

Abstract
Non-traumatic osteonecrosis of the femoral head (ONFH) often occurs after corticosteroid therapy in patients with inflammatory diseases. Recent studies suggest that toll-like receptor (TLR) signaling may contribute to the pathogenesis of inflammatory diseases, and that the reason for corticosteroid therapy for inflammatory diseases is related to the anti-inflammatory activities of corticosteroids through the reduction of NF-κB. We hypothesized that the administration of TLR ligands in combination with corticosteroid causes ONFH and that transcription factors may contribute to the pathogenesis of ONFH. The aim of the study was to evaluate (1) the incidence of ONFH in rats after the administration of TLR7 or TLR9 ligands together with methylprednisolone (MPSL) and (2) whether transcription factors contribute to the development of ONFH. Male Wistar rats (n=148) were divided into five groups as follows: Group 1: Saline+MPSL, Group 2: Imiquimod+Saline, Group 3: Imiquimod+MPSL, Group 4: CpG-C+MPSL, Group 5: Imiquimod+BAY11-7082+MPSL. As a result, ONFH was observed in 0 of 12 rats in Group 1, in 1 of 10 in Group 2, in 6 of 12 in Group 3, in 4 of 12 in Group 4, in 0 of 9 in Group 5. MPSL treatment did not significantly affect IRF7 activity, whereas NF-κB activity was significantly repressed in Group 2 and Group 3. Furthermore, the repression in interferon regulatory factor 7 (IRF7) activity by BAY11-7082 interfered with the development of ONFH simultaneously with the MPSL treatment-induced repression in NF-κB activity. In conclusion, in the present study, corticosteroid treatment after the administration of TLR7 or TLR9 ligands caused ONFH. Repression in NF-κB activity by corticosteroid treatment boosted the development of ONFH.
Subject terms: Inflammatory diseases, Osteoimmunology, Immunopathogenesis
Main
High-dose corticosteroid therapy for inflammatory diseases, such as autoimmune disease, was reported to be a risk factor of non-traumatic osteonecrosis of the femoral head (ONFH).1 On the other hand, it was reported that mega-dose corticosteroid therapy for traumatic spinal cord injury does not induce non-traumatic ONFH.2 Thus, the role of corticosteroid in the pathogenesis of non-traumatic ONFH is poorly understood.
We previously reported that the toll-like receptor (TLR) 4 signaling pathway, which induces inflammatory status, contributes to the pathogenesis of non-traumatic ONFH in rats.3, 4, 5 TLRs were identified as receptors related to the innate immune system in 1997.6 Recently, TLRs have been reported to play a crucial role in autoimmunity.7, 8 In particular, TLRs contribute to the pathogenesis of underlying diseases, such as systemic lupus erythematodes (SLE), nephrotic syndrome, polymyositis/dermatmyositis, bronchial asthma, and thrombocytopenic purpura, in patients with corticosteroid-induced ONFH.9, 10, 11, 12, 13 The signaling pathway via TLR7 or TLR9 and type I interferon may, in particular, contribute to the pathogenesis of systemic autoimmune diseases, such as SLE,9, 14 and some inhibitors such as biologics were developed to downregulate these signaling pathways for the treatment of inflammatory diseases.15, 16
The reason for using corticosteroid therapy for inflammatory diseases is related to the anti-inflammatory and immunosuppressive activities of corticosteroids,17 and it was reported that the reduction of nuclear factor kappa B (NF-κB) activity via glucocorticoid receptors has a central role in these activities.18 NF-κB and interferon regulatory factor 7 (IRF7) are signal transcription factors related to the proinflammatory response via TLR7 or TLR9 and the MyD88-dependent pathway. Thus, we hypothesized that the administration of these TLR7 or TLR9 ligands and subsequent treatment with methylprednisolone (MPSL) leads to ONFH in rats, and that these transcription factors may contribute to the pathogenesis of ONFH via the TLRs. In the present study, to verify this hypothesis, we investigated (1) the incidence of ONFH in rats after the administration of TLR7 or TLR9 ligands in combination with MPSL and (2) whether NF-κB and IRF7 contributed to the pathogenesis of ONFH after MPSL treatment.
MATERIALS AND METHODS
Animals
All experiments were conducted in accordance with the guidelines of the Ministry of Sports, Culture, Science, and Technology of Japan, and followed protocols approved by the Animal Ethics Committee of the Sapporo Medical University (#11–042). Male Wistar rats (300–350 g) were obtained from Sankyo Labo Service (Sapporo, Japan). All animals were housed in temperature- and humidity-controlled rooms with unlimited food and water and a 12-h light/dark cycle.
Experimental Groups and Protocols
Animals (n=148) were divided into five groups and treated as follows (Table 1): Saline+MPSL rats (n=12) were given saline (1.0 ml/kg subcutaneously) on Day 1 and 20 mg/kg MPSL (Sigma, St Louis, MO, USA) intramuscularly on Day 2; Imiquimod+Saline rats (n=28) were given 30 mg/kg imiquimod (Tokyo Chemical Industry, Tokyo, Japan), a ligand for TLR7,19 subcutaneously on Day 1 and saline 1.0 ml/kg intramuscularly on Day 2; Imiquimod+MPSL rats (n=52) were given 30 mg/kg imiquimod subcutaneously on Day 1 and 20 mg/kg MPSL intramuscularly on Day 2; CpG-C+MPSL rats (n=12) were given 5 mg/kg CpG-C (5′-TCGTCGAACGTTCGAGATGAT-3′), a ligand for TLR9,20 subcutaneously on Day 1 and 20 mg/kg MPSL intramuscularly on Day 2; and Imiquimod+BAY11+MPSL rats (n=44) were given 30 mg/kg imiquimod subcutaneously on Day 1 and 1, 3, 5, or 10 mg/kg BAY11-7082 (Sigma), an inhibitor of I kappaB kinase (Ikk) α and β, which has the potential to inhibit NF-κB and IRF7 activity,21 intraperitonealy with 20 mg/kg MPSL intramuscularly on Day 2. All the injections were performed at 7:00 p.m.
Table 1.
List of experimental groups
| 1 | 3 | 7 | 14 days | ||
|---|---|---|---|---|---|
| Saline+MPSL | 0 | 0 | 0 | 12 | n=12 |
| Imiquimod+saline | 6 | 6 | 6 | 10 | n=28 |
| Imiquimod+MPSL | 10 | 10 | 10 | 22 | n=52 |
| CpG-C+MPSL | 0 | 0 | 0 | 12 | n=12 |
| Imiquimod+BAY11+MPSL | 8 | 0 | 0 | 36 | n=44 |
Animals were killed at 1, 3, 7, or 14 days after the last injection. The femurs and livers were harvested and fixed with 10% formalin—0.1 M phosphate buffer (pH 7.4).
Histopathology
The bone samples were decalcified with Kalkitox (Wako Pure Chemical Industries, Osaka, Japan) and then neutralized with a 5% sodium sulfate buffer. The tissues were then processed for routine hematoxylin and eosin staining to assess the general architecture and ONFH. Osteonecrosis was defined, in the present study, as the diffuse presence of empty lacunae or pyknotic nuclei in osteocytes within the bone trabeculae, accompanied by surrounding bone marrow cell necrosis.3, 22, 23
Electrophoretic Mobility Shift Assay
NF-κB and IRF7 activity were assessed by electrophoretic mobility shift assay as described previously.24 In brief, equal amounts of liver nuclear extract (2.0 mg of protein) were incubated for 1 h at room temperature with 32P-labelled NF-κB or IRF7 consensus oligonucleotide probes (5′-AGTTGAGGGGACTTTCCCAGGC-3′ or 5′-ACTGATCGGAACCGAACGATCTATG-3′, respectively) in binding buffer (10 mM HEPES (pH 7.9), 50 mM KCl, 0.2 mM ethylenediaminetetraacetic acid, 2.5 mM dithiothreitol, 10% glycerol, and 0.05% NP-40). The DNA protein complexes were separated on 7% non-denaturing polyacrylamide gels at a constant voltage of 100 V at room temperature. The gels were then exposed to an Image Plate (Fuji Film, Tokyo, Japan) at room temperature. The radioactivity of the DNA-binding complexes in the gel was analyzed using an FLA3000 Image Analyzer (Fuji Film) and ImageQuant Software (Molecular Dynamics, Sunnyvale, CA, USA).
Statistical Analysis
Data represent mean±s.e.m. Comparisons between two groups were performed using the Fisher’s exact test or the unpaired t-test with Welch’s correction. A P-value of<0.05 was considered significant. All statistical analysis was performed using GraphPad Prism 5.0c software for Mac OS X (GraphPad Software, La Jolla, CA, USA).
RESULTS
Administration of the TLR Ligand is required for ONFH to Develop
To evaluate the incidence of ONFH, rats in the Saline+MPSL (n=12) used as a control, Imiquimod+MPSL (n=12) and CpG-C+MPSL (n=12) groups were killed 14 days after the MPSL injection. ONFH was observed in 6 of 12 rats in the Imiquimod+MPSL group and 4 of 12 rats in the CpG-C+MPSL group. By contrast, no ONFH was observed in the Saline+MPSL group (0 of 12) (Table 2). The incidence of ONFH in the Imiquimod+MPSL group was significantly higher from that in the Saline+MPSL group (P<0.01; Fisher’s exact test). Figure 1 shows the histopathological appearance of the femoral head after hematoxylin and eosin staining in the Saline+MPSL (A), Imiquimod+MPSL (B), and CpG-C+MPSL (C) rats. Normal trabeculae as well as hematopoietic and fat cells were observed in the Saline+MPSL rats (Figure 1a). In the Imiquimod+MPSL and CpG-C+MPSL group, empty lacunae and pyknotic nuclei were observed within the necrotic bone trabeculae, and bone marrow cell necrosis was observed in the medullary space across most areas of the femoral head (Figure 1b and c).
Table 2.
Incidence of ONFH within 14 days after last treatment
| 1 | 3 | 7 | 14 days | |
|---|---|---|---|---|
| Saline+MPSL | 0/12 | |||
| CpG-C+MPSL | 4/12 | |||
| Imiquimod+MPSL | 0/10 | 3/10 | 4/10 | 11/22a |
| Imiquimod+Saline | 0/6 | 1/6 | 1/6 | 1/10 b |
Fisher’s exact test.
aP=0.0026 vs Saline+MPSL.
bP=0.02 vs Imiquimod+MPSL.
Figure 1.
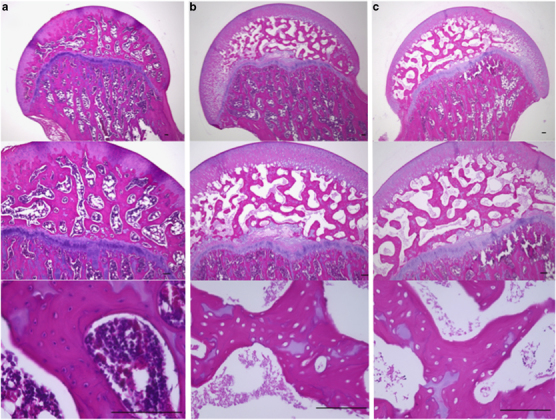
Histological appearance of the femoral head affected by osteonecrosis. Panels show hematoxylin and eosin-stained femurs. Typical images from the Saline+MPSL (a); Imiquimod+MPSL (b); and CpG-C+MPSL (c) groups are shown. The diffuse presence of empty lacunae and pyknotic osteocytic nuclei in the bone trabeculae accompanied by bone marrow cell necrosis was observed in the femoral head of Imiquuuuuimod+MPSL and CpG-C+MPSL groups. Scale bar: 100 m.
Effects of MPSL on Signal Transcription Factor Activity
To elucidate the precise time of onset of ONFH in rats and evaluate the effects of MPSL on the ONFH development, the Imiquimod+Saline (3 × n=6 and n=10) was used as a control group, and it and the Imiquimod+MPSL (4 × n=10) group (which group significantly developed ONFH compared with the Saline+MPSL group) were each divided into four equal subgroups. One subgroup from each treatment group was killd on Day 1, 3, 7, and 14, respectively, after the last treatment to detect changes that led to the development of ONFH. Table 2 shows the incidence of ONFH in each group. ONFH was observed in the Imiquimod+Saline and Imiquimod+MPSL rats killed on Day 3, 7, and 14, respectively. Figure 2 shows the histopathological appearance of the femoral head after hematoxylin and eosin staining in the Imiquimod+Saline rats with ONFH (A) and those without ONFH (B) at 3 days. Empty lacunae were observed within the necrotic bone trabeculae, and bone marrow cells showing fat cell necrosis were observed in the medullary space across most areas of the femoral head as observed in the Imiquimod+MPSL and CpG-C+MPSL rats (Figure 2a). Normal trabeculae as well as hematopoietic and fat cells were observed in the femoral head as observed in the Saline+MPSL rats (Figure 2b). In the present study, we found that ONFH occurs within 3 days after MPSL treatment in rats.
Figure 2.
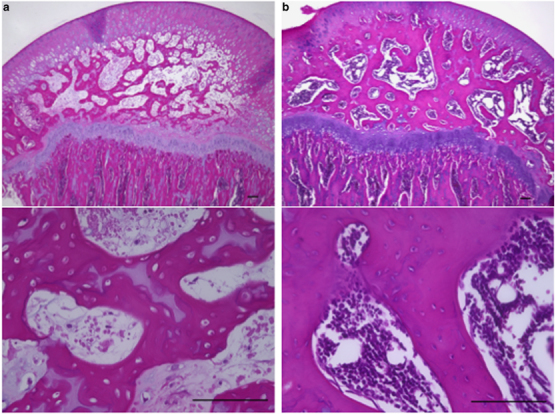
Histological appearance of the femoral head affected by osteonecrosis. Panels show hematoxylin and eosin-stained femurs. Typical images from the Imiquimod+Saline group with ONFH (a) and without ONFH (b) are shown. The diffuse presence of empty lacunae and pyknotic osteocytic nuclei in the bone trabeculae accompanied by bone marrow cell necrosis was also observed in the femoral head of rats with ONFH (a). Scale bar: 100 μm.
Figure 3 shows the activity of the transcription factors NF-κB and IRF7 in the liver. The activity of NF-κB was repressed significantly in rats treated with MPSL at 1 day after the administration of TLR ligands (Figure 3; Imiquimod+Saline vs Imiquimod+MPSL, 100±10.6 vs 69.3±4.9; P<0.01). However, MPSL treatment did not alter IRF7 activity at any phase in either group (Figure 3).
Figure 3.
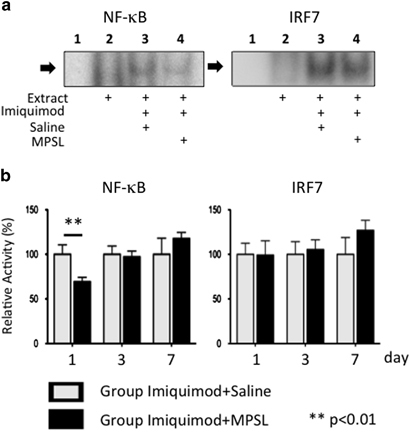
Activity of transcription factors. (a) EMSA for NF-κB and IRF7 in Imiquimod+Saline and Imiquimod+MPSL rats on Day 1. Lane 1 contains no extract. Lane 2 contains extract from untreated rat. Lane 3 contains extract from rat treated with Imiquimod+Saline. Lane 4 contains extract from rat treated with Imiquimod+MPSL. (b) Activity of NF-κB and IRF7. A repression in NF-κB activity was observed at Day 1 after corticosteroid treatment. Data represent the mean±s.e.m. **P<0.01 compared with Imiquimod+Saline rats.
Inhibition of IRF7 by BAY11-7082 Interferes with the Development of ONFH
As described above, we observed changes in transcription factor activity in the liver 24 h after MPSL treatment. We hypothesized that this change might trigger the onset of primary corticosteroid-induced ONFH, and a repression in IRF7 activity by the use of an inhibitor could interfere with the development of ONFH in spite of the simultaneous repression in NF-κB activity, resulting from MPSL treatment. To clarify this hypothesis, the Imiquimod+BAY11+MPSL (3 × n=10 and n=6 receiving 1, 3, 5, or 10 mg/kg BAY11-7082, respectively) groups were killed 14 days after MPSL treatment. Table 3 shows the incidence of ONFH in each group. Co-administration of BAY11-7082 with MPSL was found to interfere with the development ONFH in a dose-dependent manner. In the groups co-administered 5 or 10 mg/kg BAY11-7082, no ONFH was observed after MPSL treatment. The incidence of ONFH in the group co-administered 5 mg/kg BAY11-7082 was significantly less than that in the Imiquimod+MPSL group (P=0.01; Fisher’s exact test). However, 1 of 10 rats died within 7 days after the administration of 1, 3, or 5 mg/kg BAY11-7082, and 3 of 6 rats died within 7 days after the administration of 10 mg/kg BAY11-7082. Owing to the high mortality observed in the BAY11-7082 group, no further experiments were performed using these rats.
Table 3.
Incidence of ONFH 14 days after co-treatment with BAY11-7082
| Imiquimod+BAY11-7082+MPSL | |||||
|---|---|---|---|---|---|
| Imiquimod+MPSL | 1 mg/kg | 3 mg/kg | 5 mg/kg | 10 mg/kg | |
| ONFH | 11/22 | 2/9 | 2/9 | 0/9 | 0/3 |
| P-value | 0.24 | 0.24 | 0.01 | 0.23 | |
Fisher’s exact test vs Group Imiquimod+MPSL.
Figure 4 shows the activity of NF-κB and IRF7 in the liver at 24 h after MPSL treatment in Imiquimod+MPSL (n=8) and Imiquimod+BAY11+MPSL (n=8, 5 mg/kg BAY11-7082) rats. BAY11-7082 administration did not alter the relative activity of NF-κB at 1 day. However, the activity of IRF7 was repressed significantly at 1 day in rats administered BAY11-7082 (Figure 4; Imiquimod+MPSL vs Imiquimod+BAY11+MPSL, 100±9.9 vs 58.9±4.1; P=0.012).
Figure 4.
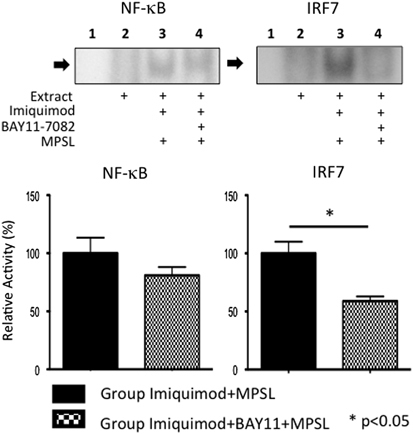
Activity of transcription factors. EMSA for NF-κB and IRF7 in Imiquimod+MPSL and Imiquimod+BAY11+MPSL rats on Day 1. Lane 1 contains no extract. Lane 2 contains extract from untreated rat. Lane 3 contains extract from rat treated with Imiquimod+MPSL. Lane 4 contains extract from rat treated with Imiquimod+BAY11+MPSL. A repression in IRF7 activity was observed at Day 1 after co-administration of BAY11-7082 and corticosteroids. Data represent the mean±s.e.m. **P<0.01 compared with Imiquimod+MPSL rats.
DISCUSSION
Fukushima et al25 conducted a nationwide epidemiologic survey in Japan and reported a high frequency of SLE, nephrotic syndrome, polymyositis/dermatomyositis, bronchial asthma, and thrombocytopenic purpura, as the underlying disease in patients with corticosteroid-induced ONFH. Recently, it was reported that innate immune signaling via TLR7 and TLR9 contributed to the pathogenesis of SLE,9 TLR2, TLR3 and TLR4 signaling contributed to the pathogenesis of nephrotic syndrome,10 TLR3, and TLR7 signaling contributed to the pathogenesis of polymyositis and dermatomyositis,11 TLR4 signaling contributed to the pathogenesis of bronchial asthma,12 and TLR4 and TLR7 signaling contributed to the pathogenesis of thrombocytopenic purpura.13 These reports showed that innate immune signaling via TLRs contributes to the pathogenesis of underlying diseases in patients with corticosteroid-induced ONFH. We previously reported that corticosteroid treatment after a lipopolysaccharide (LPS), a TLR 4 ligand, injection induces ONFH in Wistar rats, and suggested that the TLR4 signaling pathway may have a role in the pathogenesis of ONFH in rats.3, 4, 5 In the present study, we showed that the administration of TLR7 and TLR9 ligands in combination with MPSL treatment resulted in the development of ONFH in rats. In contrast, it was reported that mega-dose corticosteroid therapy, recommended under the national acute spinal cord injury protocol for spinal cord injury, resulting from trauma did not induce ONFH.2 In the present study, ONFH was not observed in the Saline+MPSL group, in which the rats did not receive the TLR ligands, indicating that corticosteroids alone failed to induce ONFH in healthy animals. There are several reports on osteonecrosis animal models using corticosteroid administration alone. Corticosteroid administration alone led to the development of ONFH in spontaneously hypertensive rats (SHRs).26 However, this treatment in the SHRs model is different from corticosteroid administration in healthy animals because of a genetic abnormality in the background of the SHRs.27 Further, it was reported that corticosteroids alone led to the development of ONFH in Leghorn chickens.28 Histopathological examination of specimens in this model showed that the subchondral bone trabeculae in the femoral head had disappeared.28 The diminution of the bone trabeculae was similar in appearance to micro-CT images of an osteoporosis animal model,29 but it was not clear whether it resulted from osteonecrosis. In addition, Yamamoto et al30 reported an osteonecrosis rabbit model in 1997. A characteristic of the rabbit model was that osteonecrosis developed in the metaphysis but not in the femoral head after the treatment of healthy rabbits with corticosteroids. On the other hand, Yamamoto et al30 also reported that corticosteroid treatment after LPS administration led to the development of osteonecrosis in the femoral head and the metaphysis in rabbits in 1995.22 These reports indicate that the administration of TLR ligands has a crucial role in the development of femoral head osteonecrosis. Therefore, present findings suggest that the administration of TLR ligands in combination with subsequent corticosteroid treatment are required to trigger the onset of primary corticosteroid-induced ONFH in rats, and that the pathogenesis of underlying diseases treated with corticosteroids contributes to the development of corticosteroid-induced ONFH. High-dose corticosteroid treatment for severe acute respiratory syndrome (SARS) infection was reported to be a risk factor of ONFH.31 SARS virus is a single-stranded RNA virus, which is one of the ligands of TLR7.32 Consequently, we believe that the pathogenesis of ONFH in Imiquimod+MPSL group resembles the pathogenesis of ONFH after corticosteroid treatment for SARS infection. Although ONFH was not found in the Saline+MPSL group rats, the fact that the Imiquimod+Saline rats, to which no alcohol or corticosteroids were given, developed ONFH at a lower rate indicates that this group resembled idiopathic ONFH. This result suggests that TLR7 signaling contributes to the development of idiopathic ONFH, and indicates that infection by a single-stranded RNA virus, which was one of the ligands for TLR7, could be a risk factor for idiopathic ONFH. Infection with human immunodeficiency virus, a single-stranded RNA virus, has already been reported as an isolated risk factor for idiopathic ONFH.33 Therefore, our results may explain the pathogenesis of idiopathic ONFH in patients infected with single-stranded RNA viruses.
Among the underlying diseases treated with corticosteroids, SLE was reported to be frequently observed in ONFH patients.25 The incidence of ONFH in SLE patients after corticosteroid therapy was reported to be 10–50%.34, 35 In the present study, 33–50% of the Imiquimod+MPSL and CpG-C+MPSL group rats developed ONFH, which is similar to the incidence of ONFH in patients with SLE.
It is well known that corticosteroids are mainly metabolized in the liver by a hepatic corticosteroid-metabolizing enzyme.36, 37 Recently, we reported that differences in liver function were observed between patients with corticosteroid-induced ONFH and those without ONFH within 1 week after corticosteroid therapy.38 Moreover, Ichiseki et al39 reported that a disturbance in oxidative stress in the liver was observed within 24 h in an oxidative stress-induced ONFH rat model. These reports suggest that various changes in the liver in the early phase after treatment contribute to the development of ONFH. In the present study, we found that ONFH occurs within 3 days after MPSL treatment in rats, suggesting that detection of the early phase disease after MPSL treatment is important in clarifying the pathogenesis of ONFH. We also found that the activity of NF-κB in the liver at 1 day was significantly repressed by corticosteroid treatment after TLR7 ligand administration. Auphan et al 17 reported that glucocorticoids are the most potent anti-inflammatory and immunosuppressive agents. Repression of NF-κB activity via glucocorticoid receptors is one critical component of the inhibitory effect that glucocorticoids have on proinflammatory and immune responses.18 Therefore, our results suggest that ONFH results from the activation of NF-κB and IRF7 via the MyD88-dependent pathway, followed by a subsequent repression in NF-κB activity by corticosteroid treatment, whereas IRF7 activity is not affected by corticosteroid treatment. Therefore, this result indicates that a repression in IRF7 activity using a specific inhibitor interferes with the development of ONFH in spite of the simultaneous repression in NF-κB activity resulting from MPSL treatment. BAY11-7082 was reported to be an Ikk α inhibitor and Ikk β inhibitor with potential to inhibit NF-κB and IRF7 activity.21 In the present study, MPSL treatment significantly repressed the activity of NF-κB in Imiquimod+MPSL compared with Imiquimod+Saline rats (Figure 3), and accordingly, the co-administration of BAY11-7082 and MPSL did not repress the activity of NF-κB in Imiquimod+BAY11+MPSL compared with Imiquimod+MPSL rats (Figure 4). In contrast, MPSL treatment did not alter the activity of IRF7 in Imiquimod+MPSL compared with Imiquimod+Saline rats (Figure 3), whereas the co-administration of BAY11-7082 and MPSL significantly repressed the activity of IRF7 in Imiquimod+BAY11+MPSL compared with Imiquimod+MPSL rats (Figure 4). And co-administration of BAY11-7082 and MPSL significantly lowered the incidence of ONFH through a repression in IRF7 activity. These findings confirmed that the effect of corticosteroid treatment on the transcription factor activity in the liver triggered the development of ONFH, and the repression in NF-κB activity by corticosteroid treatment boosted the development of ONFH.
In conclusions, the present study shows that corticosteroid treatment after the administration of TLR7 or TLR9 ligands caused ONFH in rats. Differences in IRF7 and NF-κB activity in the liver induced by corticosteroid treatment triggered the development of ONFH, although it remains unclear how these signal transductions in the liver lead to femoral head osteonecrosis. The present study suggests that the pathogenesis/condition of underlying diseases treated with corticosteroid contributes to the development of ONFH. This might help to explain why ONFH develops or does not develop after corticosteroid therapy of the same dosage. We expect that the normalization of inflammatory status with new biologics for inflammatory diseases can prevent ONFH in the near future.
Acknowledgements
This work was supported in part by the Grants-in-Aid for Young Scientists (B) (SO, 22791390) and for Scientific Research (B) (HM, 20390196) from the Japanese Society for the Promotion of Science, and A-STEP (SO, AS232Z02563G) from Japan Science and Technology Agency.
Competing interests
The authors declare no conflict of interest.
Footnotes
Osteonecrosis of the femoral head (ONFH) commonly occurs after orticosteroid therapy for inflammation. It is known that toll-like receptor (TLR) signaling contributes to inflammation, and that corticosteroids reduce levels of NF-κB. In this study, the authors found that corticosteroid treatment after administration of TLR ligands to rats causes a decrease in NF-κB activity, resulting in ONFH.
References
- 1.Abeles M, Urman JD, Rothfield NF. Aseptic necrosis of bone in systemic lupus erythematosus. Relationship to corticosteroid therapy. Arch Intern Med. 1978;138:750–754. doi: 10.1001/archinte.1978.03630290052018. [DOI] [PubMed] [Google Scholar]
- 2.Wing PC, Nance P, Connell DG. Risk of avascular necrosis following short term megadose methylprednisolone treatment. Spinal Cord. 1998;36:633–636. doi: 10.1038/sj.sc.3100647. [DOI] [PubMed] [Google Scholar]
- 3.Okazaki S, Nishitani Y, Nagoya S. Femoral head osteonecrosis can be caused by disruption of the systemic immune response via the toll-like receptor 4 signalling pathway. Rheumatology. 2009;48:227–232. doi: 10.1093/rheumatology/ken462. [DOI] [PubMed] [Google Scholar]
- 4.Tateda K, Okazaki S, Nagoya S. The suppression of TRIM21 and the accumulation of IFN-alpha play crucial roles in the pathogenesis of osteonecrosis of the femoral head. Lab Invest. 2012;92:1318–1329. doi: 10.1038/labinvest.2012.89. [DOI] [PubMed] [Google Scholar]
- 5.Okazaki S, Nagoya S, Tateda K. Weight bearing does not contribute to the development of osteonecrosis of the femoral head. Int J Exp Pathol. 2012;93:458–462. doi: 10.1111/j.1365-2613.2012.00836.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Medzhitov R, Janeway CA., Jr. Innate immunity: the virtues of a nonclonal system of recognition. Cell. 1997;91:295–298. doi: 10.1016/S0092-8674(00)80412-2. [DOI] [PubMed] [Google Scholar]
- 7.Toubi E, Shoenfeld Y. Toll-like receptors and their role in the development of autoimmune diseases. Autoimmunity. 2004;37:183–188. doi: 10.1080/08916930410001704944. [DOI] [PubMed] [Google Scholar]
- 8.Fischer M, Ehlers M. Toll-like receptors in autoimmunity. Ann N Y Acad Sci. 2008;1143:21–34. doi: 10.1196/annals.1443.012. [DOI] [PubMed] [Google Scholar]
- 9.Christensen SR, Shupe J, Nickerson K. Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity. 2006;25:417–428. doi: 10.1016/j.immuni.2006.07.013. [DOI] [PubMed] [Google Scholar]
- 10.Lichtnekert J, Vielhauer V, Zecher D. Trif is not required for immune complex glomerulonephritis: dying cells activate mesangial cells via Tlr2/Myd88 rather than Tlr3/Trif. Am J Physiol Renal Physiol. 2009;296:F867–F874. doi: 10.1152/ajprenal.90213.2008. [DOI] [PubMed] [Google Scholar]
- 11.Tournadre A, Lenief V, Miossec P. Expression of Toll-like receptor 3 and Toll-like receptor 7 in muscle is characteristic of inflammatory myopathy and is differentially regulated by Th1 and Th17 cytokines. Arthritis Rheum. 2010;62:2144–2151. doi: 10.1002/art.27465. [DOI] [PubMed] [Google Scholar]
- 12.Hammad H, Chieppa M, Perros F. House dust mite allergen induces asthma via Toll-like receptor 4 triggering of airway structural cells. Nat Med. 2009;15:410–416. doi: 10.1038/nm.1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yu H, Liu Y, Han J. TLR7 regulates dendritic cell-dependent B-cell responses through BlyS in immune thrombocytopenic purpura. Eur J Haematol. 2011;86:67–74. doi: 10.1111/j.1600-0609.2010.01534.x. [DOI] [PubMed] [Google Scholar]
- 14.Lovgren T, Eloranta ML, Bave U. Induction of interferon-alpha production in plasmacytoid dendritic cells by immune complexes containing nucleic acid released by necrotic or late apoptotic cells and lupus IgG. Arthritis Rheum. 2004;50:1861–1872. doi: 10.1002/art.20254. [DOI] [PubMed] [Google Scholar]
- 15.Yao Y, Richman L, Higgs BW. Neutralization of interferon-alpha/beta-inducible genes and downstream effect in a phase I trial of an anti-interferon-alpha monoclonal antibody in systemic lupus erythematosus. Arthritis Rheum. 2009;60:1785–1796. doi: 10.1002/art.24557. [DOI] [PubMed] [Google Scholar]
- 16.Guma M, Ronacher LM, Firestein GS. JNK-1 deficiency limits macrophage-mediated antigen-induced arthritis. Arthritis Rheum. 2011;63:1603–1612. doi: 10.1002/art.30271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Auphan N, DiDonato JA, Rosette C. Immunosuppression by glucocorticoids: inhibition of NF-kappa B activity through induction of I kappa B synthesis. Science. 1995;270:286–290. doi: 10.1126/science.270.5234.286. [DOI] [PubMed] [Google Scholar]
- 18.Flammer JR, Rogatsky I. Minireview: glucocorticoids in autoimmunity: unexpected targets and mechanisms. Mol Endocrinol. 2011;25:1075–1086. doi: 10.1210/me.2011-0068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hemmi H, Kaisho T, Takeuchi O. Small anti-viral compounds activate immune cells via the TLR7 MyD88-dependent signaling pathway. Nat Immunol. 2002;3:196–200. doi: 10.1038/ni758. [DOI] [PubMed] [Google Scholar]
- 20.Hemmi H, Takeuchi O, Kawai T. A Toll-like receptor recognizes bacterial DNA. Nature. 2000;408:740–745. doi: 10.1038/35047123. [DOI] [PubMed] [Google Scholar]
- 21.Miyamoto R, Ito T, Nomura S. Inhibitor of IkappaB kinase activity, BAY 11-7082, interferes with interferon regulatory factor 7 nuclear translocation and type I interferon production by plasmacytoid dendritic cells. Arthritis Res Ther. 2010;12:R87. doi: 10.1186/ar3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yamamoto T, Hirano K, Tsutsui H. Corticosteroid enhances the experimental induction of osteonecrosis in rabbits with Shwartzman reaction. Clin Orthop Relat Res. 1995;316:235–243. doi: 10.1097/00003086-199507000-00033. [DOI] [PubMed] [Google Scholar]
- 23.Ichiseki T. Oxidative stress by glutathione depletion induces osteonecrosis in rats. Rheumatology. 2006;45:287–290. doi: 10.1093/rheumatology/kei149. [DOI] [PubMed] [Google Scholar]
- 24.Matsumoto H, Sato Y, Azumi J. Role of endotoxin in NF-kappaB activation by ethanol in rat hepatocytes. Alcohol, Clin Exp Res. 2002;26(8 Suppl):6S–10S. doi: 10.1097/01.ALC.0000026827.79925.C9. [DOI] [PubMed] [Google Scholar]
- 25.Fukushima W, Fujioka M, Kubo T. Nationwide epidemiologic survey of idiopathic osteonecrosis of the femoral head. Clin Orthop Relat Res. 2010;468:2715–2724. doi: 10.1007/s11999-010-1292-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Murata M, Kumagai K, Miyata N. Osteonecrosis in stroke-prone spontaneously hypertensive rats: effect of glucocorticoid. J Orthop Sci. 2007;12:289–295. doi: 10.1007/s00776-007-1129-y. [DOI] [PubMed] [Google Scholar]
- 27.Higaki J, Katsuya T, Morishita R. Symposium on the etiology of hypertension—summarizing studies in 20th century. 1. Hypertension and genes. Intern Med. 2001;40:144–147. doi: 10.2169/internalmedicine.40.144. [DOI] [PubMed] [Google Scholar]
- 28.Erken HY, Ofluoglu O, Aktas M. Effect of pentoxifylline on histopathological changes in steroid-induced osteonecrosis of femoral head: experimental study in chicken. Int Orthop. 2012;36:1523–1528. doi: 10.1007/s00264-012-1497-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nam SH, Jeong JH, Che X. Topically administered Risedronate shows powerful anti-osteoporosis effect in ovariectomized mouse model. Bone. 2012;50:149–155. doi: 10.1016/j.bone.2011.10.017. [DOI] [PubMed] [Google Scholar]
- 30.Yamamoto T, Irisa T, Sugioka Y. Effects of pulse methylprednisolone on bone and marrow tissues: corticosteroid-induced osteonecrosis in rabbits. Arthritis Rheum. 1997;40:2055–2064. doi: 10.1002/art.1780401119. [DOI] [PubMed] [Google Scholar]
- 31.Lv H, de Vlas SJ, Liu W. Avascular osteonecrosis after treatment of SARS: a 3-year longitudinal study. Trop Med Int Health. 2009;14(Suppl 1):79–84. doi: 10.1111/j.1365-3156.2008.02187.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Holmes KV, Enjuanes L. Virology. The SARS coronavirus: a postgenomic era. Science. 2003;300:1377–1378. doi: 10.1126/science.1086418. [DOI] [PubMed] [Google Scholar]
- 33.Ries MD, Barcohana B, Davidson A. Association between human immunodeficiency virus and osteonecrosis of the femoral head. J Arthroplasty. 2002;17:135–139. doi: 10.1054/arth.2002.28727. [DOI] [PubMed] [Google Scholar]
- 34.Matsui M, Saito S, Ohzono K. Experimental steroid-induced osteonecrosis in adult rabbits with hypersensitivity vasculitis. Clin Orthopaedics Relat Res. 1992;277:61–72. [PubMed] [Google Scholar]
- 35.Assouline-Dayan Y, Chang C, Greenspan A. Pathogenesis and natural history of osteonecrosis. Semin Arthritis Rheum. 2002;32:94–124. doi: 10.1053/sarh.2002.3202094. [DOI] [PubMed] [Google Scholar]
- 36.Varis T, Kivisto KT, Backman JT. The cytochrome P450 3A4 inhibitor itraconazole markedly increases the plasma concentrations of dexamethasone and enhances its adrenal-suppressant effect. Clin Pharmacol Ther. 2000;68:487–494. doi: 10.1067/mcp.2000.110772. [DOI] [PubMed] [Google Scholar]
- 37.Tokuhara Y, Wakitani S, Oda Y. Low levels of steroid-metabolizing hepatic enzyme (cytochrome P450 3A) activity may elevate responsiveness to steroids and may increase risk of steroid-induced osteonecrosis even with low glucocorticoid dose. J Orthop Sci. 2009;14:794–800. doi: 10.1007/s00776-009-1400-5. [DOI] [PubMed] [Google Scholar]
- 38.Okazaki S, Nagoya S, Yamamoto M. High risk of osteonecrosis of the femoral head in autoimmune disease patients showing no immediate increase in hepatic enzyme under steroid therapy. Rheumatol Int. 2013;33:51–55. doi: 10.1007/s00296-011-2295-y. [DOI] [PubMed] [Google Scholar]
- 39.Ichiseki T, Kaneuji A, Ueda Y. Osteonecrosis development in a novel rat model characterized by a single application of oxidative stress. Arthritis Rheum. 2011;63:2138–2141. doi: 10.1002/art.30365. [DOI] [PubMed] [Google Scholar]