewis S, Montgomery AB, et al. Aztreonam 75mg powder and solvent for nebuliser solution (AZLI) in cystic fibrosis (CF) patients with chronic Burkholderia species (Burk) infection: baseline demographics and microbiology from randomized , placebo‐controlled trial [abstract]. Journal of Cystic Fibrosis 2011;10 Suppl 1:S22, Abstract no: 85. [CENTRAL: 867116; CFGD Register: PI259c ; CRS: 5500100000011258] [Google Scholar]
- Tullis E, Burns JL, Retsch‐Bogart GZ, Bresnik M, Lewis S, LiPuma J. Lung function in cystic fibrosis (CF) patients with chronic Burholderia (Burk) species infection over the course of a prospective, randomized trial of aztreonam for inhalation solution (AZLI) [abstract]. Pediatric Pulmonology 2012;47(S35):334, Abstract no: 310. [CENTRAL: 867112; CFGD Register: PI259f ; CRS: 5500100000011254] [Google Scholar]
- Tullis E, LiPuma JJ, Retsch‐Bogart G, Bresnik M, Henig N, McKevitt M, et al. Effects of continuous aztreonam for inhalation solution (AZLI) use on pathogens and antibiotic susceptibility in cystic fibrosis (CF) patients with chronic Burkholderia species infection [abstract]. Pediatric Pulmonology 2011;46 Suppl 34:305, Abstract no: 257. [CENTRAL: 867119; CFGD Register: PI259a ; CRS: 5500100000011265] [Google Scholar]
Wang 1988 {published data only}
- Wang CI, Inderlied CB, Armer C, Roldan MA, Osher AB. Comparison of the efficacy and safety of oral ciprofloxacin with that of i.v. tobrarmycin plus azlocillin and/or tobramycin plus ticarcillin in patients with cystic fibrosis [abstract]. Excerpta Medica, Asia Pacific Congress Series 1988;74:R(c)19. [CFGD Register: PI89] [Google Scholar]
Wesley 1988 {published data only}
- Wesley AW, Quested C, Edgar BW, Lennon DR. A double‐blind comparison of ceftazidime with tobramycin and ticarcillin in the treatment of exacerbations of pseudomonas chest infection in children with cystic fibrosis [abstract]. Excerpta Medica, Asia Pacific Congress Series. 1988:R(c)13. [CFGD Register: PI90]
Additional references
Aaron 2000
- Aaron SD, Ferris W, Henry DA, Speert DP, Macdonald NE. Multiple combination bactericidal antibiotic testing for patients with cystic fibrosis infected with Burkholderia cepacia. American Journal of Respiratory and Critical Care Medicine 2000;161(4 Pt 1):1206‐12. [DOI] [PubMed] [Google Scholar]
Beckman 1979
- Beckman W, Lessie TG. Response of Pseudomonas cepacia to beta‐lactam antibiotics: utilization of penicillin G as the carbon source. Journal of Bacteriology 1979;140(3):1126‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Biddick 2003
- Biddick R, Spilker T, Martin A, LiPuma JJ. Evidence of transmission of Burkholderia cepacia, Burkholderia multivorans and Burkholderia dolosa among persons with cystic fibrosis. FEMS Microbiology Letters 2003;228(1):57‐62. [DOI] [PubMed] [Google Scholar]
Bobadilla 2002
- Bobadilla JL, Macek M, Fine JP, Farrell PM. Cystic fibrosis: a worldwide analysis of CFTR mutations‐‐correlation with incidence data and application to screening. Human Mutation 2002;19(6):575‐606. [DOI] [PubMed] [Google Scholar]
Boucher 2007
- Boucher RC. Airway surface dehydration in cystic fibrosis: pathogenesis and therapy. Annual Review of Medicine 2007;58:157‐70. [DOI] [PubMed] [Google Scholar]
Burns 1996
- Burns JL, Wadsworth CD, Barry JJ, Goodall CP. Nucleotide sequence analysis of a gene from Burkholderia (Pseudomonas) cepacia encoding an outer membrane lipoprotein involved in multiple antibiotic resistance. Antimicrobial Agents and Chemotherapy 1996;40(2):307‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
CF Foundation 2009
- Patient Registry 2009 Annual Report. Cystic Fibrosis Foundation, Bethesda, Maryland 2009.
CF Trust 2004
- Report of UK Cystic Fibrosis Trust Infection Control Group. The Burkholderia cepacia complex. Suggestions for prevention and infection control. UK Cystic Fibrosis Trust 2004.
CF Trust 2008
- UK CF Registry Annual Report. UK Cystic Fibrosis Trust 2008.
CF Trust 2009
- Report of the UK Cystic Fibrosis Trust Antibiotic Group. Antibiotic treatment for cystic fibrosis. UK Cystic Fibrosis Trust 2009.
CF Trust 2010
- UK Cystic Fibrosis Trust Microbiology Laboratory Standards Working Group. Laboratory standards for processing microbiological samples from people with cystic fibrosis. UK Cystic Fibrosis Trust 2010.
Chaparro 2001
- Chaparro C, Maurer J, Gutierrez C, Krajden M, Chan C, Winton T, et al. Infection with Burkholderia cepacia in cystic fibrosis: outcome following lung transplantation. American Journal of Respiratory and Critical Care Medicine 2001;163(1):43‐8. [DOI] [PubMed] [Google Scholar]
Coenye 2001
- Coenye T, Vandamme P, Govan JR, LiPuma JJ. Taxonomy and identification of the Burkholderia cepacia complex. Journal of Clinical Microbiology 2001;39(10):3427‐36. [DOI] [PMC free article] [PubMed] [Google Scholar]
Conly 1986
- Conly JM, Klass L, Larson L, Kennedy J, Low DE, Harding GK. Pseudomonas cepacia colonization and infection in intensive care units. Canadian Medical Association Journal 1986;134(4):363‐6. [PMC free article] [PubMed] [Google Scholar]
Courtney 2004
- Courtney JM, Dunbar KE, McDowell A, Moore JE, Warke TJ, Stevenson M, et al. Clinical outcome of Burkholderia cepacia complex infection in cystic fibrosis adults. Journal of Cystic Fibrosis 2004;3(2):93‐8. [DOI] [PubMed] [Google Scholar]
De Boeck 2004
- Boeck K, Malfroot A, Schil L, Lebecque P, Knoop C, Govan JR, et al. Epidemiology of Burkholderia cepacia complex colonisation in cystic fibrosis patients. European Respiratory Journal 2004;23(6):851‐6. [DOI] [PubMed] [Google Scholar]
De Soyza 2010
- Soyza A, Meachery G, Hester KL, Nicholson A, Parry G, Tocewicz K, et al. Lung transplantation for patients with cystic fibrosis and Burkholderia cepacia complex infection: a single‐center experience. Journal of Heart and Lung Transplantation 2010;29(12):1395‐404. [DOI] [PubMed] [Google Scholar]
Duff 2002
- Duff AJ. Psychological consequences of segregation resulting from chronic Burkholderia cepacia infection in adults with CF. Thorax 2002;57(9):756‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Emerson 2002
- Emerson J, Rosenfeld M, McNamara S, Ramsey B, Gibson RL. Pseudomonas aeruginosa and other predictors of mortality and morbidity in young children with cystic fibrosis. Pediatric Pulmonology 2002;34(2):91‐100. [DOI] [PubMed] [Google Scholar]
Flume 2009
- Flume PA, Mogayzel PJ, Robinson KA, Goss CH, Rosenblatt RL, Kuhn RJ, et al. Cystic fibrosis pulmonary guidelines: treatment of pulmonary exacerbations. American Journal of Respiratory and Critical Care Medicine 2009;180(9):802‐8. [DOI] [PubMed] [Google Scholar]
Frangolias 1999
- Frangolias DD, Mahenthiralingam E, Rae S, Raboud JM, Davidson AG, Wittmann R, et al. Burkholderia cepacia in cystic fibrosis. Variable disease course. American Journal of Respiratory and Critical Care Medicine 1999;160(5 Pt 1):1572‐7. [DOI] [PubMed] [Google Scholar]
Gee 2000
- Gee L, Abbott J, Conway S, Etherington C, Webb A. Development of a disease specific health related quality of life measure for adults and adolescents with cystic fibrosis. Thorax 2000;55(11):946‐54. [DOI: 10.1136/thorax.55.11.946] [DOI] [PMC free article] [PubMed] [Google Scholar]
Govan 2007
- Govan JR, Brown AR, Jones AM. Evolving epidemiology of Pseudomonas aeruginosa and the Burkholderia cepacia complex in cystic fibrosis lung infection. Future Microbiology 2007;2(2):153‐64. [DOI] [PubMed] [Google Scholar]
Grimwood 2009
- Grimwood K, Kidd TJ, Tweed M. Successful treatment of cepacia syndrome. Journal of Cystic Fibrosis 2009;8(4):291‐3. [DOI] [PubMed] [Google Scholar]
Harrison 2007
- Harrison F. Microbial ecology of the cystic fibrosis lung. Microbiology (Reading, England) 2007;153(Pt 4):917‐23. [DOI] [PubMed] [Google Scholar]
Higgins 2002
- Higgins JPT, Thompson SG. Quantifying heterogeneity in a meta‐analysis. Statistics in Medicine 2002;21(11):1539‐58. [DOI] [PubMed] [Google Scholar]
Higgins 2003
- Higgins JPT, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analyses. BMJ 2003;327(7414):557‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011a
- Higgins JPT, Altman DG. Chapter 8: Assessing risk of bias in included studies. In: Higgins JPT, Green S (editors). Cochrane Handbook of Systematic Reviews of Interventions. Version 5.1 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Higgins 2011b
- Higgins JPT, Deeks JJ, Altman DG on behalf of the Cochrane Statistical Methods Group. Chapter 16: Special topics in statistics. In: Higgins JPT, Green S (editors). Cochrane Handbook of Systematic Reviews of Interventions. Version 5.1 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Hoogeveen 1991
- Hoogeveen AT, Keulemans J, Willemsen R, Scholte BJ, Bijman J, Edixhoven MJ, et al. Immunological localization of cystic fibrosis candidate gene products. Experimental Cell Research 1991;193(2):435‐7. [DOI] [PubMed] [Google Scholar]
Horsley 2011
- Horsley A, Webb AK, Bright‐Thomas R, Govan J, Jones AM. Can early Burkholderia cepacia complex infection in CF be eradicated with antibiotic therapy?. Frontiers in Cellular and Infection Microbiology 2011;1:18. [DOI: 10.3389/fcimb.2011.00018] [DOI] [PMC free article] [PubMed] [Google Scholar]
Isles 1984
- Isles A, Maclusky I, Corey M, Gold R, Prober C, Fleming P, et al. Pseudomonas cepacia infection in cystic fibrosis: an emerging problem. Journal of Pediatrics 1984;104(2):206‐10. [DOI] [PubMed] [Google Scholar]
Jones 2004
- Jones AM, Dodd ME, Govan JR, Barcus V, Doherty CJ, Morris J, et al. Burkholderia cenocepacia and Burkholderia multivorans: influence on survival in cystic fibrosis. Thorax 2004;59(11):948‐51. [DOI] [PMC free article] [PubMed] [Google Scholar]
Khalish 2006
- Kalish LA, Waltz DA, Dovey M, Potter‐Bynoe G, McAdam AJ, Lipuma JJ, et al. Impact of Burkholderia dolosa on lung function and survival in cystic fibrosis. American Journal of Respiratory and Critical Care Medicine 2006;173(4):421‐5. [DOI] [PubMed] [Google Scholar]
Leitao 2010
- Leitao JH, Sousa SA, Ferreira AS, Ramos CG, Silva IN, Moreira LM. Pathogenicity, virulence factors, and strategies to fight against Burkholderia cepacia complex pathogens and related species. Applied Microbiology and Biotechnology 2010;87(1):31‐40. [DOI] [PubMed] [Google Scholar]
LiPuma 1990
- LiPuma JJ, Dasen SE, Nielson DW, Stern RC, Stull TL. Person‐to‐person transmission of Pseudomonas cepacia between patients with cystic fibrosis. Lancet 1990;336(8723):1094‐6. [DOI] [PubMed] [Google Scholar]
Mahenthiralingam 2005
- Mahenthiralingam E, Urban TA, Goldberg JB. The multifarious, multireplicon Burkholderia cepacia complex. Nature Reviews 2005;3(2):144‐56. [DOI] [PubMed] [Google Scholar]
Mahenthiralingam 2008
- Mahenthiralingam E, Baldwin A, Dowson CG. Burkholderia cepacia complex bacteria: opportunistic pathogens with important natural biology. Journal of Applied Microbiology 2008;104(6):1539‐51. [DOI] [PubMed] [Google Scholar]
Matsui 1998
- Matsui H, Grubb BR, Tarran R, Randell SH, Gatzy JT, Davis CW, et al. Evidence for periciliary liquid layer depletion, not abnormal ion composition, in the pathogenesis of cystic fibrosis airways disease. Cell 1998;95(7):1005‐15. [DOI] [PubMed] [Google Scholar]
Middleton 2005
- Middleton PG, Kidd TJ, Williams B. Combination aerosol therapy to treat Burkholderia cepacia complex. European Respiratory Journal 2005;26(2):305‐8. [DOI] [PubMed] [Google Scholar]
Murray 2008
- Murray S, Charbeneau J, Marshall BC, LiPuma JJ. Impact of burkholderia infection on lung transplantation in cystic fibrosis. American Journal of Respiratory and Critical Care Medicine 2008;178(4):363‐71. [DOI] [PubMed] [Google Scholar]
Parmar 2005
- Parmar JS, Nasser S. Antibiotic allergy in cystic fibrosis. Thorax 2005;60(6):517‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
Pegues 1993
- Pegues DA, Carson LA, Anderson RL, Norgard MJ, Argent TA, Jarvis WR, et al. Outbreak of Pseudomonas cepacia bacteremia in oncology patients. Clinical Infectious Diseases 1993;16(3):407‐11. [DOI] [PubMed] [Google Scholar]
Prayle 2010
- Prayle A, Smyth AR. Aminoglycoside use in cystic fibrosis: therapeutic strategies and toxicity. Current Opinion in Pulmonary Medicine 2010;16(6):604‐10. [DOI] [PubMed] [Google Scholar]
Quittner 2009
- Quittner AL, Modi AC, Wainwright C, Otto K, Kirihara J, Montgomery AB. Determination of the minimal clinically important difference scores for the Cystic Fibrosis Questionnaire‐Revised respiratory symptom scale in two populations of patients with cystic fibrosis and chronic Pseudomonas aeruginosa airway infection. Chest 2009;135(6):1610‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Ratjen 2003
- Ratjen F, Doring G. Cystic fibrosis. Lancet 2003;361(9358):681‐9. [DOI] [PubMed] [Google Scholar]
Regan 2014
- Regan KH, Bhatt J. Eradication therapy for Burkholderia cepacia complex in people with cystic fibrosis. Cochrane Database of Systematic Reviews 2014, Issue 10. [DOI: 10.1002/14651858.CD009876.pub2] [DOI] [PubMed] [Google Scholar]
RevMan 2011 [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.1. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2011.
Riordan 1989
- Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science 1989;245(4922):1066‐73. [DOI] [PubMed] [Google Scholar]
Rosenfeld 2001
- Rosenfeld M, Gibson RL, McNamara S, Emerson J, Burns JL, Castile R, et al. Early pulmonary infection, inflammation, and clinical outcomes in infants with cystic fibrosis. Pediatric Pulmonology 2001;32(5):356‐66. [DOI] [PubMed] [Google Scholar]
Sajjan 1995
- Sajjan US, Sun L, Goldstein R, Forstner JF. Cable (cbl) type II pili of cystic fibrosis‐associated Burkholderia (Pseudomonas) cepacia: nucleotide sequence of the cblA major subunit pilin gene and novel morphology of the assembled appendage fibers. Journal of Bacteriology 1995;177(4):1030‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Tablan 1985
- Tablan OC, Chorba TL, Schidlow DV, White JW, Hardy KA, Gilligan PH, et al. Pseudomonas cepacia colonization in patients with cystic fibrosis: risk factors and clinical outcome. The Journal of Pediatrics 1985;107(3):382‐7. [DOI] [PubMed] [Google Scholar]
Tablan 1987
- Tablan OC, Martone WJ, Doershuk CF, Stern RC, Thomassen MJ, Klinger JD, et al. Colonization of the respiratory tract with Pseudomonas cepacia in cystic fibrosis. Risk factors and outcomes. Chest 1987;91(4):527‐32. [DOI] [PubMed] [Google Scholar]
Vanlaere 2008
- Vanlaere E, Lipuma JJ, Baldwin A, Henry D, Brandt E, Mahenthiralingam E, et al. Burkholderia latens sp. nov., Burkholderia diffusa sp. nov., Burkholderia arboris sp. nov., Burkholderia seminalis sp. nov. and Burkholderia metallica sp. nov., novel species within the Burkholderia cepacia complex. International Journal of Systematic and Evolutionary microbiology 2008;58(7):1580‐90. [DOI] [PubMed] [Google Scholar]
Vanlaere 2009
- Vanlaere E, Baldwin A, Gevers D, Henry D, Brandt E, LiPuma JJ, et al. Taxon K, a complex within the Burkholderia cepacia complex, comprises at least two novel species, Burkholderia contaminans sp. nov. and Burkholderia lata sp. nov. International Journal of Systematic and Evolutionary Microbiology 2009;59(Pt 1):102‐11. [DOI] [PubMed] [Google Scholar]
Vinion‐Dubiel 2004
- Vinion‐Dubiel AD, Spilker T, Dean CR, Monteil H, LiPuma JJ, Goldberg JB. Correlation of wbiI genotype, serotype, and isolate source within species of the Burkholderia cepacia complex. Journal of Clinical Microbiology 2004;42(9):4121‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Zahariadis 2003
- Zahariadis G, Levy MH, Burns JL. Cepacia‐like syndrome caused by Burkholderia multivorans. Canadian Journal of Infectious Diseases 2003;14(2):123‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
References to other published versions of this review
Horsley 2012
- Horsley A, Jones AM. Antibiotic treatment for Burkholderia cepacia complex in people with cystic fibrosis experiencing a pulmonary exacerbation. Cochrane Database of Systematic Reviews 2012, Issue 10. [DOI: 10.1002/14651858.CD009529.pub2] [DOI] [PubMed] [Google Scholar]