

Abstract
Background
This is an updated version of the original Cochrane review published in The Cochrane Library 2001, Issue 4.
Nearly a third of people with epilepsy do not have their seizures controlled with current treatments. Continuous attempts have been made to find new antiepileptic drugs based on increasing knowledge of the cellular and molecular biology involved in the genesis of epilepsy and seizures. Therefore, calcium antagonists that can alter the effects of calcium on brain cells have been investigated for their effect on epileptic seizures.
Objectives
To evaluate the effects of calcium antagonists when used as an add‐on therapy for people with drug‐resistant epilepsy.
Search methods
We searched the Cochrane Epilepsy Group Specialized Register (29 January 2013), Cochrane Central Register of Controlled Trials (CENTRAL) (The Cochrane Library 2012, Issue 12), MEDLINE (1948 to 29 January 2013) and SCOPUS (all years to 29 January 2013).
Selection criteria
Randomised placebo‐controlled or active‐controlled add‐on trials of any calcium antagonist in people with drug‐resistant epilepsy.
Data collection and analysis
Two review authors (MH and JP) independently selected trials for inclusion and extracted data. Outcomes investigated included 50% or greater reduction in seizure frequency, treatment withdrawal, adverse effects, cognition and quality of life. Analyses were by intention to treat.
Main results
Eleven trials were included with a total of 424 participants, one parallel‐group and seven cross‐over trials of flunarizine, two cross‐over trials of nimodipine and one cross‐over trial of nifedipine.
For flunarizine, the risk ratio (RR) with 95% confidence interval (CI) for a 50% or greater reduction in seizure frequency in a single parallel trial was 1.53 (95% CI 0.59 to 3.96) indicating a non‐significant advantage of flunarizine. We were unable to acquire data for this outcome from the other seven cross‐over trials. The overall RR for treatment withdrawal of flunarizine was 7.11 (95% CI 1.73 to 29.30) indicating individuals were significantly more likely to have flunarizine withdrawn than placebo. No adverse effects were associated statistically with flunarizine.
For nifedipine, we were unable to acquire the data we required for our specified outcomes.
For nimodipine, we had data only from the first treatment period from one of the two cross‐over trials (17 participants). The RR for a 50% or greater reduction in seizure frequency was 7.78 (99% CI 0.46 to 130.88) and for treatment withdrawal the RR was 2.25 (99% CI 0.25 to 20.38).
Authors' conclusions
Flunarizine may have a weak effect on seizure frequency but had a significant withdrawal rate, probably due to adverse effects, and should not be recommended for use as an add‐on treatment. Similarly, there is no convincing evidence to support the use of nifedipine or nimodipine as add‐on treatments for epilepsy.
Keywords: Humans; Anticonvulsants; Anticonvulsants/therapeutic use; Calcium Channel Blockers; Calcium Channel Blockers/adverse effects; Calcium Channel Blockers/therapeutic use; Drug Resistance; Drug Therapy, Combination; Drug Therapy, Combination/methods; Epilepsy; Epilepsy/drug therapy; Flunarizine; Flunarizine/adverse effects; Flunarizine/therapeutic use; Nifedipine; Nifedipine/therapeutic use; Nimodipine; Nimodipine/therapeutic use; Randomized Controlled Trials as Topic
Plain language summary
Calcium antagonists as an add‐on therapy for drug‐resistant epilepsy
There is no evidence to suggest that calcium antagonists have a useful effect on seizures.
Nearly a third of people with epilepsy become resistant to antiepileptic drugs. Older drugs do not prevent seizures for everyone, and they have adverse effects. A range of new drugs have been tested as 'add‐on' treatments to try and improve the results from antiepileptic drugs. The calcium antagonist drugs flunarizine, nifedipine and nimodipine can be used as an add‐on treatment. The review of trials found no evidence to show a useful effect of these particular calcium antagonist drugs on seizures. Adverse effects of the calcium antagonists reviewed included dizziness, fatigue and unsteadiness (ataxia), however the percentage of adverse events were no more significant than with placebo.
Summary of findings
Summary of findings for the main comparison. Flunarizine versus placebo for drug‐resistant epilepsy.
| Flunarizine versus placebo for drug‐resistant epilepsy | ||||||
| Patient or population: patients with drug‐resistant epilepsy Settings: Out‐patient Intervention: Flunarizine versus placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Control | Flunarizine versus placebo | |||||
| 50% or greater reduction in seizure frequency Number of seizures | 13 per 100 | 20 per 100 (8 to 51) | RR 1.53 (0.59 to 3.96) | 93 (1 study) | ⊕⊕⊕⊕ high | 1 Study did not find a significant difference in seizure reduction |
| Treatment withdrawal Number of withdrawals | 2 per 100 | 12 per 100 (3 to 49) | RR 7.11 (1.73 to 29.30) | 247 (4 studies) | ⊕⊕⊕⊝ moderate1 | 1 study found a significant difference in treatment withdrawal. 3 did not find a significant difference in treatment withdrawal. |
| Adverse effects ‐ Blurred vision No of patients with blurred vision | 6 per 100 | 26 per 100 (5 to 100) | RR 4.09 (0.85 to 19.73) | 93 (1 study) | ⊕⊕⊕⊕ high | 1 study did not find a significant difference in blurred vision. |
| Adverse effects ‐ Dizziness No of patients with dizziness | 19 per 100 | 35 per 100 (14 to 88) | RR 1.82 (0.72 to 4.61) | 93 (1 study) | ⊕⊕⊕⊕ high | 1 study did not find a significant difference in dizziness. |
| Adverse effects ‐ Fatigue No of patients with fatigue | 17 per 100 | 28 per 100 (10 to 79) | RR 1.66 (0.59 to 4.64) | 93 (1 study) | ⊕⊕⊕⊕ high | 1 study did not find a significant difference in fatigue |
| Adverse effects ‐ Irritability No of patients with irritability | 6 per 100 | 20 per 100 (4 to 100) | RR 3.07 (0.6 to 15.68) | 93 (1 study) | ⊕⊕⊕⊕ high | 1 study did not find a significant difference in Irritablity. |
| Adverse effects ‐ Vomiting No of patients with vomiting | 4 per 100 | 17 per 100 (2 to 100) | RR 4.09 (0.57 to 29.16) | 93 (1 study) | ⊕⊕⊕⊕ high | 1 study did not find a significant difference in vomiting. |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio. | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 3 studies did not report outcomes adequately and intention‐to‐treat analysis not employed adequately
Summary of findings 2. Nimodipine versus placebo for drug‐resistant epilepsy.
| Nimodipine versus placebo for drug‐resistant epilepsy | ||||||
| Patient or population: patients with drug‐resistant epilepsy Settings: Out‐patient Intervention: Nimodipine versus placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Control | Nimodipine versus placebo | |||||
| 50% or greater reduction in seizure frequency Number of seizures | 0 per 100 | 0 per 100 (0 to 0) | RR 7.78 (0.46 to 130.88) | 17 (1 study) | ⊕⊕⊕⊝ moderate1 | 1 study did not find a significant difference in seizure reduction. |
| Treatment withdrawal Number of withdrawals | 11 per 100 | 25 per 100 (3 to 100) | RR 2.25 (0.25 to 20.38) | 17 (1 study) | ⊕⊕⊕⊝ moderate1 | 1 study did not find a significant difference in treatment withdrawal. |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio. | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 This study had missing data and did not employ an intention‐to‐treat analysis, dropouts were excluded from the analysis
Background
This review is an update of a previously published review in the Cochrane Database of Systematic Reviews (The Cochrane Library 2001, Issue 4) on 'Calcium antagonists as an add‐on therapy for drug‐resistant epilepsy'. The purpose of this review was to include studies that examined the use of calcium channel antagonists in conjunction with other antiepileptic medications in order to reduce the number of seizures experienced by individuals with epilepsy refractory to first line medications.
Description of the condition
Epilepsy is a chronic neurological condition that results in recurrent spontaneous seizures. The diagnosis is primarily clinical and relies upon taking a history from the patient and eye witnesses about the nature and number of seizures; an electroencephalogram (EEG) can supplement the diagnosis (Smith 2005) and brain imaging might identify the cause. For up to 70% of patients, seizures are controlled with antiepileptic medication. A number of factors have been shown to influence prognosis including gender, treatment history, age, total number of seizures and time from first seizure (Bonnett 2012). For around 30% of patients, seizures continue despite treatment and these patients often experience adverse effects, neuropsychological problems and poor quality of life.
Description of the intervention
Epilepsy can be treated with a number of medications. They are under the umbrella name of 'antiepileptic drugs' (AEDs) but are actually drugs of differing mechanisms which have been shown to be effective in reducing the number of seizures (Nunes 2012). Calcium antagonists can be used as an add‐on treatment with AEDs. They can be administered in tablet form.
How the intervention might work
Abnormalities of the P/Q calcium channel have been implicated in epilepsy (Jouvenceau 2001) and calcium channel antagonists block these channels (Triggle 2007), potentially preventing the occurrence of seizures. Verapamil, one of the calcium antagonists, has an additional putative mechanism in that it inhibits P‐glycoprotein, which is a drug transporter that might pump antiepileptic drugs out of the brain and away from the site of action (Summers 2004).
Why it is important to do this review
Epilepsy specialists are likely to encounter patients with drug‐resistant epilepsy. As such, providing them with an up to date guide to clinical trials measuring the effect of calcium channel antagonists as add‐on therapy will help guide the decision making process.
Objectives
To evaluate the effects of calcium antagonists when used as add‐on therapy for people with drug‐resistant epilepsy.
Methods
Criteria for considering studies for this review
Types of studies
We included studies that were: (a) randomised controlled trials (RCTs) using an adequate method of randomisation; (b) double, single or unblinded trials; (c) placebo controlled; (d) parallel group or cross‐over studies; (e) active controlled i.e. 'head to head' trials.
Types of participants
Individuals of any age with drug‐resistant epilepsy.
As there is no internationally accepted definition of drug‐resistant epilepsy, for the purpose of this review we considered individuals to be drug‐resistant if they had failed to respond to a minimum of two AEDs given as monotherapy.
Types of interventions
The study treatment group received a calcium antagonist in addition to their current conventional antiepileptic drug treatment (i.e. add‐on treatment) for a minimum period of eight weeks.
The control group received a placebo in addition to their current conventional antiepileptic drug treatment for a minimum period of eight week; or
The control group received a differing calcium antagonist to their current conventional antiepileptic drug treatment for a minimum period of eight weeks.
Types of outcome measures
Primary outcomes
50% or greater reduction in seizure frequency
The proportion of individuals with a 50% or greater reduction in seizure frequency in the treatment period compared with the pre‐randomisation baseline period was chosen as our primary outcome. This outcome is commonly reported in this type of study, and can be calculated for studies that do not report this outcome provided that baseline seizure data were recorded.
Secondary outcomes
Treatment withdrawal
The proportion of individuals having treatment (calcium antagonist, placebo or other active calcium antagonist) withdrawn during the treatment period was used as a global measure of tolerability. However, in studies of short duration treatment withdrawal is more likely to be due to adverse effects than lack of efficacy.
Adverse effects
-
Proportion of individuals experiencing any of the following five adverse effects, which we considered to be common and important adverse effects of AEDs:
ataxia;
dizziness;
fatigue (asthenia);
nausea;
somnolence.
The proportion of individuals experiencing the five most common adverse effects, if different from the above (also see methods section).
Cognitive effects
The differences between overall cognition scores, including scores reported of cognitive domains such as:
attention;
executive function;
language;
memory;
visuo‐spatial.
Quality of life
The differences between overall quality of life scores, including scores reported for quality of life domains such as:
social function;
seizure worry;
emotional well‐being;
energy or fatigue;
medication effects.
Search methods for identification of studies
Electronic searches
We searched the Cochrane Epilepsy Group Specialized Register (29 January 2013). In addition, we searched the following databases.
(a) Cochrane Central Register of Controlled Trials (CENTRAL) (The Cochrane Library 2012, Issue 12 of 12) using the search strategy outlined in Appendix 1;
(b) MEDLINE (Ovid) (1948 to 29 January 2013) using the search strategy outlined in Appendix 2;
(c) SCOPUS (all years to 29 January 2013) using the search strategy outlined in Appendix 3.
There were no language restrictions.
Searching other resources
We reviewed the reference lists of included studies to search for additional reports of relevant studies.
Data collection and analysis
Selection of studies
Two review authors (MH and JP) independently assessed articles for inclusion. Any disagreements were resolved through mutual discussion; failing this, a third party opinion was sought. The same review authors independently carried out data extraction and assessed risk of bias. Again, disagreements were resolved by mutual discussion and failing this a third party opinion was sought.
Data extraction and management
The following information was extracted for each trial using a data extraction sheet.
Methodological/trial design
Method of randomisation and allocation concealment
Method of blinding
Number of people excluded from reported analyses
Duration of baseline period
Duration of treatment period
Dose(s) of each calcium antagonist tested
Individual participant/demographic information
Total number of participants allocated to each treatment group
Age and gender
Number of participants with partial or generalized epilepsy
Seizure types.
Seizure frequency during the baseline period.
Number of background drugs.
Where necessary, we contacted original authors to confirm the following information.
The method of randomisation.
The total number randomised to each group.
The number of participants in each group achieving a 50% or greater reduction in seizure frequency per treatment group.
The number of participants having treatment withdrawn post‐randomisation per treatment group.
For those excluded:
the reason for exclusion;
whether any of those excluded completed the treatment phase;
whether any of those excluded had a 50% or greater reduction in seizure frequency during the treatment phase.
Assessment of risk of bias in included studies
Two review authors independently made an assessment of the risk of bias for each trial using the Cochrane risk of bias table as described in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2010). Any disagreements were discussed and resolved. Included studies were rated as adequate, inadequate or unclear on six domains applicable to randomised controlled trials: randomisation method, allocation concealment, blinding methods, incomplete outcome data, selective outcome reporting, and other sources of bias. Summary of findings tables were created where the GRADE approach for assessing quality of evidence was employed.
Measures of treatment effect
The primary outcome of seizure reduction was presented as a risk ratio. Secondary outcomes including treatment withdrawal and adverse effects were presented as risk ratios. The secondary outcomes of cognition and quality of life were to be presented as the standardised mean difference (SMD).
Unit of analysis issues
A range of cognitive and quality of life measures were likely to be utilised in trials. If measures differed across studies, the standardised mean difference would take into account this variance. Cognitive outcome is also likely to vary and close examination of outcomes was to be carried out to ensure the appropriate combination of study data.
Dealing with missing data
Any missing data were sought from the study authors. We carried out intention‐to‐treat, best case and worst case analysis to account for any missing data. All analyses are presented in the main report.
Assessment of heterogeneity
Clinical heterogeneity was assessed by comparing the distribution of important individual participant factors among trials (for example age, seizure type, duration of epilepsy, number of AEDs taken at the time of randomisation) and trial factors (for example randomisation concealment, blinding, losses to follow‐up). Statistical heterogeneity was examined using a Chi2 test and the I2 statistic for heterogeneity and, providing no significant heterogeneity was present (P > 0.1), we employed a fixed‐effect model. In the event heterogeneity was found, a random‐effects model analysis was planned using the inverse variance method.
Assessment of reporting biases
All protocols were requested from study authors to enable comparison of outcomes of interest.
Reporting biases, such as publication bias, were examined by identifying certain aspects of each study (for example sponsors of the research, research teams involved). We intended to examine funnel plots in the event an appropriate number of studies were able to be combined.
Data synthesis
A fixed‐effect model meta‐analysis was employed to synthesise the data. Comparisons we expected to carry out included:
intervention group versus controls on seizure reduction;
intervention group versus controls on treatment withdrawal;
intervention group versus controls on adverse effects;
intervention group versus controls on cognitive outcome;
intervention group versus controls on quality of life.
Each comparison was to be stratified by type of control group, that is placebo or active control, and study characteristics to ensure the appropriate combination of study data.
Our preferred estimator was the Mantel‐Haenzsel risk ratio (RR). For the outcomes 50% or greater reduction in seizure frequency and treatment withdrawal, we used 95% confidence intervals (CIs). For individual adverse effects we used 99% CIs to make an allowance for multiple testing.
Our analyses included all participants in the treatment group to which they had been allocated. For the efficacy outcome (50% or greater reduction in seizure frequency) we undertook three analyses.
Primary (intention‐to‐treat (ITT)) analysis: participants not completing follow‐up or with inadequate seizure data were assumed non‐responders. To test the effect of this assumption, we undertook the following sensitivity analyses. Analysis by ITT was done where this was reported by the included studies.
Worst case analysis: participants not completing follow‐up or with inadequate seizure data were assumed to be non‐responders in the calcium antagonist group, and responders in the placebo group.
Best case analysis: participants not completing follow‐up or with inadequate seizure data were assumed to be responders in the calcium antagonist group, and non‐responders in the placebo group.
Data from cross‐over and parallel studies were not combined together in meta‐analyses due to the high possibility of carry‐over or period effects. Within the included cross‐over studies it was difficult to rule out the existence of this bias. Half of the studies (Battaglia 1991; Fröscher 1988; Keene 1989; Moglia 1986; Overweg 1984; Pelliccia 1993; Starreveld 1989 reported either no washout period or a very short one (two weeks), and only three of the 11 studies (Alving 1989; Battaglia 1991; Starreveld 1989) performed any statistical analysis to test for the existence of a carry‐over or period effect. Therefore, the choice was taken not to combine the data in a meta‐analysis.
Subgroup analysis and investigation of heterogeneity
Subgroup analysis was undertaken for adverse effects. We intended to investigate heterogeneity using sensitivity analysis if deemed appropriate.
Sensitivity analysis
We also intended to carry out sensitivity analysis if peculiarities were found between study quality, characteristics of participants, interventions and outcomes.
Results
Description of studies
Results of the search
The search revealed 422 records identified from the databases outlined in Electronic searches. Three hundred and two records remained after duplicates were removed, all were screened for inclusion in the review. Two hundred and eighty‐three records were excluded at this point leaving 19 full‐text articles to be assessed for eligibility. Following this seven trials were excluded (see Figure 1 and Characteristics of excluded studies for reasons for exclusion). A total of 11 studies were included in the review.
1.
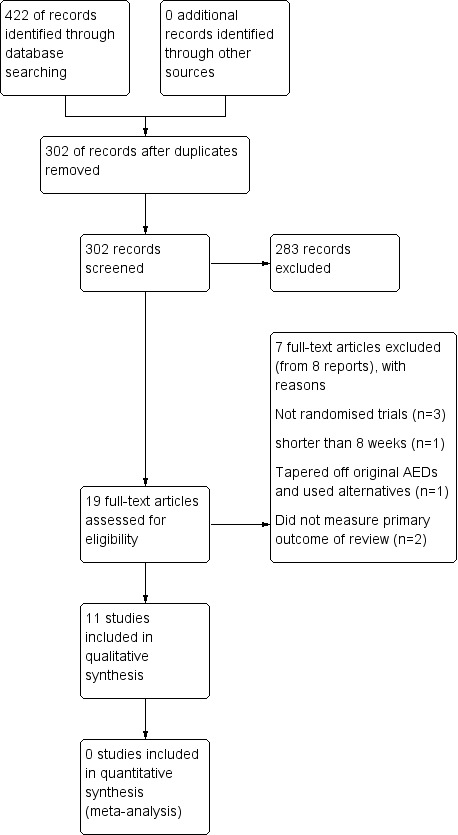
Study flow diagram.
Included studies
There were eight trials included which compared the calcium antagonist flunarizine to placebo, two trials which compared nimodipine to placebo, and one trial which compared nifedipine to placebo. Overall a total of 424 patients with drug‐resistant epilepsy took part in the 11 trials.
Eight trials (representing 363 individuals) compared flunarizine with placebo (Alving 1989; Battaglia 1991; Fröscher 1988; Keene 1989; Moglia 1986; Overweg 1984; Pledger 1994; Starreveld 1989). Pledger 1994 was a parallel‐group trial recruiting 93 individuals and provided data for all outcomes investigated in this review. The remaining seven trials were cross‐over trials, however we were unable to obtain data from the first treatment phase for 50% reduction in seizure frequency or adverse effects. Data for treatment withdrawal were available from only three of these seven cross‐over trials (Fröscher 1988; Keene 1989; Moglia 1986) (247 individuals). In view of this difficulty, we decided to summarize the reported results of all of the cross‐over trials meeting our inclusion criteria in tables and in the text of this review.
The trial comparing nifedipine to placebo (Larkin 1992) was a cross‐over trial that recruited 22 individuals. Although 50% or greater reduction in seizure frequency, treatment withdrawal and adverse effects were reported we were unable to acquire data for the first treatment period. As for the flunarizine trials, results of this trial are presented in tables.
The two trials that compared nimodipine with placebo (Larkin 1991; Pelliccia 1993) were also cross‐over trials and recruited a total of 39 individuals. We were able to obtain data for the first treatment phase from only one of these trials (Pelliccia 1993) (17 individuals) for the outcomes investigated in this review. For the other trial (Larkin 1991) we were unable to obtain data from the first treatment period. We have tabulated the reported results of both cross‐over trials.
Excluded studies
We excluded seven trials. Cavazzuti 1986 was a cross‐over trial (14 individuals) that compared flunarizine with placebo. We excluded this trial because some participants received the study medication for less than eight weeks. The other three excluded trials (195 individuals) compared nimodipine with placebo (Malashkhia 1996; Meyer 1995; Sasso 1993). One trial (Malashkhia 1996) gradually tapered off all previous AEDs then randomised individuals into phenobarbital plus nimodipine or phenobarbital alone, and the other two trials (Meyer 1995; Sasso 1993) did not investigate 50% or greater reduction in seizure frequency. Binnie 1985 did not have a randomisation process and included patients from three arms of a previous study. Handforth 1995 was an open‐label extension study and Treiman 1993 was a non‐randomised concentration‐controlled study.
Risk of bias in included studies
See Figure 2 for a summary of the risk of bias in the included studies.
2.
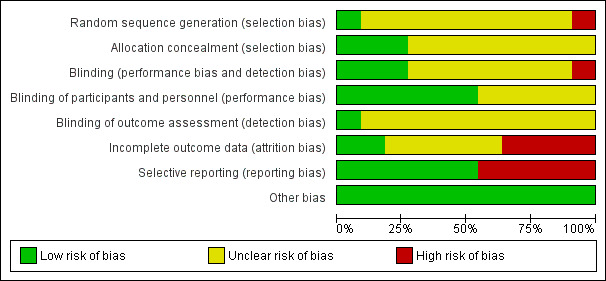
Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
Allocation
The method of randomisation used by Pledger 1994 was a computer‐generated stratified random list. Concealment of allocation was achieved by giving that list to an unblinded pharmacist, who dispensed consecutive blinded treatments accordingly. For one trial (Pelliccia 1993), individuals were allocated alternately to one treatment or the other, a method of allocation concealment that has a recognizable pattern and is not truly random. We assigned an unclear risk of bias to it.
Treatment allocation and allocation concealment methods were not described for the remaining trials (Alving 1989; Battaglia 1991; Fröscher 1988; Keene 1989; Larkin 1991; Larkin 1992; Moglia 1986; Overweg 1984; Starreveld 1989). The authors were contacted by previous writers of this review, however no response was received.
Blinding
Six of the trials were blinded by using matching placebo and active treatment (Alving 1989; Fröscher 1988; Keene 1989; Larkin 1991; Larkin 1992; Pelliccia 1993). This was graded as being adequate blinding for both key personnel and the patients involved, but no information was provided as to how outcome assessors were blinded and as such it was graded as at unclear risk of bias. The methods of blinding in the remaining trials (Battaglia 1991; Moglia 1986; Overweg 1984; Starreveld 1989) were not described and they were graded as having an unclear risk of bias for key study personnel, patients and outcome assessors.
Incomplete outcome data
Ten of the trials reported attrition but did not perform an intention‐to‐treat analysis (Alving 1989; Battaglia 1991; Fröscher 1988; Larkin 1991; Larkin 1992; Moglia 1986; Overweg 1984; Pelliccia 1993; Pledger 1994; Starreveld 1989). Certain studies (Alving 1989; Battaglia 1991; Fröscher 1988; Larkin 1991; Moglia 1986; Overweg 1984; Pelliccia 1993) lost a significant number of patients and not performing an intention‐to‐treat analysis subjected them to a high risk of bias. Larkin 1992 lost a small percentage of patients and not performing an intention‐to‐treat analysis would not have impacted the findings significantly. Two trials (Keene 1989; Pledger 1994) contained no missing data and thus intention‐to‐treat analysis was not deemed necessary, and these trials were graded as having a low risk of bias.
Selective reporting
The studies by Fröscher 1988; Keene 1989; Overweg 1984; and Starreveld 1989 did not include the first phase data from their trials thus posing a high risk of reporting bias for both primary and secondary outcomes. As for Moglia 1986, the results reported were regarding the overall incidence of seizure and certain side effects of the medication. This meant a deviation from the primary outcome 50% reduction in seizures. The secondary outcomes were incomplete and no figures were presented.
Other potential sources of bias
No other biases were detected in the included studies.
Effects of interventions
Studies comparing flunarizine to placebo
A 50% or greater reduction in seizure frequency
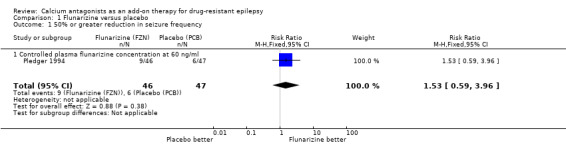
Data for this outcome were only provided from one trial (Pledger 1994).
(i) Intention‐to‐treat analysis The RR for this study was 1.53 (95% CI 0.59 to 3.96) indicating a non‐significant advantage for flunarizine.
(ii) Best case and worst case scenarios As no individuals were excluded from analyses, we did not undertake any best and worst case analyses.
We were unable to acquire the data for this outcome for the first treatment phase from the other seven cross‐over trials (Alving 1989; Battaglia 1991; Fröscher 1988; Keene 1989; Moglia 1986; Overweg 1984; Starreveld 1989). Although all the studies reported a 50% or greater reduction in seizure frequency on flunarizine compared with placebo, only two studies (Alving 1989; Overweg 1984) showed a statistically significant reduction. The remaining trials did not report a statistical analysis for this outcome. Some of the cross‐over trials reported the overall reduction in seizure frequency on flunarizine compared with placebo. In four trials no significant difference was found (Alving 1989; Battaglia 1991; Fröscher 1988; Starreveld 1989). In one cross‐over trial no statistical analysis was reported (Keene 1989) but eight people taking placebo had a 50% or greater reduction in seizure frequency compared with five on flunarizine. In the remaining trial (Moglia 1986), a statistically significant reduction (P = 0.001) in seizure frequency was found but the effect was small. For example, in individuals allocated placebo in the first treatment period and flunarizine in the second, the mean monthly seizure frequency on placebo was 5.13, which dropped to 3.56 on flunarizine. We have summarized the results of individual trials in Table 3.
1. 50% responders and treatment withdrawal outcomes of cross‐over trials.
| Trial | Participants (n) | 50% responder (n) |
| Alving 1989 (FZN) | Total randomised participants = 29, completing study = 22, having treatment withdrawn = 7, excluded from analysis = 7. | 4 participants had 50% SZ reduction on FZN. Overall no significant reduction in SZ frequency on FZN compared with PCB. |
| Battaglia 1991 (FZN) | Total randomised participants = 20, completing study = 13, having treatment withdrawn = 7, excluded from analysis = 7. | 1 participant had 50% SZ reduction on FZN. Overall no significant reduction in SZ frequency on FZN compared with PCB. |
| Fröscher 1988 (FZN) | Total randomised participants = 30, completing study = 22, having treatment withdrawn = 8, excluded from analysis = 8. | PCB‐FZN sequence (n = 13): 2 patients had 50% SZ reduction on FZN. Overall significant reduction in SZ frequency on FZN compared with PCB. // FZN‐PCB sequence (n = 9): 1 pt had 50% SZ reduction while 5 had SZ increase on FZN. Overall no significant SZ reduction on FZN compared with PCB. |
| Keene 1989 (FZN) | Total randomised participants = 34, completing study = 34, having treatment withdrawn = 0, excluded from analysis = 0. | 5 participants had 50% SZ reduction on FZN. 8 participants had 50% SZ reduction on PCB compared with baseline. |
| Moglia 1986 (FZN) | Total randomised participants = 90, completing study = 90, having treatment withdrawn = 28, excluded from analysis = 28. | 14 participants had 50% SZ reduction on FZN. Overall significant SZ reduction on FZN compared with baseline. |
| Overweg 1984 (FZN) | Total randomised participants = 33, completing study = 30, having treatment withdrawn = 3, excluded from analysis = 3. | 7 participants had 50% SZ reduction while 9 participants had SZ increase on FZN. Overall significant SZ reduction on FZN compared with PCB. |
| Starreveld 1989 (FZN) | Total randomised participants = 34, completing study = 25, having treatment withdrawn = 0, excluded from analysis = 9. | 5 participants had 50% SZ reduction on FZN. Overall no significant SZ reduction on FZN compared with PCB. |
| Larkin 1992 (NFD) | Total randomised participants = 22, completing study = 21, having treatment withdrawn = 1, excluded from analysis = 1. | 2 participants had 50% SZ reduction on NFD. 2 participants had 50% SZ reduction on PCB compared with baseline. No significant difference in SZ reduction between NFD and PCB. |
| Larkin 1991 (NMD) | Total randomised participants = 22, completing study = 17, having treatment withdrawn = 5, excluded from analysis = 5. | 7 participants had 50% reduction in partial SZ, 1 had 50% reduction in tonic‐clonic SZ on NMD. 5 participants had 50% reduction in partial SZ, 1 had 50% reduction in tonic‐clonic SZ on PCB compared with baseline. No significant difference in SZ reduction between NMD and PCB. |
FZN: flunarizine NFD: nifedipine NMD: nimodipine PCB: placebo
SZ: seizure
Treatment withdrawal
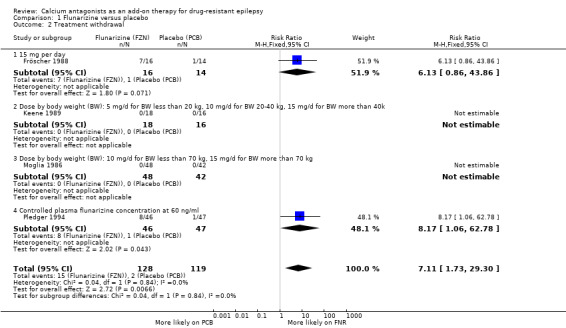
We had data for this outcome from four trials (Fröscher 1988; Keene 1989; Moglia 1986; Pledger 1994). A Chi2 test for heterogeneity suggested no significant statistical heterogeneity (Chi2 = 0.04, df = 1, P = 0.84). Individuals were significantly more likely to have flunarizine withdrawn than placebo, and the RR for treatment withdrawal was 7.11 95% CI 1.73 to 29.30). We were unable to obtain data for this outcome from the first treatment period of four cross‐over trials (Alving 1989; Battaglia 1991; Overweg 1984; Starreveld 1989).
Adverse effects
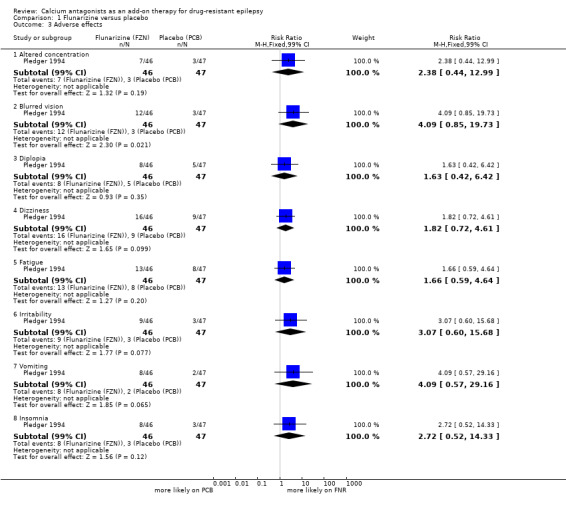
We were unable to obtain data from the first treatment period of the cross‐over trials, hence we were only able to analyse data from one trial (Pledger 1994). In addition to reports of dizziness, fatigue and nausea (vomiting), the five most common adverse effects were altered concentration, blurred vision, diplopia (double vision), irritability and insomnia, and these were included in our analysis. All these adverse effects were associated with flunarizine. However, their 99% CIs included unity indicating no statistical significance: altered concentration RR 2.38 (99% CI 0.44 to 12.99); blurred vision RR 4.09 (99% CI 0.85 to 19.73); diplopia RR 1.63 (99% CI 0.42 to 6.42); dizziness RR 1.82 (99% CI 0.72 to 4.61); fatigue RR 1.66 (99% CI 0.59 to 4.64); irritability RR 3.07 (99% CI 0.60 to 15.68); vomiting RR 4.09 (99% CI 0.57 to 29.16), and insomnia RR 2.72 (99% CI 0.52 to 14.33). In the cross‐over trials, various adverse effects were reported but none were reported as being significantly associated with flunarizine. We summarized these results in Table 4.
2. Numbers of participants experiencing adverse effects in cross‐over trials.
| Adverse effects | Alving 1989(n = 29) | Battaglia 1991(n = 20) | Fröscher 1988(n = 30) | Keene 1989(n = 34) | Moglia 1986(n = 90) | Overweg 1984(n = 23) | Starreveld 1989(n = 34) | Larkin 1992(n = 22) | Larkin 1991(n = 22) |
| Change in mood | FZN = 4 : PCB = 2 | NFD = 0 : PCB = 1 | |||||||
| Change in appetite | FZN = 7 : PCB = 6 | FZN = PCB | NFD = 0 : PCB = 1 | ||||||
| Drowsiness | FZN = 1 : PCB = 0 | FZN = 16 : PCB = 0 | FZN = 1 : PCB = 0 | FZN = PCB | NFD = 0 : PCB = 1 | ||||
| Dry mouth | NFD = 1 : PCB = 1 | ||||||||
| Fatigue | FZN = 8 : PCB = 4 | ||||||||
| Headache | FZN = 4 : PCB = 5 | FZN = PCB | NFD = 2 : PCB = 0 | ||||||
| Irritability | FZN = 9 : PCB = 5 | ||||||||
| Poor memory | NFD = 1 : PCB = 0 | ||||||||
| Tiredness | Approximately the same extent in both FZN and PCB | ||||||||
| Tremor | FZN = 3 : PCB = 2 | ||||||||
| Vertigo | FZN = PCB | ||||||||
| Weight change | FZN = PCB | ||||||||
| Other | FZN = 8 : PCB = 8 | No significant side effects were reported by any patients during the FZN phase compared with the PCB phase. | Neither NMD nor PCB significantly changed the severity of the following side effects compared with baseline period: agitation, double vision, flushing, headache, itching, nausea, palpitation, poor concentration, sedation, unsteadiness. |
FZN: flunarizine NFD: nifedipine NMD: nimodipine PCB: placebo
Study comparing nifedipine to placebo
Nifedipine was compared with placebo in one cross‐over trial (Larkin 1992) in which 22 participants were recruited. For this trial we were unable to obtain data from the first treatment period on 50% or greater reduction in seizure frequency or adverse effects. The trialists did report 50% or greater reduction in seizure frequency and no significant difference between nifedipine and placebo was found. Similarly, no adverse effects were significantly associated with nifedipine. Results are summarized in Table 3 and Table 4. No individuals had treatment withdrawn while taking nifedipine and one person had placebo withdrawn.
Studies comparing nimodipine to placebo
Of the two cross‐over trials comparing nimodipine and placebo (Larkin 1991; Pelliccia 1993), we were able to obtain data from the first treatment period for only one (Pelliccia 1993), which recruited 17 participants. Larkin 1991 reported an analysis for 50% or greater reduction in seizure frequency, adverse effects and treatment withdrawal, but data for the first treatment period were not available.
50% or greater reduction in seizure frequency
(i) Intention‐to‐treat analysis
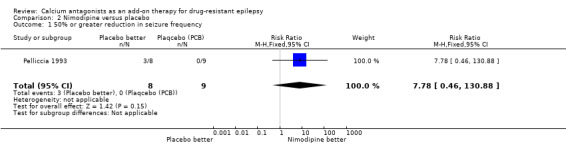
Three of eight participants allocated flunarizine and none of those allocated placebo had a 50% or greater reduction in seizure frequency. This gave a RR of 7.78 (99% CI 0.46 to 130.88) suggesting an advantage for nimodipine. However, given the small number of individuals recruited into this trial, the data were insufficient to conclude with confidence that nimodipine had an effect on seizure frequency.
This outcome was investigated in the cross‐over trial reported by Larkin 1991 and no significant difference between nimodipine and placebo was found (Table 3).
(ii) Best case and worst case scenarios
As no participants were excluded from analyses, we did not undertake any best and worst case analyses.
Treatment withdrawal
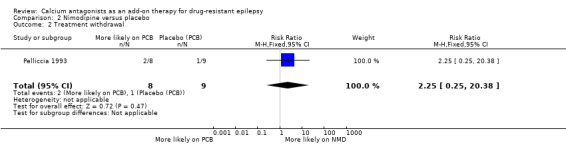
Two of eight participants allocated nimodipine and one of nine allocated placebo had treatment withdrawn. This gave a RR of 2.25 (99% CI 0.25 to 20.38) suggesting a higher but non‐significant withdrawal rate on nimodipine.
Adverse effects
The adverse effects reported in this trial were ataxia, dystonia, hypotension, irritability, somnolence and tremor. Each adverse effect was experienced by one of eight participants allocated to the nimodipine group but by none of the individuals allocated to placebo. The number of individuals recruited into this trial and the number experiencing adverse effects were too small to allow any firm conclusions about the adverse effects associated with nimodipine.
In the trial by Larkin 1991, no adverse effects significantly associated with nimodipine were reported. Results are summarized in Table 4.
Discussion
On the whole the trials included in this review were small, the largest being a parallel‐group trial (Pledger 1994) recruiting 93 participants while the remainder were two‐period cross‐over trials recruiting between 17 and 90 participants. The descriptions of important methodology in the trial reports were poor. One trial report described a method of allocation concealment which was adequate (Pledger 1994), while a second described a method which was inadequate (Pelliccia 1993) leaving the trial subject to bias. For the remaining nine trials, the method of allocation concealment was not described and the authors have not responded to our correspondence asking for clarification. We are therefore left uncertain as to whether nine of the 11 included trials had been subject to allocation bias. All the trials were double‐blinded and placebo controlled. For seven trials (Alving 1989; Fröscher 1988; Keene 1989; Larkin 1991; Larkin 1992; Pelliccia 1993; Pledger 1994) adequate blinding methods were described, whereas methods were not described for the other four trials (Battaglia 1991; Moglia 1986; Overweg 1984; Starreveld 1989).
The majority of the trials included in this review were cross‐over trials. The use of cross‐over trials in this scenario is questionable, particularly for flunarizine which has an elimination half life of around 18 days (Reynolds 1993). Furthermore, flunarizine was still detected in the serum four months after taking the last dose of this drug in four of nine participants allocated to the sequence of flunarizine then placebo in one of the trials included in this review (Fröscher 1988). It is therefore likely that the cross‐over trials included in this review have been subject to carry‐over effects. On the whole, this would lead to an underestimate of the effect of flunarizine, nifedipine and nimodipine on seizures, and an underestimate of the occurrence of their adverse effects. The analysis we proposed in this review, using the first treatment period as a parallel‐group trial, would not have been subject to this bias. However, for the majority of trials we were unable to acquire the required data.
For our primary outcome, 50% or greater reduction in seizure frequency, flunarizine was found to be significantly better than placebo in two out of seven cross‐over trials, while the parallel‐group trial found no significant difference. Due to difficulties obtaining data from the first treatment period from the cross‐over trials, we were unable to undertake a meta‐analysis. The overall results of these trials do not provide convincing evidence of an effect of flunarizine on seizure frequency, and the estimates of effect are on the whole small. Similarly we have found no convincing evidence of an effect on seizure frequency for nifedipine and nimodipine.
For our outcome treatment withdrawal, flunarizine was seven times more likely to be withdrawn than placebo. The adverse effects with flunarazine were not statistically significantly more than with placebo. As such, withdrawal could be due to the small sample size in the studies. We have insufficient data to conclude whether nifedipine or nimodipine were more likely to be withdrawn than placebo. The confidence intervals around the estimates were however wide, and there could be substantial withdrawal rates.
We found no adverse effects that were significantly associated with any of the calcium antagonists, which probably represents the small amount of data reviewed. We are, therefore, unable to describe adverse effect profiles for any of the drugs reviewed.
No data were available for quality of life and cognitive effect outcomes for any trials of flunarizine, nimodipine and nifedipine.
Quality of the evidence
We used the GRADE approach to assess quality of evidence, and this is presented in Table 1; Table 2. For the comparison of flunarizine versus placebo, the one study by Pledger 1994 for which risk ratios (RRs) were calculated was rated as high in quality of evidence as the study was a well‐conducted randomised controlled trial. For the outcome of treatment withdrawal four studies were combined and the evidence was rated as moderate as three of the four studies were at a high risk of bias for outcome reporting. The study which examined nifedipine versus placebo was downgraded to moderate for the inadequate methods used to deal with missing data.
Authors' conclusions
Implications for practice.
Our results indicate that flunarizine may have a weak effect on seizures, but it had a significant withdrawal rate which probably represents problems with tolerability. Similarly, there is insufficient evidence of effect to recommend the use of nifedipine or nimodipine as antiepileptic drugs. Since only one study was included in the meta‐analysis it would be appropriate to conclude that the evidence is inconclusive for the use of these drugs as adjunct therapy for drug‐resistant epilepsy.
Implications for research.
Given that the estimates of effect for the calcium antagonists reviewed were low, and that there are an ever increasing number of antiepileptic drugs for the clinician and patient to choose from, we do not think it is reasonable to undertake further trials with these drugs in people with epilepsy at this time. However, other calcium channel antagonists such as verapamil currently have no data on their use as adjunct therapy for drug‐resistant epilepsy. As such it would be appropriate to test them.
Most of the trials reviewed were cross‐over trials and some of the problems with this trial design have been highlighted. We would recommend future antiepileptic drug trials to be large parallel‐group trials and to examine the effects on patient‐centered outcomes such as cognition and quality of life, two important outcomes which were not investigated in any of the included trials.
What's new
| Date | Event | Description |
|---|---|---|
| 29 January 2013 | New search has been performed | Searches updated 29 January 2013; no new trials identified. |
| 22 January 2013 | New citation required but conclusions have not changed | Conclusions remain the same. |
History
Protocol first published: Issue 4, 2000 Review first published: Issue 4, 2001
| Date | Event | Description |
|---|---|---|
| 7 October 2009 | New search has been performed | Searches updated 7 October 2009; no new trials identified. |
Acknowledgements
We acknowledge Drs Rungsan Chaisewikul and Nick Baillie for conducting the previous reviews.
Appendices
Appendix 1. CENTRAL search strategy
#1 (epilep*) or (seizure*) or (convuls*)
#2 MeSH descriptor Epilepsy explode all trees
#3 MeSH descriptor Seizures explode all trees
#4 (#1 OR #2 OR #3)
#5 MeSH descriptor Calcium Channel Blockers explode all trees
#6 calcium NEXT channel NEXT blocker*
#7 (calcium NEXT antagonist*)
#8 (amlodipine) or (amrinone) or (bencyclane) or (bepridil) or (cinnarizine)
#9 (conotoxins) or (diltiazem) or (felodipine) or (fendiline) or (flunarazine)
#10 (gallopamil) or (isradipine) or (lidoflazine) or (mibefradil) or (nicardipine)
#11 (nifedipine) or (nimodipine) or (nisoldipine) or (nitrendipine) or (perhexiline)
#12 (prenylamine) or (verapamil)
#13 (#5 OR #6 OR #7 OR #8 OR #9 OR #10 OR #11 OR #12)
#14 (#4 AND #13)
Appendix 2. MEDLINE search strategy
For the most recent update of this review (January 2013) we used the following search strategy which is based on the Cochrane Highly Sensitive Search Strategy for identifying randomised trials published in Lefebvre 2011.
1. exp Calcium Channel Blockers/
2. ((calcium adj channel adj blocker$) or (calcium adj antagonist$) or amlodipine or amrinone or bencyclane or bepridil or cinnarizine or conotoxins or diltiazem or felodipine or fendiline or flunarizine or gallopamil or isradipine or lidoflazine or mibefradil or nicardipine or nifedipine or nimodipine or nisoldipine or nitrendipine or perhexiline or prenylamine or verapamil).tw.
3. 1 or 2
4. (randomised controlled trial or controlled clinical trial).pt. or (randomised or placebo or randomly).ab.
5. clinical trials as topic.sh.
6. trial.ti.
7. 4 or 5 or 6
8. exp animals/ not humans.sh.
9. 7 not 8
10. exp Epilepsy/
11. exp Seizures/
12. (epilep$ or seizure$ or convuls$).tw.
13. 10 or 11 or 12
14. 9 and 13
15. 3 and 14
Appendix 3. SCOPUS search strategy
(((TITLE‐ABS‐KEY(epilep* OR "infantile spasm")) OR ((TITLE‐ABS‐KEY(seizure* OR convuls*) AND NOT TITLE‐ABS‐KEY(*eclampsia)) AND SUBJAREA(mult OR agri OR bioc OR immu OR neur OR phar OR mult OR medi OR nurs OR vete OR dent OR heal))
OR ((TITLE‐ABS‐KEY(syndrome) AND TITLE‐ABS‐KEY(aicardi OR angelman OR doose OR dravet OR "landau kleffner" OR "lennox gastaut" OR ohtahara OR panayiotopoulos OR rasmussen OR rett OR "sturge weber" OR "unverricht lundborg" OR west)) AND SUBJAREA(mult OR agri OR bioc OR immu OR neur OR phar OR mult OR medi OR nurs OR vete OR dent OR heal))
OR (TITLE‐ABS‐KEY("ring chromosome 20" OR "R20" OR "myoclonic encephalopathy" OR "pyridoxine dependency") AND SUBJAREA(mult OR agri OR bioc OR immu OR neur OR phar OR mult OR medi OR nurs OR vete OR dent OR heal)))
AND
(((TITLE‐ABS‐KEY(trial AND random*) AND NOT KEY(nonhuman) AND NOT KEY(animals)) AND SUBJAREA(mult OR agri OR bioc OR immu OR neur OR phar OR mult OR medi OR nurs OR vete OR dent OR heal))
OR (TITLE‐ABS‐KEY((controlled OR placebo OR cluster OR "head to head" OR factorial OR crossover OR "cross over" OR "double blind*" OR "single blind*" OR "multi arm") PRE/2 (trial OR method OR procedure)) AND NOT KEY(nonhuman) AND NOT KEY(animals) AND SUBJAREA(mult OR agri OR bioc OR immu OR neur OR phar OR mult OR medi OR nurs OR vete OR dent OR heal))))
AND
((TITLE‐ABS‐KEY("calcium channel block*" OR "calcium antagonis*") AND SUBJAREA(mult OR agri OR bioc OR immu OR neur OR phar OR mult OR medi OR nurs OR vete OR dent OR heal))
OR ((TITLE‐ABS‐KEY(amlodipine OR amrinone OR bencyclane OR bepridil OR cinnarizine OR conotoxins OR diltiazem OR felodipine OR fendiline OR flunarizine OR gallopamil OR isradipine OR lidoflazine OR mibefradil OR nicardipine OR nifedipine OR nimodipine OR nisoldipine)
OR TITLE‐ABS‐KEY(nitrendipine OR perhexiline OR prenylamine OR verapamil)) AND SUBJAREA(mult OR agri OR bioc OR immu OR neur OR phar OR mult OR medi OR nurs OR vete OR dent OR heal)))
Data and analyses
Comparison 1. Flunarizine versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 50% or greater reduction in seizure frequency | 1 | 93 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.53 [0.59, 3.96] |
| 1.1 Controlled plasma flunarizine concentration at 60 ng/ml | 1 | 93 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.53 [0.59, 3.96] |
| 2 Treatment withdrawal | 4 | 247 | Risk Ratio (M‐H, Fixed, 95% CI) | 7.11 [1.73, 29.30] |
| 2.1 15 mg per day | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 6.13 [0.86, 43.86] |
| 2.2 Dose by body weight (BW): 5 mg/d for BW less than 20 kg, 10 mg/d for BW 20‐40 kg, 15 mg/d for BW more than 40k | 1 | 34 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 2.3 Dose by body weight (BW): 10 mg/d for BW less than 70 kg, 15 mg/d for BW more than 70 kg | 1 | 90 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 2.4 Controlled plasma flunarizine concentration at 60 ng/ml | 1 | 93 | Risk Ratio (M‐H, Fixed, 95% CI) | 8.17 [1.06, 62.78] |
| 3 Adverse effects | 1 | Risk Ratio (M‐H, Fixed, 99% CI) | Subtotals only | |
| 3.1 Altered concentration | 1 | 93 | Risk Ratio (M‐H, Fixed, 99% CI) | 2.38 [0.44, 12.99] |
| 3.2 Blurred vision | 1 | 93 | Risk Ratio (M‐H, Fixed, 99% CI) | 4.09 [0.85, 19.73] |
| 3.3 Diplopia | 1 | 93 | Risk Ratio (M‐H, Fixed, 99% CI) | 1.63 [0.42, 6.42] |
| 3.4 Dizziness | 1 | 93 | Risk Ratio (M‐H, Fixed, 99% CI) | 1.82 [0.72, 4.61] |
| 3.5 Fatigue | 1 | 93 | Risk Ratio (M‐H, Fixed, 99% CI) | 1.66 [0.59, 4.64] |
| 3.6 Irritability | 1 | 93 | Risk Ratio (M‐H, Fixed, 99% CI) | 3.07 [0.60, 15.68] |
| 3.7 Vomiting | 1 | 93 | Risk Ratio (M‐H, Fixed, 99% CI) | 4.09 [0.57, 29.16] |
| 3.8 Insomnia | 1 | 93 | Risk Ratio (M‐H, Fixed, 99% CI) | 2.72 [0.52, 14.33] |
1.1. Analysis.
Comparison 1 Flunarizine versus placebo, Outcome 1 50% or greater reduction in seizure frequency.
1.2. Analysis.
Comparison 1 Flunarizine versus placebo, Outcome 2 Treatment withdrawal.
1.3. Analysis.
Comparison 1 Flunarizine versus placebo, Outcome 3 Adverse effects.
Comparison 2. Nimodipine versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 50% or greater reduction in seizure frequency | 1 | 17 | Risk Ratio (M‐H, Fixed, 95% CI) | 7.78 [0.46, 130.88] |
| 2 Treatment withdrawal | 1 | 17 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.25 [0.25, 20.38] |
2.1. Analysis.
Comparison 2 Nimodipine versus placebo, Outcome 1 50% or greater reduction in seizure frequency.
2.2. Analysis.
Comparison 2 Nimodipine versus placebo, Outcome 2 Treatment withdrawal.
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Alving 1989.
| Methods | Randomised double‐blind placebo‐controlled cross‐over trial. 2 treatment sequences: placebo‐flunarizine and flunarizine‐placebo sequences. Baseline = 4 weeks. No titration period. Treatment phase 1 and 2 = 16 weeks each. | |
| Participants | Single Danish centre. Total randomised 29; all with drug‐resistant partial epilepsy; numbers of randomised patients into individual treatment sequence groups = data not available. 41% male. Age range 14 to 58 years. Other AEDs = 1 to 3. Baseline seizure frequency: data not available. | |
| Interventions | Flunarizine 15 mg per day. Placebo. |
|
| Outcomes | The following outcomes were measured: 1) Seizure incidence during flunarizine and placebo periods were measured. 2) 50% or greater reduction in seizure frequency. 3) Treatment withdrawal. 4) Side effects. 5) Neuropsychological changes. 6) Treatment preference. |
|
| Notes | We were not able to acquire the data from the first treatment phase for meta‐analysis for any outcomes. Study was funded by Janssenpharma A/S. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote ‐ "randomised" Comment ‐ No details of method of randomisation therefore uncertain risk of bias. |
| Allocation concealment (selection bias) | Unclear risk | Comment ‐ No details of allocation concealment therefore uncertain risk of bias. |
| Blinding (performance bias and detection bias) Seizure Reduction | Unclear risk | Quote ‐ "Double Blinded" Comment ‐ No details given to how blinding was achieved |
| Blinding of participants and personnel (performance bias) Seizure Reduction | Low risk | Quote ‐ "Flunarazine pills and placebo were identical" Comment ‐ Participants were adequately blinded |
| Blinding of outcome assessment (detection bias) Seizure Reduction | Unclear risk | Quote ‐ "Double Blinded" Comment ‐ No details given to how blinding was achieved |
| Incomplete outcome data (attrition bias) Seizure Reduction | Unclear risk | Comment ‐ Missing data and study attrition reported but no intention‐to‐treat analysis |
| Selective reporting (reporting bias) | Low risk | Comment ‐ Outcomes were reported clearly |
| Other bias | Low risk | None identified |
Battaglia 1991.
| Methods | Randomised double‐blind placebo‐controlled cross‐over trial. 2 treatment sequences: placebo‐flunarizine and flunarizine‐placebo sequences. Baseline = 16 weeks. No titration period. Treatment phase 1 and 2 = 16 weeks each. | |
| Participants | Single Italian centre. Total fed 20; all with drug‐resistant partial or generalized epilepsy; numbers of randomised patients into individual treatment sequence groups = data not available. 50% male. Age range = 6 to 18 years (mean 12 years 7 months). Other AEDs: most patients on 2 to 3. Baseline seizure frequency: data not available. | |
| Interventions | 1) Flunarizine 5 mg/d for age < 10 years or 10 mg/d for age > 10 years. 2) Placebo. |
|
| Outcomes | The following outcomes were measured: 1) Seizure incidence during flunarizine and placebo periods. 2) 30%, 40% and 60% greater reduction in seizure frequency. 3) Treatment withdrawal. 4) Adverse effects. |
|
| Notes | We were not able to acquire the data from the first treatment phase for meta‐analysis for any outcomes. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote ‐ "randomised" Comment ‐ No details of method of randomisation therefore uncertain risk of bias |
| Allocation concealment (selection bias) | Unclear risk | Quote ‐ "randomised" Comment ‐ No details of method of allocation concealment therefore uncertain risk of bias |
| Blinding (performance bias and detection bias) Seizure Reduction | Unclear risk | Quote ‐ "Blinded" Comment ‐ No details of method of blinding therefore uncertain risk of bias |
| Blinding of participants and personnel (performance bias) Seizure Reduction | Unclear risk | Quote ‐ "Blinded" Comment ‐ No details of method of blinding therefore uncertain risk of bias |
| Blinding of outcome assessment (detection bias) Seizure Reduction | Unclear risk | Quote ‐ "Blinded" Comment ‐ No details of method of blinding therefore uncertain risk of bias |
| Incomplete outcome data (attrition bias) Seizure Reduction | Unclear risk | Comment ‐ Missing data reported but no intention to treat analysis performed |
| Selective reporting (reporting bias) | Low risk | Comment‐ Outcomes were reported clearly |
| Other bias | Low risk | None identified |
Fröscher 1988.
| Methods | Randomised double‐blind placebo‐controlled cross‐over trial. 2 treatment sequences: placebo‐flunarizine and flunarizine‐placebo sequences. Baseline = 8 weeks. Titration period = 3 weeks. Treatment phase 1 and 2 = 16 weeks each. | |
| Participants | Single Belgian centre. Total randomised 30; all with drug‐resistant partial epilepsy. 14 to placebo‐flunarizine sequence; 16 to flunarizine‐placebo sequence. 57% male. Age range 14 to 51 years. Other AEDs: most patients on 1 to 3. Baseline seizure frequency: data not available. | |
| Interventions | 1) Flunarizine 15 mg/day 2) Placebo pill. |
|
| Outcomes | Following outcomes were reported: 1) Seizure incidence during flunarizine and placebo periods. 2) Change (%) of seizure frequency compared between the last month of each treatment phase. 3) Effect on severity and duration of seizures. 4) Treatment withdrawal. 5) Adverse effects. 6) Flunarizine plasma concentration. 7) Interaction with existing AEDs (total and free blood levels). |
|
| Notes | The first author was not able to provide data for the treatment allocation method and its concealment as well as data for the following outcomes from the first treatment phase due to those data no longer being available: (a) 50% or greater reduction in seizure frequency; (b) side effects. We used the data of treatment withdrawal from the first treatment phase for meta‐analysis. We presented the data of 50% responders and side effects from both treatment phases in Table 3 and Table 4 respectively. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote ‐ "randomised" Comment ‐ No details of method of randomisation therefore uncertain risk of bias |
| Allocation concealment (selection bias) | Unclear risk | Quote ‐ "randomised" Comment ‐ No details of method of allocation concealment therefore uncertain risk of bias |
| Blinding (performance bias and detection bias) Seizure Reduction | Unclear risk | Quote ‐ "Blinded" Comment ‐ No details of method of blinding therefore uncertain risk of bias |
| Blinding of participants and personnel (performance bias) Seizure Reduction | Unclear risk | Quote ‐ "Blinded" Comment ‐ No details of method of blinding therefore uncertain risk of bias |
| Blinding of outcome assessment (detection bias) Seizure Reduction | Unclear risk | Quote ‐ "Blinded" Comment ‐ No details of method of blinding therefore uncertain risk of bias |
| Incomplete outcome data (attrition bias) Seizure Reduction | High risk | Comment ‐ The study has two phases and seizure reduction was not provided for the first phase and the authors commented that the data was lost as such it is not possible to discern attrition rates or ITT analysis |
| Selective reporting (reporting bias) | High risk | Comment ‐ Selective outcome reporting as the authors did not report seizure reduction or side effects of AED for first phase of the study |
| Other bias | Low risk | None identified |
Keene 1989.
| Methods | Randomised double‐blind placebo‐controlled cross‐over trial. 2 treatment sequences: placebo‐flunarizine and flunarizine‐placebo sequences. Baseline = 4 weeks. No titration period. Treatment phase 1 and 2 = 12 weeks each. | |
| Participants | Single Canadian centre. Total randomised 34; all with drug‐resistant partial and generalized epilepsy; 16 to placebo‐flunarizine sequence; 18 to flunarizine‐placebo sequence. 56% male. Age range 2 to 18 years (mean 15.5 years). Other AEDs: data not available. Baseline seizure frequency: data not available. | |
| Interventions | 1) Flunarizine:
(a) 5 mg/d for BW less than 20 kg;
(b) 10 mg/d for BW 20 to 40 kg;
(c) 15 mg/d for BW more than 40 kg. 2) Placebo pill |
|
| Outcomes | Following outcomes were measured: 1) 50% or greater reduction in seizure frequency. 2) Treatment withdrawal. 3) Adverse effects. 4) Serum flunarizine level. |
|
| Notes | The first author was not able to provide data for the treatment allocation method and data for the following outcomes from the first treatment phase:
(a) 50% or greater reduction in seizure frequency;
(b) adverse effects. We used the data of treatment withdrawal from the first treatment phase for meta‐analysis. We presented the data of 50% responders and adverse effects from both treatment phases in Table 3 and Table 4 respectively. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote ‐ "randomised" Comment ‐ No details of method of randomisation therefore uncertain risk of bias |
| Allocation concealment (selection bias) | Low risk | Quote ‐ "randomised" Comment ‐ No explicit details provided in the study but authors contacted and said sealed envelopes were given to subjects |
| Blinding (performance bias and detection bias) Seizure Reduction | Low risk | Quote ‐ "Identical appearance for flunarazine and placebo pills" Comment ‐ Key study personnel were not aware which pills were active and which were placebo |
| Blinding of participants and personnel (performance bias) Seizure Reduction | Low risk | Quote ‐ "Identical appearance for flunarazine and placebo pills" Comment ‐ Participants were not aware which pills were active and which were placebo |
| Blinding of outcome assessment (detection bias) Seizure Reduction | Unclear risk | Comment ‐ No details on outcome assessors provided and how they were blinded |
| Incomplete outcome data (attrition bias) Seizure Reduction | Low risk | Comment ‐ All patients completed the study and no intention to treat analysis was needed |
| Selective reporting (reporting bias) | High risk | Comment ‐ Required primary outcomes not reported for first phase of study |
| Other bias | Low risk | None identified |
Larkin 1991.
| Methods | Randomised double‐blind placebo‐controlled cross‐over trial. 2 treatment sequences: placebo‐nimodipine and nimodipine‐placebo sequences. Baseline = 4 weeks. No titration period. Treatment phase 1 and 2 = 12 weeks each. | |
| Participants | Single British centre. Total randomised 22; all with drug‐resistant partial and generalized epilepsy; numbers of randomised participants into individual treatment sequence groups = data not available. 36% male. Age range 18 to 53 years. Other AEDs = 1 to 2. Baseline seizure frequency: data not available. | |
| Interventions | 1) Nimodipine:
(a) 90 mg/d for the initial 4 weeks;
(b) 180 mg/d for the middle 4 weeks;
(c) 270 mg/d for the final 4 weeks. 2) Placebo. |
|
| Outcomes | Outcomes reported: 1) 25% and 50% or greater reduction in generalized tonic‐clonic or partial seizures or seizure days. 2) Treatment withdrawal. 3) Adverse effects. 4) Heart rate and systolic blood pressure. 5) Drug preference. 6) Serum concentration of nimodipine and concomitant AEDs. 7) Correlations between nimodipine concentrations and changes in seizure control. |
|
| Notes | The last author was not able to provide data for the randomisation method, allocation concealment and blinding method as well as data for any outcomes from the first treatment phase because those data are no longer available. Study was funded by Bayer U.K. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote ‐ "randomised" Comment ‐ No details of method of randomisation therefore uncertain risk of bias |
| Allocation concealment (selection bias) | Unclear risk | Quote ‐ "randomised" Comment ‐ No details of method of allocation concealment therefore uncertain risk of bias |
| Blinding (performance bias and detection bias) Seizure Reduction | Low risk | Quote ‐ "Identical appearance for Nimodipine and placebo pills" Comment ‐ Key study personnel were not aware which pills were active and which were placebo |
| Blinding of participants and personnel (performance bias) Seizure Reduction | Low risk | Quote "Identical appearance for Nimodipine and placebo pills" Comment ‐ Participants were not aware which pills were active and which were placebo |
| Blinding of outcome assessment (detection bias) Seizure Reduction | Unclear risk | Comment ‐ No details on outcome assessors provided and how they were blinded |
| Incomplete outcome data (attrition bias) Seizure Reduction | High risk | Quote ‐ "17 patients out of 22 completed the study" Comment ‐ The authors reported attrition but did not perform an intention to treat analysis |
| Selective reporting (reporting bias) | Low risk | Comment ‐ Primary and secondary outcomes reported |
| Other bias | Low risk | None identified |
Larkin 1992.
| Methods | Balanced double‐blind placebo‐controlled cross‐over trial. 2 treatment sequences: placebo‐nifedipine and nifedipine‐placebo sequences. Baseline = 8 weeks. No titration period. Treatment phase 1 and 2 = 8 weeks each. | |
| Participants | Single British centre. Total randomised 22; all with drug‐resistant partial and generalized epilepsy; 12 to placebo‐nifedipine sequence; 10 to nifedipine‐placebo sequence. 55% male. Age range 17 to 22 years. Other AEDs = 1 to 4. Baseline seizure frequency: data not available. | |
| Interventions | 1) Nifedipine 40 mg/d during the initial half period then 80 mg/d during the final half period. 2) Placebo. |
|
| Outcomes | Following outcomes reported: 1) Total number of any seizures in each treatment phase. 2) Total number of partial seizures in each treatment phase. 3) 50% or greater reduction in seizure frequency. 4) Treatment withdrawal. 5) Adverse events. 6) Heart rate and blood pressure. 7) Serum nifedipine concentrations 1 hour post dosing at 4 weeks on 20 mg twice daily and at 4 weeks on 40 mg twice daily. 8) Correlation between improvement in total seizure numbers, in partial seizure numbers, and nifedipine concentration following 8 weeks of treatment. 9) Comparison of total electroencephalography scores with nifedipine and placebo. |
|
| Notes | The last author was not able to provide data for the treatment allocation method and its concealment as well as data for the following outcomes from the first treatment phase because those data are no longer available:
(a) 50% or greater reduction in seizure frequency;
(b) adverse effects. We used the data of treatment withdrawal from the first treatment phase for analysis. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote ‐ "randomised" Comment ‐ No details of method of randomisation therefore uncertain risk of bias |
| Allocation concealment (selection bias) | Unclear risk | Quote ‐ "randomised" Comment ‐ No details of method of allocation concealment therefore uncertain risk of bias |
| Blinding (performance bias and detection bias) Seizure Reduction | Unclear risk | Quote ‐ "Identical appearance for Nifdepine and placebo pills" Comment‐ Did not mention if Key study personnel were aware which pills were active and which were placebo |
| Blinding of participants and personnel (performance bias) Seizure Reduction | Low risk | Quote ‐ "Identical appearance for Nifdepine and placebo pills" Comment ‐ Patients were not aware which pills were active and which were placebo |
| Blinding of outcome assessment (detection bias) Seizure Reduction | Unclear risk | Comment ‐ No details given on how assessors were blinded |
| Incomplete outcome data (attrition bias) Seizure Reduction | Unclear risk | Quote ‐ "20 patients completed the study" This was out of total of 22 patients Comment ‐ Authors did not perform intention to treat analysis |
| Selective reporting (reporting bias) | Low risk | Comment ‐ No selective reporting of outcomes |
| Other bias | Low risk | None identified |
Moglia 1986.
| Methods | Randomised double‐blind placebo‐controlled cross‐over trial. 2 treatment sequences: placebo‐flunarizine and flunarizine‐placebo sequences. Baseline = 12 weeks. No titration period. Treatment phase 1 and 2 = 12 weeks each. | |
| Participants | Single Italian centre. Total randomised 90; all with drug‐resistant partial and generalized epilepsy; 42 to placebo‐flunarizine sequence; 48 to flunarizine‐placebo sequence. 57% male. Age range 15 to 73 years. Other AEDs: data not available. Baseline seizure frequency: data not available. | |
| Interventions | 1) Flunarizine:
(a) 10 mg/d for BW less than 70 kg;
(b) 15 mg/d for BW more than70 kg. 2) Placebo pill. |
|
| Outcomes | Outcomes Reported: 1) Incidence of seizure per month before and at the end of each treatment. 2) 50% or greater reduction in seizure frequency. 3) Treatment withdrawal. 4) Adverse effects. |
|
| Notes | The first author was not able to provide data for the treatment allocation method and its concealment as well as data for the following outcomes from the first treatment phase because those data are no longer available:
(a) 50% or greater reduction in seizure frequency;
(b) adverse effects. We used the data of treatment withdrawal from the first treatment phase for meta‐analysis. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote ‐ "Patients divided into two equal groups" Comment ‐ The study is listed as a randomised trial but there is no evidence in the paragraphs of the study, only this vague sentence |
| Allocation concealment (selection bias) | Unclear risk | Comment ‐ Unclear, no data available on concealment |
| Blinding (performance bias and detection bias) Seizure Reduction | Unclear risk | Comment ‐ No data regarding blinding was provided. The word itself is not mentioned in the study |
| Blinding of participants and personnel (performance bias) Seizure Reduction | Unclear risk | Comment ‐ No data regarding blinding was provided. The word itself is not mentioned in the study |
| Blinding of outcome assessment (detection bias) Seizure Reduction | Unclear risk | Comment ‐ No data regarding blinding was provided. The word itself is not mentioned in the study |
| Incomplete outcome data (attrition bias) Seizure Reduction | High risk | Comment ‐ Attrition rate was repoted but no intention to treat analysis performed. 28 patients dropped out from the 90 patient sample, this constitutes a high risk of attrition bias |
| Selective reporting (reporting bias) | High risk | Comment ‐ Authors did not report the required primary and secondary outcomes |
| Other bias | Low risk | None identified |
Overweg 1984.
| Methods | Randomised double‐blind placebo‐controlled cross‐over trial. 2 treatment sequences: placebo‐flunarizine and flunarizine‐placebo sequences. Baseline = 12 weeks. No titration period. Treatment phase 1 and 2 = 12 weeks each. | |
| Participants | Single Dutch centre. Total randomised 33; all with drug‐resistant partial and generalized epilepsy; numbers of randomised participants to individual treatment sequence groups not available. 55% male. All adults. Other AEDs: most patients on 1 to 3. Baseline seizure frequency: data not available. | |
| Interventions | 1) Flunarizine 10 mg per day. 2) Placebo pill. |
|
| Outcomes | Following outcomes reported: 1) Total seizure incidence during on each treatment. 2) 25% or more and 50% or greater reduction in seizure frequency. 3) Treatment withdrawal. 4) Adverse effects. 5) Blood level of concomitant AEDs. |
|
| Notes | We were not able to acquire the data from the first treatment phase for meta‐analysis for any outcomes. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote ‐ "randomised" Comment ‐ No details of method of randomisation therefore uncertain risk of bias |
| Allocation concealment (selection bias) | Unclear risk | Quote ‐ "randomised" Comment ‐ No details of method of allocation concealment therefore uncertain risk of bias |
| Blinding (performance bias and detection bias) Seizure Reduction | Unclear risk | Quote ‐ "Blinded" Comment ‐ No details of method of blinding therefore uncertain risk of bias |
| Blinding of participants and personnel (performance bias) Seizure Reduction | Unclear risk | Quote ‐ "Blinded" Comment ‐ No details of method of blinding therefore uncertain risk of bias |
| Blinding of outcome assessment (detection bias) Seizure Reduction | Unclear risk | Quote ‐ "Blinded" Comment ‐ No details of method of blinding therefore uncertain risk of bias |
| Incomplete outcome data (attrition bias) Seizure Reduction | Unclear risk | Comment ‐ Study attrition was reported but no intention to treat analysis was performed |
| Selective reporting (reporting bias) | High risk | Comment ‐ Required primary and secondary outcomes were not reported for Phase 1 of the study |
| Other bias | Low risk | None identified |
Pelliccia 1993.
| Methods | Quasi‐randomised, double‐blind, placebo‐controlled, cross‐over trial. 2 treatment sequences: placebo‐nimodipine and nimodipine‐placebo sequences. Baseline = 30 days. No titration period. Treatment phase 1 and 2 = 12 weeks each. | |
| Participants | Single Italian centre. Total randomised 17; 8 randomised into nimodipine‐placebo sequence and 9 randomised into placebo‐nimodipine sequence. All with drug‐resistant partial and generalized epilepsy. 53% male. Age range 5 to 22 years. Other AEDs: 1 to 3. Baseline seizure frequency: range 3 to 603 per month, mean 166.7 per month. | |
| Interventions | 1) Nimodipine 1.5 to 2 mg/kg/d. 2) Placebo. |
|
| Outcomes | Following outcomes reported: 1) Incidence of total, partial, generalized, absence seizures or seizure days. 2) 25% or more and 50% or greater reduction in seizure frequency. 3) Treatment withdrawal. 4) Adverse effects. 5) Blood level of concomitant AEDs. |
|
| Notes | We used the data from the first treatment phase for analysis for the outcomes of 50% or greater reduction in seizure frequency, treatment withdrawal and adverse effects. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Comment ‐ Random list generation: order of entry into trial, allocation by odd or even numbers No true “Randomisation”. There is a pattern to the selection |
| Allocation concealment (selection bias) | Low risk | Quote ‐ "sequential sealed packages." Comment ‐ adequate method of concealment chosen |
| Blinding (performance bias and detection bias) Seizure Reduction | High risk | Quote ‐ "allocation by odd or even numbers." Comment ‐ Key study personnel would be able to memorize if the patient is on an active or placebo pill if they knew what their order was (odd or even) |
| Blinding of participants and personnel (performance bias) Seizure Reduction | Low risk | Quote ‐ "Identical appearance for nimodipine and placebo" Comment ‐ Subjects unaware of the medication they were taking |
| Blinding of outcome assessment (detection bias) Seizure Reduction | Unclear risk | Comment ‐ No information given on how outcome assessors were blinded |
| Incomplete outcome data (attrition bias) Seizure Reduction | Unclear risk | Comment ‐ Attrition was reported, but no intention to treat analysis preformed.Three out of 17 patients missing from analysis |
| Selective reporting (reporting bias) | Low risk | Comment ‐ no selective outcome reporting |
| Other bias | Low risk | None identified |
Pledger 1994.
| Methods | Randomised double‐blind placebo‐controlled parallel trial. 2 treatment arms: 1 placebo and 1 flunarizine. Baseline = 12 weeks. Titration period = 1 week. Treatment phase = 24 weeks. | |
| Participants | All adults. Multicentre across USA. Total randomised 93; all with drug‐resistant partial epilepsy; 47 to placebo; 46 to flunarizine. 47% male. Age range 16 to 64 years. Other AEDs: 1 to 2. Baseline seizure frequency per week: mean = 6, median = 3.5, range = 1 to 31.8. | |
| Interventions | 1) Adjusted dose of flunarizine to keep plasma concentration at 60 ng/ml. 2) Placebo pills. |
|
| Outcomes | Following outcomes reported: 1) Seizures, seizure duration and seizure severity per cent reduction. 2) 50% or greater reduction in seizure frequency. 3) Treatment withdrawal. 4) Adverse effects. 5) Plasma concentrations of flunarizine and concomitant AEDs. |
|
| Notes | We used the data of 50% or greater reduction in seizure frequency and adverse effects for analyses. We also used the data of treatment withdrawal for meta‐analysis. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Comment ‐ Authors noted that the random sequence was stratified by a computer |
| Allocation concealment (selection bias) | Low risk | Quote "treatment allocated by the centre and kept at pharmacy separated from study site." Comment ‐ Appropriate treatment allocation sequence by authors |
| Blinding (performance bias and detection bias) Seizure Reduction | Low risk | Comment ‐ All personnel involved in the study were blinded |
| Blinding of participants and personnel (performance bias) Seizure Reduction | Low risk | Comment ‐ All personnel involved in the study were blinded |
| Blinding of outcome assessment (detection bias) Seizure Reduction | Low risk | Comment ‐ All personnel involved in the study were blinded |
| Incomplete outcome data (attrition bias) Seizure Reduction | Low risk | Comment ‐ No study attrition and no need for intention to treat analysis |
| Selective reporting (reporting bias) | Low risk | Comment ‐ no selective outcome reporting |
| Other bias | Low risk | None identified |
Starreveld 1989.
| Methods | Double‐blind placebo‐controlled cross‐over trial. Baseline = 8 weeks. No titration period. Treatment phase 1 and 2 = 16 weeks. | |
| Participants | Single Canadian centre. Total randomised 34; all with drug‐resistant partial epilepsy; numbers of randomised participants into individual treatment sequence groups not available. 38% male. Age range within 15 to 60 years. Other AEDs: most participants on 1 to 2. Baseline seizure frequency: data not available. | |
| Interventions | 1) Flunarizine 15 mg per day. 2) Placebo. |
|
| Outcomes | Following outcomes were reported: 1) Total seizure frequency on each treatment phase. 2) 25%, 25 to 49%, and 50% or greater reduction in seizure frequency. 3) Treatment withdrawal. 4) Adverse effects. 5) Serum concentration of flunarizine and concomitant AEDs. |
|
| Notes | We were not able to acquire the data from the first treatment phase for meta‐analyses for any of the outcomes. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote ‐ "randomised" Comment ‐ No details of method of randomisation therefore uncertain risk of bias |
| Allocation concealment (selection bias) | Unclear risk | Quote ‐ "randomised" Comment ‐ No details of method of allocation concealment therefore uncertain risk of bias |
| Blinding (performance bias and detection bias) Seizure Reduction | Unclear risk | Quote ‐ "Blinded" Comment ‐ No details of method of blinding therefore uncertain risk of bias |
| Blinding of participants and personnel (performance bias) Seizure Reduction | Unclear risk | Quote ‐ "Blinded" Comment ‐ No details of method of blinding therefore uncertain risk of bias |
| Blinding of outcome assessment (detection bias) Seizure Reduction | Unclear risk | Quote ‐ "Blinded" Comment ‐ No details of method of blinding therefore uncertain risk of bias |
| Incomplete outcome data (attrition bias) Seizure Reduction | High risk | Comment ‐ Missing data was mentioned but no intention to treat analysis performed |
| Selective reporting (reporting bias) | High risk | Comment‐ Did not report primary and secondary outcomes for first phase of the study |
| Other bias | Low risk | None identified |
AED: antiepileptic drug BW: body weight
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Binnie 1985 | Not RCT due to lack of randomisation. Three patient arms from a previous study were entered into this study as one arm. Placebo was not mentioned in the methods. |
| Cavazzuti 1986 | Some participants received study medication for less than 8 weeks. |
| Handforth 1995 | The paper reports results from a previous RCT and then presents the results of an open‐label extension study. |
| Malashkhia 1996 | Although this study was a randomised trial, it was not a placebo‐controlled trial and all concurrent AEDs were substituted with phenobarbitone rather than being kept unchanged. |
| Meyer 1995 | This trial did not investigate for 50% or greater reduction in seizure frequency. We were also unable to acquire the proportion of participants having treatment withdrawn and the proportion of participants experiencing individual adverse effects in individual treatment groups in the first treatment phase. |
| Sasso 1993 | This trial did not investigate for 50% or greater reduction in seizure frequency. We were also unable to acquire the proportion of participants having treatment withdrawn and the proportion of participants experiencing individual adverse effects in individual treatment groups. |
| Treiman 1993 | Pilot study to assess concentration treatment for 34 days. Not RCT, concentration‐controlled study. |
AED: antiepileptic drug
Characteristics of studies awaiting assessment [ordered by study ID]
Andrade 2012.
| Methods | Randomised double‐blind interventional controlled trial. |
| Participants | 18 to 60 year old patients with refractory epilepsy. Both genders included. |
| Interventions | Drug: Verapamil (80 mg TID). Drug: Placebo (TID). |
| Outcomes | Primary outcome measures:
|
| Notes | Taking place in Toronto. |
Differences between protocol and review
We included randomised controlled trials which examined differences between two or more active drugs i.e. head to head trials.
Contributions of authors
MH and JP were involved in all stages of conducting and writing of the review. Any disagreements were resolved by discussion with Tony Marson who supervised the review and gave comments.
Sources of support
Internal sources
No sources of support supplied
External sources
-
National Institute for Health Research, UK, Not specified.
This review presents independent research commissioned by the National Institute for Health Research (NIHR). The views expressed in this publication are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health.
Declarations of interest
None known
New search for studies and content updated (no change to conclusions)
References
References to studies included in this review
Alving 1989 {published data only}
- Alving J, Kristensen O, Tsiropoulos I, Mondrup K. Double‐blind placebo‐controlled evaluation of flunarizine as adjunct therapy in epilepsy with complex partial seizures. Acta Neurologica Scandinavica 1989;79:128‐32. [DOI] [PubMed] [Google Scholar]
Battaglia 1991 {published data only}
- Battaglia A, Ferrari AR, Guerrini R. Double‐blind placebo‐controlled trial of flunarizine as add‐on therapy in refractory childhood epilepsy. Brain and Development 1991;13(4):217‐22. [DOI] [PubMed] [Google Scholar]
Fröscher 1988 {published data only}
- Froscher W, Bulau P, Burr W, Penin H, Rao ML, Beukelaar F. Double‐blind placebo‐controlled trial with flunarizine in therapy‐resistant epileptic patients. Clinical Neuropharmacology 1988;11(3):232‐40. [DOI] [PubMed] [Google Scholar]
Keene 1989 {published data only}
- Keene D, Whiting S, Humphreys P, Jacob P. Flunarizine as a supplementary medication in refractory childhood epilepsy: a double‐blind crossover study. Canadian Journal of Neurological Sciences 1989;16(2):191‐3. [DOI] [PubMed] [Google Scholar]
Larkin 1991 {published data only}
- Larkin JG, McKee PJW, Blacklaw J, Thompson GG, Morgan IC, Brodie MJ. Nimodipine in refractory epilepsy: a placebo‐controlled, add‐on study. Epilepsy Research 1991;9:71‐7. [DOI] [PubMed] [Google Scholar]
Larkin 1992 {published data only}
- Larkin JG, Besag FMC, Cox A, Williams J, Brodie MJ. Nifedipine for epilepsy? A double‐blind, placebo‐controlled trial. Epilepsia 1992;33(2):346‐52. [DOI] [PubMed] [Google Scholar]
Moglia 1986 {published data only}
- Moglia A, Bergamasco B, Perri R, Mancia D. Flunarizine as add‐on therapy in epilepsy: crossover study versus placebo. Functional Neurology 1986;1(4):547‐50. [PubMed] [Google Scholar]
Overweg 1984 {published data only}
- Overweg J, Binnie CD, Meijer JWA, Meinardi H, Nuijten STM, Schmaltz S, et al. Double‐blind placebo‐controlled trial of flunarizine as add‐on therapy in epilepsy. Epilepsia 1984;25(2):217‐22. [DOI] [PubMed] [Google Scholar]
Pelliccia 1993 {published data only}
- Pelliccia A, Sciarretta A, Monticelli M, Porro G, Matricardi M. Nimodipine in drug‐resistant childhood epilepsy: a double‐blind, placebo‐controlled trial [Impiego della nimodipina nelle epilessia farmacoresistente: studio in doppio cieco placebo‐controllato]. Bolletino Lega Italiana Contro L'Epilessia 1993;82/83:153‐4. [Google Scholar]
Pledger 1994 {published data only}
- Pledger GW, Sackellares JC, Treiman DM, Pellock JM, Wright FS, Mikati M, et al. Flunarizine for treatment of partial seizures: results of a concentration‐controlled trial. Neurology 1994;44:1830‐6. [DOI] [PubMed] [Google Scholar]
Starreveld 1989 {published data only}
- Starreveld E, Beukelaar F, Wilson AF, McLean DR, Findlay HP. Double‐blind cross‐over placebo controlled study of flunarizine in patients with therapy resistant epilepsy. Canadian Journal of Neurological Sciences 1989;16:187‐90. [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Binnie 1985 {published data only}
- Binnie CD, de Beukelaar F, Meijer JW, Meinardi H, Overweg J, Wauquier A, van Wieringen A. Open dose‐ranging trial of flunarizine as add‐on therapy in epilepsy. Epilepsia 1985;26:424‐8. [ISSN: 0013‐9580] [DOI] [PubMed] [Google Scholar]
Cavazzuti 1986 {published data only}
- Cavazzuti GB, Galli V, Benatti A. The use of flunarizine in pediatric epilepsy. Functional Neurology 1986;1(4):551‐4. [PubMed] [Google Scholar]
Handforth 1995 {published data only}
- Handforth A, Mai T, Treiman DM. Rising dose study of safety and tolerance of flunarizine. European Journal of Clinical Pharmacology 1995;49:91‐4. [DOI] [PubMed] [Google Scholar]
Malashkhia 1996 {published data only}
- Malashkhia VY, Nadareishvili ZG, Malashkhia YA. Add‐on therapy with nimodipine in intractable epilepsy of childhood. Journal of Child Neurology 1996;11(6):500‐1. [DOI] [PubMed] [Google Scholar]
Meyer 1995 {published data only}
- Cascino GD, Meyer FB, Sharbrough FW, Ivnik RJ, Hirschorn KA, So EL, et al. Nimodipine for intractable seizures: a double‐blind placebo‐controlled crossover study. Epilepsia 1992;33 Suppl 3:119. [Google Scholar]
- Meyer FB, Cascino GD, Whisnant JP, Sharbrough FW, Ivnik RJ, Gorman DA, et al. Nimodipine as an add‐on therapy for intractable epilepsy. Mayo Clinic Proceedings 1995;70:623‐7. [DOI] [PubMed] [Google Scholar]
Sasso 1993 {published data only}
- Sasso E, Delsoldato S, Mancia D. Nimodipine as adjunctive therapy in refractory epilepsy [Effetto della nimodipina nelle epilessie farmacoresistenti]. Bolletino Lega Italiana Contro L'Epilessia 1993;82/83:155‐6. [Google Scholar]
Treiman 1993 {published data only}
- Treiman DM, Pledger GW, DeGiorgio C, Tsay JY, Cereghino JJ. Increasing plasma concentration tolerability study of flunarizine in comedicated epileptic patients . Epilepsia 1993;34:944‐53. [DOI] [PubMed] [Google Scholar]
References to studies awaiting assessment
Andrade 2012 {unpublished data only}
- Verapamil study in refractory epilepsy. http://clinicaltrials.gov/ct2/show/NCT01126307.
Additional references
Bonnett 2012
- Bonnett L, Smith CT, Smith D, Williamson P, Chadwick D, Marson AG. Prognostic factors for time to treatment failure and time to 12 months of remission for patients with focal epilepsy: post‐hoc, subgroup analyses of data from the SANAD trial. Lancet Neurology 2012;11(4):331‐40. [DOI: 10.1016/S1474-4422(12)70018-2] [DOI] [PubMed] [Google Scholar]
Higgins 2010
- Higgins J, Green S. Cochrane Handbook for Systematic Reviews of Interventions. Cochrane Handbook for Systematic Reviews of Interventions. West Sussex: John Wiley & Sons Ltd, 2010. [Google Scholar]
Jouvenceau 2001
- Jouvenceau A, Eunson LH, Spauschus A, Ramesh V, Zuberi SM, Kullmann DM, et al. Human epilepsy associated with dysfunction of the brain P/Q‐type calcium channel. Lancet 2001;358(9284):801‐7. [DOI: 10.1016/S0140-6736(01)05971-2] [DOI] [PubMed] [Google Scholar]
Nunes 2012
- Nunes VD, Sawyer L, Neilson J, Sarri G, Cross JH. Diagnosis and management of the epilepsies in adults and children: summary of updated NICE guidance. BMJ 2012;344:e281. [DOI] [PubMed] [Google Scholar]
Reynolds 1993
- Reynolds J. Martindale: The extra pharmacopoeia. 30th Edition. London: Pharmaceutical Press, 1993. [Google Scholar]
Smith 2005
- Smith SJM. EEG in the diagnosis, classification, and management of patients with epilepsy. Journal of Neurology, Neurosurgery, and Psychiatry 2005;76:ii2‐ii7. [DOI: 10.1136/jnnp.2005.069245] [DOI] [PMC free article] [PubMed] [Google Scholar]
Summers 2004
- Summers MA, Moore JL, McAuley JW. Use of verapamil as a potential P‐glycoprotein inhibitor in a patient with refractory epilepsy. The Annals of Pharmacotherapy 2004;38(10):1631‐4. [DOI: 10.1345/aph.1E068] [DOI] [PubMed] [Google Scholar]
Triggle 2007
- Triggle DJ. Calcium channel antagonists: Clinical uses ‐ past, present and future. Biochemical Pharmacology 2007;74(1):1‐9. [DOI: 10.1016/j.bcp.2007.01.016] [DOI] [PubMed] [Google Scholar]
References to other published versions of this review
Chaisewikul 2001
- Chaisewikul R, Baillie N, Marson AG. Calcium antagonists as an add‐on therapy for drug‐resistant epilepsy. Cochrane Database of Systematic Reviews 2001, Issue 4. [DOI: 10.1002/14651858.CD002750] [DOI] [PubMed] [Google Scholar]