

Abstract
Monoclonal antibodies (mAbs) developed for either the prevention or treatment of viral diseases represent a small, but valuable, class of products. Since 1985, commercial firms have initiated clinical studies involving a total of 28 mAbs. To date, one product (palivizumab) has been approved and eight candidates are currently in clinical study.
Most commercial mAbs studied as antiviral agents in the clinic have either directly or indirectly targeted human immunodeficiency virus, respiratory syncytial virus, or hepatitis C virus infections. However, the ability of mAbs to bind to specific targets and utilize various anti-infective modes of action would seem to make them well suited for the prevention and/or treatment of a wider variety of viral diseases. A number of factors, including the continuing need for innovative medicines for viral infections, the global spread of viral infections, and increased government funding for the study of pathogen countermeasures, have prompted companies to reconsider mAbs as antiviral agents. Public sector research into the use of mAbs against emerging pathogens, such as severe acute respiratory syndrome coronavirus, may have already provided candidates for further development.
Keywords: Respiratory Syncytial Virus, West Nile Virus, Rabies, Severe Acute Respiratory Syndrome, Severe Acute Respiratory Syndrome
Antibodies are produced by the immune system to combat invading organisms such as viruses. Prior to the development of monoclonal antibodies (mAbs), polyclonal antibody preparations derived from human serum were used for both prophylaxis and the treatment of a number of viral infections.[1] mAbs, which can be designed to function using various modes of action, seem to be well suited to use as antiviral interventions. However, mAbs are inconvenient to administer compared with oral antibiotics and provide protection from infection for much shorter time periods compared with vaccines. mAbs also tend to be more expensive than either antibiotics or vaccines. As a consequence, in the past mAbs have not been the interventions of choice for infectious diseases. In fact, mAbs for infectious diseases have comprised only 13% of the total number of mAbs in clinical development,[2] and only one product (palivizumab for the prevention of respiratory syncytial virus [RSV] infection in high-risk infants) is currently marketed. Due to the recent focus on emerging viral diseases such as severe acute respiratory syndrome (SARS), for which limited treatment options and no vaccines exist, this situation might be changing.
To inform future efforts in the research and development of these innovative agents, an overview of trends in the commercial development of mAbs for viral infections, with a focus on mAbs for HIV, RSV, and hepatitis C virus (HCV) infections, is provided, and the possibility of increased efforts to develop mAbs for emerging pathogens is discussed.
1. Analysis Criteria
Since it was founded in 1976, the Tufts Center for the Study of Drug Development has collected data on the development and approval of therapeutics and vaccines. Data for mAbs sponsored in clinical and preclinical study by commercial firms were collected by the survey of pharmaceutical and biotechnology firms and from information in the public domain (e.g. press releases and the medical literature). Of all commercial mAbs studied in the clinic, 28 candidates studied primarily for their ability to prevent or treat viral infections were identified (nonspecific immunomodulatory mAbs were excluded). The 28 mAbs entered clinical study between 1985 and 2005, and were composed of either a single mAb or a combination of mAbs (mAb cocktails).
As of July 2006, the development status of mAbs was that eight were in clinical studies and not yet approved in any country (five in phase I, one in phase I/II, one in phase II, and one in phase III development), one was approved in the US and other countries, and the development of 19 had been discontinued (mAbs were considered discontinued in clinical study if no studies were identified as ongoing or recently completed and the sponsoring company indicated that no additional studies were planned in the near future). The ‘preclinical’ category comprised candidates that had been tested in animals; a total of nine such antiviral mAbs sponsored at least in part by commercial firms were identified. All mAbs either originated at companies or were licensed from commercial, government, or academic sources. Data were updated with all changes noted through July 2006.
2. Antibodies as Antiviral Agents
Antibodies are complex glycoproteins produced by B-lymphocytes. The molecules bind to, and help eliminate, foreign and infectious pathogens in the body. Antibodies are Y-shaped, having two branches attached to a single stem. The tips of the branches bind to the target, while the stem, also called the crystallizable fragment (Fc), can perform several functions, including the activation of components of the human immune system. Specifically, after binding to a target, antibodies can recruit effector cells (e.g. neutrophils, macrophages, natural killer cells) or activate complement to destroy the target.[3] These two modes of action are referred to as antibody-dependent cell cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC), respectively, and are mediated through the Fc portion of the molecule. There are five classes of antibodies, the most abundant of which is IgG, and there are four isotypes (IgG1-4) within the IgG class. Human IgG isotypes vary in their ability to induce ADCC and CDC; this is most likely because of their differential affinities for immune system components.[4] Of the four isotypes, IgG1 most effectively induces ADCC and CDC.
Monoclonal versions of antibodies were originally produced from single B-lymphocyte cells. Genetic manipulation now allows genes from multiple sources of B-lymphocytes (e.g. mouse and human) to be combined, resulting in chimeric or humanized mAbs. In addition, a number of methods are currently available for the production of fully human mAbs, including the use of transgenic animals and phage display. mAbs are naturally well suited to intercepting hostile invaders and prompting responses from the immune system. However, mAbs can also simply block entry of an infectious agent into host cells by targeting human cellular components. This mode of action might be desirable against viruses that mutate rapidly and, thus, do not provide stable targets. mAbs can also be designed to pursue their quarry into the interior of cells, though this is a more complicated mode of action compared with the other mechanisms.
Between 1985 and 2005, companies sponsored clinical studies of 28 mAbs as either monotherapies or mAb cocktails for the prevention or treatment of infection with 6 viruses; these viruses were HIV (15 mAbs), RSV (4 mAbs), HCV and cytomegalovirus (3 mAbs each), hepatitis B virus (2 mAbs), and human rhinovirus (1 mAb). The candidates were in-licensed as well as self-originated. As a result, they most likely represent the most promising mAbs with commercial potential for these indications. Nearly half (46%) of the mAbs were human, a quarter were humanized mAbs, and the remainder were either murine (14%), chimeric (11%), or bispecific (4%) antibodies. The human, humanized, and chimeric mAbs were all either IgG1 (87%) or IgG4 (13%). Four product candidates were mAb cocktails. A trend towards the study of more mAbs was observed — of 24 candidates that entered clinical study in the four 5-year periods shown in figure 1, the largest number (8 mAbs) began studies between 2000 and 2004. A further four mAbs for viral infections entered clinical study in 2005.
Fig. 1.
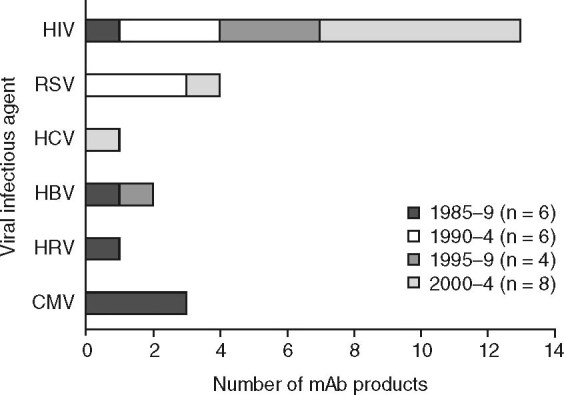
Number of commercial monoclonal antibodies (mAb) for viral infections entering clinical study between 1985 and 2004. CMV = cytomegalovirus; HBV = hepatitis B virus; HCV = hepatitis C virus; HIV = human immunodeficiency virus; HRV = human rhinovirus; RSV = respiratory syncytial virus.
3. Current Commercial Clinical Development
Of the 28 mAbs in the data set, the majority (79%) were for HIV, RSV, or HCV infections. In fact, all the mAb candidates now under clinical study are for these three infections (see table I). Most mAbs currently in studies are potential treatments for HIV infection, though none has progressed further than phase II studies. The only antiviral mAb in phase III is an anti-RSV mAb, motavizumab, which is an improved version of the approved anti-RSV agent palivizumab. Two mAbs intended as treatments for patients with HCV infection are currently at phase I. It is important to note that results from phase I and II studies, which are generally designed to assess safety and target-dosage regimens, do not provide sufficient data to be able to draw conclusions about efficacy or comparisons with study results (e.g. reduction in viral load) for other agents.
Table I.

Commercial monoclonal antibodies (mAb) for viral infections in clinical study or currently marketed
4. Monoclonal Antibodies (mAbs) for HIV Infection
HIV infection is currently a global pandemic affecting millions of people. The genetic composition of the virus is highly variable, which has greatly increased the difficulty of designing a drug that effectively treats HIV infection. There are two main types of virus, HIV-1 and HIV-2, but many genetically distinct viral subtypes. HIV-1, the most prevalent variety, can be subdivided into three groups (M, N, and O); the most prevalent of these, group M, can be further subdivided into at least nine subgroups or clades, the most wide-spread of which are clades B and C.[5] The virus itself is composed of a viral envelope of glycoproteins (gp) and a core containing the viral RNA and enzymes necessary for replication.
A clear understanding of how HIV enters cells, replicates, and then emerges has been critical to designing interventions. HIV is an RNA-based virus and as such cannot replicate without host cellular machinery. As a consequence, the virus must bind to and enter host cells. Infection is initiated by envelope protein spikes[6] and occurs via a two step procedure:
surface envelope gp120 binds to the CD4 receptor and a co-receptor such as chemokine (C-C motif) receptor 5 (CCR5);[7]
transmembrane envelope gp41 mediates fusion of the viral envelope with the cell membrane.[8]
When fusion is complete, the core of viral RNA and proteins are released into the cell. Once inside, the core disassembles and the contents then reform as a complex. Viral RNA is transcribed by reverse transcriptase into DNA as this complex travels to the nucleus of the cell. After entering the nucleus, the DNA encoding the viral protein is then incorporated into the host genome by an integrase enzyme.[9] At this point, the viral DNA can either lie dormant or the protein-production machinery generates the viral proteins from the integrated DNA, though studies suggest that HIV proteins can also be produced from non-integrated viral DNA.[10] The coordinated activity of both viral components and cellular components is needed to produce the new viral elements.[11] The RNA and proteins comprising the new viral particle are packaged and, through a procedure involving both viral and cellular components, the virus buds from the cell while maturing into a new infectious agent.[12]
The current standard of care for patients with HIV is highly active antiretroviral therapy (HAART), which comprises a drug cocktail of two nucleoside reverse transcriptase inhibitors and either a non-nucleoside reverse transcriptase inhibitor or protease inhibitor.[13] Although the therapy can suppress viral replication to low levels, the virus is not eliminated. As a consequence, life-long adherence to a drug regimen is necessary. Furthermore, viral resistance to drugs in the cocktail can occur and most of the drugs available for HAART have associated toxicities (e.g. mitochondrial dysfunction, hepatotoxicity, lipodystrophy).
Because of the limitations of the existing treatments, alternatives to the components of the currently available drug cocktails are needed. To date, a total of 15 commercially sponsored mAbs have been studied as treatments for HIV infection. The majority (67%) of candidates have targeted HIV envelope proteins — six mAbs have targeted gp120, two have targeted gp41, and two mAb cocktails have targeted both gp120 and gp41. However, only one of these mAbs (anti-gp120 mAb KD-247; Kaketsuken, Japan) remains in clinical study and as yet no efficacy data are available.
Due to issues associated with directly targeting HIV (e.g. high viral mutation rate), alternative methods to block the progression of HIV infection using mAbs have been explored. Three mAbs currently in clinical development target cellular receptors used by the virus to gain entry into lymphocytes, and so are in the entry inhibitor (EI) class of HIV drugs. One mAb (TNX-355; Tanox, Houston, TX, USA) targets CD4 while two other mAbs (PRO-140, Progenics, Tarrytown, NY, USA; CCR5mAb004, Human Genome Sciences, Rockville, MD, USA) target CCR5. The anti-CD4 mAb has undergone preliminary safety and efficacy (phase I and II) studies. In a phase II study,[14] TNX-355 was administered to HIV-1 infected patients intravenously at doses of 10 mg/kg and 15 mg/kg. The mAb was well tolerated and reduced viral load at both doses, although the reduction was greater at the 10 mg/kg dose. The US FDA requested an additional dose-finding study. TNX-355 has been given fast track designation, which entitles the sponsoring company to additional input from the FDA during the clinical study period. To date, the two anti-CCR5 mAbs have been subjected to preliminary safety studies only. One of these mAbs (PRO-140) has also been given fast track designation by the FDA.
The EI class of mAbs already faces competition from drugs with similar modes of action. One EI peptide drug (enfuvirtide; Trimeris/Hoffmann LaRoche, USA) is already approved and a small molecule EI drug (maraviroc; Pfizer, USA) is in late-stage clinical development. Enfuvirtide, a 36 amino acid synthetic peptide approved by the FDA in 2003, binds viral gp41 and disrupts the fusion of the virus to the cell membrane.[15,16] In two phase III studies in which enfuvirtide 90mg was administered twice daily, the mean viral load was reduced by 1.67 log10 and 1.43 log10 at 24 weeks.[16] Maraviroc, an orally-delivered small molecule that binds CCR5, is currently in a phase III study. The drug reduced viral load by at least 1.6 log10 at doses over 100mg administered twice daily in earlier studies. The FDA has given maraviroc fast track designation.
An alternative approach to treating HIV does not involve targeting the virus. HIV infection is characterized by the indiscriminate destruction of CD4+ lymphocytes, which are critical components of the human immune system. Further adding to the complexity of the disease, the decline in CD4+ lymphocytes cannot be predicted from the presenting RNA levels, which suggests that other factors are involved.[17] The host’s own activated immune system has been implicated in CD4+ cell loss, although the exact mechanism for this phenomenon is not well understood.[18,19] One theory suggests that the persistent decline in CD4+ lymphocytes is due partially to the action of cytotoxic T lymphocytes.[20] Therefore, blocking the activity of lymphocyte function-associated antigen-1 (LFA-1), a molecule on cytotoxic T lymphocytes, might help reduce CD4+ depletion that is not directly due to HIV. An anti-LFA-1 mAb has been administered at low doses (≤2 mg/kg) in phase I and I/II studies, although no definitive efficacy data is yet available. Since the anti-LFA-1 mAb does not affect HIV directly, it must be used in conjunction with antiviral drugs.
5. mAbs for Respiratory Syncytial Virus Infection
RSV is a common human pathogen that primarily infects respiratory epithelial cells. Like HIV and HCV, RSV is an enveloped RNA virus. The viral envelope gpF mediates fusion of the envelope with host cell membranes. In a process that also requires F proteins, infected cells subsequently fuse with each other, resulting in the formation of multi-nucleated masses called syncytia.[21] The F protein of RSV is an attractive target for therapeutic intervention, because the protein sequence is similar across viral strains. RSV causes lower respiratory tract infections in infants and children, although severe RSV disease usually develops in infants and immunosuppressed individuals. High-risk groups include infants under six months of age, premature infants with or without chronic lung disease, and infants with congenital heart disease or immunodeficiency.[22]
To date, a total of four commercially sponsored mAbs have been studied for prophylaxis or the treatment of RSV infection. All four mAbs have targeted the RSV F protein; two anti-RSV mAbs (felvizumab, HNK-20) were discontinued, one (motavizumab) is currently in phase III, and another (palivizumab) is already marketed worldwide. Interestingly, despite having the same target as palivizumab, felvizumab did not demonstrate efficacy in a phase III study. Subsequent direct comparisons between in vitro and in vivo activities of these agents revealed that palivizumab was notably more potent than felvizumab in antigen binding, virus neutralization, and fusion inhibition assays, and also displayed enhanced activity in a cotton rat model of RSV infection.[23]
Palivizumab (MedImmune, USA) was approved by the FDA in 1998, and is indicated for the prevention of serious lower respiratory tract disease caused by RSV in pediatric patients at high risk of RSV disease. Palivizumab targets an epitope in the A antigenic site of gpF of RSV and prevents viral binding to cells. The mAb is administered intramuscularly at a dose of 15 mg/kg on a monthly basis for up to 5 months. A total of 14 clinical studies involving 1281 patients were performed with palivizumab between 1994 and 1997 — 5 studies in adult volunteers, 4 prophylaxis studies conducted in pediatric patients, and 5 treatment studies in both adult and pediatric patients. In the phase III IMpact-RSV study,[24] palivizumab reduced the risk of RSV-associated hospitalizations by 55% (10.6% placebo vs 4.8% palivizumab) in high risk infants and RSV-associated hospitalizations were reduced by 78% in children with prematurity but without bronchopulmonary dysplasia.
Motavizumab, a next generation anti-RSV mAb currently in phase III studies, is derived from palivizumab and also targets the RSV gpF. The mAb has more potent anti-RSV neutralizing activity than palivizumab; it has a 70-fold greater binding affinity, 18-fold increased neutralizing activity, and is 50```-100 times more potent in a cotton rat model.[25] However, details on the efficacy of motavizumab in humans are limited. A phase III study assessing the reduction of hospitalizations due to RSV in high-risk infants has been completed.[26] In the study, motavizumab was compared with palivizumab in over 6600 children, but full results of the study have not yet been released. Preliminary results indicate that motavizumab showed non-inferiority in the primary endpoint by reducing the incidence of hospitalizations caused by RSV (1.4% motavizumab vs 1.9% palivizumab) and showed superiority in a secondary endpoint by reducing the incidence of RSV-specific medically attended outpatient lower respiratory infection (1.9% motavizumab vs 3.9% palivizumab). A number of other studies are on-going.
6. mAbs for Hepatitis C Virus Infection
Like HIV, HCV is an enveloped RNA virus that mutates rapidly and comprises numerous genetically distinct viral subtypes. Infection with HCV causes chronic inflammation of the liver (hepatitis), which can lead to reduced liver function and the need for liver transplantation. The viral genome was described in 1989,[27] but study of the virus life-cycle was hampered until systems suitable for culturing the virus in the laboratory were developed more than a decade later.[28–30] The HCV genome encodes two envelope proteins, E1 and E2, and nonstructural proteins required for replication (e.g. protease), but little is known about the virion structure.[31] HCV gains entry into hepatocytes through interaction of the viral envelope proteins with cell surface receptors.[32,33]
To date, a total of three commercially sponsored mAbs have been studied for the treatment of HCV infection. Two mAbs target the E2 envelope protein and interfere with viral entry into host cells. One of these, AB68 (XTL Biopharmaceuticals, Rehovot, Irsael), was in phase II studies, but was discontinued in favor of a mAb cocktail (XTL-6865) comprising AB68 and another anti-HCV E2 mAb, AB65. The mAb cocktail is considered a separate and distinct product from AB68. AB68 was tested as a single agent at doses of up to 480mg in a phase II study.[34] However, preclinical data suggested that the mAb cocktail might be more efficacious and so a phase I study of that product candidate was initiated. XTL-6865 has been given fast track designation by the FDA.
The third mAb, bavituximab (Peregrine Pharmaceutical, Tustin, CA, USA), is in phase I development as a treatment for HCV infection, although this mAb has the potential to target other viruses. During the process of exiting the host cell, enveloped viruses such as HCV, HIV, and RSV incorporate components of the host cell membrane into the viral envelope.[35] Bavituximab targets phosphatidylserine, which resides on the inside of cellular membranes. The mAb should, thus, bind to any extracellular virus with phosphatidylserine-modified envelope proteins without affecting host cells. Bavituximab has been administered at a dose of 3 mg/kg in a phase Ia study; the mAb showed evidence of anti-HCV activity and was well tolerated. Interestingly, the mAb may also be useful for other indications. Since phosphatidylserine is also exposed on the outside of tumor vasculature,[36,37] bavituximab is also in clinical studies as a treatment for solid tumors.
7. Commercial Preclinical Development
The type and number of mAbs that might enter clinical study sponsored by companies can be assessed, but not necessarily accurately predicted, based on commercial preclinical pipelines. A degree of unpredictability is present because preclinical mAbs developed by academic and academic institutions were excluded from the data set, but discoveries by these institutions might be licensed for commercialization in the future. As a consequence, the results discussed here have probably undercounted the total possible number of candidates to an unknown degree. Nevertheless, examination of commercial preclinical mAbs indicates that only a few new targets are of interest (table II). Furthermore, the size of the group (nine mAbs) suggests that, as a whole, the industry has dedicated few resources to candidate anti-viral mAb programs. The majority (56%) of the preclinical mAbs targeted either SARS coronavirus (SCV) or West Nile virus. The remaining four products targeted rabies (two mAbs), hepatitis B, or influenza (one mAb each) virus.
Table II.
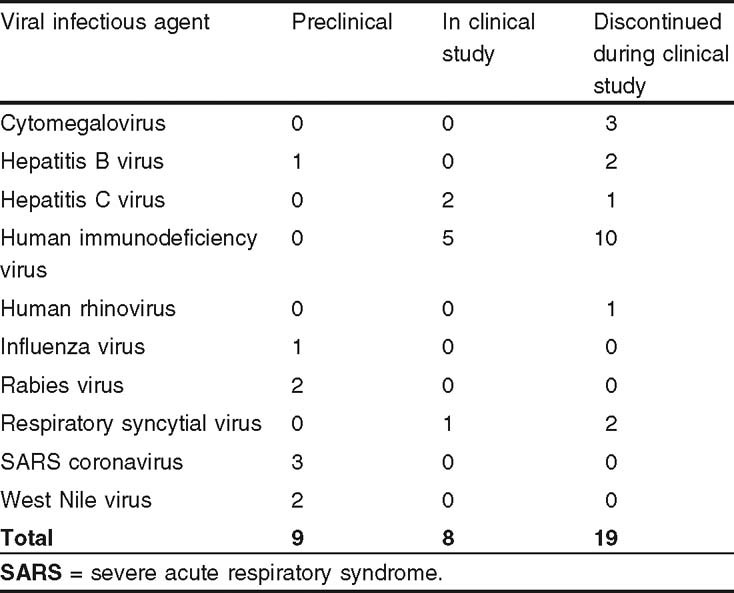
Viral infections targeted by commercial monoclonal antibodies at three development stages
8. Discussion
To date, the majority of commercial clinical development of mAbs for viral infections has focused on products that might meet existing medical needs for new therapies and provide sufficient return on investment. Opportunities of this type exist for numerous viral diseases for which (i) current treatment options are not optimal; (ii) no effective vaccines are available; and (iii) markets, at least those in the US and Europe, are defined. Examples of agents that meet such criteria are the mAbs directed against HIV, RSV, and HCV infections. Examination of the commercial preclinical pipeline suggests that similar selection criteria have been applied to the majority of candidates that might enter clinical study in the near future. However, in the case of mAbs targeting SCV, the current low incidence of new infections begs the question of whether the preclinical candidates will progress, although as a defensive public health measure, products for these infections should be available.
The current dearth of commercial research and development of mAbs for emerging pathogens might be ameliorated somewhat by:
an increased level of government funding available for the development of bioterrorism countermeasures;
an emphasis on priority pathogens by the US National Institutes of Health (NIH);
the FDA’s easing of efficacy requirements in cases where human studies would not be ethical or feasible.[2]
While these factors might indeed bolster the somewhat anemic efforts of industry, the public sector appears to have already responded to the challenge in a more vigorous way. Numerous mAbs for emerging viral diseases are in early research and preclinical stages at academic and government institutions.[38,39] Work has focused on priority pathogens such as Hanta and Ebola viruses that are easily disseminated or transmitted person-to-person, and on emerging pathogens such as Crimean-Congo hemorrhagic fever virus, rabies, and SCV. Importantly, the emphasis has been on development of human mAbs, which, along with humanized products, comprise most of the commercial mAbs now in clinical study.
Ties between the public and private sectors are notoriously intricate.[40] In fact, the preclinical mAbs for SCV and West Nile virus infections attributed here to the commercial sector were all developed with at least some input from the public sector. However, it remains to be seen whether mAbs for infections that occur at high levels only sporadically, or those that would be likely to provide poor return on investment, will be commercialized. MAbs could potentially be critical as a first response measure in the case of a public health crisis, but substantial public sector input might be required to get such products to the market. The NIH’s current emphasis on translational medicine might ease the progress of less commercially attractive products through the process of preclinical and clinical development.
Despite obstacles to the development of innovative mAbs for the prophylaxis or treatment of viral diseases, there is reason for cautious optimism. Scientific advances have uncovered potential new viral targets and mAb modes of action. New possibilities exist for designing safer and more efficacious mAbs. An example of the success potentially achievable with improved design can be found in the case of the mAbs for RSV — felvizumab did not prove efficacious, palivizumab was sufficiently efficacious to be approved, motavizumab shows some improved efficacy compared with palivizumab, and even more potent anti-RSV mAbs can be designed.[41] In addition, increased emphasis from both the public and private sectors on the study of mAbs as viral countermeasures might serve the immediate purpose of providing mAbs useful as either preventative measures or as treatments, but might also provide information potentially useful in the development of vaccines. In either case, the results could greatly benefit public health.
Acknowledgment
The Tufts Center for the Study of Drug Development (CSDD) is funded primarily by unrestricted grants from a consortium of companies within the pharmaceutical industry, but no funds were provided specifically for the preparation of this article.
The author thanks Matthew Dewitz and Julia Wenger for their assistance with data collection and colleagues at Tufts CSDD for critical review of the manuscript.
The author has no conflicts of interest that are directly relevant to the content of this article.
References
- 1.Sawyer L.A. Antibodies for the prevention and treatment of viral diseases. Antiviral Res. 2000;47:57–77. doi: 10.1016/S0166-3542(00)00111-X. [DOI] [PubMed] [Google Scholar]
- 2.Reichert J.M., Dewitz M.C. Anti-infective monoclonal antibodies: perils and promise of development. Nat Rev Drug Discov. 2006;5:191–5. doi: 10.1038/nrd1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Burton D.R. Antibodies, viruses and vaccines. Nat Rev Immunol. 2002;2:706–13. doi: 10.1038/nri891. [DOI] [PubMed] [Google Scholar]
- 4.Nimmerjahn F., Ravetch J.V. Divergent immunoglobulin G subclass activity through selective Fc receptor binding. Science. 2005;310:1510–2. doi: 10.1126/science.1118948. [DOI] [PubMed] [Google Scholar]
- 5.Wainberg M.A. HIV-1 subtype distribution and the problem of drug resistance. AIDS. 2004;18(Suppl.3):S63–8. doi: 10.1097/00002030-200406003-00012. [DOI] [PubMed] [Google Scholar]
- 6.Zhu P., Liu J., Bess J., et al. Distribution and three-dimensional structure of AIDS virus envelope spikes. Nature. 2006;441:847–52. doi: 10.1038/nature04817. [DOI] [PubMed] [Google Scholar]
- 7.Lederman M.M., Penn-Nicholson A., Cho M., et al. Biology of CCR5 and its role in HIV infection and treatment. JAMA. 2006;296:815–26. doi: 10.1001/jama.296.7.815. [DOI] [PubMed] [Google Scholar]
- 8.Biscone M., Pierson T.C., Doms R.W. Opportunities and challenges in targeting HIV entry. Curr Opin Pharmacol. 2002;2:529–33. doi: 10.1016/S1471-4892(02)00200-X. [DOI] [PubMed] [Google Scholar]
- 9.Bukrinsky M. A hard way to the nucleus. Mol Med. 2004;10:1–5. [PMC free article] [PubMed] [Google Scholar]
- 10.Wu Y. HIV-1 gene expression: lessons from provirus and non-integrated DNA. Retrovirology. 2004;1:13. doi: 10.1186/1742-4690-1-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stevens M., De Clercq E., Balzarini J. The regulation of HIV-1 transcription: molecular targets for chemotherapeutic intervention. Med Res Rev. 2006;26:595–625. doi: 10.1002/med.20081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Adamson C.S., Jones I.M. The molecular basis of HIV capsid assembly-five years of progress. Rev Med Virol. 2004;14:107–21. doi: 10.1002/rmv.418. [DOI] [PubMed] [Google Scholar]
- 13.Deeks S.G. Antiretroviral treatment of HIV infected adults. BMJ. 2006;332:1489–96. doi: 10.1136/bmj.332.7556.1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tanox, Inc. New data confirm durable response of TNX-355 regimens through 48 weeks in treatment-experienced HIV patients [press release 2006 Aug 17; online]. Available from URL: http://tanox.com/Release/Release20060817.pdf [Accessed 2007 Jan 12]
- 15.Jamjian M.C., McNicholl I.R. Enfuvirtide: first fusion inhibitor for treatment of HIV infection. Am J Health-System Pharmacy. 2004;61:1242–7. doi: 10.1093/ajhp/61.12.1242. [DOI] [PubMed] [Google Scholar]
- 16.Fung H.B., Guo Y. Enfuvirtide: a fusion inhibitor for the treatment of HIV infection. Clin Ther. 2004;26:352–78. doi: 10.1016/S0149-2918(04)90032-X. [DOI] [PubMed] [Google Scholar]
- 17.Rodriguez B., et al. Predictive value of plasma HIV RNA level on rate of CD4 T-cell decline in untreated HIV infection. JAMA. 2006;296:1498–506. doi: 10.1001/jama.296.12.1498. [DOI] [PubMed] [Google Scholar]
- 18.Derdeyn C.A., Silvestri G. Viral and host factors in the pathogenesis of HIV infection. Curr Opin Immunol. 2005;17:366–73. doi: 10.1016/j.coi.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 19.Ahr B., Robert-Hebmann V., Devaux C., et al. Apoptosis of uninfected cells induced by HIV envelope glycoproteins. Retrovirology. 2004;1:12. doi: 10.1186/1742-4690-1-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zarling J.M., Ledbetter J.A., Sias J., et al. HIV-infected humans, but not chimpanzees, have circulating cytotoxic T lymphocytes that lyse uninfected CD4+ cells. J Immunol. 1990;144:2992–8. [PubMed] [Google Scholar]
- 21.Harris J., Werling D. Binding and entry of respiratory syncytial virus into host cells and initiation of the innate immune response. Cell Microbiol. 2003;5:671–80. doi: 10.1046/j.1462-5822.2003.00313.x. [DOI] [PubMed] [Google Scholar]
- 22.Openshaw P.J., Tregoning J.S. Immune responses and disease enhancement during respiratory syncytial virus infection. Clin Microbiol Rev. 2005;18:541–55. doi: 10.1128/CMR.18.3.541-555.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Johnson S., Griego S.D., Pfarr D.S., et al. A direct comparison of the activities of two humanized respiratory syncytial virus monoclonal antibodies: MEDI-493 and RSHZl9. J Infect Dis. 1999;180:35–40. doi: 10.1086/314846. [DOI] [PubMed] [Google Scholar]
- 24.The IMpact-RSV Study Group. Palivizumab, a humanized respiratory syncytial virus monoclonal antibody, reduces hospitalization from respiratory syncytial virus infection in high-risk infants. Pediatrics. 1988;102:531–7. doi: 10.1542/peds.102.3.531. [DOI] [PubMed] [Google Scholar]
- 25.Mejias A., Chavez-Bueno S., Rios A.M., et al. Comparative effects of two neutralizing anti-respiratory syncytial virus (RSV) monoclonal antibodies in the RSV murine model: time versus potency. Antimicrob Agents Chemother. 2005;49:4700–7. doi: 10.1128/AAC.49.11.4700-4707.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.MedImmune, Inc. Numax® achieves primary endpoint in preliminary analysis of data from comparative phase 3 trial with Synagis® [press release 2006 Nov 6; online]. Available from URL: http://phx.corporate-ir.net/phoenix.zhtml?c=83037&p;=irol-investornewsArticle&ID;=926801&highlight;= [Accessed 2007 Jan 12]
- 27.Choo Q.-L., Kuo G., Weiner A.J., et al. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science. 1989;244:359–62. doi: 10.1126/science.2523562. [DOI] [PubMed] [Google Scholar]
- 28.Lohmann F., Korner J.-O., Koch U., et al. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science. 1999;285:110–3. doi: 10.1126/science.285.5424.110. [DOI] [PubMed] [Google Scholar]
- 29.Blight K.J., Kolykhalov A.A., Rice C.M. Efficient initiation of HCV RNA replication in cell culture. Science. 2000;290:1972–4. doi: 10.1126/science.290.5498.1972. [DOI] [PubMed] [Google Scholar]
- 30.Bartenschlager R. The hepatitis C virus replicon system: from basic research to clinical application. J Hepatol. 2005;43:210–6. doi: 10.1016/j.jhep.2005.05.013. [DOI] [PubMed] [Google Scholar]
- 31.Moradpour D., Blum H.E. A primer on the molecular virology of hepatitis C. Liver Int. 2004;24:519–25. doi: 10.1111/j.1478-3231.2004.0965.x. [DOI] [PubMed] [Google Scholar]
- 32.Bartosch B., Dubuisson J., Cosset F.L. Infectious hepatitis C virus pseudo-particles containing functional E1-E2 envelope protein complexes. J Exp Med. 2003;197:633–42. doi: 10.1084/jem.20021756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hsu M., Zhang J., Flint M., et al. Hepatitis C virus glycoproteins mediate pH-dependent cell entry of pseudotyped retroviral particles. Proc Natl Acad Sci US A. 2003;100:7271–6. doi: 10.1073/pnas.0832180100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.XTL Biopharmaceuticals, Ltd. XTLbio to enroll new patients in HepeX-C trial [press release 2004 Aug 20; online]. Available from URL: http://www.xtlbi-o.com/news/news_item.asp?id=93 [Accessed 2007 Jan 12]
- 35.Cantin R., Methot S., Tremblay M.J. Plunder and stowaways: incorporation of cellular proteins by enveloped viruses. J Virol. 2005;79:6577–87. doi: 10.1128/JVI.79.11.6577-6587.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ran S., Thorpe P.E. Phosphatidylserine is a marker of tumor vasculature and a potential target for cancer imaging and therapy. Int J Radiation Oncol Biol Phys. 2002;54:1479–84. doi: 10.1016/S0360-3016(02)03928-7. [DOI] [PubMed] [Google Scholar]
- 37.Beck A.W., Luster T.A., Miller A.F., et al. Combination of a monoclonal anti-phosphatidylserine antibody with gemcitabine strongly inhibits the growth and metastasis of orthotopic pancreatic tumors in mice. Int J Cancer. 2006;118:2639–43. doi: 10.1002/ijc.21684. [DOI] [PubMed] [Google Scholar]
- 38.Zhu Z., Dimitrov A.S., Chakraborti S., et al. Development of human monoclonal antibodies against diseases caused by emerging and biodefense-related viruses. Expert Rev Antiinfect Ther. 2006;4:57–66. doi: 10.1586/14787210.4.1.57. [DOI] [PubMed] [Google Scholar]
- 39.Zhang M.Y., Choudhry V., Xiao X., et al. Human monoclonal antibodies to the S glycoprotein and related proteins as potential therapeutics for SARS. Curr Opin Mol Ther. 2005;7:151–6. [PubMed] [Google Scholar]
- 40.Reichert J.M., Milne C.P. Public and private sector contributions to the discovery and development of ‘impact’ drugs. Am J Ther. 2002;9:543–55. doi: 10.1097/00045391-200211000-00016. [DOI] [PubMed] [Google Scholar]
- 41.Wu H., Pfarr D.S., Tang Y., et al. Ultra-potent antibodies against respiratory syncytial virus: effects of binding kinetics and binding valence on viral neutralization. J Mol Biol. 2005;350:126–44. doi: 10.1016/j.jmb.2005.04.049. [DOI] [PubMed] [Google Scholar]